
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A survey of non-coding RNAs in the social and predatory myxobacterium Myxococcus xanthus DK1622†
David E.
Whitworth
 * and
Martin T.
Swain
* and
Martin T.
Swain
Institute of Biological Environmental and Rural Sciences, Aberystwyth University, SY23 3DD, UK. E-mail: dew@aber.ac.uk
First published on 6th August 2020
Abstract
Prokaryotic ncRNAs are important regulators of gene expression, and can be involved in complex signalling networks. The myxobacteria are model organisms for studies into multicellular development and microbial predation, being particularly renowned for their large genomes and exceptionally sophisticated signalling networks. However, apart from two specific examples, little is known about their regulatory ncRNAs. Here, we integrate bioinformatic predictions and transcriptome sequence data to provide a comprehensive survey of the ncRNAs made by the exemplar myxobacterium M. xanthus DK1622. M. xanthus RNA-seq data from four experimental conditions was interrogated to identify transcripts mapping outside coding sequences and to known ncRNAs. The resulting 37 ncRNAs were clustered on the genome and most (30/37) were conserved across the myxobacteria. A majority of ncRNAs (22/37) were intergenic, while 13 were at least partially antisense to protein-coding genes. Predicted promoter and terminator sequences explained the start/stop sites of 18 ncRNAs. mRNA targets for the ncRNAs were predicted, including plausible candidates for a known regulatory ncRNA. 22 ncRNAs were differentially expressed by nutrient availability and expression of 25 predicted targets was found to correlate strongly with that of their regulatory ncRNAs. Sharing of predicted mRNA targets by multiple ncRNAs suggests that some ncRNAs might regulate each other within signalling networks. This genomic survey of M. xanthus ncRNA biology provides a starting point for further studies of myxobacterial ncRNAs, which are likely to have important functions in these industrially important and sophisticated organisms.
Introduction
The RNA molecules within a cell have diverse functions. Some are enzymes (e.g., RNase P), others have structural roles in macromolecular assemblies like the ribosome and SRP (signal recognition particle), some mediate molecular recognition through base–base complementarity (e.g., CRISPRs and regulatory sRNAs), others dictate the sequences of amino acids in proteins during translation, and some have multiple such functions.1–3The central role of rRNA, tRNA and mRNA in the translation of proteins was elucidated in the latter half of the previous century. Further studies of translation then led to the identification of catalytic RNAs, such as the RNA component of RNase P (RnpB), which processes tRNA gene transcripts to give mature tRNAs, and self-splicing introns.4,5 Other RNAs involved with translation were subsequently found, including Ffs, which is the RNA component of SRP for co-translational translocation of nascent proteins across membranes, and SsrA, which is a transfer-messenger RNA (tmRNA) for unstalling stalled ribosomes.6,7
In prokaryotes, the term small RNAs (sRNA) has been widely used to describe regulatory RNAs such as Ffs, SsrA and RnpB, as they are typically small (50–500 nt), and they are distinct from tRNAs, rRNAs and mRNAs.1 However, cells have also been found to contain large RNAs which are not tRNA, rRNA or mRNA, and these long transcripts became known as non-coding RNAs (ncRNA), a term which includes rRNAs, tRNAs and sRNAs.
At the beginning of this century, a role for the cryptic 6S ncRNA of Escherichia coli (SsrS) was finally elucidated. Rather than being involved with translation, SsrS was found to be a transcriptional regulator, repressing transcription of Sigma70-dependent promoters during stationary phase.8 Since that first discovery, many ncRNAs have been shown to regulate the expression of specific subsets of genes, and ncRNAs are being discovered in increasing numbers due to bioinformatics predictions and whole-transcriptome sequencing.9,10
ncRNAs can participate in functional associations with other biomolecules through two mechanisms – by binding to target proteins (e.g., within nucleoprotein complexes), and/or by base-pairing with mRNA. Regulatory ncRNAs bind to the mRNA transcripts of their target genes, and in doing so stimulate/inhibit their transcription/translation. For instance, ncRNAs can bind to the ribosome-binding site (RBS) of their target mRNA, blocking translation. Conversely, some ncRNAs bind to their target mRNAs close to the RBS, preventing formation of an inhibitory secondary structure involving the RBS, and thus stimulating translation.11 In a similar fashion, binding of an ncRNA to its target mRNA can regulate transcription of the mRNA by occluding or exposing rut sites for rho-dependent transcription termination.2 ncRNAs can also bind to ncRNAs that themselves bind to gene regulatory elements, competing with one another for binding, and forming complex gene regulatory networks.12 In addition, matchmaking between ncRNAs and their mRNA targets is often stimulated by RNA chaperone proteins such as Hfq, ProQ and CsrA.13
The myxobacteria are a family of proteobacteria that are particularly noted for their complex gene regulatory networks.14 Nevertheless, with two notable exceptions (Pxr and MsDNA), their ncRNAs have received little research attention. Myxobacteria are predators of a broad range of prey microbes, and possess personalised genomes encoding large repertoires of secondary metabolite biosynthetic gene clusters.15–17 When prey becomes scarce, a population of myxobacterial cells embark co-operatively on a developmental programme culminating in the formation of a macroscopic fruiting body, within which a subset of cells differentiate into spores.18,19 This social phenomenon requires complex regulatory systems for responding to: the nutritional status within each cell, the cell's context within the population, and its progression through the developmental programme.20
The best understood regulatory ncRNA in myxobacteria is Pxr, a gatekeeping ncRNA that is expressed when nutrients are abundant, and which prevents cells from commencing multicellular development.21 Expression of the pxr gene is under the control of the two-component system PxrR/PxrK (also known as SpdS/SpdR and MXAN_1077/MXAN_1078) and its secondary structure includes a stem-loop which is essential for function.22,23 How Pxr inhibits fruiting is currently unclear.
Another peculiar ncRNA was first described in M. xanthus, but has since been shown to occur widely across bacteria. Known as msDNA (multicopy single-stranded DNA), the msDNA gene is transcribed and the nascent MsDNA RNA folds into a structure which allows priming by reverse transcriptase from a 2′-OH group of the RNA. DNA synthesis proceeds using the MsDNA RNA strand as a template, which is then degraded by RNase H. After reverse transcription has completed, the end product is a mature molecule comprising single-stranded DNA covalently linked to a portion of the original RNA transcript.3,24 Despite being discovered more than 30 years ago and being conserved across bacteria, the function of MsDNA, and msDNA more generally, remains unknown.
