
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical binding of copperII to N-truncated Aβ4–16 peptide†
Xiangyu
Teng‡
 a,
Ewelina
Stefaniak‡
b,
Paul
Girvan
a,
Radosław
Kotuniak
b,
Dawid
Płonka
b,
Wojciech
Bal
*b and
Liming
Ying
*c
a,
Ewelina
Stefaniak‡
b,
Paul
Girvan
a,
Radosław
Kotuniak
b,
Dawid
Płonka
b,
Wojciech
Bal
*b and
Liming
Ying
*c
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK
bInstitute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland. E-mail: wbal@ibb.waw.pl
cNational Heart and Lung Institute, Imperial College London, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK. E-mail: l.ying@imperial.ac.uk
First published on 1st April 2020
Abstract
N-Truncated Aβ4–42 displays a high binding affinity with CuII. A mechanistic scheme of the interactions between Aβ4–42 and CuII has been proposed using a fluorescence approach. The timescales of different conversion steps were determined. This kinetic mechanism indicates the potential synaptic functions of Aβ4–42 during neurotransmission.
Significance to metallomicsN-Truncated Aβ4–x is abundant in both healthy and AD brains. Its Cu(II) binding affinity is three orders of magnitude stronger than well-known Aβ1–42 or Aβ1–40. Using a model peptide, Aβ4–16, we have elucidated the reaction mechanism of Cu(II) with Aβ4–x, crucial to understand the physiological role and toxicity of Aβ peptides. The presence of two kinetic intermediates prior to the formation of the tight ATCUN complex has implications for the potential function of Aβ4–42 as a Cu(II) transporter during neurotransmission. The methodology used in this work may also stimulate the research of Cu(II) interactions with other intrinsically disordered proteins (IDPs). |
The amyloid-β (Aβ) peptides associated with Alzheimer's Disease (AD) comprise a number of species. The “canonical” Aβ1–42 and Aβ1–40 peptides derived directly by proteolysis of the Amyloid Precursor Protein (APP) are complemented by N- and C-truncated species, yielded by a variety of brain proteases.1 Among them, the N-truncated Aβ4–42 has been reported as particularly abundant in the hippocampus and cortex of sporadic AD patients, as well as in healthy controls,2,3 even exceeding Aβ1–42 and Aβ1–40.4,5 Aβ1–x peptides can bind CuII using the N-terminus and H6, H13, and His14 residues.6–8 Hence, Aβ1–16 has been adopted as a common model peptide in metal binding studies. Kd in the range of 0.1 nM to 1 nM at pH 7.1–7.4 was determined for Aβ1–16 and Aβ1–40.9–11 The adventitious binding of CuII ions to Aβ1–42/40 and the concomitant generation of reactive oxygen species (ROS) via the CuII/CuI redox pair has been proposed to be the molecular basis of oxidative stress and neuronal death in AD.12 On the other hand, Aβ4–x peptides bind a CuII ion more than three orders of magnitude more strongly (Kd = 30 fM and 6.6 fM at pH 7.4 for Aβ4–16 and Aβ4–9, respectively), using their N-terminal ATCUN motif spanning the Phe4, Arg5 and His6 residues. These complexes are redox-inert and do not generate significant ROS. CuII ion transfer from Aβ1–16 to Aβ4–16 occurs upon adding the latter to the CuIIAβ1–16 solution.13 This reaction is quantitative, in agreement with the affinity difference, and fast, occurring within the sample preparation time ∼s. Such a reaction suggested that Aβ4–42 should prevail as a CuII binding Aβ species in the extracellular spaces of the brain. This finding gave rise to a hypothesis that Aβ4–42 may have a physiological role as a synaptic CuII scavenger during neurotransmission.14 However, CuII release events in glutamatergic synapses may occur on a much faster, millisecond scale. Therefore, a thorough determination of association and dissociation rate constants for the participating species is necessary to help evaluate their relevance in vivo. Such data have been obtained previously for CuIIAβ1−x complexes.15–17 Here, we studied the reaction mechanism for CuII binding to the model peptide Aβ4–16 and found that the reaction follows a hierarchical fashion, going through two intermediate states and then reaching the final tight complex.
