
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of gold–palladium core–shell nanowires towards H2O2 reduction by adjusting shell thickness†
Yongdi
Dong
a,
Qiaoli
Chen
*ab,
Xiqing
Cheng
a,
Huiqi
Li
a,
Jiayu
Chen
a,
Xibo
Zhang
a,
Qin
Kuang
 *a and
Zhaoxiong
Xie
a
*a and
Zhaoxiong
Xie
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China. E-mail: qkuang@xmu.edu.cn; qlchen@zjut.edu.cn
bCollege of Chemical Engineering, State Key Laboratory Breeding Base of Green Chemistry Synthesis Technology, Zhejiang University of Technology, Hangzhou, 310014, China
First published on 31st December 2019
Abstract
Designable bimetallic core–shell nanoparticles exhibit superb performance in many fields including industrial catalysis, energy conversion and chemical sensing, due to their outstanding properties associated with their tunable electronic structure. Herein, Au–Pd core–shell (AurichPd@AuPdrich) nanowires (NWs) were synthesized through a one-pot facile chemical reduction method in the presence of cetyltrimethyl ammonium bromide (CTAB) surfactant. The thickness of the Pd shell could be adjusted by directly controlling the Au/Pd feeding ratio while maintaining the nanowire morphology. The as-obtained Au75Pd25 core–shell NWs with a thin Pdrich shell showed significantly enhanced activities towards the reduction of hydrogen peroxide with the sensitivity reaching 338 μA cm−2 mM−1 and a linear range up to 10 mM. In sum, Pd shell thickness could be used to adjust the electronic structure, thereby optimizing the catalytic activity.
Introduction
Bimetallic nanomaterials are important in catalysis due to their unique electronic structures, which could be adjusted through their elemental configuration, morphology and size to yield excellent catalytic activities.1–3 Among various bimetallic structures, the core–shell structure is prominent as it can achieve the highest atomic utilization,4,5 integrate the functions of the shell and core,6–9 and exhibit outstanding catalytic performances.10,11 The regulation of the electronic structure and the design of an optimal composition are crucial in developing superior core–shell nanoparticle catalysts. Particularly, the electronic structure of the shell surface, where catalytic reactions directly take place, is influenced by the effect of the substrate metal and surface strain caused by the lattice mismatch between the core and shell.12 These two effects are strongly correlated to shell thickness. For example, the chemisorption properties change vastly depending on ultrathin shell (sub-monolayer, monolayer, and few layers) structures. Jakob et al. reported a single Pt monolayer on Ru(0001) with significantly decreased CO adsorption energy.13 Li et al. noticed that 0.7–0.9 layer Pd on gold nanoparticles shows the lowest overpotential for the alkaline oxygen reduction reaction due to the reduced binding strength of oxygen-containing adsorbates.14Wet chemical synthesis is commonly used in the fabrication of core–shell nanomaterials with a desired shell thickness and morphology. To this end, seed-mediated growth,15,16 galvanic replacement17 and co-reduction18 are three typical routes to achieve such core–shell structures. The former two methods usually require multiple steps. Nanoparticles synthesized in previous steps serve as nuclei for further growth and sometimes as a morphology template. The shell thickness could be regulated by the concentration of metal precursors or the number of seeds. For example, Liu et al. fabricated Au@Pd and Au@Ag@Pd core–shell nanowires (NWs) with continuous and isolated Pd sites by using Au NWs as seeds.19 Dong et al. used Te NW templates to synthesize PdM (M = Au, Pt) alloy NWs through galvanic replacement.20 However, the inhomogeneity of seeds or templates may result in inaccurate shell thickness control and unguaranteed repeatability due to deposition of the metal precursor on small seeds with higher surface energies. The problem could get worse when thin shell structures are required. In comparison, a co-reduction method is convenient and promising for large-scale synthesis. The discrepancy in reduction ability, galvanic replacement or the Kirkendall effect may lead to core–shell structures. However, the control over the morphology and shell thickness remains challenging due to the complex bimetallic growth kinetics. Only a handful of studies dealing with one-pot reduction methods for the synthesis of well-defined nanostructures with adjustable shell thickness have so far been reported.18 Au–Pd core–shell nanomaterials have wide application in analyte detection,21,22 fuel cells,23,24 and organic synthesis.25 Nishi et al. have prepared Au@Pd core–shell nanofibers with a jagged Pd shell at a redox active water/ion liquid interface through a one-step method, which showed an enhanced catalytic performance towards ethanol electroxidation.24 Li et al. reported the synthesis of serrated Au–Pd core–shell NWs with different Pd shell thicknesses via a one-pot dual capping agent-assisted method which also boosts their electrocatalytic activity towards ethanol and formic acid oxidation.23 Despite the above success, the synthetic method of Au–Pd core–shell NWs needs further simplification with unambiguous growth kinetics. Besides, a systematic study of the shell-dependent structure–activity relationship is required.
In this study, a facile one-pot synthesis method was employed to fabricate Au–Pd core–shell (AurichPd@AuPdrich) NWs with a Au-rich AuPd alloy core and a Pd-rich AuPd alloy surface. Shell thickness was adjusted by changing the ratio of the Au/Pd precursor while the nanowire morphology remained stable. Surfactant cetyltrimethyl ammonium bromide (CTAB) served as a soft template for the growth of NWs. The attachment and coalescence of small nuclei during the growth process generated abundant defects, providing highly effective catalytic sites. The great conductivity of the as-obtained NWs ensured the fast electron transport during electro-catalysis. The change in Pd shell thickness offered a continuously adjustable electronic structure to optimize the catalytic activity. Electrocatalytic H2O2 sensing, which enjoys wide application in biological, pharmaceutical, and food industries, is one of the typical reactions that are sensitive to the surface structure and composition of catalysts.26 As vital catalysts for H2O2 sensing, noble metal nanomaterials have advantages in terms of high stability and good repeatability.27,28 Thus, electrocatalytic H2O2 sensing is selected as a model reaction to evaluate the activity of Au–Pd core–shell NWs. Au75Pd25 NWs with a thin Pd shell exhibit excellent catalytic performance towards the electro-reduction of H2O2.
