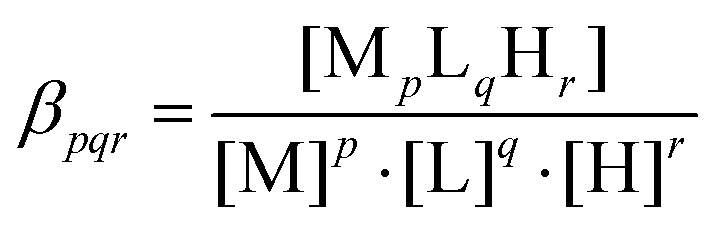DOI:
10.1039/D0MT00103A
(Paper)
Metallomics, 2020,
12, 1220-1229
Both metal-chelating and free radical-scavenging synthetic pentapeptides as efficient inhibitors of reactive oxygen species generation†
Received
16th April 2020
, Accepted 28th May 2020
First published on 2nd June 2020
Abstract
Reactive oxygen species (ROS) are major sources of oxidative stress playing prominent roles in the development of several pathologies including cardiovascular and neurodegenerative diseases or cancers. The presence of transition biometal ions, specifically copper and iron, induces ROS formation by catalyzing the reduction of molecular oxygen to superoxide anion (O2˙−), hydrogen peroxide (H2O2) and hydroxyl (HO˙) radical. To limit ROS production and their detrimental effects, we report on the synthesis, physicochemical studies and antioxidant assays of an innovative series of synthetic pentapeptides exhibiting a dual direct/indirect mode of action, both as iron(III)-chelators and as radical scavengers. These combined effects lead to a drastic reduction of in vitro reactive oxygen species production up to 95% for the more reactive hydroxyl radical.
Significance to metallomics
We report herein on a new series of peptides exhibiting iron(III)-chelation abilities from acidic to neutral pH corresponding to conditions in which redox-active iron(III) is found in vivo, such as in both the gastrointestinal tract and the lysosomal iron labile pool, and in the blood plasma, respectively. Thanks to their chelating properties and their inherent design, this series of peptides act as effective antioxidants based on a dual indirect (i.e., inhibition of the metal redox activity) and direct (i.e., hydroxyl radical scavenging) mode of action, drastically limiting the production of reactive oxygen species up to −99% for the hydroxyl radical.
|
Introduction
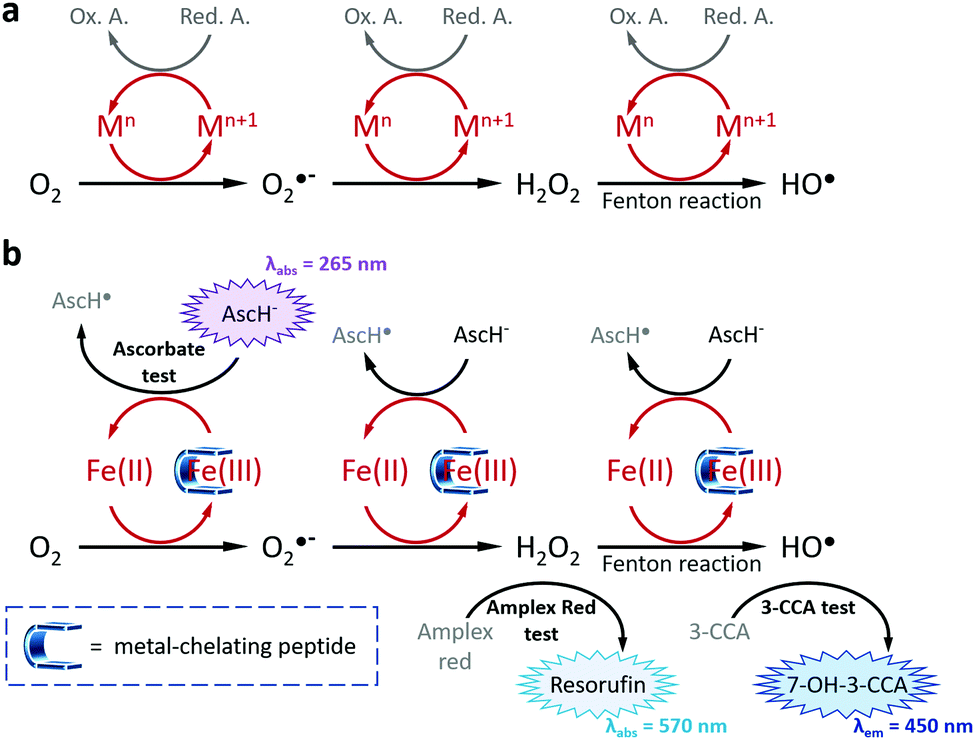
Metal ions are naturally occurring in living organisms and play pivotal roles in a wide variety of physiological processes including photosynthesis, respiration, metabolism, transmission of nervous influx or protection against pathogenic agents.1 Among the transition metal ions having in vivo roles, copper and iron are associated with several biological processes and the misregulation of their concentration, homeostasis or metabolism are at the origin of pathologies (e.g., Wilson's and Menkes diseases, anaemia, cancers).2,3 In particular, iron and copper involve the formation of reactive oxygen species (ROS) through the catalytic reduction of molecular oxygen (O2) to hydrogen peroxide (H2O2), superoxide anion (O2˙−) and hydroxyl (HO˙, via the Fenton reaction) radicals (Fig. 1a).4,5 Thanks to a precisely controlled concentration in healthy cells, ROS contribute to complex signalling pathways including metabolism, immune system regulation and proliferation.6,7 However, under endogenous and exogenous stresses (e.g., pollution, UV-irradiation, tobacco, diet) this homeostasis is dysregulated, and the subsequent overproduction of ROS induces deleterious effects to cells. This so-called oxidative stress has been reported as a source of several pathologies in humans, including respiratory, neurodegenerative, cardiovascular and digestive diseases, and even cancers.8,9 To prevent or forestall oxidative stress and its damaging effects, the intake of exogenous antioxidant supplementation has proved its effectiveness on ROS regulation.10 Among the most representative antioxidants, a large majority are natural compounds,10,11 including ascorbic acid, carotenoids, phenolic derivatives and, more recently, peptides. Bioactive peptides have been attracting growing interests in pharmaceutical, therapeutical and nutraceutical industries and researches thanks to their inherent advantages in terms of diversity, modularity, selectivity, efficiency, safety and tolerability.12,13 Thus, several antioxidant peptides have been reported, mainly identified a posteriori via LC/MS/MS analyses from hydrolysates of natural resources14,15 (e.g., milk, vegetables, cereals,16,17 algae, animals18) exhibiting antioxidant activities. However, this usual approach remains challenging and suffers from several drawbacks:14,19 time-consuming, expensive, hazardous and uncertain in terms of quantity and recovery, drastically limiting the discovery of effective antioxidant peptides. To tackle these issues, we assume that a more rational strategy is required; thus, we report herein on the design, synthesis, physicochemical studies and antioxidant assays of a new series of pentapeptides exhibiting drastic inhibition of ROS production thanks to a dual direct/indirect mode of action, i.e., both Fe(III)-chelating and radical scavenging properties.
