
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanolipogels as a cell-mimicking platform for controlled release of biomacromolecules†
Ye
Cao‡
,
Yee Shan
Wong‡
,
Amira
Ben Mabrouk
,
Vincent
Anita
,
Melvin Wen
Jie Liew
,
Yang Fei
Tan
and
Subbu S.
Venkatraman§
*
School of Materials Science and Engineering, Nanyang Technological University, Singapore. E-mail: ASSubbu@ntu.edu.sg
First published on 5th February 2020
Abstract
We present studies of protein (insulin) efflux rates from nano-sized core–shell systems with a gelled core and a lipid bilayer (nanolipogels). The efflux control mechanism is the manipulation of mesh size, and we show that diffusion control via crosslinking is the dominant mechanism for efflux control. The concept is inspired by the macromolecular crowding effect in human cells, which may be considered as a physical network of undefined mesh size. Our bio-inspired system is made of chemically crosslinked water-swellable poly(ethylene glycol) diacrylate cores, whose mesh size can be manipulated to yield a quantifiable crowding effect that then leads to predictable release rates for biomacromolecules.
Treating diseases using therapeutic biomolecules (proteins, peptides, and genes) has been fast becoming a preferred strategy, and over 200 bio-therapeutics (also called “biologics”) have already been approved.1 Therapeutic biomolecules are currently mainly administered by subcutaneous or intravenous injections. For chronic conditions, the cost and inconvenience to the patient can be formidable;2 thus sustained-release bio-therapeutics are of great benefit to patients. The key factors to successful protection and prolonged action of singularly administered therapeutic biomolecules include the ability to encapsulate sufficient quantities of biologics into carriers and to thus protect the biologics from harsh environments and sustain the release over several days while maintaining their bioactivity.3 Very few such sustained-release systems have been approved to date mainly because it is challenging to load carriers with sufficiently high amounts of biologics while still having to modulate their release, especially initial burst release, upon administration for prolonged therapeutic effects.4
Among several US Food and Drug Administration (FDA)-approved nanocarriers, lipid-based nanocarriers such as liposomes, solid lipid nanoparticles, and micelles are the dominant types.5,6 Liposomes can deliver both hydrophobic and hydrophilic drugs (including proteins, peptides, and small interfering RNA) in their lipid bilayers and aqueous cores, respectively.6–8 Doxorubicin encapsulated inside a PEGylated liposome was the first liposomal nanocarrier approved by the FDA.9 Although liposomal nanocarriers have the above-mentioned advantages, they are not ideal candidates for protein and peptide delivery, because of their low encapsulation efficiencies, high burst release, and insignificant sustained-release.4
To overcome these drawbacks, we use the concept of the macromolecular crowding (MMC) phenomenon in cellular cytoplasm to develop hybrid nanocarriers termed nanolipogels (NLGs). In nature, cells are densely packed by various biological macromolecules in the cytosol, which is covered with lipid membranes; the combination slows down the efflux of biomacromolecules from cells. The current literature on macromolecular mobility and barriers for diffusion has focused on the crowdedness of the cytoplasm and the effect of crowding on protein diffusion within various cells and simply indicated that macromolecular crowding of cellular cytoplasm delays the molecular diffusion in live cells.10–13 To be more specific, there is no quantification of the effect of macromolecular crowding, nor is there a definition of what constitutes this diffusional barrier. To the best of our knowledge, there are no studies demonstrating the extent to which macromolecular crowding affects protein release in nanosized entities, or on how the understanding can be used for nanocarrier drug delivery systems. This is because the crowding entity is not defined; we hypothesize that it is a physical network of macromolecules which are packed (“crowded”; concentrations range from 300–400 mg mL−1 of macromolecules) inside the cell so that there are entanglements between the macromolecules, leading to a physical network that offers a diffusional barrier. It is also believed that the membrane offers a secondary barrier to the efflux of macromolecules from within the cell, via negligible partitioning of the macromolecule into the bilayer.