Experimental
Chemicals
Chloroauric acid hydrate (HAuCl4·4H2O, analytical grade), L-ascorbic acid (AA, analytical grade), sodium bromide (NaBr, analytical grade), cetyltrimethyl ammonium bromide (CTAB, analytical grade) and octadecyltrimethyl ammonium bromide (OTAB, analytical grade) were all purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Palladium(II) chloride (PdCl2, analytical grade) was provided by Shanghai Fine Chemical Materials Institute. Phosphate buffer solution (PBS) was prepared with tablets obtained from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). 30% w/w hydrogen peroxide (H2O2, analytical grade) was received from Guangzhou Jinhuada Chemical Reagent Co. Ltd. H2PdCl4 solution was prepared by dissolving PdCl2 in HCl solution followed by dilution with ultrapure water. All reagents were used as received without further purification. All aqueous solutions were prepared with ultrapure water with a resistivity of 18.2 MΩ cm−1.Synthesis of Au–Pd bimetallic NWs
In a typical synthesis, 5 mL solution containing 20 mM CTAB, 0.6 mM HAuCl4 and 0.2 mM H2PdCl4 (feeding ratio of HAuCl4 to H2PdCl4 was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to a vessel placed in a water bath at 30 °C for 15 minutes. The obtained solution was then vigorously shaken for 3–4 seconds, and freshly prepared AA (0.1 mL, 0.1 M) aqueous solution was quickly added. Next, the reaction solution was left undisturbed for 2 hours at 30 °C to allow the growth process to take place. Several seconds after the addition of the AA solution, the solution color turned from orange to light brown and further changed to dark turbid brown after 30 minutes. The final black gray products aggregated and settled down at the vessel bottom. The products were collected by centrifugation (3000 rpm, 3 minutes) and then washed three times with ultrapure water. Au–Pd NWs with tunable Pd shell thickness were obtained by changing the ratio of HAuCl4 and H2PdCl4 while keeping the total molar concentration of metal precursors at 0.8 mM. For convenience, the as-synthesized NWs were denoted as AuxPdy according to the feeding molar ratio of Au and Pd precursors (x + y = 100, where x and y represent the feeding molar percentages of Au and Pd, respectively). For temperature-controlled synthesis experiments, the reaction solution was first kept at 30 °C and immediately switched to 6 °C, 30 °C and 50 °C after the addition of AA solution.
:1) was added to a vessel placed in a water bath at 30 °C for 15 minutes. The obtained solution was then vigorously shaken for 3–4 seconds, and freshly prepared AA (0.1 mL, 0.1 M) aqueous solution was quickly added. Next, the reaction solution was left undisturbed for 2 hours at 30 °C to allow the growth process to take place. Several seconds after the addition of the AA solution, the solution color turned from orange to light brown and further changed to dark turbid brown after 30 minutes. The final black gray products aggregated and settled down at the vessel bottom. The products were collected by centrifugation (3000 rpm, 3 minutes) and then washed three times with ultrapure water. Au–Pd NWs with tunable Pd shell thickness were obtained by changing the ratio of HAuCl4 and H2PdCl4 while keeping the total molar concentration of metal precursors at 0.8 mM. For convenience, the as-synthesized NWs were denoted as AuxPdy according to the feeding molar ratio of Au and Pd precursors (x + y = 100, where x and y represent the feeding molar percentages of Au and Pd, respectively). For temperature-controlled synthesis experiments, the reaction solution was first kept at 30 °C and immediately switched to 6 °C, 30 °C and 50 °C after the addition of AA solution.
Characterization
The morphologies and crystal structures of Au–Pd core–shell NWs were investigated by scanning electron microscopy (SEM, Hitachi S4800) and high-resolution transmission electron microscopy (HRTEM, JEM2100) at an acceleration voltage of 200 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were carried out on an FEI TECNAI F30 microscope operating at 300 kV. All TEM samples were prepared by depositing a drop of the diluted solution on a copper grid coated with a carbon film. The crystal phases of the products were determined by powder X-ray diffraction (PXRD) using a Rigaku Ultima IV X-ray diffractometer with Cu Kα radiation. The surface elemental compositions of NWs were identified by X-ray photoelectron spectrometry (XPS, PHI QUANTUM-2000) using a monochromatic magnesium X-ray source. The binding energies were calibrated with respect to the signal of carbon 1s (binding energy of 284.8 eV). All measurements were performed at 25.0 °C (±0.1 °C).Electrochemical testing
The electrochemical measurements were performed on a CHI 1070b analyzer (CHI Instruments, Chenhua Co., Shanghai, China). A conventional three-electrode cell was used. A Ag/AgCl electrode was employed as the reference, a platinum mesh as the counter electrode, and glassy carbon (GC) loaded with catalysts as the working electrode. All potentials were reported relative to the reversible hydrogen electrode (RHE).First, Au–Pd core–shell NW samples (2 mg) together with 5 μL Nafion (5%) were dispersed in 0.5 mL ethanol to obtain a Au–Pd catalyst suspension at a concentration of 4 mg mL−1. The suspension was then ultrasonicated for 0.5 hours and the as-obtained catalyst suspension (3 μL) was deposited onto a 3 mm glassy carbon electrode previously polished and washed carefully.
The electrochemical H2O2 reduction tests were carried out in 0.025 M PBS (pH 6.8) at a scan rate of 50 mV s−1. The coated electrode was scanned by cyclic voltammetry (CV) between 0.2 and 1.2 V vs. RHE until a stable CV curve was achieved. Under the same potential range, linear sweep voltammetry curves (LSVs) were recorded after the addition of H2O2 (increased by 1 mM). A current density of 0.97 V vs. RHE around the reduction peak was selected for calibration of current density vs. H2O2 concentration. The current–time curves were obtained at 0.97 V vs. RHE under constant stirring. The modified electrode was washed with pure water and then dried in air before each measurement.
For in situ surface structure information, the CV curves of the as-synthesized Au–Pd core–shell NWs were collected in 0.5 M H2SO4 electrolyte between −0.05 and ∼1.64 V vs. RHE.
