
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a 96-well plate sample preparation method for integrated N- and O-glycomics using porous graphitized carbon liquid chromatography-mass spectrometry†
Tao
Zhang‡
 *a,
Katarina
Madunić‡
a,
Stephanie
Holst
a,
Jing
Zhang
b,
Chunsheng
Jin
c,
Peter
ten Dijke
b,
Niclas G.
Karlsson
c,
Kathrin
Stavenhagen
a and
Manfred
Wuhrer
*a
*a,
Katarina
Madunić‡
a,
Stephanie
Holst
a,
Jing
Zhang
b,
Chunsheng
Jin
c,
Peter
ten Dijke
b,
Niclas G.
Karlsson
c,
Kathrin
Stavenhagen
a and
Manfred
Wuhrer
*a
aCenter for Proteomics and Metabolomics, Leiden University Medical Center, Leiden, The Netherlands. E-mail: t.zhang@lumc.nl; m.wuhrer@lumc.nl
bDepartment of Cell and Chemical Biology and Oncode Institute, Leiden University Medical Center, The Netherlands
cDepartment of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Acedemy, University of Gothenburg, Sweden
First published on 13th April 2020
Abstract
Changes in glycosylation signatures of cells have been associated with pathological processes in cancer as well as infectious and autoimmune diseases. The current protocols for comprehensive analysis of N-glycomics and O-glycomics derived from cells and tissues often require a large amount of biological material. They also only allow the processing of very limited numbers of samples at a time. Here we established a workflow for sequential release of N-glycans and O-glycans based on PVDF membrane immobilization in 96-well format from 5 × 105 cells. Released glycans are reduced, desalted, purified, and reconstituted, all in 96-well format plates, without additional staining or derivatization. Glycans are then analyzed with porous graphitized carbon nano-liquid chromatography coupled to tandem mass spectrometry using negative-mode electrospray ionization, enabling the chromatographic resolution and structural elucidation of glycan species including many compositional isomers. The approach was demonstrated using glycoprotein standards and further applied to analyze the glycosylation of the murine mammary gland NMuMG cell line. The developed protocol allows the analysis of N- and O-glycans from relatively large numbers of samples in a less time consuming way with high repeatability. Inter- and intraday repeatability of the fetuin N-glycan analysis showed two median intraday coefficients of variations (CVs) of 7.6% and 8.0%, and a median interday CV of 9.8%. Median CVs of 7.9% and 8.7% for the main peaks of N- and O-glycans released from the NMuMG cell line indicate a very good repeatability. The method is applicable to purified glycoproteins as well as to biofluids and cell- or tissue-based samples.
1. Introduction
Protein glycosylation is involved in many biological processes such as cellular signaling, eliciting of immune responses and cancer progression.1,2 Glycosylation studies focus on defining the diversity of glycan structures carried by specific glycoproteins or whole cells facilitating the understanding of the contribution of glycans in biological processes and the development of diseases. Due to the structural complexity and heterogeneity of glycans in complex biological samples, the available methodologies often have difficulties to achieve a comprehensive characterization, and require large amounts of biological material.3Mass spectrometry (MS)-based glycomics has become one of the most powerful methods for analyzing glycans released from glycoproteins and has vastly benefitted from rapid advances in sample preparation, chromatographic separation, MS methodology and data processing.4 Importantly, MS structural analysis enhanced by powerful separation may provide detailed information and deeper understanding of glycan expression at high sensitivity. However, the low ionization efficiency, sialic acid decay and fucose migration in MS-based glycomics often pose problems in glycan characterization.5–8 To address these challenges, derivatization and/or labeling followed by additional purification steps are employed. However, these methodologies often lack sufficient chromatographic separation resulting in failure to differentiate isomeric glycan species.
Another challenge in the field is the increasing need for high-throughput sample preparation for glycan analysis, due to a vast increase of sample numbers and complexity required in functional glycomics, systems biology, and clinical applications. Currently, there are several high-throughput approaches for N-glycan analysis (mainly glycoprotein),9–11 while less methods are available for O-glycomics.12 Therefore, there is a high demand for high-throughput, reproducible and robust analytical methods for integrated N- and O-glycomics. Scaling up the conventional glycomics analysis to higher-throughput approaches especially with respect to the sample preparation workflow is of great importance for ensuring sufficient sample size providing more reliable results.
Here, we present an integrated N- and O-glycomics approach for sequential release of N- and O-glycans from biological samples based on protein immobilization on polyvinylidene fluoride (PVDF) membrane filter plates in 96-well format. The approach is based on methodology developed by Packer and coworkers.13,14 who have pioneered the analysis of glycan alditols by porous graphitized carbon nano-liquid chromatography coupled to a tandem mass spectrometer (PGC nano-LC-ESI-MS/MS) using negative electrospray ionization.13 Powerful separation capacity enabling discrimination between glycan isomers is a major advantage of PGC chromatography.15,16 Taking their methodology as a starting point,13 considerable modifications were introduced with respect to sample preparation in order to allow a repeatable and less time consuming analysis in a higher-throughput manner. The method was demonstrated by analysis of glycans released from glycoprotein standards, and finally applied to the characterization of a murine breast cancer cell line. The method is applicable to a range of complex biological samples, including biofluids, cell lines and tissues.
2. Experimental
2.1 Materials
Ammonium bicarbonate (ABC), Dowex cation-exchange resin (50W-X8), trifluoroacetic acid (TFA), fetuin from fetal bovine serum (FBS), hydrochloric acid (HCl), DL-dithiothreitol (DTT), ammonium acetate, sodium chloride (NaCl), and sodium borohydride (NaBH4) were obtained from Sigma-Aldrich (Steinheim, Germany). 8 M guanidine hydrochloride (GuHCl), Dulbecco's modified Eagle's medium (DMEM) and FBS were purchased from Thermo Fisher Scientific. Peptide N-glycosidase F (PNGase F) was purchased from Roche Diagnostics (Mannheim, Germany). Glacial acetic acid and potassium hydroxide (KOH) were purchased from Honeywell Fluka. Solid phase extraction (SPE) bulk sorbent Carbograph was obtained from Grace Discovery sciences (Columbia, USA). HPLC SupraGradient acetonitrile (ACN) was obtained from Biosolve (Valkenswaard, The Netherlands) and other reagents and solvents such as methanol, ethanol, and 2-propanol were purchased from Merck (Darmstadt, Germany). MultiScreen® HTS 96 multiwell plates (pore size 0.45 μm) with high protein-binding membrane (hydrophobic Immobilon-P PVDF membrane) and 96-well PP Microplate were purchased from Millipore (Amsterdam, The Netherlands), conical 96-well Nunc plates from Thermo Scientific (Roskilde, Denmark). 96-well PP filter plate from Orochem Technologies (Naperville, IL, USA). Ultrapure water was used for the all preparations and washes, generated from a Q-Gard 2 system (Millipore).2.2 Cell culture and cell lysis
Mouse mammary gland (NMuMG) epithelial cell lines were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured as previously described.17 In brief, the cells were cultured in DMEM, supplied with 100 U mL−1 penicillin–streptomycin and 10% FBS at 37 °C in 5% CO2 and harvested after reaching 80% confluency. Pre-warmed trypsin was added to detach the cells. After detaching more than 90% of the cells, the equivalent of 2 volumes of pre-warmed complete DMEM was added to quench the enzyme activity. The cells were collected from the flask by washing with pre-warmed phosphate buffered saline (PBS) for 3 times. Thereafter, the cells were transferred to a 15 mL conical tube and centrifuged at 1 × 103 rpm for 5 min. Cells were washed with PBS to remove the medium for 3 times by resuspension and centrifugation. The cells were counted using the Countess® Automated Cell Counter. The cells were washed twice with 5 mL of 1x PBS, aliquoted to 2.0 × 106 cells per mL of 1× PBS and pelleted by centrifuging 3 min at 1500 × g. Finally, the supernatant was removed, and the cell pellets were stored in the freezer at −20 °C.Mechanical cell lysis was performed in water as described previously.18 Briefly, cell pellets with approximately 2 × 106 cells were resuspended in 100 μL of water by pipetting for 30 seconds and vortexed for 30 seconds followed by sonication for 60 min in an ultrasound bath at room temperature (RT).
2.3 Glycoprotein and cell lysate immobilization, denaturation and reduction
The 96-well plates with hydrophobic Immobilon-P PVDF membrane were preconditioned with 200 μL 70% ethanol followed by equilibration 3 times with 200 μL water. Purified glycoproteins, biofluidics and cell lysates (derived from cell lines, organoids and tissue samples) can be applied to the PVDF-membrane directly. In this work, either 2 μL (20 μg in water) of fetuin aliquot or 25 μL of cell lysate (approximately 5 × 105 cells) were applied per well followed by horizontal shaking for 15 min. Protein denaturation was achieved by applying 75 μL of denaturation mix (72.5 μL 8 M GuHCl and 2.5 μL 200 mM DTT) in each well, followed by shaking for 15 min and incubating at 60 °C in a moisture box for 30 min. Subsequently the unbound material was removed by washing three times with water each time followed by centrifugation at 500 × g for 1 min. Any residual liquid was subsequently removed from the membrane by pipetting and discarded.2.4 Enzymatic N-glycan release, reduction and purification
The N-glycan release was performed as previously described18 with small modifications. PNGase F (2 U of enzyme diluted with water to 15 μL) was added to each well followed by 15 min shaking at RT. The plate was incubated in a humidified box for 15 min at 37 °C. Subsequently, another 15 μL of water was added in each well. The plate was incubated overnight in a humidified plastic box at 37 °C to avoid evaporation of the digestion solution. Released N-glycans were recovered from the PVDF plate by centrifugation and washing three times with 30 μL of water. Subsequently the glycosylamine forms of the released N-glycans were hydrolyzed by adding 20 μL of 100 mM ammonium acetate (pH 5), incubated at RT for 1 h, and dried in a SpeedVac concentrator 5301 (Eppendorf, Hamburg, Germany) at 35 °C. Collected N-glycans were then reduced and desalted followed by PGC cleanup using a 96-well plate based protocol adapted from Jensen, et al.13 For the reduction, 20 μL of 1 M NaBH4 in 50 mM KOH was added to each well followed by incubation for 3 hours at 50 °C in a humidified plastic box. To neutralize and quench the reaction, 3 μL of glacial acetic acid was added to each sample. Desalting of the samples was performed using a strongly acidic cation exchange resin Dowex 50W-X8 which was self-packed into 96-well filter plates. Briefly, 100 μL of the resin slurry in methanol (50/50, v/v) was added to each well in the filter plate. The columns were preconditioned by 3 × 100 μL of 1 M HCl, followed by 3 × 100 μL methanol and 3 × 100 μL water each time removed by centrifuging at 500 × g. The samples containing N-glycans were loaded onto the columns and eluted two times with 40 μL of water followed by centrifugation at 500 × g. The combined flow-through and wash were collected and dried in a SpeedVac concentrator at 35 °C. The remaining borate was removed by several rounds of co-evaporation using 100 μL methanol in the SpeedVac concentrator at 35 °C. SPE cleaning step was performed by packing 60 μL (approximately 6 mg) of bulk sorbent carbograph slurry in methanol into a 96-well filter plate. The columns were preconditioned by 3 × 100 μL of 80% acetonitrile in water containing 0.1% TFA, and 3 × 100 μL water with 0.1% TFA. After loading the sample, the columns were washed three times with 80 μL of 0.1% TFA, followed by N-glycan elution by 3 × 40 μL of 60% acetonitrile in water containing 0.1% TFA. Samples were dried in a SpeedVac concentrator directly in polymerase chain reaction (PCR) plates for injection and re-dissolved in 10 μL of water prior to PGC nano-LC-ESI-MS/MS analysis.2.5 O-Glycan release and purification
After removal of N-glycans, the O-glycans were released from the same PVDF membrane immobilized sample via reductive β-elimination. Briefly, 50 μL of 0.5 M NaBH4 in 50 mM KOH was applied onto each PVDF membrane well after rewetting with 3 μL of methanol. Plates were shaken for 15 min on a horizontal shaker and incubated in a humidified plastic box for 16 h at 50 °C. After incubation and cooling to RT, released O-glycans were recovered by centrifugation at 1000 × g for 2 min into 96-well collection plates. The wells were rewetted by 3 μL of methanol and washed three times with 50 μL of water with 10 min incubation steps on a horizontal shaker prior to centrifugation at 500 × g for 2 min. Prior to desalting, the collected samples were concentrated to approximately 30 μL under vacuum in a SpeedVac concentrator at 35 °C for 2 h. Subsequently, 3 μL of glacial acetic acid was added to neutralize the reaction followed by brief centrifugation to collect the sample at the bottom of the well. The following high throughput desalting and PGC SPE purification were performed as described in the N-glycan preparation section under 2.4. The purified O-glycan alditols were re-suspended in 10 μL of water prior to PGC nano-LC-ESI-MS/MS analysis.2.6 Analysis of released N- and O-glycan alditols using PGC nano-LC-ESI-MS/MS
The analysis of N-glycans and O-glycans was performed on an Ultimate 3000 UHPLC system (Dionex/Thermo) equipped with a Hypercarb PGC trap column (5 μm Hypercarb Kappa, 32 μm × 30 mm, Thermo Fisher Scientific, Waltham, MA) and a Hypercarb PGC nano-column (3 μm Hypercarb 75 μm × 100 mm, Thermo Fisher Scientific, Waltham, MA) coupled to an amaZon ETD speed ion trap (Bruker, Bremen, Germany). Mobile phase A consisted of 10 mM ABC, while mobile phase B was 60% (v/v) acetonitrile/10 mM ABC. To analyze glycans, 1 μL of sample prepared from glycoprotein fetuin standard (31.25 picomole) and 3 μL of each sample derived from cell line NMUMG (1.5 × 105 cells) was injected and trapped on the trap column using a 6 μL min−1 loading flow in 2% buffer B for N-glycan and 1% buffer B for O-glycans for 5 min. Separation was achieved with a multi-step gradient of B: 2–9% in 1 min and 9–49% in 80 min for N-glycan and 1–52% over 72 min for O-glycans followed by a 10 min wash step using 95% of B at a flow of rate of 0.6 μL min−1. The column was held at a constant temperature of 45 °C. Ionization was achieved using the CaptiveSpray nanoBooster source (Bruker) with a capillary voltage of 1000 V applied, dry gas temperature of 280 °C at 5 L min−1 and nebulizer at 3 pound per square inch (psi). Acetonitrile enriched dopant nitrogen was used. MS spectra were acquired within an m/z range of 500–1850 for N-glycans and 380–1850 for O-glycans in enhanced mode, smart parameter setting (SPS) was set to m/z 1200 for N-glycans and m/z 900 for O-glycans; ion charge control (ICC) to 4 × 103 and maximum acquisition time to 200 ms. MS/MS spectra were generated using collision-induced dissociation over an m/z range from 100 to 2500 on the top three most abundant precursors, applying an isolation width of 3 Thomson. The fragmentation cut-off was set to 27% with 100% fragmentation amplitude using the Enhanced SmartFrag option from 30–120% in 32 ms and ICC was set to 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
2.7 Data processing
Glycan structures were assigned based on glycan composition obtained from accurate mass, relative PGC elution position, MS/MS fragmentation pattern in negative-ion mode and general glycobiological knowledge,15,19 with help of Glycoworkbench20 and Glycomod21 software tools. Extracted ion chromatograms were used to integrate area under the curve (AUC) for each individual glycan isomer using Compass Data Analysis software v.5.0. The most abundant peaks in the glycan profile were manually picked and integrated. Relative quantitation of individual glycans was performed on the total area of all included glycans within one sample normalizing it to 100%. The raw data is available in GlycoPost repository: [https://glycopost.glycosmos.org/preview/7996273655e60ec00bb52c].3. Results and discussion
3.1 Integrated N- and O-glycan sample preparation in 96-well plate format
A high-throughput sample preparation in 96-well plate format was established for integrated N- and O-glycomics, and combined with in-depth characterization of glycans using PGC nano-LC-ESI-MS/MS in negative ion mode, which can be applied for the analysis of purified glycoproteins and biological samples containing complex mixtures of glycoproteins (Fig. 1). This approach is based on the sequential structural analysis of N- and O-glycans released from glycoproteins developed by Jensen, et al.13 and a high-throughput N-glycan preparation workflow that was recently reported by our group.18 The highlights of the adapted workflow for combined N- and O-glycomics include high-throughput approach in 96-well plate format for the entire sample preparation, integration of N-glycan and O-glycan analysis, and powerful isomer separation on PGC for in-depth characterization of glycans. Importantly, large numbers of samples can be analyzed in the same batch with potential for method transfer to a liquid-handling robotic workstation prior to glycan analysis by PGC nano-LC-ESI-MS/MS.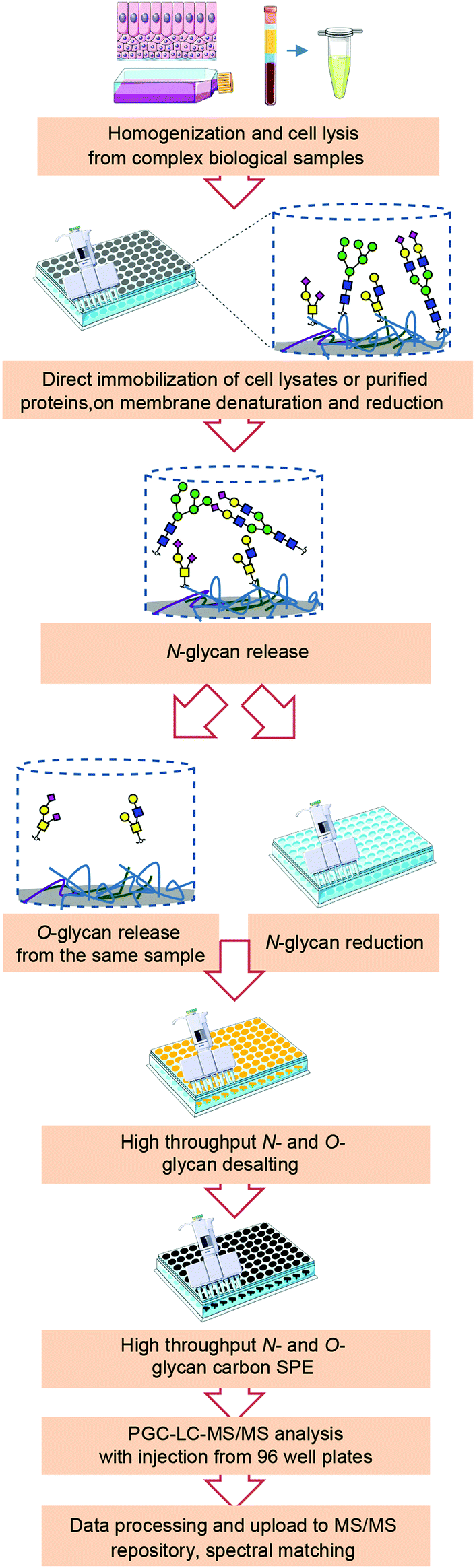 | ||
| Fig. 1 Workflow for integrated N- and O-glycomics based on a 96-well plate sample preparation and analysis on PGC nano-LC-ESI-MS/MS. Glycoproteins were extracted from biological samples by cell lysis and immobilized on PVDF membrane. Proteins were denatured and reduced by guanidine HCl and DTT without detergent followed by enzymatic N-glycan release by PNGase F. The released N-glycans were collected and reduced. O-Glycans were then released from the same sample by reductive β-elimination. Reduced glycans were further desalted, purified, reconstituted and injected all in 96-well format plates, and subjected to PGC nano-LC-ESI-MS/MS analysis in negative ion mode. | ||
The current protocols for glycomics analysis of cells often require a large quantity of biological material (4–10 × 106 cells),11,22,23 and therefore are of limited use to decipher the glycosylation of cells that are available in minor amounts. Upon considerable modifications, with this approach we were able to analyze N- and O-glycosylation derived from 5 × 105 cells. Our aim was to provide a high throughput workflow suitable for cell glycosylation profiling ensuring sufficient material for glycomics analysis assuming different glycosylation characteristics of different biological samples. Therefore, we have not attempted to test the lowest cell amount necessary for the analysis of glycans derived from NMuMG cell line, as this would be specific for this cell line. However, PGC-MS based glycomics workflows have recently undergone major improvements with respect to sensitivity.24–26 Using post-column make-up flow (PCMF) for enhancing sensitivity has shown potential to allow glycomics analysis from minor amounts of biological material such as rare cell populations as well as patient derived materials.
As shown in Fig. 1, (glyco)proteins from complex biological samples such as cell pellets from cultured cells, organoids or tissue should first be extracted by cell lysis. For tough or fibrous tissues, homogenization is often needed to assist extraction of glycoproteins prior to cell lysis. Taking into consideration the compatibility with MS, cells were lysed mechanically in water or lysis buffer without any detergents. Next, glycoproteins in solution or in cell lysates were then applied and immobilized on 96-well PVDF membrane filter plates followed by denaturation and reduction to achieve efficient and complete release of glycans. To avoid nonspecific binding of PNGase F to the PVDF membrane, the effect of blocking agent PVP-40 was tested, however it showed no benefit on the overall N-glycan signal when cell lysates from 5 × 105 cells were used, likely due to high amounts of protein in the samples which presumably already largely occupied the PVDF membrane pores (data not shown). After removing the denaturation and reduction agents, N-glycans were released from immobilized glycoproteins on PVDF membrane using 2 U of PNGase F, as illustrated in Fig. 1. To ensure the efficient generation of reducing-end glycans and subsequent quantitative reduction, glycosylamines resulting from the release deamidation were hydrolysed using acidic buffer (pH = 5). The released N-glycans were collected and further subjected to reduction, desalting, and PGC solid-phase extraction (SPE) clean-up all in 96-well plate format to ensure efficient sample-throughput. Subsequently, O-glycans were released by reductive β-elimination from the same wells and purified in a high-throughput manner following the same procedure together with the released N-glycans. Thereafter, the released N-glycan and O-glycan alditols were separated on PGC chromatographic column and analyzed with an ion trap mass spectrometer in negative ionization mode.
Most methodologies employ sialic acid derivatization for their stabilization and chemical labeling to increase ionization efficiency.6,18 In addition, the fucose migration in positive mode often poses a problem for structural analysis.8,27 Working in negative ionization mode has key advantages to address these challenges due to its high ionization efficiency especially for sialylated glycans, stability of sialic acids and no migration of fucose. More importantly, negative ionization mode fragmentation allows in-depth structural identification of glycan structures,14,16,28–30 since additional structural information can be obtained from cross-ring fragments providing diagnostic ions for the characterization of glycan linkages.28 MS/MS spectra can be made publicly available by exporting from the DataAnalysis software using Glycoworkbench workspace and uploading to Unicarb DR repository following a standardized bioinformatics infrastructure,31 serving as an open resource for automated N- and O-glycan identification via spectral matching.
3.2 Application of the workflow for the release of N- and O-glycans from bovine fetuin standard
The established approach was evaluated using bovine fetuin standard, which contains highly sialylated N-glycans32,33 and O-glycans34,35 differing in the number of sialic acids as well as their linkages. To assess the technical variation of glycan profiles, aliquots of 20 μg fetuin were applied to PVDF membrane wells in three technical replicates. N-Glycans and O-glycans were prepared following this approach and analyzed by PGC nano-LC-ESI-MS/MS in negative ion mode. Glycan structures and linkages were assigned following known MS/MS fragmentation patterns in negative-ion mode,15,19 known retention behaviour of different sialic acid isomers on PGC column32 and general glycobiological knowledge.Within this analysis, 30 different N-glycan structures were detected of which 24 passed the quantification criteria (signal-to-noise ratio (S/N) ≥ 9). As expected, sialylated N-glycans were detected as dominant glycan types, with different number of sialic acids and their linkages. The profiles obtained by our analysis (Fig. 2A) are very similar to other published characterizations of bovine fetuin N-glycans.32,33,36 Relative quantitation was performed on the top 13 most abundant N-glycans within one sample normalized to 100%. Details on glycan structures and relative abundance are given in Table S1, ESI.† Inter- and intraday repeatability of the fetuin N-glycan analysis showed two median intraday coefficients of variations (CVs) of 7.6% and 8.0%, and a median interday CV of 9.8% within six technical replicates distributed into two plates over 1 month. The highest intraday CV for the relative quantification of N-glycans released from fetuin was 15.7%, indicating the good repeatability of the workflow (Fig. 2B). Our results showed good reduction efficiency, without detection of unreduced species in the top 10 most abundant N-glycans released from fetuin and with less than 2% of unreduced species in the top 10 most abundant N-glycans released from NMuMG cells (data not shown).
 | ||
| Fig. 2 Analysis of N-glycans and O-glycans derived from bovine fetuin standard. (A) Combined extracted ion chromatograms (EIC) of N-glycans released from bovine fetuin standard. Blue square: N-acetylglucosamine, green circle: mannose, yellow circle: galactose, red triangle: fucose, right pointing pink diamond: α2,6-linked N-acetylneuraminic acid, left pointing pink diamond: α2,3-linked N-acetylneuraminic acid, H: hexose, N: N-acetylhexosamines, S: N-acetylneuraminic acid. (B) Inter- and intraday repeatability of the fetuin N-glycan analysis based on relative quantification of top 13 most abundant N-glycans. Inter- and intraday repeatability of the fetuin N-glycan analysis showed two median coefficients of variation (CV) of 7.6% and 8.0% within three technical replicates form the same plate, and a median CV of 9.8% within six technical replicates distributed into two plates over 1 month (displayed as mean relative abundance plus standard deviation; CVs of each glycans were listed on the top of the bar; intraday n = 3, interday n = 6, independent two different plates over 1 month). More details displayed in Table S1, ESI.† (C) Combined EICs of 5 O-glycans released from bovine fetuin standard, in which the top three most abundant O-glycans account for 98% of the relative abundance. Blue square: N-acetylglucosamine, yellow circle: galactose, pink diamond: N-acetylneuraminic acid, H: hexose, N: N-acetylhexosamines, S: N-acetylneuraminic acid. (D) Inter- and intraday repeatability of the top 3 most abundant O-glycans released from fetuin after removing N-glycans. (displayed as mean relative abundance plus standard deviation; CVs of each glycans were listed on the top of the bar; intraday n = 3, interday n = 6, derived from two different plates performed over 1 month). More details displayed in Table S2, ESI.† | ||
The powerful separation capacity of PGC chromatography in N- and O-glycans has been widely demonstrated by previous work, enabling separation of glycan isomers based on glycan structure and linkage specificity.28,32,37–39Fig. 2A shows four glycan isomers with the same composition containing five hexoses, four N-acetylhexosamines and two N-acetylneuraminic acids (H5N4S2) (blue trace) which are separated due to different sialic acid linkage combinations. First glycan isomer with two α2,6-linked N-acetylneuraminic acids elutes at 27.2 min, followed by the two coeluting isomers at 32.9 min, which have both one α2,6- and one α2,3-linked N-acetylneuraminic acids, carried on different arms. Lastly, the same glycan with two α2,3-linked N-acetylneuraminic acids elutes at 37.3 min. From this result, we can confirm the previous reports that glycans containing α2,6-linked sialic acid residues elute significantly earlier than the α2,3-linked counterparts.32 While the order of isomer elution is in line with previous reports, we observe slightly different relative retention of e.g. later eluting monosialylated compared to disialylated diantennary N-glycan.
Next to N-glycans, three O-glycans were also detected which were released and prepared from fetuin after N-glycan removal (Fig. 2C), showing the feasibility of integrated N- and O-glycan sample preparation in a 96-well plate format. Fig. 2D shows the relative quantification and repeatability data for the three glycans which are sialylated core 1 and core 2 structures. The median CVs for two intra- and interday repeatability of all quantified structures were 1.9%, 2.3%, and 8.9%, respectively (Table S2, ESI†).
3.3 Application of the workflow for the release N- and O-glycans from murine NMuMG cells
To demonstrate that this established integrated N- and O-glycomics approach is applicable for complex biological samples, N- and O-glycans were prepared from murine NMuMG cell line following the method described in Fig. 1. For complex biological samples, cell lysis was required to extract glycoproteins from the cell membrane. In this study, we utilized mechanical cell lysis in water via sonication for 60 min in an ultrasound bath. In addition, lysis buffer without detergent can be used as an alternative.N-Glycan (Fig. 3A) and O-glycan (Fig. 3B) alditols were prepared from 5 × 105 cells in a high-throughput manner following our approach. N-Glycan alditols were analyzed using PGC nano-LC-ESI-MS/MS platform which revealed 94 different N-glycan structures in a single analysis, including 3 paucimannose, 16 oligommanose, 11 hybrid and 64 complex glycans (Table S3, ESI†). Additional low abundant glycans may be present but were not considered further. The composition and proposed structures of N-glycans were assigned on the basis of general knowledge on N-glycan biosynthesis in mice, previous publications on N-glycan fragmentation in negative ion mode40–44 as well as using automated spectral matching tool available online via Unicarb DB. Fig. 3A shows the combined extracted ion chromatograms of the 25 most abundant N-glycan structures. The N-glycan profile obtained from 2 mg of total proteins extracted from NMuMG cell lines has been reported previously, which revealed only 27 N-glycan structures by MALDI-TOF/TOF-MS analysis after acetohydrazide derivatizaton of sialic acids.22 With this approach we were able to confirm all of the N-glycans observed in the previous work,22 but also revealed a large number of additional glycans including paucimannose, phosphorylated oligomannose and more sialylated N-glycans (Fig. 3A). Phosphorylated oligomannose N-glycans were characterized by a decreased retention time on PGC compared to unmodified ones. (Fig. S1, ESI†). In contrast, addition of a sulfate group often results in an increased retention on PGC (unpublished data). Notably, powerful isomer separation on PGC vastly contributed to the higher number of identified glycans, compared to MALDI-TOF/TOF-MS analysis. In agreement with literature,32 we overall confirmed the earlier elution of oligomannose N-glycans, and an increasing retention of N-glycans with increasing numbers of sialic acids and antennae.
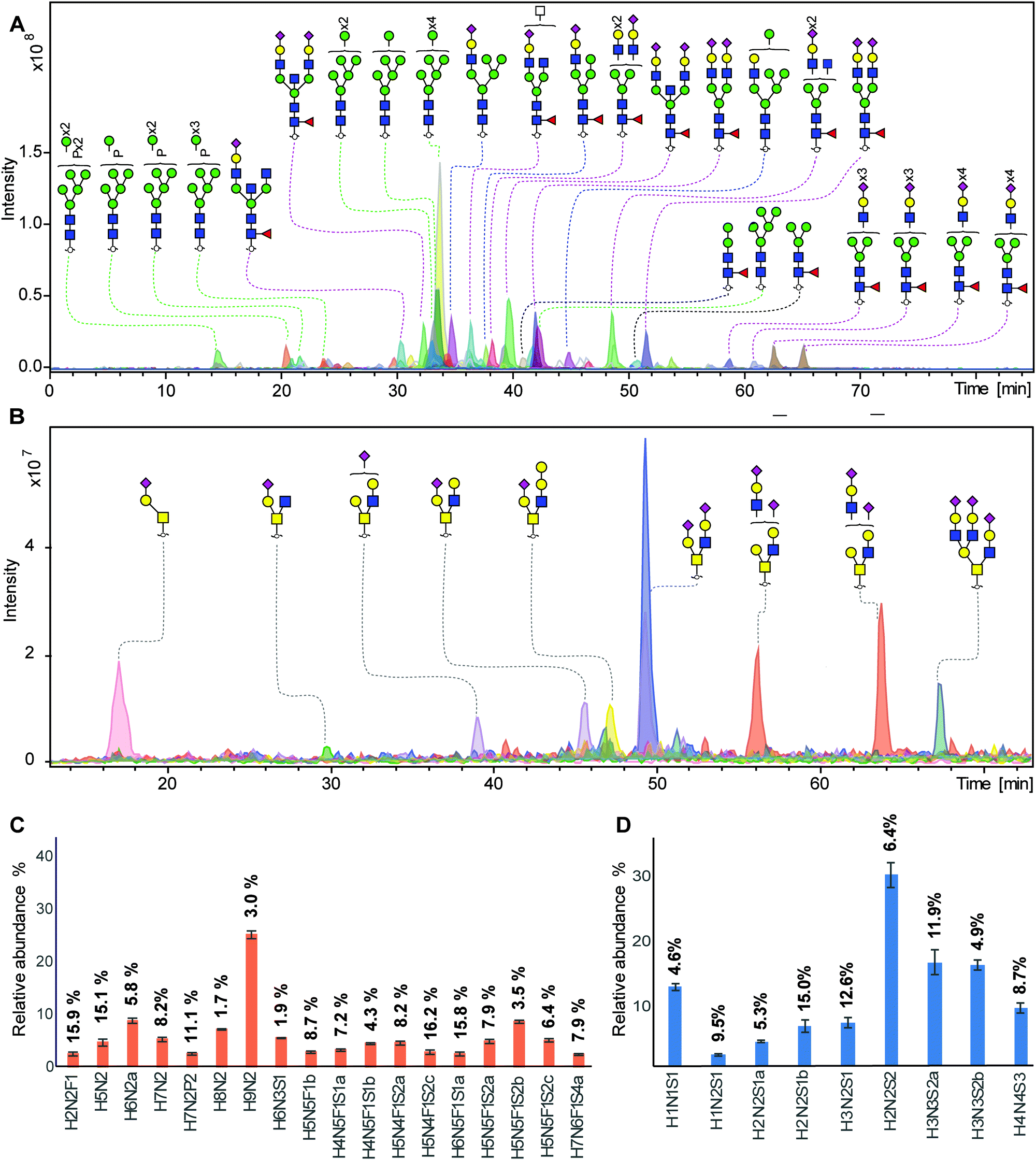 | ||
| Fig. 3 Integrated N- and O-glycans analysis derived from NMuMG cells following the 96-well plate sample preparation method and analyzed using PGC nano-LC-ESI-MS/MS. (A) Combined EICs of N-glycans released from 5 × 105 NMuMG cells. (Black line: paucimannose; green line: oligomannose; blue line: hydride; red line; complex type). Blue square: N-acetylglucosamine, green circle: mannose, yellow circle: galactose, red triangle: fucose, pink diamond: N-acetylneuraminic acid, transparent square: N-acetylhexosamine, P: phosphate, H: hexose, N: N-acetylhexosamines, S: N-acetylneuraminic acid. (B) Combined EICs of O-glycans released from NMuMG cells after removing N-glycans. (C) Relative quantification of top 18 most abundant N-glycans. (displayed as mean relative abundance plus standard deviation; CVs of each glycans were listed on the top of the bar; n = 3). More details displayed in Table S4, ESI.† (D) Relative quantification of top 9 most abundant O-glycans. (displayed as mean relative abundance plus standard deviation; CVs of each glycans were listed on the top of the bar; n = 3). More details displayed in Table S5, ESI.† | ||
Relative quantitation of the top 18 most abundant glycans was performed on the total area of the selected glycans within one sample normalized to 100%. The relative abundance and corresponding standard deviation are shown in Fig. 3C and Table S4, ESI.† The robustness of the method was confirmed by a median CV of 7.9% as well as the highest CV of 16.2% within the top 18 glycan species. Specifically, 13 out of 18 the main peaks showed a CV lower than 10%, indicating a very good repeatability. In contrast to N-glycans, mucin-type O-glycans do not share a common core beyond the innermost GalNAc, resulting in a vast structural diversity as well as the presence of multiple isomers. Released O-glycan analysis from the same 5 × 105 NMuMG cells revealed 11 O-glycans (Fig. 3B). All O-glycans in NMuMG cell line were mono-and disialylated core 1 and core 2 structures. The power of isomer separation of PGC can be displayed by the two glycan isomers with composition H2N2S1 at m/z 1040.4 (Fig. 3B, purple trace), which carry the same H2N2 neutral core structure but with different linkage of sialic acid or sialic acid linked to different galactoses, as well as two different core 2 glycan isomers with composition H3N3S2 at m/z 1696.5 (Fig. 3B, orange trace). The distribution of the O-glycans was determined by relative quantification, revealing the dominance of disialylated core 2 O-glycans (Fig. 3D and Table S5, ESI†). The technical variability of O-glycans derived from cell lines was determined in three technical replicates (Fig. 3D) with CVs less than 14.9%.
4. Conclusions
In summary, we present an integrated approach for N- and O-glycomics from purified proteins and complex biological samples combining sequential release of N- and O-glycans based on protein immobilization on PVDF membrane filter plates in 96-well format and structural elucidation based on PGC nano-LC-ESI-MS/MS using negative electrospray ionization, inspired by the methodology developed by Packer and coworkers.13,14 Here, we provide a method with high repeatability for combined N- and O-glycomics of biological samples, in which glycans were prepared in a higher-throughput and less time consuming manner as compared to previous workflows.13,14 Still, this method requires multiple manual handling steps and several sample transfers between different 96-well plates. The transfer of this sample preparation method to a liquid-handling robotic workstation should be feasible. The method was successfully applied for N- and O-glycomics of NMuMG cells as an example of complex biological samples. In the future, the method may be used to handle larger numbers of samples for a comprehensive glycomics analysis in an in-depth manner in the context of functional glycomics, systems biology, and clinical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank A. L. Hipgrave Ederveen, C. A. M. Koeleman and L. de Neef for technical support, and Y. Mohammed for bioinformatics support. This work was supported by the European Commission's Horizon 2020 programme “GlyCoCan” project, grant number 676421.References
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS PubMed
.
- J. G. Rodrigues, M. Balmana, J. A. Macedo, J. Pocas, A. Fernandes, J. C. M. de-Freitas-Junior, S. S. Pinho, J. Gomes, A. Magalhaes, C. Gomes, S. Mereiter and C. A. Reis, Cell. Immunol., 2018, 333, 46–57 CrossRef CAS PubMed
- T. Song, D. Aldredge and C. B. Lebrilla, Anal. Chem., 2015, 87, 7754–7762 CrossRef CAS PubMed
- S. Gaunitz, G. Nagy, N. L. Pohl and M. V. Novotny, Anal. Chem., 2017, 89, 389–413 CrossRef CAS PubMed
- S. Holst, B. Heijs, N. de Haan, R. J. van Zeijl, I. H. Briaire-de Bruijn, G. W. van Pelt, A. S. Mehta, P. M. Angel, W. E. Mesker, R. A. Tollenaar, R. R. Drake, J. V. Bovee, L. A. McDonnell and M. Wuhrer, Anal. Chem., 2016, 88, 5904–5913 CrossRef CAS PubMed
- G. S. Kammeijer, I. Kohler, B. C. Jansen, P. J. Hensbergen, O. A. Mayboroda, D. Falck and M. Wuhrer, Anal. Chem., 2016, 88, 5849–5856 CrossRef CAS PubMed
- C. W. Sutton, J. A. O'Neill and J. S. Cottrell, Anal. Biochem., 1994, 218, 34–46 CrossRef CAS PubMed
- M. Wuhrer, C. A. Koeleman, C. H. Hokke and A. M. Deelder, Rapid Commun. Mass Spectrom., 2006, 20, 1747–1754 CrossRef CAS PubMed
- I. Trbojevic-Akmacic, M. Vilaj and G. Lauc, Expert Rev. Proteomics, 2016, 13, 523–534 CrossRef CAS PubMed
- A. Shubhakar, K. R. Reiding, R. A. Gardner, D. I. Spencer, D. L. Fernandes and M. Wuhrer, Chromatographia, 2015, 78, 321–333 CrossRef CAS PubMed
- S. Holst, G. W. van Pelt, W. E. Mesker, R. A. Tollenaar, A. I. Belo, I. van Die, Y. Rombouts and M. Wuhrer, Methods Mol. Biol., 2017, 1503, 185–196 CrossRef CAS PubMed
- M. Kotsias, R. P. Kozak, R. A. Gardner, M. Wuhrer and D. I. R. Spencer, PLoS One, 2019, 14, e0210759 CrossRef CAS PubMed
- P. H. Jensen, N. G. Karlsson, D. Kolarich and N. H. Packer, Nat. Protoc., 2012, 7, 1299–1310 CrossRef CAS PubMed
- C. Ashwood, B. Pratt, B. X. MacLean, R. L. Gundry and N. H. Packer, Analyst, 2019, 144, 3601–3612 RSC
- N. G. Karlsson, N. L. Wilson, H. J. Wirth, P. Dawes, H. Joshi and N. H. Packer, Rapid Commun. Mass Spectrom., 2004, 18, 2282–2292 CrossRef CAS PubMed
- A. V. Everest-Dass, J. L. Abrahams, D. Kolarich, N. H. Packer and M. P. Campbell, J. Am. Soc. Mass Spectrom, 2013, 24, 895–906 CrossRef CAS PubMed
- E. Piek, A. Moustakas, A. Kurisaki, C. H. Heldin and P. ten Dijke, J. Cell Sci., 1999, 112(Pt 24), 4557–4568 CAS
- S. Holst, A. I. Belo, E. Giovannetti, I. van Die and M. Wuhrer, Sci. Rep., 2017, 7, 16623 CrossRef PubMed
- N. G. Karlsson, B. L. Schulz and N. H. Packer, J. Am. Soc. Mass Spectrom, 2004, 15, 659–672 CrossRef CAS PubMed
- A. Ceroni, K. Maass, H. Geyer, R. Geyer, A. Dell and S. M. Haslam, J. Proteome Res., 2008, 7, 1650–1659 CrossRef CAS PubMed
- C. A. Cooper, E. Gasteiger and N. H. Packer, Proteomics, 2001, 1, 340–349 CrossRef CAS PubMed
- Z. Tan, W. Lu, X. Li, G. Yang, J. Guo, H. Yu, Z. Li and F. Guan, J. Proteome Res., 2014, 13, 2783–2795 CrossRef CAS PubMed
- H. Hamouda, M. Kaup, M. Ullah, M. Berger, V. Sandig, R. Tauber and V. Blanchard, J. Proteome Res., 2014, 13, 6144–6151 CrossRef CAS PubMed
- H. Hinneburg, S. Chatterjee, F. Schirmeister, T. Nguyen-Khuong, N. H. Packer, E. Rapp and M. Thaysen-Andersen, Anal. Chem., 2019, 91, 4559–4567 CrossRef CAS PubMed
- H. Hinneburg, P. Korac, F. Schirmeister, S. Gasparov, P. H. Seeberger, V. Zoldos and D. Kolarich, Mol. Cell. Proteomics, 2017, 16, 524–536 CrossRef CAS PubMed
- T. Nguyen-Khuong, A. Pralow, U. Reichl and E. Rapp, Glycoconjugate J., 2018, 35, 499–509 CrossRef CAS PubMed
- E. Mucha, M. Lettow, M. Marianski, D. A. Thomas, W. B. Struwe, D. J. Harvey, G. Meijer, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2018, 57, 7440–7443 CrossRef CAS PubMed
- C. Ashwood, C. H. Lin, M. Thaysen-Andersen and N. H. Packer, J. Am. Soc. Mass Spectrom, 2018, 29, 1194–1209 CrossRef CAS PubMed
- D. J. Harvey and J. L. Abrahams, Rapid Commun. Mass Spectrom., 2016, 30, 627–634 CrossRef CAS PubMed
- J. L. Abrahams, M. P. Campbell and N. H. Packer, Glycoconjugate J., 2018, 35, 15–29 CrossRef CAS PubMed
- M. A. Rojas-Macias, J. Mariethoz, P. Andersson, C. Jin, V. Venkatakrishnan, N. P. Aoki, D. Shinmachi, C. Ashwood, K. Madunic, T. Zhang, R. L. Miller, O. Horlacher, W. B. Struwe, Y. Watanabe, S. Okuda, F. Levander, D. Kolarich, P. M. Rudd, M. Wuhrer, C. Kettner, N. H. Packer, K. F. Aoki-Kinoshita, F. Lisacek and N. G. Karlsson, Nat. Commun., 2019, 10, 3275 CrossRef PubMed
- G. Palmisano, M. R. Larsen, N. H. Packer and M. Thaysen-Andersen, RSC Adv., 2013, 3, 22706–22726 RSC
- E. D. Green, G. Adelt, J. U. Baenziger, S. Wilson and H. Van Halbeek, J. Biol. Chem., 1988, 263, 18253–18268 CAS
- M. Windwarder and F. Altmann, J. Proteomics, 2014, 108, 258–268 CrossRef CAS PubMed
- Y. Goso, T. Sugaya, K. Ishihara and M. Kurihara, Anal. Chem., 2017, 89, 8870–8876 CrossRef CAS PubMed
- M. Nakano, R. Saldanha, A. Gobel, M. Kavallaris and N. H. Packer, Mol. Cell. Proteomics, 2011, 10, M111 009001 CrossRef PubMed
- K. Stavenhagen, D. Kolarich and M. Wuhrer, Chromatographia, 2015, 78, 307–320 CrossRef CAS PubMed
- L. R. Ruhaak, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2009, 394, 163–174 CrossRef CAS PubMed
- M. Pabst, J. S. Bondili, J. Stadlmann, L. Mach and F. Altmann, Anal. Chem., 2007, 79, 5051–5057 CrossRef CAS PubMed
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 631–646 CrossRef CAS PubMed
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 622–630 CrossRef CAS PubMed
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 647–659 CrossRef CAS PubMed
- D. J. Harvey, M. Edgeworth, B. A. Krishna, C. Bonomelli, S. A. Allman, M. Crispin and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2014, 28, 2008–2018 CrossRef CAS PubMed
- J. H. Chik, J. Zhou, E. S. Moh, R. Christopherson, S. J. Clarke, M. P. Molloy and N. H. Packer, J. Proteomics, 2014, 108, 146–162 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1: Relative quantification of N-glycans released from fetuin standard using a 96-well plate sample preparation method and PGC nano-LC-ESI-MS/MS. Table S2: Relative quantification of O-glycans released from fetuin standard using a 96-well plate sample preparation method and PGC nano-LC-ESI-MS/MS. Table S3: N-glycans released from NMuMG cell line using a 96-well plate sample preparation method and PGC nano-LC-ESI-MS/MS. Table S4: Relative quantification of N-glycans released from NMuMG cells using a 96-well plate sample preparation method and PGC nano-LC-ESI-MS/MS. Table S5: Relative quantification of O-glycans released from NMuMG cells using a 96-well plate sample preparation method and PGC nano-LC-ESI-MS/MS. Fig. S1: Elution pattern of N-glycans with and without phosphate on PGC. See DOI: 10.1039/c9mo00180h |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2020 |