
Protein glycosylation in Leishmania spp.†
Simon Ngao
Mule‡
a,
Joyce Silva
Saad‡
a,
Livia Rosa
Fernandes
a,
Beatriz S.
Stolf
b,
Mauro
Cortez
b and
Giuseppe
Palmisano
 *a
*a
aGlycoProteomics Laboratory, Department of Parasitology, Institute of Biomedical Sciences, University of Sao Paulo, Avenida Lineu Prestes 1374, Butantã, Sao Paulo - 05508-000, Brazil. E-mail: palmisano.gp@usp.br; Tel: +55(11) 3091-3899
bDepartment of Parasitology, Institute of Biomedical Sciences, University of Sao Paulo, Sao Paulo, Brazil
First published on 24th July 2020
Abstract
Protein glycosylation is a co- and post-translational modification that, in Leishmania parasites, plays key roles in vector–parasite–vertebrate host interaction. In the mammalian host, Leishmania protein glycosylation is involved in virulence, host cell invasion, and immune evasion and modulation. The Leishmania glycocalyx is composed by a dense array of glycoconjugates including lipophosphoglycan, glycoinositolphospholipids, glycoproteins and proteophosphoglycans which varies in composition between Leishmania species and developmental stages. The current knowledge on Leishmania protein glycosylation is quite limited. The development of novel analytical tools to characterize the Leishmania glycoproteome and the expanding toolbox to modulate the parasite glycocode will help in deciphering the processes involved in Leishmania–host interaction. This review will recapitulate the current knowledge of Leishmania protein glycosylation, and glycan structures reported, and the potential application of mass spectrometry-based analysis for system-wide Leishmania glycoproteome and glycome analysis.
Introduction
Leishmania biology and life cycle
Leishmania spp. are unicellular protozoan parasites belonging to the Trypanosomatidae family, and are the etiological agents of leishmaniasis. Of the 53 known Leishmania species, 20 are pathogenic to humans.1 Leishmaniasis is prevalent in the tropical, subtropical and Mediterranean basin affecting over 98 countries,2 with over 350 million people at risk of infection.1,3 The dipteran sandfly insects of the genus Phlebotomus and Lutzomyia of the Old and New Worlds, respectively, are the biological vectors of Leishmania parasites.4 Organ transplant,5,6 blood transfusion7 and in rare cases congenital transmission8,9 may also contribute to leishmaniasis transmission. The World Health Organization (WHO) estimates that 0.7–1 million new cases and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 deaths are reported each year,10 while current infection cases are estimated at 12 million. Leishmaniasis has varying clinical pathology and disease outcomes, depending on the infecting Leishmania spp. and host factors including the immune status.11 Infected individuals can be asymptomatic or may present one of the two main clinical forms: cutaneous/tegumentary and visceral leishmaniasis, which account for approximately 0.7–1.2 and 0.2–0.4 million new cases each year, respectively.12 Cutaneous leishmaniasis (CL) is further subdivided into localized disease, the most common form, characterized by localized ulcerative lesions at or near the site of the bite, and the mucocutaneous form, in which the parasites reach the mucosa of the upper respiratory tract affecting the nasal cavity and often the oral cavity.13 Other rare forms of CL include disseminated leishmaniasis (DL)14 and diffuse cutaneous leishmaniasis (DCL).15,16 Visceral leishmaniasis occurs through the metastasis of infected cells from the bite site to visceral organs such as spleen, liver and bone marrow, causing hepatosplenomegaly, pancytopenia, thrombocytopenia, and anemia due to marrow suppression,17 and is fatal if left untreated. Post-kalazar dermal leishmaniasis (PKDL) occurs in clinically cured patients of visceral leishmaniasis in East Africa and the Indian subcontinent, where L. donovani is the causative agent.18 Clinical diagnosis is complicated and lacks specificity due to the broad clinical spectrum of leishmaniasis and closely related diseases, which usually co-exist in endemic regions.19,20 Few chemotherapeutic agents exist, but are limited by high costs, toxicity, and treatment failures due to resistance.21,22 As leishmaniasis is endemic mostly in developing countries, there is little interest by pharmaceutical companies and public health authorities to invest in drug and vaccine development, research, prevention, or control strategies.23,24 Rightly, the inclusion of leishmaniasis as a neglected tropical disease (NTD) by WHO is commendable for it has allowed improved focus on surveillance, technical and financial support for control programs, monitoring disease trends, prevention, promotion of research and the use of safe and affordable drugs.10
000–30000 deaths are reported each year,10 while current infection cases are estimated at 12 million. Leishmaniasis has varying clinical pathology and disease outcomes, depending on the infecting Leishmania spp. and host factors including the immune status.11 Infected individuals can be asymptomatic or may present one of the two main clinical forms: cutaneous/tegumentary and visceral leishmaniasis, which account for approximately 0.7–1.2 and 0.2–0.4 million new cases each year, respectively.12 Cutaneous leishmaniasis (CL) is further subdivided into localized disease, the most common form, characterized by localized ulcerative lesions at or near the site of the bite, and the mucocutaneous form, in which the parasites reach the mucosa of the upper respiratory tract affecting the nasal cavity and often the oral cavity.13 Other rare forms of CL include disseminated leishmaniasis (DL)14 and diffuse cutaneous leishmaniasis (DCL).15,16 Visceral leishmaniasis occurs through the metastasis of infected cells from the bite site to visceral organs such as spleen, liver and bone marrow, causing hepatosplenomegaly, pancytopenia, thrombocytopenia, and anemia due to marrow suppression,17 and is fatal if left untreated. Post-kalazar dermal leishmaniasis (PKDL) occurs in clinically cured patients of visceral leishmaniasis in East Africa and the Indian subcontinent, where L. donovani is the causative agent.18 Clinical diagnosis is complicated and lacks specificity due to the broad clinical spectrum of leishmaniasis and closely related diseases, which usually co-exist in endemic regions.19,20 Few chemotherapeutic agents exist, but are limited by high costs, toxicity, and treatment failures due to resistance.21,22 As leishmaniasis is endemic mostly in developing countries, there is little interest by pharmaceutical companies and public health authorities to invest in drug and vaccine development, research, prevention, or control strategies.23,24 Rightly, the inclusion of leishmaniasis as a neglected tropical disease (NTD) by WHO is commendable for it has allowed improved focus on surveillance, technical and financial support for control programs, monitoring disease trends, prevention, promotion of research and the use of safe and affordable drugs.10
The life cycle of Leishmania parasites alternates between sandfly insect vectors and mammalian hosts, including humans (Fig. 1). During a blood meal, infected female phlebotomine insects transmit the non-replicative infective metacyclic promastigotes to the bite site in the host. This mobile and flagellated form is phagocytosed by immune cells, such as macrophages and neutrophils.25,26 In the macrophages, the main Leishmania host cells, metacyclic promastigotes transform into amastigotes inside parasitophorous vacuoles, where they multiply and subsequently are released upon host cell rapture, infecting other cells.27 When a phlebotomine insect takes a blood meal from an infected vertebrate host, cells containing amastigotes or free amastigotes are ingested. Inside the insect's gut, amastigotes transform into procyclic promastigotes, which multiply and migrate to the anterior midgut (stomodeal valve) of the insect where they undergo metacyclogenesis, differentiating into metacyclic infective forms, which are passed to a new mammalian host during a blood meal through regurgitation.27 Metacyclic promastigotes that are not regurgitated during a blood meal may undergo re-differentiation into retroleptomonad promastigotes, which replicate inside the vector, increasing the haptomonad promastigotes attached to the sandfly anterior midgut.28
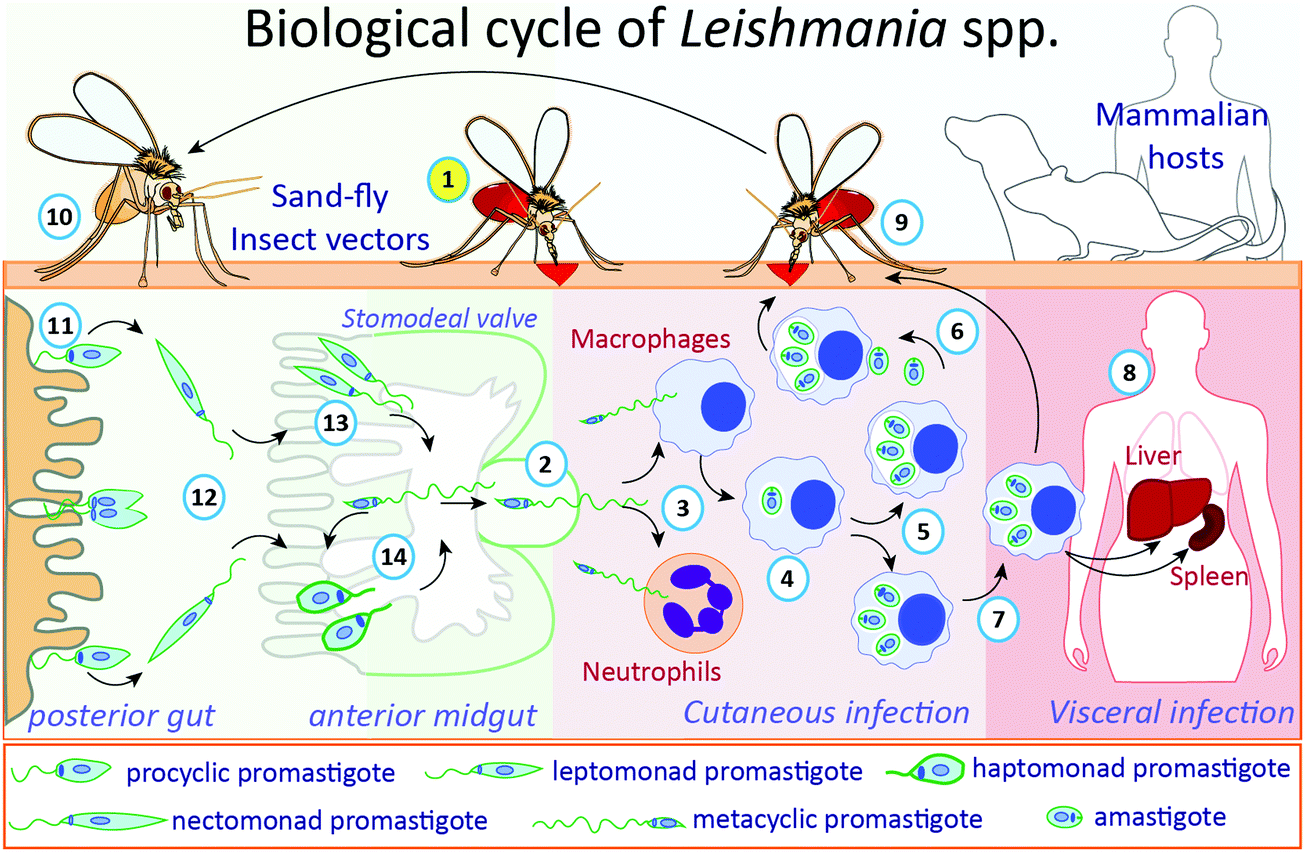 | ||
| Fig. 1 The biological cycle of Leishmania spp. During a blood meal (1, in yellow circle), infected female phlebotomine insects regurgitate blood, transmitting, and releasing the non-replicative but infective metacyclic promastigotes (2) into the mammalian host. These flagellated forms are phagocytosed by macrophages and neutrophils (3). In macrophages, parasites transform into infective amastigotes (4), which multiply by binary fission (5), following the exit from ruptured cells and infect other neighbor cells (6). For Leishmania species associated with visceral infection, different organs can be reached, such as liver or spleen, by infected macrophages or released amastigotes (7), where parasites will replicate and establish the infection (8). When a phlebotomine insect takes a blood meal from an infected mammalian host (9), surrounded cells containing-amastigotes or released free amastigotes will be ingested. Inside the insect's gut (10), amastigotes will transform into procyclic promastigotes in the posterior gut (11), which will multiply and migrate (12) to the anterior midgut (stomodeal valve) of the insect (13) where they will differentiate into metacyclic infective forms, establishing the process called metacyclogenesis. Metacyclic promastigotes that are not regurgitated during the blood meal undergo re-differentiation into retro-leptomonad promastigotes, which replicate inside the vector, increasing the haptomonad promastigotes attached to the anterior sandfly midgut (14). | ||
Chemotherapy of leishmaniasis
The chemotherapy for leishmaniasis counts on few drugs: pentavalent antimonials, pentamidine, amphotericin B and miltefosine (Table 1). Most of them are considerably toxic, have high cost and, except for miltefosine, must be administered by the parenteral route.22 Despite their toxicity and narrow therapeutic window, pentavalent antimonials (meglumine antimoniate and sodium stibogluconate) are still the first line drugs for cutaneous leishmaniasis in most endemic countries and for visceral leishmaniasis in regions such as Latin America and East Africa.22,29 The mechanism of action of pentavalent antimonials (SbV) is not completely understood. Some studies suggest that SbV acts as a prodrug that is reduced by the macrophage or by the amastigote to SbIII, more toxic and active against Leishmania.30 Others support the direct involvement of SbV in parasite death.31 SbV was shown to inhibit Leishmania type I DNA topoisomerase. It also forms complexes with ribonucleosides, and these complexes may either inhibit Leishmania purine transporters or be internalized and inhibit the purine salvage pathway. This process leads to the depletion of ATP and GTP (revised in ref. 31). The drug also affects the immune system. It was shown to damper the activity of protein tyrosine phosphatases, increasing cytokine responses, and to augment the phagocytic capacity of monocytes and neutrophils and the production of superoxide anion by phagocytes (revised in ref. 31). SbIII interacts with sulfhydryl-containing biomolecules such as thiols, peptides, proteins and enzymes. It affects parasite's redox equilibrium by two mechanisms: by interacting with trypanothione reductase, leading to accumulation of disulfide forms of glutathione and trypanothione, and by increasing the efflux of trypanothione and glutathione, increasing the levels of reactive oxygen species (ROS).32 Besides, SbIII induces parasite death by oligonucleosomal DNA fragmentation.33| Drug | Mechanisms of action | Ref. |
|---|---|---|
| Pentavalent antimonials (SbV) (meglumine antimoniate and sodium stibogluconate) | Inhibition of type I DNA topoisomerase | 31–33 |
| Inhibition of purine transporters or of purine salvage pathway, depleting ATP and GTP | ||
| Increase in cytokine responses, in phagocytic capacity of monocytes and neutrophils and in the production of superoxide anion by phagocytes | ||