Results and discussion
Characterization of typical Au–Pd bimetallic core–shell NWs
As shown in Fig. 1a and b, the typical Au75Pd25 sample prepared at a feeding ratio of HAuCl4 to H2PdCl4 of 75:25 contained uniform NWs with an average diameter of 40 ± 1 nm (lower left inset of Fig. S1a, ESI†) and a length of several micrometers. The polycrystalline structure of NWs was confirmed by selected area electron diffraction (SAED) (upper right inset of Fig. S1a, ESI†). The low magnification TEM image (Fig. 1b) revealed the presence of kinks and twists in the wavy NWs. The magnified TEM images (Fig. 1c and S1b–d, ESI†) suggested the presence of structural defects, such as twin boundaries, grain boundaries, and stacking faults in NWs. Such boundaries might be caused by attachment and fusion between the nanoparticles during the growth process.
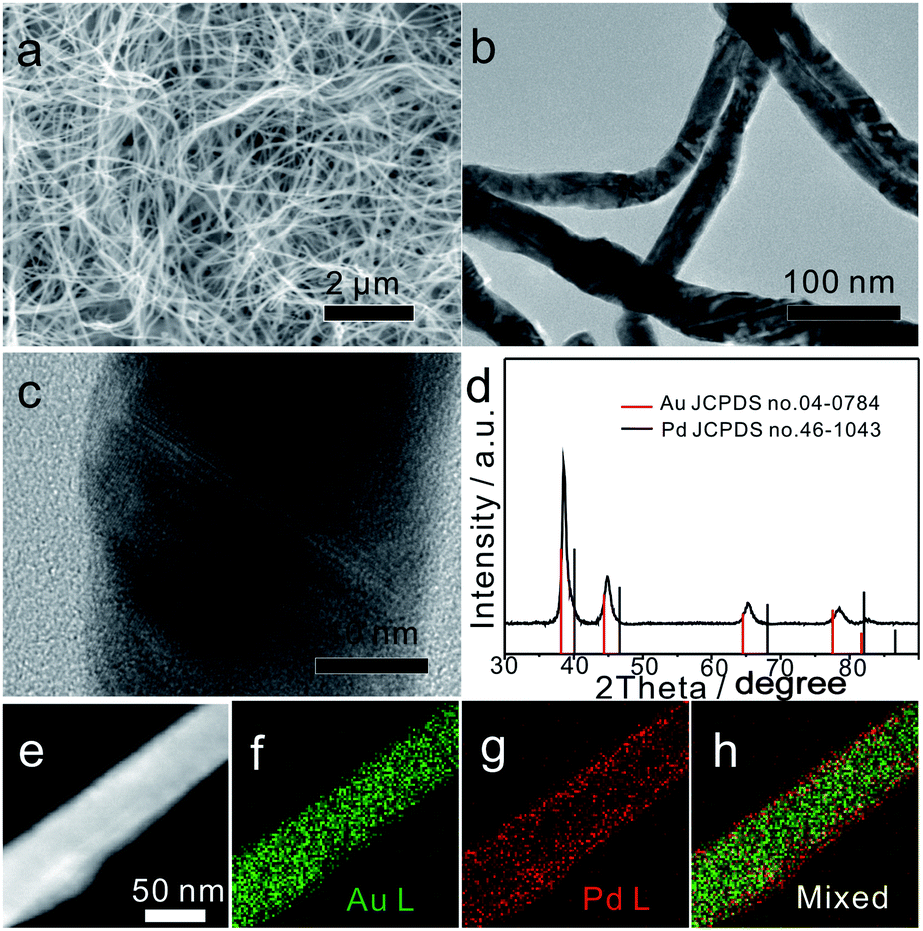 | ||
| Fig. 1 (a and b) SEM and TEM images of Au75Pd25 NWs. (c) Enlarged TEM image of an individual Au75Pd25 NW. (d) PXRD pattern of Au75Pd25 NWs, and (e–h) HAADF-STEM image of Au75Pd25 NWs and the corresponding elemental EDS mapping. | ||
To further analyze the phase structure and composition of NWs, techniques such as PXRD, HAADF-STEM, EDS mapping and XPS were employed. The PXRD pattern (Fig. 1d) showed four characteristic diffraction peaks at 38.35°, 44.61°, 64.93° and 77.96°, corresponding to the (111), (200), (220) and (311) planes of the face-centered cubic (fcc) structure, respectively. Note that all diffraction peaks were slightly shifted to the right side when compared with the standard peaks of bulk Au, indicating that the major phase of Au75Pd25 NWs was made of the Au-rich AuPd alloy. The average Pd content in the NWs (23% calculated from EDS, Fig. S2b, ESI†) was basically consistent with the feeding ratio of Pd (25%), meaning equivalent reduction of Au and Pd precursors. The HAADF-STEM image combined with EDS mapping results (Fig. 1e–h) depicted a clear core–shell structure with a thin outer shell rich in Pd. Hence, the major phase detected by XRD can be assigned to the core since the Pd-rich shell looked thin in Au75Pd25 NWs. The surface states of Au75Pd25 NWs were further investigated by XPS (Fig. S3, ESI†). The peaks at 84 eV and 335 eV in XPS spectra were assigned to Au and Pd, respectively. The Au 4f spectrum revealed typical Au(0) double spin orbits without obvious oxidation states. The signal of the Pd 3d spectrum was split into two pairs of spin–orbits. The major part was assigned to Pd(0) species and minor parts with higher binding energy were attributed to Pd(II), which, in turn, were related to surface oxidation of Pd. Surface elemental distributions calculated from XPS indicated a Au/Pd atomic ratio of 10:90, further confirming a surface enriched with Pd in Au75Pd25 NWs. In sum, Au75Pd25 NWs possessed a core–shell structure with a Au-rich AuPd alloy core and a Pd-rich AuPd alloy shell (AurichPd@AuPdrich). For simplification, the Pd-rich shell is denoted as Pd shell in the subsequent sections.
