DOI:
10.1039/C9NA00578A
(Paper)
Nanoscale Adv., 2020,
2, 239-248
Au/NiFe2O4 nanoparticle-decorated graphene oxide nanosheets for electrochemical immunosensing of amyloid beta peptide†
Received
12th September 2019
, Accepted 4th November 2019
First published on 5th November 2019
Abstract
In the present work, an electrochemical immunosensor has been fabricated for the detection of amyloid beta peptide (βA1-–42) based on a gold nanoparticle/nickel ferrite decorated graphene oxide-chitosan nanocomposite (Au/NiFe2O4@GO-Ch) modified glassy carbon electrode (GCE) as an effective sensing platform. βA1–42 has been analyzed as a potential biomarker for its application in Alzheimer's disease monitoring. The combination of highly conducting Au and NiFe2O4 nanoparticles on two-dimensional GO nanosheets provides an excellent platform for sensitive and selective sensing applications. A miniaturized Au/NiFe2O4@GO-Ch/GCE immunosensor was prepared by immobilization of βA antibody onto Au//NiFe2O4@GO-Ch/GCE via carbodiimide coupling. Various characterization techniques were utilized in the study to estimate the morphological and electronic attributes of the components used to fabricate the immunosensor. Differential pulse voltammetry (DPV) was performed to study the amperometric response of the developed immunosensor as a function of βA1–42 concentration. The DPV results confirmed that the immunosensor detected βA1–42 selectively and demonstrated a wide linear range from 1 pg mL−1 to 1 ng mL−1 and a detection limit of 3.0 pg mL−1. Furthermore, the immunosensor also indicated its clinical viability by detecting βA1–42 in cerebrospinal fluid.
1 Introduction
Normally an average adult human brain contains 100 billion neuron or nerve cells.1 Each neuron constantly processes and transmits information to others as electrical and chemical signals via neurotransmitters. Alzheimer's disease (AD) disrupts the communication among neurons, which leads to memory loss and dementia. Currently, the number of people with AD is estimated to be around 50 million worldwide, and according to the prediction of WHO this number will increase to 82 million in 2030 and 152 million in 2050. A hallmark lesion that is associated with AD is the abnormal structure of amyloid beta plaques present in the brain. These amyloid beta plaques are formed by the accumulation of a small peptide amyloid beta (βA) that is derived by proteolytic cleavage of amyloid beta protein (APP) by β- and γ-secretase from the C-terminal region of amyloid precursor protein.2 Thus, the βA monomer and its aggregates have been recognized not only as a therapeutic target but also as a diagnostic marker.3 Among βA isoforms that are present in AD, βA1–42 is generally considered as the most pathogenic.4 Therefore, developing a sensitive biosensing platform for βA is of paramount importance towards early diagnosis and treatment of AD. To date, various analytical techniques have been explored for detecting βA, such as enzyme-linked immunosorbent assay (ELISA),5 photoelectrochemical immunosensors,6 surface plasma resonance (SPR) sensors,7 fluorescence8 and surface-enhanced Raman spectroscopy9etc. However, laborious and complicated fabrication procedures are required for the aforementioned methods. Nevertheless, electrochemical methods have received keen interest in this perspective, owing to their advantages such as high sensitivity, simple instrumentation, rapid response and low cost, which in turn allow point-of-care analysis.
Graphene oxide (GO) because of its 2D structure and fascinating chemical and physical attributes has created enormous interest in the field of electrochemistry. It possesses sp2 bonded carbons packed in a honeycomb lattice and has a large surface area, good electrocatalytic properties and strong mechanical strength.10 Because of these unique properties, GO is proven to be useful for the development of novel sensing systems.
With the advancement of nanotechnology, nanomaterials have gained tremendous interest in sensing applications to improve its quality. Nowadays, magnetic nanoparticles are widely used in sensing applications as they improve the selectivity and sensitivity of sensors in detecting various analytes.11–14 Among them, nickel ferrite (NiFe2O4) has received tremendous interest because of its unique electrical and magnetic properties, biocompatibility, low toxicity, super-paramagnetic properties, ease of preparation and high absorption properties. Thus, decorating GO with NiFe2O4 results in a hybrid material with improved electrochemical characteristics. Additionally, the adhesion and structural stability of the nanocomposite can be further improved by incorporating chitosan.15 Incorporation of metal nanoparticles further enhances the electrochemical sensing performance. In particular, gold nanoparticles (AuNPs) are intensively studied owing to their pertinent properties such as enhanced electrocatalytic activity, high surface to volume ratio, biocompatibility, excellent adsorption capacities, and brilliant electron-transfer capacity.16
In the view of above, we propose an Au/NiFe2O4 nanoparticle decorated 2D GO-based electrochemical immunosensor with enhanced electrochemical sensing performance for determining βA1–42 peptide in the physiological range. The nanocomposite modification provides improvised electrochemical characteristics and permits effective functionalization of antibodies specific to βA1–42 peptide over the electrode surface. The obtained immunosensing performance towards determining βA1–42 peptide is highly promising in terms of selectivity and sensitivity. To further assess its clinical viability, the immunosensor has also been tested in cerebrospinal fluid so that the outcome of the detection technique has a potential usage to decide therapeutics in AD patients.
2 Experimental
2.1 Materials
Nickel(II)chloride hexahydrate (NiCl2·6H2O), iron(III)chloride hexahydrate (FeCl3·6H2O), trisodium acetate (Na3C6H5O7), chitosan (Ch), graphene powder, sulfuric acid (H2SO4), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), hydrochloric acid (HCl), gold(III) chloride trihydrate (HAuCl4·3H2O), sodium citrate tribasic dihydrate 99%, bovine serum albumin (BSA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), sodium phosphate monobasic (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), potassium chloride (KCl), potassium ferrocyanide [K4Fe(CN)6] and potassium ferricyanide [K3Fe(CN)6] were procured from Sigma Aldrich, USA. Amyloid β peptide (1–42) (human) and amyloid β antibody were obtained from Abcam and ethanol was purchased from Merck, India. All the chemicals were used without further purification. Deionized (DI) water from a Millipore system (∼18.2 MΩ cm) was used throughout the experiment.
2.2 Instrumentation
X-ray diffraction (XRD) analysis was performed using an X-ray diffractometer (Rigaku, Japan). The analyses were performed in the scanning range of 2θ = 10–60° and at a scanning speed of 2.0° min−1. Electron microscopes were used to carry out the morphological studies. A high-resolution transmission electron microscope (HRTEM JEOL, JEMCXII, Japan) was used to visualize the physical attributes of nanocomposites and their components at an operating voltage of 200 kV after coating the sample on a carbon-coated copper grid with 400 mesh (Ted-Pella Inc.). A Thermo Fisher Scientific (UK) ESCALAB Xi+ X-ray photoelectron spectrometer (XPS) was used in the study of the elemental state of the nanoparticles.
2.3 Preparation of nickel ferrite (NiFe2O4) nanoparticles
NiFe2O4 nanoparticles were synthesized by a hydrothermal method.17 For this, 1 g of FeCl3·6H2O and 2 g of NiCl2·6H2O were mixed together in double distilled water followed by the addition of 80 mg of trisodium acetate as a chelating agent. This mixture was then stirred for about 1 h and shifted to a Teflon-lined hydrothermal reactor. The temperature was maintained at 180 °C for 12 h. After the completion of the process, the black precipitate was filtered off, washed with deionized water and ethanol several times and dried in an oven at 70 °C for 3 h. The as-prepared NiFe2O4 nanoparticles were then added to chitosan (5 mg mL−1) solution and stirred for 1 h. The mixture was sonicated further for another 2–3 h to obtain NiFe2O4-Ch nanocomposites.
2.4 Preparation of nickel ferrite/graphene oxide nanocomposites (NiFe2O4@GO-Ch)
For the preparation of graphene oxide (GO), a modified Hummer's method was adopted.18 In this method, 2 g of graphitic powder was mixed with H2SO4 under constant stirring for about 1 h, followed by the addition of 5 g of KMnO4 to this mixture. The temperature was maintained below 20 °C with vigorous stirring with a stir bar for about 4 h. The temperature was then increased and maintained up to 35 °C. After this, the mixture was diluted by pouring 70 mL of water and stirred about 1 h. To this solution, 35% H2O2 was added until the colour turned from brown to brilliant yellow. Then, the mixture was filtered and washed using 1 M HCl and water. Lastly, the synthesized GO was dried in an oven at 80 °C. NiFe2O4@GO-Ch nanocomposites were prepared by adding GO in NiFe2O4-Ch solution followed by ultrasonication for 2 h.