In this study, we designed a NLG system that mimics the MMC effect via chemical crosslinking, with a synthetic membrane bilayer to gauge the relative contributions of diffusion and partitioning for the efflux of insulin from within the core nanogel (Fig. 1). Thus insulin was employed as a model protein drug and encapsulated into a poly(ethylene glycol) diacrylate (PEGDA) based NLG to mimic the reservoir-membrane system (Fig. 1A). PEGDA served as a crowding agent and the idea was to use crosslink density variations (“mesh size” of network) to mimic molecular crowding and then control diffusion in order to gauge the role of diffusion in insulin efflux at the nanoscale.
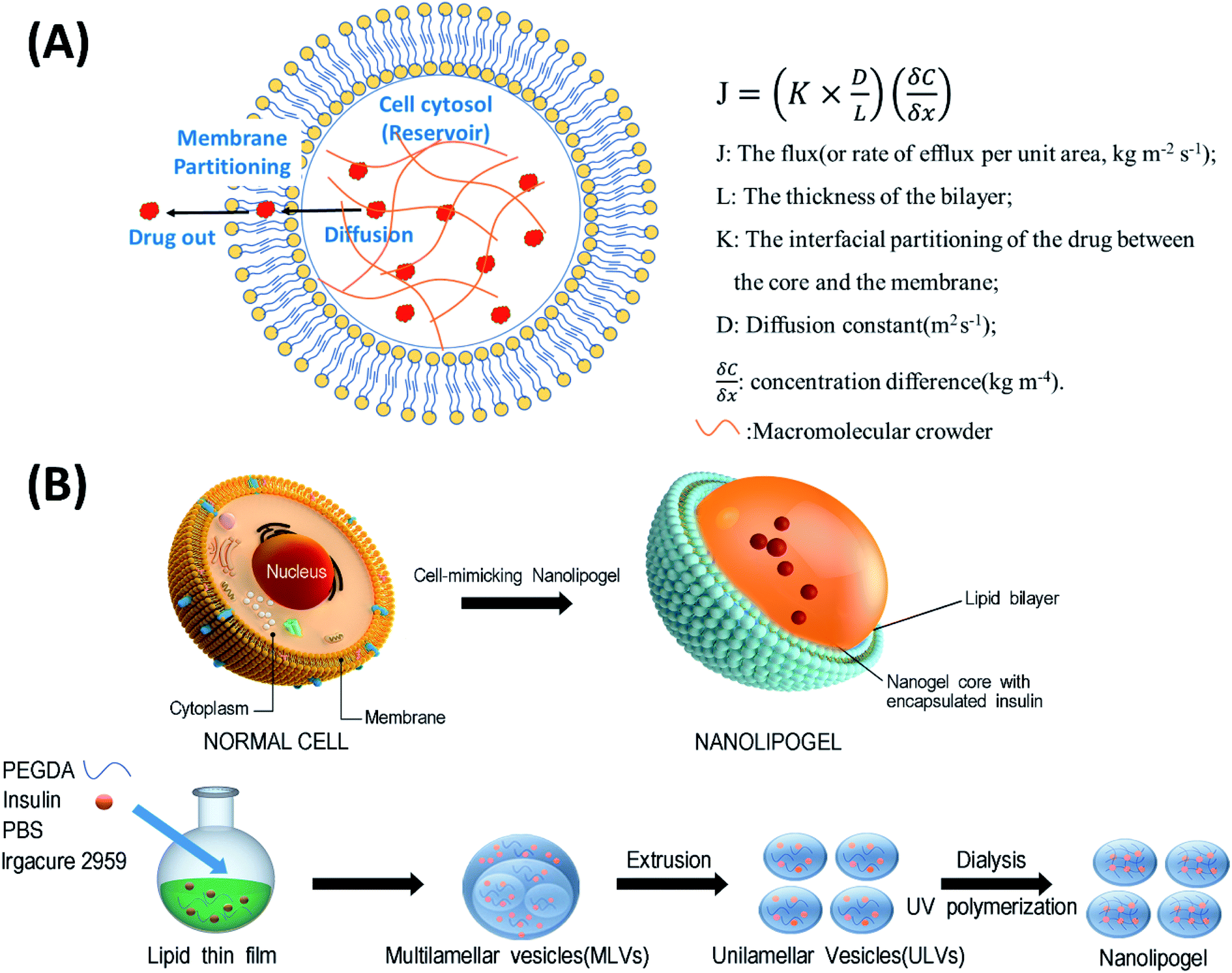 | ||
| Fig. 1 (A) Schematic representation of a cell-mimicking nanolipogel with the macromolecular crowding effect in the core and membrane structure. In a steady state, the drug flux can be estimated using the Fickian diffusion equation after modeling the cell as a reservoir-membrane system. (B) The fabrication process of a cell-mimicking NLG. Multilamellar vesicles (MLVs) and unilamellar vesicles (ULVs). | ||
It should be noted in this context that at the micron-scale, protein efflux from microparticles has been demonstrated to be controllable with solid microparticles such as poly(lactide-co-glycolide) or PLGA; here the control is entirely through a diffusional barrier provided by the sub-Tg matrix of PLGA. In fact at the micron scale, sufficient diffusional control can be obtained without the use of a bilayer coating. This is not possible at the nanolevel, because the diffusional path lengths are significantly reduced, and hence the perceived need for a coating layer significantly different chemically from the semi-aqueous core. To the best of our knowledge there is no study that sheds light on the relative extents of diffusional control and partition control at the nanoscale, for core–shell systems.