Shell thickness control of Au–Pd bimetallic core–shell NWs
The ratio of HAuCl4 and H2PdCl4 was changed to regulate the shell thickness of the core–shell structure. Feeding only HAuCl4 solution led to the formation of major products with multi-connected short NWs attached in random directions (Fig. S4, ESI†). The supply of only H2PdCl4 led to no formation of NWs but the formation of irregular particles. The tuning of the Au/Pd feeding ratio from 85:15 to 50:50 yielded products with wavy NW morphologies. The EDX results (Fig. S2, ESI†) showed that the average Au/Pd molar ratio of products was dependent on the feeding ratio.
The compositions of Au–Pd NWs obtained at different feeding ratios were systematically investigated by PXRD (Fig. 2). The major diffraction peaks were located near the standard peaks of bulk Au, revealing that the major component of Au–Pd NWs is a Au-rich AuPd alloy. According to the calculations with regard to the {111} peak from 36° to 42° based on Vegard's law, which describes a linear relationship between the crystal lattice constant and the composition in a continuous substitutional solid solution,32 the content of Pd in bimetallic core–shell NWs increased from 9% for Au85Pd15 NWs to 16% for Au75Pd25 NWs. The rise in Pd feeding ratios to 50:50 led to the generation of a new peak at 40° related to nearly pure Pd with an fcc phase accompanying the peak assigned to the Au-rich AuPd alloy. This may be caused by the increase in epitaxial growth or self-nucleation of Pd at high Pd feeding ratios. When combined with TEM data, the above XRD analysis suggested that the major Au-rich AuPd alloy phase could be assigned to the core of Au–Pd NWs, and the peak of Pd in Au50Pd50 NWs could be attributed to the thick Pd shell.
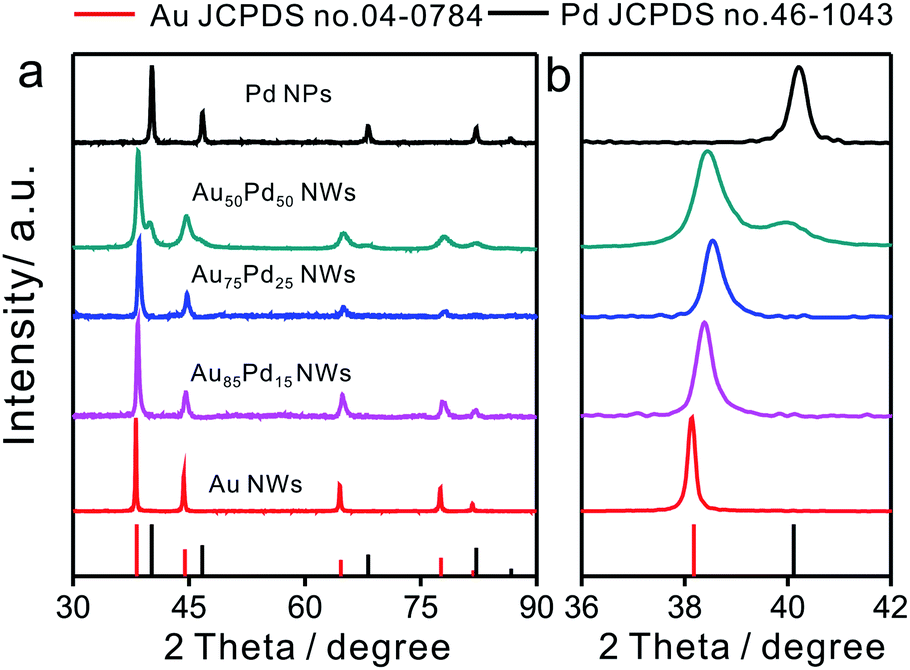 | ||
| Fig. 2 (a) XRD patterns of Au–Pd NWs with different Au/Pd feeding ratios and (b) the enlarged pattern of the (111) peak from 36 to 42 degrees. | ||
As shown in TEM images (Fig. 1b and 3a, b), the morphology of NWs obtained at different Au/Pd feeding ratios (85:15, 75:25, and 50:50) remained similar. The mixed EDS mapping images of all three NWs (Fig. 1h and 3c, d) showed core–shell structure features. Of note, the Pd-rich surface of Au85Pd15 NWs can be considered an incomplete Pd shell. For Au75Pd25 NWs, the Pd shell looked more complete and dense. As for Au50Pd50 NWs, a thicker Pd shell was formed. The elemental line profiles (Fig. S5, ESI†) also confirmed the increase in shell thickness with Pd content. The Pd shell thicknesses of Au75Pd25 and Au50Pd50 NWs were estimated to about 2 and 10 nm, respectively. It should be stated that the Pd distribution was uneven, making the calculation of accurate thickness difficult. The shell constituents were further explored by XPS. The detection depth of XPS was 1–2 nm, corresponding to about 5–10 atom layer thickness of Au and Pd. Hence, the elemental distribution of Pd near the surface region could be provided. From the XPS data (Table 1), the surface Pd content appears to be much higher than the total Pd content in Au–Pd core–shell NWs. The Pd content near the surface rose from 75% for Au85Pd15 NWs to 90% for Au75Pd25 NWs, reaching the saturation value around 91% for Au50Pd50 NWs. The CV fingerprints also elucidated the difference in the outermost shell layers of Au–Pd NWs. In Fig. 3e, the two reduction peaks at 1.13 V and 0.58 V belonged to the reduction of surface Au and Pd oxides, respectively. The Au85Pd15 NWs and Au75Pd25 NWs showed both reduction peaks, suggesting that the outermost surfaces contain Au and Pd. The Pd contents in the outermost surfaces of Au85Pd15 NWs and Au75Pd25 NWs were calculated from CV fingerprints as 73% and 85%, respectively. The Au50Pd50 NWs showed typical Pd-like CV features, meaning that the outermost surfaces are almost fully covered by the Pd shell. These results were consistent with the XRD and EDS mapping data.