| Interaction with trypanothione reductase, leading to accumulation of disulfide forms of glutathione and trypanothione | ||
| Increase in the efflux of trypanothione and glutathione, increasing reactive oxygen species levels | ||
| Induction of oligonucleosomal DNA fragmentation | ||
| Pentamidine | Decrease in ornithine decarboxylase and spermine synthase, leading to a decrease in polyamines | 34–36 |
| Enlargement of mitochondria and disintegration of the kinetoplast structure | ||
| Inhibition of phosphohydrolytic activity of nucleoside triphosphate diphosphohydrolase (NTPDase) | ||
| Amphotericin B (AmB) | Binding to ergosterol-related sterols, creating pores in the plasma membrane that lead to exchange of ions across the surface and consequent cell death | 29 |
| Induction of oxidative stress | ||
| Miltefosine | Decrease in intracellular choline, affecting cell membrane composition | 22 and 29 |
| Induction of mitochondrial depolarization, reduction in cytochrome-c oxidase activity and decrease of intracellular ATP levels, leading to cell death | ||
Pentamidine interferes with the synthesis of polyamines by decreasing ornithine decarboxylase and spermine synthase. It also probably competes with polyamines for binding to nucleic acid, particularly kinetoplast DNA.34 In fact, the drug induces changes in the kinetoplast and mitochondria, with enlargement of mitochondria and disintegration of the kinetoplast structure.35 Besides, it was shown to inhibit phosphohydrolytic activity of nucleoside triphosphate diphosphohydrolase (NTPDase).36
Amphotericin B (AmB) was first employed as an antifungal agent and later used to treat leishmaniasis due to its ability to bind to ergosterol-related sterols, the main membrane sterols in both organisms (revised in ref. 29). Its interaction with these lipids creates a pore, allowing the exchange of ions across the surface and consequent cell death. AmB also induces oxidative stress.29
Miltefosine binds to the plasma membrane and is internalized either by the endocytic pathway or by complex formed by the miltefosine transporter (MT) and its non-catalytic subunit Ros3 (revised in ref. 22). This complex is responsible for phosphocholine accumulation, and by decreasing intracellular choline, the drug inhibits phospholipid metabolism and phosphatidylcholine and phosphatidylethanolamine synthesis, affecting cell membrane composition (revised in ref. 22). Miltefosine treatment also induces mitochondrial depolarization, a reduction in cytochrome-c oxidase activity and a decrease of intracellular ATP levels, leading to cell death.22,29
Treatment failure has been reported for all drugs mentioned. Numerous facts account for failure, including parasite resistance to the drug and host factors. Combination therapies employing two or more drugs with different mechanisms of action may reduce the development of resistance.29 Besides, new drugs must be tested. Although there have been some advances in the treatment of visceral leishmaniasis and some novel compounds are currently in pre-clinical and clinical phases for this disease form, there were few advances in drug research and development for cutaneous leishmaniasis, the most common disease form.37
The role of glycans in Leishmania biology
Glycans may occur as free oligosaccharides or as simple (e.g. monosaccharides such as the addition of O-linked GlcNAc to nucleocytoplasmic proteins) or complex (oligosaccharides such as complex and/or hybrid N-linked glycans) structures covalently attached to different macromolecules such as proteins and lipids to form glycoproteins, proteoglycans or proteophosphoglycans, and glycolipids, respectively.38 During the intricate life cycle of Leishmania spp. between the invertebrate insect vector and vertebrate hosts, glycoconjugates play crucial roles in the interaction and survival of the parasite. The surface membrane of Leishmania spp. is covered by a dense layer of glycoconjugates collectively termed glycocalyx, which play different roles in the parasites’ survival, infectivity, virulence and establishment of disease. The Leishmania spp. surface membrane glycoconjugates include lipophosphoglycans (LPGs), glycoinositolphospholipids (GIPLs), glycoproteins and proteophosphoglycans (PPGs), and seminal reviews and studies have been published describing their molecular and functional characterization.39–55 These Leishmania spp. glycoconjugate structures are been summarized in Fig. 2.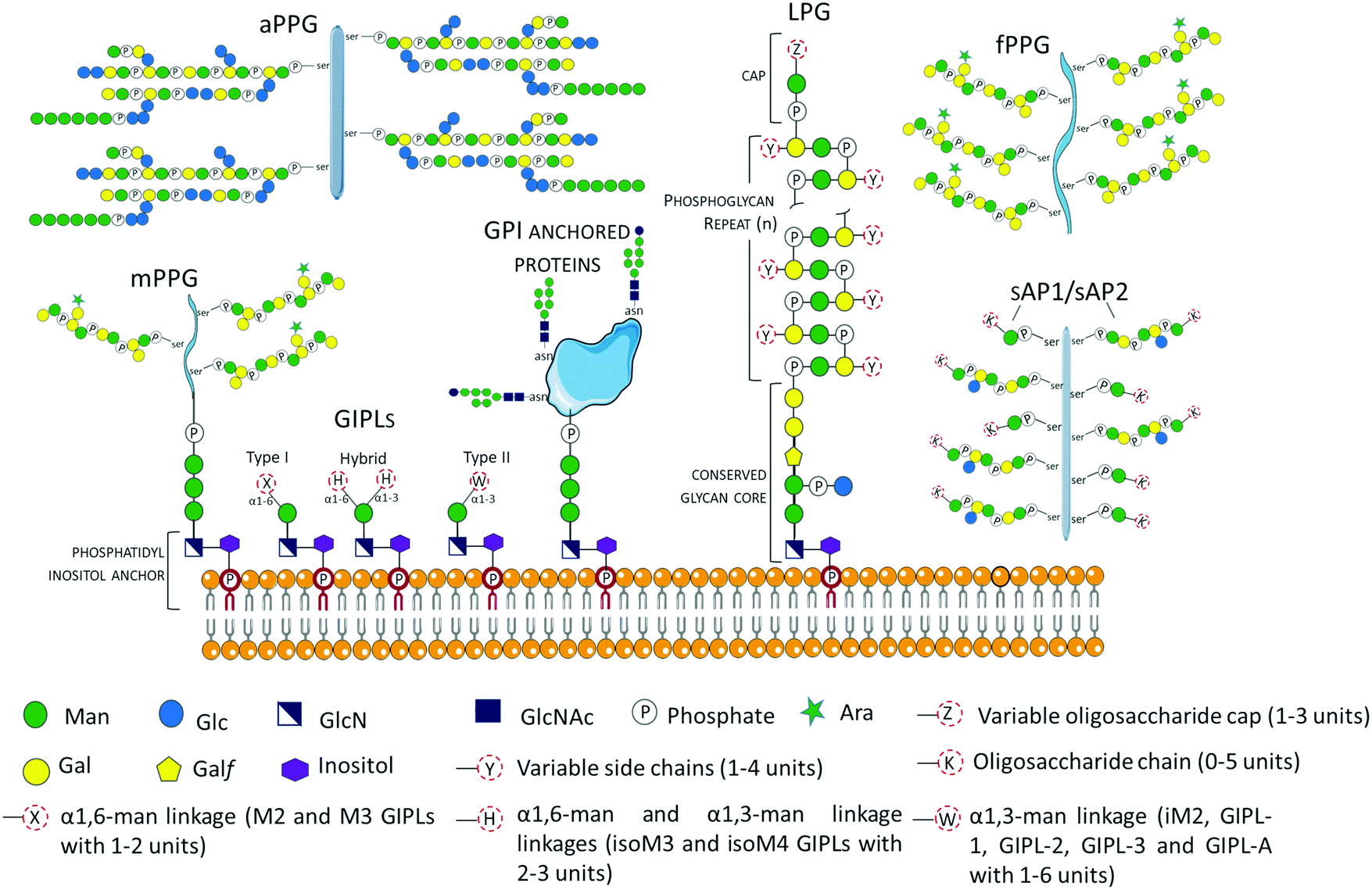 | ||
| Fig. 2 Schematic representation of glycoconjugates in Leishmania spp. The glycoconjugates include the GPI anchored lipophosphoglycans (LPGs)39,40,43,55 and membrane proteins (e.g. gp63),57 and the free low molecular weight glycoinositolphospholipids (GIPLs).41–43,56,58 Proteophosphoglycans (PPGs)45,46,57,59,60 predominantly expressed in promastigote life stage can be bound to the membrane (mPPGs), or they can be secreted (sAPs and fPPGs). A structurally different aPPG is predominantly expressed in the amastigote life stage. | ||
Glycosyl-phosphatidylinositols (GPIs) anchor LPGs, cell membrane glycoproteins, proteophosphoglycans (PPGs) and glycoinositolphospholipids (GIPLs) to the plasma membrane.43 GPI anchors are ubiquitously conserved in other protozoan and higher eukaryotic cells, and have the general structure composed of a conserved backbone of ethanolamine-phosphate-Manα1-2Manα1-6Manα1-4GlcNα1-6myo-inositol.43 GIPLs are the free (unattached to phosphoglycans or proteins), low molecular weight GPIs, with a conserved Manα1-4GlcN linked to an alkyl-acylglycerol through a phosphatidylinositol (PI) residue.41,43,53,56,57 GIPLs are the most abundant glycoconjugates in both promastigote and amastigote life stages,42,43 and show species and stage-specific modulation.42,53,56,58,61 Based on monosaccharide substitutions in glycan moiety, GIPLs are classified in three types43,56,57 (Fig. 2). Type I GIPLs have α1,6-mannose linked to Manα1-4GlcN, and are structurally similar to protein GPI anchors. Type II GIPL glycan moiety is structurally related to LPG with α1,3-mannose residue linked to Manα1-4GlcN. Hybrid-type GILPs have both α1,6-mannose and α1,3-mannose linked to the Manα1-4GlcN motif.43,44 GIPLs play important roles in macrophage infectivity,62 and modulation of the innate immune system by inhibition of cytokines and nitrite production.53 In L. mexicana, GIPLs were shown to be essential for growth.49,63
In Leishmania spp. promastigote life stage, LPGs are predominantly expressed on the flagellar and cell surface.39,64,65 The general structure of the LPG molecule is composed of terminating oligosaccharide cap structures, a repeating phosphorylated saccharide region attached to a lyso-alkyl phosphatidylinositol (lysoalkyl-PI) lipid anchor through a phosphorylated hexasaccharide glycan core (Fig. 2).39,40,66 The role of LPG in host–parasite interaction has been extensively reported to be an important virulence factor with different roles depending on the Leishmania species.67,68Leishmania spp. promastigote stage surface membrane is predominantly covered by species-specific and growth stage specific lipophosphoglycans (LPGs).39,41,66L. major amasigotes express a structurally, biochemically and antigenically distinct LPG from the promastigote life stage parasites.69,70 LPG from L. donovani was shown to be a potent inhibitor of PKC activity in vitro.71 During infection, PKC is involved in the generation of macrophage's oxidative burst, which leads to the destruction of invading microbes. In infections with L. mexicana, LPG differentially regulates PKCa in macrophages derived from BALB/c or C57BL/6 mice by inhibiting or stimulating their activities, respectively.72 Promastigote LPGs from L. amazonensis play an immune modulatory role by the induction of neutrophil extracellular traps (NETs).73 Inside the macrophage, LPG inhibits maturation of the phagosome at the early stage of infection,74 allowing the parasite's survival within the vacuole until their differentiation into amastigotes. In addition, L. amazonensis LPG has been demonstrated to induce the activation of the promotor for protein kinase R (PKR), whose expression favors infection by the induction of IFN-1.75 LPG also plays a vital role in the parasite–vector interaction. Indeed, LPG has been shown to be involved in the protection of promastigotes from the insect's digestive system, as demonstrated by the inability of L. donovani LPG2 mutants deficient in LPG and other phosphoglycans to survive in the hydrolytic environment of the insect midgut.76 Besides, LPG has been demonstrated to be involved in the binding of promastigotes to epithelial cells in the insect midgut, preventing the parasites from being excreted with the blood meal.76,77 Structural and functional details of LPG have been extensively reviewed.39,54,76,78
Among Leishmania spp. glycoconjugates with protein components include glycoproteins and proteophosphoglycans (PPGs). Protein glycosylation is a co- and post-translational modification in which glycan macromolecules are covalently attached to specific amino acid residues. Depending on the amino acid residue attachment of the glycan moiety, two major classes of protein glycosylation have been reported; N-linked glycosylation and O-linked glycosylation where the glycans are attached to asparagine (Asn) and serine (Ser)/threonine (Thr) amino acid side chains, respectively.79 In Leishmania parasites, N-linked glycosylation has been associated with virulence, immune evasion, host–cell interaction (including, adhesion and invasion), host immune modulation and structural integrity of proteins.80–85 The highly O-glycosylated proteophosphoglycans play crucial roles in the parasites’ complement activation, inhibition of lysis by serum, and prevention of opsonization of amastigote life stages.86 In addition, O-GlcNAc, a unique type of protein O-glycosylation of nucleocytoplasmic proteins, was described in Leishmania spp. gp96/92,87 and has been proposed to exert glycan-dependent signaling similar to protein phosphorylation such as transcriptional activation, nuclear transport and degradation of proteins.88,89
One of the most studied Leishmania glycoproteins is gp63, also known as leishmanolysin, a 60–66 kDa zinc-metalloprotease recognized as a major surface antigen and the most abundant surface protein in promastigotes90–92 and also an important virulence factor.93 Gp63, a predominant N-glycosylated surface protein is present in both promastigote and amastigote stages, and confers protection from phagolysosomal degradation in the host macrophages.94,95 In addition, gp63 is involved in the attachment of promastigotes to macrophages,83,84,96 and in the cleavage of C3b to iC3b, avoiding parasite lysis by the complement cascade.97 The generated iC3b acts in the opsonization of the parasite, crucial for internalization by the macrophage receptor CR3.97,98 Fibronectin-like properties of GP63 have also been demonstrated, with studies showing the interaction of gp63 with β1 integrins, receptors for fibronectin.96,99,100 Moreover, gp63 mediates the degradation of components of the extracellular matrix or subcutaneous tissue.101
In Uniprot database,102 gp63 protein from different Leishmania spp. show varying numbers of N-linked glycosylation sites: L. mexicana (P43150) has 8 sites, L. tropica (Q8MNZ1) has 7, L. guyanensis (Q00689) has 6, L. major (P08148) has 3, L. amazonensis (Q27673) and L. chagasi (P15706) have 2 sites each, and L. donovani (P23223) has 1 sites, indicating a species-specific N-linked glycosylation pattern. Based on sequence alignment analysis (Fig. 3), shared and specie-specific N-linked glycosylation sites were identified. All Leishmania spp. gp63 amino acid sequences, with the exception of L. mexicana, have a conserved N-glycosite at position 304 suggesting a conserved role. At position 401, L. mexicana, L. tropica, L. guyanensis and L. chagasi gp63 share a conserved N-linked glycosylation site. At position 411, L. mexicana, L. tropica, L. guyanensis, L. major and L. amazonensis gp63 glycoprotein share a conserved N-glycosylation site, which is absent for L. chagasi and L. donovani. The glycosylation sites on gp63 glycoprotein indicate that Leishmania parasites have shaped their protein glycosylation sites in a species-specific manner.