2.5 Preparation of Au nanoparticles (AuNPs)
The standard citrate reduction method was used to prepare AuNPs, as described in our previous study.19 Briefly, the reduction of HAuCl4 (0.5 mM, 100 mL H2O) has been initiated by the addition of citrate solution (50 mM) by bringing the solution to a reflux condition with constant stirring. The solution turned to pale yellow to deep red after adding citrate solution, indicating particle formation. The solution was then allowed to reflux for another 20 min and then cooled to room temperature.
2.6 Preparation of gold nanoparticle/nickel ferrite decorated graphene oxide-chitosan (Au/NiFe2O4@GO-Ch) nanocomposites
The as-prepared NiFe2O4@GO-Ch nanocomposites and AuNPs were mixed in equal proportions. After this, the mixture was sonicated for 1 h in an ultrasonic bath. The prepared Au/NiFe2O4@GO-Ch nanocomposite was then stored in a glass vial for further study.
2.7 Preparation of the working electrode using the Au/NiFe2O4@GO-Ch nanocomposite
A potentiostat/galvanostat model GAMRY Reference 3000 was used for carrying out the electrochemical experiments. A conventional three-electrode system consisting of the Au/NiFe2O4@GO-Ch modified glassy carbon electrode (GCE) (diameter of 3 mm) as the working electrode, a platinum wire as the counter electrode and Ag/AgCl as the reference electrode was used. Prior to modification, the bare GCE was hand-polished successively with 0.3 μm and 0.5 μm micropolish alumina (Buehler, USA) using polishing cloth to obtain a cleaned and polished surface.20 It was then washed with ethanol and water in a sonication bath and dried at room temperature. A 10 μL Au/NiFe2O4@GO-Ch nanocomposite was dropped onto the GCE surface and dried at room temperature for 24 h. The as-prepared Au/NiFe2O4@GO-Ch modified GC electrode is denoted as the Au/NiFe2O4@GO-Ch/GCE. The various electrochemical analyses carried out in the present study include cyclic voltammetry (CV), differential pulse voltammetry (DPV) and electrochemical impedance spectroscopy (EIS).
2.8 Fabrication of the immunosensor
Once the modified electrode was ready, it was subjected to immobilization of βA antibody (anti βA) via the formation of carbodiimide bonds by using EDC/NHS as a cross-linker.21,22 The cross-linker covalently binds to the carboxylic group of GO at one end and to an amino group of βA antibody at the other. In this method, the Au/NiFe2O4@GO-Ch modified GCE was soaked in a solution comprising 0.4 M EDC and 0.1 M NHS for 1 h to activate the nanocomposite system. A 10 μL solution of βA antibody was drop-cast onto the EDC-NHS functionalized Au/NiFe2O4@GO-Ch/GC electrode for immobilizing the antibodies on the surface of the electrode. It was incubated at 4 °C for 12 h to obtain the anti βA-Au/NiFe2O4@GO-Ch modified GCE. After rinsing with PBS, 10 μL of 5 mg mL−1 BSA solution was deposited on the anti βA- Au/NiFe2O4@GO-Ch/GCE to block the non-specific binding sites. Then, the electrode was further incubated in different concentrations of βA1–42 peptide and the corresponding alterations in the current response were obtained using the DPV measurements. The variations in the current response were then correlated with the various concentrations of βA1–42 by using Origin 8.5 Pro analytical software. The performance characteristics were evaluated by statistical analysis using Microsoft Excel, 2019.
3 Results and discussion
3.1 Synthesis and characterization
AuNPs are synthesized by citrate reduction of HAuCl4 in water. Here, citric acid plays the role of both reducing and stabilizing agents. The particle formation of AuNPs is a multistep process that is initiated by the formation of small gold nuclei. As the reaction progresses, these small nuclei assemble into Au particles at the nanoscale. The reaction conditions were optimized to obtain nanosized AuNPs. GO nanosheets are synthesized from graphitic powder by using a modified Hummer's method. Herein, the bulk graphite is converted into a sulfuric acid-graphitic intercalation compound (H2SO4-GIC) by exposing it to an acidic medium, which is thereafter oxidized.23 Then, the oxidized GIC is converted to GO nanosheets. GO nanosheets prepared by such a chemical method consist of various oxygen-containing functional groups such as carboxyl, carbonyl, hydroxyl, epoxy, etc. NiFe2O4 nanoparticles are prepared via the hydrothermal route. The process utilizes water under high-temperature and high-pressure conditions. Under these conditions, the properties of water and related parameters for hydrothermal syntheses, such as metal oxide solubility and reaction rates, can be controlled by manipulating temperature and pressure. All these nanomaterials possess distinctive functions and are therefore used in the fabrication of the Au/NiFe2O4@GO-Ch nanocomposite. AuNPs possessed high catalytic and electrochemical properties and excellent conductivity, while NiFe2O4 is chemically stable and non-toxic and provides tunable electron transport properties with fast analytical response.24 GO provides 2D high surface area with oxygen functional groups allowing opportunities for integrating it with nanoparticles GO along with Ch provides the site for antibody immobilization by virtue of the surface functional groups. Ch also possesses high film forming ability. Therefore, inclusion of all these nanomaterials to fabricate a unique nanocomposite is noteworthy towards the achievement of high electrochemical properties.
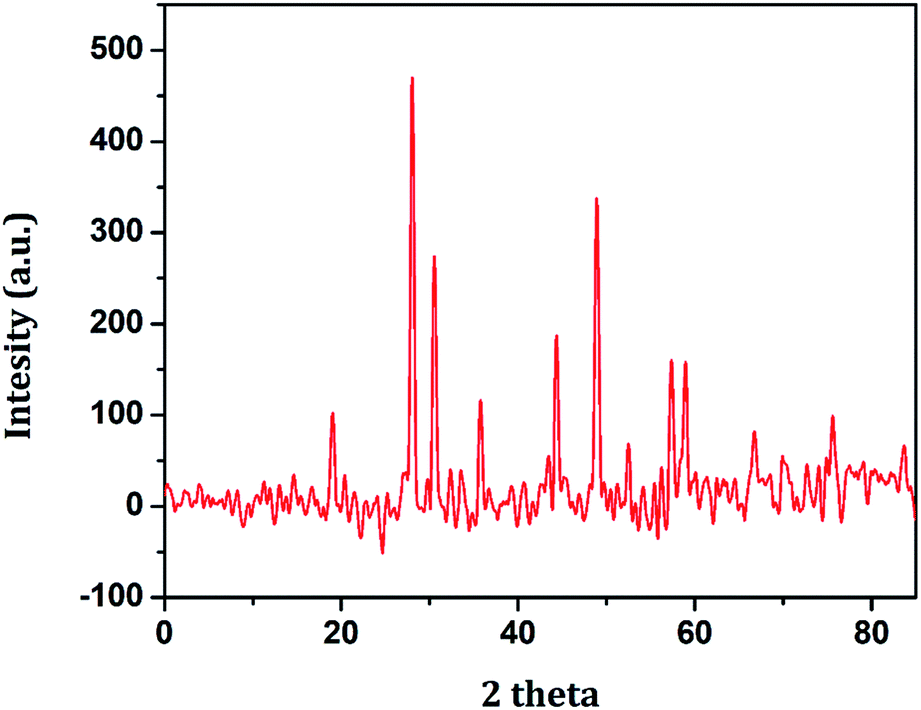
The various individual and synergistic components of the nanocomposite were characterized by using different spectroscopic techniques. The structural identity and the crystalline phase present in the as-prepared NiFe2O4 nanoparticles were examined by XRD, the results of which are shown in Fig. 1. The XRD peaks 18.9°, 29.3°, 35.7°, 44.5°, 49.1°, 57.4° and 60.1° are attributed to the (111), (220), (311), (400), (422), (511) and (440) crystal planes of NiFe2O4, respectively. The diffraction peaks were confirmed by indexing the data obtained from ICDD-PDF-2 (Card no 77-1716).