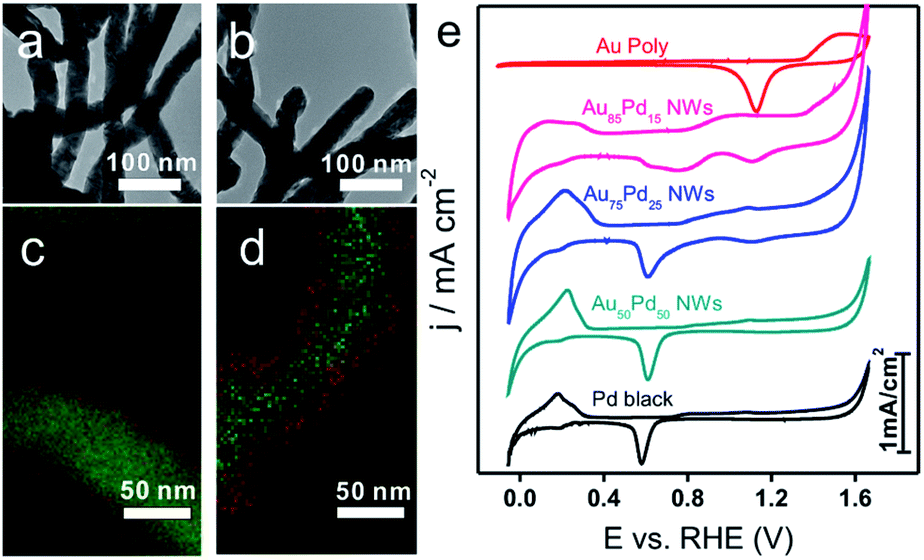 | ||
| Fig. 3 (a and b) TEM images of Au85Pd15 and Au50Pd50 NWs. (c and d) Corresponding EDS mapping images (green represents Au and red is Pd). (e) CV curves of a Au polycrystalline electrode, Au–Pd NWs with different Au/Pd ratios, and Pd black in 0.5 M H2SO4. | ||
| Name | Pd feeding ratio | EDX | XRDa | XPS | Electrochemical methodb | Shell morphology |
|---|---|---|---|---|---|---|
| a Calculated from Vegard's law, the lattice parameter of an AuPd alloy (aAuPd) is linearly related to the Pd percentage (x) in the alloy: aAuPd = xapd + (1 − x)aAu.29 b Calculated from the charge associated with the reduction of Au(QAu–O) and Pd(QPd–O) surface oxides. Pd% = (QPd–O/420)/(QAu–O/400 + QPd–O/420).30,31 | ||||||
| Au85Pd15 NWs | 15 | 16 | 9 | 75 | 73 | Incomplete Pd shell |
| Au75Pd25 NWs | 25 | 25 | 16 | 90 | 85 | ca. 2 nm thin Pd shell |
| Au50Pd50 NWs | 50 | 49 | 10 | 91 | 100 | ca. 10 nm thick Pd shell |
Growth mechanism of Au–Pd bimetallic core–shell NWs
In the presence of CTAB, AuCl4− and PdCl42− would be transformed into AuBr4− and PdBr42−, respectively. Since the standard electrode potential of AuBr4−/Au (0.854 V) was higher than that of PdBr42−/Pd (0.591 V), the Au precursor could thermodynamically be reduced ahead of Pd precursors. Note that the core and shell of the as-prepared Au–Pd NWs were both made of the AuPd alloy, despite the great difference in the ratios. In particular, the thickness of the Pd-rich shell can be controlled by simply tuning the feeding ratio of the precursors. Clearly, the formation mechanism of the AurichPd@AuPdrich core–shell structure differed from previously reported heterogeneous Au@Pd core–shell structures33 or homogeneous alloy structures34 prepared by one-step routes. Here, Au–Pd core–shell NWs were first formed via co-reduction of Au precursors along with Pd precursors. Subsequently, the remaining Pd atoms were epitaxially deposited on the Au–Pd core surface due to a small lattice mismatch under suitable reaction kinetics. During the formation process, CTA+ played important roles in controlling the growth kinetics of NWs. Without CTA+, the solution of PdBr42− (20 mM NaBr and 0.8 mM H2PdCl4) would turn black in only 5 minutes after AA addition. In contrast, the Pd growth solution incubated with CTAB (20 mM CTAB and 0.8 mM H2PdCl4) remained unchanged for 24 hours. The high viscosity of the CTA+ solution blocked the diffusion and aggregation of atoms, limiting the growth rate and providing mild growth conditions for the formation of the AurichPd@AuPdrich structure.The soft template theory could account for the formation of polycrystalline NWs.35 Surfactants could form worm-like assemblies as soft templates for the growth of NWs. To reveal the growth mechanism of the as-prepared NWs, the effect of CTAB was specifically investigated. Theoretically, spherical micelles would form once the concentration of CTAB was beyond the critical micelle concentration (CMC) (0.92 mM at 25 °C for CTAB),36 and rod-like micelles37 or a hexagonal liquid crystal phase38 would form at higher CTAB concentrations. The addition of salt may weaken the electrostatic repulsions between hydrophilic ionic groups, facilitating the transformation of spherical micelles into worm-like micelles at low CTAB concentrations.39 Thus, the conformation of CTAB micelles in the reaction solution was regulated by tuning the concentration of NaBr (Fig. 4). At low CTAB concentrations (1 mM and 4 mM), worm-like micelles could not be formed and the products aggregated to yield irregular nanocrystals. After the addition of 3 mM or 16 mM NaBr, the NWs were formed due to the emerged worm-like CTAB micelles. Temperature control experiments (Fig. S6, ESI†) also confirmed the template effect of CTAB micelles. At low temperatures (6 °C), the final products looked like plates, similar to lamellar-shaped CTAB micelles.38 At high temperatures, the products were polyhedral nanocrystals thanks to the reduced quantity of micelles. Furthermore, OTAB was applied to replace CTAB (Fig. S7, ESI†). The final products were NWs, implying the ubiquitous nature of the template effect of alkyl ammonium bromide surfactant.