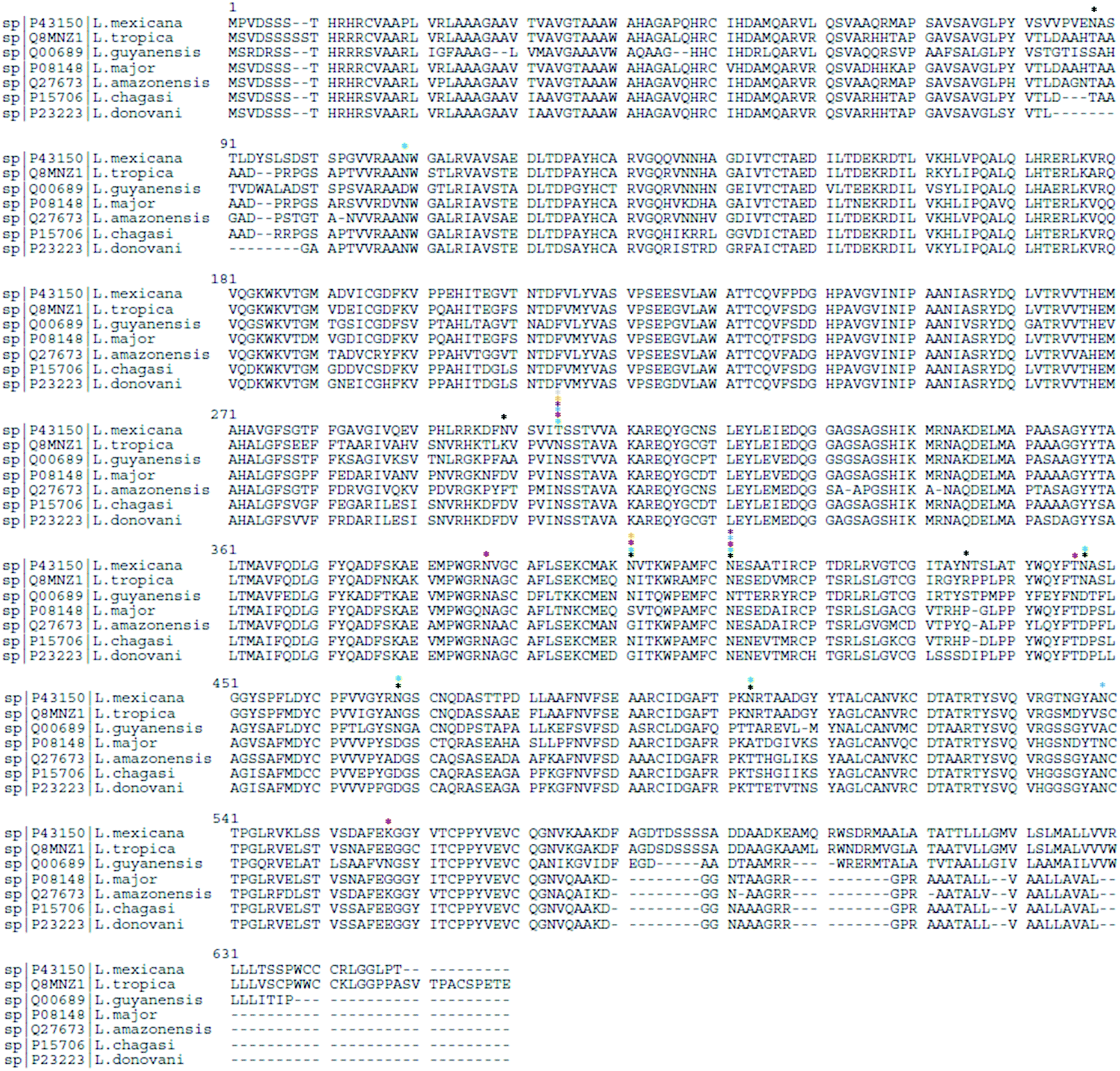 | ||
| Fig. 3 Sequence alignment analysis of gp63 from different Leishmania spp. highlighting conserved and species-specific N-glycosites (N[X]S/T/C, where X is any amino acid except P). The N-glycosylation sites for L. mexicana (*), L. tropica (*), L. guyanensis (*), L. major (*), L. amazonensis (*), L. chagasi (*) and L. donovani (*) are highlighted. | ||
Three N-glycosylation sites were reported on L. major gp63 protein by gene transfection into gp63 deficient L. amazonensis promastigotes and characterization by site-specific mutagenesis.94 Structural analysis of released gp63 N-linked glycans from L. amazonensis revealed 4 major biantennary N-linked oligomannose structures, with no evidence of hybrid or complex N-glycan structures.103 As shown in Table S1 (ESI†), there is species-specific N-linked protein glycosylation on gp63, as evidenced in GlyConnect.104 In L. donovani and L. major promastigote life stage, 2 N-linked oligosaccharides structures (Man6GlcNAc2 and GlcMan6GlcNAc2) were reported by Funk and colleagues, who showed that the glycosylation was not consistent between the two species in the amastigote life stage, with L. donovani lacking N-linked glycans, indicating a specie and life stage-specific protein N-linked glycosylation.105
Tunicamycin, a nucleoside analog inhibitor of GlcNAc-1-P-transferase (ALG7, Fig. 4) which transfers N-acetylglucosamine-1-phosphate to dolichol monophosphate,106 has been used by different research groups to establish the functions of protein N-linked glycosylation in Leishmania spp. L. chagasi promastigotes expressing deglycosylated gp63 surface proteins from tunicamycin-resistant population showed that deglycosylated form of gp63 was proteolytically inactive compared to the glycosylated form.107 This strain was able to bind to the receptor for the iC3b fragment of complement, CR3, but not to the mannose receptor indicating a selective N-linked glycan-dependent binding.107 On the contrary, treatment of L. major promastigotes with tunicamycin and the deglycosylation of gp63 from L. major and L. amazonensis promastigotes demonstrated that deglycosylation did not interfere with the proteolytic activity of this membrane protease,108 suggesting that gp63 folding, function, signaling to the plasma membrane, and resistance to proteolysis were not dependent on its N-linked glycosylation.108,109 These conflicting results could arise from differences in degrees of deglycosylation of gp63,94 differences between Leishmania species, enzymatic assays, and the limited site-specific knowledge on gp63 glycosylation modulation upon tunicamycin and/or glycosidase treatment.
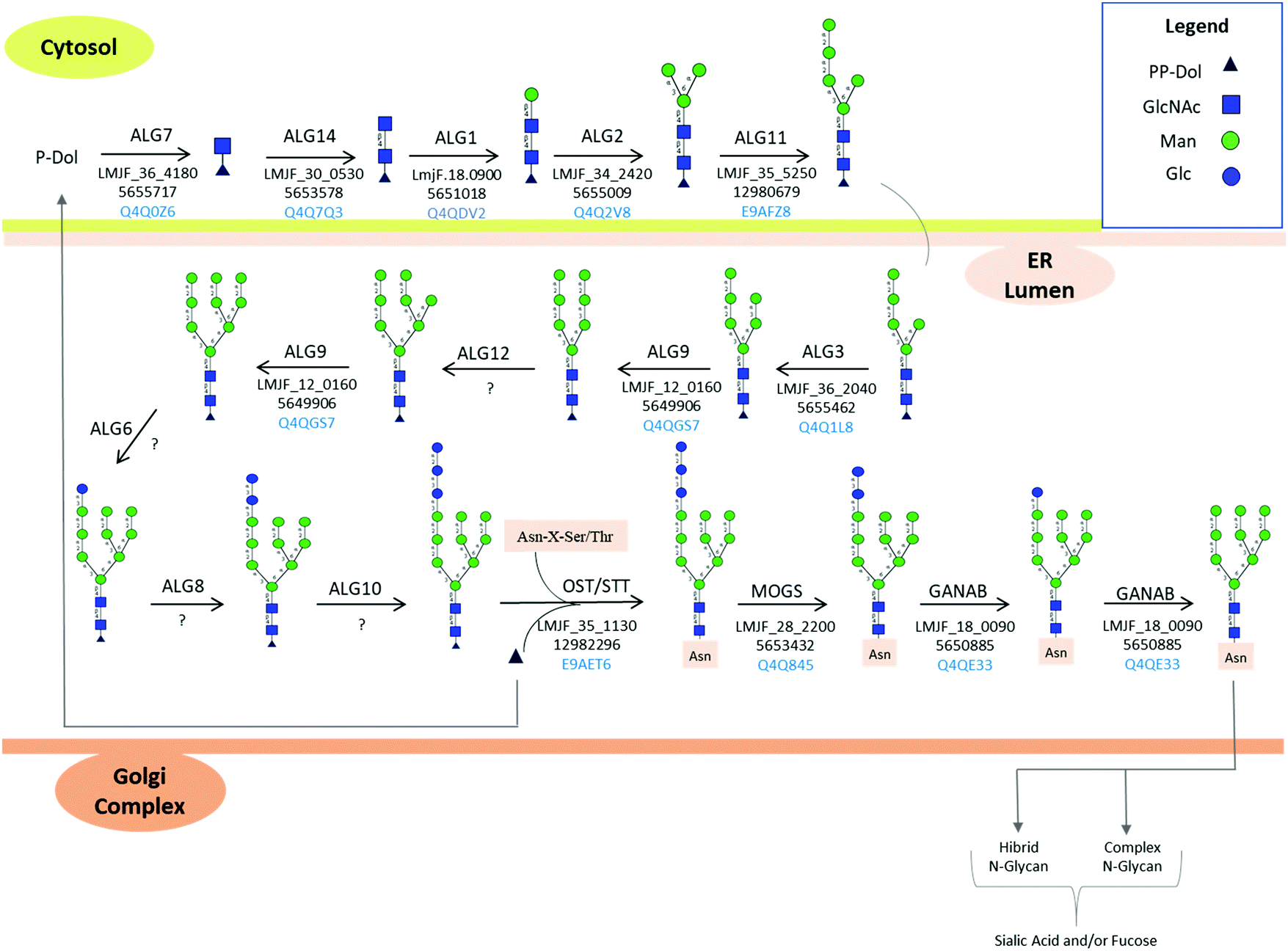 | ||
| Fig. 4 Protein N-glycosylation biosynthesis in Leishmania spp. according to KEGG pathway. L. major, was used as a model to illustrate the N-glycan biosynthetic pathway. The spaces filled by “?” indicate enzymes not mapped in the Leishmania spp. genome. Unusual biosynthetic and structural features of N-linked protein glycosylation in Leishmania spp. are reported in Box 1. ER: endoplasmic reticulum. | ||
Proteophosphoglycans (PPGs) are modified by phosphoglycosylation of phosphoglycan chains through the unusual Manα1-PO(4)-Ser linkage.46 Filamentous (fPPG), membrane (mPPG) and secreted acid phosphatases (SAP) are primarily synthesized by the promasigote life stage, while the non-filamentous (aPPG) is secreted in the amastigote life stage.45 In the sandfly, PPGs play important roles in parasite–vector interaction, including insect gut colonization by conferring resistance to insect digestive enzymes.110,111 fPPGs aid in the efficient transmission of the parasites to the mammalian hosts through plug formation.112 In the mammalian host, PPGs increase macrophage recruitment to the site of the bite, augmenting the activity of arginase generating polyamides involved in the growth of the parasite, which reduces L-arginine for the production of nitric oxide (NO) by inducible nitric oxide synthase (iNOS).113 Inside the macrophages, secreted amastigote PPGs play a key role in the enhancement of the maturation of parasitophorous vacuoles.59,114
In this review, protein N- and O-linked glycosylation in Leishmania spp. will be presented, with a focus on the biosynthetic pathways, cellular functions and analytical tools to characterize the glycoconjugates. At present, there is a paucity of system-wide glycoproteome and/or glycome studies on LC-MS/MS-based approaches in Leishmania parasites. An in-depth understanding of protein glycosylation would offer unprecedented opportunities for the identification and/or development of next generation glycoprotein-based therapeutics and/or in the uncovering of putative novel drug target candidates. Indeed, glycopeptide-based therapeutics have been developed and used in the treatment of metabolic disorders, development of antivirals, and the shaping of vaccines for viruses and cancer (recently reviewed115). Unusual and/or unique features between the parasite and mammalian host glycosylation pathways are highlighted here. Moreover, we have used literature mining to assemble the Leishmania spp. glycoproteins described so far.
Protein glycosylation landmarks in Leishmania spp.
The surface coat of L. tarentolae promastigote stage was detected by Strauss in 1971 using electron microscopy in the presence of rabbit antiserum.116 In 1973, lectin agglutination assays with concanavalin A and phytohemagglutinin-P (PHA-P) specific for terminal α-D-glucose/α-D-mannose and N-acetyl-D-galactosamine sugar moieties, respectively, on trypsinized and non-trypsinized promastigote samples confirmed that the surface of L. donovani promastigotes was decorated by sugar molecules.117 Pre-treatment of Leishmania parasites with dextranase and α-amylase enzymes showed reduced agglutination by both Con A and PHA-P lectins, confirming that the polysaccharides contained D-glucose like units with α-1,6 and α-1,4 glyosidic linkages.117 During the same year, Dwyer and colleagues studied the surface coats of L. donovani in the promastigote and amastigote stages by cytochemical techniques coupled to light and electron microscopy.118 Using alcian blue-lanthanum nitrate and ruthenium dyes which precipitate and stain acid mucopolysaccharides, they were able to stain the parasites’ surface coats and established that the cell surface (and flagellar tip for promastigotes) were covered with acid polysaccharide moieties in the different stages of development.A year later, lectin agglutination assays on L. braziliensis promastigote and amastigote stages were carried out to determine the terminal sugar moieties on the polysaccharide chains on the parasite's surface in the different life stages.119 Agglutination reactions with Con A specifically recognized by α-D-glucose and α-D-mannose terminal sugars and Ricinus communis agglutinin (RCA) recognized by α-D-galactose terminal sugars confirmed the presence of polysaccharide complexes with glucose/mannose and/or galactose terminal residues in both life stages.120 Wheat germ agglutinin (WGA) specific for N-acetylglucosamine and sialic acid, soybean agglutinin (SBA), and PHA-P specific for N-acetyl-D-galactosamine were negative in both promastigote and amastigote stages, suggesting that the polysaccharides attached to the surface of the parasites lacked these terminal sugar moieties on their cell surfaces, or their presence in undetectable amounts. Interestingly, RCA agglutination was negative for non-infective promastigotes with high passage numbers in cultures, suggesting loss of or processing of D-galactose terminal residues of the carbohydrate chains. Lectin agglutination assays of surface radioiodinated proteins extracted from L. tropica promastigotes using detergents showed that concanavalin A agglutinin, but not RCA, WGA, SBA and UEA (Ulex europaeus agglutinin I), agglutinated with the surface components, suggesting the presence of surface glycoproteins with mannose and/or glucose residues.121 Taken together, these early studies using lectin agglutination assays and glycan-specific stains demonstrated that protein glycosylation is species and strain-specific and modulated during Leishmania spp. life stages.
