
Effects of mercuric chloride on spatial memory deficit-induced by beta-amyloid and evaluation of mitochondrial function markers in the hippocampus of rats
Ladan
Behzadfar
a,
Shokoufeh
Hassani
ab,
Hossein
Feizpour
c,
Nakisa
Abbasian
a,
Armin
Salek Maghsoudi
 a,
Ghorban
Taghizadeh
d,
Jalal
Pourahmad†
*e and
Mohammad
Sharifzadeh†
*a
a,
Ghorban
Taghizadeh
d,
Jalal
Pourahmad†
*e and
Mohammad
Sharifzadeh†
*a
aDepartment of Toxicology and Pharmacology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran. E-mail: msharifzadeh@tums.ac.ir; Tel: +98(21) 6695-9095
bToxicology and Diseases Group, The Institute of Pharmaceutical Sciences (TIPS), Tehran University of Medical Sciences, Tehran, Iran
cDepartment of Cardiology, Cardiovascular Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
dRehabilitation Research Center, Department of Occupational Therapy, School of Rehabilitation Sciences, Iran University of Medical Sciences, Tehran, Iran
eDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran. E-mail: jpourahmadjaktaji@utoronto.ca; Tel: +98(21) 88200090
First published on 8th November 2019
Abstract
Mercury is a highly poisonous heavy metal abundantly found in the environment in its inorganic form. Although evidence have been provided about the possible role of inorganic mercury in the pathology of Alzheimer's disease (AD), its effect on cognitive and mitochondrial functions have not yet been completely understood. Thus, the purpose of the present study was to examine the effects of the chronic exposure to mercuric chloride (0.4, 0.8 and 1.6 mg kg−1 per day for 3 weeks) through drinking water (by gavage) on spatial learning and memory and hippocampal mitochondrial function in beta-amyloid treated rats (1 μg per μL per side, intrahippocampally). The acquisition and retention of spatial memory were evaluated by the Morris water maze (MWM) test. Several parameters of hippocampal mitochondrial function were also measured. The results indicated that mercury impaired spatial learning and memory as well as aggravated Aβ-induced memory impairments in a concentration-dependent manner. Furthermore, mercury exposure resulted in a significant increase in ROS generation, MMP collapse, mitochondrial swelling, glutathione oxidation, lipid peroxidation, and outer membrane damage. In addition, a reduced cytochrome c oxidase (complex IV) activity and elevated ADP/ATP ratio in the rats' hippocampus was also observed. The findings of the current study revealed that chronic mercury exposure led to mitochondrial dysfunction, which resulted in spatial memory impairments. The results also showed that mercury can exacerbate the toxic effects of Aβ on spatial memory and hippocampal mitochondrial function.
Significance to metallomicsExposure to even a small amount of mercury may cause serious health problems and is a threat to the nervous, digestive and immune system. The small amount of this metal also has toxic effects on the lungs, kidneys, skin, and eyes. As mercuric chloride is a very poisonous form of mercury, chronic exposure to the various amounts of it can permanently damage the brain and cause Alzheimer's disease. |
Introduction
Mercury (hydrargyrum or Hg) is an extremely hazardous heavy metal for humans and other organisms with well-known neurotoxic effects.1,2 Mercury can be found in various forms, including organic mercury (methylmercury, MeHg), inorganic mercury (IM), mercuric ions (Hg+ and Hg++), and elemental mercury or metallic mercury (Hg0). The most common form of toxic mercury in humans is MeHg. However, intestinal microflora can slowly metabolize MeHg into inorganic mercury.3Human populations throughout the world are exposed to environmental inorganic mercury, which induces toxic effects.4 It can enter the air through mining the deposits of mercury ores. Coal-fired power plants are the biggest industrial sources of inorganic mercury emission. Factories that use mercury can also release inorganic mercury into the water. So, populations living downstream of mining areas or near dams or factories with large scale projects that concentrate mercury in the environment may be exposed to inorganic mercury.5
Despite the low lipid solubility of inorganic mercury, it can enter the central nervous system (CNS) and cause different neurologic disorders.6 However, the precise mechanism of inorganic mercury accumulation in the brain following chronic exposure is not well known. Szumafiska et al. suggested that the impaired activity of Na/K ATPase in the microvessels of the cerebral cortex can be a possible pathway for inorganic mercury absorbance.7
Alzheimer's disease (AD) is a prevalent neurodegenerative disorder and the most common form of dementia in the aging population throughout the world.8,9 Although many studies have been conducted on AD, the exact mechanism of its pathology is not clear; however, two different accumulations (i.e., intracellular neurofibrillary tangles and senile plaques in the extracellular matrix) have been identified in the brain.10 Senile plaques are formed by the extracellular accumulation of β-amyloid (Aβ).10,11 Aβ is the production of an amyloid precursor protein (APP) cleavage by β-secretase (beta-site APP cleaving enzyme 1, BACE1) and γ-secretase enzymes.12,13 Moreover, various proteases such as the endothelin-converting enzyme, insulin-degrading enzyme and neprilysin degrade the Aβ.14,15 Animal models have shown that brain neprilysin plays a leading role in Aβ degradation.16,17 Reduced neprilysin expression and enhanced APP expression have also been observed in the hippocampus of patients with early-onset familial AD.15,17 Recently, many metals have been suggested as risk factors related to the AD pathogenesis, and the concentrations of different metals, including aluminum, iron and copper, were evaluated in brains affected by AD.18,19 Some studies have found a link between mercury exposure and neurological degeneration,20 oxidative stress and cell cytotoxicity.21,22 A marked increase in mercury concentrations in AD brains was shown in some autopsy investigations.23,24In vitro studies have shown that all pathological alterations induced by AD were increased by a subsequent chronic exposure to inorganic mercury.25 The extremely high affinity of inorganic mercury for selenoproteins indicated that it resulted in neurodegenerative diseases through the disruption of redox regulation.26–28 The mitochondrion is an important source for the formation of reactive oxygen species (ROS).29,30 It was found that elevated ROS generation resulted in mitochondrial and cellular dysfunction, leading to behavioral impairments.31 Some studies have shown that mercury exposure caused the induction of a mitochondrial permeability transition pore (MPTP) and decreased the activity of mitochondrial enzymes.32 The MPTP was induced by the permeability transition pore (PTP) opening, which is located in the inner mitochondrial membrane. The increased permeability of the inner mitochondrial membrane causes the collapse of mitochondrial membrane potential (MMP) and finally increased the ADP/ATP ratio in the rat brain.32,33 An important role of brain mitochondria in the plasticity needed for spatial learning and memory has been reported previously.34,35 Therefore, it was hypothesized that mercury leads to the impairment of spatial learning and memory via hippocampal mitochondrial dysfunction. Nevertheless, this hypothesis needs strong validation and not enough research has been done on the mechanistic connection between memory impairments and mitochondrial dysfunction caused solely by mercury administration and with Aβ.
Hence, we evaluated the effects of chronic exposure to mercuric chloride through drinking water (alone and in combination with Aβ) on spatial learning and memory. We also investigated the possible role of hippocampal mitochondrial dysfunction in the effects of mercuric chloride on spatial learning and memory.
Materials and methods
Materials
HgCl2 was bought from Merck (Darmstadt, Germany). 4-2-Hydroxyethyl-1-piperazineethanesulfonic acid (HEPES), dimethyl sulfoxide (DMSO), rotenone (Rot), thiobarbituric-acid (TBA), D-mannitol, Tris–HCl, 2′,7′ dichlorodihydrofluorescein diacetate (DCFH-DA), sucrose, sodium-succinate, Na2HPO4, KCl, MgCl2, Coomassie blue, Rhodamine123 (Rh123), xylazine, ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetra acetic acid (EGTA), ketamine and amyloid-beta (1-42) were purchase from Sigma Aldrich Chemical Co. (St. Louis, MO, USA).Animals
Male albino-Wistar rats, weighing 220–250 g, were used in our investigation. All of the animals were kept in cages with easy access to water and food until surgery. They were also kept at room temperature (20–22 °C) with a controlled humidity on a 12 hour light–dark cycle. In the current study, eight groups of rats (n = 7 for each group, 8 × 7 = 56 rats) were classified into two categories: without amyloid-beta and with amyloid-beta.(ii) Group that received 0.4 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
(iii) Group that received 0.8 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
(iv) Group that received 1.6 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
(vi) Group with injected Aβ (1 μg per μL per side) intra-CA1 bilaterally once, and then after recovery received 0.4 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
(vii) Group with injected Aβ (1 μg per μL per side) intra-CA1 bilaterally once, and then after recovery received 0.8 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
(viii) Group with injected Aβ (1 μg per μL per side) intra-CA1 bilaterally once, and then after recovery received 1.6 mg kg−1 Hg2+ daily in drinking water by gavage for 21 days.
All animal experiments were done based on guidelines from the Ethical Committee for Laboratory Animals Care and Usage, Tehran University of Medical Sciences. We endeavored to reduce any pain as much as possible to the animals and use the least number of rats.
Preparation of beta-amyloid peptide 1-42 (Aβ1-42)
To prepare the aliquots of amyloid-beta, Aβ was dissolved in phosphate buffer saline (PBS 0.1 M) to yield a 200 μg μL−1 concentration and then incubated for five days at 37 °C. Aliquots were stored at −20 °C until use. On the day of trial, PBS was added to the solution as a diluent in order to obtain an absolute Aβ concentration of 1 μg μL−1.Stereotaxic surgery
To anesthetize the rats, 20 mg kg−1 xylazine combined with 100 mg kg−1 ketamine was injected intra-peritoneally. Then, the rats were put in a stereotaxic device (Stoelting, USA). In the scalp region, an incision was placed on the midline. In the intrahippocampal CA1 region, the Aβ injection was performed based on the rat brain atlas of Paxinos and Watson, (coordinates: 3.2 mm posterior and 2.4 mm lateral to the bregma, 2.7 mm from the ventral to skull surface36). Microinjections were administered using a 10 μL Hamilton syringe (0.5 μL min−1) and the needle was kept in the tissue for one minute.Spatial learning and memory assessment
The Morris water maze (MWM) test was used to measure spatial learning and memory. The test apparatus consisted of a black circular pool (diameter: ∼136 cm, depth: ∼60 cm), a platform (10 cm in diameter) and four equal quadrants. The temperature of the water was about 25 ± 2 °C and the water depth was 40 cm. The platform was submerged 1 cm beneath the water surface in the middle of the target quadrant (north–west). All of the rats participated in a four-day training session that included one block of four trials each day. In each trial, the rats were randomly placed in one of the four quadrants and allowed to swim freely to find the platform. If the rats did not find the submerged platform within the allocated time (i.e. 90 s), the examiner guided them to the platform. All of the training sessions were recorded by a camera that was positioned over the pool. Then, they were allowed to rest for about 20 s on the platform. Spatial learning was measured by different parameters, including swimming speed, escape latency (time to reach the platform) and traveled distance (path length to reach the platform). In order to evaluate the spatial memory, the probe test was performed after training completion, which measured the time that each rat spent in the target quadrant. The target platform was removed and the rats were placed in the quadrant opposite to the target quadrant and then permitted to swim in the pool for 90 s. Ethovision software (Noldus Information Technology, Wageningen, Netherlands) was used to calculate the different spatial learning and memory parameters of the MWM.Hippocampal mitochondrial function
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and the aforementioned layer was discarded. Next, the mitochondrial pellet was washed and then suspended in a solution of the isolation medium. Finally, it was centrifuged for approximately 10 min at 10000g. Mitochondrial suspensions were prepared in Tris buffer (0.05 M Tris–HCl, 0.25 M sucrose, 2.0 mM MgCl2, 20 mM KCl and 1.0 mM Na2HPO4, pH of 7.4) at 4 °C, except for the mitochondrial samples that were used to assay the ROS content, MMP and mitochondrial swelling, which were respectively incubated in the respiration buffer (10 mM Tris, 0.32 mM sucrose, 20 mM Mops, 0.5 mM MgCl2, 50 μM EGTA, 5 mM sodium succinate and 0.1 mM KH2PO4), MMP assay buffer (68 mM D-mannitol, 220 mM sucrose, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 5 mM sodium succinate, 50 μM EGTA, 10 mM HEPES and 2 μM rotenone) and swelling buffer (3 mM HEPES, 70 mM sucrose, 230 mM mannitol, 5 mM succinate, 2 mM Tris-phosphate and 1 μM of rotenone). The protein concentrations were measured using the Coomassie blue protein-binding procedure.37,38 All of the steps were conducted on ice to ensure the quality of the mitochondrial preparation.
000g (10 min at 4 °C). The upper layer was removed and then neutralized with KOH (4 M). ATP and ADP were measured according to the standard curve in order to calculate the ratio.
000g and the aforementioned layer was discarded. Next, the mitochondrial pellet was washed and then suspended in a solution of the isolation medium. Finally, it was centrifuged for approximately 10 min at 10000g. Mitochondrial suspensions were prepared in Tris buffer (0.05 M Tris–HCl, 0.25 M sucrose, 2.0 mM MgCl2, 20 mM KCl and 1.0 mM Na2HPO4, pH of 7.4) at 4 °C, except for the mitochondrial samples that were used to assay the ROS content, MMP and mitochondrial swelling, which were respectively incubated in the respiration buffer (10 mM Tris, 0.32 mM sucrose, 20 mM Mops, 0.5 mM MgCl2, 50 μM EGTA, 5 mM sodium succinate and 0.1 mM KH2PO4), MMP assay buffer (68 mM D-mannitol, 220 mM sucrose, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 5 mM sodium succinate, 50 μM EGTA, 10 mM HEPES and 2 μM rotenone) and swelling buffer (3 mM HEPES, 70 mM sucrose, 230 mM mannitol, 5 mM succinate, 2 mM Tris-phosphate and 1 μM of rotenone). The protein concentrations were measured using the Coomassie blue protein-binding procedure.37,38 All of the steps were conducted on ice to ensure the quality of the mitochondrial preparation.
000g (10 min at 4 °C). The upper layer was removed and then neutralized with KOH (4 M). ATP and ADP were measured according to the standard curve in order to calculate the ratio.
Mercury concentration measurements
The mercury concentration was determined in the hippocampus using cold vapor atomic absorption spectrometry (Semi-automated Mercury Analyzer, model Hg-201, Sanso Seisakusho Co. Ltd, Tokyo, Japan). Succinctly, a homogenized sample (150 mg of wet weight) was digested by adding 2 mL of nitric acid and perchloric acid 1 + 1 (HNO3–HClO4) and 5 mL of sulfuric acid (H2SO4) to a sample digestion bottle. Then, the sample was subjected to 30 min of heat treatment on a hot plate (200–230 °C), after which the final volume was achieved using double-distilled deionized water.Statistical analysis
The mean value of each spatial learning parameter (i.e., swimming speed, traveled distance and escape latency) was calculated over four trials from the four training days. The main and interaction effects of the Aβ and mercury doses on the spatial memory and hippocampal mitochondrial function were analyzed using a two-way analysis of variance (ANOVA). The Bonferroni post hoc test was used for multiple comparisons and the P-value was set at 0.05.Results
Spatial learning and memory
The main effect of the mercury doses without amyloid-beta was significant for both the mean of the traveled distance (iii: P < 0.05) and escape latency (ii P < 0.01) over the 4 training days in the MWM compared to that for the control group (i). In the amyloid-beta groups, the mean of both the traveled distance and escape latency were significant (viii: P < 0.05) over the 4 training days of the MWM compared to that for the group that just received Aβ (v) (Fig. 1A and B). There wasn’t any significant difference between all eight groups in the mean of the velocity during the 4 training days of the MWM (Fig. 1C).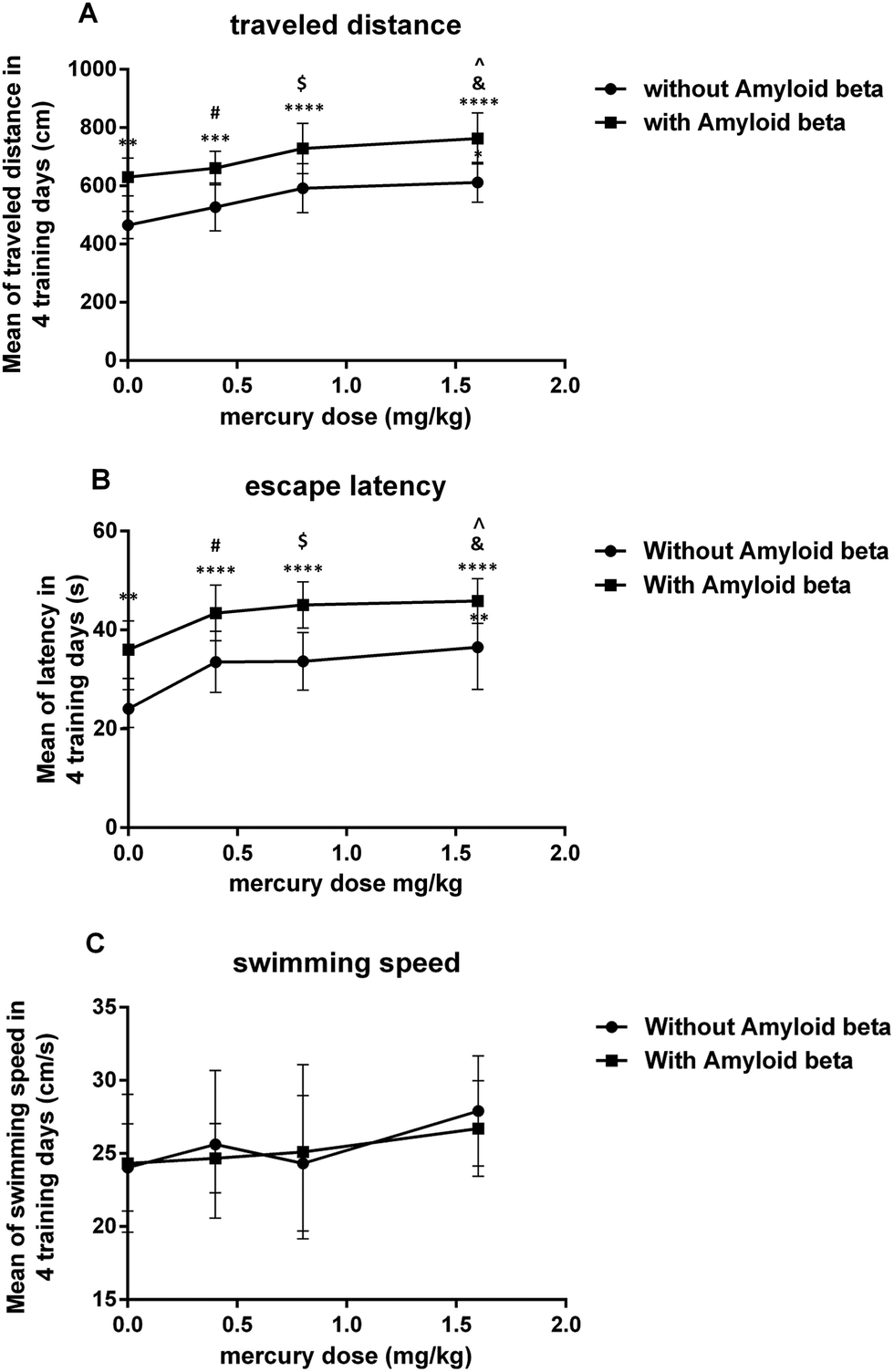 | ||
| Fig. 1 Plot on the effect of mercury doses without Aβ and with Aβ on the mean of the traveled distance (A), escape latency (B) and swimming speed (C) for 4 days in the MWM test. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared i with the control (i) group. #P < 0.05 compared ii with the vi group, $P < 0.05 compared iii with the vii group and &P < 0.05 compared iv with the viii group. ^P < 0.05 compared with just the Aβ (v) group. | ||
The results of the probe trial test indicate that this parameter was significantly reduced in group iii (P < 0.05) and group iv (P < 0.01) compared with the control group. Moreover, in with amyloid-beta groups, there was a significant difference in vii (P < 0.05) and viii (P < 0.01) groups compared with just the Aβ injected group (Fig. 2).
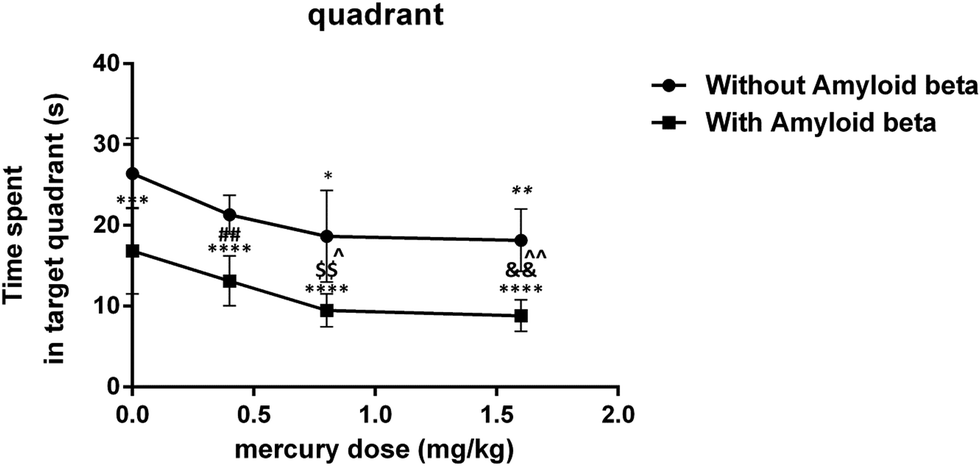 | ||
| Fig. 2 Plot on the effect of mercury doses without Aβ and with Aβ on the time spent in the target quadrant of the probe trial test for MWM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared i with the control (i) group. ##P < 0.01 compared ii with the vi group, $$P < 0.01 compared iii with the vii group and &&P < 0.01 compared iv with the viii group. ^P < 0.05, ^^P < 0.01 compared with just the Aβ (v) group. | ||
Hippocampal mitochondrial function
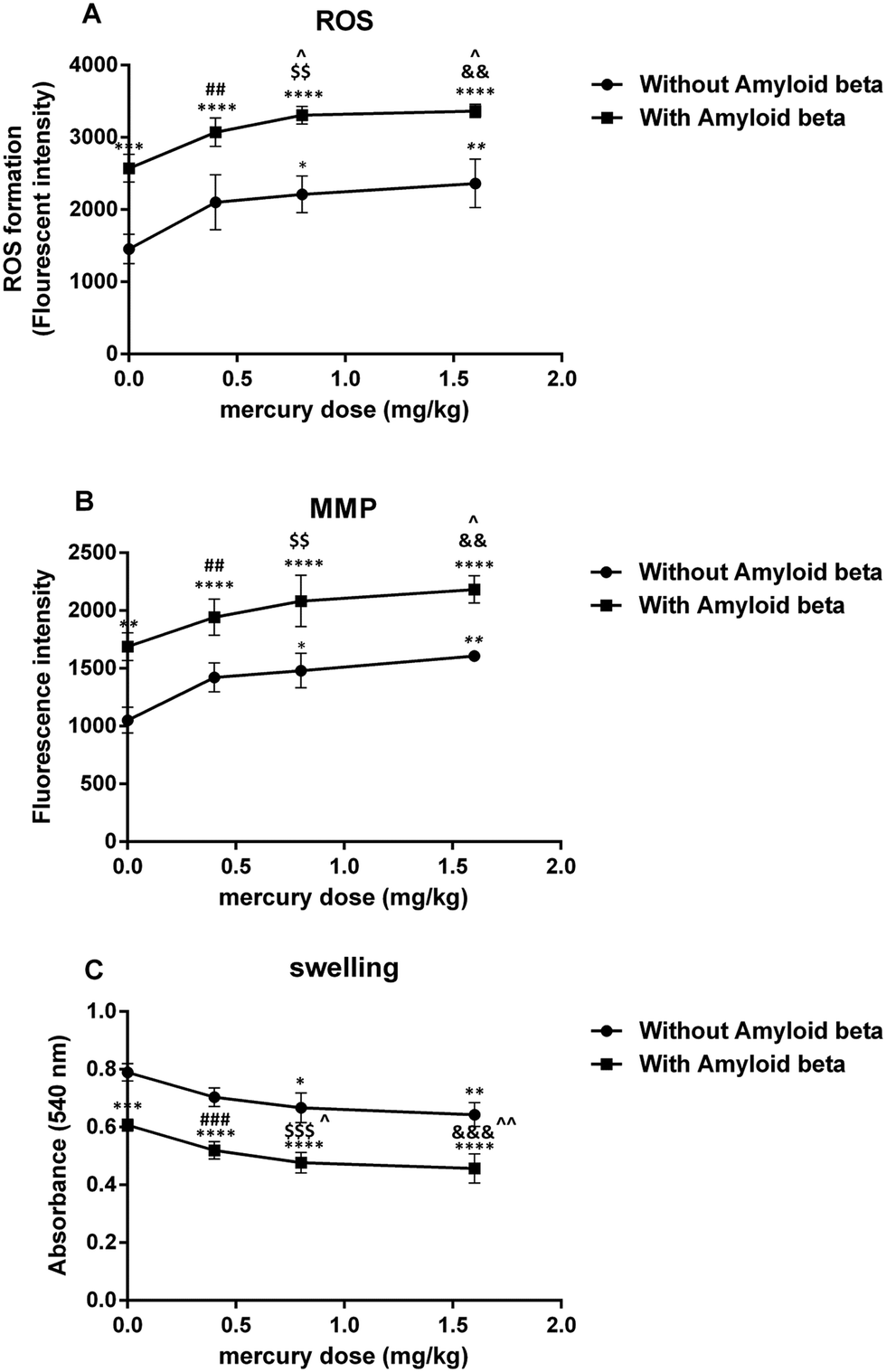 | ||
| Fig. 3 Plot on the effect of mercury doses without Aβ and with Aβ on ROS formation (A), MMP (B) and swelling (C). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with the control (i) group. ##P < 0.01, ###P < 0.001 compared ii with the vi group, $$P < 0.01, $$$P < 0.001 compared iii with the vii group and &&P < 0.01, &&&P < 0.001 compared iv with the viii group. ^P < 0.05, ^^P < 0.01 compared with just the Aβ (v) group. | ||
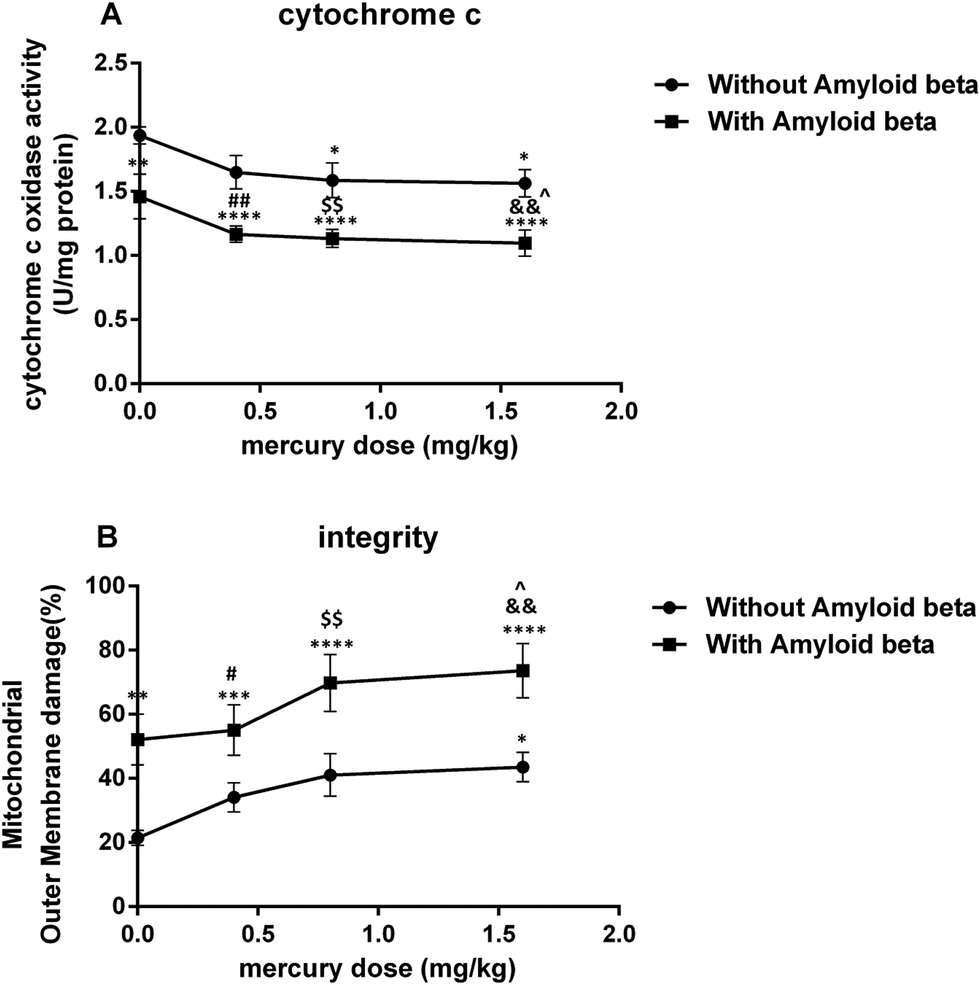 | ||
| Fig. 4 Plot on the effect of mercury doses without Aβ and with Aβ on cytochrome c oxidase activity (A) and mitochondrial outer membrane damage (B). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with the control (i) group. #P < 0.05, ##P < 0.01 compared ii with the vi group, $$P < 0.01 compared iii with the vii group and &&P < 0.01 compared iv with the viii group. ^P < 0.05 compared with just the Aβ (v) group. | ||
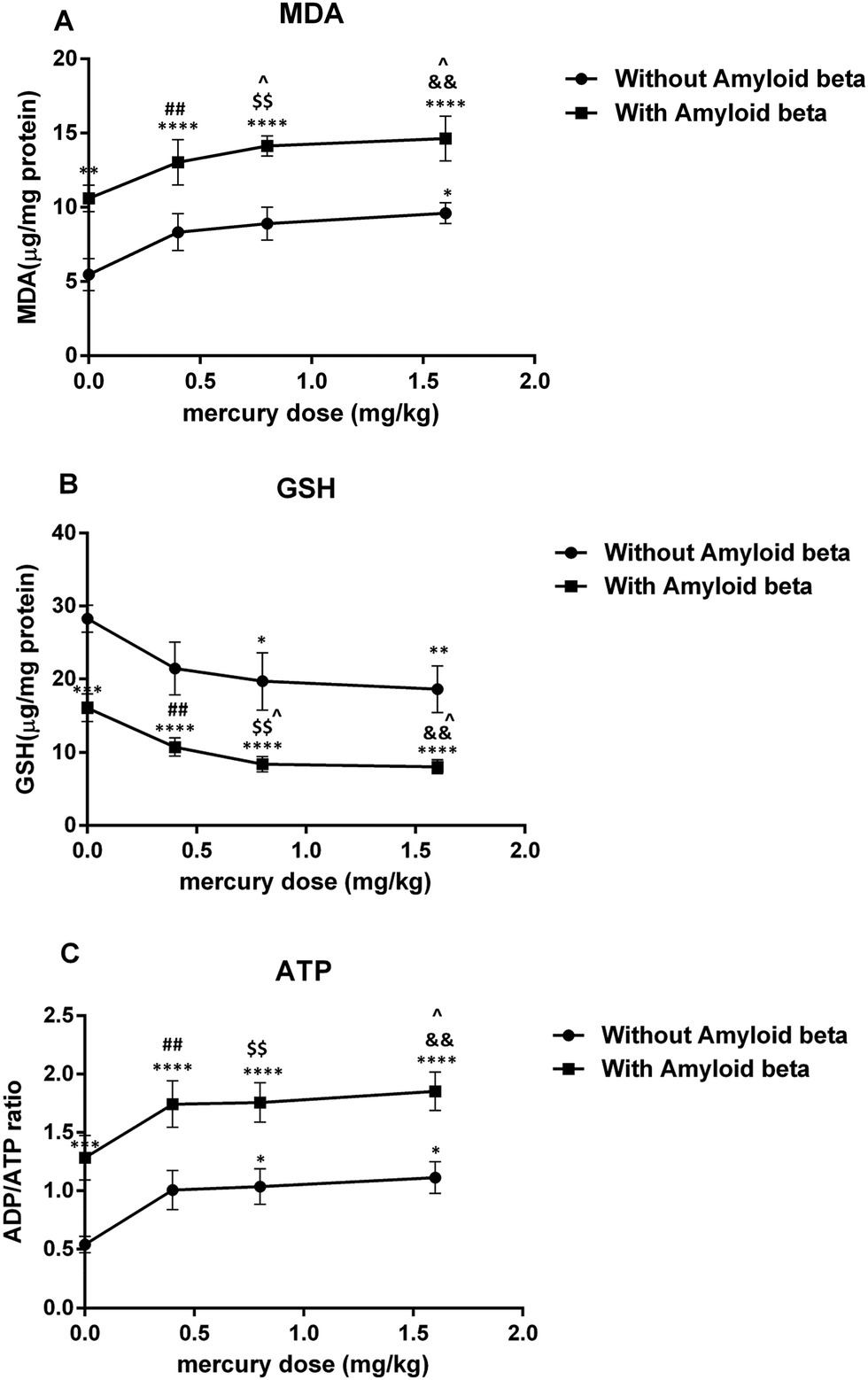 | ||
| Fig. 5 Plot on the effect of mercury doses without Aβ and with Aβ on lipid peroxidation (A), the GSH content (B) and the ADP/ATP ratio (C). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 compared with the control (i) group. ##P < 0.01 compared ii with the vi group, $$P < 0.01 compared iii with the vii group and &&P < 0.01 compared iv with the viii group. ^P < 0.05 compared with just the Aβ (v) group. | ||
Hippocampal mercury concentration
As shown in Table 1, the Hg concentrations in the hippocampus samples were significantly higher in the mercury administered groups (i.e., mercury, 0.4 mg kg−1 (alone and with Aβ: P < 0.05), 0.8 mg kg−1 (alone and with Aβ: P < 0.01) and 1.6 mg kg−1 (alone and with Aβ: P < 0.001)), in comparison with the control group.| Groups | Hippocampal mercury concentration (μg g−1 wet weight) |
|---|---|
| The mercury concentration was determined by a cold vapor atomic absorption spectrophotometer as described in the Materials and methods section. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group. The values are shown as mean ± SD for the seven rats in each group. | |
| Control | 0.042 ± 0.01 |
| Hg 0.4 mg kg−1 | 0.053 ± 0.02* |
| Hg 0.8 mg kg−1 | 0.064 ± 0.03** |
| Hg 1.6 mg kg−1 | 0.094 ± 0.04*** |
| Aβ 1 μg per μL per side | 0.044 ± 0.03 |
| Aβ + Hg 0.4 mg kg−1 | 0.055 ± 0.03* |
| Aβ + Hg 0.8 mg kg−1 | 0.065 ± 0.04** |
| Aβ + Hg 1.6 mg kg−1 | 0.097 ± 0.03*** |
Discussion
The results of the present study indicated that chronic exposure to mercuric chloride (3 weeks) through drinking water caused spatial learning and memory impairments and hippocampal mitochondrial dysfunction. Furthermore, mercuric chloride exacerbated the Aβ-induced impairments of spatial learning and memory as well as hippocampal mitochondrial dysfunction. As far as we know, our study is the first study to investigate the effects of chronic mercury exposure alone and with amyloid-beta on spatial learning and memory as well as hippocampal mitochondrial function. Several studies have shown that mercuric chloride toxicity in AD could depend on its ability to misfold and aggregate amyloid-beta proteins. In vitro studies by Song and Choi demonstrated that the treatment of PC12 cells with 10, 100 and 1000 nM HgCl2 for 48 hours significantly increased Aβ accumulation in a dose and time-dependent manner. They suggested that mercuric chloride induced Aβ accumulation via APP overproduction and NEP reduction.47 The study by Olivieri et al. on SHSY5Y neuroblastoma cells also suggested the role of inorganic mercury in the pathophysiological mechanisms of AD. They showed that the administration of 50 mg L−1 (180 nM) of HgCl2 caused cellular GSH reduction, induced the release of Aβ peptides (Aβ40 and Aβ42) and significantly increased Tau phosphorylation, thus suggesting a possible link between AD and inorganic mercury.21 Meleleo et al. suggested that HgCl2 interacted directly with Aβ in a dose-dependent manner. Their experimental data showed that mercuric chloride induced a decrease in the Aβ42 channel frequency and also the formation of large and amorphous aggregates of Aβ42.48 Teixeira et al. found short- and long-term memory impairments after the chronic administration of mercuric chloride (0.375 mg kg−1 per day for 45 days) and they also postulated that chronic low doses of inorganic mercury accumulated in the brain hippocampus and cortex.49 Mello-Carpes et al. reported that long-term exposure (30 and 60 days) to low concentrations of mercuric chloride (1st dose: 4.4 μg kg−1 and later dose: 0.07 μg kg−1 per day, intramuscular) caused memory impairments in object recognition and inhibitory avoidance tests.50Considering the important role of mitochondria in memory function, a possible mechanism of mercury-induced memory impairment may be mitochondrial dysfunction, which was observed in the present study. The mitochondrial electron transport chain (ETC) complexes (I and III) have a major role in ROS generation and their dysfunction leads to oxidative stress.51 Their important role has been shown in the pathogenesis of different brain disorders and injuries.52 Our findings showed that mercury increased ROS generation in the hippocampal mitochondria of Aβ treated rats. This was in accordance with Belyaeva et al.'s findings, which showed the main role of mitochondrial ETC (motets) in mercury-induced neurotoxicity.53 They suggested that the incubation of the PC12 cell line with Hg2+ induced intracellular ROS production, inhibited respiratory function, decreased MMP and caused cell death in a dose and time-dependent manner. The study by Olivieri et al. on SHSY5Y neuroblastoma cells also suggested the role of inorganic mercury in the pathophysiological mechanisms of AD.21 They showed that the administration of 50 mg L−1 (180 nM) of HgCl2 caused cellular GSH reduction as well as a marked increase in the release of Aβ into the cell culture and Tau phosphorylation. In addition, Lund et al. showed that HgCl2 (1.5 mg kg−1, intra-peritoneally) increased H2O2 production in the mitochondrial ETC of rat kidney, especially in the ubiquinone-cytochrome b region.54
MMP has an important role in regulating mitochondrial function and its collapse is the main stimulus for cell death. Our study indicated that the decrease in MMP after a chronic exposure to a high dose of mercury in groups that received either mercury alone or a combination with Aβ. This result was in agreement with the study by Palmeira et al., which reported decreased MMP in association with intracellular GSH and ATP levels following mercuric chloride exposure (1, 5 and 10 μM) in freshly isolated rat liver mitochondria, leading to a concentration and time-dependent cell death.32
GSH, the main antioxidant system for mitochondria against various reactive oxygen species and hydrogen peroxide, has an important role in the maintenance of the sulfhydryl groups in its reduced form.54 Thus, the depletion of the reduced GSH form in the mitochondria led to a severe deficit in their protective activity against oxidative damage and then finally an increase in lipid peroxidation. Based on our data, chronic mercury exposure resulted in enhanced GSH oxidation and lipid peroxidation in hippocampal mitochondria. In addition, the reduced form of GSH was needed for thiol group maintenance of the proteins in the mitochondrial membrane.55 When the thiol groups were oxidized, conformational changes in the pore complex caused a mitochondrial permeability transition56 (MPT). Therefore, the mercury-induced ROS formation may have led to GSH oxidation and lipid peroxidation, which impaired the integrity of the mitochondrial outer membrane, and opened the MPT pores, which are important factors in necrosis and apoptosis.57,58 Nevertheless, our findings differed from the study by Berntssen et al., which did not show any significant effects of feeding with 100 mg kg−1 inorganic mercury for four months on the neural glutathione peroxidase and superoxide dismutase activity, as well as the post-feeding behavior and TBARS in Atlantic salmon parr.59 This discrepancy may have been due to the different animal species used in this study compared to our own.
The results of the present study demonstrated that mercury caused hippocampal mitochondrial swelling, which was exacerbated in the Aβ-administered groups. This result was in accordance with previous in vitro studies that demonstrated mercury-induced mitochondrial swelling in kidney cortical cells.53 Several factors such as sulfhydryl reactive compounds, heavy metals and oxidants are capable of inducing a permeability transition.60 Therefore, it was suggested that the mercury-induced and enhanced mitochondrial swelling observed in the current study may have been caused by mitochondrial outer membrane damage, which was observed in both, the mercury and mercury plus Aβ treated groups.
In addition, it was suggested that the mercury-induced lipid oxidation in the hippocampal mitochondria (due to respiratory chain failure, increased ROS production and the GSH reduction) may have resulted in mitochondrial outer membrane damage and a reduced cytochrome c oxidase activity.
It is well-known that mitochondrial function is necessary for ATP formation by oxidative phosphorylation. Indeed, ATP operates as a switch between necrosis and apoptosis. Apoptosis needs ATP and the reduction of ATP causes cell necrosis.61 We found that mercury decreased ATP generation, increased the ADP/ATP ratio in hippocampal mitochondria and augmented the ADP/ATP ratio in Aβ-treated groups. As suggested by Pereira et al., this may have occurred due to mitochondrial ETC inhibition and the opening of the MPT pore.62 When the MPT pore opened, an unlimited number of protons moved through the inner membrane and finally causes oxidative phosphorylation uncoupling.63 Under this condition, the remaining ATP was consumed for MMP maintenance, which led to more depletion of the ATP content and increased ROS formation.63
The inactivation of cyt-reductive enzymes relied on GSH. The change in the ADP/ATP ratio suggested an impairment in oxidative phosphorylation and a marked effect of Aβ on this phenomenon. In conclusion, chronic exposure to inorganic Hg, despite its low liposolubility, promoted the deposition of Hg in the hippocampus of rats. A probable mechanism is that inorganic Hg promoted an interference in the enzymatic activity and affected ionic movement in Na+/K+ ATPase in the cerebral cortex layers, which could have explained their accumulation in these brain regions. Another hypothesis is the use of amino acid transporters, particularly the cysteine transporter, to cross the blood-brain barrier.64 The extraordinarily high affinity of mercuric chloride for selenium and selenoproteins disrupted the cellular redox balance, especially in the hippocampal neurons. Hg inactivated methionine synthase and the resultant decrease in the methylation of phosphatase 2 A (PP2A) promoted tau hyperphosphorylation and Aβ secretion. The accumulation of Aβ interfered with apolipoprotein e receptor (ApoER2)-mediated selenoprotein P (SelP) uptake, further limiting selenium availability and creating a self-reinforcing pathological cycle. The accumulation of mercury in the brain affected the ability of SelP to fully buffer its toxicity, therefore contributing to oxidative stress and apoptosis.65,66 The immunohistochemical analyses of Aragao et al. revealed that chronic exposure to HgCl2 promoted tissue changes in the CA1, CA3, and hilus hippocampal regions. The reduction of glial fibrillary acidic protein (GFAP), the main protein constituent in the astrocyte cytoskeleton, due to HgCl2 neurotoxicity showed a disorder in the mnemonic neural processes because these glia cells are auxiliary to the neurons and act in metabolic processes, synaptic transmission and local homeostasis.
Chronic exposure to HgCl2 demonstrated neuronal cell death in the hippocampal areas. The mechanism for this process may have been cytotoxicity or apoptosis, which was triggered by oxidative stress.67,68
Conclusion
The present study showed that chronic mercury exposure (3 weeks) through drinking water led to enhanced ROS generation, followed by MMP reduction, mitochondrial swelling, mitochondrial outer membrane damage, an elevated ADP/ATP ratio and finally reduced cytochrome c oxidase activity in a dose-dependent manner. Because mitochondria play an important role in the synaptic plasticity required for learning and memory, mercury-induced mitochondrial dysfunction in the hippocampus might be a possible mechanism for mercury-induced impairments in learning and memory. Moreover, mercury might worsen the damaging effects of amyloid-beta in the hippocampal mitochondria and spatial learning and memory in Aβ-administered animals.Abbreviations
| MWM | Morris water maze test |
| Aβ | Beta-amyloid peptide 1-42 |
| ROS | Reactive oxygen species |
| MMP | Mitochondrial membrane potential |
| IH | Intrahippocampally |
| MTPT | Mitochondrial permeability transition pore |
| PTP | Permeability transition pore |
| ATP | Adenosine triphosphate |
| ETC | Electron transport chain |
| DCFH-DA | Dichlorodihydrofluorescein diacetate |
| DCF | Dichlorofluorescin |
| MDA | Malondialdehyde |
| TBA | Thiobarbituric acid |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by grants (No. 29528-33-02-94) from the Tehran University of Medical Sciences.References
- T. W. Clarkson, The three modern faces of mercury, Environ. Health Perspect., 2002, 110, 11–23 CrossRef CAS PubMed.
- T. W. Clarkson, L. Magos and G. J. Myers, The toxicology of mercury—current exposures and clinical manifestations, N. Engl. J. Med., 2003, 349, 1731–1737 CrossRef CAS PubMed.
- R. A. Bernhoft, Mercury toxicity and treatment: a review of the literature, J. Environ. Public Health, 2012, 460508 Search PubMed.
- L. R. Goldman, M. W. Shannon and A. A. O. Pediatrics, Committee on Environmental Health Technical report: mercury in the environment: implications for pediatricians, Pediatrics, 2001, 108, 197–205 CrossRef CAS PubMed.
- D. Obrist, J. L. Krik, L. Zhang, M. Jiskra and N. E. Selin, A review of global environmental mercury processes in response to human and natural perturbations: Changes of missions, J. Clim., 2018, 47(2), 116–140 Search PubMed.
- T. W. Clarkson and L. Magos, The toxicology of mercury and its chemical compound, Crit. Rev. Toxicol., 2006, 36, 609–662 CrossRef CAS PubMed.
- G. Szumańska, R. Gadamski and J. Albrecht, Changes of the Na/K ATPase activity in the cerebral cortical microvessels of rat after single intraperitoneal administration of mercuric chloride: histochemical demonstration with light and electron microscopy, Acta Neuropathol., 1993, 86, 65–70 CrossRef PubMed.
- M. Goedert and M. G. Spillantini, A century of Alzheimer's disease, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- C. Qiu, D. De Ronchi and L. Fratiglioni, The epidemiology of the dementias: an update, Current Opinion in Psychiatry, 2007, 20, 380–385 CrossRef PubMed.
- R. A. Armstrong, The pathogenesis of Alzheimer's disease: a reevaluation of the “amyloid cascade hypothesis”, J. Alzheimer's Dis., 2011, 630865 CAS.
- M. P. Mattson, Pathways towards and away from Alzheimer's disease, Nature, 2004, 430, 631 CrossRef CAS PubMed.
- M. Carter, G. Simms and D. Weaver, The development of new therapeutics for Alzheimer's disease, Clin. Pharmacol. Ther., 2010, 88, 475–486 CrossRef CAS PubMed.
- K. Nägga, J. Gottfries, K. Blennow and J. Marcusson, Cerebrospinal fluid phospho-Tau, total Tau and β-Amyloid 1-42 in the differentiation between Alzheimer's disease and vascular dementia, Dementia Geriatr. Cognit. Disord., 2002, 14, 183–190 CrossRef PubMed.
- H. Guan, Y. Liu, A. Daily, S. Police, M. H. Kim, S. Oddo, F. M. LaFerla, J. R. Pauly, M. P. Murphy and L. B. Hersh, Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer's disease, J. Neurosci. Res., 2009, 87, 1462–1473 CrossRef CAS.
- J. S. Miners, Z. Van Helmond, K. Chalmers, G. Wilcock, S. Love and P. G. Kehoe, Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy, J. Neuropathol. Exp. Neurol., 2006, 65, 1012–1021 CrossRef CAS.
- N. Iwata, S. Tsubuki, Y. Takaki, K. Watanabe, M. Sekiguchi, E. Hosoki, M. Kawashima-Morishima, H.-J. Lee, E. Hama and Y. Sekine-Aizawa, Identification of the major Aβ 1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition, Nat. Med., 2000, 6, 143 CrossRef CAS.
- N. Iwata, S. Tsubuki, Y. Takaki, K. Shirotani, B. Lu, N. P. Gerard, C. Gerard, E. Hama, H.-J. Lee and T. C. Saido, Metabolic regulation of brain Aβ by neprilysin, Science, 2001, 292, 1550–1552 CrossRef CAS PubMed.
- A. D. Armendariz, M. Gonzalez, A. V. Loguinov and C. D. Vulpe, Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App, Physiol. Genomics, 2004, 20, 45–54 CrossRef CAS.
- M. Lovell, J. Robertson, W. Teesdale, J. Campbell and W. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- C. C. Leong, N. I. Syed and F. L. Lorscheider, Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury, NeuroReport, 2001, 12, 733–737 CrossRef CAS PubMed.
- G. Olivieri, C. Brack, F. Müller-Spahn, H. Stähelin, M. Herrmann, P. Renard, M. Brockhaus and C. Hock, Mercury induces cell cytotoxicity and oxidative stress and increases β-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells, J. Neurochem., 2000, 74, 231–236 CrossRef CAS.
- G. Olivieri, M. Novakovic, E. Savaskan, F. Meier, G. Baysang, M. Brockhaus and F. Müller-Spahn, The effects of β-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and β-amyloid secretion, Neuroscience, 2002, 113, 849–855 CrossRef CAS.
- L. Björkman, B. F. Lundekvam, T. Lægreid, B. I. Bertelsen, I. Morild, P. Lilleng, B. Lind, B. Palm and M. Vahter, Mercury in human brain, blood, muscle and toenails in relation to exposure: an autopsy study, Environ. Health, 2007, 6, 30 CrossRef.
- W. Ehmann, W. Markesbery, M. Alauddin, T. Hossain and E. Brubaker, Brain trace elements in Alzheimer's disease, Neurotoxicology, 1986, 7, 195–206 CAS.
- J. Mutter, A. Curth, J. Naumann, R. Deth and H. Walach, Does inorganic mercury play a role in Alzheimer's disease? A systematic review and an integrated molecular mechanism, J. Alzheimer's Dis., 2010, 22, 357–374 CAS.
- B. Carlson, H. Mix, Y. Zhang, K. Saira, R. Glass, M. Berry, V. Gladyshev and D. hatfield, Biosynthesis of selenocysteine on its tRNA in eukaryotes, PLoS Biol., 2007, 5, e4 Search PubMed.
- R. F. Burk and K. E. Hill, Selenoprotein P—expression, functions, and roles in mammals, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1441–1447 CrossRef CAS.
- S. Yoneda and K. T. Suzuki, Equimolar Hg-Se complex binds to selenoprotein P, Biochem. Biophys. Res. Commun., 1997, 231, 7–11 CrossRef CAS PubMed.
- J. Pourahmad and M. J. Hosseini, Application of isolated mitochondria in toxicological and clinical studies, Iran. J. Pharm. Res., 2012, 11, 703–704 Search PubMed.
- A. Navarro and A. Boveris, Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function, Front. Biosci., 2007, 12, 1154–1163 CrossRef CAS.
- A. Navarro, M. A. J. S. S. D. Pino, C. Gómez, J. L. Peralta and A. Boveris, Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice, Am. J. Physiol., 2002, 282, R985–R992 CAS.
- C. M. Palmeira, and V. t. M. Madeira, Mercuric chloride toxicity in rat liver mitochondria and isolated hepatocytes, Environ. Toxicol. Pharmacol., 1997, 3, 229–235 CrossRef CAS.
- P. Bernardi, The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death, Biochim. Biophys. Acta, Bioenerg., 1996, 1275, 5–9 CrossRef.
- D. Tanaka, K. Nakada, K. Takao, E. Ogasawara, A. Kasahara, A. Sato, H. Yonekawa, T. Miyakawa and J.-I. Hayashi, Normal mitochondrial respiratory function is essential for spatial remote memory in mice, Mol. Brain, 2008, 1, 21 CrossRef.
- L. Behzadfar, M. Abdollahi, O. Sabzevari, R. Hosseini, A. Salimi, P. Naserzadeh, M. Sharifzadeh and J. Pourahmad, Potentiating role of copper on spatial memory deficit induced by beta amyloid and evaluation of mitochondrial function markers in the hippocampus of rats, Metallomics, 2017, 9(7), 969–970 RSC.
- G. Paxinos and C. Watson, The rat brain in stereotaxic coordinates: hard cover edition, Elsevier, 2006 Search PubMed.
- M. Ghazi-Khansari, A. Mohammadi-Bardbori and M. J. Hosseini, Using Janus green B to study paraquat toxicity in rat liver mitochondria: role of ACE inhibitors (thiol and nonthiol ACEi), Ann. N. Y. Acad. Sci., 2006, 1090, 98–107 CrossRef CAS.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- X. Gao, C. Y. Zheng, L. Yang, X. C. Tang and H. Y. Zhang, Huperzine A protects isolated rat brain mitochondria against β-amyloid peptide, Free Radicals Biol. Med., 2009, 46, 1454–1462 CrossRef CAS.
- M. Talari, E. Seydi, A. Salimi, Z. Mohsenifar, M. Kamalinejad and J. Pourahmad, Dracocephalum: novel anticancer plant acting on liver cancer cell mitochondria, BioMed Res. Int., 2014, 892170 Search PubMed.
- A. Baracca, G. Sgarbi, G. Solaini and G. Lenaz, Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F0 during ATP synthesis, Biochim. Biophys. Acta, Bioenerg., 2003, 1606, 137–146 CrossRef CAS.
- Y. Zhao, L. Ye, H. Liu, Q. Xia, Y. Zhang, X. Yang and K. Wang, Vanadium compounds induced mitochondria permeability transition pore (PTP) opening related to oxidative stress, J. Inorg. Biochem., 2010, 104, 371–378 CrossRef CAS.
- T. P. A. Devasagayam, K. K. Boloor and T. Ramasarma, Methods for estimating lipid peroxidation: An analysis of merits and demerits, Indian J. Biochem. Biophys., 2003, 40(5), 300–308 CAS.
- C. Sadegh and R. P. Schreck, The spectroscopic determination of aqueous sulfite using Ellman's reagent, MURJ, 2003, 8, 39–43 Search PubMed.
- A. Hosseini, A. M. Sharifi, M. Abdollahi, R. Najafi, M. Baeeri, S. Rayegan, J. Cheshmehnour, S. Hassani, Z. Bayrami and M. Safa, Cerium and yttrium oxide nanoparticles against lead-induced oxidative stress and apoptosis in rat hippocampus, Biol. Trace Elem. Res., 2015, 164, 80–89 CrossRef CAS.
- R. Dringen, Metabolism and functions of glutathione in brain, Prog. Neurobiol., 2000, 62, 649–671 CrossRef CAS.
- J.-W. Song and B.-S. Choi, Mercury induced the accumulation of amyloid beta (Aβ) in PC12 cells: the role of production and degradation of Aβ, Toxicol. Res., 2013, 29, 235 CrossRef CAS.
- D. Meleleo1, G. Notarachille, V. Mangini and F. Arnesano, Concentration-dependent effects of mercury and lead on Aβ42: possible implications for Alzheimer's disease, Eur. Biophys. J., 2019, 48(2), 173–187 CrossRef.
- F. Teixeira, R. Fernandes, P. Farias-Junior, N. Costa, L. Fernandes, L. Santana, A. Silva-Junior, M. Silva, C. Maia and R. Lima, Evaluation of the effects of chronic intoxication with inorganic mercury on memory and motor control in rats, Int. J. Environ. Res. Public Health, 2014, 11, 9171–9185 CrossRef CAS.
- P. B. Mello-Carpes, W. Barros, S. Borges, N. Alves, D. Rizzetti, F. M. Pecanha, D. V. Vassallo, G. A. Wiggers and I. Izquierdo, Chronic exposure to low mercury chloride concentration induces object recognition and aversive memories deficits in rats, Int. J. Dev. Neurosci., 2013, 31, 468–472 CrossRef CAS.
- G. Barja, Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity, J. Bioenerg. Biomembr., 1999, 31, 347–366 CrossRef CAS.
- V. Adam-Vizi, Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non–electron transport chain sources, Antioxid. Redox Signaling, 2005, 7, 1140–1149 CrossRef CAS.
- E. A. Belyaeva, T. V. Sokolova, L. V. Emelyanova and I. O. Zakharova, Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: effects of cadmium, mercury, and copper, Sci. World J., 2012, 136063 Search PubMed.
- B.-O. Lund, D. M. Miller and J. S. Woods, Mercury-induced H2O2 production and lipid peroxidation in vitro in rat kidney mitochondria, Biochem. Pharmacol., 1991, 42, S181–S187 CrossRef CAS.
- Q. Zhong, D. A. Putt, F. Xu and L. H. Lash, Hepatic mitochondrial transport of glutathione: studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells, Arch. Biochem. Biophys., 2008, 474, 119–127 CrossRef CAS.
- A. J. Kowaltowski, L. E. Netto and A. E. Vercesi, The Thiol-specific antioxidant enzyme prevents mitochondrial permeability transition evidence for the participation of reactive oxygen species in this mechanism, J. Biol. Chem., 1998, 273, 12766–12769 CrossRef CAS.
- J.-S. Kim, L. He and J. J. Lemasters, Mitochondrial permeability transition: a common pathway to necrosis and apoptosis, Biochem. Biophys. Res. Commun., 2003, 304, 463–470 CrossRef CAS.
- F. M. Rubino, Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms, Toxics, 2015, 3(1), 20–62 CrossRef CAS.
- M. H. Berntssen, A. Aatland and R. D. Handy, Chronic dietary mercury exposure causes oxidative stress, brain lesions, and altered behaviour in Atlantic salmon (Salmo salar) parr, Aquat. Toxicol., 2003, 65, 55–72 CrossRef CAS.
- R. W. Thompson, H. L. Valentine and W. M. Valentine, Cytotoxic mechanisms of hydrosulfide anion and cyanide anion in primary rat hepatocyte cultures, Toxicology, 2003, 188, 149–159 CrossRef CAS.
- Y. Eguchi, S. Shimizu and Y. Tsujimoto, Intracellular ATP levels determine cell death fate by apoptosis or necrosis, Cancer Res., 1997, 57, 1835–1840 CAS.
- C. V. Pereira, A. C. Moreira, S. P. Pereira, N. G. Machado, F. S. Carvalho, V. A. Sardão and P. J. Oliveira, Investigating drug-induced mitochondrial toxicity: a biosensor to increase drug safety?, Curr. Drug Saf., 2009, 4, 34–54 CrossRef CAS.
- J. S. Armstrong, The role of the mitochondrial permeability transition in cell death, Mitochondrion, 2006, 6, 225–234 CrossRef CAS.
- R. A. Bernhoft, Mercury Toxicity and Treatment: A Review of the Literature, J. Environ. Public Health, 2012, 460–508 Search PubMed.
- E. Sontag, V. Nunbhakdi-Craig, J.-M. Sontag, R. Diaz-Arrastia, E. Ogris, S. Dayal, S. Lentz, E. Arning and T. Bottiglieri, Protein phosphatase 2A mathyltransferase links homocysteine metabolis with tau and amyloid precursor protein regulation, J. Neurosci., 2007, 27, 2751–2759 CrossRef CAS PubMed.
- U. Schweizer, A. U. Brauer, J. K. Ohrle, R. Nitsch and N. E. Savaskan, Selenium and brain function: a poorly recognized liaison, Brain Res. Rev., 2004, 45, 164–178 CrossRef CAS PubMed.
- R. M. Ransohoff and M. A. Brown, Innate immunity in the central nervous system, J. Clin. Invest., 2012, 122(4), 1164–1171 CrossRef CAS.
- C. C. Bridges and R. K. Zalups, Mechanisms involved in the transport of mercuric ions in target tissues, Arch. Toxicol., 2017, 91(1), 63–81 CrossRef CAS.
Footnote |
| † These authors are the co-corresponding authors. |
| This journal is © The Royal Society of Chemistry 2020 |