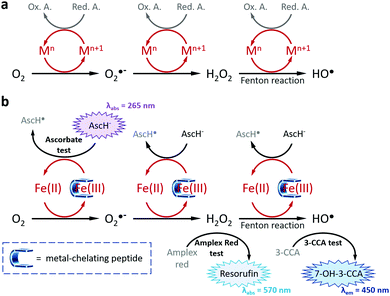 |
| | Fig. 1 (a) Schematic representation of transition metal-catalysed ROS formations (M = Fe, Cu; Red. A. = Reducing Agent, Ox. A. = Oxidized Agent) and (b) of the strategy used in this work to inhibit ROS formations via, inter alia, the chelation of Fe(III). The three tests allowing the measure of the antioxidant properties of the metal-chelating peptides are depicted with their corresponding absorbance or emission wavelengths. | |
Results and discussion
Design and synthesis of antioxidant peptides
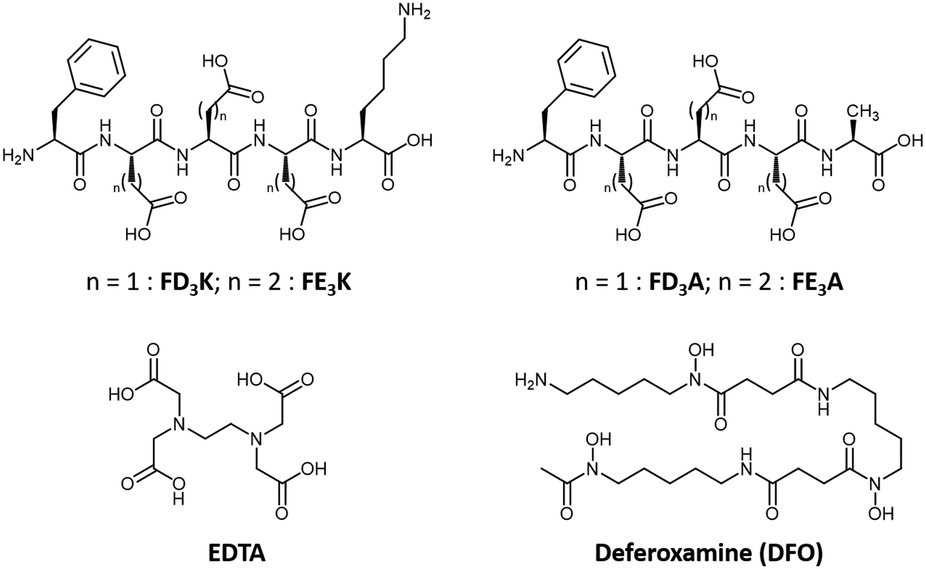
The indirect mode of action is based on metal chelation, an approach that has demonstrated its effectiveness on reduction of ROS production and on ROS-mediated pathologies.10,20–22 Among the most studied aforementioned metal-chelating antioxidants, we can mention polyphenols23 including flavonoids24,25 (e.g., quercetin,26 epigallocatechin-3-gallate20,27), resveratrol28 or curcumin,29 capsaicin,30 quinolines (e.g., clioquinol,21 8-hydroxyquinoline derivatives20,31), and the siderophores deferiprone32 and deferoxamine (this latter will be further discussed below). While metal-chelating peptides have been widely studied in the last decades for their copper-binding properties,33,34 Fe(III) has hardly been considered, with only few examples of peptides35 and peptide derivatives functionalized with well-known iron-chelating chemical moieties (e.g., EDTA, hydroxamate).36–38 Besides meeting this challenge, we selected Fe(III) as a target metal ion instead of Fe(II), because of its: 1/unusual +III oxidation state (only shared with Al3+, Cr3+ and Co3+ ions in a biological context),39 2/stability in aerobic conditions, 3/specific coordination properties (i.e., while Cu(I), Cu(II) and Fe(II) are classified as soft acid and borderline, respectively, Fe(III) is a hard acid).40 Based on the Pearson's HSAB theory, we developed a series of four peptides comprised of acidic amino acid (i.e., aspartates (D), or glutamates (E)) triads acting as hard Lewis bases (Fig. 2). In C-terminal position, two amino acids were considered: alanine (A, uncharged, hydrophobic) and lysine (K, bringing an additional positive charge to improve water-solubility). The direct antioxidant mode of action, i.e., radical scavenging, originates from the phenylalanine (F) introduced at the N-terminal position. Already reported as a biomarker of oxidative stress,41,42 the sensitivity of phenylalanine to ROS is harnessed herein to entrap the hydroxyl radicals produced at the contiguous catalytic metal centre during the Fenton reaction. Thus, peptides have been synthesized by solid-phase peptide synthesis using a Fmoc/tBu strategy, purified by RP-HPLC and fully characterized by HPLC, ESI-HRMS and 1H, 13C and 2D NMR (see ESI† for details).
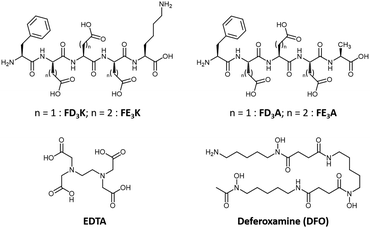 |
| | Fig. 2 Chemical structures of the four peptides studied (top), the negative control EDTA (bottom left) and the positive control DFO (bottom right). | |
Evaluation of the iron(III)-chelation properties
Potentiometric studies were carried out on the synthesised compounds, in the presence and absence of Fe(III). Such titrations were essential to both access the species distributions as a function of pH and to obtain characteristic dissociation constants of the peptides and stability constants of iron(III) complexes. The deprotonation constants of the ligands are listed in Table 1. These ligands have five or six protonable groups: four carboxyl groups and one or two amino groups. Only the deprotonations of the three side chain carboxyl functions were measurable in the studied pH range (pH 2–11), since the terminal COOH probably has a pK < 2. The COOH deprotonation processes are overlapping, thus their pK values cannot be assigned to the distinct donor groups. The basicity of the terminal amino groups corresponds well to the values measured for other ligands.43,44 Potentiometric titration, CD and 1H/13C NMR measurements were carried out in order to obtain information on the Fe(III)-pentapeptide complexes. Stability constants and pK values can be found in Table 2. In the case of FE3A and FD3A precipitation occurred at a low pH (<5), therefore the constants have a greater error.
Table 1 Overall protonation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β HiL) and deprotonation constants (pK) of the four studied peptides (T = 298 K, I = 0.1 M NaNO3 (standard deviations are in parentheses))
β HiL) and deprotonation constants (pK) of the four studied peptides (T = 298 K, I = 0.1 M NaNO3 (standard deviations are in parentheses))
| logβ |
FD3K |
FE3K |
FD3A |
FE3A |
| HL |
10.98 (3) |
10.82 (3) |
7.77 (1) |
7.66 (2) |
| H2L |
18.62 (3) |
18.32 (3) |
12.55 (3) |
12.73 (6) |
| H3L |
23.25 (4) |
23.18 (5) |
16.47 (5) |
16.92 (7) |
| H4L |
26.99 (5) |
27.27 (5) |
20.11 (5) |
20.97 (9) |
| H5L |
30.70 (5) |
31.10 (6) |
— |
— |
| pK |
|
|
|
|
| COOH |
3.71 (5) |
3.83 (6) |
3.64 (5) |
4.05 (9) |
| COOH |
3.74 (5) |
4.09 (5) |
3.92 (5) |
4.19 (7) |
| COOH |
4.63 (4) |
4.86 (5) |
4.78 (3) |
5.07 (6) |
| NH3+ (Nterm) |
7.64 (3) |
7.50 (3) |
7.77 (1) |
7.66 (2) |
| ε-NH3+ (Lys) |
10.98 (3) |
10.82 (3) |
— |
— |
Table 2 Stability constants (logβ) and derived data (pK) for Fe(III) complexes of the four studied peptides (T = 298 K, I = 0.1 M NaNO3 (standard deviations are in parentheses))
|
|
FD3K |
FE3K |
Peptide protonation state (in bold coordinated to Fe(III)) |
Global charge |
FD3A |
FE3A |
Peptide protonation state (in bold coordinated to Fe(III)) |
| logβ |
logβ |
| FeH3L |
29.61 (2) |
30.02 (3) |
3 COO
−, ε-NH3+ (Lys), NH3+ (Nterm) |
+2 |
— |
— |
|
| FeH2L |
26.25 (3) |
26.25 (5) |
4 COO
−, ε-NH3+ (Lys), NH3+ (Nterm) |
+1 |
17.61 (13) |
— |
3 COO
−, NH3+ (Nterm) |
| FeHL |
22.17 (3) |
21.87 (5) |
4 COO
−
, OH
−, ε-NH3+ (Lys), NH3+ (Nterm) |
0 |
14.53 (7) |
13.82 (11) |
4 COO
−, NH3+ (Nterm) |
| FeL |
17.08 (3) |
16.88 (4) |
4 COO
−
, 2 OH
−, ε-NH3+ (Lys), NH3+ (Nterm) |
−1 |
10.19 (11) |
9.23 (19) |
4 COO
−
, OH
−, NH3+ (Nterm) |
| FeH−1L |
9.54 (5) |
— |
4 COO
−, 2 OH−, ε-NH3+ (Lys), NH2 (Nterm) |
−2 |
4.72 (13) |
4.23 (18) |
4 COO
−
, 2 OH
−, NH3+ (Nterm) |
| pK |