In 1981, Chang demonstrated L. donovani surface glycoproteins’ role in macrophage binding by pretreatment of promastigotes with enzymes which cleaved off sialic acids (neuraminidase), mannoses (α-mannosidase), GlcNAc (α-N-acetylglucosaminidase) and glucose (β-glucosidase) moieties from the parasites surface, which led to a reduction in the binding of the parasites to hamster peritoneal macrophages.81 In 1984, Parodi and Martin-Berrientos studied the glycoprotein assembly in Leishmania parasites looking at the lipid-linked oligosaccharide (LLO) molecules transferred to asparagine (Asn) residues during N-glycosylation in L. mexicana.122 They showed that, as opposed to most eukaryotic systems which transfer Glc3Man9GlcNAc2 from the carrier lipid to a nascent polypeptide at the asparagine side chain,123L. mexicana LLO carrier was attached to Man6GlcNAc2 oligosaccharide chain which migrated equally to a Man6GlcNAc2 standard by paper chromatography. In addition, paper chromatography analysis of glycoproteins labeled with [U-14C]-glucose showed that processing of the transferred Man6GlcNAc2 occurred, with a glucose moiety being transiently added to yield GlcMan6GlcNAc2, but the glycan structure on mature glycoproteins lacks glucose.122 During this study, no complex and hybrid-type glycan structures were detected.
In 1984, functional studies by Dagger and colleagues on N-linked protein glycosylation inhibition using tunicamycin revealed the involvement of protein N-linked glycosylation in growth and cellular morphology in L. braziliensis promastigotes124 and macrophage infection by L. donovani in 1985 by Nolan and Farrell.125 Studies on L. amazonensis showed growth inhibition of promastigotes in the presence of tunicamycin.126 In 1987, Lovalace and Gottlieb demonstrated the effect of N-linked glycoprotein inhibition using tunicamycin in the growth of L. donovani promastigotes, and its role in secreted acid phosphatase activity,127 which did not interfere with the secretion of the enzyme. Later that year, Bates and Dwyer128 arrived at the same conclusion by showing the presence of N-linked glycans on mature acid phosphatase in L. donovani promastigotes using tunicamycin and N-glycosidase F treatment, and that the glycosylation of the enzyme did not inhibit its secretion and processing. In 1988, Bates and colleagues applied lectin binding assays using agarose-conjugated lectin beads on secreted proteins from metabolically labeled L. donovani promastigotes cultured in vitro in RE-III medium lacking bovine serum albumin.129 Out of 40 electrophoretically distinct bands corresponding to secreted proteins, half were modified with carbohydrate moieties with terminal mannose residues, as demonstrated by positive binding to concanavalin A and Lens culinaris lectins.129 The relevance of protein N-glycosylation as a biochemical basis for Leishmania virulence was demonstrated by Kink and Chan in 198893 who showed decreased virulence of L. amazonensis of tunicamycin treated promastigotes as compared to tunicamycin-resistant parasites consistent with wild type parasites.
O-Linked fucosylated glycoconjugates in Leishmania parasites have been reported by Guo and colleagues working on L. major.130 Using cryo-electron microscopy coupled with lectin binding using biotinylated Ulex europaeus agglutinin I (UEA-I) specific for O-fucose,131 they demonstrated that fucose was present in various cellular compartments including the endoplasmic reticulum (ER), cytosol, mitochondria, Golgi apparatus and parasite surface.130 Non-permeabilized cells bound to fucose-specific fluorescent UEA-I, as confirmed by flow cytometry, albeit with lower reactivity compared to mutant parasites ectopically overexpressing genes coding for enzymes which catalyze the synthesis of GDP-fucose.130 At least five genes involved in the fucosylation/arabinosylation have been mapped in the Leishmania genome. This study shed new light on the importance of GDP-fucose and fucosylated glycoconjugates for the viability of L. major.
The reported glycans and the methodologies used in their characterization are summarized in Table 2.
| Glycan type | Leishmania sp. | Glycans | Methodology | Ref. |
|---|---|---|---|---|
| N-Glycan | L. donovani promastigotes | Terminal α-1,4 and α-1,6-glucan linked D-glucose; N-acetyl-D-galactosamine | Lectin agglutination assays (ConA and PHA-P lectins); inhibition assays | 117 |
| L. braziliensis promastigote and amastigotes | Terminal α-D-mannose and/or α-D-glucose and α-D-galactose | Lectin agglutination assays (ConA and RCA); inhibition assays with MAM, glucose and D galactose | 119 | |
| L. tropica | Terminal α-D-mannose residues | Lectin agglutination assays on radioiodinated proteins (ConA); inhibition assays | 121 | |
| L. mexicana promastigotes | Man6GlcNAc2; GlcMan6NAc2 (transient) | Paper chromatography oligosaccharides released by Endo H treatment of proteins from [U-14C] glucose labelled cells | 122 | |
| L. donovani promastigotes | Secreted soluble glycoproteins with terminal mannose and galactose (some linked subterminally to N-acetyl galactosamine residues i.e. Gal-β(1–3)-GalNAc) | Lectin affinity chromatography (ConA+, LCA, PNA, RCA); inhibition assays | 129 | |
| L. mexicana amazonensis | Glc1Man6GlcNAc2 | High resolution gel-permeation chromatography, exoglycosidase digestion, acetolysis fragmentation and methylation analysis | 103 | |
| Man6GlcNAc2 | ||||
| Man5GlcNAc2 | ||||
| Man4GlcNAc2 | ||||
| O-Glycan | L. major | O-GlcNAc on gp96/92 | Lectin affinity chromatography (WGA+), transfer of radiolabelled [3H]UDP-galactose to terminal GlcNAc; anhydrous hydrogen fluoride deglycosylation, β elimination, β-galactosidase treatment | 87 |
| L. donovani | Fucoglycoconjugates | ITRAQ LC-MS/MS | 158 | |
| L. major | Fucoglycoconjugates | Cryo-electron mcroscopy; lectin binding assays (UEA I) | 130 | |
N-Linked glycosylation biosynthesis in Leishmania spp.
N-Linked protein glycosylation in eukaryotes involves the covalent attachment of sugar molecules to asparagine residues within the canonical sequon (AsnXxxSer/Thr/Cys; where Xxx ≠ Pro). Unlike gene transcription and protein translation, protein glycosylation undergoes a non-template driven biosynthesis pathway, which is determined by the expression and activity of the glycosylation machinery enzymes, the nature of the proteins undergoing glycosylation, the availability of sugar donors and the cellular metabolism.132 The en bloc transfer of oligosaccharides from a lipid-linked oligosaccharide (LLO) donor to asparagine side chains of nascent proteins in the ER and the subsequent processing of the attached oligosaccharide in the ER and Golgi apparatus is a conserved and well-orchestrated process catalyzed by finely tuned biosynthetic enzymes. Glycosyltransferases, α-glycosidases, and α-mannosidases in the ER and cis-Golgi transfer or cleave off glycan residues. Most of these transmembrane enzymes are located in the ER and Golgi apparatus.133 Four types of mature N-glycans exist in higher eukaryotes depending on their structure: high mannose, hybrid, complex, and paucimannose types. The high mannose, hybrid, and complex types share a common core termed pentasaccharide/trimannosylchitobiose core composed of Manα1,3(Manα1,6)Manβ1,4GlcNAcβ1,4GlcNAcβ (M3).134 In particular, high mannose N-glycans contain only mannose attached to the core. Complex N-glycans have all mannose residues after the core removed by enzymatic processing and replaced to form antenna structures. Hybrid N-glycans are incompletely processed and have at least one antenna containing only mannose residues.135 Paucimannosidic structures contain a monosaccharide composition less than or equal to the N-glycan trimannosylchitobiose core, i.e., Man1–3Fuc0–1GlcNAc2.136 Paucimannose glycans have been reported in several eukaryotes ranging from plants, protists, and vertebrates.136 A deep glycoprotein-centric analysis of Trypanosoma cruzi following enrichment of glycopeptides revealed that Man3GlcNAc2 was present in the epimastigote and trypomastigote life stages.137T. brucei transferrin receptor and class 2 variant surface glycoproteins have been identified with the Man3GlcNAc2 paucimannose structure.138,139 In Leishmania, high mannose (Man5–6GlcNAc2) and an unusual terminally glucosylated glycan structure (GlcMan6GlcNAc2) have been reported.103,122 Man4GlcNAc2, also identified in Leishmania mature proteins, is of unknown biosynthetic origin.136 No report on paucimannose structures on Leishmania glycoproteins is available in the literature.N-Linked glycosylation biosynthetic process begins in the cytosolic side of the ER membrane, where glycosyltransferases add sugar molecules to a growing LLO chain.140 The N-glycan biosynthetic pathway for L. major is shown in Fig. 4, according to KEGG pathway.141 Trypanosomatid dolichol chain is shorter with 10–12 isoprene units compared to mammalian LLO (18–21), plant and fungi (15–16) dolichol.142 The lipid-linked oligosaccharide (LLO) carrier, known as dolichol (Dol, a polyprenol lipid), already phosphorylated to dolichyl diphosphate (Dol-PP),143 acts as the acceptor molecule for N-acetylglucosamine phosphate through a reaction catalyzed by the enzyme UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase to form N-acetyl-D-glucosaminyldiphosphodolichol. This enzyme is coded by the NAGT gene, and its inhibition by treatment of Leishmania parasites using tunicamycin,107 corroborates the expression of this enzyme in Leishmania parasites.
β-1,4-N-Acetylglucosaminyltransferase adds the second N-acetyl glucosamine to the growing LLO chain, to form N,N′-chitobiosyldiphosphodolichol [(GlcNAc)2(PP-Dol)]. Dolichyl-P-Man:GDP-ManGlcNAc2-PP-dolichyl beta-1,4-mannosyltransferase adds a mannose sugar to the chitobiosyldiphosphodolicol to form beta-D-mannosyldiacetylchitobiosyldiphosphodolichol [(Man)(GlcNAc)2(PP-Dol)]. A second mannosyltransferase enzyme, termed dolichyl-P-Man:GDP-Man1GlcNAc2-PP-dolichyl alpha-1,3-mannosyltransferase (alpha-1,3/alpha-1,6-mannosyltransferase) adds the second and third mannose sugars to the growing LLO chain, resulting in alpha-D-mannosyl-beta-D-mannosyl-diacetylchitobiosyldiphosphodolichol (Man)2(GlcNAc)2(PP-Dol) and (Man)3(GlcNAc)2(PP-Dol), respectively. Alpha-1,2-mannosyltransferase adds the fourth and fifth mannose sugar molecules, forming (Man)4(GlcNAc)2(PP-Dol) and (Man)5(GlcNAc)2(PP-Dol), respectively. Dolichyl-P-Man:GDP-Man5GlcNAc2-PP-dolichyl alpha-1,3-mannosyltransferase adds the sixth mannose sugar to form (Man)6(GlcNAc)2(PP-Dol). Paper chromatography analysis of dolichol-P-P bound oligosaccharides isolated from L. mexicana after incubation with radiolabelled glucose ([U-14C] glucose) did not show glucose incorporation into the oligomannose chain, while the largest oligomannose structure identified on the dolichol-P-P was (Man)6(GlcNAc)2.122 The terminal Man (and Glc in mammals) are essential in the correct folding of the newly glycosylated proteins and ensure glycoprotein quality control in the ER.132 The transfer of unglucosylated oligosaccharides in Leishmania parasites is an unusual feature in trypanosomatids compared to the canonical biosynthesis of N-linked glycosylation of higher animals, fungi and plants, which occurs in the ER lumen.
Oligosaccharyltransferase (OST) enzyme (dolichyl-diphosphooligosaccharide–protein glycosyltransferase) catalyzes the en bloc transfer of the oligosaccharide from the PP-Dol to an asparagine side chain of the acceptor protein to form (Man)6(GlcNAc)2(Asn).122 Kinetoplastids encode a single-subunit OST enzyme, while higher eukaryotes encode OSTs with different complexities varying among species. STT3 is the catalytic subunit, and in Trypanosoma brucei and L. major, a single subunit is encoded144,145 from several copies of the gene. In the parasitic protozoa L. major, four STT3 paralogues, but no homologs to the other OSTase subunits are encoded in the genome.146
Subsequently, a glucose molecule is transiently transferred from UDP-Glc to the newly glycosylated protein to form (Glc)(Man)6(GlcNAc)2 by a glucose transferase enzyme.122 The addition of a glucose residue to (Man)6(GlcNAc)2(Asn) to form (Glc)(Man)6(GlcNAc)2Asn has been proposed to protect the newly glycosylated proteins from α-mannosidase hydrolysis of the proteins during transit and modification in the ER. Analysis of L. mexicana parasites after a 2 h incubation with radiolabeled glucose [U-14C] resulted in glycoproteins with Man6GlcNAc2, suggesting that the added glucose is transiently removed after its addition in the ER.122 However, the presence of (Glc)(Man)6(GlcNAc)2 sugars on the main cell surface glycoprotein (gp63) suggests that, even though the glucose is susceptible to glucosidase II enzyme activity, the addition of the glucose molecule (glucosylation) is not necessarily transient.103 Glucosylation of smaller molecular weight oligomannose structures was not identified, suggesting that the addition of a glucose moiety was specific and restricted to (Man)6(GlcNAc)2 substrate.103 The predominant oligosaccharides identified in L. major promastigote and amastigote stages after the enzymatic release of gp63 were (Glc)(Man)6(GlcNAc)2 and (Man)6(GlcNAc)2,108 Table S1 (ESI†). The binding of human mannose-binding protein (MBP) to live promastigotes of L. major and L. mexicana, and to purified LPG immobilized on microwells using I-labelled MBP also indicated the presence of molecules with terminal mannose.147 GIPLs have also been demonstrated to bind to MBP, indicating the presence of terminal mannose sugars.
As noted before, hybrid, complex and paucimannose-type glycan glasses have not been characterized in Leishmania spp.103 In addition, high mannose-type glycans with only one glucose molecule and 4–6 mannoses [(Glc)(Man)6(GlcNAc)2 and (Man)4–6(GlcNAc)2] have been characterized on mature Leishmania spp. proteins.103 Genome mapping of Leishmania spp. enzymes involved in N-linked protein glycosylation is shown in Fig. 4. Based on KEGG PATHWAY database,141 glycans comprised of up to (Glc)1–3(Man)9(GlcNAc)2 are reported. However, no evidence of bi and triglucosylated high mannose together with hybrid, complex and paucimannose glycan structures are reported in the literature so far.