As illustrated in Fig. 1B, the preparation process of NLGs is divided into four steps: (1) lipid thin film preparation, (2) hydration of the thin film with the PEGDA monomer, insulin and Irgacure 2959, (3) extrusion of multilamellar vesicles (MLVs) to unilamellar vesicles (ULVs), and (4) dialysis (removal of unencapsulated insulin and PEGDA) and UV photopolymerization. As mentioned above, different amounts of PEGDA were used in the lipid-film hydrating solution in order to achieve NLGs with different macromolecular crowding effects. The morphology and core structure of NLGs formed using different amounts of PEGDA were analyzed by cryo-transmission electron microscopy (TEM), dynamic light scatting (DLS) and static light scatting (SLS). All NLG formulations under study (10%, 30%, and 50%) showed the formation of spherical nanostructures (Fig. 2A) under cryo-TEM. Furthermore, DLS measurements showed that all NLGs exhibited a similar hydrodynamic size of ∼180 nm and neutral zeta potential. The polydispersity indexes (PDIs) of these samples were less than 0.15, indicating homogeneous and narrow size distribution. The DLS and zeta potential of all NLGs are similar to those of liposomes fabricated under the same conditions (190.3 nm, PDI 0.05) (Table S1, ESI†). In terms of long-term stability of the obtained NLGs, the average size, size distribution, and zeta potential of the NLGs remained unaltered for at least 28 days, when stored at 4 °C, as proved by DLS measurements (Table S3, ESI†).
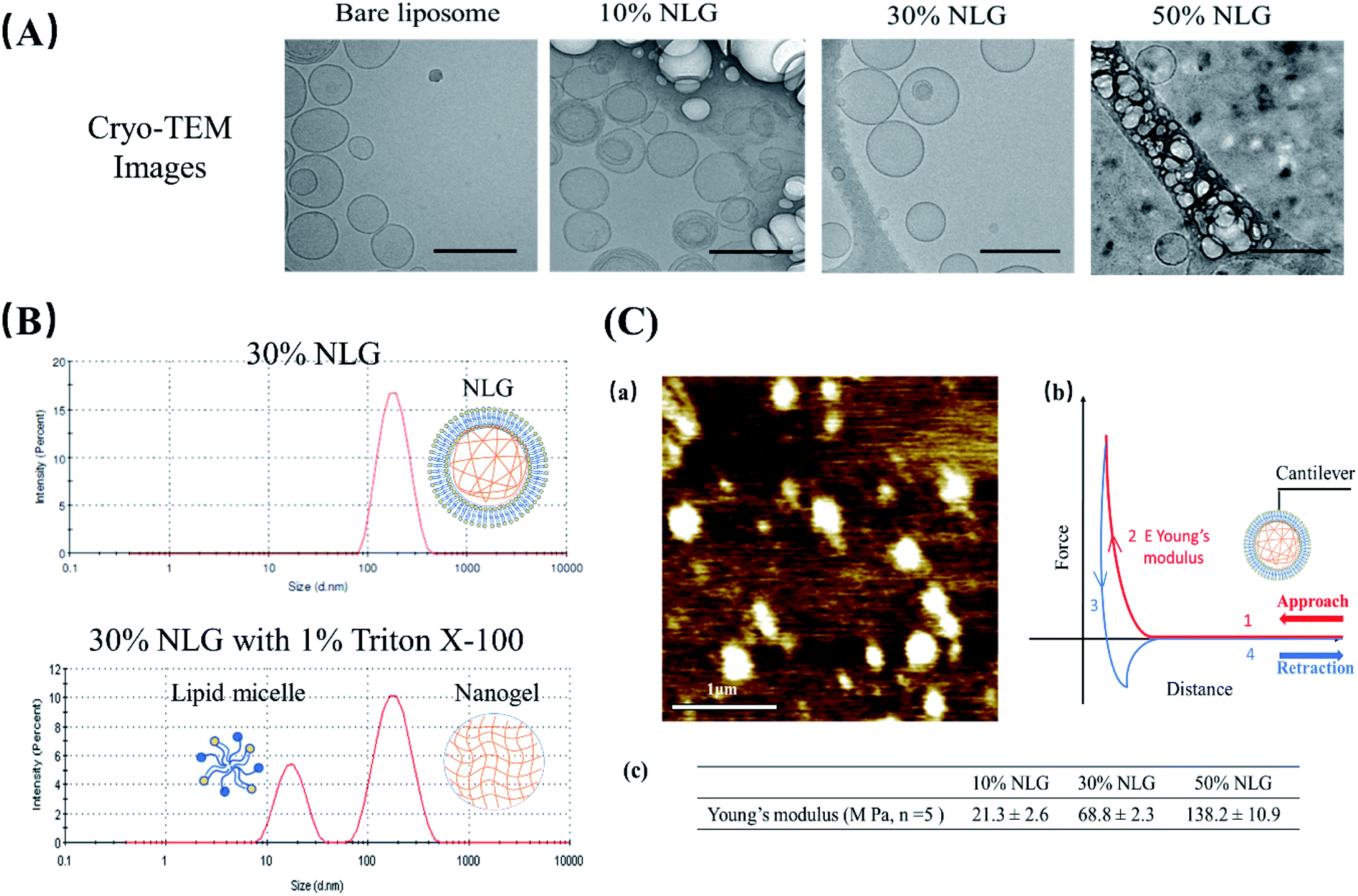 | ||
| Fig. 2 (A) Cryo-TEM images of bare liposomes, 10% NLGs, 30% NLGs, and 50% NLGs (scale bar: 200 nm). (B) DLS results of 30% NLG and 30% NLG with 1% Triton X-100. (C) (a) Morphology of 30% NLG characterized by AFM in liquid mode (scale bar: 1 μm); (b) representative force–distance curve for Young's modulus measurement, where the cantilever probe is brought down into contact (curve 1), and cantilever indents the sample causing cantilever deflection (curve 2) and then retracted from the surface (curve 3) until the cantilever is fully detached (curve 4). Young's modulus E was determined from the indentation part of the approach curve (2);14 (c) Young's modulus results of 10%, 30% and 50% NLGs. | ||