FD3K |
FE3K |
Deprotonated group |
FD3A |
FE3A |
Deprotonated group |
| pK (FeH3L/FeH2L) |
3.36 |
3.77 |
COOH |
— |
— |
|
| pK (FeH2L/FeHL) |
4.08 |
4.38 |
H2O |
3.08 |
— |
COOH |
| pK (FeHL/FeL) |
5.09 |
4.99 |
H2O |
4.34 |
4.59 |
H2O |
| pK (FeL/FeH−1L) |
7.54 |
— |
NH3+ (Nterm) |
5.47 |
5.00 |
H2O |
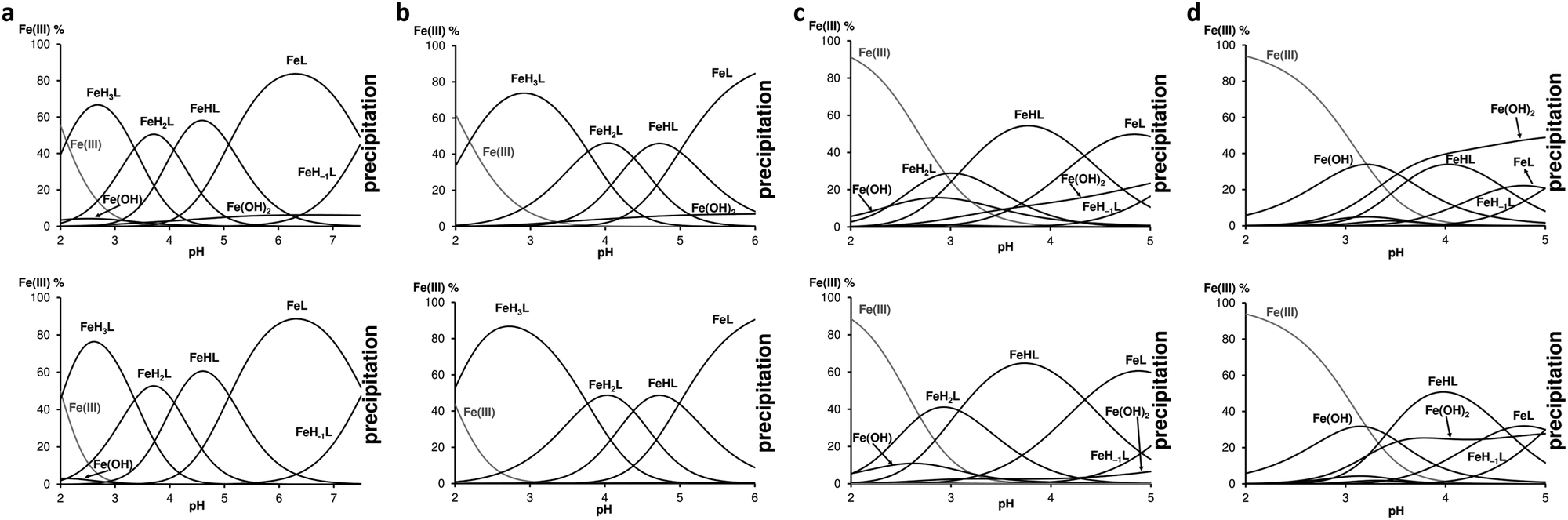
The chelation of Fe(III) by three of the four peptides (except for FE3A) started at pH 2 and only monocomplexes were formed. For FE3A the complexation started at higher pH, near pH 3. The species distribution curves of the Fe(III):ligand systems in 1:1 and 1:2 ratio are depicted in Fig. 3 and allow to observe significant differences between the peptides in terms of chelation ability: while FD3K is effective up to pH 7.6, FE3K releases iron beyond pH 6, FD3A and FE3A do the same from pH 5. Analysis by 1H NMR measurements of the solution after complete precipitation showed that only the free peptides were kept in solution. Based on the spectroscopic information discussed later, the metal ion is coordinated by 3 COO− groups in FeH3L species (lysine-containing peptides) or in FeH2L (FD3A) (Table 2), and the next deprotonation step can be attributed to the fourth COOH deprotonation and its coordination to Fe(III). This statement is also supported by the downshifted values of the carbonyl deprotonation constants. Indeed, for the sake of example, pKFD3K(H3L/H2L) = 4.63 corresponding to the deprotonation of the fourth carboxylic acid in the free FD3K peptide is downshifted to pK(FeH3L/FeH2L) = 3.36 in the presence of one equivalent of Fe(III), indicating a metal chelation via the carboxylate groups (i.e., aspartate or glutamate side chains and C-terminal carboxylate). To explain the further steps above pH 3, consecutive deprotonations of coordinated water molecules is suggested to form firstly {4COO−, OH−} coordination environment in FeHL species in the case of peptides containing lysine, and in FeL species in the case of alanine-containing ligands (Table 2). The deprotonation of the second coordinated water gave a {4COO−, 2OH−} coordination sphere around the iron(III) ion with the ligands FD3K and FE3K, but led to the iron precipitation with FD3A and FE3A. Interestingly, the primary amines from both the lysine and the N-terminal are not involved in the chelation, evidenced by similar dissociation constant both in the presence or in the absence of Fe(III) (pK = 7.64 and 7.54 for FD3K, respectively). Moreover, iron precipitation started with deprotonation of NH3+ (N-term) in the case of lysine-containing FD3K peptide.
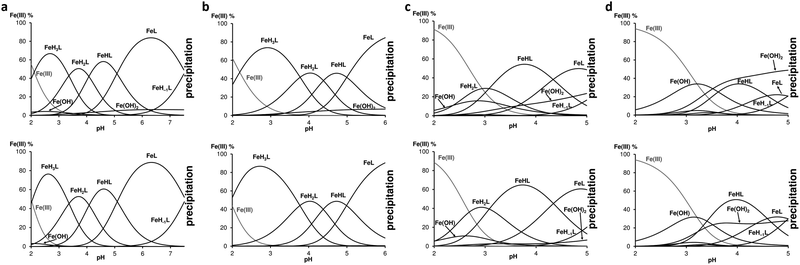 |
| | Fig. 3 Speciation diagrams of (a) Fe(III)-FD3K 1:1 (top) and 1:2 (bottom) systems, (b) Fe(III)-FE3K 1:1 (top) and 1:2 (bottom) systems, (c) Fe(III)-FD3A 1:1 (top) and 1:2 (bottom) systems and (d) Fe(III)-FE3A 1:1 (top) and 1:2 (bottom) systems. For all the experiments [peptide]tot = 2 mM, T = 298 K, I = 0.1 M NaNO3. | |
As a standard for measuring and comparing the effectiveness of potential chelating agents for a metal ion, the concentration of the uncomplexed aqueous ion in solution can be calculated under specific conditions. Noted as pM = −log[Mfree], this value provides more relevant information than logKML since it takes into account the ligand basicity, the pH, the ligand:metal ratio and the metal ion hydrolysis. The pM values are calculated for FD3K, FE3K, EDTA and DFO at 100 μM concentration with 1:1 ligand-to-Fe(III) ratio at pH 6.0 and 7.4, and tabulated in Table 3. The higher the pM value, the greater the affinity of the ligand to the iron(III) under these specific conditions used for the measurement of antioxidant activity (vide infra). The data show that the pentapeptides are not as strong complexing agents than EDTA46 or DFO45 (Fig. 2) because they leave more free Fe(III) in solution regardless of the pH. Precipitation as iron(III) hydroxide is produced if the pM value is lower than the limit set by the Ksp of Fe(OH)3.
Table 3 Free iron(III) concentrations at equilibrium (M) and pM (= −log[Mfree]) values calculated from ligand protonation and complex stability constants for 100 μM ligands and Fe(III) at pH 6.0 and 7.4 at T = 298 K, I = 0.1 M NaNO3
|
|
Free iron(III) concentration (M) |
pM value |
| pH 6 |
pH 7.4 |
pH 6 |
pH 7.4 |
| DFO45 |
1.22 × 10−16 |
6.12 × 10−20 |
15.91 |
19.21 |
| EDTA46 |
5.84 × 10−16 |
3.00 × 10−18 |
15.23 |
17.52 |
| FD3K |
9.67 × 10−11 |
1.52 × 10−13 |
10.01 |
12.82 |
| FE3K |
9.06 × 10−11 |
— |
10.04 |
— |
| Fe(III) alone47 |
3.16 × 10−15 |
2.00 × 10−19 |
14.50 |
18.70 |
Extrapolating these potentiometric data to a in vivo context, we can first assume that such pentapeptides will be able to chelate iron(III) ions in acidic and slightly acidic biological media. For instance, they could chelate dietary non-heme iron in the gastrointestinal tract (pH from 2 to 8), mainly in the duodenum and the jejunum where iron is present in its ferric form, and subsequently influence iron absorption.48,49 The pentapeptides could also interact with iron(III) ions from the labile iron pool, a pool of chelatable and redox-active iron playing pivotal roles in the formation of ROS, and in particular from the lysosomal labile pool in which iron is in its free iron(III) form and in an acidic (pH ≈ 4.5–5) environment.50,51 In parallel, potentiometric measurements also revealed that only one peptide, namely FD3K, forms a stable complex at pH 7.4, corresponding to the pH of the blood plasma (also referred as “physiological pH”). In the plasma of healthy individuals iron is sequestrated by circulating transferrin forming highly stable complexes (logK ≈ 23)52 which drastically limit the possibilities of chelation by FD3K. However, in the case of long-term transfusions for patients with anemia caused by several genetic disorders (e.g., thalassemia, sickle cell disease), a systemic iron overload is observed, surpassing the total iron binding capacity (i.e., saturating transferrin).53,54 In these conditions, labile plasma iron is circulating and a chelator, such as our pentapeptide FD3K could entrap the residual labile iron and limit its detrimental effects.