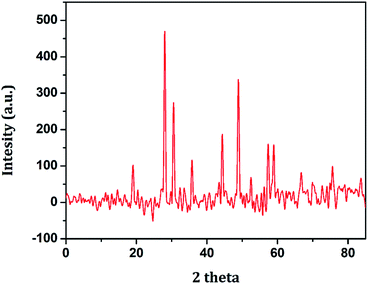 |
| | Fig. 1 XRD pattern of NiFe2O4 nanoparticles. | |
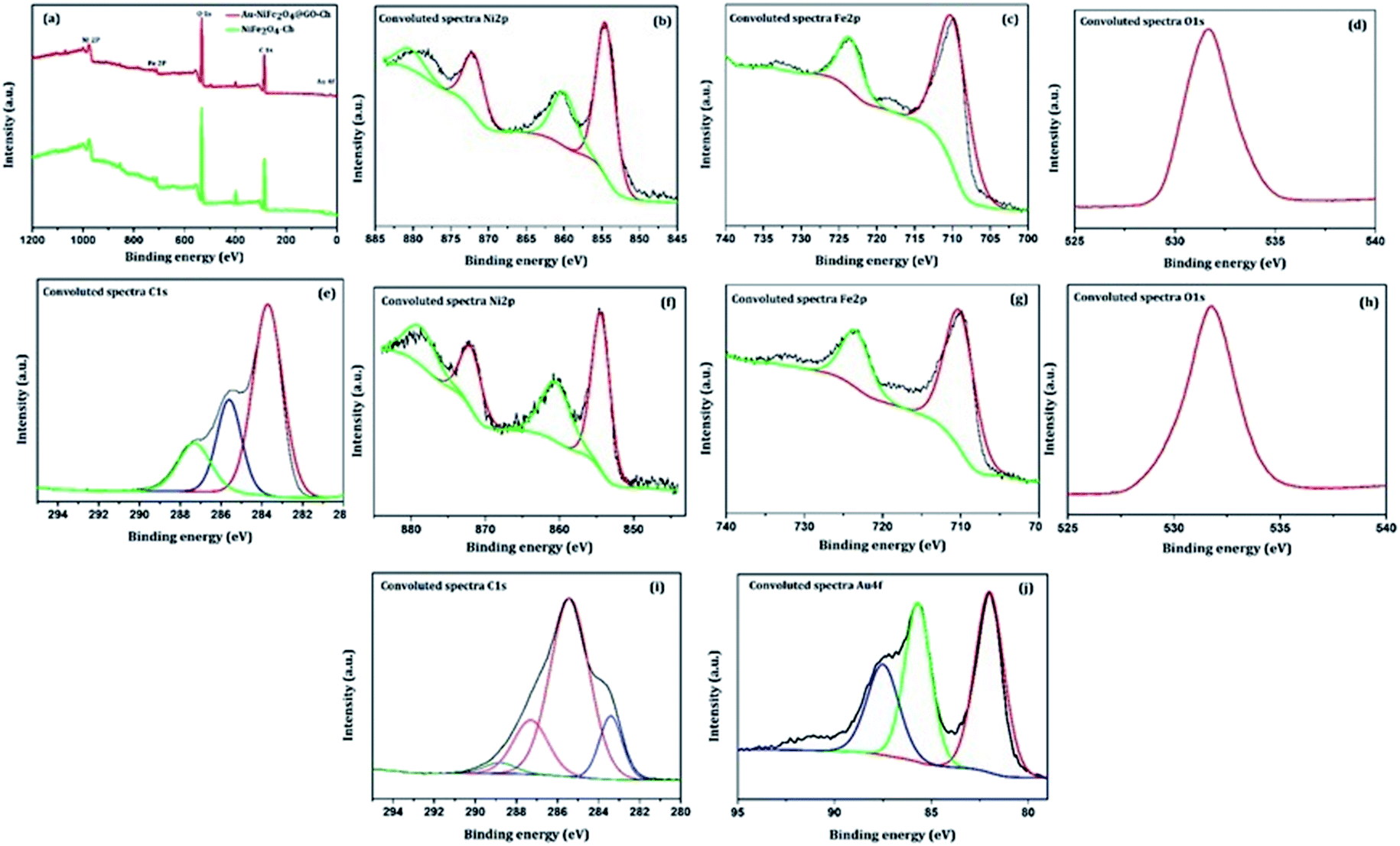
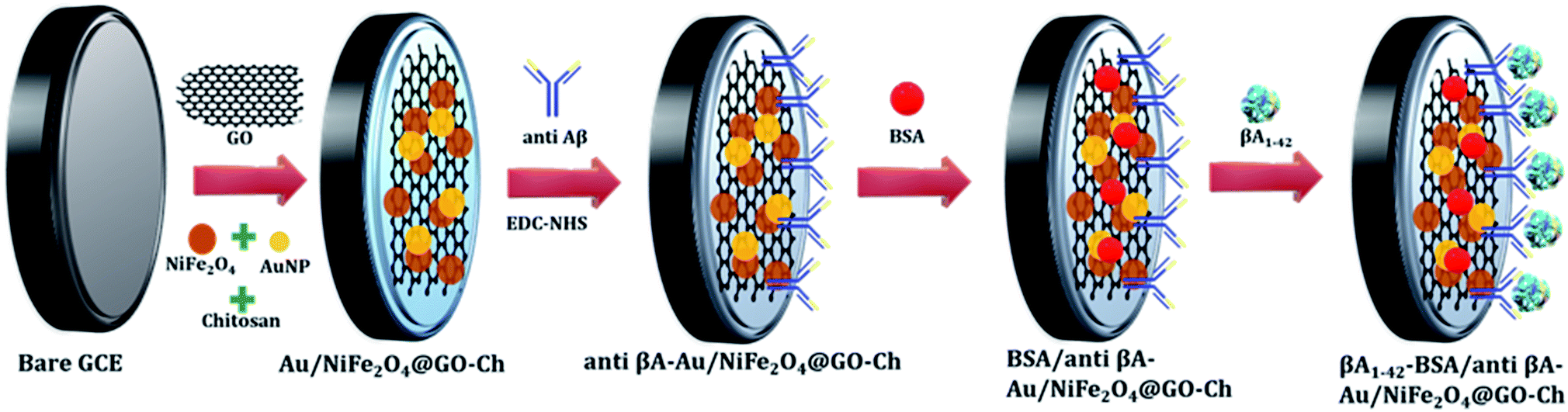
The XPS technique was further used to investigate the surface chemical composition and structural evolution of the as-synthesized NiFe2O4-Ch and Au/NiFe2O4@GO-Ch nanocomposites. Typical XPS survey spectra corresponding to NiFe2O4-Ch and Au/NiFe2O4@GO-Ch are shown in Fig. 2(a). As expected, the peaks of Ni 2p, Fe 2p, O 1s and C 1s for both NiFe2O4-Ch and Au/NiFe2O4@GO-Ch are revealed in the XPS spectra. The deconvoluted peaks of Ni2p orbital of NiFe2O4-Ch and Au/NiFe2O4@GO-Ch nanocomposites are shown in Fig. 2(b) and (f). In NiFe2O4-Ch, the peaks at binding energies of 854.6 eV and 872.3 eV correspond to Ni 2p3/2 and Ni 2p1/2, respectively. The shake-up satellite peaks around 860.2 eV and 880.7 eV exhibited high energy binding sites of Ni 2p3/2 and Ni 2p1/2. The asymmetric Ni 2p3/2 peak exhibits the presence of Ni2+ cations at the octahedral site (48.84%) and tetrahedral site (20.04%), respectively. After ultrasonication of NiFe2O4 nanoparticles with GO and Au nanoparticles, there is a shift in the binding energy at 854.3 eV for Ni 2p3/2 and 871.16 eV for Ni 2p1/2 in Au/NiFe2O4@GO-Ch compared to NiFe2O4-Ch, which is due to the strong coupling between NiFe2O4, GO and Au nanoparticles during the ultrasonic treatment. The deconvolution of the Fe 2p peaks is shown in Fig. 2(c) and (g), which reveals two peaks at 710.2 eV and 723.6 eV. They are assigned to Fe 2p3/2 and Fe 2p1/2 respectively, which suggests the existence of Fe(III) cations in the nanocomposite. On the other hand, no shift in the binding energy of Fe 2p peaks was observed after the modification of NiFe2O4-Ch to Au/NiFe2O4@GO-Ch nanocomposites. Here the Fe 2p3/2 and Fe 2p1/2 peaks contributed 78.77% and 21.23%, respectively. As shown in Fig. 2(d) and (h), the O 1s of NiFe2O4-Ch showed a peak at 531.6 eV, corresponding to the lattice oxygen in the Ni/Fe–O framework, while a positive shift of about 0.2 eV was observed for the same peak in the Au/NiFe2O4@GO-Ch nanocomposite. Fig. 2(e) shows the C 1s spectrum of NiFe2O4-Ch that can be resolved into three Gaussian peaks, which is due to the presence of chitosan in the composite. As shown in the figure, the C 1s spectra with binding energy peaks around 283.70 eV (59.39%), 285.56 eV (24.12%) and 287.31 eV (16.49%) are assigned to the C–C/C–H bond, C–NH/C–NH2 bond C–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–OH bond, respectively. However, the C 1s spectra of Au/NiFe2O4@GO-Ch can be fitted into four peaks at 283.38 eV (13.68%), 285.47 eV (65.51), 287.37 eV (17.65%), and 288.86 eV (3.16%), which indicates the presence of the sp2 carbon peak (CC), epoxide group (C–O–C), carbonyl group (CO), and carboxyl group (–COOH), respectively. These peaks are originated due to the presence of GO in the nanocomposite [Fig. 2(i)]. As shown in Fig. 2(j), the binding energy in Au exhibits two major peaks at 82.0 eV and 85.7 eV corresponding to the Au 4f7/2 and Au 4f5/2 signals, respectively, which confirms the presence of AuNPs in the hybrid. Furthermore, one characterization peak is also observed at 87.47 eV, which might be due to the formation of Au+ in the synthesis process of AuNPs. The TEM analysis was carried out to gain more insight into the morphology and microstructure of the as-synthesized NiFe2O4 nanoparticles, NiFe2O4@GO-Ch, and Au/NiFe2O4@GO-Ch nanocomposite. Fig. 3(a) and (b) show the successful synthesis of spherical NiFe2O4 nanoparticles. In Fig. 3(c), the HRTEM image of NiFe2O4 further shows the presence of lattice fringes with a spacing of 0.294 nm corresponding to the (220) plane. In Fig. 3(d) and (e), GO displays two dimensional nanosheets with an ultrathin wrinkle and ripple like morphology over which NiFe2O4 nanoparticles were distributed with high density. This combination of the ultrathin sheet-like structure of GO and highly conductive NiFe2O4 possesses fast electron transfer and more active area for the electrocatalytic reaction. Moreover, the TEM image shown in Fig. 3(f) indicates the presence of AuNPs over the GO and NiFe2O4 nanocomposite. The AuNP