To confirm the nanogel formation, DLS was employed to evaluate the formation of the nanogel core inside NLGs. Triton X-100 was employed to solubilize the lipid bilayers.15,16 After adding 1% Triton X-100 to NLG solution, a micelle peak (∼10 nm) and a separate nanogel peak (100–200 nm) were observed as shown in Fig. 2B. The micelle is formed by lipids stripped from the NLG cores; thus the observed bimodal structures indicated the existence of a nanogel-core interior surrounding by a liposomal vesicle. In combination with TEM observation, the structural morphology of NLGs in aqueous solutions was studied by determining their Rg/Rh ratios (Rg: radius of gyration; Rh: hydrodynamic radius) as shown in Table S2 (ESI†). Rg/Rh defined as the shape factor (ρ) is a sensitive tool for monitoring the structures of the nanoparticles by reflecting the radial density distribution of the particle.17 Theoretically, the ρ values for a uniform solid sphere, vesicle, and random coil are 0.775, 1.0, and 1.5, respectively.18,19 In our study, the ρ values for bare liposomes and NLGs were calculated to be 0.98 and 1.05 respectively, corresponding to the vesicle structure. Collectively, these results show the following:
(1) Our system is a core–shell system, with a spherical gel core and a synthetic bilayer “membrane”; although we cannot categorically state that the surface coverage by the bilayer is “complete”, it is likely to be so, especially in view of the size stability of these core–shell particles (ESI, Table S3†);
(2) The bilayer lipids are certainly integrated with the gel core, because if they were free-floating, they would form micelles, which is not shown by the DLS data (single peak at about 100 nm and no micelle peak (∼10 nm)).
To further probe the crosslinking density of the hydrogel core inside the NLG, the Rg, Rh and ρ values of the nanogels were measured after lipid bilayer removal (Table S2 and Fig. S1, ESI†). Our results show that the Rg decreases with increasing PEGDA concentration, indicating an increase in crosslinking density.20,21 The ρ values of nanogels after bilayer removal are in the range between 0.62 and 0.91 for various PEGDA concentrations suggesting a spherical geometry.22 Compared to the relatively homogeneous crosslinked structure of 10% and 30% nanogels (NGs) (ρ > 0.775), the lower ρ value of 50% NG suggests having a “corona”-type structure, that is, a denser or highly crosslinked core surrounded with a less dense layer.22–24 This observation is in agreement with the mesh size data as shown in Table S1 (ESI†) when the PEGDA concentration increases with reducing mesh size corresponding to a tighter hydrogel network. NLG with 50% PEGDA has, as expected, the smallest mesh size at 0.86 nm. As the PEGDA concentration decreased, the mesh size increased to 1.21 and 2.55 nm, respectively. Small peptides, such as insulin with a monomeric radius of 1.3 nm,25 can be easily encapsulated into the nanogel core of NLGs, and their release may be modulated at these mesh sizes (crosslink densities or macromolecular crowding). Therefore, we selected insulin as a model drug to study the effects of diffusion control over the release.
Subsequently, we assessed the morphology and elasticity of NLGs using contact mode Atomic Force Microscopy (AFM) for mechanical properties to control the stability, size, shape, and fusion of the liposomes as well as the drug release profile.26Fig. 2C(a) shows the morphology of NLGs adsorbed on a PEI coated mica surface in a liquid environment. This image consisted of spherical particles on a flat surface. The average diameter of the NLGs was measured to be 235 ± 53.8 nm. Fig. 2C(b) shows the typical force–distance curve for Young's modulus measurement. Young's modulus was estimated using the Hertz equation. The results in Fig. 2C(c) showed that the elasticity of 50% NLG (138 MPa) was the highest, while those of 10% and 30% NLG were 21.3 MPa and 68.8 MPa, respectively. The results, therefore, revealed that 50% NLG had the highest elasticity because of its higher crosslinking density. These results correlate with the study conducted by Peng Guo et al. that Young's modulus of alginate-core NLG increased with increasing crosslinking density.27
Next, we encapsulated insulin into NLGs and investigated the drug encapsulation and release ability of our engineered NLGs. As shown in Table S1 (ESI†), increasing the PEGDA amount decreased the encapsulation efficiency (EE) but enhanced the loading efficiency (LE). It was observed that during the NLG fabrication, increasing PEGDA amount in the hydration buffer made it increasingly difficult to hydrate the lipid thin film and extrude MLVs, presumably due to the higher viscosity. Thus, it resulted in a lipid loss and subsequent less amount of liposomal formation, especially for the NLG formulation with 50% PEGDA. Nonetheless, we have indicated the successful encapsulation of insulin into the engineered NLGs with a high loading efficiency (up to 27.8%). For release kinetic studies, the insulin release from bare liposomes, uncrosslinked NLGs, and NLGs was monitored over a period of 28 days under physiologically relevant in vitro conditions (pH 7.4 and 37 °C) as shown in Fig. 3A.