In order to provide a genome-wide view of the production of ncRNAs by M. xanthus, RNA-seq data from four experimental conditions were interrogated, identifying distributions of transcripts that mapped between protein coding sequences (CDSs). In this fashion 37 ncRNAs were identified and further characterised. We also integrated knowledge on previously identified ncRNAs and bioinformatics predictions to give a comprehensive overview of ncRNAs in M. xanthus DK1622, providing a starting point for further studies of these enigmatic entities.
Experimental procedures
Identification of ncRNAs in experimental datasets
ncRNAs were identified from the RNA-seq transcriptome datasets described by Livingstone et al. (2018b) using the ToRNAdo script, which has previously been applied to characterise staphylococcal ncRNAs.25,26 ToRNAdo maps sequencing coverage to distributions with peaks above a threshold, and calls each distribution an RNA. Annotated RNA and protein-coding genes are then excluded, and each remaining ncRNA is outputted with start/stop positions, the genomic strand transcribed and maximum peak height. ToRNAdo also identifies ncRNAs as intragenic (found within and antisense to a gene), intergenic (found between genes), and mixed (partially antisense to a gene).Transcriptome sequence datasets were available for M. xanthus DK1622 incubated under four experimental conditions: nutrient-free buffer (STARVED, n = 4), nutrient broth (FED, n = 3), buffer with live E. coli prey (LIVE, n = 4) and buffer with pre-killed E. coli prey (DEAD, n = 4). See Livingstone et al. for full technical details.27 In short, liquid cultures of predator and prey were grown separately in a medium which sustained growth of both organisms (LBCY).27 Mid-exponential phase cells were harvested and resuspended in either buffer (TM) or LBCY, either in pure culture, or mixed with the other organism. After 4 hours of unshaken incubation at 30 °C, cells were harvested for RNA extraction, ribodepletion, cDNA synthesis, library preparation and sequencing, as described previously.27
If ncRNAs identified by ToRNAdo in one replicate overlapped ncRNAs in another replicate, the records were integrated such that the ncRNA was ascribed the first start site, the last stop site, and the maximum peak height of any constituent ncRNA. For each experimental condition, if an ncRNA was absent from at least 1 replicate, it was excluded from further analysis. sRNA genes already in the DK1622 genome annotation were not detected by ToRNAdo, and were therefore manually incorporated into the ncRNA dataset. Their relative expression levels in different experimental conditions were taken from Livingstone et al.27 ncRNAs were also discarded from the dataset if they were on the same strand and within 10 nt of the 5′ end of a CDS as they likely represented 5′-untranslated regions (5′-UTRs) of mRNAs. Also excluded were the non-coding and leader sequences of polycistronic rRNA gene transcripts.
Characterisation of ncRNAs
Identification of homologues of ncRNAs in myxobacteria was achieved by querying against the Refseq myxobacterial genomes in the ‘Myxococcales’ taxon within the NCBI nr database, using BLASTn with an e-value cut-off 0.1 to maximise the sensitivity of homologue identification. Location of ncRNAs and other genes around the genome were visualised using the Circos platform.28Putative promoters and terminators were predicted using PePPER, queried with regions of genomic DNA extending at least 1 kb beyond the start and stop sites of each ncRNA.29 Terminators were often identified as nested sequences centred on the same genome position and these were manually curated to remove redundancy.
Regulatory ncRNA target gene predictions
mRNA targets were predicted for intergenic ncRNAs and the intergenic portions of mixed ncRNAs using CopraRNA.30 Firstly, BLASTn was used to identify homologues of ncRNAs in myxobacterial genomes as described above. For every ncRNA, five homologues were included in CopraRNA queries: M. xanthus DK1622 (NC_008095), Myxococcus macrosporus HW-1 (NC_015711), Myxococcus stipitatus DSM14675 (NC_020126), Myxococcus fulvus 124B02 (NZ_CP006003), and Corallococcus coralloides DSM2259 (NC_017030). These five organisms were chosen as the four phylogenetically closest organisms to M. xanthus for which there was a RefSeq genome available. For all ncRNAs except Mxs009, Mxs015, Mxs021, Mxs030 and Mxs037, at least 3 homologues could be found from amongst the five organisms, allowing CopraRNA prediction of targets. Mxs028 and Mxs035 were not found in Corallococcus coralloides, therefore their queries comprised four Myxococcus spp. homologues. All other queries contained homologues from all five organisms.Differential expression analysis
For the four experimental conditions employed, the total maximum peak heights of the full ToRNAdo output was used for normalisation, allowing comparison of relative abundance of individual ncRNAs between conditions. The relative abundances of normalised maximum peak heights for each ncRNA under all four experimental conditions were compared to the corresponding normalised expression values for their putative target mRNAs (or normalised peak heights if their predicted target was an ncRNA), and correlation coefficients calculated. Differential expression of ncRNAs was defined as log2(maximum peak height under condition 1/maximum peak height under condition 2). A cut-off for differential expression was applied to the ncRNAs, such that only values with a greater than two-fold change were considered. Heatmaps of normalised expression values were generated using Heatmapper with complete linkage clustering of Spearman rank correlated distances.31Prediction of ncRNAs
ncRNAs were identified in Myxococcus spp. genomes using RNAz 2.1, which searches for conserved DNA sequences in non-coding parts of genomes, predicts the secondary structure, and calculates its thermodynamic stability.32 The input for RNAz was a multiple whole genome alignment performed using progressive Mauve, with M. xanthus DK1622 as the reference genome. (The resulting XMFA alignment is not a file format supported by RNAz. It was therefore converted to a ClustalW file format which is supported by RNAz using the script provided in the Mauve package).33 The alignment was then processed using the windowing approach described in the RNAz manual. Genomes included in the alignment were Myxococcus fulvus 124B02 (NZ_CP006003), Myxococcus hansupus mixupus (NZ_CP012109), Myxococcus macrosporus HW-1 (NC_015711), M. macrosporus DSM 14697 (NZ_CP022203), Myxococcus stipitatus DSM 14675 (NC_020126), and M. xanthus DK1622 (NC_008095). RNAz output was filtered with a RNA class probability cut-off of 0.9, as suggested in the RNAz manual. To visualize the ncRNAs predicted by RNAz in a genome browser, the unfiltered RNAz output was processed using custom scripts to extract ncRNA sequences in fasta format. BLASTn was used to locate these within the M. xanthus DK1622 genome and a GFF format file was generated, including Z-scores and probabilities for each prediction.Results
Compiling the ncRNAs of M. xanthus DK1622
To investigate the possibility of further ncRNAs in M. xanthus, we interrogated DK1622 transcriptomic data for evidence of transcription of non-coding regions of the genome.