immobilized GO/NiFe2O4 nanoarchitecture could promote the electron-transfer process by improving the electrical conductivity for enhancing the electrochemical detection potential. Furthermore, the elemental compositions of the nanocomposite and its synergistic constituents were also examined by energy dispersive X-ray (EDX) analysis (Fig. S1†). Additionally, the EDX analyses were also done at random areas of the nanocomposite to assess its uniformity (Fig. S2 and Table S1†). The results show the successful fabrication of the Au/NiFe2O4@GO-Ch nanocomposite system. Thus, all the spectroscopic and microscopic analyses are conclusive of the fabrication process of the nanocomposite. The fabrication process of the immunosensor is shown in Scheme 1.
O/C–OH bond, respectively. However, the C 1s spectra of Au/NiFe2O4@GO-Ch can be fitted into four peaks at 283.38 eV (13.68%), 285.47 eV (65.51), 287.37 eV (17.65%), and 288.86 eV (3.16%), which indicates the presence of the sp2 carbon peak (CC), epoxide group (C–O–C), carbonyl group (CO), and carboxyl group (–COOH), respectively. These peaks are originated due to the presence of GO in the nanocomposite [Fig. 2(i)]. As shown in Fig. 2(j), the binding energy in Au exhibits two major peaks at 82.0 eV and 85.7 eV corresponding to the Au 4f7/2 and Au 4f5/2 signals, respectively, which confirms the presence of AuNPs in the hybrid. Furthermore, one characterization peak is also observed at 87.47 eV, which might be due to the formation of Au+ in the synthesis process of AuNPs. The TEM analysis was carried out to gain more insight into the morphology and microstructure of the as-synthesized NiFe2O4 nanoparticles, NiFe2O4@GO-Ch, and Au/NiFe2O4@GO-Ch nanocomposite. Fig. 3(a) and (b) show the successful synthesis of spherical NiFe2O4 nanoparticles. In Fig. 3(c), the HRTEM image of NiFe2O4 further shows the presence of lattice fringes with a spacing of 0.294 nm corresponding to the (220) plane. In Fig. 3(d) and (e), GO displays two dimensional nanosheets with an ultrathin wrinkle and ripple like morphology over which NiFe2O4 nanoparticles were distributed with high density. This combination of the ultrathin sheet-like structure of GO and highly conductive NiFe2O4 possesses fast electron transfer and more active area for the electrocatalytic reaction. Moreover, the TEM image shown in Fig. 3(f) indicates the presence of AuNPs over the GO and NiFe2O4 nanocomposite. The AuNP immobilized GO/NiFe2O4 nanoarchitecture could promote the electron-transfer process by improving the electrical conductivity for enhancing the electrochemical detection potential. Furthermore, the elemental compositions of the nanocomposite and its synergistic constituents were also examined by energy dispersive X-ray (EDX) analysis (Fig. S1†). Additionally, the EDX analyses were also done at random areas of the nanocomposite to assess its uniformity (Fig. S2 and Table S1†). The results show the successful fabrication of the Au/NiFe2O4@GO-Ch nanocomposite system. Thus, all the spectroscopic and microscopic analyses are conclusive of the fabrication process of the nanocomposite. The fabrication process of the immunosensor is shown in Scheme 1.
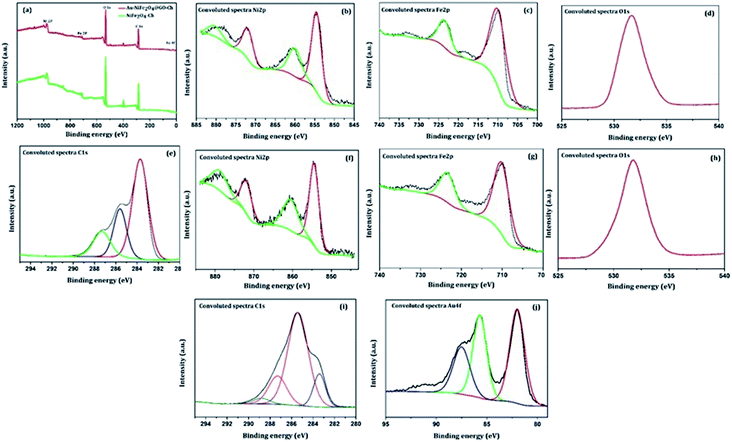 |
| | Fig. 2 (a) XPS survey spectra of NiFe2O4-Ch and Au/NiFe2O4@GO-Ch nanocomposites; (b) Ni 2p (c) Fe 2p (d) O 1s (e) C 1s XPS profile of the NiFe2O4-Ch nanocomposite; (f) Ni 2p, (g) Fe 2p (h) O 1s (i) C 1s (j) Au 4f XPS profile of the Au/NiFe2O4@GO-Ch nanocomposite. | |
 |
| | Fig. 3 (a and b) TEM images of NiFe2O4 nanoparticles; (c) HRTEM images of NiFe2O4 nanoparticles; (d and e) TEM images of NiFe2O4@GO-Ch; (f) TEM images of Au/NiFe2O4@GO-Ch. | |
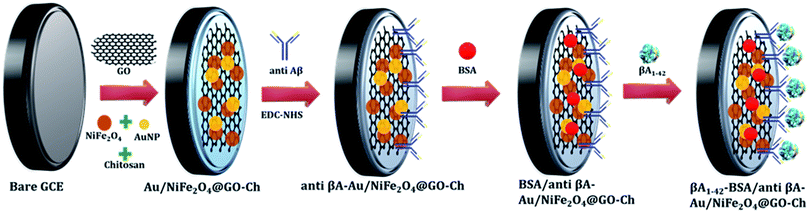 |
| | Scheme 1 Schematic representation of fabrication of the immunosensor. | |
3.2 Electrode modification analysis using voltammetric measurements
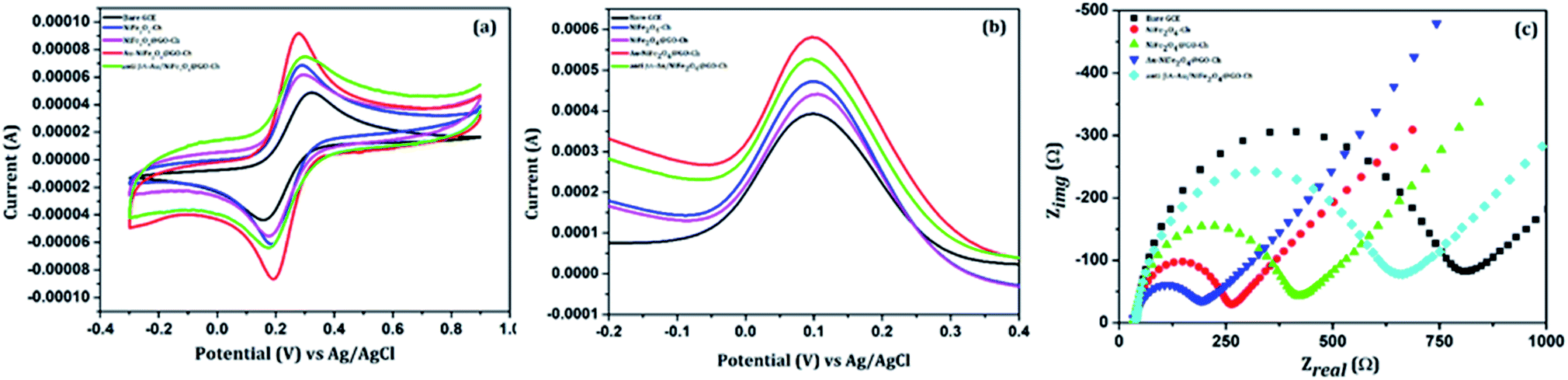
The electrochemical performance of the stepwise immobilization process of the immunosensor was initially evaluated by CV and DPV measurements. First the CV curve was used to study the interface properties of different modified electrodes, as shown in Fig. 4(a), in 100 mM PBS (pH = 7.4) containing 5 mM [Fe(CN)6]3−/4−, at a scan rate of 20 mV s−1, and in a potential range between −0.3 V and 0.9 V. At the bare GCE, a pair of redox waves was measured with a peak-to-peak separation ΔEp(Epa − Epc) of 165 mV. But the modification of the GCE with NiFe2O4-Ch led to a remarkable decrease of the value to 105 mV, which was accompanied by the enhancement of the current response. Such a behaviour indicates the improvement of surface area and conductive properties due to the electrode modification. Modification of the NiFe2O4-Ch/GCE with GO increased the ΔEp value to 121 mV along with a decrement in the peak current. The reason behind the effect was the less conductive nature of GO. But, the thin layered high surface area nanosheets of GO have the potential to make the electrode surface strongly hydrophilic for facilitating biochemical functionalization, which can enhance the sensing performance of the immunosensor. Nevertheless, the current response of the nanocomposite was improved by incorporating AuNPs on the surface of the NiFe2O4@GO-Ch/GCE. The Au/NiFe2O4@GO-Ch/GCE exhibited a low ΔEp of 88 mV with an increased current response (anodic peak current (Ipa)/cathodic peak current (Ipc) ∼ 1), indicating the excellent conductivity and rate of electron transfer between [Fe(CN)6]3−/4− and the electrode. The improved functionality of the nanocomposite can also be inferred from Fig. S3,† which compares the electrochemical characteristics of its individual/synergistic components. After the evaluation of electrochemical properties, we were interested in immobilizing anti βA onto the Au/NiFe2O4@GO-Ch/GCE surface. Due to this, the redox peak current (Ipa & Ipc) decreased and peak separation potential increased to 131 mV at the anti βA-Au/NiFe2O4@GO-Ch/GCE, the reason being the presence of bulky groups over the surface of the Au/NiFe2O4@GO-Ch/GCE, which inhibits the electron transfer between the electrode and the solution, resulting in a considerable decrement in the peak current.
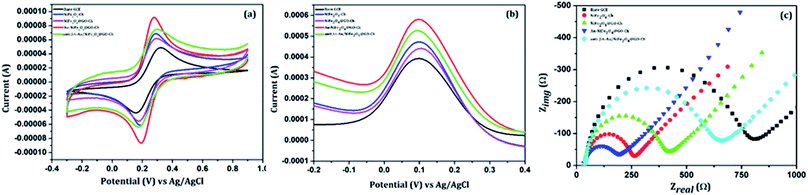 |
| | Fig. 4 Comparison of the bare GCE, NiFe2O4-Ch, NiFe2O4@GO-Ch, Au/NiFe2O4@GO-Ch and anti βA-Au/NiFe2O4@GO-Ch: (a) cyclic voltammetry (b) differential pulse voltammetry and (c) Nyquist plot. | |
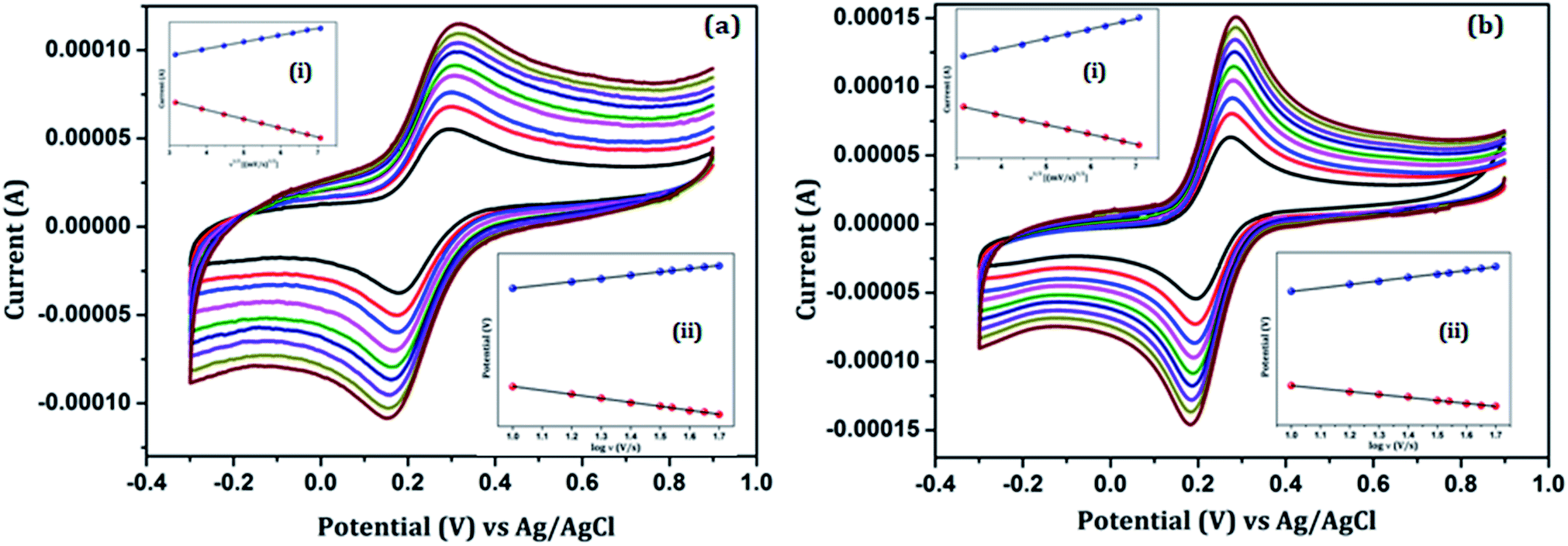
Fig. 5 shows the effect of the scan rate on the redox peak current responses of the (a) Au/NiFe2O4@GO-Ch/GCE and (b) anti βA-Au/NiFe2O4@GO-Ch/GCE in 100 mM PBS (pH = 7.4) containing 5 mM [Fe(CN)6]3−/4− and at a scan rate ranging from 10 mV s−1 to 50 mV s−1. A systematic increase in Ipa and Ipc as well as ΔEp was observed with the increase in the scan rate. Inset (i) of Fig. 5(a) and (b) describes the plot of the peak currents (Ipa & Ipc) as a function of the square root of the scan rate (υ1/2). A linear behavior was observed which corresponds to the regression eqn (i)–(iv). These results indicate a diffusion-controlled electron transfer process that leads to the spontaneous transfer of the electro-active species from the high to low concentration regions. The regression equations for the linear variations of Ipa and Ipc with υ1/2 for the Au/NiFe2O4@GO-Ch and anti βA-Au/NiFe2O4@GO-Ch/GCE are given as follows:
| | | (a) Ipa (Au/NiFe2O4@GO-Ch/GCE) = 2.29 × 10−5 × υ1/2 + 4.186 × 10−5, R2 = 0.999 | (i) |
| | | (b) Ipc (Au/NiFe2O4@GO-Ch/GCE) = −2.26 × 10−5 × υ1/2 − 3.784 × 10−5, R2 = 0.997 | (ii) |
| | | (c) Ipa (anti βA-Au/NiFe2O4@GO-Ch/GCE) = −1.38 × 10−5 × υ1/2 + 1.57 × 10−5, R2 = 0.9996 | (iii) |
| | | (d) Ipc (anti βA-Au/NiFe2O4@GO-Ch/GCE) = −1.83 × 10−5 × υ1/2 − 2.04 × 10−5, R2 = 0.9991 | (iv) |
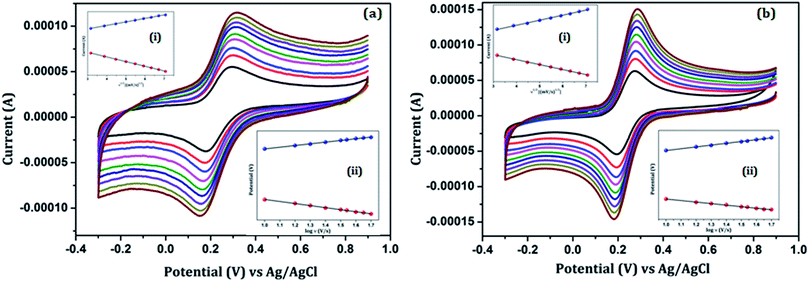 |
| | Fig. 5 Cyclic voltammetric variation of (a) Au/NiFe2O4@GO-Ch/GCE and (b) anti βA-Au/NiFe2O4@GO-Ch/GCE with a scan rate in 100 mM PBS (pH = 7.4) containing 5 mM [Fe(CN)6]3−/4−. The inset shows the variation of (i) current with the square root of the scan rate and (ii) potential with log of scan rate. | |
The diffusion coefficient of the modified electrode was estimated using the well-known Randal–Sevcik equation as follows:
| | | Ip = (2.69 × 105)α1/2n3/2ACD1/2υ1/2 | (v) |
where
Ip is the peak current,
α is the electron transfer coefficient,
υ is the scan rate in V s
−1 (20 mV s
−1),
A is the area,
C is the concentration of the redox species, and
n is the number of transferred electrons. The derived diffusion coefficients were calculated to be 2.26 × 10
−4 cm
2 s
−1 for the Au/NiFe
2O
4@GO-Ch/GCE and 7.8 × 10
−5 cm
2 s
−1 for the anti βA-Au/NiFe
2O
4@GO-Ch/GCE. Further, the surface coverage of the immunosensor was calculated by using the Brown–Anson model:
In the above equation, n is the number of electrons transferred, F is the Faraday constant, S is the concentration of the surface (mol cm−2), A is the surface area (0.3 cm2) of the electrode, υ is the scan rate (10 mV s−1), R is the gas constant and T is the absolute temperature. Taking the average of both the cathodic and anodic results the surface concentration of the antibody on the electrode is found to be 1.86 × 10−12 mol cm−2.
The electron charge transfer and the charge transfer rate could also be calculated from the CV technique using Laviron's equation:
| | Epa = Eo + (2.3RT/1 − α)nF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logυ logυ | (vii) |
| | | Epc = Eo + 2.3RT/αnFlogυ | (viii) |
| | | logks = αlog(1 − α) + (1 − α) logα − log(RTlnFυ) − α (1 − α)nFΔEp/2.3RT | (ix) |
where,
ks is the heterogeneous electron transfer rate constant,
α is the charge transfer coefficient,
υ is the scan rate,
R is the gas constant,
T is the absolute temperature,
F is the Faraday constant, Δ
Ep is the peak-to-peak separation and
Eo is the apparent formal potential. The
ks and
α value can be estimated from the intercept and slope of the linear plot of
Epversus log
υ, as shown in inset (ii) of
Fig. 5(a) and (b), and was found to be 0.936 s
−1 and 0.6 for the Au/NiFe
2O
4@GO-Ch/GCE and 0.85 s
−1 & 0.4 for the anti βA/Au/NiFe
2O
4@GO-Ch/GCE, respectively.
Based on the electroconductivity studies of different modified electrodes, it can be concluded that the as-synthesized Au/NiFe2O4@GO-Ch modified GCE gave favourable results regarding peak to peak separation, current response, and electron transfer kinetics. Thus, the Au/NiFe2O4@GO-Ch modified GCE is a suitable platform for effective electrochemical sensing of βA1–42.
To further confirm the successful immobilization of the antibodies on the surface of the electrode, DPV measurements were conducted in 100 mM PBS (pH = 7.4) containing 5 mM [Fe(CN)6]3−/4−. The changes in the GCE's amperometric responses with the modification process can be inferred from Fig. 4(b). The concluded results of DPV measurements conform to the outcomes of the CV analyses.
3.3 Electrode modification analysis using impedance measurements
EIS is an effective technique for probing the interfacial attributes of the electrode with surface modifications. The EIS measurements were performed on the bare GCE and the modified GCE. Fig. 4(c) shows the Nyquist plot of modified electrodes for EIS analysis. The semicircle portion of the Nyquist curve in the high frequency region is associated with the electron transfer process and its diameter reflects the charge transfer resistance, RCT, whereas the linear part in the low frequency region is related to the diffusion limited step of the electron transfer process. The EIS analyses showed the notable differences in the values of RCT amongst the variedly modified electrode surfaces and its comparison to the bare GCE endorses the surface modification. The charge transfer resistance (RCT) value of the bare GCE is 615.74 Ω which is decreased to 197.28 Ω after the deposition of NiFe2O4-Ch on the GCE, indicating that the conductive nature of NiFe2O4 facilitates the rate of charge transfer. The RCT value was increased to 314.36 Ω after modification of the NiFe2O4-Ch/GC electrode with GO. This can be attributed to its less conductive nature due to the presence of oxygenated functional groups. But, after functionalizing the NiFe2O4@GO-Ch electrode with AuNPs, the RCT value decreased dramatically to 121.78 Ω, representing the enhancement in the conductive properties of the nanocomposite. However, the immobilization of the antibodies on the surface of the Au/NiFe2O4@GO-Ch/GCE results in an increase in the value of RCT to 486.86 Ω, showing the hindrance in the electron transfer between the probe and the electrolyte solution.
3.4 Voltammetric determination of βA1–42
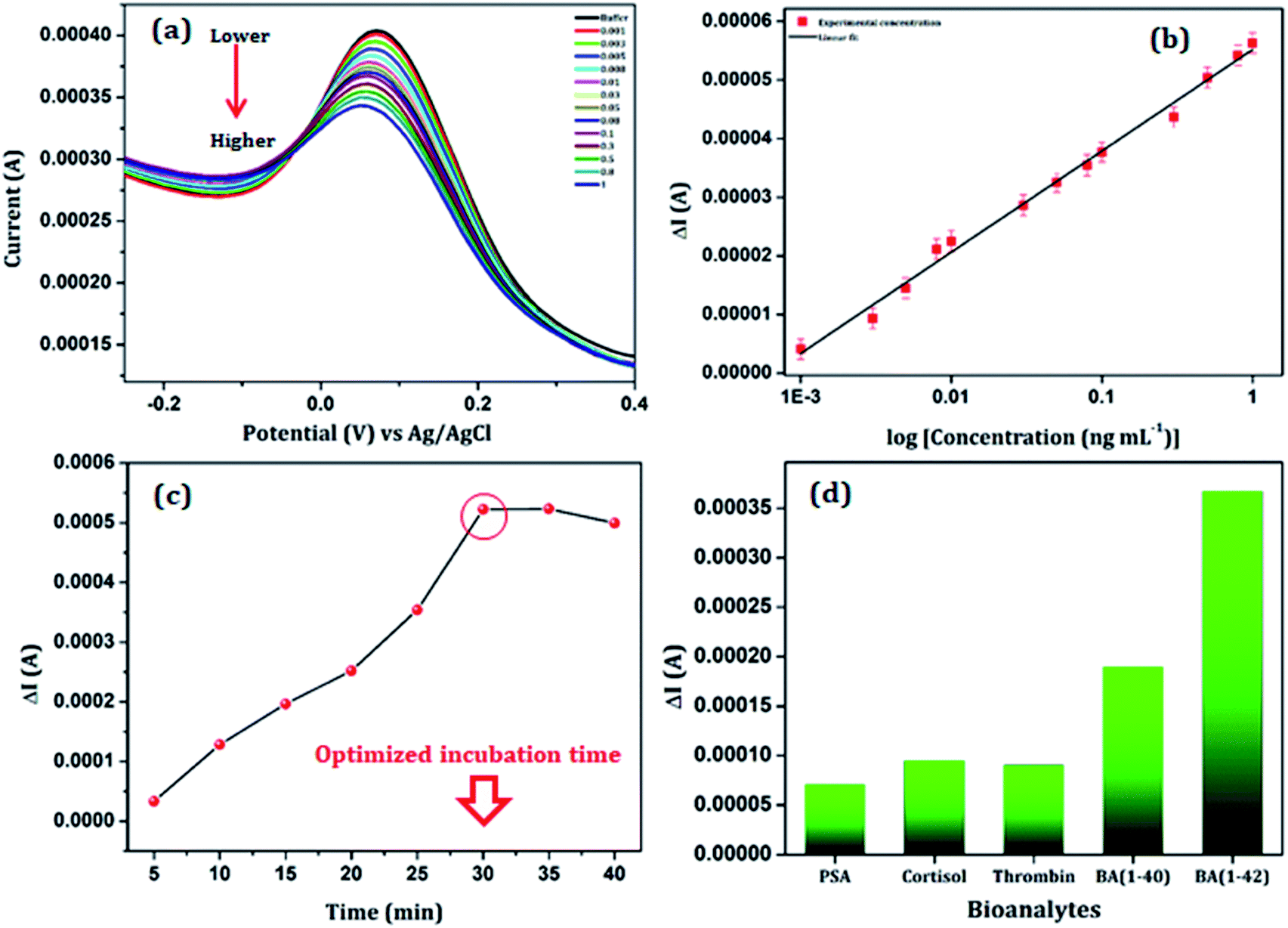
The sensing performance of the anti βA functionalized Au/NiFe2O4@GO-Ch/GCE was studied as a function of different βA1–42 concentrations varying from 1 pg mL−1 to 1 ng mL−1, using the DPV technique in 100 mM PBS (pH = 7.4), containing 5 mM [Fe(CN)6]3−/4− as redox moieties. The response current decreased linearly with the increase in the βA1–42 concentration (as shown in Fig. 6(a). The decrease in the current response can be related to the fact that there is a formation of an antigen–antibody immunocomplex at the surface of the working electrode. The as-formed immunocomplex hinders the transfer of electrons from the electrode to the solution and results in a decrease in the current response with increasing βA1–42 concentration. Fig. 6(b) shows the calibration curve of the DPV measurement based on the βA1–42 concentration. It showed a linear characteristic for the change in current versus the log of concentration in the range of 0.001–1.00 ng mL−1 with a R2 value of 0.99. The detection limit of the sensor was calculated to be 3 pg mL−1 by using the formula 3α/m, where α and m are the standard deviation and slope of the calibration curve, respectively. Furthermore, a comparative study has been undertaken with the reported literature for the detection of βA and is shown in Table 1. The survey highlighted a significant sensitivity and ultralow detection limit of the immunosensor. This could attribute to the outstanding performance of the Au/NiFe2O4@GO-Ch nanocomposite, which had an impressive impact on the selective detection of βA1–42.
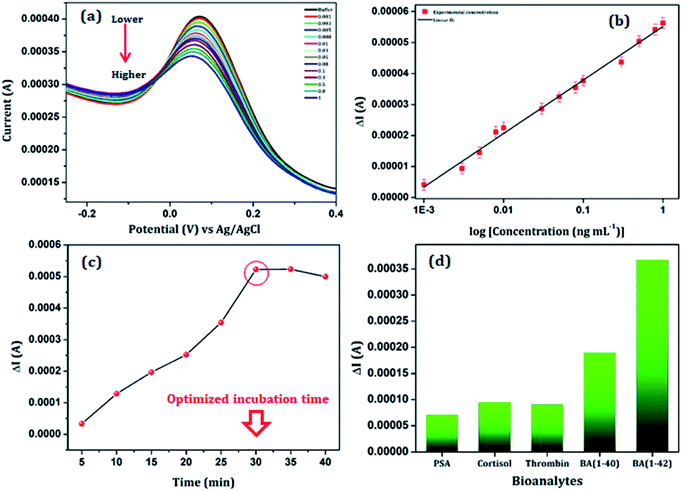 |
| | Fig. 6 (a) Amperometric current response of anti βA-Au/NiFe2O4@GO-Ch in the presence of varying concentration of βA1–42, (b) calibration curve showing linearity between change in current and βA1–42 concentration (detection range of 1 pg mL−1 to 1 ng mL−1), (c) effect of incubation time on detection of βA1–42 using an anti βA-Au/NiFe2O4@GO-Ch sensing platform, (d) comparison of current responses of the anti βA-Au/NiFe2O4@GO-Ch sensor towards βA1–42 and other biomolecules. | |
Table 1 Performance comparison of the immunosensor for βA detection
| Electrode |
Detection technique |
Mechanism |
Linear range |
LOD |
Ref. |
| SPCEs–NPAu |
Electrochemical |
Enzyme based |
0.5–500 ng mL−1 |
0.1 ng mL−1 |
25
|
| AuNPs@C/N-Ab(1−42) |
Colorimetric |
Colour change |
7.5–500 nM |
2.3 nM |
26
|
| Streptavidin-QDs |
Fluorescence |
Fluorescence intensity |
23–456.2 pg mL−1 |
7.6 pg mL−1 |
27
|
| AuNPs/MPA/Au electrode |
Electrochemical |
Block effect of electron transfer |
10−1000 pg mL−1 |
5.2 pg mL−1 |
28
|
| HRP–Au–gelsolin |
Electrochemical |
Enzyme based |
0.1–50 nM |
28 pM |
29
|
| Cu-BTC/Tb |
Fluorescence |
Intensity based |
1−550 nM |
0.3 nM |
30
|
| GNRs-Ru(bpy)32+ |
Electro-chemiluminescence |
Electro-chemiluminescence intensity |
1.0 × 10−5 – 100 ng mL−1 |
4.2 × 10−6 ng mL−1 |
31
|
| DTSP-SAM/IDE |
Electrochemical |
Block effect of electron transfer |
10 pM to 100 nM |
10 pM |
32
|
| SAM/ICE |
Electrochemical |
Block effect of electron transfer |
10−3 – 103 ng mL−1 |
100 pg mL−1 |
33
|
| Au/NiFe2O4@GO-Ch/GCE |
Electrochemical |
Block effect of electron transfer |
1 pg mL−1 to 1 ng mL−1 |
3 pg mL−1 |
This work |
3.5 Evaluation of the incubation time, selectivity and stability of the immunosensor
Optimization of the incubation time for the immobilization of βA1–42 on the surface of the anti βA-Au/NiFe2O4@GO-Ch/GCE is essential for a proper binding between the bio-recognition element and the targeted marker at a saturation level. Thus the optimized incubation time was investigated by placing the as-prepared immunosensor in 5 pg mL−1 of βA1–42 solution in 100 mM PBS (pH = 7.4) with 5 mM [Fe(CN)6]3−/4− for various time intervals starting from 5 min to 40 min. The variation in the current response (ΔI) with respect to the time required for the binding of anti βA with βA1–42 is shown in Fig. 6(c). A relatively high signal was produced after 30 min of incubation (at a temperature of 25 °C). However, there was a steady state change in current response after this, suggesting the accomplishment of a saturation level in forming the immunocomplex. Therefore, the incubation time for immunosensing was selected to be 30 min for the reported βA1–42 assay.
To study the selectivity of the electrode towards βA1–42, a series of bioanalytes (each 100 pg mL−1) were selected such as βA1-40, prostate specific antigens, cortisol and thrombin. The DPV measurements showed that the current variations (ΔI) in the presence of βA1–42 were considerably higher as compared to the other bioanalytes under the same experimental conditions and analyte concentrations [Fig. 6(d)]. The variation in the current response (ΔI) is found to be larger in the presence of βA1–42 than other interfering substances, implicating high selectivity of the immunosensor.
In addition, the long-term stability of the immunosensor was further examined. In this perspective, three working electrodes were fabricated freshly and placed in a refrigerator for 20 days at 4 °C. The results of intermittent measurement of immunosensors in every four days showed only a reasonably small decrement of the peak currents. They retained 90% of their initial response even after a storage interval of 20 days. Furthermore, three electrodes were fabricated in a paralleled fashion to detect 5 pg mL−1 βA1–42 and an RSD value of 3.5% was achieved. These results confirmed good stability and a satisfactory reproducibility of the immunosensor.
3.6 Sensing of βA1–42 in cerebrospinal fluid
For clinical utilization, it is important to highlight the analytical performance of a biosensor in real samples compared to the buffer without losing efficiency. In this regard, the developed method was utilized for determining βA1–42 in cerebrospinal fluid (CSF) by using the anti βA-Au/NiFe2O4@GO-Ch/GCE immunosensor. Furthermore, the functional estimation was done by examining the immunosensor to detect various known concentrations of βA1–42 in the CSF sample. Subsequently, DPV measurement was carried out by spiking five standard concentrations of βA1–42 into the CSF sample and the obtained data are summarized in Table 2. As shown in the table, the recoveries and RSDs for spiked samples were analyzed, and at all concentrations, the observed recovery was found to be greater than 96%. These results proved the feasibility and veracity of the anti βA-Au/NiFe2O4@GO-Ch/GCE immunosensor for βA1–42 detection.
Table 2 Determination of βA1–42 in cerebrospinal fluid based on the DPV technique
| Sl no. |
Added βA1–42 (pg mL−1) |
Detected βA1–42 (pg mL−1) |
Recovery (%) |
RSDa (%) |
|
Relative standard deviation (RSD) for recovery (n = 3).
|
| 1 |
8 |
7.7 |
96.4 |
3.4 |
| 2 |
50 |
50.2 |
100.5 |
3.6 |
| 3 |
100 |
99.4 |
99.4 |
4.2 |
| 4 |
500 |
500.4 |
100.07 |
4.8 |
4 Conclusions
In conclusion, an efficient and ultrasensitive electrochemical immunosensor was developed using Au and NiFe2O4 nanoparticle decorated GO nanosheets and was successfully applied for the sensitive and selective detection of βA1–42 in buffer and in a real sample (CSF). The Au/NiFe2O4@GO-Ch nanocomposite enhanced the electrochemical performance of the immunosensor. Further, the immunosensor exhibited favourable sensitivity towards βA1–42 with a wide linear range from 1 pg mL−1 to 1 ng mL−1 and an ultralow detection limit of 3.0 pg mL−1. It also displayed superior characteristics in terms of sensitivity, reproducibility, stability and selectivity. These advantages of the method provide a perspective for expanding the applications of Au/NiFe2O4@GO-Ch based nanocomposites in βA1–42 detection, which can be beneficial for early diagnosis and treatment of AD.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors express thanks to The Director (CSIR-NEIST) for his encouragement in this work. CSIR-Advanced Analytical Facility for NE (CAAF-NE), CSIR-NEIST, Jorhat is acknowledged for the SEM/EDS analysis. RD and HSD acknowledge the DBT, India for financial support through grant no. BT/PR16223/NER/95/494/2016. SG acknowledges SERB, DST, India for financial support (Grant no-PDF/2016/003142).
References
- S. Herculano-Houzel, Front. Hum. Neurosci., 2009, 3, 31 CrossRef PubMed.
- R. J. O'Brien and P. C. Wong, Annu. Rev. Neurosci., 2011, 34, 185–204 CrossRef PubMed.
- Y. Xing, X. Z. Feng, L. Zhang, J. Hou, G. C. Han and Z. Chen, Int. J. Nanomed., 2017, 12, 3171 CrossRef CAS PubMed.
-
L. F. Hernández-Zimbrón, E. Gorostieta-Salas, M. L. Díaz-Hung, R. Pérez-Garmendia, G. Gevorkian and H. Quiroz-Mercado, Update on Dementia, 2016 Search PubMed.
- L. Song, D. R. Lachno, D. Hanlon, A. Shepro, A. Jeromin, D. Gemani, J. A. Talbot, M. M. Racke, J. L. Dage and R. A. Dean, Alzheimer's Res. Ther., 2016, 8, 58 CrossRef PubMed.
- Y. Wang, D. Fan, G. Zhao, J. Feng, D. Wei, N. Zhang, W. Cao, B. Du and Q. Wei, Biosens. Bioelectron., 2018, 120, 1–7 CrossRef CAS PubMed.
- M. I. Aguilar and D. H. Small, Neurotoxic. Res., 2005, 7, 17–27 CrossRef CAS PubMed.
- J. Hatai, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2017, 139, 2136–2139 CrossRef CAS PubMed.
- I. H. Chou, M. Benford, H. T. Beier, G. L. Coté, M. Wang, N. Jing, J. Kameoka and T. A. Good, Nano Lett., 2008, 8, 1729–1735 CrossRef PubMed.
- I. Tiwari, M. Singh, C. M. Pandey and G. Sumana, RSC Adv., 2015, 5, 67115–67124 RSC.
- C. Cristea, M. Tertis and R. Galatus, Nanomaterials, 2017, 7, 119 CrossRef PubMed.
- Y. Jia, Y. Peng, J. Bai, X. Zhang, Y. Cui, B. Ning, J. Cui and Z. Gao, Sens. Actuators, B, 2018, 254, 629–635 CrossRef CAS.
- D. Quesada-González and A. Merkoçi, Chem. Soc. Rev., 2018, 47, 4697–4709 RSC.
- J. Singh, A. Roychoudhury, M. Srivastava, V. Chaudhary, R. Prasanna, D. W. Lee, S. H. Lee and B. D. Malhotra, J. Phys. Chem. C, 2013, 117, 8491–8502 CrossRef CAS.
- C. Sun, Y. Zou, D. Wang, Z. Geng, W. Xu, F. Liu and J. Cao, Sensors, 2018, 18, 1865 CrossRef PubMed.
- G. Bharath, A. Naldoni, K. H. Ramsait, A. Abdel-Wahab, R. Madhu, E. Alsharaeh and N. Ponpandian, J. Mater. Chem. A, 2016, 4, 6385–6394 RSC.
- S. Gogoi and N. Karak, RSC Adv., 2016, 6, 94815–94825 RSC.
- B. Paulchamy, G. Arthi and B. D. Lignesh, J. Nanomed. Nanotechnol., 2015, 6, 1 Search PubMed.
- R. Devi, S. Gogoi, S. Barua, H. S. Dutta, M. Bordoloi and R. Khan, Food Chem., 2019, 276, 350–357 CrossRef CAS PubMed.
- N. G. Mphuthi, A. S. Adekunle, O. E. Fayemi, L. O. Olasunkanmi and E. E. Ebenso, Sci. Rep., 2017, 7, 43181 CrossRef PubMed.
- Y. Wang, D. Fan, G. Zhao, J. Feng, D. Wei, N. Zhang, W. Cao, B. Du and Q. Wei, Biosens. Bioelectron., 2018, 120, 1–7 CrossRef CAS PubMed.
- Y. Zhou, H. Zhang, L. Liu, C. Li, Z. Chang, X. Zhu, B. Ye and M. Xu, Sci. Rep., 2016, 6, 35186 CrossRef CAS PubMed.
- A. A. Ensafi, F. Saeid, B. Rezaei and A. R. Allafchian, Anal. Methods, 2014, 6, 6885–6892 RSC.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS PubMed.
- E. C. Rama, M. B. González-García and A. Costa-García, Sens. Actuators, B, 2014, 201, 567–571 CrossRef CAS.
- T. Hu, S. Lu, C. Chen, J. Sun and X. Yang, Sens. Actuators, B, 2017, 243, 792–799 CrossRef CAS.
- M. Tang, J. Pi, Y. Long, N. Huang, Y. Cheng and H. Zheng, Spectrochim. Acta, Part A, 2018, 201, 82–87 CrossRef CAS PubMed.
- P. Carneiro, J. Loureiro, C. Delerue-Matos, S. Morais and M. do Carmo Pereira, Sens. Actuators, B, 2017, 239, 157–165 CrossRef CAS.
- Y. Yu, X. Sun, D. Tang, C. Li, L. Zhang, D. Nie, X. Yin and G. Shi, Biosens. Bioelectron., 2015, 68, 115–121 CrossRef CAS PubMed.
- B. Liu, H. Shen, Y. Hao, X. Zhu, S. Li, Y. Huang, Q. Peng and M. Xu, Anal. Chem., 2018, 90, 12449–12455 CrossRef CAS PubMed.
- H. Ke, H. Sha, Y. Wang, W. Guo, X. Zhang, Z. Wang, C. Huang and N. Jia, Biosens. Bioelectron., 2018, 100, 266–273 CrossRef CAS PubMed.
- A. Kaushik, P. Shah, P. K. Vabbina, R. D. Jayant, S. Tiwari, A. Vashist, A. Yndart and M. Nair, Anal. Methods, 2016, 8, 6115–6120 RSC.
- H. T. N. Le, J. Park, S. R. Chinnadayyala and S. Cho, Biosens. Bioelectron., 2019, 144, 111694 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00578a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Satyabrat
Gogoi
ab,
Satyabrat
Gogoi