O-Linked glycan biosynthesis
While N-glycans are transferred en bloc from pre-synthesized oligosaccharide precursors to newly synthesized polypeptide chains, stepwise addition of individual monosaccharide units to the hydroxyl group of a serine/threonine amino acid side chain defines O-glycosylation.148O-Glycans vary depending on the connecting sugar. Generally, the term O-glycosylation refers to mucin-type O-glycosylation, which is the modification of proteins at Ser/Thr hydroxyl group by GalNAc monosaccharides with α-linkages.149 This modification involves the transfer of N-acetylgalactsamine (GalNAc) from UDP-GalNAc to a serine or threonine amino acid residue to form (GalNAc)(Ser/Thr). Mannose-type O-glycosylation (O-mannosylation) is another type of protein O-glycosylation whose initial step involves the transfer of a mannose sugar from mannose-P-dolichol to a Ser/Thr residue by the enzyme dolichyl-phosphate-mannose-protein mannosyltransferase to form (Man) (Ser/Thr).150 Mucin and mannan-type protein O-glycosylation have not been evidenced in Leishmania spp.46Another type of O-glycosylation, termed phosphoglycosylation, involves the addition of phosphoglycans to a polypeptide at a serine residue forming a phosphodiester bond with the sequence R-Manα1-PO4-Ser.151,152 Proteophosphoglycan biosynthesis and functional characterization have been reported in Leishmania spp.47,153 and reviewed elsewhere,46 and their structures are summarized in Fig. 2.
O-GlcNAc modification, also known as cytosolic O-glycosylation, involves the attachment of O-linked GlcNAc sugars to Ser/Thr hydroxyl groups, forming (GlcNAc)(Ser/Thr).154 Despite the fact that pathways that utilize UDP-GlcNAc have not been described in Leishmania spp.,155,156 two cytosolic glycoproteins, gp96/92 were described by Handman and colleagues in 1993.87 The O-GlcNac modified proteins were detected by WGA lectin affinity chromatography in various Leishmania species and their corresponding gene and transcript characterized by southern and northern blotting techniques, respectively.87 The gene coding for the protein was termed 1–3B, a 3 kb single-copy gene, while the transcripts for this protein were 7.5 and 4.0 kb, respectively. The O-linked N-acetylglucosamine glycoprotein was expressed in both promastigote, and amastigote stages of the Leishmania species assayed but varied in molecular weight between the species.87 In addition, these proteins shared the peptide backbone and appeared to be highly conserved in Leishmania, Leptomonas, and Crithidia protozoa, suggesting a vital conserved function. This glycoprotein lacked any N-linked glycosylation, as confirmed by the lack of effect after digestion with N-glycanase or treatment with tunicamycin. The cytosolic water-soluble pg96/92 glycoprotein contained unusual glycan composition of glucosamine, as confirmed by WGA lectin binding characteristics.
Protein O-fucosylation is a protein modification which occurs within consensus sequences on serine or threonine amino acid residues, and is catalyzed by protein O-fucosyltransferases.157 The directly linked fucose attached to the modified protein can be elongated by other glycans.157 In Leishmania spp., O-fucosylated glycoconjugates were first described by Rosenzweig and colleagues in 2008 by employing iTRAQ-LC-MS/MS on L. donovani clone 1SR from promastigote and amastigote life stages.158 Four fucosylated proteins were identified in this study from the soluble protein fraction: chaperonin HSP60, HSP70-related protein 1, β-tubulin and kinesin k39, putative.
Sialylated glycoproteins in Leishmania spp.
The sialic acid molecule is a 9 carbon polyhydroxyl amino keto sugar of N- and O-substituted derivatives of neuraminic acid (2-keto-3-deoxy-5-acetamido-D-glycero-D-galacto-nonulosonic acid), a monosaccharide commonly referred to as N-acetylneuraminic acid (Neu5Ac). Sialic acids (Sias) are terminally located on oligosaccharide chains, which decorate cell membranes and secreted macromolecules (e.g., serum glycoproteins). Commonly occurring derivatives of sialic acids include Neu5Ac, Neu5Gc, and the O-acetylated sialoglycoconjugates, which are acetylated at C-7/8/9 to form N-acetyl 7/8/9 O-acetylneuraminic acid, respectively.159,160 Over 50 different derivatives of N-acetyl neuraminic acid molecules161 occur naturally in a diverse range of living organisms from the Animalia kingdom, ranging from microbes (viruses, pathogenic bacteria, and protozoa), higher invertebrates (starfish) and vertebrates (from echinoderms to higher vertebrates).160 Microorganisms usually have a different pathway from eukaryotes for the de novo biosynthesis of sialic acids (e.g., E. coli K1 and Neisseria meningitides),162 but some microbes utilize truncated sialylation pathways (e.g., Neisseria gonorrhoeae)161 for the biosynthesis of sialic acids. Many biological and structural functions have been attributed to sialic acids in different cells and tissues. In higher eukaryotes, sialic acids decorate virtually all cell-surface glycans and serum glycoconjugates terminally,159 where they contribute to the structural integrity of cell membranes, recognition functions by receptor molecules and in the masking of recognition sites by target molecules ligands.163
Leishmania spp. and T. cruzi protozoan parasites lack the endogenous sialic acid biosynthetic pathways.164,165T. cruzi incorporates sialic acid sugar molecules on their surface glycans by scavenging from host cell glycoconjugates by the function of trans-sialidase enzymes (a modified α(2,3) sialidase) through a glycosyl-transfer reaction. Other trypanosomatids, including T. brucei and T. congelense, also express the trans-sialidase enzyme.166Trypanosoma rangeli and T. vivax express a sialidase enzyme without the trans-sialylation function, while Leishmania parasites do not code for either the sialidase or trans-sialidase enzymes.166 However, sialic acids have been demonstrated on the glycosylated surface membrane (glycoproteins and lipophosphoglycans) in L. donovani promastigotes and amastigote life stages,166 and have been implicated in parasite–host interaction, infectivity and immune evasion.164,167 The presence of sialic acids in L. donovani promastigotes has also been demonstrated by Chava et al.168 by employing fluorometric HPLC, GC-MS, lectin binding and ELISA to identify and characterize different sialic acid derivatives and their linkages transferred from the culture medium to the promastigote surface membrane. 9-O-Acetylated sialic acids and α2-3 and α2-6 linked sialoglycans were reported.168L. major lacks the trans-sialidase enzyme, and the subsequent α2-3 linked sialic acids, which are the products of the trans-sialidase enzyme. Transgenic L. major expressing recombinant TS from T. cruzi was shown to be more virulent in genetically susceptible mice in vivo, suggesting that the expression of T. cruzi TS conferred virulence to L. major.169 The interaction of L. donovani sialic acids and siglecs (sialic acid-specific receptors on haematopoetic cells) has shown that sialic acids play a key role in their cell–cell interaction, adherence, virulence, proliferation of amastigotes in macrophage phagolysosomes, and immune evasion.170 A total of 14 siglecs have been reported on mammalian immune cells.171 The interaction between sialic acids and siglec 1 enhances adherence to macrophages and their subsequent uptake by the cells through phagocytosis. Siglec 5 interaction with Sias leads to an upregulation of an acid phosphatase expressed by the host cells (SHP-1), downregulation of MAPKs (p38, ERK, and JNK), PI3K/Akt pathways followed by the reduced translocation of p65 subunit of NF-κβ to the nucleus from cytosol in the downstream signaling pathways leading to suppression of effector functions of innate immune response.170 See reviews on trypanosomatid sialoglycosylation.168,172,173
Box 1. Unusual features of Leishmania glycosylationi. Leishmania parasites, as all other trypanosomatids, synthesize a truncated dolicholpyrophosphate-linked precursor (Man6GlcNAc2) compared to the common LLO for most eukaryotes, Glc3Man9GlcNAc2. In addition, trypanosomatids transfer unglucosylated oligosaccharides from LLOs to nascent proteins.142ii. After the transfer of Man6GlcNAc2 to protein chains, minor trimming occurs in Leishmania parasites, with the most common modification being restricted to the addition of unusual α1-3 glucosyl residues.103,105,174 iii. UDP-galactopyranose mutase (UGM) and galactofuranosyl transferases (GalfTs), enzymes in the biosynthesis of galactofuranose pathway and attachment to cell surfaces, respectively, lack mammalian homologs, and have been proposed as promising drug targets.175 iv. Sialic acid derivatives Neu5Gc and 9-O-acetylated Neu5Gc could be candidates for sialoglycotherapeutics.176 |
Advances in mass spectrometry-based analysis of protein glycosylation in Leishmania
Carbohydrate moieties attached to proteins or lipids have received increased attention with respect to their structure and function. This has been facilitated by the speed and sensitivity of improved and novel analytical methodologies.177 The complexity of glycan structures and their subsequent conformations present both unprecedented opportunities and challenges in the study of their structures, biosynthesis, and functions.177 Glycoproteomics, defined as the system-wide site-specific characterization of protein glycosylation,177 faces many analytical challenges that have made it very difficult to comprehensively perform the structural, biosynthesis and regulation studies of the glycoproteome.132 These challenges include the multiple layers of structural diversity that form a spectrum of the chemically similar repertoire of glycans present on variant synthesized glycoproteins (termed glycoforms).178 Glycomics, defined as the study of the complete set of glycans and glycoconjugates synthesized by a cell or organism in different conditions(glycome),179 also faces enormous challenges arising from the complexity of sugar molecules expressed in a cell or organism.38 Attempts to address these challenges are being sought by new advances in mass spectrometry, genetics, and cell biology studies.38Proteomic approaches (the large scale characterization of proteins in a cell line, tissue and/or organism to gain a global and integrated overview as opposed to the individual information of all the proteins in a cell, tissue or organism180) have been utilized in the study of Leishmania parasite and leishmaniasis, and their applications to Leishmania biology and clinical implications recently reviewed.181–183 These large scale characterization techniques utilize mass-spectrometry based approaches. The large scale study of proteins, glycoproteins, and glycans using mass spectrometry-based tools, both on the parasite and the host in response to infection, offers unprecedented opportunities to uncover novel drug/vaccine candidates and to help elucidate mechanisms of infection, parasite survival, disease diagnosis, and biomarkers. We reviewed mass spectrometry-based studies on Leishmania parasites with a strict focus on the system-wide analysis of Leishmania glycoproteins and glycans by LC-MS/MS.
Glycoproteomic and glycomic-centric studies that have utilized mass spectrometry techniques to characterize Leishmania parasites or clinical samples from patients diagnosed with leishmaniasis to gain an in-depth understanding of how these parasites survive and interact with their vectors or hosts, and/or changes during the course of an infection are few, and highlighted herein. At present, two studies have been reported on the modulation of protein glycosylation in Leishmania infected patients looking at the host glycosylation. Bag and colleagues analyzed plasma proteins from healthy, and L. donovani infected (VL) patients to elucidate disease-associated alterations and host response to infection.184 Two-dimensional fluorescent difference gel electrophoresis combined with MALDI-TOF MS allowed the detection of 39 differentially expressed spots assigned to 10 proteins with different heterogeneity. Alpha-1-antitrypsin, alpha-1-B glycoprotein, and amyloid-A1 precursor, fibrinogen gamma-B chain precursor were upregulated, while vitamin-D binding protein, alipoprotein-A-I, and transthyretin downregulated. Multi-lectin affinity purification of glycoproteins in VL, healthy endemic and non-endemic controls revealed the up-regulation of μ1-B glycoprotein and haptoglobin precursor, allele-1,2. No information on the glycosylation sites and glycan composition was described. Protein–protein interaction analysis of differentially regulated proteins/glycoproteins revealed biological associations pointing to cellular mechanisms employed by the parasites to cause disease, from the regulation of innate immune response to inhibition of proteases.184
The second glycoproteome-centric study was focused on immunoglobulin G Fc changes during infection with L. chagasi.185 The antibodies were affinity-purified from serum or plasma on protein G monolithic plates and digested with trypsin. The resulting glycopeptides were purified and desalted by reverse-phase solid-phase extraction and analyzed by MALDI-TOF MS. N-Glycosylation of antibodies from VL patients were profoundly altered as compared to antibodies collected from uninfected individuals in endemic and non-endemic regions. Fc galactosylation, sialylation, and bisection of IgG1 and IgG2 and 3 Fc regions in VL patients was lower while fucosylation were upregulated compared to asymptomatic individuals and controls. Moreover, IgG1 and IgG2 and -3 Fc galactosylation and sialylation was negatively correlated with clinical severity. Treatment of VL patients with pentavalent antimony and/or amphotericin B changed IgG Fc N-glycosylation.185 Taken together, these results offer new insights into the regulatory role of antibodies in immune responses elicited during Leishmania spp. parasite infection.
A study by Rosenzweig et al., utilized ITRAQ LC-MS/MS to characterize the post translational modifications of L. donovani promastigote and amastigote life stages.158 In addition to the previously mentioned fucosylated proteins identified in this study, they were able to identify one asparagine-linked hexosylated peptide.
An example of analytical approaches to characterize glycoproteins and glycans in Leishmania spp. are summarized in Fig. 5. Mass spectrometry-based analysis of intact glycopeptides involves the enzymatic hydrolysis of the parasites protein lysate using trypsin or a combination of different proteolytic enzymes (step 1–4). Glycopeptide enrichment strategies such as HILIC, TiO2, lectins and others allow the separation of a glycopeptide enriched fraction (step 5). Glycopeptides can be analyzed directly (step 6) or treated with PNGase F to cleave the sugar moiety and generate a N-linked glycosylation site signature (step 7 and 8) before mass spectrometry analysis using dedicated chromatographic and glycopeptide fragmentation strategies (step 9–12). Intact glycopeptides can be identified in specific software such as Byonic, pGlyco, Mascot, Protein Prospector, sugar QB and others.137,186–193 Intact glycopeptide quantification can be performed by matching spectral features using MaxQuant and Proteome Discoverer (step 13–14).177,193,194 The identification of N-linked glycosylation sites in PNGase F-treated glycopeptides can be identified by the conversion of asparagine to aspartic acid signature during peptide sequencing.195–197
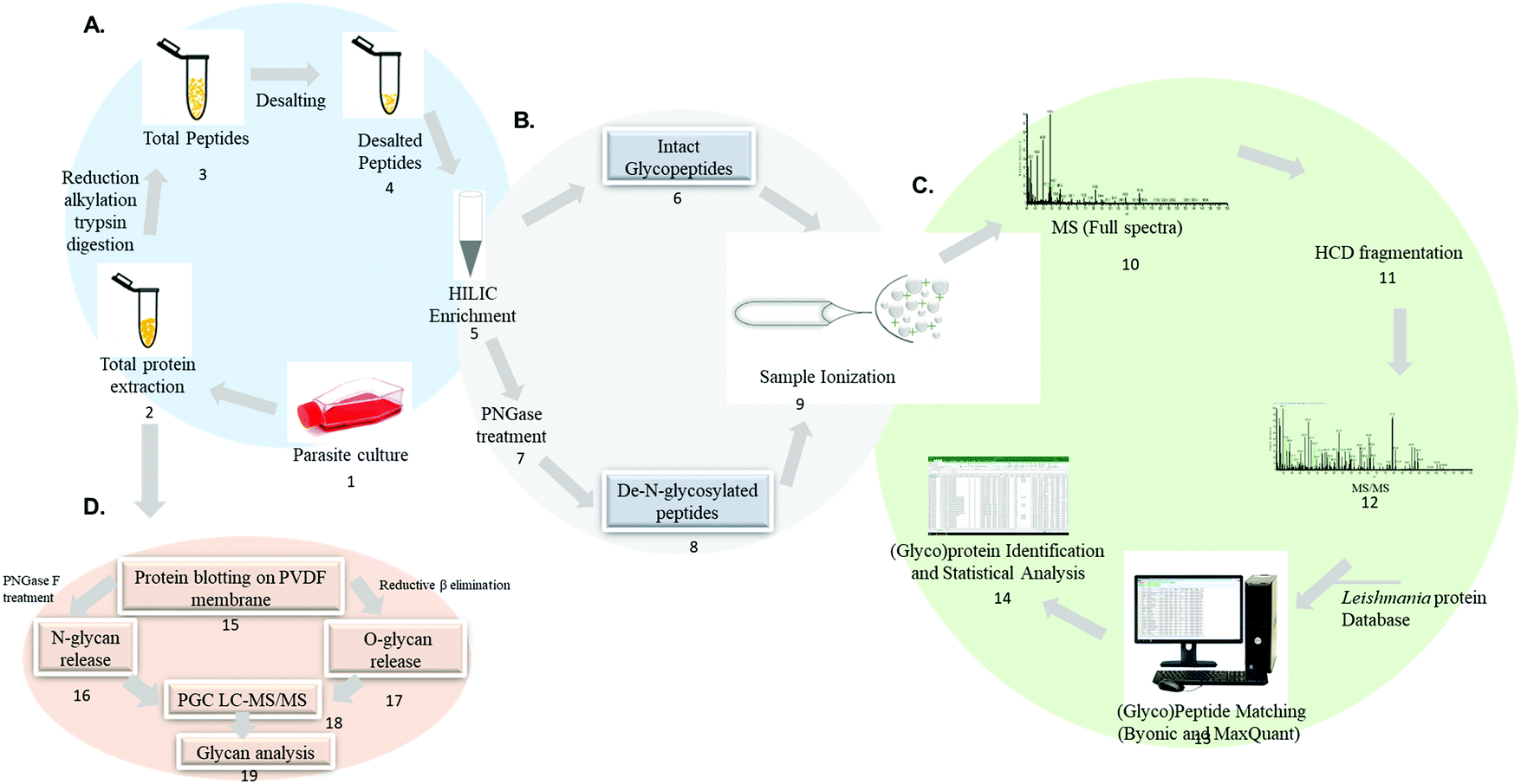 | ||
| Fig. 5 Mass spectrometry-based analytical workflow envisioned for Leishmania spp. large-scale glycoproteome, deglycoproteome and glycome analysis. Proteins are extracted from parasite cell cultures, clinical isolates or biological fluids and digested into peptides (1–4). Glycopeptides are enriched from a complex mixture using specific affinity strategies (5). Intact glycopeptides can be directly analyzed by LC-MS/MS (6) or deglycosylated using PNGaseA/F and the formerly glycosylated peptides are analyzed by LC-MS/MS (7–8) followed by data and bioinformatic analysis (9–14). The analysis of intact glycopeptides and formerly glycosylated peptides will elucidate the glycoproteome and deglycoproteome, respectively. Extracted proteins can be alternatively blotted on PVDF membrane and subjected to enzymatic and chemical release of N and O-glycans (15–17). The released glycans are subsequently analyzed by PGC LC-MS/MS to uncover the glycome (18–19). | ||
In particular, the large scale identification of N-linked glycosylation sites allows to define the deglycoproteome of a biological system. The most common strategy to identify and quantify N-linked glycosylation sites in large scale involves: (1) a protein or peptide-centric enrichment strategy, (2) followed by peptide: N-glycosidase A or F (PNGase A/F) cleavage and (3) mass spectrometry analysis. Glycoproteins and/or glycopeptides can be enriched from a complex mixture using different affinity strategies such as hydrophilic interaction chromatography (HILIC), titanium dioxide (TiO2), graphite, hydrazide chemistry, boronic acid and lectins.197–203 Subsequently, PNGase A/F, a glycosylasparaginase, is used to cleave GlcNAc-Asn linkage releasing N-linked oligosaccharides and converting the asparagine to aspartic acid.204 The formerly-glycosylated peptides are analyzed by LC-MS/MS and the data are searched including asparagine deamidation as variable modification. The asparagine to aspartic acid conversion is monitored during MS analysis due to a mass increment of ΔM = 0.9840 Da. It should be noted that asparagine to (iso)aspartic acid conversion can be derived from chemical deamidation which can happen spontaneously during sample preparation.205–207 To improve the confidence in the N-linked glycosylation site assignment, the enzymatic release of oligosaccharides by PNGaseF/A is performed in heavy-oxygen water (H218O). Under these conditions, the incorporation of H218O will lead to a mass increment of ΔM = 2.9890 Da on the corresponding asparagine residue reducing the false N-linked glycosylation site assignment.208 The deglycoproteome strategy has improved the current knowledge on N-linked glycosylation sites in different biological systems and constitutes a useful technique to be applied to Leishmania spp.
The technological advances in the field of glycomics have helped improve deciphering the complexity and heterogeneity of the glycome. One approach for glycomic analysis relies on protein blotting on PVDF membrane, N and O-linked glycan release and PGC LC-MS/MS analysis for structural characterization (step 15–19).209,210 Other analytical approaches for glycan release and MS analysis have been reported.211–216 Glycan-centric approach is not able to provide information such as the carrier protein and the site-specific attachment of each glycan.177 Thus, glycoproteomics offer more information regarding the protein carriers, glycan attachment sites and the structure, and occupancy of the glycan.132
Conclusion
The first efforts in characterizing Leishmania protein glycosylation have been important to build up the current knowledge on these biomolecules. However, protein glycosylation in Leishmania still deserves more investigation. The protein and glycan diversity of this parasite has been associated with specific species and developmental stages. Moreover, functional studies have highlighted the importance of these biomolecules in host–Leishmania interaction. Unusual glycan features represent valuable therapeutic, diagnostic, and vaccine opportunities for this disease. A detailed characterization of Leishmania spp. glycoproteome is needed and will benefit from advanced analytical LC-MS and computational strategies. We foresee the importance of studying the glycoproteome changes in: (1) different Leishmania species, (2) along the parasite life cycle, (3) parasite clinical isolates (4) the invertebrate vector during infection, (5) non-human hosts, and (6) patients’ tissue biopsies and biofluids.As shown in Fig. 3, Leishmania parasites have shaped their protein glycosylation sites in a species-specific manner. Knowing the glycan macro- and micro-heterogeneity for the different Leishmania species will help direct functional studies for each protein and species. Moreover, it will be possible to correlate the glycan profile of a certain species with the clinical phenotype.
The glycosylation profiles of Leishmania life stages shown in Fig. 1 can modulate parasite–host interaction. Due to that, measuring the glycosylation enzymes expression and activity, activated sugars availability, substrate concentration and the metabolic status of the parasites will offer a complete picture of the biosynthetic steps regulating the glycome of each life stage.
Since different cellular and phenotypic differences have been found between laboratory cultured Leishmania parasites and clinical isolates, evaluation of their protein glycosylation profiles will be important to understand the biology of this parasite to validate biological, diagnostic and therapeutic findings obtained in the laboratory cell cultures.
While this review has been focused on the parasite glycosylation, a comprehensive characterization of the hosts during infection is important. Development of alternative vector control strategies based on modulation of its glycosylation profile might impact vector competence and disease transmission. Moreover, the development of vaccines and chemotherapeutic treatments based on glycans can help in in veterinary and human medicine, supporting the concept of ‘one health’ to eradicate the disease. Deep characterization of protein glycosylation profiles of biopsies and biofluids will help in improving leishmaniasis disease diagnosis and prognosis, allowing a better stratification of the patients to deliver a proper therapeutic solution with the aim of developing personalized therapy.
Due to that, the elucidation of the hidden complexity of the Leishmania and host protein glycosylation will allow researchers to tailor more specific and less toxic chemotherapies and more sensitive and accurate diagnostic tools, calling attention to the importance of glycans in parasite biology and its applications in disease control.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described in the literature mentioned in this review and authored by SNM, JSS, LRF and GP has been partly supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). GP is supported by FAPESP (2018/15549-1, 2020/04923-0). SNM is supported by FAPESP (2017/04032-5). JSS is supported by FAPESP (2016/23689-2). LRF is supported by CAPES.References
- M. Akhoundi, K. Kuhls, A. Cannet, J. Votypka, P. Marty, P. Delaunay and D. Sereno, PLoS Neglected Trop. Dis., 2016, 10, e0004349 CrossRef PubMed.
- A. Oryan and M. Akbari, Asian Pac. J. Trop. Med., 2016, 9, 925–932 CrossRef CAS PubMed.
- World Health Organization, Leishmaniasis – The disease and its epidemiology, https://www.who.int/leishmaniasis/disease_epidemiology/en/.
- R. P. Lane, Medical Insects and Arachnids, Springer, Netherlands, 1st edn, 1993, ch. Sandflies (Phlebotominae) DOI:10.1007/978-94-011-1554-4.
- S. Antinori, A. Cascio, C. Parravicini, R. Bianchi and M. Corbellino, Lancet Infect. Dis., 2008, 8, 191–199 CrossRef.
- F. Clavijo Sanchez, T. Vazquez Sanchez, M. Cabello Diaz, V. E. Sola Moyano, C. Jironada Gallego and D. Hernandez Marrero, Transplant. Proc., 2018, 50, 581–582 CrossRef CAS PubMed.
- C. Cohen, F. Corazza, P. De Mol and D. Brasseur, Lancet, 1991, 338, 128 CrossRef CAS.
- C. K. Meinecke, J. Schottelius, L. Oskam and B. Fleischer, Pediatrics, 1999, 104, e65 CrossRef CAS PubMed.
- I. A. Eltoum, E. E. Zijlstra, M. S. Ali, H. W. Ghalib, M. M. Satti, B. Eltoum and A. M. el-Hassan, Am. J. Trop. Med. Hyg., 1992, 46, 57–62 CrossRef CAS PubMed.
- World Health Organization, Leishmaniasis, https://www.who.int/news-room/fact-sheets/detail/leishmaniasis, accessed 2, March, 2020.
- H. W. Murray, J. D. Berman, C. R. Davies and N. G. Saravia, Lancet, 2005, 366, 1561–1577 CrossRef CAS.
- J. Alvar, I. D. Velez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CrossRef CAS.
- C. V. David and N. Craft, Dermatol. Ther., 2009, 22, 491–502 CrossRef PubMed.
- M. L. Turetz, P. R. Machado, A. I. Ko, F. Alves, A. Bittencourt, R. P. Almeida, N. Mobashery, W. D. Johnson, Jr. and E. M. Carvalho, J. Infect. Dis., 2002, 186, 1829–1834 CrossRef PubMed.
- G. Grimaldi, Jr. and R. B. Tesh, Clin. Microbiol. Rev., 1993, 6, 230–250 CrossRef PubMed.
- J. Convit, M. E. Pinardi and A. J. Rondon, Trans. R. Soc. Trop. Med. Hyg., 1972, 66, 603–610 CrossRef CAS PubMed.
- J. van Griensven and E. Diro, Infect. Dis. Clin. N. Am., 2012, 26, 309–322 CrossRef PubMed.
- E. E. Zijlstra, A. M. Musa, E. A. Khalil, I. M. el-Hassan and A. M. el-Hassan, Lancet Infect. Dis., 2003, 3, 87–98 CrossRef CAS PubMed.
- S. Singh, Indian J. Med. Res., 2006, 123, 311–330 Search PubMed.
- F. Chappuis, S. Sundar, A. Hailu, H. Ghalib, S. Rijal, R. W. Peeling, J. Alvar and M. Boelaert, Nat. Rev. Microbiol., 2007, 5, 873–882 CrossRef CAS PubMed.
- S. L. Croft and V. Yardley, Curr. Pharm. Des., 2002, 8, 319–342 CrossRef CAS PubMed.
- S. R. B. Uliana, C. T. Trinconi and A. C. Coelho, Parasitology, 2018, 145, 464–480 CrossRef PubMed.
- G. Yamey and E. Torreele, Br. Med. J. (Clin. Res. Ed.), 2002, 325, 176–177 CrossRef PubMed.
- G. Yamey, Br. Med. J. (Clin. Res. Ed.), 2002, 324, 698 CrossRef PubMed.
- N. Peters and D. Sacks, Immunol. Rev., 2006, 213, 159–179 CrossRef CAS PubMed.
- N. C. Peters, J. G. Egen, N. Secundino, A. Debrabant, N. Kimblin, S. Kamhawi, P. Lawyer, M. P. Fay, R. N. Germain and D. Sacks, Science, 2008, 321, 970–974 CrossRef CAS PubMed.
- D. E. Teixeira, M. Benchimol, J. C. Rodrigues, P. H. Crepaldi, P. F. Pimenta and W. de Souza, PLoS Pathog., 2013, 9, e1003594 CrossRef PubMed.
- P. A. Bates, Nat. Microbiol., 2018, 3, 529–530 CrossRef CAS PubMed.
- A. Ponte-Sucre, F. Gamarro, J. C. Dujardin, M. P. Barrett, R. López-Vélez, R. García-Hernández, A. W. Pountain, R. Mwenechanya and B. Papadopoulou, PLoS Neglected Trop. Dis., 2017, 11, e0006052 CrossRef PubMed.
- D. Sereno, M. Cavaleyra, K. Zemzoumi, S. Maquaire, A. Ouaissi and J. L. Lemesre, Antimicrob. Agents Chemother., 1998, 42, 3097–3102 CrossRef CAS PubMed.
- F. Frézard, C. Demicheli and R. R. Ribeiro, Molecules, 2009, 14, 2317–2336 CrossRef.
- S. Wyllie, M. L. Cunningham and A. H. Fairlamb, J. Biol. Chem., 2004, 279, 39925–39932 CrossRef CAS PubMed.
- D. Sereno, P. Holzmuller, I. Mangot, G. Cuny, A. Ouaissi and J. L. Lemesre, Antimicrob. Agents Chemother., 2001, 45, 2064–2069 CrossRef CAS PubMed.
- M. Basselin, M. A. Badet-Denisot, F. Lawrence and M. Robert-Gero, Exp. Parasitol., 1997, 85, 274–282 CrossRef CAS PubMed.
- M. Basselin, M. A. Badet-Denisot and M. Robert-Gero, Acta Trop., 1998, 70, 43–61 CrossRef CAS.
- A. Maia, G. N. Porcino, M. L. Detoni, L. R. Quellis, N. B. Emídio, D. G. Marconato, W. F. Messias, L. L. Soldati, P. Faria-Pinto, P. Capriles, E. S. Coimbra, M. J. Marques and E. G. Vasconcelos, Exp. Parasitol., 2019, 200, 1–6 CrossRef CAS PubMed.
- D. Caridha, B. Vesely, K. van Bocxlaer, B. Arana, C. E. Mowbray, S. Rafati, S. Uliana, R. Reguera, M. Kreishman-Deitrick, R. Sciotti, P. Buffet and S. L. Croft, Int. J. Parasitol., 2019, 11, 106–117 Search PubMed.
- R. D. Cummings and J. M. Pierce, Chem. Biol., 2014, 21, 1–15 CrossRef CAS PubMed.
- S. J. Turco and A. Descoteaux, Annu. Rev. Microbiol., 1992, 46, 65–94 CrossRef CAS PubMed.
- M. J. McConville, J. E. Thomas-Oates, M. A. Ferguson and S. W. Homans, J. Biol. Chem., 1990, 265, 19611–19623 CAS.
- M. J. McConville, Cell Biol. Int. Rep., 1991, 15, 779–798 CrossRef CAS.
- M. J. McConville and J. M. Blackwell, J. Biol. Chem., 1991, 266, 15170–15179 CAS.
- M. J. McConville and M. A. Ferguson, Biochem. J., 1993, 294(Pt 2), 305–324 CrossRef CAS.
- M. A. Ferguson, Philos. Trans. R. Soc., B, 1997, 352, 1295–1302 CrossRef CAS.
- T. Ilg, E. Handman and Y. D. Stierhof, Biochem. Soc. Trans., 1999, 27, 518–525 CrossRef CAS PubMed.
- T. Ilg, Parasitol. Today (Personal Ed.), 2000, 16, 489–497 CrossRef CAS.
- T. Ilg, P. Overath, M. A. Ferguson, T. Rutherford, D. G. Campbell and M. J. McConville, J. Biol. Chem., 1994, 269, 24073–24081 CAS.
- A. Descoteaux and S. J. Turco, Biochim. Biophys. Acta, 1999, 1455, 341–352 CrossRef CAS.
- M. J. McConville and A. K. Menon, Mol. Membr. Biol., 2000, 17, 1–16 CrossRef CAS PubMed.
- A. Guha-Niyogi, D. R. Sullivan and S. J. Turco, Glycobiology, 2001, 11, 45r–59r CrossRef CAS PubMed.
- L. Mendonca-Previato, A. R. Todeschini, N. Heise and J. O. Previato, Curr. Opin. Struct. Biol., 2005, 15, 499–505 CrossRef CAS PubMed.
- N. M. Novozhilova and N. V. Bovin, Biochemistry, 2010, 75, 686–694 CAS.
- R. R. Assis, I. C. Ibraim, F. S. Noronha, S. J. Turco and R. P. Soares, PLoS Neglected Trop. Dis., 2012, 6, e1543 CrossRef CAS PubMed.
- C. L. Forestier, Q. Gao and G. J. Boons, Front. Cell. Infect. Microbiol., 2014, 4, 193 Search PubMed.
- Y. Cabezas, L. Legentil, F. Robert-Gangneux, F. Daligault, S. Belaz, C. Nugier-Chauvin, S. Tranchimand, C. Tellier, J. P. Gangneux and V. Ferrieres, Org. Biomol. Chem., 2015, 13, 8393–8404 RSC.
- M. J. McConville, T. A. Collidge, M. A. Ferguson and P. Schneider, J. Biol. Chem., 1993, 268, 15595–15604 CAS.
- R. R. de Assis, I. C. Ibraim, P. M. Nogueira, R. P. Soares and S. J. Turco, Biochim. Biophys. Acta, 2012, 1820, 1354–1365 CrossRef PubMed.
- M. J. McConville and A. Bacic, J. Biol. Chem., 1989, 264, 757–766 CAS.
- T. Ilg, Y. D. Stierhof, M. J. McConville and P. Overath, Eur. J. Cell Biol., 1995, 66, 205–215 CAS.
- T. Ilg, D. Craik, G. Currie, G. Multhaup and A. Bacic, J. Biol. Chem., 1998, 273, 13509–13523 CrossRef CAS PubMed.
- P. Schneider, L. F. Schnur, C. L. Jaffe, M. A. Ferguson and M. J. McConville, Biochem. J., 1994, 304(Pt 2), 603–609 CrossRef CAS PubMed.
- K. A. Yoneyama, A. K. Tanaka, T. G. Silveira, H. K. Takahashi and A. H. Straus, J. Lipid Res., 2006, 47, 2171–2178 CrossRef CAS PubMed.
- S. C. Ilgoutz, J. L. Zawadzki, J. E. Ralton and M. J. McConville, EMBO J., 1999, 18, 2746–2755 CrossRef CAS PubMed.
- D. L. King, Y. D. Chang and S. J. Turco, Mol. Biochem. Parasitol., 1987, 24, 47–53 CrossRef CAS PubMed.
- P. F. Pimenta, E. M. Saraiva and D. L. Sacks, Exp. Parasitol., 1991, 72, 191–204 CrossRef CAS PubMed.
- S. J. Turco, Exp. Parasitol., 1990, 70, 241–245 CrossRef CAS PubMed.
- G. F. Spath, L. Epstein, B. Leader, S. M. Singer, H. A. Avila, S. J. Turco and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9258–9263 CrossRef CAS PubMed.
- T. Ilg, EMBO J., 2000, 19, 1953–1962 CrossRef CAS PubMed.
- S. J. Turco and D. L. Sacks, Mol. Biochem. Parasitol., 1991, 45, 91–99 CrossRef CAS PubMed.
- T. A. Glaser, S. F. Moody, E. Handman, A. Bacic and T. W. Spithill, Mol. Biochem. Parasitol., 1991, 45, 337–344 CrossRef CAS PubMed.
- T. B. McNeely and S. J. Turco, Biochem. Biophys. Res. Commun., 1987, 148, 653–657 CrossRef CAS PubMed.
- J. Delgado-Dominguez, H. Gonzalez-Aguilar, M. Aguirre-Garcia, L. Gutierrez-Kobeh, M. Berzunza-Cruz, A. Ruiz-Remigio, M. Robles-Flores and I. Becker, Parasite Immunol., 2010, 32, 440–449 CrossRef CAS PubMed.
- A. B. Guimarães-Costa, M. T. Nascimento, G. S. Froment, R. P. Soares, F. N. Morgado, F. Conceição-Silva and E. M. Saraiva, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6748–6753 CrossRef PubMed.
- M. Desjardins and A. Descoteaux, J. Exp. Med., 1997, 185, 2061–2068 CrossRef CAS PubMed.
- C. Vivarini Ade, M. Pereira Rde, K. L. Teixeira, T. C. Calegari-Silva, M. Bellio, M. D. Laurenti, C. E. Corbett, C. M. Gomes, R. P. Soares, A. M. Silva, F. T. Silveira and U. G. Lopes, FASEB J., 2011, 25, 4162–4173 CrossRef PubMed.
- D. L. Sacks, G. Modi, E. Rowton, G. Spath, L. Epstein, S. J. Turco and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 406–411 CrossRef CAS PubMed.
- A. Dostalova and P. Volf, Parasites Vectors, 2012, 5, 276 CrossRef CAS PubMed.
- D. L. Sacks, Infect. Agents Dis., 1992, 1, 200–206 CAS.
- A. Varki, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2nd edn, 2009 Search PubMed.
- M. Olivier, V. D. Atayde, A. Isnard, K. Hassani and M. T. Shio, Microbes Infect., 2012, 14, 1377–1389 CrossRef CAS PubMed.
- K. P. Chang, Mol. Biochem. Parasitol., 1981, 4, 67–76 CrossRef CAS PubMed.
- D. M. Dwyer, Exp. Parasitol., 1977, 41, 341–358 CrossRef CAS PubMed.
- D. G. Russell and H. Wilhelm, J. Immunol., 1986, 136, 2613–2620 CAS.
- C. S. Chang and K. P. Chang, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 100–104 CrossRef CAS PubMed.
- J. M. Blackwell, R. A. Ezekowitz, M. B. Roberts, J. Y. Channon, R. B. Sim and S. Gordon, J. Exp. Med., 1985, 162, 324–331 CrossRef CAS PubMed.
- C. Peters, M. Kawakami, M. Kaul, T. Ilg, P. Overath and T. Aebischer, Eur. J. Immunol., 1997, 27, 2666–2672 CrossRef CAS PubMed.
- E. Handman, L. D. Barnett, A. H. Osborn, J. W. Goding and P. J. Murray, Mol. Biochem. Parasitol., 1993, 62, 61–72 CrossRef CAS PubMed.
- M. D. Roos and J. A. Hanover, Biochem. Biophys. Res. Commun., 2000, 271, 275–280 CrossRef CAS PubMed.
- J. A. Hanover, FASEB J., 2001, 15, 1865–1876 CrossRef CAS PubMed.
- D. Fong and K. P. Chang, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 7366–7370 CrossRef CAS PubMed.
- J. Bouvier, R. J. Etges and C. Bordier, J. Biol. Chem., 1985, 260, 15504–15509 CAS.
- R. Etges, J. Bouvier and C. Bordier, J. Biol. Chem., 1986, 261, 9098–9101 CAS.
- J. A. Kink and K. P. Chang, Mol. Biochem. Parasitol., 1988, 27, 181–190 CrossRef CAS PubMed.
- B. S. McGwire and K. P. Chang, J. Biol. Chem., 1996, 271, 7903–7909 CrossRef CAS PubMed.
- G. Chaudhuri, M. Chaudhuri, A. Pan and K. P. Chang, J. Biol. Chem., 1989, 264, 7483–7489 CAS.
- A. Brittingham, G. Chen, B. S. McGwire, K. P. Chang and D. M. Mosser, Infect. Immun., 1999, 67, 4477–4484 CrossRef CAS PubMed.
- A. Brittingham, C. J. Morrison, W. R. McMaster, B. S. McGwire, K. P. Chang and D. M. Mosser, J. Immunol., 1995, 155, 3102–3111 CAS.
- D. M. Mosser and P. J. Edelson, J. Immunol., 1985, 135, 2785–2789 CAS.
- F. S. Rizvi, M. A. Ouaissi, B. Marty, F. Santoro and A. Capron, Eur. J. Immunol., 1988, 18, 473–476 CrossRef CAS PubMed.
- K. P. Soteriadou, M. S. Remoundos, M. C. Katsikas, A. K. Tzinia, V. Tsikaris, C. Sakarellos and S. J. Tzartos, J. Biol. Chem., 1992, 267, 13980–13985 CAS.
- B. S. McGwire, K. P. Chang and D. M. Engman, Infect. Immun., 2003, 71, 1008–1010 CrossRef CAS PubMed.
- UniProt, Universal Protein Resource (UniProt), https://www.uniprot.org/, accessed 5, March, 2020.
- R. W. Olafson, J. R. Thomas, M. A. Ferguson, R. A. Dwek, M. Chaudhuri, K. P. Chang and T. W. Rademacher, J. Biol. Chem., 1990, 265, 12240–12247 CAS.
- D. Alocci, J. Mariethoz and A. Gastaldello, J. Proteome Res., 2019, 18, 664–677 CrossRef CAS PubMed.
- V. A. Funk, J. E. Thomas-Oates, S. L. Kielland, P. A. Bates and R. W. Olafson, Mol. Biochem. Parasitol., 1997, 84, 33–48 CrossRef CAS.
- A. Heifetz, R. W. Keenan and A. D. Elbein, Biochemistry, 1979, 18, 2186–2192 CrossRef CAS PubMed.
- M. E. Wilson and K. K. Hardin, J. Immunol., 1990, 144, 4825–4834 CAS.
- V. A. Funk, A. Jardim and R. W. Olafson, Mol. Biochem. Parasitol., 1994, 63, 23–35 CrossRef CAS PubMed.
- V. A. Funk, PhD thesis, University of Victoria, 1995.
- D. L. Sacks, G. Modi, E. Rowton, G. Späth, L. Epstein, S. J. Turco and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 406–411 CrossRef CAS PubMed.
- N. Secundino, N. Kimblin, N. C. Peters, P. Lawyer, A. A. Capul, S. M. Beverley, S. J. Turco and D. Sacks, Cell. Microbiol., 2010, 12, 906–918 CrossRef CAS PubMed.
- Y. D. Stierhof, P. A. Bates, R. L. Jacobson, M. E. Rogers, Y. Schlein, E. Handman and T. Ilg, Eur. J. Cell Biol., 1999, 78, 675–689 CrossRef CAS.
- M. Rogers, P. Kropf, B. S. Choi, R. Dillon, M. Podinovskaia, P. Bates and I. Muller, PLoS Pathog., 2009, 5, e1000555 CrossRef PubMed.
- B. J. Mengeling, S. M. Beverley and S. J. Turco, Glycobiology, 1997, 7, 873–880 CrossRef CAS PubMed.
- M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 1235681 CrossRef PubMed.
- P. R. Strauss, J. Protozool., 1971, 18, 147–156 CrossRef CAS PubMed.
- D. M. Dwyer, Science, 1974, 184, 471–473 CrossRef CAS PubMed.
- D. M. Dwyer, S. G. Langreth and N. K. Dwyer, Z. Parasitenkd., 1974, 43, 227–249 CrossRef CAS.
- K. Dawidowicz, A. G. Hernandez, R. B. Infante and J. Convit, J. Parasitol., 1975, 61, 950–953 CrossRef CAS PubMed.
- I. J. Goldstein, C. M. Reichert, A. Misaki and P. A. Gorin, Biochim. Biophys. Acta, 1973, 317, 500–504 CrossRef CAS.
- P. R. Gardiner and D. M. Dwyer, Mol. Biochem. Parasitol., 1983, 8, 283–295 CrossRef CAS.
- A. J. Parodi, J. Martin-Barrientos and J. C. Engel, Biochem. Biophys. Res. Commun., 1984, 118, 1–7 CrossRef CAS.
- S. C. Hubbard and R. J. Ivatt, Annu. Rev. Biochem., 1981, 50, 555–583 CrossRef CAS PubMed.
- F. Dagger, C. Ayesta and A. G. Hernandez, Biol. Cell, 1984, 50, 173–189 CrossRef CAS PubMed.
- T. J. Nolan and J. P. Farrell, Mol. Biochem. Parasitol., 1985, 16, 127–135 CrossRef CAS PubMed.
- C. S. Chang, T. J. Inserra, J. A. Kink, D. Fong and K. P. Chang, Mol. Biochem. Parasitol., 1986, 18, 197–210 CrossRef CAS PubMed.
- J. K. Lovelace and M. Gottlieb, Mol. Biochem. Parasitol., 1987, 22, 19–28 CrossRef CAS PubMed.
- P. A. Bates and D. M. Dwyer, Mol. Biochem. Parasitol., 1987, 26, 289–296 CrossRef CAS PubMed.
- P. A. Bates, M. Gottlieb and D. M. Dwyer, Exp. Parasitol., 1988, 67, 199–209 CrossRef CAS.
- H. Guo, N. M. Novozhilova and G. Bandini, J. Biol. Chem., 2017, 292, 10696–10708 CrossRef CAS PubMed.
- S. Sughii, E. A. Kabat and H. H. Baer, Carbohydr. Res., 1982, 99, 99–101 CrossRef CAS.
- M. Thaysen-Andersen, N. H. Packer and B. L. Schulz, Mol. Cell. Proteomics, 2016, 15, 1773–1790 CrossRef CAS PubMed.
- M. A. Kukuruzinska and K. Lennon, Crit. Rev. Oral Biol. Med., 1998, 9, 415–448 CrossRef CAS PubMed.
- R. Kornfeld and S. Kornfeld, Annu. Rev. Biochem., 1985, 54, 631–664 CrossRef CAS PubMed.
- J. Zaia, Chem. Biol., 2008, 15, 881–892 CrossRef CAS PubMed.
- H. C. Tjondro and I. Loke, Biol. Rev. Cambridge Philos. Soc., 2019, 94, 2068–2100 CrossRef PubMed.
- M. J. Alves, R. Kawahara, R. Viner, W. Colli, E. C. Mattos, M. Thaysen-Andersen, M. R. Larsen and G. Palmisano, J. Proteomics, 2017, 151, 182–192 CrossRef CAS PubMed.
- S. E. Zamze, D. A. Ashford, E. W. Wooten, T. W. Rademacher and R. A. Dwek, J. Biol. Chem., 1991, 266, 20244–20261 CAS.
- A. Mehlert, M. R. Wormald and M. A. Ferguson, PLoS Pathog., 2012, 8, e1002618 CrossRef CAS PubMed.
- M. Bosch, S. Trombetta, U. Engstrom and A. J. Parodi, J. Biol. Chem., 1988, 263, 17360–17365 CAS.
- KEGG, N-Glycan biosynthesis – Leishmania major, https://www.genome.jp/kegg-bin/show_pathway?lma00510, accessed March, 2020.
- P. Low, G. Dallner, S. Mayor, S. Cohen, B. T. Chait and A. K. Menon, J. Biol. Chem., 1991, 266, 19250–19257 CAS.
- P. Burda and M. Aebi, Biochim. Biophys. Acta, 1999, 1426, 239–257 CrossRef CAS.
- A. Jinnelov, L. Ali and M. Tinti, J. Biol. Chem., 2017, 292, 20328–20341 CrossRef CAS PubMed.
- F. P. Nasab, B. L. Schulz, F. Gamarro, A. J. Parodi and M. Aebi, Mol. Biol. Cell, 2008, 19, 3758–3768 CrossRef CAS PubMed.
- K. Hese, C. Otto, F. H. Routier and L. Lehle, Glycobiology, 2009, 19, 160–171 CrossRef CAS PubMed.
- P. J. Green, T. Feizi, M. S. Stoll, S. Thiel, A. Prescott and M. J. McConville, Mol. Biochem. Parasitol., 1994, 66, 319–328 CrossRef CAS PubMed.
- P. Van den Steen, P. M. Rudd, R. A. Dwek and G. Opdenakker, Crit. Rev. Biochem. Mol. Biol., 1998, 33, 151–208 CrossRef CAS PubMed.
- C. R. Bertozzi, A. Varki and J. Esko, et al., Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1999 Search PubMed.
- S. Strahl-Bolsinger, M. Gentzsch and W. Tanner, Biochim. Biophys. Acta, 1999, 1426, 297–307 CrossRef CAS.
- D. P. Mehta, M. Ichikawa, P. V. Salimath, J. R. Etchison, R. Haak, A. Manzi and H. H. Freeze, J. Biol. Chem., 1996, 271, 10897–10903 CrossRef CAS PubMed.
- P. A. Haynes, Glycobiology, 1998, 8, 1–5 CrossRef CAS.
- T. Ilg, Y. D. Stierhof, D. Craik, R. Simpson, E. Handman and A. Bacic, J. Biol. Chem., 1996, 271, 21583–21596 CrossRef CAS.
- C. R. Bertozzi, A. Varki and J. Esko, et al., Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 1999, ch. 14 Search PubMed.
- T. Naderer, E. Wee and M. J. McConville, Mol. Microbiol., 2008, 69, 858–869 CrossRef CAS PubMed.
- T. Naderer, J. Heng and M. J. McConville, PLoS Pathog., 2010, 6, e1001245 CrossRef CAS PubMed.
- B. C. Holdener and R. S. Haltiwanger, Curr. Opin. Struct. Biol., 2019, 56, 78–86 CrossRef CAS PubMed.
- D. Rosenzweig, D. Smith, P. J. Myler, R. W. Olafson and D. Zilberstein, Proteomics, 2008, 8, 1843–1850 CrossRef CAS PubMed.
- R. Schauer, Glycoconjugate J., 2000, 17, 485–499 CrossRef CAS PubMed.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439–469 CrossRef CAS PubMed.
- E. Vimr and C. Lichtensteiger, Trends Microbiol., 2002, 10, 254–257 CrossRef CAS PubMed.
- R. Schauer, Adv. Carbohydr. Chem. Biochem., 1982, 40, 131–234 CrossRef CAS PubMed.
- S. Kelm and R. Schauer, Int. Rev. Cytol., 1997, 175, 137–240 CAS.
- A. K. Chava, M. Chatterjee, G. J. Gerwig, J. P. Kamerling and C. Mandal, Biol. Chem., 2004, 385, 59–66 CAS.
- J. O. Previato, A. F. Andrade, M. C. Pessolani and L. Mendonca-Previato, Mol. Biochem. Parasitol., 1985, 16, 85–96 CrossRef CAS PubMed.
- M. Engstler, R. Schauer and R. Brun, Acta Trop., 1995, 59, 117–129 CrossRef CAS PubMed.
- M. Chatterjee, A. K. Chava, G. Kohla, S. Pal, A. Merling, S. Hinderlich, U. Unger, P. Strasser, G. J. Gerwig, J. P. Kamerling, R. Vlasak, P. R. Crocker, R. Schauer, R. Schwartz-Albiez and C. Mandal, Glycobiology, 2003, 13, 351–361 CrossRef CAS PubMed.
- A. K. Chava, S. Bandyopadhyay, M. Chatterjee and C. Mandal, Glycoconjugate J., 2004, 20, 199–206 CrossRef CAS PubMed.
- M. Belen Carrillo, W. Gao, M. Herrera, J. Alroy, J. B. Moore, S. M. Beverley and M. A. Pereira, Infect. Immun., 2000, 68, 2728–2734 CrossRef CAS PubMed.
- S. Roy and C. Mandal, PLoS Neglected Trop. Dis., 2016, 10, e0004904 CrossRef PubMed.
- M. K. O'Reilly and J. C. Paulson, Trends Pharmacol. Sci., 2009, 30, 240–248 CrossRef PubMed.
- A. Ghoshal and C. Mandal, Mol. Biol. Int., 2011, 2011, 532106 Search PubMed.
- T. Souto-Padron, An. Acad. Bras. Cienc., 2002, 74, 649–675 CrossRef PubMed.
- A. J. Parodi, Parasitol. Today (Personal Ed.), 1993, 9, 373–377 CrossRef CAS.
- M. Oppenheimer, A. L. Valenciano and P. Sobrado, Enzyme Res., 2011, 2011, 415976 Search PubMed.
- A. Ghoshal, S. Mukhopadhyay and C. Mandal, Mini-Rev. Med. Chem., 2008, 8, 358–369 CrossRef CAS PubMed.
- M. Thaysen-Andersen and N. H. Packer, Biochim. Biophys. Acta, 2014, 1844, 1437–1452 CrossRef CAS PubMed.
- R. Kornfeld and S. Kornfeld, Annu. Rev. Biochem., 1976, 45, 217–237 CrossRef CAS PubMed.
- N. H. Packer, C. W. von der Lieth, K. F. Aoki-Kinoshita, C. B. Lebrilla, J. C. Paulson, R. Raman, P. Rudd, R. Sasisekharan, N. Taniguchi and W. S. York, Proteomics, 2008, 8, 8–20 CrossRef CAS PubMed.
- P. R. Graves and T. A. Haystead, Microbiol. Mol. Biol. Rev., 2002, 66, 39–63 CrossRef CAS PubMed ; table of contents.
- P. S. Veras and J. P. Bezerra de Menezes, Int. J. Mol. Sci., 2016, 17(8), 1270 CrossRef PubMed.
- S. Sundar and B. Singh, Expert Rev. Proteomics, 2018, 15, 371–390 CrossRef CAS PubMed.
- J. Capelli-Peixoto, S. N. Mule, F. T. Tano, G. Palmisano and B. S. Stolf, Proteomics Clin Appl, 2019, 13, e1800136 CrossRef PubMed.
- A. K. Bag, S. Saha, S. Sundar, B. Saha, A. Chakrabarti and C. Mandal, Proteome Sci., 2014, 12, 48 CrossRef PubMed.
- L. G. Gardinassi, V. Dotz, A. Hipgrave Ederveen, R. P. de Almeida, C. H. Nery Costa, D. L. Costa, A. R. de Jesus, O. A. Mayboroda, G. R. Garcia, M. Wuhrer and I. K. de Miranda Santos, mBio, 2014, 5, e01844 CrossRef CAS PubMed.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS PubMed.
- J. Stadlmann, D. M. Hoi, J. Taubenschmid, K. Mechtler and J. M. Penninger, Proteomics, 2018, 18, e1700436 CrossRef PubMed.
- R. C. Bollineni, C. J. Koehler, R. E. Gislefoss, J. H. Anonsen and B. Thiede, Sci. Rep., 2018, 8, 2117 CrossRef PubMed.
- Z. Darula, R. J. Chalkley, P. Baker, A. L. Burlingame and K. F. Medzihradszky, Eur. J. Mass Spectrom., 2010, 16, 421–428 CrossRef CAS PubMed.
- W. F. Zeng, M. Q. Liu, Y. Zhang, J. Q. Wu, P. Fang, C. Peng, A. Nie, G. Yan, W. Cao, C. Liu, H. Chi, R. X. Sun, C. C. Wong, S. M. He and P. Yang, Sci. Rep., 2016, 6, 25102 CrossRef CAS PubMed.
- B. L. Parker, M. Thaysen-Andersen, N. Solis, N. E. Scott, M. R. Larsen, M. E. Graham, N. H. Packer and S. J. Cordwell, J. Proteome Res., 2013, 12, 5791–5800 CrossRef CAS PubMed.
- H. Hu, K. Khatri, J. Klein, N. Leymarie and J. Zaia, Glycoconjugate J., 2016, 33, 285–296 CrossRef CAS PubMed.
- M. Bern, Y. J. Kil and C. Becker, Current protocols in bioinformatics, 2012, ch. 13, Unit 13.20 Search PubMed.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS PubMed.
- G. Palmisano, B. L. Parker, K. Engholm-Keller, S. E. Lendal, K. Kulej, M. Schulz, V. Schwammle, M. E. Graham, H. Saxtorph, S. J. Cordwell and M. R. Larsen, Mol. Cell. Proteomics, 2012, 11, 1191–1202 CrossRef PubMed.
- B. Kuster and M. Mann, Anal. Chem., 1999, 71, 1431–1440 CrossRef CAS PubMed.
- H. Zhang, X. J. Li, D. B. Martin and R. Aebersold, Nat. Biotechnol., 2003, 21, 660–666 CrossRef CAS PubMed.
- A. J. Alpert, J. Chromatogr., 1988, 444, 269–274 CrossRef CAS PubMed.
- H. Kaji, H. Saito, Y. Yamauchi, T. Shinkawa, M. Taoka, J. Hirabayashi, K. Kasai, N. Takahashi and T. Isobe, Nat. Biotechnol., 2003, 21, 667–672 CrossRef CAS PubMed.
- M. R. Larsen, S. S. Jensen, L. A. Jakobsen and N. H. Heegaard, Mol. Cell. Proteomics, 2007, 6, 1778–1787 CrossRef CAS PubMed.
- S. Mysling, G. Palmisano, P. Højrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef CAS PubMed.
- G. Palmisano, S. E. Lendal and M. R. Larsen, Methods Mol. Biol., 2011, 753, 309–322 CrossRef CAS PubMed.
- M. Thaysen-Andersen, M. R. Larsen, N. H. Packer and G. Palmisano, RSC Adv., 2013, 3, 22683–22705 RSC.
- S. Hirani, R. J. Bernasconi and J. R. Rasmussen, Anal. Biochem., 1987, 162, 485–492 CrossRef CAS PubMed.
- R. Tyler-Cross and V. Schirch, J. Biol. Chem., 1991, 266, 22549–22556 CAS.
- P. M. Angel, J. M. Lim, L. Wells, C. Bergmann and R. Orlando, Rapid Commun. Mass Spectrom., 2007, 21, 674–682 CrossRef CAS PubMed.
- G. Palmisano, M. N. Melo-Braga, K. Engholm-Keller, B. L. Parker and M. R. Larsen, J. Proteome Res., 2012, 11, 1949–1957 CrossRef CAS PubMed.
- B. Küster and M. Mann, Anal. Chem., 1999, 71, 1431–1440 CrossRef PubMed.
- C. Ashwood, B. Pratt, B. X. MacLean and R. L. Gundry, Analyst, 2019, 144, 3601–3612 RSC.
- P. H. Jensen, N. G. Karlsson, D. Kolarich and N. H. Packer, Nat. Protoc., 2012, 7, 1299–1310 CrossRef CAS PubMed.
- S. Yang and H. Zhang, Curr. Protoc. Chem. Biol., 2014, 6, 191–208 CrossRef PubMed.
- L. Krishnamoorthy and L. K. Mahal, ACS Chem. Biol., 2009, 4, 715–732 CrossRef CAS PubMed.
- A. Shajahan, C. Heiss, M. Ishihara and P. Azadi, Anal. Bioanal. Chem., 2017, 409, 4483–4505 CrossRef CAS PubMed.
- M. Wuhrer, Glycoconjugate J., 2013, 30, 11–22 CrossRef CAS PubMed.
- L. R. Ruhaak and G. Xu, Chem. Rev., 2018, 118, 7886–7930 CrossRef CAS PubMed.
- N. J. Agard and C. R. Bertozzi, Acc. Chem. Res., 2009, 42, 788–797 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mo00043d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |