
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin-based smart materials: a roadmap to processing and synthesis for current and future applications
Adrian
Moreno
 and
Mika H.
Sipponen
*
and
Mika H.
Sipponen
*
Department of Materials and Environmental Chemistry, Stockholm University, SE-10691, Stockholm, Sweden. E-mail: mika.sipponen@mmk.su.se
First published on 22nd June 2020
Abstract
Biomass-derived materials are green alternatives to synthetic plastics and other fossil-based materials. Lignin, an aromatic plant polymer, is one of the most appealing renewable material precursors for smart materials capable of responding to different stimuli. Here we review lignin-based smart materials, a research field that has seen a rapid growth during the last five years. We describe the main processing and chemical synthesis routes available for the fabrication of lignin-based smart materials, and focus on their use as sensors, biomedical systems, and shape-programmable materials. In addition to benchmarking their performance to the state of the art fossil counterparts, we identify challenges and future opportunities for the development of lignin-based smart materials towards new high-performance applications.
 Adrian Moreno | Adrian Moreno received his BSc and MSc in chemistry from the Universitat Rovira i Virgili (Spain). He obtained his PhD in 2019 working on Single Electron Transfer-Living Radical Polymerization (SET-LRP) and the design of stimuli-responsive polymers under the supervision of Prof. Gerard Lligadas and Prof. Marina Galià. During his PhD, he did an exchange in the group of Prof. Virgil Percec (University of Pennsylvania, USA). Currently, he holds a Postdoctoral Fellow position in the Sustainable Materials Chemistry (SUSMATCHEM) group at Stockholm University with Assist. Prof. Mika H. Sipponen. His research interests lie in the development of novel lignin-based functional materials. |
 Mika H. Sipponen | Mika H. Sipponen received his MSc and DSc (Tech.) degrees in Chemical Technology from Aalto University, Finland in 2010 and 2015. During 2016–2019, he worked as an Academy of Finland Postdoctoral Researcher on lignin-based functional materials with Prof. Monika Österberg (Aalto University) with a research stay in University of Rome Tor Vergata, Italy (Prof. Claudia Crestini). Since September 2019, he is an Assistant Professor in Materials Chemistry in the Department of Materials and Environmental Chemistry at Stockholm University. His Sustainable Materials Chemistry (SUSMATCHEM) group develops novel lignin-based processes and materials following the principles of green chemistry. |
1. Introduction
The development of materials from renewable resources via sustainable methodologies is one of the most promising solutions to reduce exploitation of fossil resources that is linked to increasing greenhouse gas emissions and climate change.1,2 In addition to synthetic polymers of renewable origin such as poly(lactide) (PLA), poly(ε-caprolactone) and poly(3-hydroxybutyrate) (PHB),3–5 abundant and renewable plant biomass and its natural polymeric components are attractive material precursors.6,7 Next to cellulose, which is the most important natural polymer and constituent of plant biomass, lignin has been considered “the black sheep” and a low-value byproduct that is mostly combusted in lignocellulosic biorefineries and chemical pulping processes.8–10 This disdain over lignin is not warranted based on its natural functions such as adding strength and rigidity to the plant cells walls, enabling transport of water, and protecting from pathogens and insects.11,12Lignin is synthetized mainly from p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol monomers (Fig. 1a) via an enzyme-initiated dehydrogenation, radical coupling, and dimerization reactions, resulting in an amorphous and three-dimensional (3D) material with both ether and carbon–carbon bonds.13,14 It is impossible to give an exact molecular structure of lignin because various different isolation processes cause structural changes and insertion of new functional groups. Furthermore, lignins are usually referred to as polymers, but isolated technical lignins can also be viewed as mixtures of oligomers. For instance, lignin obtained from the kraft pulping process has an average degree of polymerization of 30 compared to a DP of about 6000 of a polystyrene.
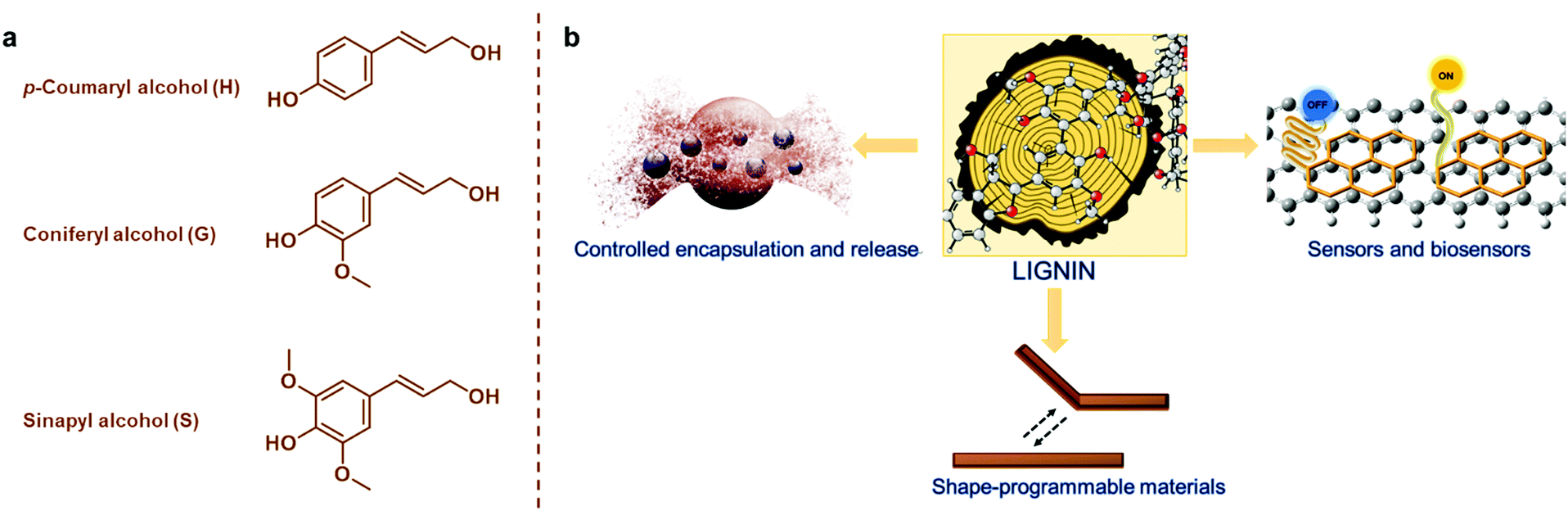 | ||
| Fig. 1 (a) Monomeric precursors of lignin. (b) An overview of lignin-based smart materials covered in the present review. | ||
In recent years, there have been widespread efforts to upgrade lignin towards advanced engineered materials.15 In fact, lignin is an aromatic biopolymer gifted with numerous attractive properties such as high (>60 atom%) carbon content, high thermal stability, biodegradability, antioxidant activity, absorbance of UV irradiation, and antimicrobial activity.16–19 Beyond conventional bulk and low-cost materials such as concrete plasticizers,20–22 the development of lignin-based smart materials is now emerging as the next generation of value-added applications. This new class of smart materials has the ability to convert specific stimuli to defined outputs and change or activate a functional property of the material (Fig. 1b). It has become important to provide control over these stimuli-responsive properties, and this often requires modification or insertion of new functional groups capable of changing one of their properties in a spatiotemporal fashion in response to external stimuli such as temperature, pH, light or electricity among others.
There are several excellent reviews dedicated to the synthesis and applications of lignin-based materials23–31 but none of these have identified lignin-based smart materials as an emerging research field. Therefore, in this review we visit the recently published literature on the synthesis and applications of lignin-based smart materials, and make a critical comparison to the state of the art benchmark materials prepared largely from fossil-based precursors. We begin with a brief roadmap to different chemical and processing routes and continue with an analysis of lignin-based smart materials for (1) sensing and biosensing, (2) controlled encapsulation and release, especially in biomedical systems, and (3) shape memory programmable materials. Finally, we conclude with challenges and perspectives for these advanced materials in current and future applications.
2. Roadmap to lignin-based smart materials
There are many different routes to obtain lignin polymers, oligomers, and dimers/monomers from plant biomass and their conversion to smart materials (Fig. 2). Low molecular weight phenolic molecules can be produced from lignin by cleavage of inter-unit linkages, among which aryl ether linkages (β-O-4′) typically account more than 50% of bonds formed during the polymerization process. Other relevant linkages include resinol (β–β), phenylcoumaran (β-5′), biphenyl (5-5′), diphenyl ether (4-O-5′) and diphenyl methane (β-1), which are significantly more complicated to degrade.32 The chemical structure of lignin is strongly related to its botanical origin and the isolation process, and recent progress in different analytical techniques have made analysis of the chemical bonds a less daunting task.33,34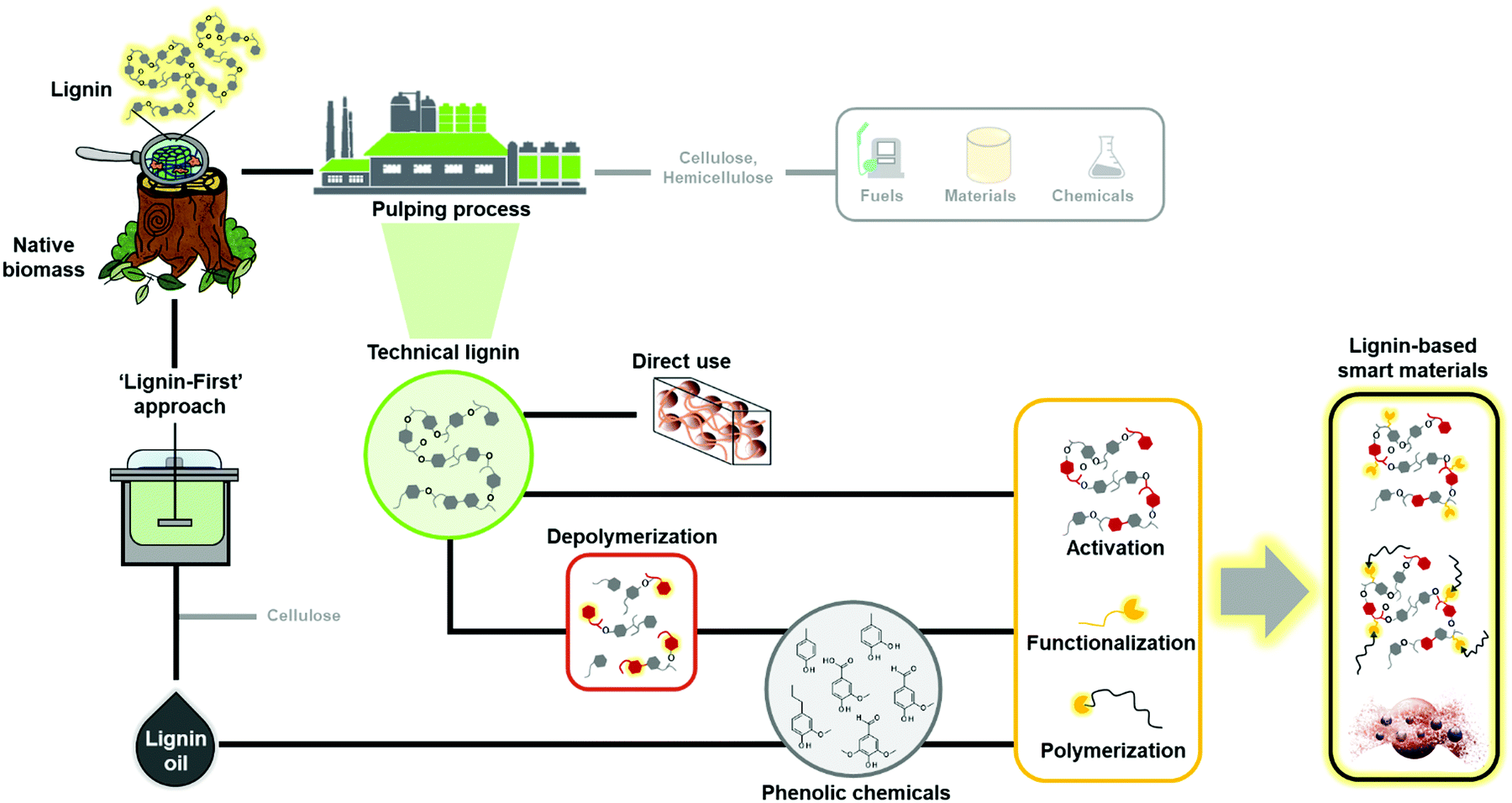 | ||
| Fig. 2 Roadmap from native lignocellulosic biomass to lignin-based smart materials. | ||
The pulp and paper industry is the primary source of technical lignin. As reviewed elsewhere,35,36 lignin is dismantled from cellulosic fibers by thermochemical treatments, resulting in depolymerization and solubilization of lignin, while at the same time non-native “condensed” bonds are formed at the reactive sites. The most relevant technical lignins for material applications are lignosulfonates (LS), kraft lignin (KL), organosolv lignin (OSL) and soda lignin (SL).37–39 It is important to mention that the selection of the type of lignin is strongly related to the desired application. For instance, less pure preparations might be readily accessible to lignin-derived plastics40,41 or composites,21,22,30 whereas highly purified or fractionated lignins should be considered for high-performance materials such as biomedical applications.42,43
The most straightforward way is to use crude technical lignins to enhance properties of existing polymeric materials.44,45 For instance, lignin has been used as an antioxidant and UV-shielding additive in plastics,46,47 and stabilizer and flame retardant agent in fillers.48,49 However, in most of these applications only a minor amount of lignin can be blended due to its inferior mechanical properties and low compatibility with many polymers.25 Chemical modification of lignin has been postulated as the main alternative to improve its material properties for polymers. These processes increase the reactivity of lignin by targeting its functional groups such as hydroxyl, methoxyl, carbonyl, or carboxyl groups through chemical modification processes such as hydoxyalkylation, esterification, and amination, among others (Fig. 3a).23,26,50–58 Many of these chemical modifications have attempted to convert lignin macromolecules into macromonomers and subsequently graft classical monomers or polymers, and in this way, synthetize lignin-based functional polymers.23,26–28
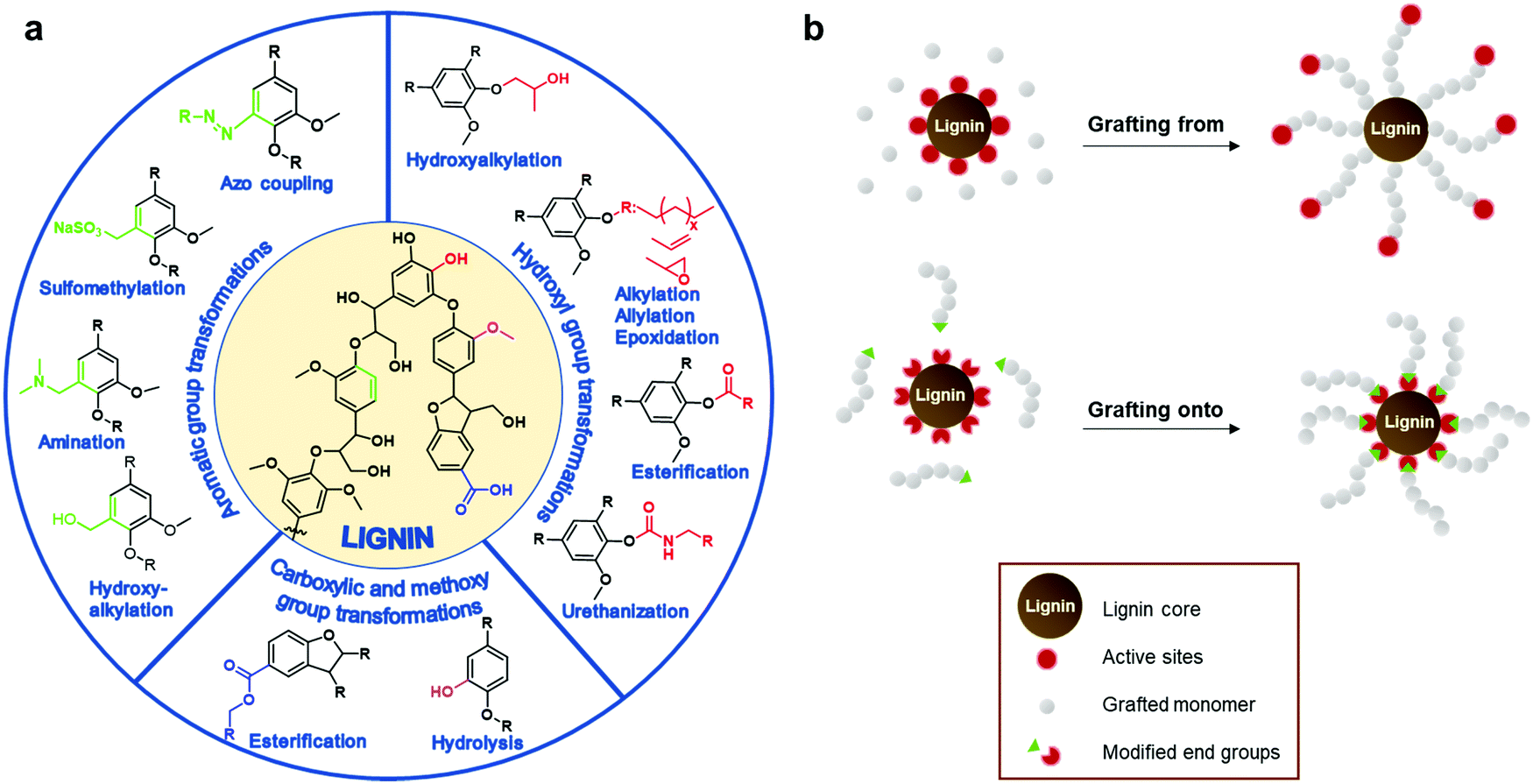 | ||
| Fig. 3 (a) Summary of chemical transformation commonly employed to increase the chemical functionality of lignin. (b) General strategies to synthetize lignin-based graft polymers. | ||
Well-defined lignin graft copolymers can be elaborated by two main methodologies: “grafting from” and “grafting to” (Fig. 3b). In the “grafting from” technique, lignin is used as a macromonomer and usually monomers react with functional groups on lignin (e.g. hydroxyl groups or alkyl halide esters) in a way that the growing polymer chain is assembled on the lignin core. Among the different “grafting from” polymerizations, atom transfer radical polymerization (ATRP) and ring opening polymerization (ROP) have been considered the most powerful methodologies to synthetize, in a controlled manner, well defined lignin-based polymers for multiple applications ranging from biobased composites to gene delivery systems.59–62 In the case of the “grafting to” technique, a previously synthetized polymer chain is anchored to the lignin core through a functional chain end group which serves as a reactive point towards lignin.23,63–66 This technique presents some advantages such as the ability to tune the length of the grafted chains prior to the grafting step and the possibility to conduct a complete characterization prior to the grafting step. Among the different reactions to perform the coupling between lignin and the synthetized polymer chain, “click” chemistry reactions, as is the case of Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC), have emerged as the preferred option due to the formation of the desired lignin copolymers in a simple and quantitative way.67,68
The recent years have revealed that many different types of technical lignins are suitable precursors for the synthesis of a wide range of materials.23,24,35,36,69,70 In contrast, the greatest hurdle in the production of aromatic chemicals is the recalcitrance of lignin resulting from the pulping processes. To circumvent these barriers, catalytic fragmentation following so-called “lignin first” concepts have shown much greater yields and selectivity of aromatic compounds, but these processes use costly metal catalyst (e.g. Ni, Co or Rh) that are difficult to recycle from the heterogeneous reaction mixtures.71–73 The resulting lignin oil fraction is enriched in low molecular weight aromatic monomers and dimers. The use of heteroatom-containing reagents for the cleavage of lignin was recently reviewed.74 This approach offers the possibility to generate lignin-derived heteroatom-containing compounds of interest to many applications such as pharmaceutical precursors (e.g. aniline) or hydrogen storage materials (e.g. lithium phenolates and phenoxides). Other common routes to obtain aromatic chemicals from lignin is through fragmentation or depolymerization processes, which involve pyrolysis, oxidation, hydrogenation, gasification, and microbial conversion.16,75–77 These low molar mass compounds include phenol, vanillin, guaiacol, p-cresol, and catechols among others, many of which are useful starting chemicals for the design and synthesis of novel biobased polymers for different applications, through different polymerization routes including polycondensation,78,79 acyclic diene metathesis (ADMET),80 and controlled radical polymerization techniques.81,82 Nevertheless, it is important to keep in mind that while a wide range of aromatic chemical products can be obtained from lignin, to date only vanillin is produced on a commercial scale (17.000 ton per year) via an alkaline oxidation method.83
Regardless of the molecular weight or source of lignin, chemical modification is an inevitable step to smart materials. One of the routes is to activate or functionalize isolated lignin or its fractions to generate responsive molecules for smart materials (Fig. 2). Another, hitherto less studied approach is to functionalize lignin-derived monomers and use them to synthesize polymers or to modify technical lignins of higher molecular weight. The latter example comes with an added benefit of lower environmental penalty compared to the sole use of petroleum-based chemicals as sources of reactive groups. However, the chemical functionalities offered by non-functionalized lignin are limited in terms of polymerization or grafting sites.
3. Applications and materials chemistry of lignin-based smart materials
Lignin-based smart materials possess the ability to sense external stimuli and translate it into an observable response based on physiochemical changes.27 A wide range of different stimuli are available such as pH, temperature, mechanical force or electric field and their development is usually motivated and driven by inspiration from nature.84,85 The following sections are dedicated to the recent developments of lignin in smart materials and their selected applications.3.1 Sensor and biosensing applications
Sensors are an important class of self-integrated devices with the ability to receive specific inputs from a surrounding media and translate them into output signals that can be transformed into a readable result. A biosensor is a device designed to be able to detect biological species of interest even in the presence of other interfering species. Likewise, lignin-based sensors have attracted considerable attention due to their ability to detect and generate quantifiable signals from complex mixtures (Table 1).| Type of sensor | Type of lignin material | Field of application | Ref. |
|---|---|---|---|
| Biosensor | LCQDs | Bioimaging and biological labelling | 43, 86, 88 and 90 |
| Biosensor | LCQDs | Drug release and bioimaging | 87 |
| Chemical sensor | LCQDs | Fe3+ detection and bioimaging | 89 |
| Chemical sensor | LCQDs | Metal-ion detection and bioimaging | 91 |
| Chemical sensor | LGQDs | Detection of H2O2 | 107 |
| Immunosensor | Lignin/peptide-based gold electrode | Detection of antibody (Ab) | 110 |
| Biocatalytic sensor | Silica/lignin hybrid electrode | Detection of glucose | 111 |
| Biocatalytic sensor | Magnetite/lignin hybrid electrode | Detection of glucose | 112 |
| Pressure sensor | Polydimethysiloxane–lignin composite | Detection of pulse rates and muscle movement | 120 |
| Pressure sensor | Lignin-based hydrogel | Detection of pulse rates and force requirement | 122 |
| Humidity sensor | Graphene oxide–lignin composite | Detection of humidity moisture | 121 |
| Chemical sensor | Lignin-based polymeric composite | Detection of chromate ions | 123 and 124 |
| Biosensor | Lignin–rhodamine polymer | Bioimaging and biological labelling | 127 |
| Chemical sensor | Lignin–porphyrin polymer | Detection of heavy metal ions | 129 |
| Chemical sensor | Lignin nanoparticles | Detection of formaldehyde | 133 |
| Chemical sensor | Lignin–silver nanoparticles (AgLNPs) | Detection of Hg2+ and Ni2+ | 135, 139 and 140 |
LCQDs are characterized by a low cytotoxicity and a good biocompatibility, but perhaps one of the most appealing features is their low cost-production from non-food resources and relative easy manufacturing process in comparison to carbon quantum dots (CQDs) based on precursors such as citric acid,93,94 gelatins95 and chitosan96,97 among others,98,99 which generally require more complex reaction conditions than LCQDs.
Chen et al. were the first to report the synthesis of LCQDs via hydrothermal processes using hydrogen peroxide (H2O2) as an oxidizing agent.86 However, these early LCQDs were irregular and weakly luminescent when used for biological labeling of tumorous cells (HeLa cell line). Later, LCQDs with potential theranostic applications were also developed by Rai et al. via microwave assisted depolymerization of sodium lignosulfonate followed by a carbonization process assisted by microwave irradiation.87 After the synthesis, the LCQDs were reduced by sodium borohydride (NaBH4) to lignin-based carbon quantum dots (r-LCQDs). The resulting r-LCQDs showed the ability to adsorb and release curcumin as a model drug compound at neutral pH (drug loading efficiency was 67.4% and 82% of curcumin released after 72 hours), and bio-imaging studies showed that r-LCQDs could penetrate into tumorous cells (A549 and SW480 cell lines) via simple diffusion trough the cell membrane. This work reflects the potential of these systems for therapeutic and cancer diagnosis applications. However, the attractiveness of this approach suffers from the use of NaBH4 that is an expensive, harmful and highly sensitive reducing agent. Therefore, it is important to note that greener reducing agents such as ascorbic acid or even lignin itself should be considered for future applications.29,100,101
Xue et al. reported the synthesis of hybrid LCQDs by one pot hydrothermal treatment of lignin with different molar ratios of citric acid and ethanediamine.88 The resulting LCQDs showed promising results for bio-imaging applications. However, the long reaction times and energy-consuming fabrication process make them less appealing from a synthetic point of view. Later, Shi et al. reported a facile synthesis of LCQDs by the introduction of secondary amine groups into lignin through Mannich and Michael addition reactions followed by carbonization, milling and filtration.89 The resulting LCQDs showed an excellent performance for the detection of iron ions (Fe3+) in a wide range of concentrations (1–100 nM) and lower detection limits (8 nM) compared to those of other CQDs based on foodstuff precursors as is the case of citric acid (0.3 μM)102 or red lentils (0.10 μM)103 among others104,105 (Fig. 4a and b). Moreover, fluorescence imaging demonstrated excellent image quality and biocompatibility of the LCQDs uptaken by biological cells.89
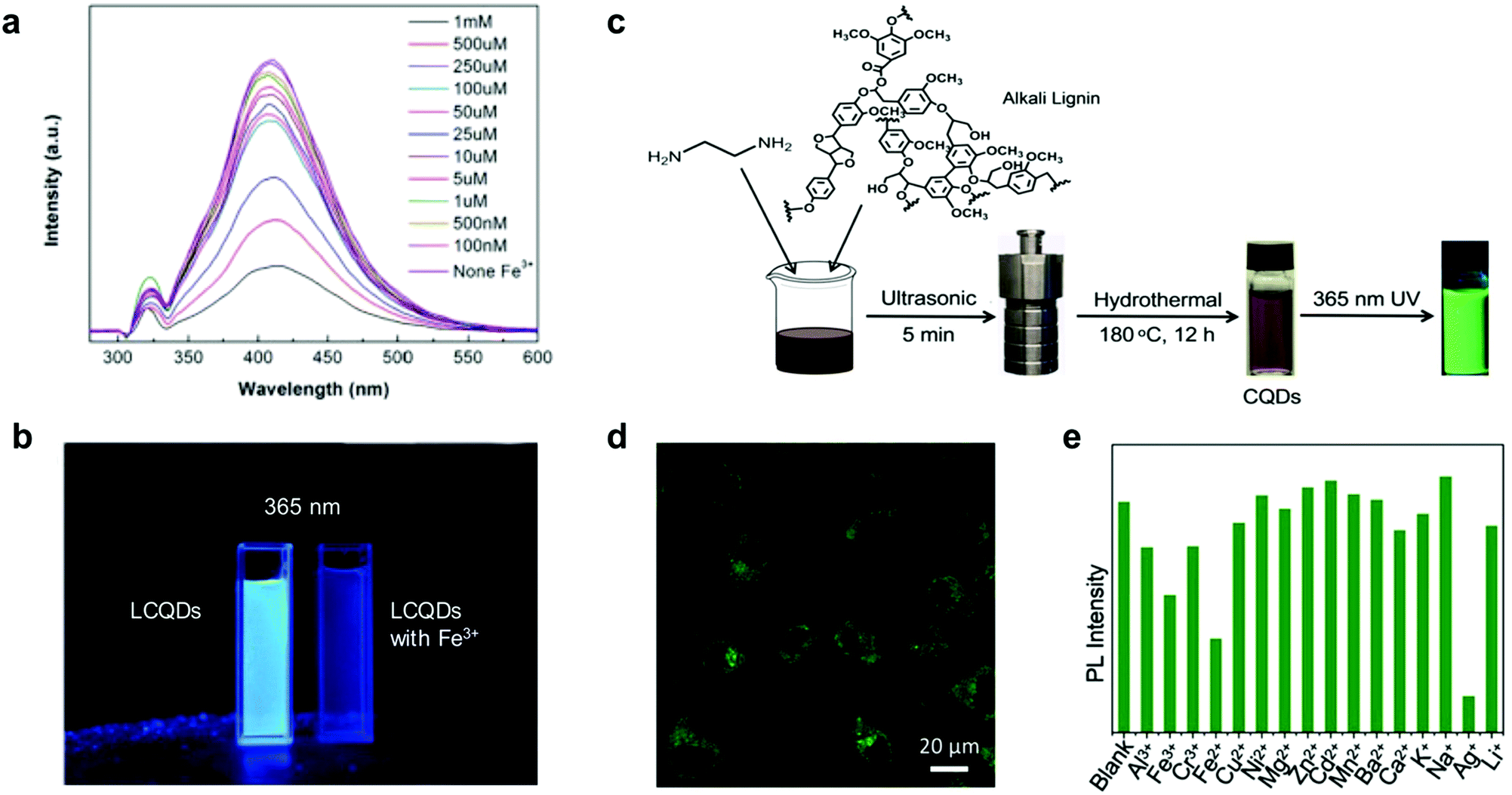 | ||
| Fig. 4 Use of LCQDs for sensing applications: (a) fluorescence change of LCQD with different aqueous Fe3+ concentrations. (b) LCQDs without and with Fe3+ under 365 nm UV light irradiation. Adapted with permission from ref. 89. Copyright © 2019, Elsevier Ltd. (c) Schematic representation of the one-pot synthesis of LCQDs; (d) confocal fluorescence image at 488 nm excitation wavelength of LCQDs in the presence of HeLa cells; (e) sensitivity and selectivity of LCQDs for a variety of different metal ions. Adapted with permission from ref. 91. Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
It is also important to point out that heteroatom doping with elements such as nitrogen or boron is a viable strategy towards increasing the quantum yields (QY) associated with high fluorescence response.88,89 For instance, Ding et al. prepared blue fluorescent LCQDs from kraft lignin via a two-step process involving sonication in nitric acid followed by hydrothermal treatment.90 However, the prepared LCQDs contained a large number of defects which inhibits their use in certain applications such as in bioimaging where broad absorption and high QY are required. More recently, Zhang et al. also reported the preparation and LCQDs with bright green fluorescence from kraft lignin by a simple one-pot method.91 In this case the prepared LCQDs revealed notable luminescence properties to be applied in cell imaging applications and also as ion detection sensors, with high sensitivity towards silver ion (Ag+) (Fig. 4c–e), comparable to those reported previously for other CQDs based on foodstuff precursors such as broccoli.106
In a different approach, Niu et al. reported the use of cellulolytic enzymatic lignin (CEL) for the synthesis of LCQDs via π–π electric interactions induced molecular aggregation using ethanol as solvent.43 This methodology enables the synthesis of LCQDs without hydrothermal carbonization by the aggregation induced emission (AIE) effect. The LCQDs exhibited one- and two-photon fluorescence emission (320 and 800 nm) and biocompatibility with HeLa cells. These properties of the LCQDs together with their absorption of near-infrared (NIR) light make these materials promising for bio-imaging applications.
Synthesis of lignin-based graphene quantum dots (LGQDs) was recently demonstrated by using recyclable ortho-aminobenzenesulfonic acid as solvent to prepare lignin nanoparticles (LNPs) followed by their transformation into LGQDs by hydrothermal carbonization.107 The LGQDs were applied as sensitive probes for the detection H2O2, a common oxidant in biological systems, due to their high UV absorbance and good biocompatibility properties. The LGQDs exhibited efficient fluorescence quenching in response H2O2 concentrations as low as 0.13 nM, representing a significant improvement compared to those reported previously for graphene QDs (0.15–3 μM).108,109 Such a high sensitivity towards H2O2 was rationalized later through a detailed density functional theory (DFT) study, which revealed that the sulfonyl group localized in the middle of LGQDs serves as the specific binding site towards H2O2.107
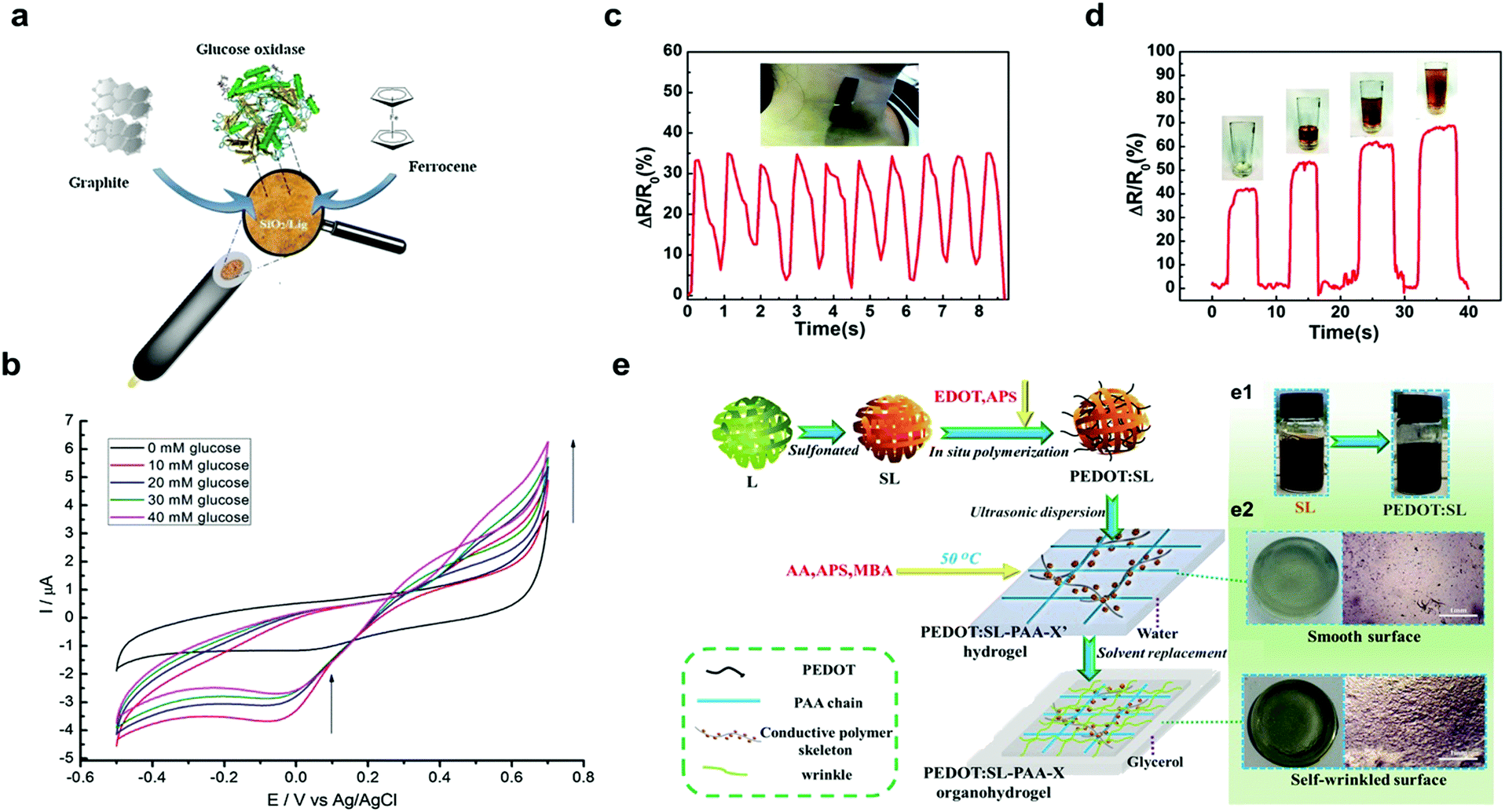 | ||
| Fig. 5 Lignin-based materials as biosensors: (a) schematic representation of preparation of SiO2–graphite–ferrocene–lignin hybrid electrode; (b) cyclic voltammetry signals of GOx–SiO2–lignin biosensor (from 10 mM to 40 mM) with an increase in oxidation current (from 0.4 V up to 0.7 V) with increasing glucose concentration. Adapted with permission from ref. 111. Copyright © 2018, Elsevier Ltd. Performance of lignin/polydimethylsiloxane composites as sensitive flexible pressure sensors: (c) measurement of arterial pulse; (d) measurement of the power required to pick up a cup. Adapted with permission from ref. 120. Copyright © 2018, Royal Society of Chemistry. Lignin-based hydrogel as a wearable sensor: (e) schematic synthesis of lignin–PEDOT based organohydrogel applied as a wearable sensor; (e1) color change of the solution during the synthesis of lignin–PEDOT based organohydrogel (e2) macro- and surface morphology of the lignin–PEDOT based organohydrogel. Adapted with permission from ref. 122. Copyright © 2019, Elsevier Ltd. | ||
Chemical modification of lignin has also proved to be an efficient approach to synthetize stimuli-responsive materials for sensing applications. In this sense, Xue et al. developed a ratiometric fluorescent pH-sensitive probe by grafting rhodamine B to sodium lignosulfonate.127 The resulting modified lignin showed a rapid response based on a conformational change in the Rhodamine moiety to pH variations (from 4.6 to 6.2 with a pKa of 5.35) which was then exploited for the detection of acidic organelles. The biological studies showed good biocompatibility of the resulting probe and the ability to differentiate normal cells (HL-7702) from cancer cells (SMMC-7721, HepG2). Although these pH-sensitive probes showed potential in sensor applications, the possibility to target the delivery of drugs to determined sites128 by chemical modification should be considered for potential nanotheranostic applications as has been previously demonstrated with LCQDs.87 On the same stream, kraft lignin have also been used as a template to graft porphyrins, and generate lignin–porphyrin polymers (Al-CTPP) with an extension of UV-vis absorbance and photoluminescence regions of lignin together with an enhancement of solvent compatibility of porphyrin in water-dominant environments at different pH.129 In addition, these lignin–porphyrin polymers showed linear correlations between the absorbance of Al-CTPP and concentration of several heavy metals ions (Mn2+, Ni2+, Co2+, Cu2+ and Zn2+) suggesting their potential as heavy metal sensor.
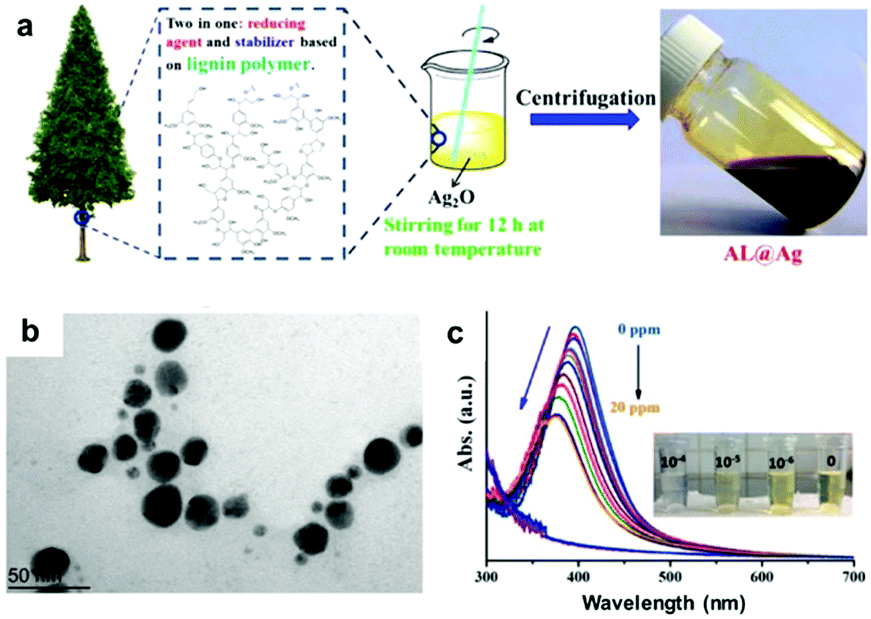 | ||
| Fig. 6 LNPs as sensor materials: (a) schematic preparation of alkali lignin-coated silver NPs (AL@Ag); (b) a TEM image of AL@Ag particles; (c) UV-vis spectra of AL@Ag particles as sensors for Hg2+ (from 0 to 20 ppm of Hg2+) with a decreasing UV-absorption with increasing Hg2+ concentrations, and photographs of the color change process demonstrating the detection Hg2+ at low concentration (10−4 M). Adapted with permission from ref. 135. Copyright © 2018, American Chemical Society. | ||
Despite the above examples on the use of LNPs for sensing applications, most of the lignin-based sensor materials presented herein have been designed using crude technical lignins.43,86,123,124 The main disadvantage of these crude lignins arises from their complex and irregular structure, which often comes in form of a structurally and physicochemical heterogeneous mixture of substances that can jeopardize the final applications. For instance, in the case of LCQDs the structural defects can hamper the fluorescence response90 and in lignin-based sensors the extent of structural regularity influences the reproducibility and quality of the final materials.124 Therefore, we urge to exploit the use of LNPs for the development of lignin-based sensors because these colloidal particles offer the possibility to control the shape, size, and surface topology depending the final application.132 Moreover, LNPs also hold a great potential to immobilize “target” biomolecules via non-covalent interactions and development of new lignin-based biosensor systems in the future.141
3.2 Biomedical applications
Lignin-based smart materials predesigned for biomedical applications have mainly been based on lignin-based polymers. The grafting polymer is selected to give responsiveness to temperature and pH, the two by far the most well-studied and understood responses in biological systems.142–144 For instance, thermoresponsive lignin-based materials usually incorporate in their structure polymers with lower critical solution temperature (LCST), which is the lower boundary for demixing via coil-globule transition mechanism.144 Below the LCST, the polymer chains exist as random coils due to mainly hydrogen bond interactions between polymers and water, forming one homogeneous mixed phase. Above the LCST, the phase separation occurs due to a collapse of the polymeric chains to globular structures. This temperature transition has been exploited in loading and delivery of active compounds or to change the morphology of lignin-based hydrogels. In the case of pH as target stimuli, lignin-based polymers either act as sacrificially solubilized polymers or incorporate polymers with pH-responsive compounds that have ionizable functional groups capable of donating or accepting protons upon environmental pH changes.142 This ionization process promotes an imbalance between the hydrophobic/hydrophilic ratio of the material, which disrupts the stability of the nanoassemblies and enhances the release of active compounds.| Carrier material | Loaded substance | Stimuli-response | EE [%] | Ref. |
|---|---|---|---|---|
| LNPs-lignin nanoparticles; LNCs-lignin nanocapsules; lig-His-lignin modified with histidine. | ||||
| LNPs | Budesonide | pH | 35 | 151 |
| LNCs | Coumarin-6 | pH | 84 | 153 |
| Lignin-g-PEG-PHIS NPs | BZL | pH | 50–57 | 150 |
| lig-His NPs | HCPT | pH | 15 | 154 |
| Succinylated lignin | 4-Acetomidophenol | pH | n.a. | 155 |
| Lignin-g-PMMA | IBU | pH | 84 | 156 |
| Lignin-g-PDEAEMA NPs | Decane | CO2/N2 gas | n.a. | 149 |
| Lignin-g-PNIPAM NPs | trans-RSV | T | n.a | 144 |
Chemical modification of lignin instead of the polymer grafting strategy has also been demonstrated to be a useful route towards pH-responsive LNPs for drug delivery applications.153–155 For instance, Chen et al. synthesized pH-responsive lignin-based nanocapsules with the ability to load and release hydrophobic model compounds.153 In their approach, lignosulfonate was modified with allyl groups in a classical esterification reaction followed by the preparation of the nanocapsules via an interfacial miniemulsion reaction, in which the modified lignin reacted with an acid-labile thiol-based crosslinking agent at the interface of miniemulsion droplets to form the nanocapsules through a thiol–ene coupling reaction (Fig. 7a and b). The resulting capsules exhibited a particle size ranging from 100 to 400 nm and their potential application as nanocarriers was evaluated using coumarin-6 as a hydrophobic model compound. The entrapment efficiency (EE) was determined to be 84%, demonstrating a high loading concentration of this system (0.713 mmol g−1 based on the weight of the modified lignosulfonate). In addition, release studies from coumarin-6 loaded nanocapsules under neutral (pH = 7.4) and slightly acidic (pH = 4.0) conditions revealed a higher release rate under acidic (60%) compared to neutral (40%) conditions after 48 hours. The difference in the release rates suggests hydrolytic response of the acid-labile β-thiopropionate cross-linkages, which makes these lignin-based nanocapsules promising nanocarriers for drug delivery systems. Later, Zhao et al. reported the synthesis of lignin-histidine (L-His) conjugates through the amination reaction between lignin and histidine.154 After the conjugation, L-His showed the ability to self-assemble in water to form homogenous nanoparticles with an average diameter of 30–40 nm, encapsulate hydroxycamptothecin (HCPT) as a drug model with moderate EE values (40%) and triggered release in different media. In addition, carboxylation of lignin has also proved to be an efficient approach for the fabrication of pH-responsive LNPs for controlled release.155 Figueiredo et al. reported the synthesis of complex pH-responsive LNPs for drug delivery and biomedical applications.150 In a first stage, kraft lignin was carboxylated in order to obtain carboxylated lignin nanoparticles (CLNPs) with an increased amount of carboxyl groups as reactive sites for grafting PEG, polyhistidine (PHIS) and a cell-penetrating peptide via EDC/NHS peptide coupling chemistry. The resulting nanoparticles showed an increased circulation in the bloodstream and also pH-responsive behavior. Cytotoxicity studies revealed a high biocompatibility with > 80% of cell viability and the ability to load (with an EE corresponding to 50%) and release benzazulene (BZL) as a model drug compound under physiological (pH = 7.4) and acidic (pH = 5.5) conditions with higher release rates in acidic media. Furthermore, in vivo studies also proved the high selectivity of this system through a lower release of BZL in physiological environment, causing less toxic effects to the normal cells than to the targeted tumorous cells.
 | ||
Fig. 7 Lignin-based smart materials for loading and release applications: (a) appearance of a miniemulsion after 3 min of sonication with 0.76, 0, 0.38, and 0 wt% of sodium dodecyl sulfate (SDS) as surfactant; (b) lignin nanocapsules with broken shells produced during the emulsification process. Adapted with permission from ref. 153. Copyright © 2016, American Chemical Society. (c) Cycle of N2/CO2-triggered emulsified/demulsified lignin-g-PEAEMA Pickering emulsion; (d) optical microscope and TEM images of a Pickering emulsion prepared using lignin-g-PEAEMA with an aqueous/oil volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Adapted with permission from ref. 149. Copyright © 2014, Royal Society of Chemistry. (e) Optical micrographs of droplets from lignin-g-PNIPAM NPs stabilized emulsions (0.1 wt%) at 25 °C and 45 °C; (f) cumulative thermal-controlled release of trans-RSV. Adapted with permission from ref. 144. Copyright © 2019, American Chemical Society. :1. Adapted with permission from ref. 149. Copyright © 2014, Royal Society of Chemistry. (e) Optical micrographs of droplets from lignin-g-PNIPAM NPs stabilized emulsions (0.1 wt%) at 25 °C and 45 °C; (f) cumulative thermal-controlled release of trans-RSV. Adapted with permission from ref. 144. Copyright © 2019, American Chemical Society. | ||
pH-Responsive lignin-based micellar nanoassemblies with the ability to load ibuprofen (IBU) at high EE (84%), have also been recently reported by grafting methacrylic acid (MMA) to lignin through a free radical polymerization process.156 The resulting lignin-g-PMMA was able to self-assemble into micelles (diameter approximately 100 nm) and control the release of IBU. In vitro release experiments showed only 16% release of IBU in a simulated gastric fluid (pH 1.5), while in a neutral simulated intestinal fluid (pH 7.4) an accumulated release of 81% was determined. This tunable release of IBU can be rationalized by the ionization of acidic groups at low pH, which protects the corona of the micellar nanoassemblies and avoids the release of the active compound at low range of pH values. In vivo studies furthermore demonstrated a certain inhibitory effect towards proliferation of colon cancer cells (NIH-3T3), while presenting low cytotoxicity to normal cells. Similar pH-responsive release processes have been observed in alkali lignins by the of ionization of carboxylic groups.151,155 However, it is important to mention that this release behavior also depends on the solubility properties of the loaded compounds as in the case of coumarin-6 that was released more efficiently under mildly acidic conditions compared to neutral pH.153
Qian et al. also developed another type of smart LNPs by grafting dimethylaminoethyl acrylate (DEAEMA) monomer into a kraft lignin core by ATRP.149 The resulting lignin-g-PDEAEMA copolymer could be easily dispersed in water to form LNPs upon CO2 gas bubbling, and to initiate aggregation and eventual precipitation by N2 gas bubbling. This CO2/N2 switching ability is conferred by the presence of DEAEMA based on a reversible protonation of amine groups catalyzed by the acidic CO2, and is tailored by the graft density and chain length of the DEAEMA units. These modified LNPs showed a promising potential as surfactants for Pickering emulsions. Stable Pickering emulsion were prepared using decane as an oil phase with only 1 wt% of LNPs; the emulsions remained stable for more than one month without any phase separation and exhibited a gas triggered de-emulsification/re-emulsification behavior. CO2/N2 bubbling cycles demonstrated that bubbling with CO2 allowed a complete emulsion breaking with the consequent phase separation, while N2 bubbling resulted in re-emulsification of the mixture (Fig. 7c and d). The authors claimed that such smart lignin surfactants could be of great potential as drug delivery systems, but no drug release and cytotoxicity studies were demonstrated. More recently, Dai et al. reported the synthesis of thermoresponsive lignin nanoparticles by grafting NIPAM into lignin via ATRP and self-assembly in water, successively.144 These copolymer nanoparticles were used as smart surfactants to stabilize trans-resveratrol (trans-RSV)-containing palm oil in water Pickering emulsion droplets. Combining the thermoresponsive and UV resistance properties in lignin-g-PNIPAM NPs, this stimuli-responsive Pickering emulsion system showed an improved performance on the controlled release and light stability of trans-RSV in comparison to other systems based on polymeric nanoparticles157,158 (Fig. 7e and f). Additionally, these Pickering emulsions showed remarkably good biocompatibility (around 100% cell viability of A549 cells at a wide range of concentrations), improved solubility and antiradical efficiency as compared to free trans-RSV in water. The inherent combination of lignin properties (UV-protection) together with the thermoresponsive behavior of NIPAM could be of renewed interest for drug delivery systems for the storage of light-sensitive and poorly water-soluble drugs. In the same vein, Peng et al. also harnessed the UV-shielding properties of lignin to fabricate avermectin-containing emulsions using microspheres with tunable surface charge and composed of sodium lignosulfonate and cetyltrimethylammonium bromide (CTAB).159 The resulting emulsions showed sustained release behavior (ranging from 56–80% in 60 h), and an improved light-storage stability of 2.18–2.96 times higher in comparison to commercial avermectin emulsifiers.
As has been discussed above, lignin-based smart material for drug loading and release applications have mainly been based on the development of lignin-based polymers with the ability to form pH-responsive LNPs.150,153–155 Indeed, pH is without any doubt one of the most attractive stimuli since different parts of the body and cellular compartments present some variations in pH values which could be exploited to trigger selective release of drugs.160 However, there are also many other stimuli in biological systems which can be exploited as is the case of redox potential,161 endogenous gases162 or certain enzymes,163 which have been already integrated and employed in classical stimuli-responsive polymers,147 while literature is lacking such reports on lignin-based smart materials. Therefore, we urge to expand the development lignin-based polymeric materials integrating other attractive stimuli or even the combination of them in order to generate materials with synergistic simultaneous action as in the case of their predecessor polymeric materials.164–166 Another relevant issue is that the systems described above are based on covalent functionalization of lignin with the final aim to introduce stimuli-responsive behavior and control over the release properties. However, non-covalent functionalization is also an appealing approach in terms of synthetic preparation and potential reversibility of functionalization, and therefore should also be considered as an efficient alternative for the design of stimuli-responsive materials. For instance, LNPs with negative surface charge have been used to adsorb cationic polymers such as poly(diallyldimethylammonium chloride) (PDADMAC),167,168 chitosan169 and water-soluble cationized lignin.170 The resulting LNPs with a cationic surface charge have been demonstrated as efficient supports for enzyme immobilization and biocatalysis,168,171,172 but could be also considered as a promising platform for the future development of smart confined biocapsules not only for drug release applications but also for the design or more complex materials such as bio-inspired nanomotors, already developed with conventional polymeric materials.173,174
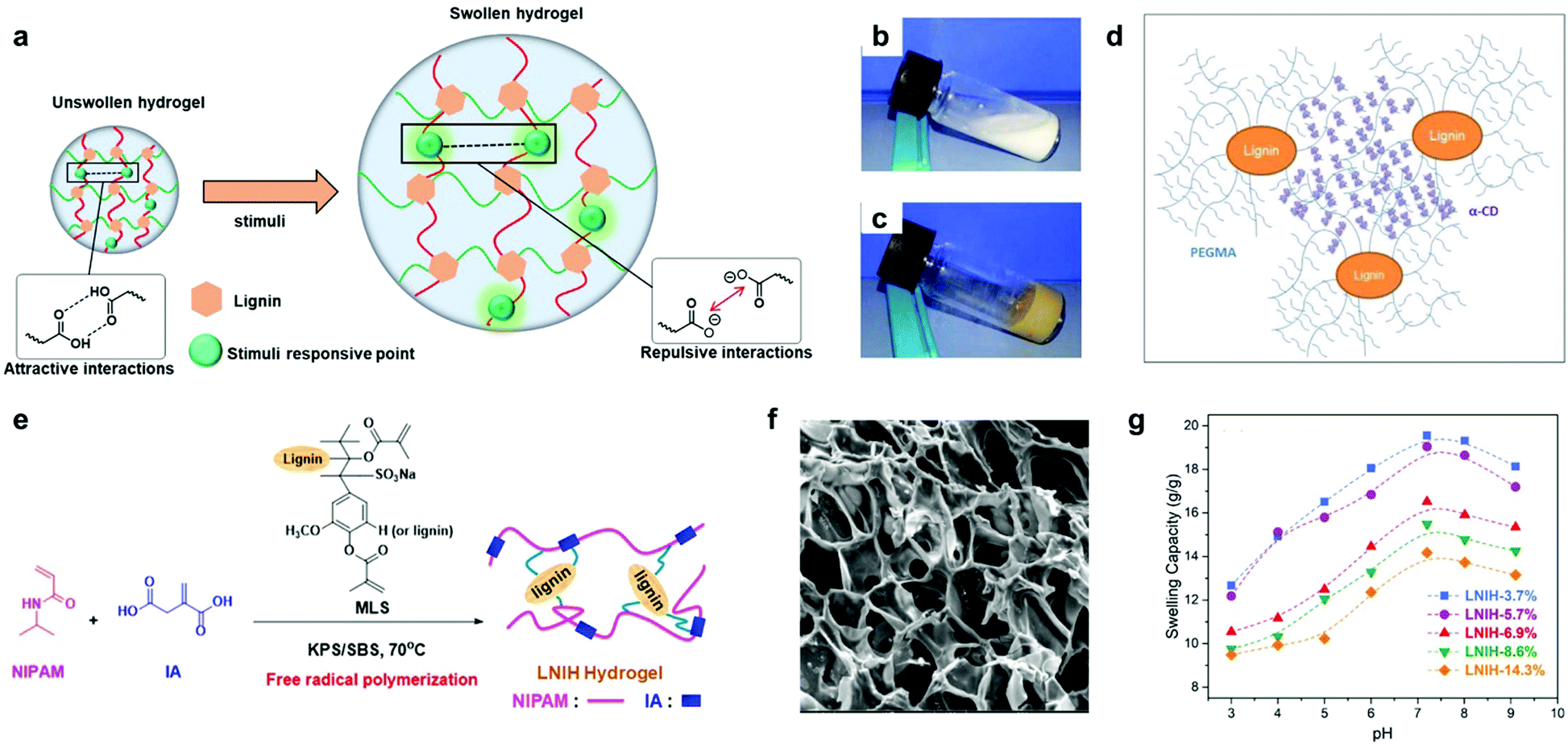 | ||
| Fig. 8 Lignin-based smart hydrogels: (a) depiction of swelling behavior of lignin-based smart hydrogels upon the application of a specific stimuli (pH-dependent lignin-based hydrogels is shown as example, where the stimuli applied corresponds to pH variation). (b) photographs of the inclusion complex suspension of PEG 10/10 (10% PEGMA + 10% α-cyclodextrin); (c) digital images of lignin-based hydrogel (2% lignin-g-PEGMA + 10% α-CD); (d) schematic representation for the proposed structure of a lignin-based supramolecular hydrogel by inclusion complexation between lignin-g-PEGMA and α-CD. Adapted with permission from ref. 176. Copyright © 2015, American Chemical Society. (e) Synthetic approach for the lignin-based hydrogels via free radical polymerization. Note that the lignin structural model MLS should be considered only as an illustration. (f) SEM image of lignin-g-PNIPAM hydrogel containing lignin in a 14.3% wt; (g) swelling capacities of lignin-g-PNIPAM with increasing content of lignin (from 3.7 wt% to 14.3 wt%) at different pH values at 25 °C. Adapted with permission from ref. 142. Copyright © 2018, American Chemical Society. | ||
More recently, temperature- and pH-responsive lignin-based hydrogels have also been synthetized via free radical polymerization.142,143 Jin et al. reported the preparation of a lignin-based hydrogel by the copolymerization of NIPAM, itaconic acid and methacrylate functionalized sodium lignosulfonate.142 The prepared hydrogels showed a reduction in the swelling capacity (from 31.6 to 19.1 g g−1) with the increase of lignin content (from 3.7% to 14.3%), revealing a lignin-dependent water absorption. In addition, phase transition temperature values around the body temperature together with a broad pH responsive range (from pH 3 to 9) make them interesting candidates for drug release applications (Fig. 8e–g). However, the poor control over the material structure generally resulting from free radical polymerization methodologies and the difficult reproducibility in the synthesis could be critical barriers to overcome when scaling up the production.
Although frequently neglected, another important consideration about these lignin nanomaterials is their biodegradability. While many of the reports focus strictly their attention on the cytotoxicity and biocompatibility studies, the biodegradability of these materials remains largely unexplored. It is well known that lignin can be partially decomposed by enzymes,16,184 but most of these smart materials described above are based on chemical modification of lignin, which probably disrupts the degradability process. For instance, cross-linked lignin materials are likely drastically less biodegradable and the absence of phenolic hydroxyl groups hinders accessibility of these primary sites in enzymatic degradation processes. Therefore, more data should be collected regarding the biodegradability of lignin-based smart materials and the nature of the degradation products.
3.3 Shape-programmable materials
Shape memory materials are capable of fixing temporally “programmed” shapes and returning to the original shape by the application of external stimuli such as temperature,185 light,186 electricity,187 moisture188 or magnetism.189 These materials are widespread in the field of polymer science due to their attractive properties for biomedical applications,85 such as polymeric-based actuators which have been used as artificial muscles for the fabrication of blood vessel connections or microvalves to regulate the urinary incontinence, among others.190–192 Lignin by itself displays a high glass transition temperature (Tg) (from 90 °C to 145 °C, depending on the source and type of isolation process), owing to its rigid aromatic backbone and strong intra and intermolecular interactions (mainly hydrogen bonding and π–π stacking interactions). When lignin is modified for use as a macromonomer, this inherent nature usually leads to materials with thermosetting characteristics, which could be further designed to act as shape memory materials for smart applications. In this context, Xu et al. reported the synthesis of lignin-based thermosets exhibiting tunable mechanical properties and shape memory behaviour.193 These thermosets were synthetized in a simple approach using crude kraft lignin without any chemical modification, citric acid as a cross-linking agent and PEG as a reactive diluent and soft segment. PEG facilitated the cross-linking reaction and balanced the final mechanical properties by increasing the dispersion of all the components, but also provided materials with excellent shape memory characteristics such as 99% recovery rate and 95% fixity ratio, which indicate how well the shape is recovered and the ability to fix the mechanical deformation, respectively. These values are comparable to those reported for other thermoset composites based on epoxy resins or polybenzoxazines.194 Although this approach is attractive in terms of synthetic preparation and low cost-production, there still remains a major concern related to the difficult recyclability of thermosetting materials, since most of them are disposed of via incineration or landfilling.To overcome the rigidity issue of lignin, the incorporation of vitrimer chemistry into the preparation of thermosets has gained attention.195,196 Incorporation of dynamic covalent linkages that can be stimulated by heat or UV light into cross-linked structures results in rearrangements in the material topology, imparting improved malleability and recyclability (Fig. 9a).197,198 In this stream, Zhang et al. reported the first example of lignin-based vitrimer material with shape memory behavior by the reaction of ozonated lignin with sebacic acid derived epoxy in the presence of a zinc catalyst (Fig. 9b).199 Zn2+ ions catalyzed transesterification reactions in the cross-linked network via associative exchange pathway (Fig. 9a), which resulted in a thermoset material with shape-memory, self-healing and malleability properties (Fig. 9c). Moreover, these lignin-based vitrimers revealed interesting features as adhesives when adhered to aluminum sheets, with a lap-shear strength value of 6.3 MPa comparable to epoxy-based adhesives such as the commercial epoxy glue with a lap-shear strength of 8 MPa,200 or bisphenol A -based epoxy adhesives with a lap-shear strength values of 4–6 MPa201 (Fig. 9d).
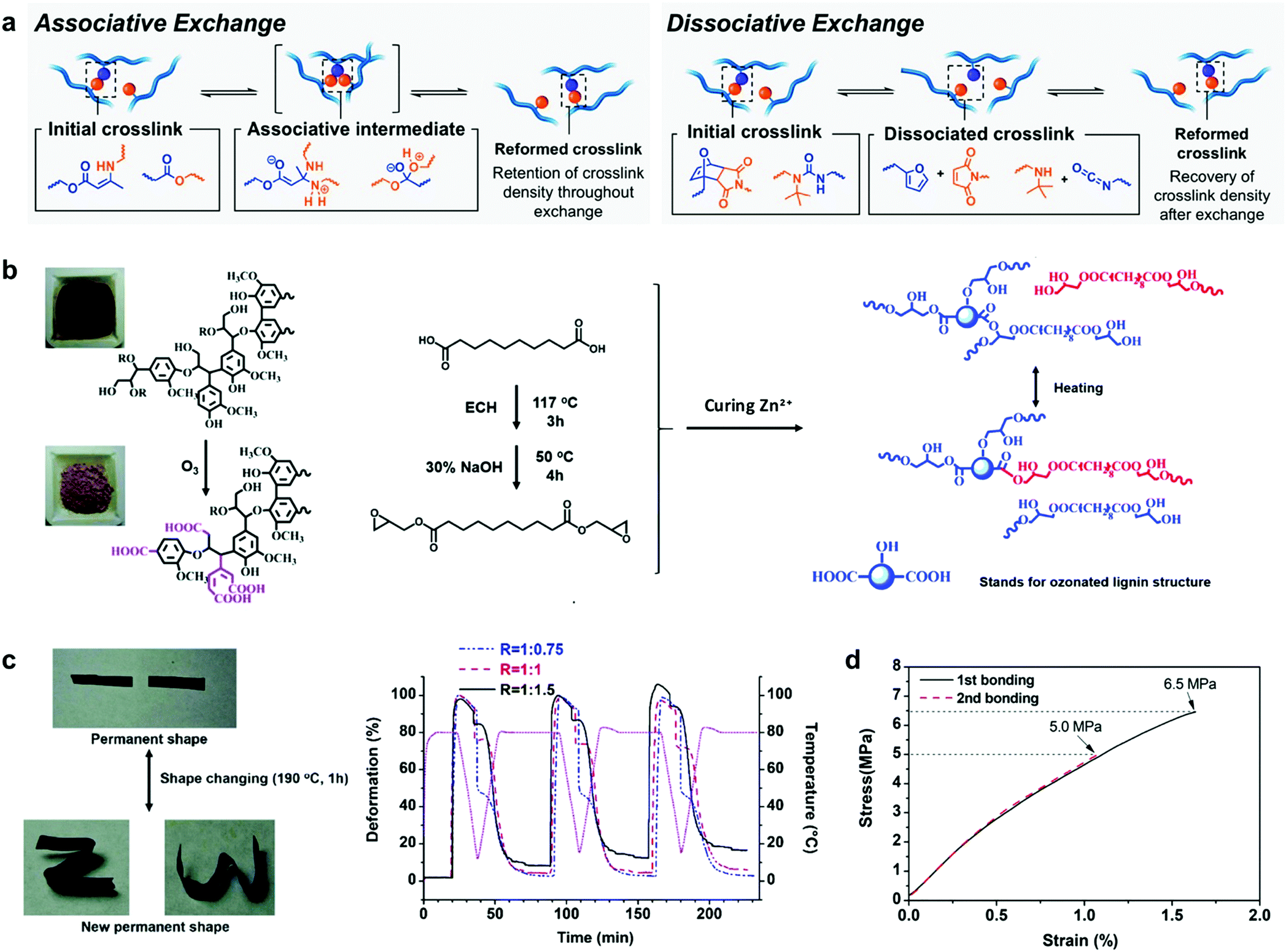 | ||
| Fig. 9 Lignin-based shape-programmable materials: (a) depiction of associative and dissociative bond exchange pathways for covalent adaptable networks by vitrimer chemistry. Adapted with permission from ref. 195. Copyright © 2019, American Chemical Society. Lignin-based vitrimer material with shape-memory behavior: (b) schematic diagram of the preparation of lignin-based vitrimer and its reconstruction via transesterification reactions; (c) shape changing and consecutive shape memory cycles for different compositions of the lignin-based vitrimer material; (d) the lap shear test of lignin-based vitrimer as an adhesive on coarsened aluminum sheets with different bonding times. Adapted with permission from ref. 199. Copyright © 2018, Royal Society of Chemistry. | ||
However, perhaps one of the main disadvantages of this approach relies in the narrow scope of functional groups presents in lignin, mainly limited to hydroxyl and phenolic groups. This fact could restrict the introduction of a wide palette of interesting dynamic covalent linkages based for instance on amino functional groups and urethane chemistry.198,202 Chemical modification of lignin (Fig. 3a) could of course be a possibility to increase the scope of dynamic covalent linkages, albeit at a cost of an additional synthetic step.
Pioneering works in the development of these lignin-based materials were reported by McDonald's group, who synthesized thermally-stimulable shape memory elastomers from lignin and glycerol–adipic acid based hyperbranched prepolymers.203 In the first step, hyperbranched prepolymers were synthetized by melt polycondensation reaction of glycerol, adipic acid and the addition of di- and tri-functional amines as cross-linking points. After that, the prepolymers were reacted with soda lignin via bulk polycondensation reaction to afford the corresponding shape memory elastomers. The obtained elastomers presented tunable Tg as well as shape transition temperature values ranging from −10 °C to 40 °C by varying the amount of the amine additives. Dynamic mechanical analysis (DMA) via a stress controlled cyclic thermomechanical test revealed that the materials exhibited a dual shape memory behavior with 94% recovery rate and 99% fixity rate. Later, the same group reported the preparation of lignin-based elastomeric materials with triple shape memory by the polymerization of soda lignin with a new hyperbranched poly(ester-amine-amide) containing amine cores and amide linkages.204 The presence of a broadened Tg in these materials revealed a mixed phase by the coexistence of two networks within lignin (polyester-amine and polyester-amine-amide) comprising covalent and physical cross-linked points, which were responsible for the triple shape memory behavior. As in the previous case, the shape memory behavior of these materials was evaluated by a stress-controlled cyclic thermomechanical test, which confirmed a reasonable triple shape memory behavior (fixity rate reached 41% and 92%, while recovery rates reached 40% and 86%, respectively for the 1st and 2nd shape recovery stage). These results were explained on the ground of the combined effect of increased covalent cross-linking and dissociation of self-complementary physical hydrogen bonding present in the network. Interestingly, it was also found that the first recovery stage was associated to the lignin–poly(ester-amine) network and the second one to the lignin(ester-amide) segments, which indicates the important role of physical cross-linking to obtain these types of materials. Although these were the first examples of lignin-based elastomeric shape memory materials with multiple shape memory behavior, the properties are far behind those of conventional polymeric materials (fixing and recovery rate up to 95%).209,210 In this sense, increasing the polymer compatibility and dispersibility of lignin seems to be crucial aspects, in addition to the use of sacrificial bonds capable to dissipate energy, eliminate stress, and promote the orientation of chain segments, which are all appealing strategies towards the development of future materials.208,211
More recently, Liu et al. reported the synthesis of lignin-cross-linked polycrapolactone (PCL) copolymers as elastomeric materials with shape memory behavior using lignin as a core segment for the cross-linking.205 This approach was based on a thiol–ene coupling reaction between a thiol terminated four-arm PCL with an alkene-modified kraft lignin. The prepared lignin-copolymers revealed tunable melting temperatures (Tm) by the variation of lignin content (from 10 to 40 wt%). Moreover, the authors claimed that the shape memory process is efficient and the recovery is immediate which could make these materials interesting candidates for biomedical applications, in which self-expansion times of less than 60 seconds are required.212 However, the reported phase transition temperature was found to be 80 °C, which is unfit for biomedical applications, in which activating temperatures are instead governed by the thermoregulations of the human body.213
Lignin-based thermoplastic elastomers (Lig-TPEs) with shape memory behavior combine both thermoplastic and elastomeric properties, together with a stimuli responsive behavior.25,41 In this context, Nguyen et al. recently reported the preparation of Lig-TPEs based on acrylonitrile–butadiene–lignin composites (ABL) with a high lignin content (40–60 wt %), via cross-linking of lignin with an acrylonitrile-butadiene rubber (NBR) by a thermal compounding process.206 The resulting ABL materials showed excellent shape memory behavior as a consequence of chemical cross-links in the inherent structure of NBR and the intrinsic networked structures of lignin, which complemented physical cross-links formed by hydrogen bonds within lignin and nitrile rubber molecules (Fig. 10a and b). In addition, the authors also reported the preparation of a programmable and switchable conducting material by depositing silver (Ag) nanoparticles on the surface of an ABL specimen. After the coating process, this new material could be shape-programmed to respond to an external stress at ambient temperature for multiple times, revealing an increase in the resistance from 0.46 kΩ to 2.86 kΩ during the process (Fig. 10d). Moreover, after the applied external force was switched off, the shape-programmed sample recovered its original shape and resistance (0.47 kΩ) which was associated to a reversible breaking of particle percolation (Fig. 10c and e). These new shape-programmable conductive materials with the ability to sense changes in the resistance induced by changes in applied stress could be of potential interest for the development of new motion sensors such as human motion tracking sensors.214 However, further work is required to engineer the Tg values for these materials (ranging from −2.5 to 11 °C) closer to ambient temperatures to broaden their stress–strain sensing window for potential applications. In this context, improvement of thermomechanical properties of these ABL materials (increasing of 18 °C in Tg values and more than 230% increasing in the storage modulus (E′)) were later reported by the same group, applying a thermal annealing process, which enhanced the crosslink density by the generation of radicals from lignin.207 It is important to mention that the obtained materials still maintained good shape memory behavior and therefore this approach could be considered of interest to tune the thermomechanical properties of these Lig-TPEs and make them more attractive for a wide range of applications where high Tg values are required.
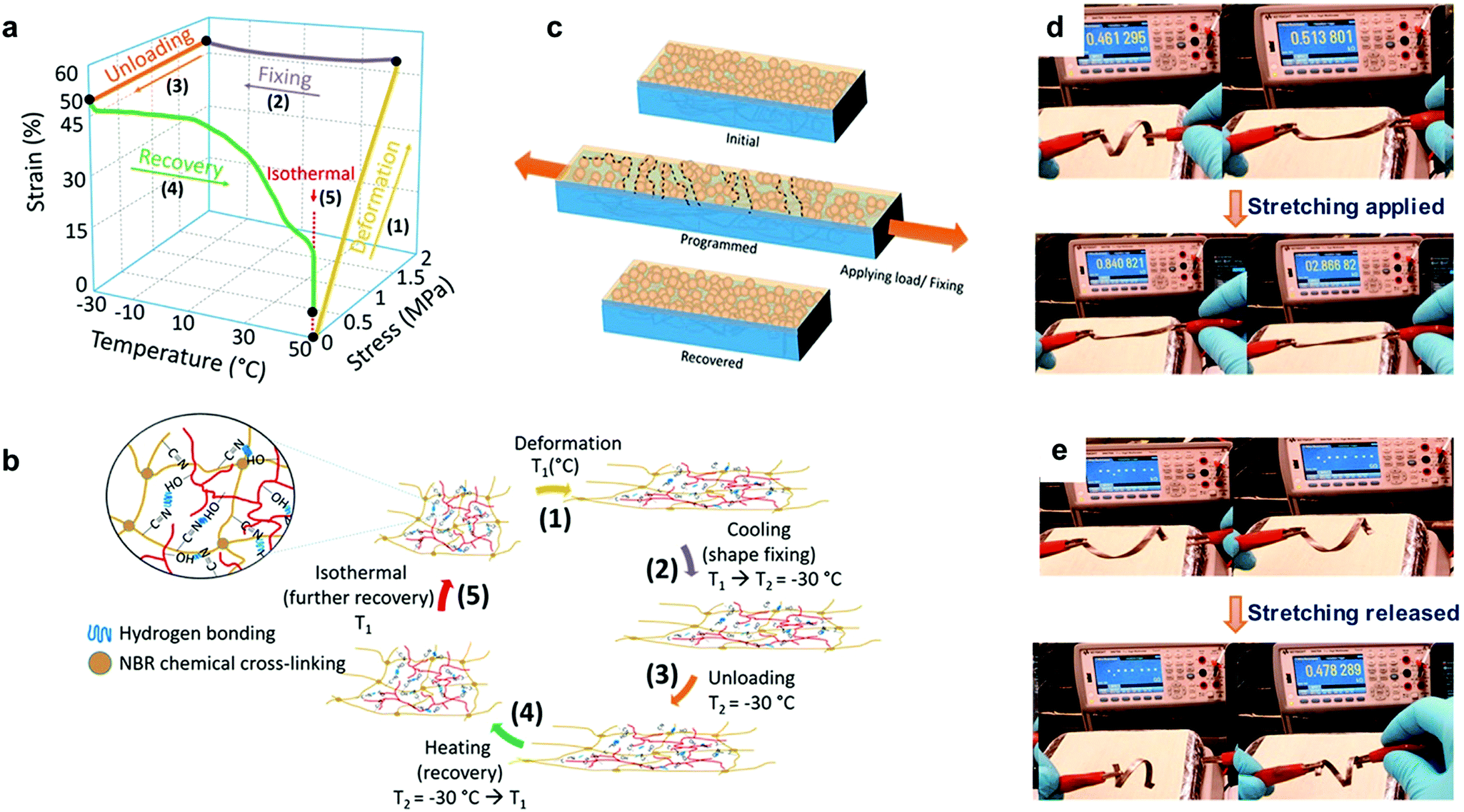 | ||
Fig. 10 Lig-TPEs as shape programmable material: (a) three-dimensional graph of one cycle of deformation, fixing and recovery in ABL material; (b) the corresponding programmed shape recovery of ABL networks with a magnified view showing a network structure of a nitrile–butadiene elastomer (NBR) and lignin in the presence of hydrogen bonds formed by –OH and –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups; (c) principle of switchable and programmable electrical conductivity of a silver nanoparticle layer assembled on a shape-memory substrate; (d and e) initial, after fixing/shape programming, and after restoring shape and resistance of the shape-programmed electrically conducting material. Adapted with permission from ref. 206. Copyright © 2018, American Chemical Society. N groups; (c) principle of switchable and programmable electrical conductivity of a silver nanoparticle layer assembled on a shape-memory substrate; (d and e) initial, after fixing/shape programming, and after restoring shape and resistance of the shape-programmed electrically conducting material. Adapted with permission from ref. 206. Copyright © 2018, American Chemical Society. | ||
Another approach to improve the mechanical properties and shape memory behavior of Lig-TPEs was reported by Huang et al. who used sacrificial bonds based on the presence of Zn-mediated coordination bonds in the interface of lignin and a non-polar elastomeric polyolefin (Fig. 11a and b).208 The presence of these sacrificial bonds was found to promote the dispersion of lignin with a particle size around 200 nm, but also to improve the interfacial interaction of lignin and polyolefin elastomeric matrix, which results in a more accessible orientation of the chain segments during the stretching with the consequent improvement on shape-memory characteristics of the material (fixing rate and recovery rate values up to 87%). Indeed, the synergistic coordination effect of lignin promoted a higher energy dissipation together with an improvement of toughness and strength of these Lig-TPEs, rendering them comparable to commercial TPEs such as polyolefin blends-based materials in which the plastic phase comes from petroleum-derived sources.215,216 Overall, harnessing intrinsic material properties of lignin holds many opportunities for future development of high-performance and cost-effective TPEs.
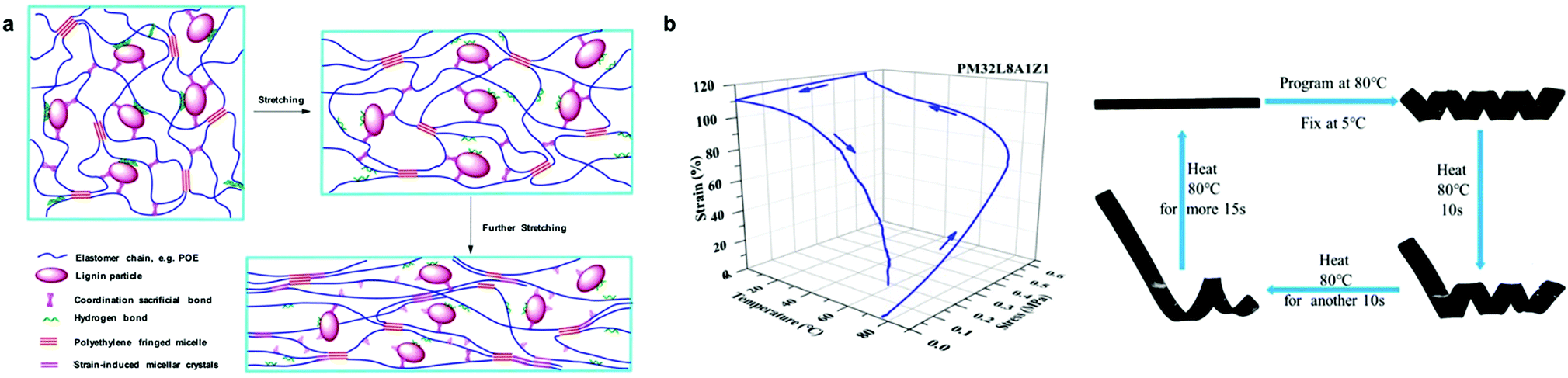 | ||
| Fig. 11 Lig-TPEs as shape programmable material incorporating sacrificial bonds: (a) schematic representation of deformation mechanism of Lig-TPEs; (b) stress-controlled program and photographs of the shape memory performance of Lig-TPEs. Adapted with permission from ref. 208. Copyright © 2019, American Chemical Society. | ||
The development of lignin-based materials with “programmable” shape memory behavior is a fascinating field with multiple potential applications as has been described above. However, it is also important to note that all of the materials presented above are capable of responding to temperature as the only stimulus. In that context, light-driven lignin-based shape memory materials appear as an appealing option in the horizon because light stimuli offer flexibility in terms of spatiotemporal control. One could envision that the development of these materials will logically require the use of light-responsive monomers (e.g. azobenzenes) able to produce changes in response to light via isomerization processes among others.217,218 However, it must be noted that some properties that make lignin attractive for other applications such as a high UV-absorbance,144,156 can play a negative role by the partial or total inhibition of these light-induced processes and affect the final performance of the lignin-based actuator. Therefore, the design of lignin-based smart materials with programmable shape memory behavior need not only focus on the “conception” of the stimuli integration, but also consider the potential interaction of lignin with the applied stimuli and the expected response of the material.
Last but not least, the development of materials with the ability to respond to multiple stimuli, and mimic artificial muscles capable of converting energy from external stimuli to mechanical forces are intriguing challenges that could be addressed with lignin-based shape-programmable materials in the near future. First steps to address these issues have already been taken, with the rational design of lignin-based hydrogel actuators sensitive to pH. The performance of these materials is based on macroscopic changes such as swelling and shrinking upon pH changes. Recently, Dai et al. reported a simple approach consisting of a one-step crosslinking reaction of industrial kraft lignin with poly(ethylene glycol)diglycidyl ether acting as a crosslinker under basic conditions.219 The resulting lignin-based hydrogel showed relatively fast (1 min) mechanical actuation by transition between softening/enhancement and straight/bending shapes by alternately immersing the hydrogel in 0.1 M HCl and 0.1 M KOH. Additionally, an intelligent hook and flow control valve based on this system was developed to control the filtration of solutions demonstrating a broader potential of this material. This approach illustrates that a good rational design of low cost lignin-based smart material can also open new avenues for the development of lignin-based actuators.
4. Perspective for future development
Lignin-based smart materials have emerged as worthy options for advanced biomaterials because of their intrinsic functional properties and green carbon footprint. Many of the examples discussed in this review demonstrate that lignin-based smart materials stand comparison to conventional stimuli-responsive polymers in sensors, biomedical applications, and shape memory materials. It is thus well rationalized to foresee that lignin-based smart materials will continue to see a rapid expansion towards new materials and applications. It is also important to note that in many of the examples described in this review the stimuli-responsive capabilities are not provided directly by the nature of lignin (e.g. lignin-based polymers). This fact can induce a thinking that in some cases the role of lignin is merely secondary and could be eventually easily replaced. However, it is unfair to judge the contribution of lignin in these materials based only on this angle. In most of the cases, a global vision of these systems demonstrated that a rational combination of lignin properties with stimuli-responsive materials can offer the possibility to fabricate systems for a more advanced performance, as is the case of lignin-based smart polymeric materials for drug delivery and light stabilization. We also envision that in a near future the development of all-lignin-based smart materials will be a reality by exploiting the inherent properties of lignin in their different shapes and sizes. For instance, LNPs hold the potential to enable reversible aggregation processes in different solution media which could be used to design new sensors based on aggregation induced emission (AIE) effect.In fact, there are many prospective materials that can be prepared from lignin precursors such as (nano)materials programmed for degradation, bio-mimicking materials, multi-stimuli responsive systems, and other engineered stimuli-responsive materials (Fig. 12). Future challenges for lignin-based smart materials include gaining a better understanding of structure, isolation, and predictable batch to batch properties of lignin, transformation to “standardized” molecular and nano/micro building blocks, and structure–function relationship in applications. It will also be important to demonstrate scale-up and proof of concept of lignin-based materials to attract industrial activity in this new material domain.
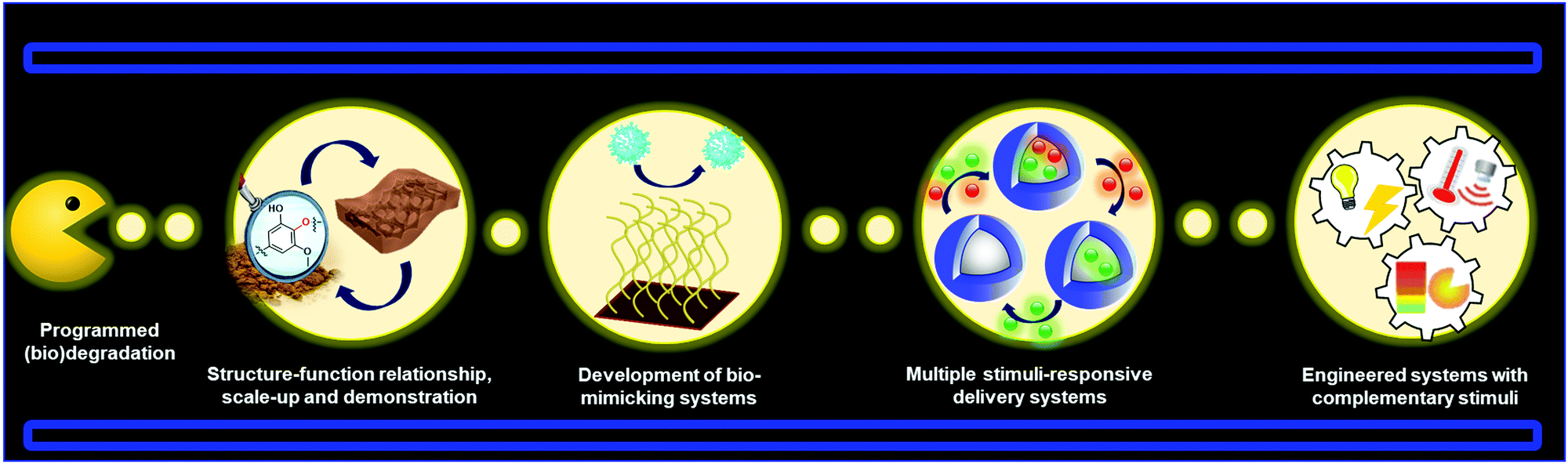 | ||
| Fig. 12 Future challenges and prospective new frontiers for lignin-based materials. | ||
Many prospective applications are linked to the development of lignin-based materials with the ability to interact or mimic biological systems such as selective membranes. These materials are typically composed of biomolecules such as proteins linked to other components that lead to changes in the macroscopic conformation. Many bio-mimicking systems developed so far involve the use of synthetic polymeric nanoparticles,220 which could instead be formed from lignin.221 For drug delivery systems it will be important to synthesize materials carrying more than one active substance with potential for site-specific and controlled sequential or simultaneous release. This also includes the possibility to fabricate LNPs with programmable shape memory behavior, which could lead to the preparation of multicompartment capsules (MCCs) mimicking biological cells,222,223 for instance through phase-separation processes with the inherent possibility to encapsulate several active compounds in the different compartments. Last but not least, as has been discussed in this review, there is potential for future work to address the introduction of multiple and external stimuli in an “intelligent” and predictable manner to generate a new generation of advanced materials. These materials will consist of engineered signaling chains that allow for switching “on” or “off” the corresponding lignin-based device, after the application of a programmable series of stimuli, including potential cascade processes already presented in polymeric systems.224–226 In summary, the combination of the lucrative properties of lignin nano- and hybrid materials together with the increasing diversity of responsive materials foretell that lignin-based smart materials will be a vibrant area to explore in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Department of Materials and Environmental Chemistry (MMK) for financing this work through the start-up grant awarded to MHS.Notes and references
- Declaration of the United Nations Conference on Environment and Development, Rio de Janeiro, Brazil, June 3-14, 1992. http://www.unesco.org/education/pdf/RIO_E.PDF, accessed, January, 10, 2020.
- J. K. Hammitt, R. J. Lempert and M. E. Schlesingert, Nature, 1992, 357, 315–318 CrossRef.
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- D. Ghosh, D. Dasgupta, D. Agrawal, S. Kaul, D. K. Adhikari, A. K. Kurmi, P. K. Arya, D. Bangwal and M. S. Negi, Energy Fuels, 2015, 29, 3149–3157 CrossRef CAS.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- N. Lavoine, I. Desloges, A. Dufresne and J. Bras, Carbohydr. Polym., 2012, 90, 735–764 CrossRef CAS PubMed.
- N. M. L. Hansen and D. Plackett, Biomacromolecules, 2008, 9, 1493–1505 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- K. V. Sarkanen and C. H. Ludwig, Lignins: Occurence, Formation, Structure and Reactions, Wiley-Interscience, New York, 1971, pp. 19–38 Search PubMed.
- D. M. O’Malley, R. Whetten, W. Bao, C.-L. Chen and R. R. Sederoff, Plant J., 1993, 4, 751–757 CrossRef.
- Y. Wang, M. Chantreau, R. Sibout and S. Hawkins, Front. Plant Sci., 2013, 4, 220 Search PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- D. Yiamsawas, G. Baier, E. Thines, K. Landfester and F. R. Wurm, RSC Adv., 2014, 4, 11661–11663 RSC.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- W. Yang, E. Fortunati, D. Gao, G. M. Balestra, G. Giovanale, X. He, L. Torre, J. M. Kenny and D. Puglia, ACS Sustainable Chem. Eng., 2018, 6, 3502–3514 CrossRef CAS.
- E. Larrañeta, M. Imízcoz, J. X. Toh, N. J. Irwin, A. Ripolin, A. Perminova, J. Domínguez-Robles, A. Rodríguez and R. F. Donnelly, ACS Sustainable Chem. Eng., 2018, 6, 9037–9046 CrossRef PubMed.
- M. Yousuf, A. Mollah, P. Palta, T. R. Hess, R. K. Vempati and D. L. Cocke, Cem. Concr. Res., 1995, 25, 671–682 CrossRef CAS.
- S. Tan, D. Liu, Y. Qian, J. Wang, J. Huang, C. Yi, X. Qiu and Y. Qin, Holzforschung, 2019, 73, 485–491 CAS.
- A. Hasan and P. Fatehi, Appl. Clay Sci., 2018, 158, 72–82 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Prog. Mater. Sci., 2018, 93, 233–269 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Polymers, 2019, 11, 1176 CrossRef PubMed.
- T. M. Budnyak, A. Slabon and M. H. Sipponen, ChemSusChem, 2020 DOI:10.1002/cssc.202000216.
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C. A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 CAS.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. De Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- S. P. S. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed.
- X. Pan, C. Arato, N. Gilkes, D. Gregg, W. Mabee, K. Pye, Z. Xiao, X. Zhang and J. Saddler, Biotechnol. Bioeng., 2005, 90, 473–481 CrossRef CAS PubMed.
- A. M. Galleti and C. Antonetti, Separation of Cellulose Hemicellulose, and Lignin Existing Technologies and Prespectives, Biorefinery: From Biomass to Chemicals and Fuels, De Gruyter, 2012, pp. 101–123 Search PubMed.
- T. Saito, J. H. Perkins, D. C. Jackson, N. E. Trammel, M. A. Hunt and A. K. Naskar, RSC Adv., 2013, 3, 21832–21840 RSC.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Green Chem., 2012, 14, 3295–3303 RSC.
- M. Witzler, A. Alzagameem, M. Bergs, B. El Khaldi-Hansen, S. E. Klein, D. Hielscher, B. Kamm, J. Kreyenschmidt, E. Tobiasch and M. Schulze, Molecules, 2018, 23, 1–22 CrossRef PubMed.
- N. Niu, Z. Ma, F. He, S. Li, J. Li, S. Liu and P. Yang, Langmuir, 2017, 33, 5786–5795 CrossRef CAS PubMed.
- M. Canetti and F. Bertini, Compos. Sci. Technol., 2007, 67, 3151–3157 CrossRef CAS.
- Y. Zhao, A. Tagami, G. Dobele, M. E. Lindström and O. Sevastyanova, Polymers, 2019, 11, 538 CrossRef PubMed.
- X. Zhang, W. Liu, W. Liu and X. Qiu, Int. J. Biol. Macromol., 2020, 142, 551–558 CrossRef CAS PubMed.
- Q. Xing, P. Buono, D. Ruch, P. Dubois, L. Wu and W. J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 4147–4157 CrossRef CAS.
- L. Liu, M. Qian, P. Song, G. Huang, Y. Yu and S. Fu, ACS Sustainable Chem. Eng., 2016, 4, 2422–2431 CrossRef CAS.
- J. Zhang, E. Fleury, Y. Chen and M. A. Brook, RSC Adv., 2015, 5, 103907 RSC.
- K. Huang, S. Ma, S. Wang, Q. Li, Z. Wu, J. Liu, R. Liu and J. Zhu, Green Chem., 2019, 21, 4964–4970 RSC.
- A. Duval and L. Avérous, Green Chem., 2020, 22, 1671–1680 RSC.
- K. A. Y. Koivu, H. Sadeghifar, P. A. Nousiainen, D. S. Argyropoulos and J. Sipilä, ACS Sustainable Chem. Eng., 2016, 4, 5238–5247 CrossRef CAS.
- M. Zieglowski, S. Trosien, J. Rohrer, S. Mehlhase, S. Weber, K. Bartels, G. Siegert, T. Trellenkamp, K. Albe and M. Biesalski, Front. Chem., 2019, 7, 562 CrossRef CAS PubMed.
- R. J. Li, J. Gutierrez, Y. L. Chung, C. W. Frank, S. L. Billington and E. S. Sattely, Green Chem., 2018, 20, 1459–1466 RSC.
- X. Zhao, Y. Zhang, L. Wei, H. Hu, Z. Huang, M. Yang, A. Huang, J. Wu and Z. Feng, RSC Adv., 2017, 7, 52382–52390 RSC.
- M. K. R. Konduri and P. Fatehi, ACS Sustainable Chem. Eng., 2015, 3, 1172–1182 CrossRef CAS.
- G. J. Jiao, P. Peng, S. L. Sun, Z. C. Geng and D. She, Int. J. Biol. Macromol., 2019, 127, 544–554 CrossRef CAS PubMed.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- X. Liu, H. Yin, Z. Zhang, B. Diao and J. Li, Colloids Surf., B, 2015, 125, 230–237 CrossRef CAS PubMed.
- Y. S. Kim and J. F. Kadla, Biomacromolecules, 2010, 11, 981–988 CrossRef CAS PubMed.
- P. Olsén, M. Jawerth, M. Lawoko, M. Johansson and L. A. Berglund, Green Chem., 2019, 21, 2478–2486 RSC.
- Y. Sun, L. Yang, X. Lu and C. He, J. Mater. Chem. A, 2015, 3, 3699–3709 RSC.
- A. L. Korich, A. B. Fleming, A. R. Walker, J. Wang, C. Tang and P. M. Iovine, Polymer, 2012, 53, 87–93 CrossRef CAS.
- H. Liu and H. Chung, ACS Sustainable Chem. Eng., 2017, 5, 9160–9168 CrossRef CAS.
- Y. Zhang, J. Liao, X. Fang, F. Bai, K. Qiao and L. Wang, ACS Sustainable Chem. Eng., 2017, 5, 4276–4284 CrossRef CAS.
- P. Buono, A. Duval, L. Averous and Y. Habibi, Polymer, 2017, 133, 78–88 CrossRef CAS.
- L. Yuan, Y. Zhang, Z. Wang, Y. Han and C. Tang, ACS Sustainable Chem. Eng., 2019, 7, 2593–2601 CrossRef CAS.
- H. Chung, A. Al-Khouja and N. R. Washburn, in Green Polymer Chemistry: Biocatalysis and Materials II, ed. H. N. Cheng, R. A. Gross and P. B. Smith, ACS Symposium Series, American Chemical Society, Washington, D. C., 2013, vol. 1144, pp. 373–391 Search PubMed.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- Y. Matsushita, J. Wood Sci., 2015, 61, 230–250 CrossRef CAS.
- H. Ma, H. Li, W. Zhao, L. Li, S. Liu, J. Long and X. Li, Green Chem., 2019, 21, 658–668 RSC.
- Y. Liu, C. Li, W. Miao, W. Tang, D. Xue, J. Xiao, T. Zhang and C. Wang, Green Chem., 2020, 22, 33–38 RSC.
- S. Rautiainen, D. Di Francesco, S. N. Katea, G. Westin, D. N. Tungasmita and J. S. M. Samec, ChemSusChem, 2019, 12, 404–408 CrossRef CAS.
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748–3763, 10.1039/d0cs00078g.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- X. Huang, C. Atay, J. Zhu, S. W. L. Palstra, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2017, 5, 10864–10874 CrossRef CAS PubMed.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- L. Mialon, A. G. Pemba and S. A. Miller, Green Chem., 2010, 12, 1704–1706 RSC.
- M. Firdaus and M. A. R. Meier, Eur. Polym. J., 2013, 49, 156–166 CrossRef CAS.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- A. L. Holmberg, J. F. Stanzione, R. P. Wool and T. H. Epps, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS.
- E. A. B. da Silva, M. Zabkova, J. D. Araújo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef.
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- A. Lendlein and O. E. C. Gould, Nat. Rev. Mater., 2019, 4, 116–133 CrossRef.
- W. Chen, C. Hu, Y. Yang, J. Cui and Y. Liu, Materials, 2016, 9, 184 CrossRef PubMed.
- S. Rai, B. K. Singh, P. Bhartiya, A. Singh, H. Kumar, P. K. Dutta and G. K. Mehrotra, J. Lumin., 2017, 190, 492–503 CrossRef CAS.
- B. Xue, Y. Yang, Y. Sun, J. Fan, X. Li and Z. Zhang, Int. J. Biol. Macromol., 2019, 122, 954–961 CrossRef CAS PubMed.
- Y. Shi, X. Liu, M. Wang, J. Huang, X. Jiang, J. Pang, F. Xu and X. Zhang, Int. J. Biol. Macromol., 2019, 128, 537–545 CrossRef CAS PubMed.
- Z. Ding, F. Li, J. Wen, X. Wang and R. Sun, Green Chem., 2018, 20, 1383–1390 RSC.
- B. Zhang, Y. Liu, M. Ren, W. Li, X. Zhang, R. Vajtai, P. M. Ajayan, J. M. Tour and L. Wang, ChemSusChem, 2019, 12, 4202–4210 CrossRef CAS PubMed.
- M. J. Molaei, Anal. Methods, 2020, 12, 1266–1287 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- X. Zhai, P. Zhang, C. Liu, T. Bai, W. Li, L. Dai and W. Liu, Chem. Commun., 2012, 48, 7955–7957 RSC.
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, Carbon, 2013, 60, 421–428 CrossRef CAS.
- X. Liu, J. Pang, F. Xu and X. Zhang, Sci. Rep., 2016, 6, 1–8 CrossRef CAS.
- J. Song, L. Zhao, Y. Wang, Y. Xue, Y. Deng, X. Zhao and Q. Li, Nanomaterials, 2018, 12, 1043 CrossRef PubMed.
- V. N. Mehta, S. Jha, H. Basu, R. K. Singhal and S. K. Kailasa, Sens. Actuators, B, 2015, 213, 434–443 CrossRef CAS.
- X. Yang, Y. Zhuo, S. Zhu, Y. Luo, Y. Feng and Y. Dou, Biosens. Bioelectron., 2014, 60, 292–298 CrossRef CAS PubMed.
- L. Minati and A. Del Piano, C, 2019, 5(4), 77 CAS.
- S. Cailotto, E. Amadio, M. Facchin, M. Selva, E. Pontoglio, F. Rizzolio, P. Riello, G. Toffoli, A. Benedetti and A. Perosa, ACS Med. Chem. Lett., 2018, 9, 832–837 CrossRef CAS PubMed.
- K. K. Chan, C. Yang, Y. H. Chien, N. Panwar and K. T. Yong, New J. Chem., 2019, 43, 4734–4744 RSC.
- Z. M. S. H. Khan, R. S. Rahman, J. M. Shumaila, S. Islam and M. Zulfequar, Opt. Mater., 2019, 91, 386–395 CrossRef CAS.
- V. Raveendran, A. R. Suresh Babu and N. K. Renuka, RSC Adv., 2019, 9, 12070–12077 RSC.
- Y. Chen, X. Sun, W. Pan, G. Yu and J. Wang, Front. Chem., 2020, 7, 911 CrossRef PubMed.
- N. Arumugam and J. Kim, Mater. Lett., 2018, 219, 37–40 CrossRef CAS.
- R. Wang, G. Xia, W. Zhong, L. Chen, L. Chen, Y. Wang, Y. Min and K. Li, Green Chem., 2019, 21, 3343–3352 RSC.
- J. Ju and W. Chen, Anal. Chem., 2015, 87, 1903–1910 CrossRef CAS PubMed.
- Y. Jiang, Y. Li, Y. Li and S. Li, Anal. Methods, 2016, 8, 2448–2455 RSC.
- B. M. Cerrutti, M. L. Moraes, S. H. Pulcinelli and C. V. Santilli, Biosens. Bioelectron., 2015, 71, 420–426 CrossRef CAS PubMed.
- A. Jędrzak, T. Rębiś, Ł. Klapiszewski, J. Zdarta, G. Milczarek and T. Jesionowski, Sens. Actuators, B, 2018, 256, 176–185 CrossRef.
- A. Jędrzak, T. Rębiś, M. Nowicki, K. Synoradzki, R. Mrówczyński and T. Jesionowski, Appl. Surf. Sci., 2018, 455, 455–464 CrossRef.
- L. Lu, B. Liu, Z. Zhao, C. Ma, P. Luo, C. Liu and G. Xie, Biosens. Bioelectron., 2012, 33, 216–221 CrossRef CAS PubMed.
- F. Kheiri, R. E. Sabzi, E. Jannatdoust, E. Shojaeefar and H. Sedghi, Biosens. Bioelectron., 2011, 26, 4457–4463 CrossRef CAS PubMed.
- N. Gan, X. Du, Y. Cao, F. Hu, T. Li and Q. Jiang, Materials, 2013, 6, 1255–1269 CrossRef CAS PubMed.
- Z. Zhang, Y. Cong, Y. Huang and X. Du, Micromachines, 2019, 10, 397 CrossRef PubMed.
- M. Zhou, L. Shang, B. Li, L. Huang and S. Dong, Biosens. Bioelectron., 2008, 24, 442–447 CrossRef CAS PubMed.
- E. Turkmen, S. Z. Bas, H. Gulce and S. Yildiz, Electrochim. Acta, 2014, 123, 93–102 CrossRef CAS.
- M. Şenel, Mater. Sci. Eng., C, 2015, 48, 287–293 CrossRef PubMed.
- B. Wang, T. Shi, Y. Zhang, C. Chen, Q. Li and Y. Fan, J. Mater. Chem. C, 2018, 6, 6423–6428 RSC.
- C. Chen, X. Wang, M. Li, Y. Fan and R. Sun, Sens. Actuators, B, 2018, 255, 1569–1576 CrossRef CAS.
- Q. Wang, X. Pan, C. Lin, D. Lin, Y. Ni, L. Chen, L. Huang, S. Cao and X. Ma, Chem. Eng. J., 2019, 370, 1039–1047 CrossRef CAS.
- F. A. C. Faria, D. V. Evtuguin, A. Rudnitskaya, M. T. S. R. Gomes, J. A. B. P. Oliveira, M. P. F. Graça and L. C. Costa, Polym. Int., 2012, 61, 788–794 CrossRef CAS.
- A. Rudnitskaya, D. V. Evtuguin, L. C. Costa, M. P. Pedro Graça, A. J. S. Fernandes, M. Rosario Correia, M. T. Teresa Gomes and J. A. B. P. Oliveira, Analyst, 2013, 138, 501–508 RSC.
- K. M. Zia, H. N. Bhatti and I. Ahmad Bhatti, React. Funct. Polym., 2007, 67, 675–692 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- Y. Xue, W. Liang, Y. Li, Y. Wu, X. Peng, X. Qiu, J. Liu and R. Sun, J. Agric. Food Chem., 2016, 64, 9592–9600 CrossRef CAS PubMed.
- A. Moreno, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Polym. Chem., 2019, 10, 5215–5227 RSC.
- H. Y. Tse, S. C. Cheng, C. S. Yeung, C. Y. Lau, W. H. Wong, C. Dong and S. Y. Leu, Green Chem., 2019, 21, 1319–1329 RSC.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- S. Iravani and R. S. Varma, Green Chem., 2020, 22, 612–636 RSC.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- Z. Ma, C. Liu, N. Niu, Z. Chen, S. Li, S. Liu and J. Li, ACS Sustainable Chem. Eng., 2018, 6, 3169–3175 CrossRef CAS.
- S. Singha, Y. W. Jun, J. Bae and K. H. Ahn, Anal. Chem., 2017, 89, 3724–3731 CrossRef CAS PubMed.
- Y. Xue, X. Qiu, Z. Liu and Y. Li, ACS Sustainable Chem. Eng., 2018, 6, 7695–7703 CrossRef CAS.
- M. Kaushik and A. Moores, Green Chem., 2016, 18, 622–637 RSC.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- N. A. Azmi, S. H. Ahmad and S. C. Low, RSC Adv., 2018, 8, 251–261 RSC.
- G. Milczarek, T. Rebis and J. Fabianska, Colloids Surf., B, 2013, 105, 335–341 CrossRef CAS PubMed.
- Z. Shen, Y. Luo, Q. Wang, X. Wang and R. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 16147–16155 CrossRef CAS PubMed.
- T. Leskinen, J. Witos, J. J. Valle-Delgado, K. Lintinen, M. Kostiainen, S. K. Wiedmer, M. Österberg and M. L. Mattinen, Biomacromolecules, 2017, 18, 2767–2776 CrossRef CAS PubMed.
- C. Jin, W. Song, T. Liu, J. Xin, W. C. Hiscox, J. Zhang, G. Liu and Z. Kong, ACS Sustainable Chem. Eng., 2018, 6, 1763–1771 CrossRef CAS.
- A. Zerpa, L. Pakzad and P. Fatehi, ACS Omega, 2018, 3, 8233–8242 CrossRef CAS PubMed.
- L. Dai, Y. Li, F. Kong, K. Liu, C. Si and Y. Ni, ACS Sustainable Chem. Eng., 2019, 7, 13497–13504 CrossRef CAS.
- Y. Wang, M. S. Shim, N. S. Levinson, H. W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- B. Diao, Z. Zhang, J. Zhu and J. Li, RSC Adv., 2014, 4, 42996–43003 RSC.
- Y. Qian, Q. Zhang, X. Qiu and S. Zhu, Green Chem., 2014, 16, 4963–4968 RSC.
- P. Figueiredo, C. Ferro, M. Kemell, Z. Liu, A. Kiriazis, K. Lintinen, H. F. Florindo, J. Yli-Kauhaluoma, J. Hirvonen, M. A. Kostiainen and H. A. Santos, Nanomedicine, 2017, 12, 2581–2596 CrossRef CAS PubMed.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- M. S. Alqahtani, A. Alqahtani, A. Al-Thabit, M. Roni and R. Syed, J. Mater. Chem. B, 2019, 7, 4461–4473 RSC.
- N. Chen, L. A. Dempere and Z. Tong, ACS Sustainable Chem. Eng., 2016, 4, 5204–5211 CrossRef CAS.
- J. Zhao, D. Zheng, Y. Tao, Y. Li, L. Wang, J. Liu, J. He and J. Lei, Biochem. Eng. J., 2020, 156, 107526 CrossRef CAS.
- M. Pishnamazi, H. Hafizi, S. Shirazian, M. Culebras, G. M. Walker and M. N. Collins, Polymers, 2019, 11, 1059 CrossRef CAS PubMed.
- L. Cheng, B. Deng, W. Luo, S. Nie, X. Liu, Y. Yin, S. Liu, Z. Wu, P. Zhan, L. Zhang and J. Chen, J. Agric. Food Chem., 2020, 68, 5249–5258 CrossRef CAS PubMed.
- T. P. Li, W. P. Wong, L. C. Chen, C. Y. Su, L. G. Chen, D. Z. Liu, H. O. Ho and M. T. Sheu, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- K. E. Washington, R. N. Kularatne, M. C. Biewer and M. C. Stefan, ACS Biomater. Sci. Eng., 2018, 4, 997–1004 CrossRef CAS.
- R. Peng, D. Yang, X. Qiu, Y. Qin and M. Zhou, Int. J. Biol. Macromol., 2020, 151, 421–427 CrossRef CAS PubMed.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- L. Liu, L. Rui, Y. Gao and W. Zhang, Polym. Chem., 2015, 6, 1817–1829 RSC.
- J. Hu, M. R. Whittaker, Y. Li, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 2407–2415 RSC.
- P. Bilalis, D. Skoulas, A. Karatzas, J. Marakis, A. Stamogiannos, C. Tsimblouli, E. Sereti, E. Stratikos, K. Dimas, D. Vlassopoulos and H. Iatrou, Biomacromolecules, 2018, 19, 3840–3852 CrossRef CAS PubMed.
- A. Moreno, J. C. Ronda, V. Cádiz, M. Galià, G. Lligadas and V. Percec, Biomacromolecules, 2019, 20, 3200–3210 CrossRef CAS PubMed.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- M. Lievonen, J. J. Valle-Delgado, M. L. Mattinen, E. L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- D. Piccinino, E. Capecchi, L. Botta, P. Bollella, R. Antiochia, M. Crucianelli and R. Saladino, Catal. Sci. Technol., 2019, 9, 4125–4134 RSC.
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 320 CrossRef PubMed.
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- M. H. Sipponen, M. Farooq, J. Koivisto, A. Pellis, J. Seitsonen and M. Österberg, Nat. Commun., 2018, 9, 2300 CrossRef PubMed.
- E. Capecchi, D. Piccinino, B. M. Bizzarri, D. Avitabile, C. Pelosi, C. Colantonio, G. Calabrò and R. Saladino, Biomacromolecules, 2019, 20, 1975–1988 CrossRef CAS PubMed.
- J. Ou, K. Liu, J. Jiang, D. A. Wilson, L. Liu, F. Wang, S. Wang, Y. Tu and F. Peng, Small, 2020, 1906184, 1–16 Search PubMed.
- I. Ortiz-Rivera, M. Mathesh and D. A. Wilson, Acc. Chem. Res., 2018, 51, 1891–1900 CrossRef CAS PubMed.
- Q. Feng, F. Chen and H. Wu, BioResources, 2011, 6, 4942–4952 CAS.
- D. Kai, Z. W. Low, S. S. Liow, A. Abdul Karim, H. Ye, G. Jin, K. Li and X. J. Loh, ACS Sustainable Chem. Eng., 2015, 3, 2160–2169 CrossRef CAS.
- Lignin and Lignans Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press, New York, 2010 Search PubMed.
- Chemical Modification, Properties and Usage of Lignin, ed. T. Q. Hu, Kluwer Academic/Plenum, New York, 2002 Search PubMed.
- K. Lintinen, Y. Xiao, R. Bangalore Ashok, T. Leskinen, E. Sakarinen, M. Sipponen, F. Muhammad, P. Oinas, M. Österberg and M. Kostiainen, Green Chem., 2018, 20, 843–850 RSC.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- H. Li, Y. Deng, B. Liu, Y. Ren, J. Liang, Y. Qian, X. Qiu, C. Li and D. Zheng, ACS Sustainable Chem. Eng., 2016, 4, 1946–1953 CrossRef CAS.
- D. Tian, J. Hu, J. Bao, R. P. Chandra, J. N. Saddler and C. Lu, Biotechnol. Biofuels, 2017, 10, 1–11 CrossRef PubMed.
- N. Bensabeh, A. Moreno, A. Roig, M. Rahimzadeh, K. Rahimi, J. C. Ronda, V. Cádiz, M. Galià, V. Percec, C. Rodriguez-Emmenegger and G. Lligadas, ACS Sustainable Chem. Eng., 2020, 8, 1276–1284 CrossRef CAS.
- R. Hilgers, G. Van Erven, V. Boerkamp, I. Sulaeva, A. Potthast, M. A. Kabel and J. P. Vincken, Green Chem., 2020, 22, 1735–1746 RSC.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
- Y. Wang, W. Tian, J. Xie and Y. Liu, Micromachines, 2016, 8, 145 CrossRef PubMed.
- J. Mendez, P. K. Annamalai, S. J. Eichhorn, R. Rusli, S. J. Rowan, E. J. Foster and C. Weder, Macromolecules, 2011, 44, 6827–6835 CrossRef CAS.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540–3545 CrossRef CAS PubMed.
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS.
- W. Small IV, P. Singhal, T. S. Wilson and D. J. Maitland, J. Mater. Chem., 2010, 20, 3356–3366 RSC.
- Q. Shi, H. Liu, D. Tang, Y. Li, X. J. Li and F. Xu, NPG Asia Mater., 2019, 11, 64 CrossRef.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 13456–13463 CrossRef CAS.
- P. Mora, H. Schäfer, C. Jubsilp, S. Rimdusit and K. Koschek, Chem. – Asian J., 2019, 14, 4129–4139 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- F. Gamardella, F. Guerrero, S. De la Flor, X. Ramis and A. Serra, Eur. Polym. J., 2020, 122, 109361 CrossRef.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- C. Borsellino, G. Di Bella and V. F. Ruisi, Int. J. Adhes. Adhes., 2009, 29, 36–44 CrossRef CAS.
- S. G. Prolongo, G. Del Rosario and A. Ureña, Int. J. Adhes. Adhes., 2006, 26, 125–132 CrossRef CAS.
- F. Gamardella, S. De la Flor, X. Ramis and A. Serra, React. Funct. Polym., 2020, 151, 104574 CrossRef CAS.
- H. Li, G. Sivasankarapillai and A. G. McDonald, Ind. Crops Prod., 2015, 67, 143–154 CrossRef CAS.
- G. Sivasankarapillai, H. Li and A. G. McDonald, Biomacromolecules, 2015, 16, 2735–2742 CrossRef CAS PubMed.
- H. Liu, N. Mohsin, S. Kim and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2121–2130 CrossRef CAS.
- N. A. Nguyen, K. M. Meek, C. C. Bowland, S. H. Barnes and A. K. Naskar, Macromolecules, 2018, 51, 115–127 CrossRef CAS.
- N. A. Nguyen, K. M. Meek, C. C. Bowland and A. K. Naskar, Polymer, 2019, 160, 210–222 CrossRef CAS.
- J. Huang, W. Liu and X. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 6550–6560 CrossRef CAS.
- Y. Bai, X. Zhang, Q. Wang and T. Wang, J. Mater. Chem. A, 2014, 2, 4771–4778 RSC.
- Z. Ding, L. Yuan, T. Huang, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2019, 58, 8734–8742 CAS.
- J. Liu, S. Wang, Z. Tang, J. Huang, B. Guo and G. Huang, Macromolecules, 2016, 49, 8593–8604 CrossRef CAS.
- L. Xue, S. Dai and Z. Li, Biomaterials, 2010, 31, 8132–8140 CrossRef CAS PubMed.
- R. Dhanasekaran, S. Sreenatha Reddy, B. Girish Kumar and A. S. Anirudh, Mater. Today Proc., 2018, 5, 21427–21435 CrossRef CAS.
- R. Liu, X. Kuang, J. Deng, Y. C. Wang, A. C. Wang, W. Ding, Y. C. Lai, J. Chen, P. Wang, Z. Lin, H. J. Qi, B. Sun and Z. L. Wang, Adv. Mater., 2018, 30, 1–8 Search PubMed.
- M. Fasihi and H. Mansouri, J. Appl. Polym. Sci., 2016, 133, 44068 CrossRef.
- A.-V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. H. J. Schenning, Nat. Commun., 2016, 7, 11976 CrossRef PubMed.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- L. Dai, M. Ma, J. Xu, C. Si, X. Wang, Z. Liu and Y. Ni, Chem. Mater., 2020, 32, 4324–4330 CrossRef CAS.
- G. Yilmaz and C. R. Becer, Polym. Chem., 2015, 6, 5503–5514 RSC.
- O. Cusola, O. J. Rojas and M. B. Roncero, ACS Appl. Mater. Interfaces, 2019, 11, 45226–45236 CrossRef CAS PubMed.
- A. X. Lu, H. Oh, J. L. Terrell, W. E. Bentley and S. R. Raghavan, Chem. Sci., 2017, 8, 6893–6903 RSC.
- Y. Huang, L. Deng, P. Ju, L. Huang, H. Qian, D. Zhang, X. Li, H. A. Terryn and J. M. C. Mol, ACS Appl. Mater. Interfaces, 2018, 10, 23369–23379 CrossRef CAS PubMed.
- M. Wang, Y. Zhai, H. Ye, Q. Lv, B. Sun, C. Luo, Q. Jiang, H. Zhang, Y. Xu, Y. Jing, L. Huang, J. Sun and Z. He, ACS Nano, 2019, 13, 7010–7023 CrossRef CAS PubMed.
- X. Liu, P. Formanek, B. Voit and D. Appelhans, Angew. Chem., Int. Ed., 2017, 56, 16233–16238 CrossRef CAS PubMed.
- P. Schattling, F. D. Jochum and P. Theato, Chem. Commun., 2011, 47, 8859–8861 RSC.
| This journal is © The Royal Society of Chemistry 2020 |