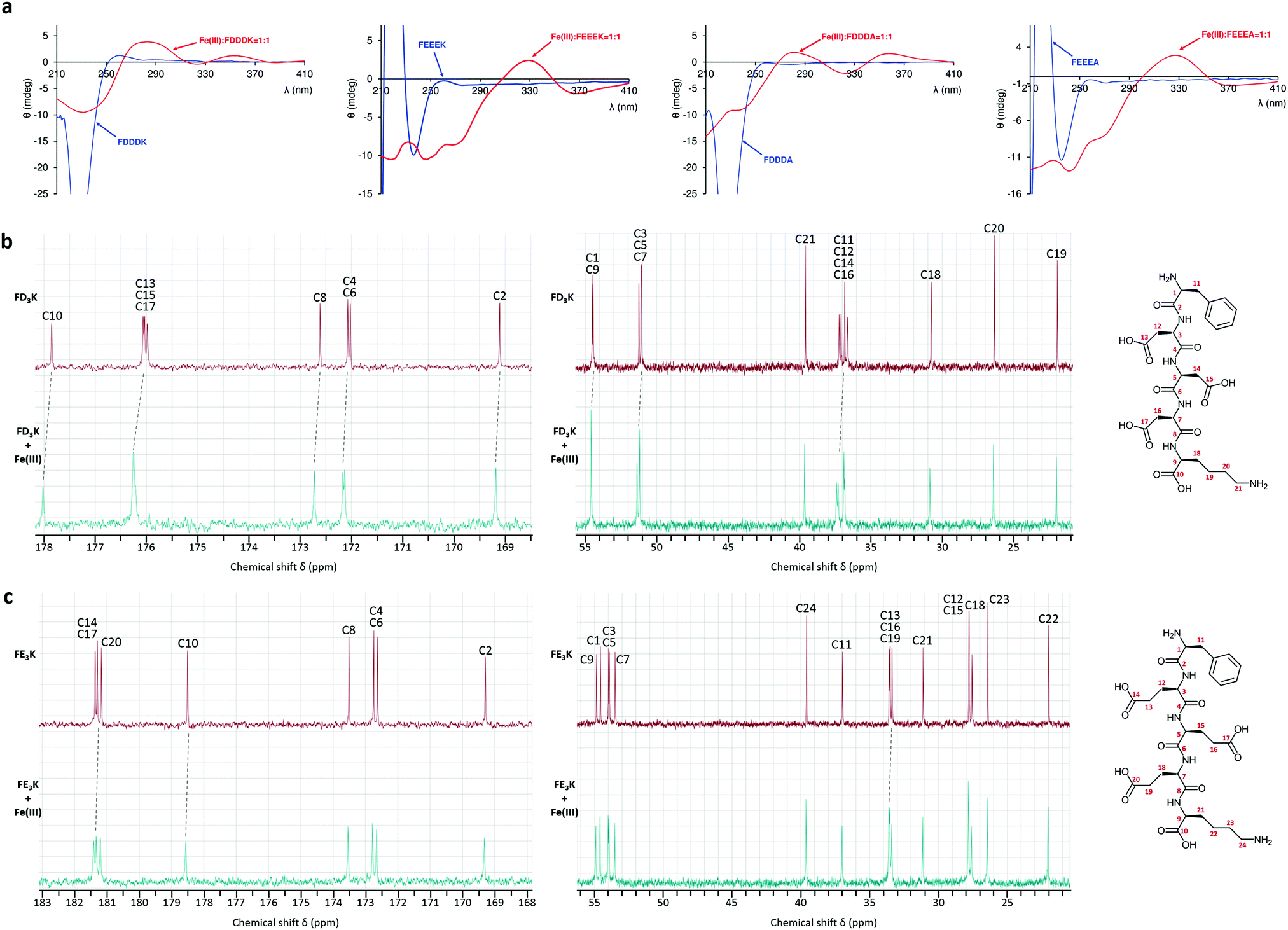
To gain further insights into the chelation behaviors of the four pentapeptides, circular dichroism and paramagnetic 1H and 13C NMR measurements were carried out. The experiments endorsed the potentiometric results and confirmed the chelation of the peptides through their carboxylate functions. The CD spectra were recorded at different pH with the ligands alone and also in the presence of iron(III) ion in ratio M:L = 1:1 (Fig. 4a and Fig. S1, S2, ESI†). The free FD3K ligand had a positive or a negative band with variable intensity below 250 nm in function of pH (Fig. S1, ESI†). Starting from acidic pH, with subsequent deprotonation of the side chain carboxyl groups, the negative CD band changed sign indicating conformational changes in the structure. When all the carboxyl groups are deprotonated, the CD band changed sign again following the deprotonation of terminal amino group (Fig. S1, right, ESI†). When 1 eq. iron(III) was added to the solution, the CD spectrum was already different at pH 2, the Cotton effect appeared above 250 nm (Fig. 4a and Fig. S2, left, ESI†), especially the appearance of a charge transfer band at 290 nm or at 330 nm due to the interaction between iron(III) and the carboxylate groups from aspartic acid containing peptides or glutamic acid containing ligands, respectively (Fig. 4a).55 With increasing pH, the CD spectra indicated considerable changes compared to the free peptide, the intensity of the charge transfer band diminished with the deprotonation of coordinated water molecules (Fig. S2, right, ESI†). In order to confirm the iron(III) ion coordination environment, paramagnetic NMR experiments were performed in the presence of a high excess of FD3K and FE3K peptides. Indeed, the presence of unpaired electron(s) of metal ion gives rise to large shifts and/or broadening effects on the NMR resonances of nuclei in close proximity. In our case, in the presence of weak donor atoms like carboxylates, the iron(III) ion is in high spin state (S = 5/2), which has a relatively long electron relaxation time (τs of the order of 10−9–10−11 s) because the electron relaxation mechanisms are relatively inefficient.56 The spectral resolution is also affected by the exchange rate between the free ligand and the ligand bound to the paramagnetic metal ion. Thus, due to the labile character of iron(III), at low M:L ratio (1:6) the 1H NMR signals were too broad to be attributable (Fig. S5 and S6, ESI†). The line broadening effects are less significant for 13C nucleus, which makes NMR detection more advantageous for the identification of residue near the paramagnetic centre.57 Thus, on the 13C spectra (Fig. 4b and c) the most affected signals were assigned to the carbon atoms of four carboxylate groups (C10, C13, C15 and C17 for FD3K; C10, C14, C17 and C20 for FE3K). Signals of α-carbon relative to carboxylate seemed to be also affected (C9, C12, C14, C16 for FD3K; C13, C16, C19 for FE3K). No carbon atom signals from the N-terminal phenylalanine or from the side chain of lysine were broadened confirming that no NH2 group are present in the iron(III) coordination environment.
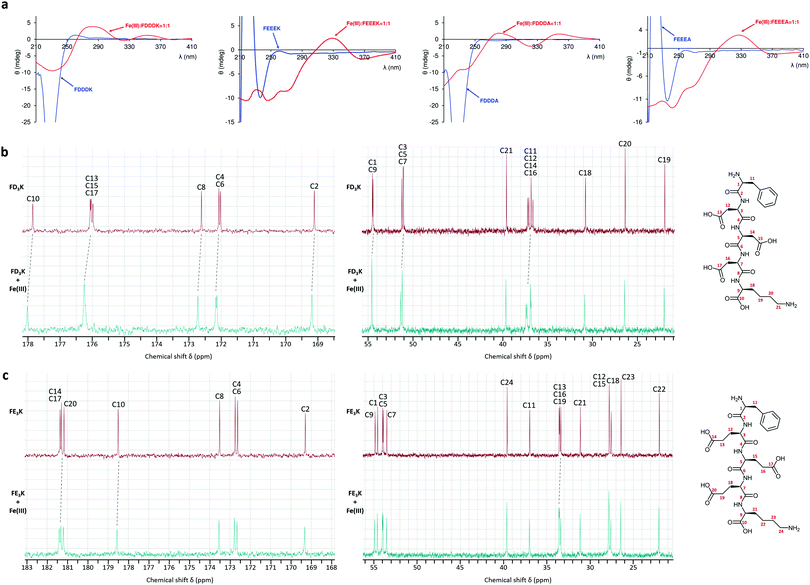 |
| | Fig. 4 (a) Circular dichroism spectra of the free ligand (blue lines) and iron(III) complexes (red lines) in ratio M:L = 1:1 at pH 3. 13C NMR spectra of (b) FD3K and (c) FE3K alone (in red) and in the presence of Fe(III) (in cyan) in ratio M:L = 1:6 and 1:5, respectively. | |
Antioxidant properties
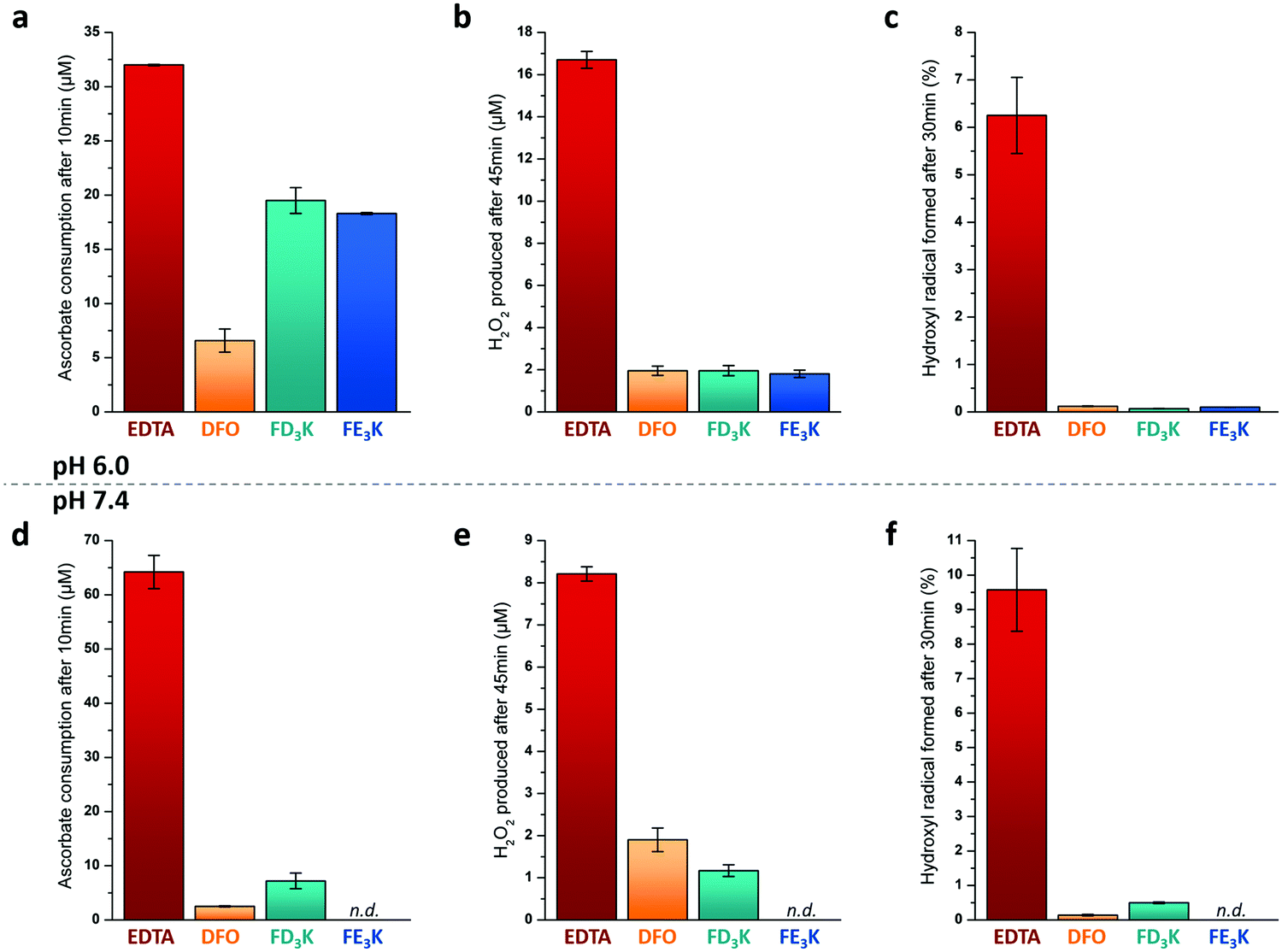
To assess the antioxidant properties of the two more efficient Fe(III) chelators FD3K and FE3K, a global approach considering several aspects of the antioxidant process, namely the metal redox activity and the formation of different types of ROS (Fig. 1b), have been used. For comparison purpose, we selected two reference iron(III)-chelating ligands ensuring iron solubility in a wide range of pH and offering opposite antioxidant properties. EDTA (for ethylenediaminetetraacetic acid, Fig. 2) has been chosen as a negative control based on its weak antioxidant capabilities well-documented since the 1980's,58–61 while deferoxamine (also termed DFO, Fig. 2), an approved drug used for half a century to treat iron overload and reported as an efficient in vitro59–61 and in vivo antioxidant,62 has been considered as our positive control. Thus, antioxidant experiments were firstly carried out at pH 6.0 to ensure the stability of the two Fe(III)-pentapeptide complexes (vide supra). To evaluate the reducibility of Fe(III) to Fe(II), we performed the ascorbate test63,64 in which the consumption of ascorbate (AscH−, playing the role of reducing agent, Fig. 1), is monitored as a function of time by UV-vis spectroscopy (λabs = 265 nm). While no variation was observed in the absence of metal ion after 10 min (Table S1 and Fig. S7, ESI†), consumption of AscH− occurred in the presence of Fe(III) with a maximum observed for the negative control EDTA with 32 μM AscH− consumed (Fig. 5a). The use of FD3K or FE3K induces a decrease of this value to 19.5 μM and 18.3 μM, corresponding to a reduction of 39% and 43%, respectively, compared to EDTA. These data highlight the ability of the two pentapeptides to inhibit the redox activity of Fe(III), even if less efficient than the positive control DFO (6.6 μM AscH− consumed, 79% reduction relative to EDTA). To evaluate the impact of this limited Fe(III) redox activity on the production of ROS, two different assays were considered for two different ROS species (i.e., H2O2 and HO˙, Fig. 1b). Firstly, the production of H2O2 was monitored by UV-vis spectroscopy due to its ability to oxidize Amplex Red to the coloured Resorufin (λabs = 570 nm) in the presence of horseradish peroxidase (HRP).65,66 As depicted in Fig. 5b, the presence of FD3K and FE3K induces a strong decrease of H2O2 formation by 88% and 89%, corresponding to 2.0 μM and 1.8 μM H2O2, respectively, compared to the negative control EDTA (16.7 μM H2O2 formed). Interestingly, both pentapeptides exhibit similar high efficiencies than the positive control DFO (1.9 μM H2O2 produced). Secondly, the formation of the more reactive hydroxyl radical was studied using the 3-CCA (for coumarin-3-carboxylic acid) assay in which 3-CCA reacts with HO˙ to generate the fluorescent 7-OH–3-CCA (λex = 395 nm, λem = 450 nm) followed spectrofluorimetrically.67 Once again, the two peptides strongly inhibit the HO˙ production with a decrease of 98% (FD3K) and 99% (FE3K), similar to DFO (Fig. 5c). Altogether, these data confirm the high antioxidant efficiency of FD3K and FE3K thanks to their inhibition of both Fe(III) redox activity and ROS productions. However, these peptides have only a moderate binding ability as previously discussed by comparing pM values relative to EDTA and DFO (Table 3). Interestingly, EDTA has a very good complexation capacity but is not able to protect iron(III) from reduction. These known properties of the EDTA–Fe(III) complex can have different origins such as its unusual hepta-coordinated structure68 and the N/O-type donor atoms, which are less suitable to hard iron(III) ion than the purely O-donor atoms as in DFO, or in the FD3K and FE3K peptides. These observations highlight the importance of both the binding groups and the geometry around the metal ion, which strongly impact the antioxidant properties. Concerning FD3K and FE3K peptides, the nature of the acidic amino acid (i.e., aspartate or glutamate) does not significantly affect the global antioxidant properties; however, the Fe(III)–FD3K complex has a better stability in a broader range of pH (vide supra, Fig. 3a and b), offering better opportunities for biomedical applications. Thus, antioxidant studies were carried out on this peptide at physiological pH (≈ pH 7.4). The ascorbate test (Fig. 5d) reveals a strong inhibition of the redox activity when Fe(III) is chelated by FD3K compared to EDTA with a decrease of 89% of the AscH− consumption. The reduction of H2O2 and HO˙ productions by 86% and 95% (Fig. 5e and f), respectively, is also highly significant, close to the DFO efficiency (−76% for H2O2, −98% for HO˙). Thus, FD3K has confirmed its antioxidant properties both at pH 6.0 and at physiological pH through its ability to chelate Fe(III), reducing the metal redox activity and, in fine, the ROS formation.
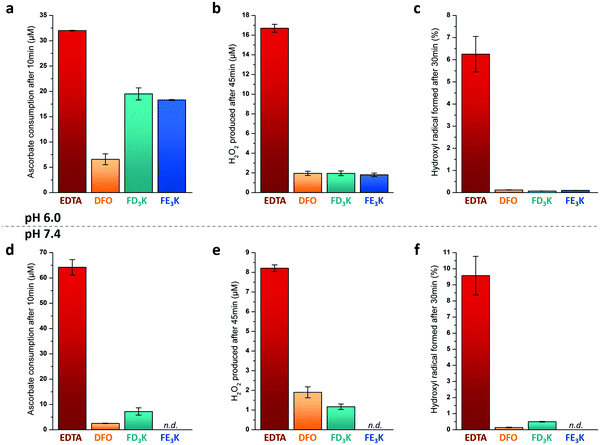 |
| | Fig. 5 (a and d) Consumed AscH− concentrations after 10 min reaction at pH 6.0 (a) or pH 7.4 (d) (50 mM HEPES buffer). [AscH−]0 = [Ligand]0 = 100 μM and [Fe(III)]0 = 100 μM. (b and e) Produced H2O2 concentrations after 45 min reaction at pH 6.0 (b) or pH 7.4 (e) in water. [AscH−]0 = 200 μM, [Ligand]0 = 50 μM and [Fe(III)]0 = 50 μM. (c and f) Formed HO˙ concentrations after 30 min reaction at pH 6.0 (c) or pH 7.4 (f) in water. [AscH−]0 = 125 μM, [Ligand]0 = 50 μM and [Fe(III)]0 = 50 μM in the case of peptides (FD3K, FE3K) and DFO. [AscH−]0 = 12.5 μM, [Ligand]0 = 5.0 μM and [Fe(III)]0 = 5.0 μM in the case of EDTA. All experiments were carried out at 298 K. n.d. for not determined. | |
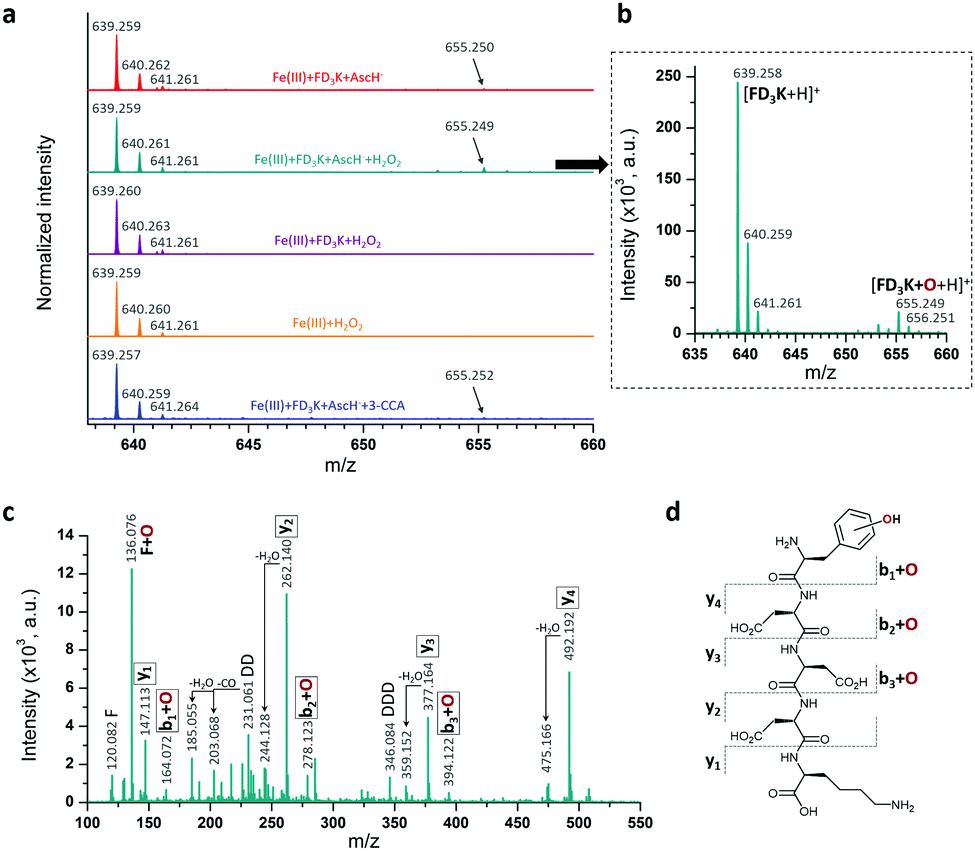
Besides this indirect antioxidant mode of action, peptides have also been designed as radical scavengers to entrap the radicals formed at the metal centre (direct antioxidant mode of action), thanks to the phenylalanine moiety grafted at the N-term.69 To study this effect, a series of mass spectroscopy analyses were performed (Fig. 6a). The MS spectrum of a solution containing Fe(III)/AscH−/FD3K (Fig. 6a, in red) reveals the presence of the peptide at m/z = 639.259 ([FD3K + H]+) and of another peak at m/z = 655.250 corresponding to [FD3K + H + O]+, i.e., a mono-oxidized form of the peptide. To improve the signal and to determine which ROS triggers the oxidation, an H2O2 supplementation has been added to the Fe(III)/AscH−/FD3K mixture (Fig. 6a, in dark cyan) in order to favour the in situ formation of HO˙. In these conditions, the [FD3K + H + O]+/[FD3K + H]+ signal intensity ratio increased, confirming the involvement of HO˙ in the oxidation process. The oxidized peptide at m/z = 655.249 has been subsequently analysed by HRMS/MS (Fig. 6c) and the fragmentation confirms the formation of an oxidized phenylalanine, i.e., a hydroxyl-phenylalanine, as previously reported.41,42 To discard the possibility of an H2O2-induced oxidation, two control conditions were analysed: one with a Fe(III)/FD3K/H2O2 mixture (i.e., without the reducing agent AscH−) and one with a FD3K/H2O2 mixture. For both, no detectable peak corresponding to the oxidized peptide has been observed: FD3K is therefore insensitive to H2O2. As a further control experiment, 3-CCA (the reactant used to evaluate the amount of HO˙ radicals formed, vide supra) was added to the Fe(III)/AscH−/FD3K mixture, and the corresponding HRMS analysis (Fig. 6a, in blue) reveals the presence of the oxidized peptide. Such results highlight that once formed at the metal centre, hydroxyl radicals are both reacting with surrounding molecules (3-CCA in this case) and entrapped by the peptide (i.e., thanks to the hydroxylation of the phenylalanine), confirming the dual mode of action of this latter. Thus, these data highlight the ability of the peptide to act as a HO˙ radical scavenger, due to the hydroxylation of the phenylalanine moiety.
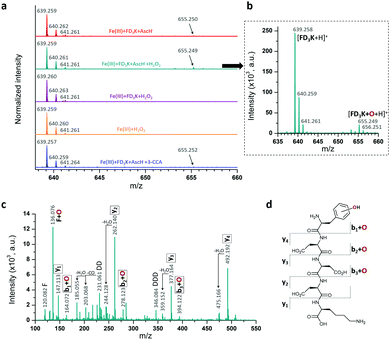 |
| | Fig. 6 (a) HRMS spectra of FD3K oxidation by HO˙ radical. Red spectrum: [Fe(III)] = [FD3K] = [AscH−] = 1 mM, dark cyan spectrum: [Fe(III)] = [FD3K] = [AscH−] = [H2O2] = 1 mM (a zoom is provided in b), purple spectrum: [Fe(III)] = [FD3K] = [H2O2] = 1 mM, orange spectrum: [Fe(III)] = [H2O2] = 1 mM, blue spectrum: [Fe(III)] = [FD3K] = [AscH−] = [3-CCA] = 1 mM. (c) HRMS/MS spectrum at m/z = 655.249 and (d) Proposed fragmentation schema of oxidized FD3K peptide. | |
Conclusion
In summary, we reported on the very first Fe(III) peptide chelators exhibiting antioxidant properties thanks to a dual indirect (i.e., inhibition of the metal redox activity) and direct (i.e., HO˙ radical scavenging via phenylalanine hydroxylation) mode of action. In particular, we highlighted that the FD3K pentapeptide strongly inhibits both the iron redox activity (up to −89%) and the ROS production (−86% and −95% for H2O2 and HO˙, respectively) at physiological pH, rivalling with deferoxamine (DFO), a clinically used iron chelator and antioxidant standard.59,60,62 Contrasting with the fastidious and hazardous methods commonly used,14–16,18 this work demonstrates the relevance and the efficiency of a more rational approach to discover new antioxidant peptides. We assume that a comprehensive overview of the peptide properties from the physicochemical key parameters (e.g., metal/ligand stoichiometry, chemical groups involved) to the impact on the iron redox activity and on the ROS formation is required to decipher and understand the parameters governing the antioxidant peptide capabilities, paving the way to the development of more efficient compounds.
Materials and methods
Generalities
Reagents, chemicals and solvents were purchased from Sigma-Aldrich, except Fmoc-L-Glu(OtBu)-OH, Fmoc-L-Asp(OtBu)-OH, Fmoc-L-Phe-OH and preloaded Wang resins from Iris Biotech, sodium hydroxide from VWR. All reagents and chemicals were used without further purification. Peptides were synthesized on an automated ResPep XL synthesizer from Intavis AG, and subsequently purified by semi-preparative reversed-phase HPLC using a Macherey-Nagel Nucleosil 100-5 C18 250 × 21 mm column on a Waters HPLC system (equipped with a Waters 600 pump, a Waters 600 controller and a Waters 486 tunable absorbance detector). Analytical reversed-phase HPLC were carried out using a Macherey-Nagel Nucleosil 100-5 C18 250 × 4.6 mm column on a Shimadzu Prominence UFLC (equipped with a LC20AB solvent delivery, SPD-M20A diode array detector and a CTO-20AC column oven). NMR spectra (1H, 13C, HMQC, HMBC, COSY) were recorded at 298 K on a Bruker DRX 400 spectrometer on the NMR Platform of the Jean Barriol Institute (Université de Lorraine, Nancy, France). All analysed samples were aqueous solution at known pH, 1,4-dioxane was used as external standard in capillary (1H δ = 3.75 ppm, 13C δ = 67.2 ppm, dissolved in D2O). For 1H NMR spectra, a WATERGATE pulse sequence was used for water suppression. High resolution ESI-MS analyses were performed on a Bruker Daltonics MicrOTOFQ mass spectrometer provided by the MassLor Platform (Université de Lorraine, Nancy, France). Circular dichroism spectra were recorded on a MOS-450 spectrometer from BioLogic, coupled with a Metrohm 716 titrator combined with a Fisher Bioblock electrode for pH adjustments. For potentiometric measurements, a Dosimat 715 and a pH-meter equipped with a semi-micro combined electrode from Metrohm were used. UV-vis spectra were recorded on a PerkinElmer Lambda1050 spectrophotometer, room temperature fluorescence emission spectra were recorded on a Fluorolog-3 spectrofluorometer from Horiba Scientific.
Peptide synthesis
The four peptides were synthesized at a 400 μmol-scale using a Fmoc/tBu strategy and double couplings for each amino acid. The experimental conditions for each coupling were: Fmoc-amino acid (6 equivalents), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium tetrafluoroborate (HBTU, 5 equivalents), and 4-methylmorpholine (NMM, 10 equivalents) in dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP), coupling time = 40 minutes. Fmoc-deprotection steps were carried out using a 20% piperidine solution in DMF (3 × 15 min), and final cleavages were achieved using a trifluoroacetic acid/triisopropylsilane/water (92,5/5/2,5) mixture (2 hours). The crude peptides were precipitated from cold diethylether (−20 °C), centrifuged, washed with cold diethylether, dried under reduced pressure, resolubilized in water and finally, lyophilized. Then, peptides were dissolved in solvent A and purified by semi-preparative HPLC using solvent A (95% water, 5% acetonitrile, 0.1% trifluoracetic acid) and solvent B (100% acetonitrile, 0.1% trifluoroacetic acid) with a 10 min isocratic elution (100% A) followed by a 45 min linear gradient (up to 95% B) with a UV detection at 214 nm. The resulting solutions were evaporated under reduced-pressure and double-lyophilized. Purity of each peptide was evaluated by analytical reversed-phase HPLC using solvent A′ (95% water, 5% acetonitrile, 0.1% formic acid) and solvent B′ (100% acetonitrile, 0.1% formic acid) with a 5 min isocratic elution (100% A′) followed by a 25 min linear gradient (0% to 40% solvent B′) and a 10 min linear gradient (40% to 100% B′) with a UV detection at 214 nm. All the chemical characterizations (NMR, HRMS and HPLC) are provided in ESI.†
Potentiometric measurements
The pH-potentiometric titrations were investigated in the pH range 2.0–11.2 and 2.0–7.5 for the ligands and iron(III)-containing systems, respectively (I = 0.1 M NaNO3 and T = 298.0 ± 0.1 K). A Dosimat 715 (Metrohm) automatic burette and pH-meter equipped with a semi-micro combined electrode (Metrohm) was used for the titration. The initial concentration of peptides was 2 × 10−3 M, using 1:1 and 1:2 metal-to-ligand ratios. The titrations were performed with carbonate free stock solution of sodium hydroxide at known concentration. During the titration argon was bubbled through the samples to ensure the absence of oxygen and carbon dioxide. The recorded pH readings were converted to hydrogen ion concentration as described by Irving et al.70 Protonation constants of the ligands and the overall stability constants (logβpqr) of the complexes were calculated from at least 3 independent titrations (ca. 70 data points per titration) by means of the general computational programs, PSEQUAD71 and SUPERQUAD72 using eqn (1) and (2).| | 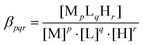 | (2) |
Antioxidant assays
MES (50 mM, pH 6.0) and HEPES (50 mM, pH 7.4) buffers were treated on Chelex100 to remove trace metal contaminants from the solution and prevent unwanted reactions before the different tests. In Amplex Red and 3-CCA tests, the buffers were replaced with water to determine the buffer effect on these reactions. Indeed, some organic buffer (HEPES, TRIS) show efficient radical scavenging activity preventing a correct measurement of H2O2 and HO˙ radicals.73 Thus, high-purity water treated on Chelex100 was used and the pH was adjusted by addition of 0.1 M NaOH. The stock solution at 2 mM of iron(III)–nitrate was prepared from analytical grade reagent in an acidic media (0.1 M HNO3) to keep iron(III) ion in solution, its exact concentration was checked spectrophotometrically using complexometric titration. Peptides (FD3K and FE3K), EDTA and DFO ligand stock solutions at 2 mM were prepared using MES or HEPES buffer depending on the pH applied. To perform the tests, a semi-micro quartz cell with 1 cm optical pathway was used, the sample holder in the spectrophotometer was thermostated at 25 °C by a Peltier temperature controller. Note: NBT (for nitroblue tetrazolium) assays were performed to measure the superoxide anion radical formation; however, despites our many efforts no convincing conditions can be found because of interfering side effects (see ESI† for a deeper discussion) and the NBT assay was therefore discarded.
Ascorbate tests
For a standard assay, a 5 mM ascorbate stock solution was freshly prepared using MES or HEPES buffer depending on the pH applied. The absorbance of ascorbate was followed over 10 min at 265 nm (ε = 14500 M−1 cm−1), and at least 3 independent measurements were carried out. In a total volume of 1 mL, the final concentration of the components was 100 μM with 1:1:1 AscH−:ligand:Fe(III) ratio. The pH was checked at the end of each assay. The reaction was triggered by the addition of ascorbate. As reference reaction, AscH− oxidation by ligands alone was also studied. The initial ascorbate oxidation rate was calculated from the slope of [AscH−] = f(t).
Amplex red assay
For this assay, all components except the iron(III) ion and Amplex Red reagent, were dissolved in 50 mM MES or 50 mM HEPES buffers or in Milli-Q water. The Amplex Red reagent was dissolved in DMSO:buffer mixture at 1:4 ratio. When the buffers were replaced with water to determine the buffer effect on this reaction, high-purity water was used and the pH was adjusted by addition of 0.1 M NaOH. The H2O2 production was detected by Amplex Red reagent in the presence of horseradish peroxidase (HRP, Scheme S2, ESI†) forming the compound Resorufin with λabs = 570 nm.74 A working solution was first prepared containing 0.8 U mL−1 HRP and 200 μM Amplex Red reagent in a light-protected tube. 50 μL of this solution was placed in the total volume of 1 mL reaction mixture. The final concentration of the AscH− was 200 μM, and the ligand (peptides, EDTA or DFO):Fe(III):AscH− ratio was 1:1:4. The AscH− was the last reagent added thus initiating the reaction between O2 and Fe(III), the pH was checked at the end of each reaction. A negative control containing only the working solution in buffer was necessary in order to monitor the background absorbance of the Amplex Red itself. The Resorufin formation was monitored by visible absorption spectroscopy at 570 nm at RT over 45 min (Fig. S9, ESI†). To determine the H2O2 concentration, a standard calibration curve was used. To obtain this calibration curve, a 20 mM hydrogen peroxide solution was previously titrated by permanganometry and then diluted successively.
Coumarin-3-carboxyilic acid (3-CCA) assay
The MES and HEPES buffers disturbed the fluorescent spectra and also had efficient HO˙ radical scavenging activity, thus pure water treated on Chelex100 was used as a solvent and the pH was adjusted by adding 0.1 M NaOH. Thus, all the reagents were dissolved in Milli-Q water except of the 3-CCA, since it was only soluble in phosphate buffer (50 mM, pH 6.0 and pH 7.4). The concentration of 3-CCA stock solution was 5 mM. In the total volume of 2 mL, the final concentrations of the ascorbate and 3-CCA were both 125 μM, and the ratio of the reaction mixture was ligand (peptides, EDTA or DFO):Fe(III):AscH− = 1:1:2.5. The pH was checked at the end of the measurement. In the case of EDTA, the amount of all components was 10 times less than that of the other ligand systems. For the greater reproducibility of the measured data, each measurement was started by monitoring a blank sample (without any ascorbate) for 5 min, then the ascorbate was added to the sample. The reaction between 3-CCA and hydroxyl radical generating the fluorescent compound 7-OH-CCA was followed over 30 min at 25 °C at λem = 450 nm (λex = 395 nm) by spectrofluorometry (Scheme S3 and Fig. S10, ESI†). The HO˙ concentration was determined in different systems (at pH 6 and 7.4 with different ligands) by a standard calibration curve using commercially available 7-OH-CCA.
HRMS and HRMS/MS measurements
In order to investigate FD3K ligand oxidation by HO˙ radical, the following reaction mixtures were prepared at room temperature and analysed after 18 h by HRMS and HRMS/MS measurements: in a total volume of 1 mL at pH 6.0 in water (pH was adjusted by 0.1 M NaOH), the concentrations of FD3K, Fe(III), 3-CCA, AscH− and H2O2 were all adjusted equal to 1 mM.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the “Impact Biomolecules” project of the “Lorraine Université d'Excellence” (Investissements d’avenir – ANR-15-IDEX-04-LUE) for financial support. The authors thanks L. Saidi for preliminary works, P. Lemiere, S. Parant and M. Achard for technical help.
Notes and references
-
R. Crichton, in Biological Inorganic Chemistry, ed. R. Crichton, Academic Press, 3rd edn, 2019, ch. 1, pp. 1–18 Search PubMed.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS PubMed.
- C. R. Ferreira and W. A. Gahl, Transl. Sci. Rare Dis., 2017, 2, 101–139 Search PubMed.
- F. Collin, Int. J. Mol. Sci., 2019, 20, 2407 CrossRef PubMed.
- B. Halliwell, J. Neurochem., 2006, 97, 1634–1658 CrossRef CAS PubMed.
- K. M. Holmström and T. Finkel, Nat. Rev. Mol. Cell Biol., 2014, 15, 411–421 CrossRef PubMed.
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453 CrossRef CAS PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- Z. Liu, Z. Ren, J. Zhang, C.-C. Chuang, E. Kandaswamy, T. Zhou and L. Zuo, Front. Physiol., 2018, 9, 477 CrossRef PubMed.
- A. M. Pisoschi and A. Pop, Eur. J. Med. Chem., 2015, 97, 55–74 CrossRef CAS PubMed.
- M. Wojcik, I. Burzynska-Pedziwiatr and L. Wozniak, Curr. Med. Chem., 2010, 17, 3262–3288 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorgan. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- N. J. Kang, H.-S. Jin, S.-E. Lee, H. J. Kim, H. Koh and D.-W. Lee, Crit. Rev. Environ. Sci. Technol., 2020, 50, 72–103 CrossRef CAS.
- B. H. Sarmadi and A. Ismail, Peptides, 2010, 31, 1949–1956 CrossRef CAS PubMed.
- R. Esfandi, M. E. Walters and A. Tsopmo, Heliyon, 2019, 5, e01538 CrossRef PubMed.
- L. L. S. Canabady-Rochelle, K. Selmeczi, S. Collin, A. Pasc, L. Muhr and S. Boschi-Muller, Food Chem., 2018, 239, 478–485 CrossRef CAS PubMed.
- J. Bechaux, P. Gatellier, J.-F. Le Page, Y. Drillet and V. Sante-Lhoutellier, Food Funct., 2019, 10, 6244–6266 RSC.
- E. C. Y. Li-Chan, Curr. Opin. Food Sci., 2015, 1, 28–37 CrossRef.
- A. Carocci, A. Catalano, M. S. Sinicropi and G. Genchi, Biometals, 2018, 31, 715–735 CrossRef CAS PubMed.
- P. Poprac, K. Jomova, M. Simunkova, V. Kollar, C. J. Rhodes and M. Valko, Trends Pharmacol. Sci., 2017, 38, 592–607 CrossRef CAS PubMed.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Metallomics, 2015, 7, 1459–1476 RSC.
- N. R. Perron and J. L. Brumaghim, Cell Biochem. Biophys., 2009, 53, 75–100 CrossRef CAS PubMed.
- M. Samsonowicz, E. Regulska and M. Kalinowska, Chem. – Biol. Interact., 2017, 273, 245–256 CrossRef CAS PubMed.
- M. M. Kasprzak, A. Erxleben and J. Ochocki, RSC Adv., 2015, 5, 45853–45877 RSC.
- D. Xu, M.-J. Hu, Y. Q. Wang and Y. L. Cui, Molecules, 2019, 24, 1123 CrossRef CAS PubMed.
- M. B. H. Youdim, Exp. Neurobiol., 2010, 19, 1–14 CrossRef PubMed.
- L. Belguendouz, L. Fremont and A. Linard, Biochem. Pharmacol., 1997, 53, 1347–1355 CrossRef CAS PubMed.
- A. Shakeri, Y. Panahi, T. P. Johnston and A. Sahebkar, BioFactors, 2019, 45, 304–317 CrossRef CAS PubMed.
- J. M. Lü, P. H. Lin, Q. Yao and C. Chen, J. Cell. Mol. Med., 2010, 14, 840–860 CrossRef PubMed.
- H. Zheng, L. M. Weiner, O. Bar-Am, S. Epsztejn, Z. I. Cabantchik, A. Warshawsky, M. B. H. Youdim and M. Fridkin, Bioorg. Med. Chem., 2005, 13, 773–783 CrossRef CAS PubMed.
- R. C. Hider, S. Roy, Y. M. Ma, X. Le Kong and J. Preston, Metallomics, 2011, 3, 239–249 RSC.
-
E. Farkas and I. Sóvágó, in Amino Acids, Peptides and Proteins, The Royal Society of Chemistry, 2017, vol. 41, pp. 100–151 Search PubMed.
- I. Sóvágó, K. Várnagy, N. Lihi and Á. Grenács, Coord. Chem. Rev., 2016, 327–328, 43–54 CrossRef.
- N. Lihi, M. Lukács, M. Raics, G. Szunyog, K. Várnagy and C. Kállay, Inorgan. Chim. Acta, 2018, 472, 165–173 CrossRef CAS.
- L. R. Perez and K. J. Franz, Dalton Trans., 2010, 39, 2177–2187 RSC.
- T. Zheng and E. M. Nolan, Metallomics, 2012, 4, 866–880 RSC.
- C. Kurth, H. Kage and M. Nett, Org. Biomol. Chem., 2016, 14, 8212–8227 RSC.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- A. Galano and A. Cruz-Torres, Org. Biomol. Chem., 2008, 6, 732–738 RSC.
- B. R. Ipson and A. L. Fisher, Ageing Res. Rev., 2016, 27, 93–107 CrossRef CAS PubMed.
- C. A. Damante, K. Ősz, Z. Nagy, G. Pappalardo, G. Grasso, G. Impellizzeri, E. Rizzarelli and I. Sóvágó, Inorg. Chem., 2008, 47, 9669–9683 CrossRef CAS PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15, 1214–1218 CrossRef CAS PubMed.
- E. Farkas, É. A. Enyedy and H. Csóka, Polyhedron, 1999, 18, 2391–2398 CrossRef CAS.
-
A. E. Martell, R. M. Smith, A. E. Martell and R. M. Smith, in Critical Stability Constants, ed. A. E. Martell and R. M. Smith, Springer, 1982 Search PubMed.
- A. Stefánsson, Environ. Sci. Technol., 2007, 41, 6117–6123 CrossRef PubMed.
- B. K. Fuqua, C. D. Vulpe and G. J. Anderson, J. Trace Elem. Med. Biol., 2012, 26, 115–119 CrossRef CAS PubMed.
- S. Gulec, G. J. Anderson and J. F. Collins, Am. J. Physiol.: Gastrointest. Liver Physiol., 2014, 307, G397–G409 CrossRef CAS PubMed.
- H. Lv and P. Shang, Metallomics, 2018, 10, 899–916 RSC.
- T. Kurz, J. W. Eaton and U. T. Brunk, Int. J. Biochem. Cell Biol., 2011, 43, 1686–1697 CrossRef CAS PubMed.
- R. B. Martin, J. Savory, S. Brown, R. L. Bertholf and M. R. Wills, Clin. Chem., 1987, 33, 405–407 CrossRef CAS.
- Y. Kohgo, K. Ikuta, T. Ohtake, Y. Torimoto and J. Kato, Int. J. Hematol., 2008, 88, 7–15 CrossRef CAS PubMed.
- Z. I. Cabantchik, Front. Pharmacol., 2014, 5, 45 Search PubMed.
- H. Drechsel and G. Jung, J. Pept. Sci., 1998, 4, 147–181 CrossRef CAS PubMed.
-
I. Bertini, C. Luchinat, G. Parigi and E. Ravera, in NMR of Paramagnetic Molecules, ed. I. Bertini, C. Luchinat, G. Parigi and E. Ravera, Elsevier, 2nd edn, 2017, ch. 7, pp. 175–253 Search PubMed.
- S. Balayssac, I. Bertini, C. Luchinat, G. Parigi and M. Piccioli, J. Am. Chem. Soc., 2006, 128, 15042–15043 CrossRef CAS PubMed.
- L. T. Dalvi, D. C. Moreira, R. Andrade, J. Ginani, A. Alonso and M. Hermes-Lima, Spectrochim. Acta, Part A, 2017, 173, 910–917 CrossRef CAS PubMed.
- V. A. Roginsky, T. K. Barsukova, G. Bruchelt and H. B. Stegmann, Biochim. Biophys. Acta, Gen. Subj., 1997, 1335, 33–39 CrossRef CAS.
- R. T. Dean and P. Nicholson, Free Radical Res., 1994, 20, 83–101 CrossRef CAS PubMed.
- S. Singh and R. C. Hider, Anal. Biochem., 1988, 171, 47–54 CrossRef CAS PubMed.
- P. Holden and L. S. Nair, Tissue Eng., Part B, 2019, 25, 461–470 CrossRef CAS PubMed.
- E. Atrián-Blasco, M. Del Barrio, P. Faller and C. Hureau, Anal. Chem., 2018, 90, 5909–5915 CrossRef PubMed.
- R. R. Grinstead, J. Am. Chem. Soc., 1960, 82, 3464–3471 CrossRef CAS.
- W. G. Gutheil, M. E. Stefanova and R. A. Nicholas, Anal. Biochem., 2000, 287, 196–202 CrossRef CAS PubMed.
- T. Brumbarova, C. Le and P. Bauer, Bio-Protoc., 2016, 6, e1999 Search PubMed.
- Y. Manevich, K. D. Held and J. E. Biaglow, Radiat. Res., 1997, 148, 580–591 CrossRef CAS PubMed.
- X. Solans, M. Font Altaba and J. Garcia-Oricain, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 635–638 CrossRef.
-
J. L. Hougland, J. Darling and S. Flynn, in Molecular Basis of Oxidative Stress, Chemistry, Mechanics and Disease Pathogenesis, ed. F. A. Villamena, 1st edn, 2013, ch. 3, pp. 71–92 Search PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
-
L. Zekany and I. Nagypal, in Computational Methods for the Determination of Formation Constants, ed. D. J. Leggett, Springer, 1985, ch. 8, pp. 291–353 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, J. Chem. Soc., Dalton Trans., 1985, 1195–1200 RSC.
- M. Hicks and J. M. Gebicki, FEBS Lett., 1986, 199, 92–94 CrossRef CAS.
- M. Zhou, Z. Diwu, N. Panchuk-Voloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162–168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mt00103a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Marie-Christine
Averlant-Petit
c,
Marie-Christine
Averlant-Petit