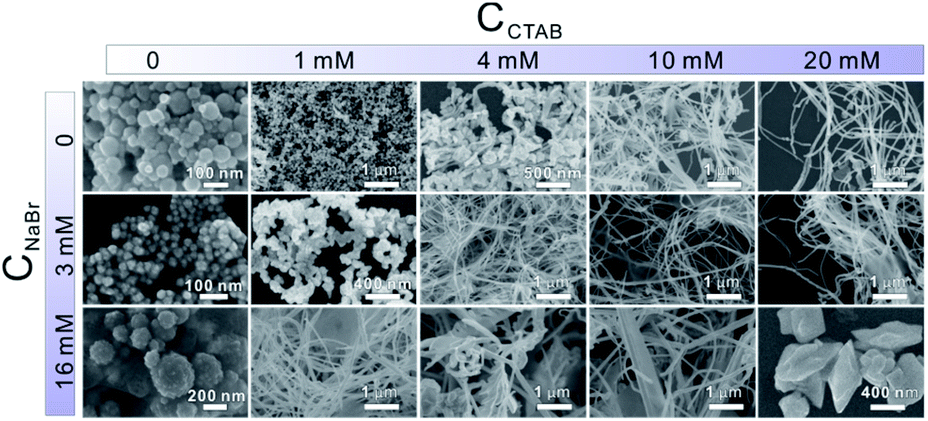 | ||
| Fig. 4 SEM images of products with different CTAB concentrations (from left to right, CCTAB in the growth solution is varied from 0 to 20 mM) and NaBr concentrations (from top to bottom, CNaBr is changed from 0 to 16 mM). | ||
The proposed growth mechanism is illustrated in Scheme 1. The reaction of CTAB solution (20 mM) with HAuCl4 and H2PdCl4 led to the conversion into long worm-like micelles in the solution due to the salt effect as mentioned before.40 In aqueous solution, the worm-like micelles possessed a hydrophobic internal structure with a positively charged surface. Metal precursors became concentrated in the Stern layer of worm-like micelles due to a strong polarized electric field,41 further reducing to small nuclei aggregated along the worm-like micelle template. After the initial formation of NWs, extended NW growth took place through attachment and fusion with ripening.
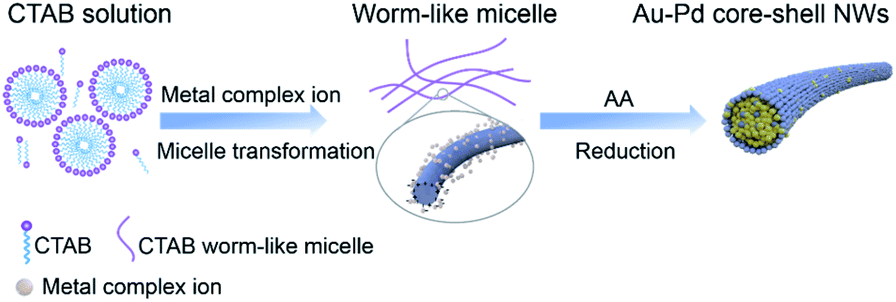 | ||
| Scheme 1 Schematic representation of the growth mechanism of NWs. | ||
H2O2 electrocatalytic detection
Due to the importance of H2O2 sensing in biological, pharmaceutical, and food industries, the development of efficient H2O2 sensors with high sensitivity, selectivity, and stability is attractive to researchers. Au/Pd nanomaterials were reported as promising nanocatalysts for electrocatalytic H2O2 sensing.42,43 To examine the catalytic properties of Au–Pd core–shell NWs, H2O2 detection experiments were carried out in 0.025 M PBS solution. Linear scan voltammograms of Au–Pd core–shell NW modified electrodes were recorded from 1.2 to 0.2 V as a function of H2O2 concentration (Fig. 5a and S8a–d, ESI†). Au75Pd25 NWs showed higher reduction current density than other samples, indicating excellent catalytic activity toward H2O2. From the calibration curves (Fig. 5b and S8e–h, ESI†), one could notice that pure Au NWs had the lowest sensitivity of 37 μA cm−2 mM−1 towards H2O2. Thus, Au was inert towards H2O2 reduction, consistent with other reported studies.44–46 Au–Pd NWs displayed improved sensitivities towards the reduction of H2O2. Au85Pd15 NWs with an incomplete Pd outer shell displayed a sensitivity of 269 μA cm−2 mM−1 towards the reduction of H2O2 while Au75Pd25 NWs with a thin Pd shell exhibited a sensitivity of 338 μA cm−2 mM−1. Au50Pd50 NWs with a thick Pd shell presented two separated linear ranges with a sensitivity of 540 μA cm−2 mM−1 in the range of up to 2 mM and 162 μA cm−2 mM−1 in the range of 2–10 mM. Therefore, Au75Pd25 NWs showed the best sensitivity among all samples in the range up to 10 mM. Apart from the sensitivity, Au75Pd25 NWs also displayed remarkable selectivity towards H2O2 in the presence of interferents, such as ascorbic acid (AA), dopamine (DA), uric acid (UA), and glucose (Glu) (Fig. 5c). The detection limit (Fig. 5d) was a little higher than the values reported in other studies.47 Table S1 in the ESI† shows the comparison of the performance of Au–Pd core–shell NWs towards H2O2 sensing with that of other Au–Pd nanomaterials.16,45,47–51 The Au–Pd NWs in this work show an enhanced sensitivity compared to pure Au and Pd,45,48 and are comparable with other Au–Pd nanomaterials in terms of sensitivity and linear range.16,47 Besides, Au–Pd NWs can even surpass some hybrid nanomaterials in sensitivity due to the higher electronic conductivity of NWs and their unique core shell structure.49,50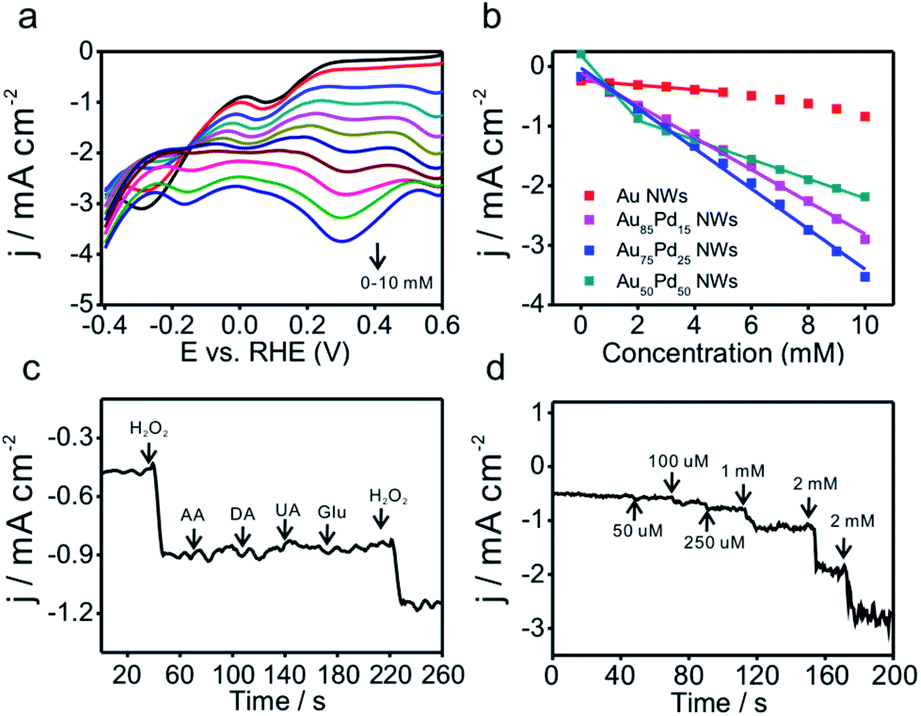 | ||
| Fig. 5 (a) LSV curves of Au75Pd25 NWs in 0.025 M PBS with increasing H2O2 concentrations. (b) Calibrated curves of current density versus concentration of H2O2 for different samples. (c) I–t curves of Au75Pd25 NWs upon addition of 1 mM H2O2, 1 mM AA, 1 mM DA, 1 mM UA, 5 mM Glu, and 1 mM H2O2 at 0.97 V. (d) I–t curves at 0.97 V at specific concentrations of H2O2. | ||
The mechanism of direct electrochemical reduction of H2O2 on the Pd surface has widely been investigated in previous studies.16,52,53 H2O2 can be decomposed to surface adsorbed hydroxide (OHad), which would further be reduced to water. The reaction intermediate OHad played important roles in the catalytic process. Moderate binding energy of OHad on sensors led to high H2O2 reduction activity. Au with weak oxyphilicity54 would allow easy desorption of H2O2 from the surface, leading to poor catalytic activities. Although Pd should be better in reducing H2O2 than Au, the strong binding energy between Pd and OH would lead to the occupation of the active sites of Pd by OHad, inhibiting the catalytic reaction. The formation of the alloy core–shell structure combining Pd with Au could optimize the OH binding energy and thus the catalytic activity. Here, Au75Pd25 NWs achieved the best activity. Their OHad binding strength evaluated by an electrochemical method (Fig. S9, ESI†) (surface oxide reduction peak) could be located at an optimal position.14 For Au–Pd core–shell NWs with different Pd shell thicknesses the surface Pd oxide reduction peaks were located between Au (1.13 V) and Pd (0.58 V), indicating the moderate OH binding ability of Pd. The Pd surface redox peaks of the three NWs became broader than that of Pd black. Hence, the NWs possessed multiple Pd surface states with different OH affinities. In sum, the redox behavior of surface Pd was directly related to the thickness of the Pd shell and the corresponding surface Pd content. The thin shell thickness yielded Au75Pd25 NWs with moderate OH binding ability while keeping the Pd dominated surface. The synergistic effect of both features contributed to enhanced activities.
Conclusions
Au–Pd bimetallic (AurichPd@AuPdrich) NWs with adjustable shell thickness were successfully synthesized by a simple one-pot wet chemical method. The intrinsic redox properties of the metal precursors led to the formation of core–shell structures. Besides, CTAB played a vital role in controlling the reduction rate and growth of NWs. Au75Pd25 NWs with a thin Pd-rich shell showed the best electrocatalytic performance towards H2O2 reduction. In sum, the present work deepens the understanding of the structure–function relationship of bi-metallic NCs (especially the shell-associated electronic effect). More importantly, the proposed synthetic method looks promising for fabrication of novel core–shell nanostructures to yield catalytic materials with high reactivity and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0206500 and 2017YFA0206801), the National Basic Research Program of China (2015CB932301), and the National Natural Science Foundation of China (21671163, 21773190, 21905247, 21931009, and 21721001).Notes and references
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- P. Liu and J. K. Nørskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 RSC.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- Z. Cao, Q. Chen, J. Zhang, H. Li, Y. Jiang, S. Shen, G. Fu, B. A. Lu, Z. Xie and L. Zheng, Nat. Commun., 2017, 8, 15131 CrossRef PubMed.
- J. M. Yan, X. B. Zhang, T. Akita, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5326–5327 CrossRef CAS PubMed.
- H. Yang, L. Q. He, Y. W. Hu, X. Lu, G. R. Li, B. Liu, B. Ren, Y. Tong and P. P. Fang, Angew. Chem., Int. Ed., 2015, 54, 11462–11466 CrossRef CAS PubMed.
- H. Huang, L. Zhang, Z. Lv, R. Long, C. Zhang, Y. Lin, K. Wei, C. Wang, L. Chen, Z. Y. Li, Q. Zhang, Y. Luo and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 6822–6828 CrossRef CAS PubMed.
- A. M. El-Toni, M. A. Habila, J. P. Labis, A. L. ZA, M. Alhoshan, A. A. Elzatahry and F. Zhang, Nanoscale, 2016, 8, 2510–2531 RSC.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- A. Schlapka, M. Lischka, A. Gross, U. Kasberger and P. Jakob, Phys. Rev. Lett., 2003, 91, 016101 CrossRef CAS PubMed.
- X. Huang, A. J. Shumski, X. Zhang and C. W. Li, J. Am. Chem. Soc., 2018, 140, 8918–8923 CrossRef CAS PubMed.
- X. Yuan, L. Zhang, L. Li, H. Dong, S. Chen, W. Zhu, C. Hu, W. Deng, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2019, 141, 4791–4794 CrossRef CAS PubMed.
- M. Jamal, M. Hasan, A. Mathewson and K. M. Razeeb, J. Electrochem. Soc., 2012, 159, 825–829 CrossRef.
- S. Guo, S. Dong and E. Wang, Chem. Commun., 2010, 46, 1869–1871 RSC.
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS PubMed.
- R. Liu, L. Q. Zhang, C. Yu, M. T. Sun, J. F. Liu and G. B. Jiang, Adv. Mater., 2017, 29, 1604571–1604577 CrossRef PubMed.
- C. Zhu, S. Guo and S. Dong, Adv. Mater., 2012, 24, 2326–2331 CrossRef CAS PubMed.
- X. Chen, H. Pan, H. Liu and M. Du, Electrochim. Acta, 2010, 56, 636–643 CrossRef CAS.
- C. Li, Y. Su, X. Lv, Y. Zuo, X. Yang and Y. Wang, Sens. Actuators, B, 2012, 171, 1192–1198 CrossRef.
- Y. L. Zhang, W. J. Shen, W. T. Kuang, S. Guo, Y. J. Li and Z. H. Wang, ChemSusChem, 2017, 10, 2375–2379 CrossRef CAS PubMed.
- Y. Zhang, N. Nishi and T. Sakka, Electrochim. Acta, 2019, 325, 134919 CrossRef CAS.
- P.-P. Fang, A. Jutand, Z.-Q. Tian and C. Amatore, Angew. Chem., Int. Ed., 2011, 50, 12184–12188 CrossRef CAS PubMed.
- W. Chen, S. Cai, Q. Q. Ren, W. Wen and Y. D. Zhao, Analyst, 2012, 137, 49–58 RSC.
- X. Chen, G. Wu, Z. Cai, M. Oyama and X. Chen, Microchim. Acta, 2013, 181, 689–705 CrossRef.
- H. Liu, L. Weng and C. Yang, Microchim. Acta, 2017, 184, 1267–1283 CrossRef CAS.
- C. Hahn, D. N. Abram, H. A. Hansen, T. Hatsukade, A. Jackson, N. C. Johnson, T. R. Hellstern, K. P. Kuhl, E. R. Cave, J. T. Feaster and T. F. Jaramillo, J. Mater. Chem. A, 2015, 3, 20185–20194 RSC.
- L. L. Fang, Q. Tao, M. F. Li, L. W. Liao, D. Chen and Y. X. Chen, Chin. J. Chem. Phys., 2010, 23, 543–548 CrossRef CAS.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CAS.
- L. Vegard, Zeitschrift für Physik, 1921, 5, 17–26 CrossRef CAS.
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS PubMed.
- J. Zhang, C. Hou, H. Huang, L. Zhang, Z. Jiang, G. Chen, Y. Jia, Q. Kuang, Z. Xie and L. Zheng, Small, 2013, 9, 538–544 CrossRef CAS PubMed.
- M. P. Pileni, Nat. Mater., 2003, 2, 145–150 CrossRef CAS PubMed.
- S. Angal and E. Harris, Protein Purification Applications: A Practical Approach, IRL Press at Oxford University Press, 1990. p. 71 Search PubMed.
- C. S. Ma, G. Z. Li, Y. M. Xu, H. Q. Wang and X. F. Ye, Colloids Surf., A, 1998, 143, 89–94 CrossRef CAS.
- X. Auvray, C. Petipas, R. Anthore, I. Rico and A. Lattes, J. Phys. Chem., 1989, 93, 7458–7464 CrossRef CAS.
- T. Imae, R. Kamiya and S. Ikeda, J. Colloid Interface Sci., 1985, 108, 215–225 CrossRef CAS.
- X. Gao, F. Lu, B. Dong, Y. Liu, Y. Gao and L. Zheng, Chem. Commun., 2015, 51, 843–846 RSC.
- C. A. Bunton, F. Nome, F. H. Quina and L. S. Romsted, Acc. Chem. Res., 1991, 24, 357–364 CrossRef CAS.
- Z. Li, R. Li, T. Mu and Y. Luan, Chem.–Eur. J., 2013, 19, 6005–6013 CrossRef PubMed.
- F. Yang, K. Cheng, T. Wu, Y. Zhang, J. Yin, G. Wang and D. Cao, J. Power Sources, 2013, 233, 252–258 CrossRef CAS.
- Y. H. Won, K. Huh and L. A. Stanciu, Biosens. Bioelectron., 2011, 26, 4514–4519 CrossRef CAS PubMed.
- S. Guo, D. Wen, S. Dong and E. Wang, Talanta, 2009, 77, 1510–1517 CrossRef CAS PubMed.
- M. Zheng, P. Li, C. Yang, H. Zhu, Y. Chen, Y. Tang, Y. Zhou and T. Lu, Analyst, 2012, 137, 1182–1189 RSC.
- T. Han, Y. Zhang, J. Xu, J. Dong and C.-C. Liu, Sens. Actuators, B, 2015, 207, 404–412 CrossRef CAS.
- F. Jiang, R. Yue, Y. Du, J. Xu and P. Yang, Biosens. Bioelectron., 2013, 44, 127–131 CrossRef CAS PubMed.
- T. D. Thanh, J. Balamurugan, S. H. Lee, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2016, 85, 669–678 CrossRef CAS PubMed.
- X. Y. Li and X. Z. Du, Sens. Actuators, B, 2017, 239, 536–543 CrossRef CAS.
- L. Shang, B. Zeng and F. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 122–128 CrossRef CAS PubMed.
- S. Bozkurt, B. Tosun, B. Sen, S. Akocak, A. Savk, M. F. Ebeoglugil and F. Sen, Anal. Chim. Acta, 2017, 989, 88–94 CrossRef CAS PubMed.
- D. X. Cao, L. M. Sun, G. L. Wang, Y. Z. Lv and M. L. Zhang, J. Electroanal. Chem., 2008, 621, 31–37 CrossRef CAS.
- V. Tripkovic, H. A. Hansen, J. Rossmeisl and T. Vegge, Phys. Chem. Chem. Phys., 2015, 17, 11647–11657 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00726a |
| This journal is © The Royal Society of Chemistry 2020 |