 | ||
| Fig. 3 (A) Cumulative insulin release from bare liposomes, uncrosslinked NLGs and crosslinked NLGs (PEGDA concentration used is 50%). (B) The insulin release profile of NLGs with various PEGDA concentrations (10%, 30%, and 50%) and schematic graph of drug release from NLGs with different mesh sizes. (C) The model parameters for the releases of NLGs based on the Ritger–Peppas equation. (D) Calculated mesh sizes for the 3 networks compared to insulin size. | ||
The fastest release of insulin was observed from bare liposomal nanocarriers with 90% release in the first day and complete release on day 3. Under physiologically relevant in vitro release conditions, that is close to the isoelectric point of human recombinant insulin being studied here (pH 7), insulin is weakly protonated and more likely to partition across the neutrally charged EggPC lipid membrane, resulting in rapid insulin release from the liposomal nanocarriers. This shows that partitioning is not the rate-controlling mechanism for efflux, at least for insulin.
In contrast, NLGs that have an additional element in their structure, the nanogel core (un-crosslinked and crosslinked), can significantly suppress the initial burst and prolong the release time as compared to conventional liposomes. As compared to simple liposomes that display a burst release of insulin, the NLGs provide a more controlled release of the insulin with a lower initial burst and longer release time. These dual-structure NLGs have a distinct lipid bilayer encompassing a hydrogel core and the rate of drug release can readily be tailored by changing the crowding agent concentration or mesh size of the nanogel core.
In general, nanogels consist of crosslinked polymeric networks with open spaces (or “meshes”) between the network polymer chains; and drug release from hydrogels may proceed through 3 mechanisms: simple diffusion, swelling of the core, or degradation of the network. In all our release experiments, the nanogel core did not swell if the membrane bilayer is kept intact; there was no measurable degradation of the network over 28 days (this is also additional proof of an integral bilayer). Therefore, the observed release kinetics reflect the effect of mesh size on molecular diffusion through the network. The mesh size determines how drugs diffuse through a hydrogel as it controls not only the diffusional path length (the NLG may be considered a porous network) but also steric interactions between the drugs and the polymer network.28 The interactions become important when the mesh size approaches the molecular size of the diffusant: more on this below.
The initial burst release of insulin from uncrosslinked NLGs is reduced to 45% in the first day, with an average time to 50% release (“half-life of release”) of about 4 days. The slower drug release kinetics from uncrosslinked NLGs is attributed to the presence of the PEGDA polymer chains within the liposomal core, resulting in a significant drag on the diffusing drug (a small “crowding” effect) and increasing the path length for insulin transport.28 Once the PEGDA precursor solution is crosslinked, the crosslinked NLGs further slow down the insulin release, resulting in a drastic reduction in the initial burst to about 15% and a sustained release of 45% up to day 28. Furthermore, to demonstrate the impact of crosslink density in the NLGs on the release profile of insulin, we compared the release of insulin from NLGs with different PEGDA concentrations, as shown in Fig. 3B. Clearly, the crosslinking density of the PEGDA nanogels within the lipid bilayer has a profound influence on the release of insulin. The higher the PEGDA concentration of the nanogel core (i.e. higher crosslink density) in NLGs, the slower the drug release; in 28 days a release of 94% for 10% NLGs and 90% for 30% NLGs was achieved, while only 60% of the insulin was released for 50% NLGs.
In order to gain more insight into the release mechanism, the experimental release data of NLGs with different PEGDA concentrations were fitted to the Ritger–Peppas equation:28–30Mt/M∞ = ktn, where Mt is the mass of the drug released at time t, M∞ is the total mass of the drug released, k is a kinetic constant, and n is the diffusional exponent. For Ritger–Peppas models, n ≤ 0.5 represents the Fickian diffusion release from spheres, 0.5 < n < 1.0 for non-Fickian release (anomalous) and n = 1.0 for surface erosion. The model parameters for the release of various NLGs are shown in Fig. 3C. The diffusional exponents, n, for 10%, 30% and 50% NLGs were 0.505, 0.237, and 0.389, respectively, a mix of Fickian (10% PEGDA) and non-Fickian (30%, 50% PEGDA) diffusional release.
In our study, NLGs with increasing PEGDA crowding concentration display a higher crosslink density in the nanogel core, which leads to a smaller mesh size, and correspondingly to a lower rate of drug release. Similar behavior was observed by Censi et al. in methacrylate triblock copolymer hydrogels, which showed a lower release of model proteins (lysozyme/BSA/IgG) with increasing polymer concentration from 20% to 35%.31 To put these mesh sizes into context, the model drug insulin has a hydrodynamic radius of 1.3 nm.32 When the mesh size approaches the protein size of 30% NLG (Fig. 3B, schematic graph), the effect of steric hindrance and/or interaction on protein diffusion becomes prominent, resulting in slow protein diffusion and extended release. In the table in Fig. 3 above, at 30% and 50% PEGDA concentrations, the mesh size is similar to or less than the insulin size for the dimer (1.2 nm). The transition from Fickian to non-Fickian behaviour starts around the same mesh size (1.3 to 1.1 nm) and below.
As compared to the NLGs reported previously, our NLG platforms can significantly prolong the protein release time by controlling the mesh size of the hydrogel core. For instance, a NLG made of a PLA–PEG–PLA polymeric nanogel core released 80% of IL-2 (Mw 15.5 kDa) in 4 days,33 whereas the time for 50% release of lysozyme (Mw 14.4 kDa) for polyglycerol-based NLGs is 3 days.34 In comparison, the macromolecular crowding achieved in our PEGDA-based NLGs offers the possibility of longer-term sustained delivery of insulin and similar molecules. The mesh size is a tunable parameter to be adjusted based on diffusant size.
Conclusions
Cell-mimicking, PEGDA-based NLGs are a promising controlled-release system for biologics, such as proteins/peptides/siRNA. We show here that release profiles may be modulated by controlling the diffusion of the biomolecule (insulin) inside the gel core, and the release correlates with the mesh size (the degree of MMC) of the gel network. Our insulin-encapsulated NLGs also exhibit high encapsulation efficiencies. These systems have the potential to expand their application to other peptides and proteins, as well as to siRNAs. The concept of NLGs is not new and a few studies have already reported on the promise of controlled release. However, a systematic understanding of the control parameters is lacking, and our studies are the first step to addressing this. In this paper, we systematically characterized PEGDA based NLGs by several analytical techniques and demonstrated that manipulation of the nanogel mesh size is the key approach to controlled release. Other factors that remain to be studied include bilayer membrane composition and transition temperatures, addition of cholesterol (which is a component of human cell membranes), and increasing interaction between the network and the biologic species. Future studies will address the quantification of the effects of these variables.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of a grant from the Nanyang Technological University-Northwestern Nanomedicine project. We thank Dr Yee Yan Tay from the Facility for Analysis Characterisation Testing and Simulation for assistance with Cryo TEM. And we also would like to thank Dr Lexie Xin Xu for the help with the liquid mode AFM.References
- M. R. Turner and S. V. Balu-Iyer, J. Pharm. Sci., 2018, 107, 1247–1260 CrossRef CAS PubMed.
- J. A. Sequeira, A. C. Santos, J. Serra, C. Estevens, R. Seiça, F. Veiga and A. J. Ribeiro, Expert Opin. Drug Delivery, 2019, 16, 143–151 CrossRef CAS PubMed.
- C. Dumont, S. Bourgeois, H. Fessi and V. Jannin, Int. J. Pharm., 2018, 541, 117–135 CrossRef CAS PubMed.
- C. Ye and S. Venkatraman, Ther. Delivery, 2019, 10, 269–272 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- A. D. Mali and R. S. Bathe, Asian J. Pharmaceut. Res., 2015, 5, 151–157 CrossRef.
- H. Mishra, V. Chauhan, K. Kumar and D. Teotia, J. Drug Delivery Ther., 2018, 8, 400–404 CAS.
- A. Gabizon and F. Martin, Drugs, 1997, 54, 15–21 CrossRef CAS PubMed.
- G. Rivas and A. P. Minton, Trends Biochem. Sci., 2016, 41, 970–981 CrossRef CAS.
- H. Soleimaninejad, M. Z. Chen, X. Lou, T. A. Smith and Y. Hong, Chem. Commun., 2017, 53, 2874–2877 RSC.
- D. Gnutt and S. Ebbinghaus, Biol. Chem., 2016, 397, 37–44 CAS.
- S. Smith, C. Cianci and R. Grima, J. R. Soc., Interface, 2017, 14, 20170047 CrossRef PubMed.
- J. J Roa, G. Oncins, J. Diaz, F. Sanz and M. Segarra, Recent Pat. Nanotechnol., 2011, 5, 27–36 CrossRef PubMed.
- R. Ramanathan, Y. Jiang, B. Read, S. Golan-Paz and K. Woodrow, Acta Biomater., 2016, 36, 122–131 CrossRef CAS PubMed.
- S. Petralito, R. Spera, S. Pacelli, M. Relucenti, G. Familiari, A. Vitalone, P. Paolicelli and M. A. Casadei, React. Funct. Polym., 2014, 77, 30–38 CrossRef CAS.
- J. Hotz and W. Meier, Langmuir, 1998, 14, 1031–1036 CrossRef CAS.
- Z. L. Yao and K. C. Tam, Polymer, 2012, 53, 3446–3453 CrossRef CAS.
- F. Yuen and K. C. Tam, J. Appl. Polym. Sci., 2013, 127, 4785–4794 CrossRef CAS.
- S. J. Rukmani, P. Lin, J. S. Andrew and C. M. Colina, J. Phys. Chem. B, 2019, 123, 4129–4138 CrossRef CAS PubMed.
- X. Liu, B. J. Bauer and R. M. Briber, Macromolecules, 1997, 30, 4704–4712 CrossRef CAS.
- E. Kokufuta, K. Ogawa, R. Doi, R. Kikuchi and R. S. Farinato, J. Phys. Chem. B, 2007, 111, 8634–8640 CrossRef CAS PubMed.
- D. Kuckling, C. D. Vo, H. J. P. Adler, A. Völkel and H. Cölfen, Macromolecules, 2006, 39, 1585–1591 CrossRef CAS.
- G. R. Deen, T. Alsted, W. Richtering and J. S. Pedersen, PCCP Phys. Chem. Chem. Phys., 2011, 13, 3108–3114 RSC.
- A. Oliva, J. Fariña and M. a. Llabrés, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 749, 25–34 CrossRef CAS.
- O. Et-Thakafy, N. Delorme, C. Gaillard, C. Mériadec, F. Artzner, C. Lopez and F. Guyomarc'h, Langmuir, 2017, 33, 5117–5126 CrossRef CAS PubMed.
- P. Guo, D. Liu, K. Subramanyam, B. Wang, J. Yang, J. Huang, D. T. Auguste and M. A. Moses, Nat. Commun., 2018, 9, 130 CrossRef.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- C. Salome Amarachi, G. Onunkwo and I. Onyishi, Kinetics and Mechanisms of Drug Release from Swellable and Non Swellable Matrices: A Review, 2013 Search PubMed.
- N. A. Peppas and J. J. Sahlin, Int. J. Pharm., 1989, 57, 169–172 CrossRef CAS.
- R. Censi, T. Vermonden, M. J. van Steenbergen, H. Deschout, K. Braeckmans, S. C. De Smedt, C. F. van Nostrum, P. di Martino and W. E. Hennink, J. Controlled Release, 2009, 140, 230–236 CrossRef CAS PubMed.
- R. Pohl, R. Hauser, M. Li, E. De Souza, R. Feldstein, R. Seibert, K. Ozhan, N. Kashyap and S. Steiner, J. Diabetes Sci. Technol., 2012, 6, 755–763 CrossRef PubMed.
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, P. Licona Limon, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed.
- J. N. Lockhart, D. B. Beezer, D. M. Stevens, B. R. Spears and E. Harth, J. Control. Release, 2016, 244, 366–374 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00093k |
| ‡ These authors contributed equally. |
| § Materials Science and Engineering, National University of Singapore. E-mail: subbu@nus.edu.sg. |
| This journal is © The Royal Society of Chemistry 2020 |