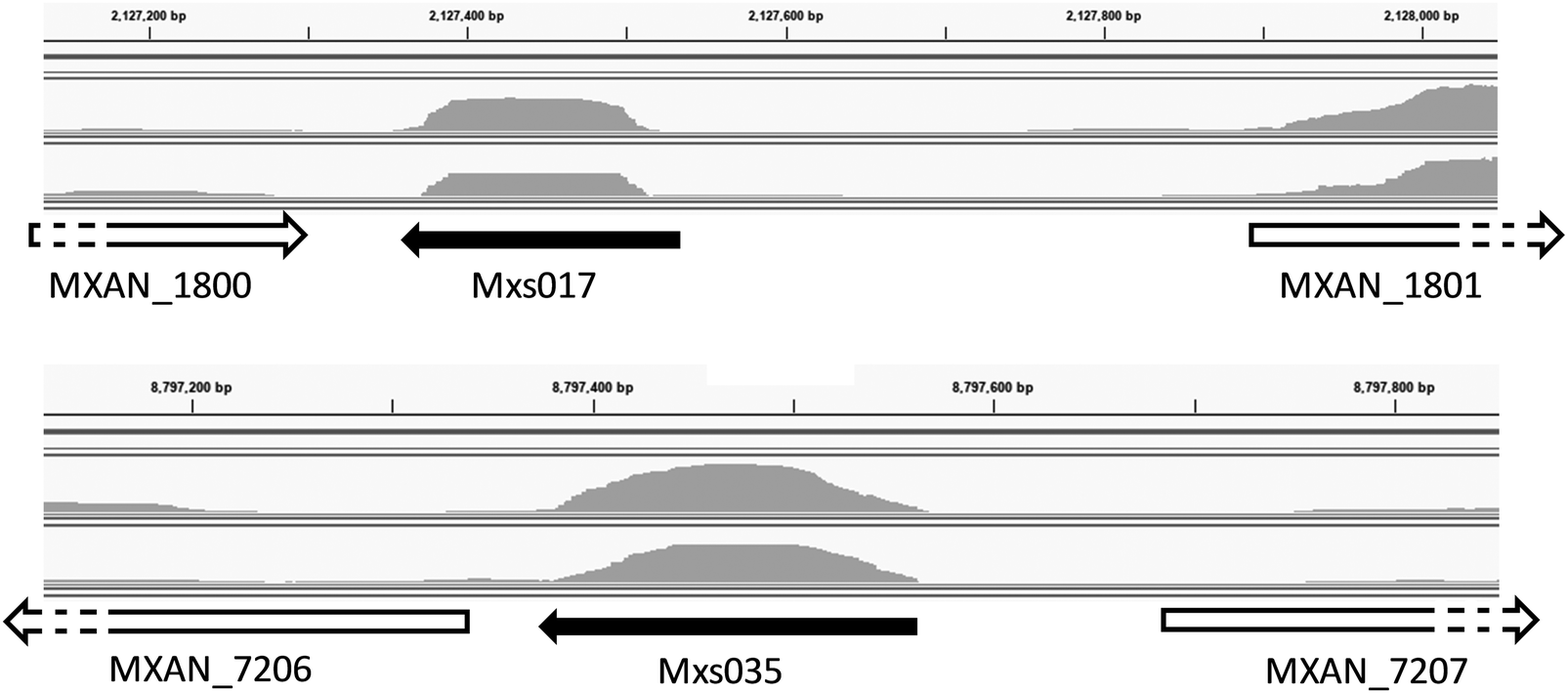 | ||
| Fig. 1 Mapped transcripts for two example ncRNAs. ncRNAs are shown as black arrows, and surrounding (protein-coding) genes as white arrows, pointing in the direction of transcription. Grey areas above each arrow show the sequencing coverage for two replicates of the experimental condition STARVED. | ||
To avoid potential confusion, hereafter the term ‘RNA-seq ncRNAs’ is used to refer to these 37 ncRNAs and not rRNA, tRNAs, or CRISPRs. This term includes 33 ncRNAs identified by ToRNAdo and the four ncRNAs annotated as sRNAs in the DK1622 genome, expression of which was observed in the transcriptome data published of Livingstone et al.27
Characteristics of M. xanthus DK1622 ncRNAs
| Name | Start | Stop | Strand | Gene organisation | Evidence | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mxs001 | 25629 | 25787 | + | Intergenic | E | H | P | T | ||||
| Mxs002 | 126508 | 126773 | − | Intergenic | E | H | Z | |||||
| MsDNA | 164715 | 165079 | − | Intergenic | C | E | H | L | P | Z | ||
| Mxs004 | 253126 | 253484 | − | 3′-Convergent | E | H | ||||||
| Mxs005 | 376445 | 376902 | − | Antisense | E | H | ||||||
| Mxs006 | 376926 | 377874* | − | Antisense | E | H | ||||||
| Mxs007 | 378695 | 381637 | − | Antisense | E | H | ||||||
| Mxs008 | 410130 | 411387 | − | 3′-Convergent | C | E | H | P | T | |||
| Mxs009 | 664956 | 665146 | − | Antisense | C | E | H | |||||
| Mxs010 | 918237* | 920929 | − | Complex | C | E | H | T | ||||
| Pxr | 1258187 | 1258294 | + | Intergenic | C | E | H | L | T | Z | ||
| Mxs012 | 1272544 | 1272957* | − | 5′-Divergent | C | E | H | |||||
| Mxs013 | 1273789 | 1276608 | + | Complex | C | E | H | |||||
| Mxs014 | 1359323 | 1359532 | + | Antisense | E | H | ||||||
| Mxs015 | 1412778 | 1413296 | + | 3′-Convergent | C | E | P | |||||
| Mxs016 | 2110024 | 2110189 | − | Intergenic | E | P | ||||||
| Mxs017 | 2127372 | 2127516 | − | Intergenic | C | E | H | T | ||||
| Ffs | 2270094 | 2270275 | − | Intergenic | A | C | E | H | T | Z | ||
| Mxs019 | 2270165 | 2270237 | + | Antisense | E | H | Z | |||||
| SsrA | 2426740 | 2427105 | + | Intergenic | A | C | E | H | P | T | ||
| Mxs021 | 2515172 | 2515241 | − | Intergenic | E | |||||||
| Mxs022 | 3115293 | 3115978 | − | Antisense | C | E | H | |||||
| Mxs023 | 3156166 | 3156740 | − | Complex | C | E | H | |||||
| Mxs024 | 3293009 | 3293469* | − | Antisense | C | E | H | |||||
| Mxs025 | 3293491 | 3294438* | − | Antisense | E | H | ||||||
| Mxs026 | 3295164 | 3298106 | − | Antisense | C | E | H | |||||
| SsrS | 3493000 | 3493254 | − | Intergenic | A | E | H | P | ||||
| Mxs028 | 3709758 | 3710025 | − | Intergenic | C | E | H | |||||
| Mxs029 | 5368332 | 5368824 | + | Antisense | E | H | ||||||
| Mxs030 | 5660826* | 5662584 | − | 3′-Convergent | C | E | ||||||
| Mxs031 | 5893476* | 5898981 | + | Complex | C | E | H | P | ||||
| RnpB | 7099786 | 7100181 | + | Intergenic | A | E | H | Z | ||||
| Mxs033 | 7099791 | 7100180* | − | Antisense | E | H | ||||||
| Mxs034 | 7779037 | 7779318 | + | Antisense | C | E | H | |||||
| Mxs035 | 8797382 | 8797562* | − | Intergenic | C | E | H | Z | ||||
| Mxs036 | 8961532* | 8966713 | + | Complex | C | E | H | P | ||||
| Mxs037 | 9117630 | 9117821 | − | Intergenic | E | H | T | |||||
BLASTn searches found that more than 80% (30) of the 37 ncRNAs had homologues across the myxobacteria. Mxs028 and Mxs035 were only present in Myxococcus spp. genomes, Mxs009 and Mxs037 were only present in M. xanthus and M. macrosporus, while Mxs15, Mxs21 and Mxs30 were only present in M. xanthus. Conservation across the myxobacteria suggests a biological role for those ncRNAs, while the less-conserved ncRNAs are presumably either clade-specific regulators, or are the result of spurious transcription.34 Only Ffs, SsrA, SsrS, RnpB were found to have homologues in Rfam.35
For genes involved in fruiting body formation, different categories of regulators are distributed unevenly around the chromosome.36 Therefore, the location of ncRNA genes in the genome was determined. The RNA-seq ncRNAs were distributed around the DK1622 genome in a non-uniform fashion (unlike that seen for tRNAs and rRNA genes), with ncRNA genes falling into clusters separated by large stretches of genome entirely lacking ncRNA genes (Fig. 2). 18 of the 37 ncRNAs (49%) were found in just 19% of the genome (from 8.80 to 1.42 Mbp – i.e., 1.76 Mbp spanning the chromosomal origin). Other ‘hotspots’ were apparent between 2.11 and 2.52 Mbp (6 ncRNAs in 4% of the genome) and between 3.11 and 3.71 Mbp (7 ncRNAs in 7% of the genome). Conversely, the region from 3.8 to 8.80 Mbp (55% of the genome) had just 6 ncRNA genes (16%). There was also a slight bias towards ncRNAs being expressed from the negative strand (25 of 37).
 | ||
| Fig. 2 Circos diagram of ncRNAs mapped onto the 9.14 Mbp M. xanthus DK1622 genome (outer ring). The red, green and blue rings indicate genes for regulatory ncRNAs, rRNAs/tRNAs, and CRISPRs, respectively. For each coloured ring, the outer leaflet indicates genes on the forward (+) strand while the inner leaflet shows those on the reverse (−) strand. A–D highlight the four rRNA gene clusters. | ||
Antisense RNAs were located entirely within another gene. Eight of the 13 were encoded antisense to other RNA genes (16S and 23S rRNA genes, ffs and rnpB), but five were antisense to CDSs (IF-3, pdhC and three hypothetic proteins MXAN_4364, MXAN_RS05615 and MXAN_RS30530.
Intergenic ncRNA genes had no overlaps with CDSs. Mixed ncRNAs partially overlapped at least one CDS, having at least one antisense and at least one intergenic region. Four were 3′-convergent with their antisense CDS, one (Mxs012) was 5′-divergent from its antisense CDS, three (Mxs009, Mxs014 and Mxs034) were antisense (with extensions (<25 bp) at one or both ends of their antisense genes), and the remaining had complex relationships with other genes, being antisense to two or more genes). Gene organisation for all RNA-seq ncRNAs is shown schematically in Fig. 3.
 | ||
| Fig. 3 Genome organisation of ncRNAs. In each panel (A–I), the ncRNA in question is shown as a black arrow with CDSs as white arrows, all reflecting the direction of transcription (double-headed arrows represent genes which are found in either orientation for different ncRNAs). Dark grey arrows represent RNA genes, light grey arrows represent genes which are either CDSs or RNA genes for different ncRNAs. | ||
For most ncRNAs there was remarkable consistency in the start/stop positions of the same ncRNAs identified under different experimental conditions. Of the 66 start or stop sites, 42 had standard deviations of less than 10 nt. However some start/stop sites were very variable (five 5′-ends, five 3′-ends), with standard deviations in their positions of more than 100 nt (Table 1). For the 5′-ends this large deviation could represent alternative promoters, or spurious transcriptional initiation. Intriguingly, all five ncRNAs with variable 5′-ends are large antisense ncRNAs for rRNA genes, making spurious initiation a more likely explanation than alternative promoters.
Predicted promoters were found at the beginning of Mxs001, Mxs008, Mxs015, Mxs016, SsrA, and SsrS, and at the start of a putative MXAN_0141 > MsDNA > MXAN_0140 operon (Table 1). Predicted terminators were located at the 3′ ends of Mxs001, Mxs008, Mxs010, Pxr, Mxs017, Ffs, SsrA and Mxs037 (Table 1). (Mxs001, Mxs008, and SsrA have both terminators and promoters identified).
Of the five ncRNAs with variable 3′-ends, three lacked predicted terminators. However, the mxs031 gene had a terminator coincident with its furthermost 3′-end, implying that a large proportion of its transcription complexes halt transcription before reaching the terminator. Conversely, the mxs010 gene has a terminator coincident with its shortest 3′-ends, suggesting that the predicted terminator is weak and may not efficiently halt transcription complexes.
The small number of predicted promoters is probably because most ncRNAs are expressed using other Sigma factors, rather than the σ70-dependent class on which PePPER is based. This supposition is supported by the large numbers of sigma factors (55) identified in the DK1622 genome by the P2TF database.38
mRNA targets of regulatory ncRNAs
CopraRNA was used to predict mRNA targets for intergenic ncRNAs and the intergenic portions of mixed ncRNAs. Output from CopraRNA is provided as a.zip file (Supplemental File 5, ESI†), including mRNA region plots, ncRNA region plots and lists of the top 200 predicted targets. Lists of CopraRNA predicted targets were then filtered to retain only those predictions with p values <0.00001 and false discovery rates of <0.1.
The two approaches combined gave 67 predictions for 32 of the 37 ncRNAs (Supplemental File 4, ESI†). Predicted targets were defined as cis or trans depending on whether they were in the same genomic location as their regulatory ncRNA. Predictions based on antisense complementarity identified 32 cis targets. CopraRNA predictions included 7 cis predictions (presumably due to overlap between an ncRNA and the region upstream of the translational start site of its neighbouring genes investigated by CopraRNA), but also provided 30 trans predictions. Looking at COG categories and annotations, there seemed to be no enrichment of particular functional classes, as might be expected given the diversity of ncRNAs and their likely roles.
Interestingly, one of the predicted trans-targets of Pxr is actA mRNA (MXAN_3213). ActA is needed for fruiting body formation upon starvation – deletion of the actA gene blocks developmental progression.39 In the presence of nutrients Pxr prevents development, and a plausible mechanism by it which it might do so, is through inhibiting the expression of actA. Pxr is known to exert its regulatory effects through a (terminator-like) stem-loop denoted SL3.22 The CopraRNA prediction of interaction between actA mRNA and Pxr is via SL3, consistent with Pxr regulating expression of actA (Fig. 4).
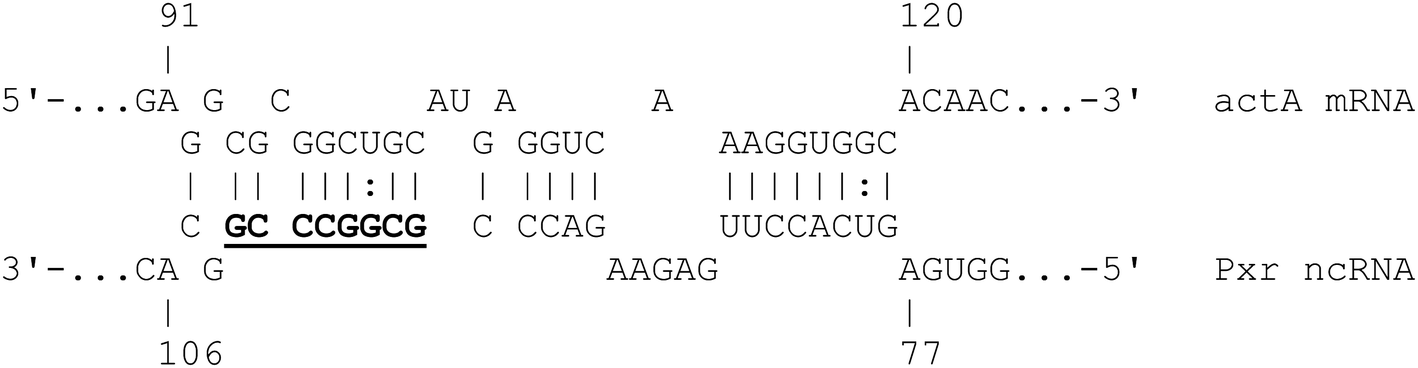 | ||
| Fig. 4 Predicted base-pairing between the actA mRNA and Pxr ncRNA. The conserved residues in the Pxr stem-loop 3 (SL3) which is required for Pxr function are highlighted in bold and underlined. | ||
Some predicted trans targets were shared by more than one ncRNA. MXAN_0702 (encoding a hypothetical protein) is targeted by Mxs013 and Mxs028, and MXAN_1951 (also encoding a hypothetical protein) is targeted by Mxs016 and Mxs028, suggesting that they may form a regulatory network in vivo.
 | ||
| Fig. 5 Heatmap of RNA-seq ncRNA relative expression under four experimental conditions: Fed = nutrient broth, Starved = nutrient-free buffer, Live = buffer with live E. coli prey and Dead = buffer with pre-killed E. coli prey, as described by Livingstone et al.27 | ||
Where expression data was available from more than one condition, the relative expression of ncRNAs was assessed comparing experimental conditions. In conditions DEAD and FED, DK1622 cells are actively feeding, and comparing those conditions with condition STARVED, most ncRNAs (28 of the 31 with non-zero expression in condition STARVED) were found to be differentially expressed more than two-fold (Supplemental File 6, ESI†).
The relative expression of ncRNAs under the four experimental conditions was compared with the relative expression of predicted targets under the same conditions (Supplemental File 6, ESI†). In many cases (33 of 65) the expression of an ncRNA was found to significantly correlate with that of a predicted target, supporting the target prediction. Most correlations were positive (30 of 33), 13 involved trans targets and 20 involved cis targets.
Pxr expression was induced more than 16-fold when fed compared to starved, as expected. Of the six predicted trans targets of Pxr, five were repressed by nutrients (four by more than 2-fold), while one (MXAN_0569) was induced more than 8-fold.
Discussion
The number and nature of DK1622 ncRNAs
Interrogation of transcriptome datasets identified 37 ncRNAs, including 13 antisense ncRNAs, which were expressed in cultures of M. xanthus, (4 of which were previously annotated in the DK1622 genome). The numbers of ncRNAs identified in other bacteria is dependent on whether an experimental or bioinformatics approach is taken, and what selection/filtering criteria are applied.37 Nevertheless, it would seem that most bacteria encode in the order of 100 ncRNA, with between 1/3 and 2/3 of those being antisense ncRNAs.37 The proportion of RNA-seq ncRNAs in DK1622 which are antisense (35%), mirrors the situation in other bacteria, and as usual, they are longer than intergenic ncRNAs (averaging 847 nt and 211 nt respectively).The relatively small number of ncRNAs, 37, is likely due to the stringent filtering we applied. We deliberately excluded 5-UTRs, ultimately because RNAs with 5′-UTRs have 3-regions which are translated, and thus by definition aren’t ncRNAs. Our merger of small ncRNAs into larger ncRNAs when the two were fused in other experimental conditions/replicates also reduced the number of apparent ncRNAs. Perhaps more importantly, we also applied a screen to retain only ncRNAs which were present in every replicate of an experimental condition. Applying this filter resulted in the loss of 174 candidate ncRNAs, albeit representing just <7% of the total ncRNA signal. Many of the candidate ncRNAs lost at this stage were present in n − 1 replicates of an experimental condition, and often in more than one experimental condition, suggesting that they may represent real but relatively low abundance ncRNAs.
The identification of ncRNAs is also highly dependent on genome annotation of CDSs. Comparing between the original DK1622 annotation from 2012, with a reannotation in 2017, around one fifth of candidate ncRNAs were discarded, as they could be mapped onto new CDSs (generally encoding hypothetical proteins averaging just 118 amino acyl residues in length), or they were found to be 5′-UTRs of CDSs whose predicted translational start site had been reannotated further upstream. It may well transpire in coming years that our approach has falsely identified transcripts as ncRNAs which turn out to encode proteins, but we may also have excluded true ncRNAs because they coincide with mis-annotated CDSs. It is also quite possible that there may be DK1622 ncRNAs which are conditionally expressed (as indeed we have shown many are), and which have remained undetected as they are not expressed in any of our four experimental conditions.
By assessing the conservation of predicted RNA secondary structures, RNAz identified 1582 putative ncRNAs in the M. xanthus DK1622 genome. Seven of those putative ncRNAs predictions mapped onto RNA-seq ncRNAs. Presumably the remaining 1575 putative ncRNAs predicted by RNAz are either false positives or conditionally-expressed. As they were not observed experimentally, and because of their large number, a detailed analysis of the 1575 remaining ncRNA predictions was not undertaken.
Start, stops, promoters and terminators
Comparing between experimental conditions (and between replicates), the start and stop sites of the majority of ncRNAs were remarkably consistent. In some cases putative promoter and terminator sequences could be identified at the ends of ncRNAs, agreeing with the experimentally inferred start/stop sites (Table 1). However, for the majority of ncRNAs, no candidate promoters or terminators could be identified. Promoter prediction algorithms rely on experimentally derived promoter sequences, and usually focus on promoters that are recognised by RNA polymerase holoenzyme containing the housekeeping Sigma factor σ70.40 According to the P2TF database, Myxococcus spp. genomes encode an average of 57 Sigma factors, with DK1622 encoding 55, suggesting that a large proportion of the DK1622 ncRNAs might be transcribed from condition-dependent non-σ70 promoters.38 The ncRNAs with the most variable 5-transcriptional start sites were all antisense to highly-expressed rRNA genes, suggesting that transcription initiation of these ncRNAs occurs non-specifically.The location of terminators can be important as regulatory ncRNAs often exert effects on their target mRNAs in a mechanism mediated by the terminator-binding RNA chaperone Hfq.2 However, M. xanthus does not have an Hfq homologue, or homologues of the other major RNA chaperones ProQ and CsrA. Nevertheless, the M. xanthus DK1622 genome does encode six homologues of CspA, which has RNA chaperone activity.13
Predicted mRNA targets
Prediction of the regulatory targets of antisense ncRNAs is trivial. However, currently available algorithms for the identification of the regulatory targets of trans-acting ncRNAs are prone to false positives.10 The performance of CopraRNA is enhanced compared to other algorithms as it looks for complementarity between ncRNAs and potential targets, and then factors in patterns of sequence conservation in both the ncRNA and its potential targets.32CopraRNA outputs the most probable 200 candidate mRNA targets, and provides p-values and FDRs as a measure of confidence in the predicted targets. We again applied a very conservative set of criteria to CopraRNA output to minimise false-positive predictions. Nevertheless, some of the predictions will be false positives. For instance querying CopraRNA with Ffs, SsrA and SsrS generated predicted targets, despite these ncRNAs not having mRNA targets.
Encouragingly, expression of many of the CopraRNA predicted target mRNAs, correlated with those of their regulatory ncRNAs. The function of only one DK1622 regulatory ncRNA has been elucidated (Pxr), which prevents fruiting body formation when nutrients are present. Intriguingly, a high confidence predicted target mRNA for Pxr (actA), is known to be a positive regulator of fruiting, and the predicted ActA mRNA-Pxr interaction involves the region of Pxr shown to be required for its function.23,39 It is therefore tempting to speculate that Pxr expression under nutrient-rich conditions prevents ActA production, impeding fruiting, and that upon starvation, loss of Pxr allows ActA production to stimulate fruiting.
Several predicted ncRNA targets are likely to be involved in predation and/or the response to changes in nutrient availability, being differentially expressed during predation/starvation. Some have annotated functions which suggest plausible mechanisms by which they might play roles in nutrition/predation. For instance, predicted targets for Mxs010 include an M10 peptidase, those for Mxs013 and Mxs016 include sugar kinases/phosphatases, predicted targets of Mxs010 and Mxs016 include transporters, while Mxs017, Mxs022 and Mxs023 likely target metabolic processes, including pyruvate dehydrogenase. It would be interesting to determine whether different ncRNAs and targets are expressed if M. xanthus feeds upon different prey organisms, or whether the generalist nature of M. xanthus predation is mirrored by a common set of ncRNAs being expressed during predation, regardless of prey species.
Are the RNA-seq ncRNAs functional?
In Table 1 the RNA-seq ncRNA of DK1622 are presented. Their proposal is based on experimental observation of non-coding transcripts, but in most cases several other lines of evidence support the hypotheses that each ncRNA has a biological function, including sequence conservation, predicted terminator and promoter sequences, and correlated expression of putative target mRNAs. For some ncRNAs, there is little supporting evidence that the ncRNA has a biological function, beyond the experimental observation of its existence (e.g. Mxs021). In other cases we can be very confident that the ncRNA has a role in some yet undetermined biological process (e.g., Mxs008 and Mxs017).Other ncRNAs give cause to doubt their biological role, despite having some supporting evidence. For instance, eight ncRNAs are antisense to the 16S and 23S rRNA genes which are found in four gene clusters in the DK1622 genome. In two cases (Mxs031 and Mxs036) an ncRNA spans the entire gene cluster. For the other gene clusters there is an ncRNA antisense to the 23S rRNA gene (Mxs007 and Mxs026) and two tandem pairs of ncRNAs antisense to the 16S rRNA gene (Mxs005/Mxs006 and Mxs024/Mxs025). All eight ncRNAs show sequence conservation across the myxobacteria, but of course they would do, as they are complementary to highly conserved rRNA genes. The ncRNAs have no identifiable promoters (despite being expressed under vegetative conditions and thus likely to be σ70-dependent), but have highly variable start sites, suggesting non-specific initiation of their transcription.
It has been proposed that antisense rRNA transcripts might have biological functions, however, it is also clear that antisense transcription is a pervasive by-product of transcriptional activity.41,42 Are the observed anti-rRNA ncRNAs a non-functional by-product of the particularly copious transcription of their antisense rRNA genes? If this were the case, it might explain why expression of the anti-rRNA ncRNAs correlates positively rather than negatively with the expression of their antisense rRNA genes. This might also be the case for ncRNAs encoded antisense to high-expressed genes (e.g. Mxs019 and Mxs033, which are antisense to Ffs and RnpB respectively).
The DK1622 ncRNAome
Here we provide evidence for the existence of 37 ncRNAs in M. xanthus DK1622. Given the likely importance of ncRNAs in regulating diverse aspects of myxobacterial biology, this characterisation provides an evidence-based framework for further investigations, including molecular genetics experiments as well as genome-wide association studies.43,44Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
We would like to thank Pavelas Sazinas for applying the ToRNAdo algorithm to our RNA-seq datasets.References
- G. Storz, J. Vogel and K. M. Wassarman, Regulation by small RNAs in bacteria: expanding frontiers, Mol. Cell, 2011, 43, 880–891 CrossRef CAS PubMed.
- J. Chen, T. Morita and S. Gottesman, Regulation of Transcription Termination of Small RNAs and by Small RNAs: Molecular Mechanisms and Biological Functions, Front. Cell. Infect. Microbiol., 2019, 9, 201 CrossRef CAS PubMed.
- A. Dhundale, B. Lampson, T. Furuichi, M. Inouye and S. Inouye, Structure of msDNA from Myxococcus xanthus: evidence for a long, self-annealing RNA precursor for the covalently linked, branched RNA, Cell, 1987, 51, 1105–1112 CrossRef CAS PubMed.
- S. Altman, Ribonuclease P: an enzyme with a catalytic RNA subunit, Adv. Enzymol. Relat. Areas Mol. Biol., 1989, 62, 1–36 CAS.
- R. B. Waring, J. A. Ray, S. W. Edwards, C. Scazzocchio and R. W. Davies, The Tetrahymena rRNA intron self-splices in E. coli: in vivo evidence for the importance of key base-paired regions of RNA for RNA enzyme function, Cell, 1985, 40, 371–380 CrossRef CAS PubMed.
- M. A. Poritz, H. D. Bernstein, K. Strub, D. Zopf, H. Wilhelm and P. Walter, An E. coli ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle, Science, 1990, 250, 1111–1117 CrossRef CAS PubMed.
- K. C. Keiler, P. R. Waller and R. T. Sauer, Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA, Science, 1996, 271, 990–993 CrossRef CAS PubMed.
- A. E. Trotochaud and K. M. Wassarman, 6S RNA function enhances long-term cell survival, J. Bacteriol., 2004, 186, 4978–4985 CrossRef CAS PubMed.
- W. Li, X. Ying, Q. Lu and L. Chen, Predicting sRNAs and their targets in bacteria, Genomics, Proteomics Bioinf., 2012, 10, 276–284 CrossRef CAS PubMed.
- J. Hör, S. A. Gorski and J. Vogel, Bacterial RNA Biology on a Genome Scale, Mol. Cell, 2018, 70, 785–799 CrossRef PubMed.
- K. Papenfort and C. K. Vanderpool, Target activation by regulatory RNAs in bacteria, FEMS Microbiol. Rev., 2015, 39, 362–378 CrossRef CAS PubMed.
- L. Bossi and N. Figueroa-Bossi, Competing endogenous RNAs: a target-centric view of small RNA regulation in bacteria, Nat. Rev. Microbiol., 2016, 14, 775–784 CrossRef CAS PubMed.
- A. P. Quendera, A. F. Seixas, R. F. Dos Santos, I. Santos, J. P. N. Silva, C. M. Arraiano and J. M. Andrade, RNA-Binding Proteins Driving the Regulatory Activity of Small Non-coding RNAs in Bacteria, Front. Mol. Biosci., 2020, 7, 78 CrossRef PubMed.
- D. E. Whitworth, Genome-wide analysis of myxobacterial two-component systems: genome relatedness and evolutionary changes, BMC Genomics, 2015, 16, 780 CrossRef PubMed.
- J. Muñoz-Dorado, F. J. Marcos-Torres, E. García-Bravo, A. Moraleda-Muñoz and J. Pérez, Myxobacteria: Moving, Killing, Feeding, and Surviving Together, Front. Microbiol., 2016, 7, 781 CrossRef PubMed.
- P. G. Livingstone, R. M. Morphew and D. E. Whitworth, Myxobacteria Are Able to Prey Broadly upon Clinically-Relevant Pathogens, Exhibiting a Prey Range Which Cannot Be Explained by Phylogeny, Front. Microbiol., 2017, 8, 1593 CrossRef PubMed.
- P. G. Livingstone, R. M. Morphew and D. E. Whitworth, Genome Sequencing and Pan-Genome Analysis of 23 Corallococcus spp. Strains Reveal Unexpected Diversity, With Particular Plasticity of Predatory Gene Sets, Front. Microbiol., 2018, 9, 3187 CrossRef PubMed.
- D. Kaiser, M. Robinson and L. Kroos, Myxobacteria, polarity, and multicellular morphogenesis, Cold Spring Harbor Perspect. Biol., 2010, 2, a000380 CrossRef PubMed.
- P. Cao, A. Dey, C. N. Vassallo and D. Wall, How Myxobacteria Cooperate, J. Mol. Biol., 2015, 427, 3709–3721 CrossRef CAS PubMed.
- L. Kroos, Highly Signal-Responsive Gene Regulatory Network Governing Myxococcus Development, Trends Genet., 2017, 33, 3–15 CrossRef CAS PubMed.
- Y. T. Yu, X. Yuan and G. J. Velicer, Adaptive evolution of an sRNA that controls Myxococcus development, Science, 2010, 328, 993 CrossRef CAS PubMed.
- Y. N. Yu, M. Kleiner and G. J. Velicer, Spontaneous Reversions of an Evolutionary Trait Loss Reveal Regulators of a Small RNA That Controls Multicellular Development in Myxobacteria, J. Bacteriol., 2016, 198, 3142–3151 CrossRef CAS PubMed.
- Y. N. Yu, E. Cooper and G. J. Velicer, A conserved stem of the Myxococcus xanthus sRNA Pxr controls sRNA accumulation and multicellular development, Sci. Rep., 2017, 7, 15411 CrossRef PubMed.
- M. Inouye and S. Inouye, msDNA and bacterial reverse transcriptase, Annu. Rev. Microbiol., 1991, 45, 163–186 CrossRef CAS PubMed.
- P. Sazinas, The application of high-throughput sequencing to study the genome composition and transcriptional response of Haemophilus influenzae, PhD Thesis, University of Warwick, UK, 2016.
- G. M. M. Hermansen, P. Sazinas, D. Kofod, A. Millard, P. S. Andersen and L. Jelsbak, Transcriptomic profiling of interacting nasal staphylococci species reveals global changes in gene and non-coding RNA expression, FEMS Microbiol. Lett., 2018, 365, fny004 CrossRef PubMed.
- P. G. Livingstone, A. D. Millard, M. T. Swain and D. E. Whitworth, Transcriptional changes when Myxococcus xanthus preys on Escherichia coli suggest myxobacterial predators are constitutively toxic but regulate their feeding, Microb. Genomes, 2018, 4, 2 Search PubMed.
- M. Krzywinski, J. Schein, I. Birol, J. Connors, R. Gascoyne, D. Horsman, S. J. Jones and M. A. Marra, Circos: an information aesthetic for comparative genomics, Genome Res., 2009, 19, 1639–1645 CrossRef CAS PubMed.
- A. de Jong, H. Pietersma, M. Cordes, O. P. Kuipers and J. Kok, PePPER: a webserver for prediction of prokaryote promoter elements and regulons, BMC Genomics, 2012, 13, 299 CrossRef CAS PubMed.
- P. R. Wright, J. Georg, M. Mann, D. A. Sorescu, A. S. Richter, S. Lott, R. Kleinkauf, W. R. Hess and R. Backofen, CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains, Nucleic Acids Res., 2014, 42(Web Server issue), W119–W123 CrossRef CAS PubMed.
- S. Babicki, D. Arndt, A. Marcu, Y. Liang, J. R. Grant, A. Maciejewski and D. S. Wishart, Heatmapper: web-enabled heat mapping for all, Nucleic Acids Res., 2016, 44(W1), W147–W153 CrossRef CAS PubMed.
- A. R. Gruber, S. Findeiß, S. Washietl, I. L. Hofacker and P. F. Stadler, RNAz 2.0: improved noncoding RNA detection, Pac. Symp. Biocomput., 2010, 2010, 69–79 Search PubMed.
- A. C. Darling, B. Mau, F. R. Blattner and N. T. Perna, Mauve: multiple alignment of conserved genomic sequence with rearrangements, Genome Res., 2004, 14, 1394–1403 CrossRef CAS PubMed.
- J. T. Wade and D. C. Grainger, Spurious transcription and its impact on cell function, Transcription, 2018, 9, 182–189 CrossRef CAS PubMed.
- I. Kalvari, J. Argasinska, N. Quinones-Olvera, E. P. Nawrocki, E. Rivas, S. R. Eddy, A. Bateman, R. D. Finn and A. I. Petrov, Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families, Nucleic Acids Res., 2017, 46, D335–D342 CrossRef PubMed.
- A. R. Evans and D. E. Whitworth, Correlations between the role, sequence conservation, genomic location and severity of phenotype in myxobacterial developmental genes, FEMS Microbiol. Lett., 2010, 312, 40–45 CrossRef CAS PubMed.
- M. K. Thomason and G. Storz, Bacterial antisense RNAs: How many are there and what are they doing?, Annu. Rev. Genet., 2010, 44, 167–188 CrossRef CAS PubMed.
- P. Ortet, G. De Luca, D. E. Whitworth and M. Barakat, P2TF: a comprehensive resource for analysis of prokaryotic transcription factors, BMC Genomics, 2012, 13, 628 CrossRef CAS PubMed.
- T. M. Gronewold and D. Kaiser, act operon control of developmental gene expression in Myxococcus xanthus, J. Bacteriol., 2002, 184, 1172–1179 CrossRef CAS PubMed.
- M. Di Salvo, E. Pinatel, A. Talà, M. Fondi, C. Peano and P. Alifano, G4PromFinder: an algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs, BMC Bioinf., 2018, 19, 36 CrossRef PubMed.
- J. F. Garcia-Mazcorro and J. R. Barcenas-Walls, Thinking beside the box: Should we care about the non-coding strand of the 16S rRNA gene?, FEMS Microbiol. Lett., 2016, 363, fnw171 CrossRef PubMed.
- R. Raghavan, D. B. Sloan and H. Ochman, Antisense transcription is pervasive but rarely conserved in enteric bacteria, mBio, 2012, 3, e00156-12 CrossRef PubMed.
- D. E. Whitworth, Interspecies conflict affects RNA expression, FEMS Microbiol. Lett., 2018, 365, fny096 CrossRef PubMed.
- D. Sutton, P. G. Livingstone, E. Furness, M. T. Swain and D. E. Whitworth, Genome-Wide Identification of Myxobacterial Predation Genes and Demonstration of Formaldehyde Secretion as a Potentially Predation-Resistant Trait of Pseudomonas aeruginosa, Front. Microbiol., 2019, 10, 2650 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental File 1: ncRNAs in the M. xanthus DK1622 genome annotation. Supplemental File 2: Putative ncRNAs identified in the M. xanthus DK1622 genome by RNAz. Supplemental File 3: ncRNAs identified in the M. xanthus DK1622 transcriptome. Supplemental File 4: ncRNA target prediction. Supplemental File 5: CopraRNA output of predicted mRNA targets for intergenic ncRNAs. Supplemental File 6: Normalised expression of ncRNAs and predicted target mRNAs under four experimental conditions. See DOI: 10.1039/d0mo00068j |
| This journal is © The Royal Society of Chemistry 2020 |