First, we studied the effect of N-truncation on the CuII binding kinetics. 20 nM Aβ labelled by HiLyte Fluor 488 on lysine 16 (FRHDSGYEVHHQK-HiLyte 488) was reacted with 400 nM CuII under various HEPES concentrations in order to obtain the HEPES-independent CuII binding rate constant (kon). The results are shown in Fig. 1a. The intercept of the fitted curve (Fig. 1b) was used to determine kon, which is 2.0(1) × 108 M−1 s−1, 2.5 times slower than the value for Aβ1–16.17
 | ||
| Fig. 1 Kinetics of CuII binding to Aβ4–16. (a) Representative raw traces of Aβ (20 nM) with CuII (400 nM) under various concentrations of HEPES. The experiments were performed in 50 mM HEPES and 100 mM NaCl buffer solution at 298 K (pH 7.5). (b) HEPES dependence of kon. The HEPES independent kon value is 2.0(1) × 108 M−1 s−1. (c) Kinetics of dissociation of CuIIAβ4–16 assisted by varying concentrations of EDTA, monitored by UV-vis absorbance. The experiments were performed for 1 mM CuIIAβ4–16 at 298 K (pH 7.5). (d) Empirical fit to derive the EDTA-independent koff. | ||
k
off was determined for the reaction of a CuII complex of unlabelled Aβ4–16 with an excess of EDTA. The estimated value is ∼5 × 10−5 s−1, which divided by kon proposed here gives Kd ∼ 250 fM. EDTA is a stronger CuII chelator than Aβ4–16, with a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β of 18.7, which can be recalculated into a conditional constant CK of 16.0 at pH 7.5.18 This value is sufficiently higher than that of CuIIAβ4–16, 13.53, to assure full CuII transfer, as demonstrated in Fig. 1c. The reaction was carried out for a range of EDTA/peptide ratios between 2 and 120. Pseudo-1st order kinetics for the CuII transfer reaction was observed for all experiments. The non-linear response of koff to EDTA required the EDTA-independent koff value to be determined by the extrapolation of the empirical exponential fit to these data, as shown in Fig. 1d.
β of 18.7, which can be recalculated into a conditional constant CK of 16.0 at pH 7.5.18 This value is sufficiently higher than that of CuIIAβ4–16, 13.53, to assure full CuII transfer, as demonstrated in Fig. 1c. The reaction was carried out for a range of EDTA/peptide ratios between 2 and 120. Pseudo-1st order kinetics for the CuII transfer reaction was observed for all experiments. The non-linear response of koff to EDTA required the EDTA-independent koff value to be determined by the extrapolation of the empirical exponential fit to these data, as shown in Fig. 1d.
To gain a glimpse of a possible reaction mechanism of CuII binding to N-truncated Aβ4–16, we performed binding experiments at a 1:1 mixing ratio of Aβ to CuII with increasing concentration. In such experiments, the effect of the second CuII binding can be ignored, as the relevant logK is as low as 6.7.13 The raw traces are shown in Fig. 2a. We noticed that the reaction process is becoming concentration independent after ∼2 s (results from the fit are summarized in Table S1, ESI†). Thus we infer the existence of an intramolecular process following the initial CuII binding.
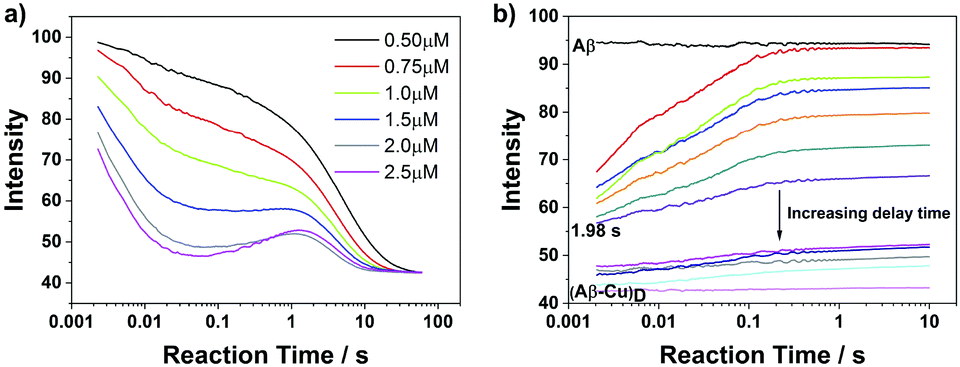 | ||
| Fig. 2 (a) Raw traces of CuII binding measurements with a series of 1:1 concentration ratio HiLyte 488 labelled Aβ and CuII showing the evidence of intermediate species formation. The experiment was performed in 50 mM HEPES and 100 mM NaCl buffer solution at 298 K (pH 7.5). (b) Kinetics of HiLyte 488 labelled Aβ/CuII interactions measured by double mixing stopped flow. Raw traces showing the change in amplitude as the delay time between mixing of an equal concentration of CuII and Aβ (2 μM) and addition of EDTA is increased from 50 ms to 1 min. The experiment was performed in 50 mM HEPES and 100 mM NaCl buffer solution at 298 K (pH 7.5). | ||
Next, a double mixing stopped flow technique was employed to further explore the potential intermediate complexes formed after the initial CuII binding. This technique was successfully applied to probe the interconversion between component I and component II CuII coordination species of Aβ1–16 and Aβ1–40.17 2 μM Aβ4–16 and 2 μM CuII were mixed in a delay loop and after various delay times the reaction was “frozen” by adding an excess of EDTA (Fig. 2b). Taking advantage of the disparities in reactivity of different CuIIAβ4–16 species with EDTA, the time evolution of the population of individual species could be resolved and analyzed, enabling us to depict details of the binding process.
As shown in Fig. 2b, the amplitude of fluorescence recovery strongly depends on the delay time, indicating that a much more inert (less reactive towards EDTA) complex (“dark” complex) formed after around 2 s. We equate this end complex, (Aβ–Cu)D, to the very stable ATCUN-type CuIIAβ4–16 complex reported previously.13 Furthermore, because the reaction rate is concentration independent after 2 s as mentioned above, we propose that a peptide conformational rearrangement process leading to this final complex must occur at around 2 s.
In order to describe the whole process of CuII binding of N-truncated Aβ4–16, we hypothesized a reaction scheme as shown in Fig. 3a. The individual amplitudes of the two phases in Fig. 2b were determined by a global fit, which were further fitted by the scheme with KinTek software to validate it (Fig. 3b). The amplitudes indicate the amounts of two intermediates, Species I and Species II, at different reaction process stages, and could be fitted well by the predicted mechanism, with fitted rate constants listed in Table 1. A corresponding free energy landscape illustration of CuII binding with Aβ4–16 is shown in Fig. 3c.
 | ||
| Fig. 3 (a) Reaction mechanism of CuII binding to Aβ4–16 and formation of the high affinity CuIIAβ4–16 complex, (Aβ–Cu)D (CuII binding site shown above). (b) Fitting of the individual amplitudes at different CuII binding process stages of the two phases by the predicted reaction mechanism. (c) Proposed free energy landscape of CuII binding to Aβ4–16. “Dark” complex refers to the very stable ATCUN-type CuIIAβ4–16 complex. | ||
| k +1 | k −1 | k +2 | |
|---|---|---|---|
| k value/s−1 | 4.10(1) | 10.34(2) | 3.31(4) |
Finally, the activation energy of the (Aβ–Cu)D complex was determined to be 64(3) kJ mol−1 (Fig. 4) by performing a series of double mixing experiments at different temperatures (raw data shown in Fig. S1, ESI†).
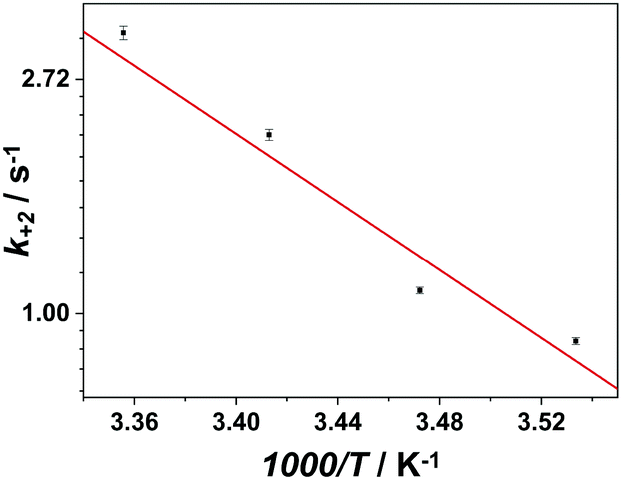 | ||
| Fig. 4 Arrhenius plot for the switching rate constant k+2. The switching activation energy determined is 64(3) kJ mol−1. | ||
The chemical properties of ATCUN CuII complexes of Aβ4–x peptides, such as high thermodynamic stability, absence of ROS production due to their resistance to oxidation and reduction, reluctance of copper to transfer to metallothionein-3 (MT3) and easy sequestering of CuII from Aβ1–x, gave rise to a concept that Aβ4–x peptides (full-length Aβ4–42 and its C-truncated analogs) may serve as guardians of synaptic function, by sequestering excess CuII ions released during neurotransmission in glutamatergic pathways.14,19 The key unsolved issue is how these exchange-inert complexes relay copper back to neurons to maintain the proper copper cycling. Furthermore, CuII-free Aβ4–42 can be neurotoxic by forming oligomeric species.20 Detailed knowledge on mechanisms of CuII association with and dissociation from Aβ4–x peptides, represented here by Aβ4–16, is thus crucial to understand the physiology and toxicity of these Aβ peptides.
The discovery of long-lived kinetic intermediates in the formation of the ATCUN complex of Aβ4–16 is a game changer in the above considerations. The lifetimes of Species I and Species II complexes are comparable to the intervals between pulses of neurotransmitter release in glutamatergic neuronal pathways.21 Therefore, these complexes may well contribute to the biological activity of Aβ4–42, and of putative short peptidic fragments generated by neprilysin cleavage, such as Aβ4–9.22,23 There is only one way in which four nitrogen ligands of the ATCUN motif can be arranged around the CuII ion, and so it is reasonable to assume that the intermediate species contain the coordinatively unsaturated CuII. Such species have been implicated in the reverse reaction of CuII dissociative transfer from CuIIAβ4–16 to MT3, to explain the catalytic effect of glutamate,24 but it has not been observed directly. The Species I and in particular the longer-lived Species II complex may be the actual species able to move copper around during neurotransmission. The fact that the CuIIAβ1−x complex, although so much weaker, was formed 2.5 times faster, prompts further research into possible synaptic roles of CuII interactions with various Aβ species.
Furthermore, the observed hierarchical binding of CuII to Aβ4–16 resembles the kinetics of the binding of many intrinsically disordered proteins (IDPs).25 The methodology used in this study may be applicable to the fundamental understanding of the emerging “coupled binding and folding” paradigm.26
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Leverhulme Trust grant RPG-2015-345 to LY and the Biotechnology and Biosciences Research Council (UK) grant BB/R022429/1 to LY, and the National Science Centre in Poland: PRELUDIUM Grant No. 2016/21/N/NZ1/02785 and ETIUDA Grant No. 2018/28/T/NZ1/00452, to ES, and OPUS Grant No. 2018/29/B/ST4/01634 to WB. The equipment used was sponsored in part by the Centre for Preclinical Research and Technology (CePT) under award number POIG.02.02.00-14-024/08-00, a project co-sponsored by European Regional Development Fund and Innovative Economy, the National Cohesion Strategy of Poland.Notes and references
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CrossRef PubMed.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef PubMed.
- E. Portelius, N. Bogdanovic, M. K. Gustavsson, I. Volkmann, G. Brinkmalm, H. Zetterberg, B. Winblad and K. Blennow, Acta Neuropathol., 2010, 120, 185–193 CrossRef PubMed.
- G. Antonios, N. Saiepour, Y. Bouter, B. C. Richard, A. Paetau, A. Verkkoniemi-Ahola, L. Lannfelt, M. Ingelsson, G. G. Kovacs, T. Pillot, O. Wirths and T. A. Bayer, Acta Neuropathol. Commun., 2013, 1, 56 CrossRef PubMed.
- T. A. Bayer and O. Wirths, Acta Neuropathol., 2014, 127, 787–801 CrossRef PubMed.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Angew. Chem., 2009, 121, 9437–9440 ( Angew. Chem., Int. Ed. , 2009 , 48 , 9273–9276 ) CrossRef.
- B. Alies, H. Eury, C. Bijani, L. Rechignat, P. Faller and C. Hureau, Inorg. Chem., 2011, 50, 11192–11201 CrossRef PubMed.
- E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 375, 38–55 CrossRef PubMed.
- B. Alies, E. Renaglia, M. Rózga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501–1508 CrossRef PubMed.
- T. R. Young, A. Kirchner, A. G. Wedd and Z. Xiao, Metallomics, 2014, 6, 505–517 RSC.
- A. Conte-Daban, V. Borghesani, S. Sayen, E. Guillon, Y. Journaux, G. Gontard, L. Lisnard and C. Hureau, Anal. Chem., 2017, 89, 2155–2162 CrossRef PubMed.
- C. Cheignon, M. Jones, E. Atrián-Blasco, I. Kieffer, P. Faller, F. Collin and C. Hureau, Chem. Sci., 2017, 8, 5107–5118 RSC.
- M. Mital, N. E. Wezynfeld, T. Frączyk, M. Z. Wiloch, U. E. Wawrzyniak, A. Bonna, C. Tumpach, K. J. Barnham, C. L. Haigh, W. Bal and S. C. Drew, Angew. Chem., 2015, 127, 10606–10610 ( Angew. Chem., Int. Ed. , 2015 , 54 , 10460–10464 ) CrossRef.
- E. Stefaniak and W. Bal, Inorg. Chem., 2019, 58, 13561–13577 CrossRef CAS PubMed.
- P. Girvan, T. Miyake, X. Teng, T. Branch and L. Ying, ChemBioChem, 2016, 17, 1732–1737 CrossRef CAS PubMed.
- T. Branch, M. Barahona, C. A. Dodson and L. Ying, ACS Chem. Neurosci., 2017, 8, 1970–1979 CrossRef CAS PubMed.
- T. Branch, P. Girvan, M. Barahona and L. Ying, Angew. Chem., 2015, 127, 1243–1246 ( Angew. Chem., Int. Ed. , 2015 , 54 , 1227–1230 ) CrossRef.
- J. Felcman and J. J. da Silva, Talanta, 1983, 30, 565–570 CrossRef CAS PubMed.
- N. E. Wezynfeld, E. Stefaniak, K. Stachucy, A. Drozd, D. Płonka, S. C. Drew, A. Krężel and W. Bal, Angew. Chem., 2016, 128, 8375–8378 ( Angew. Chem., Int. Ed. , 2016 , 55 , 8235–8238 ) CrossRef.
- J. Dunys, A. Valverde and F. Checler, J. Biol. Chem., 2018, 293, 15419–15428 CrossRef CAS PubMed.
- W. Goch and W. Bal, PLoS One, 2017, 12, e0170749 CrossRef PubMed.
- M. Mital, W. Bal, T. Frączyk and S. C. Drew, Inorg. Chem., 2018, 57, 6193–6197 CrossRef CAS PubMed.
- K. Bossak-Ahmad, M. Mital, D. Płonka, S. C. Drew and W. Bal, Inorg. Chem., 2019, 58, 932–943 CrossRef CAS PubMed.
- A. Santoro, N. Wezynfeld, E. Stefaniak, A. Pomorski, D. Płonka, A. Krezel, W. Bal and P. Faller, Chem. Commun., 2018, 54, 12634–12637 RSC.
- K. Sugase, H. J. Dyson and P. E. Wright, Nature, 2007, 447, 1021–1025 CrossRef.
- S. Gianni, J. Dogan and P. Jemth, Curr. Opin. Struct. Biol., 2016, 36, 18–24 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00299e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |