
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolution of self-healing elastomers, from extrinsic to combined intrinsic mechanisms: a review
Saul
Utrera-Barrios
 ,
Raquel
Verdejo
,
Miguel A.
López-Manchado
and
Marianella
Hernández Santana
*
,
Raquel
Verdejo
,
Miguel A.
López-Manchado
and
Marianella
Hernández Santana
*
Institute of Polymer Science and Technology (ICTP-CSIC), Juan de la Cierva, 3, 28006, Madrid, Spain. E-mail: marherna@ictp.csic.es
First published on 15th July 2020
Abstract
The evolution of self-healing polymers has resulted in a myriad of healing designs that have given way to complex systems capable of supporting multiple cycles, among other features. This progression enables us to propose the implementation of a timeline that classifies self-healing polymers in generations based on the healing mechanism, and correlated with historical development. The first generation employed the encapsulation of external healing agents; the second one, based on intrinsic mechanisms, applied reversible chemistries; and the third generation was inspired by natural examples such as plants and human skin, in which the healing agent is embedded in vascular networks. Despite great efforts and, with a few exceptions, polymers with high healing efficiency and high mechanical performance are not common. To improve this situation, a combination of different mechanisms is currently emerging, giving birth to the fourth generation of self-healing materials. This article, focused on self-healing elastomers, provides a rigorous overview of this new generation, in which the combination of covalent bonds and non-covalent interactions provides an optimal balance between mechanical performance and repairability. The implementation of this concept leads to materials with real commercial potential in functional applications, such as coatings, sensors, actuators, controlled release of drugs, seals, gaskets, hoses, and even high-performance applications such as tires and railway components.
 Saul Utrera-Barrios | Saul Utrera-Barrios (SUB) is a Materials Engineer (Simon Bolivar University, Venezuela) with a MSc in Plastics and Rubber (Menendez Pelayo International University UIMP – Spanish National Research Council CSIC, Spain) and a PhD candidate in Advanced Chemistry (Complutense University of Madrid, Spain). Since 2018, he has developed his experimental work at the Institute of Polymer Science and Technology (ICTP-CSIC) in the area of elastomer-based composite materials with self-healing capability, under the supervision of Dr Marianella Hernández Santana and Prof. Miguel Ángel López Manchado. His scientific main interests include the development, characterization, and production of elastomer-based composite materials and intrinsic self-healing concepts. |
 Raquel Verdejo | Raquel Verdejo (RV) is Senior Research Scientist at the Institute of Polymer Science and Technology (ICTP-CSIC). She did her PhD in Metallurgy and Materials at the University of Birmingham (UK) and later joined Imperial College London. She returned to Spain thanks to a Juan de la Cierva and afterwards held a prestigious Ramón y Cajal position, getting tenure in 2010. She is currently the Academic Director of the Master of High Specialization in Plastics and Rubber (UIMP-CSIC). Her main line of research is focused on the development of polymer composites and nanocomposites with special emphasis on polymer foams. |
 Miguel A. López-Manchado | Miguel A. López-Manchado (MALM) is Research Professor at ICTP-CSIC. He got his PhD in Chemistry at the Complutense University of Madrid in 1997. From 1998–2000, he did a postdoctoral stay at University of Perugia. He got a permanent position as Tenured Researcher at CSIC in 2006. He is author of more than 160 publications in scientific journals and books. His research activity is focused on the processing and characterization of composite materials and nanocomposites. |
 Marianella Hernández Santana | Marianella Hernández Santana (MHS) is Materials Engineer (Simon Bolivar University, Venezuela) with a MSc in Macromolecular Physical-Chemistry (Louis Pasteur University, France) and a PhD in Chemical Sciences (Complutense University of Madrid, Spain). She joined Prof. van der Zwaag's group (Delft University of Technology, Netherlands) as a Marie Curie Fellow developing self-healing elastomers. Later she returned to Spain to work at the Institute of Polymer Science and Technology (ICTP-CSIC). Since 2019, she holds a Ramon y Cajal position. Her research lines are based on the implementation of circular economy principles, favoring the use of recycled and self-healing materials. |
1. Introduction
The history of humankind has been classified in periods according to the use given to materials to meet their basic needs. In all cases, the discovery of new materials and the advancement in the manipulation of those already known implied a real revolution in social structures. With the impressive development attained between the 20th and the 21st century, humanity began to require new materials with properties on demand. Among these properties, self-healing has arisen as an unstoppable development in the last 20 years.Self-healing materials are attractive in the current environmental context, where there is a need for reducing waste and extending the lifetime of products. Besides, in the framework of the circular economy,1,2 certain polymers, such as elastomers or thermosets, are inappropriate because, despite great efforts,3,4 their reprocessing remains a challenge. Due to their importance in industrial applications, it has become mandatory to make them compatible with the circular economy model. Precisely, one possible path to achieve it is by conferring them with self-healing capability.
One can find in the literature some important reviews on the development of self-healing materials.5–7 However, there are just a few in the field of elastomers8–11 and to the best of our knowledge there is no specific one considering the combination of healing moieties, a new trend that has shown exponential growth in recent years. This review initially presents a brief overview of the evolution of self-healing, divided into generations, and later compiles the advances in the last five years in the field of self-healing rubbers. We define four generations and structure the review based on the nature of the involved bonds and interactions: non-covalent, covalent or combinations between them. The review ends with a final outlook and some future perspectives.
2. A brief history of self-healing polymers
2.1. Self-healing key concepts
Inspired by nature, self-healing materials have the ability to repair or restore damage, replicating mechanisms found in living organisms such as plants and human skin. To ensure the success of self-healing, three concepts have been defined by van der Zwaag:12 (a) localization, (b) temporality and (c) mobility. We include a fourth key concept: (d) mechanism (Fig. 1), in order to classify the different generations of self-healing materials.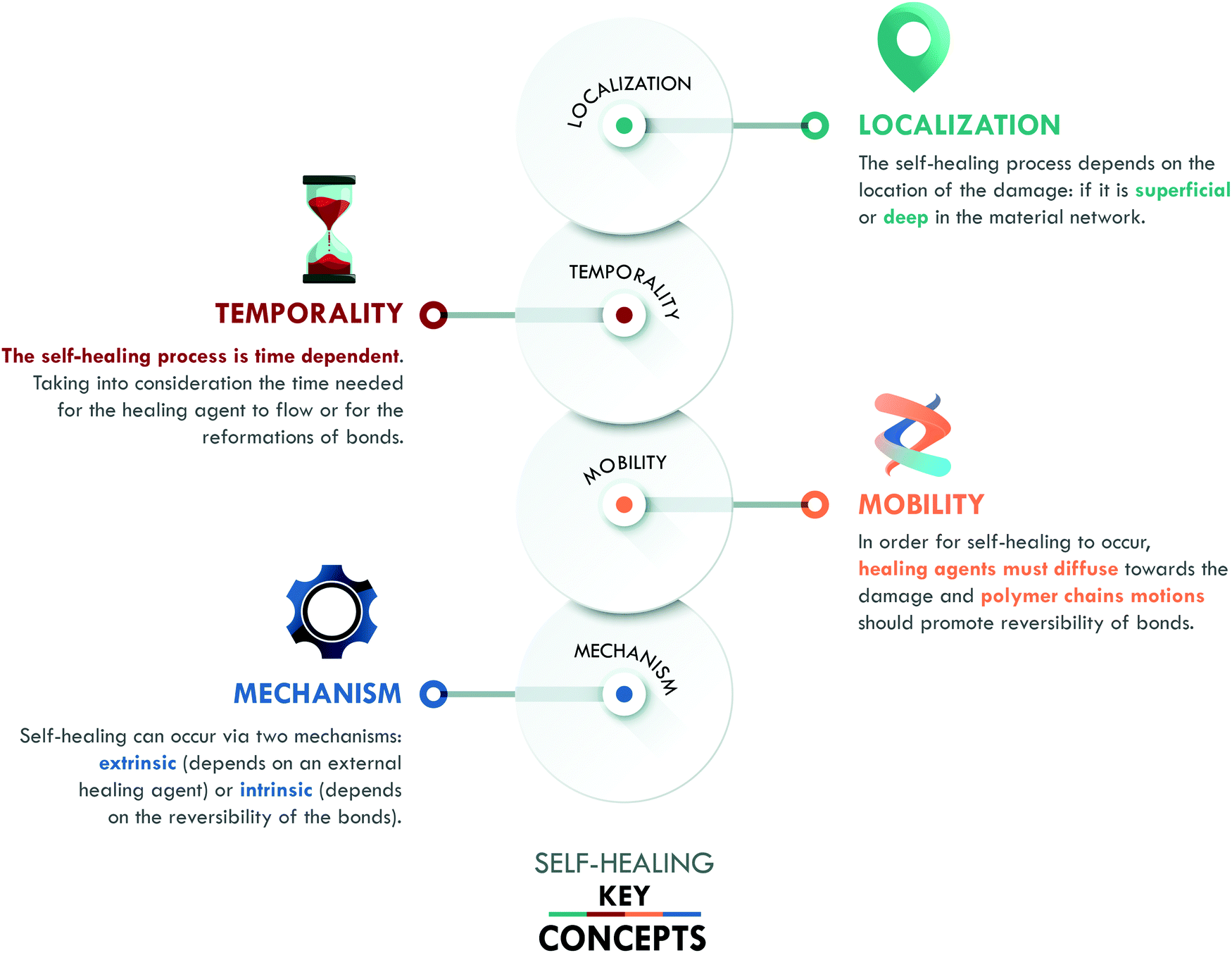 | ||
| Fig. 1 Self-healing key concepts. | ||
The concept of localization refers to the position and/or scale of the damage in the material. It can be superficial, such as scratches, (micro) cracks, or cuts; it can be deep, such as the propagation of surface damage, fiber debonding or delamination, ending up in catastrophic damage; or it can be molecular scale damage, e.g. breakage of the material network.5 The localization and scale of these types of damage play an essential role when considering the self-healing capability of the material. The aim is to reach one single protocol that assures achieving healing at all scales; however, specific protocols can be designed for particular types of damage according to the intended application of the material.
The second factor, temporality, is given by the time gap between the damage event and its repair. Even in nature, self-healing is time-dependent, not instantaneous. The target is to minimize the time for healing to occur. One way to reduce this time is by conferring mobility to the material, the third key concept. Mobility promotes the diffusion of the healing agent to the damage area, as well as the reformation of the broken bonds. This concept is key to optimize others, e.g. if the mobility of the agent is not adequate, it will not flow towards the damage or it will do so slowly.
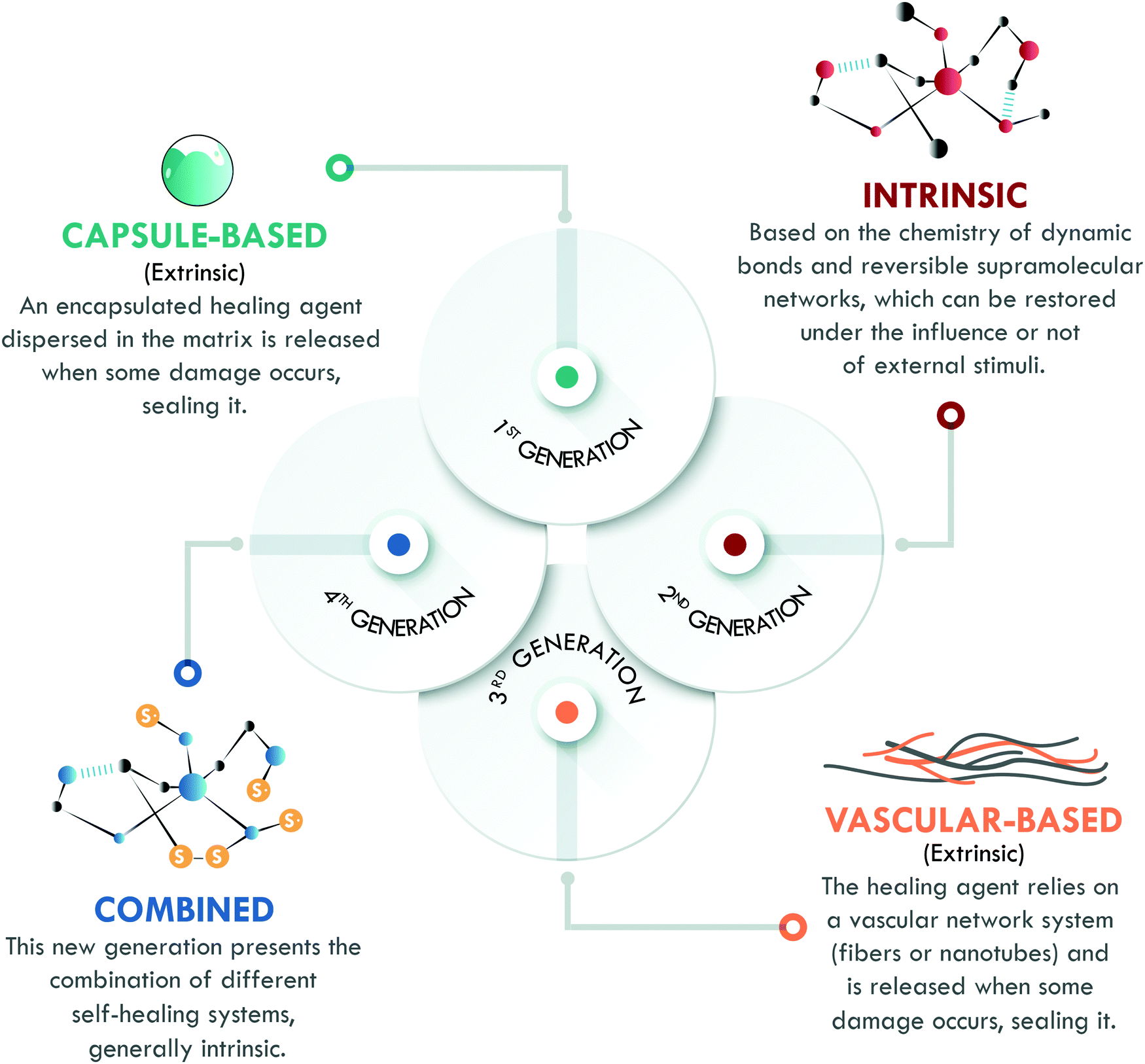
The last key concept in self-healing is the mechanism. This concept enables the classification of self-healing materials into two families: extrinsic and intrinsic. Extrinsic self-healing materials are those in which the process depends on an external agent, normally dispersed in the form of capsules or vascular systems. These agents are released to seal the damage and do not specifically interact with the matrix. On the other hand, intrinsic self-healing materials are those in which the reversible bonds present in the material can be restored after a damage event.5 In the case of polymers, extrinsic systems have widely been used in thermosets, mainly in epoxy resins,13–15 while intrinsic mechanisms have widely been considered in elastomers, such as silicones,16 polyurethanes,17 and general purpose rubbers.18–20 At this point, it is important to clarify that the current use of the terms intrinsic and extrinsic in the field of self-healing, to classify materials according to the type of mechanism involved, differs from their traditional use to designate physical quantities. In chemistry, intrinsic describes properties independent of size, shape, and quantity (e.g. density and refractive index, among others); meanwhile, extrinsic refers to dependent properties (such as weight and volume). The authors recognize that the use of these terms has some limitations, despite their extensive validation and widespread use in the self-healing field.6,7,10,11,21 All of the above has motivated us to propose a new classification, based on the self-healing mechanism and historical development, which enables organizing self-healing materials into four generations (Fig. 2).
 | ||
| Fig. 2 Generations of self-healing materials according to the healing mechanism involved. | ||
Although it was not the first published mechanism (as we will later explain), the first generation of self-healing materials was based on extrinsic mechanisms and employed encapsulated external healing agents. This generation had the disadvantage of only supporting a single cycle of self-healing.6 To overcome this limitation, the second generation of self-healing polymeric materials emerged, based on intrinsic mechanisms, using the chemistry of reversible bonds; however, the self-healing capability and mechanical performance of the materials were in compromise: the increase of one meant the decrease of the other. The intrinsic approach has been studied in all kinds of polymers, with special emphasis in the field of elastomers.7,10 Later, further development of extrinsic systems was initiated through healing agents confined in vascular networks, giving way to the third generation of self-healing polymeric materials, with strong inspiration from nature. These systems have been extensively studied in thermosets, but their application in elastomers remains limited except for a few reports on silicones.22
Finally, the fourth generation is currently growing fast and aims to overcome the different drawbacks of the previous generations. Hence, its objective is to develop a polymer with excellent mechanical properties, and high healing efficiency and resistance to multiple damage cycles through the combination of different healing mechanisms. However, the road to this point has been long and there are still some challenges to overcome.
2.2. Timeline of self-healing polymers
According to the literature, in the 1970s, Malinskii et al.23–25 presented one of the first studies on polymer self-healing, specifically in poly(vinyl acetate) (PVAc). Later, Jud et al.26 and Wool et al.27,28 deepened the study about healing of cracks in poly(methyl methacrylate) (PMMA), polystyrene (PS) and hydroxy-terminated polybutadiene (HTPB). Nevertheless, all these studies and those in the following years were based on chain interdiffusion,29 a highly known concept in polymers, which only requires a temperature slightly higher than the glass transition temperature (Tg) of the material to occur. Ellul et al.30 presented the concept of self-adhesion in butyl rubber (IIR), as an essential preliminary step to ensure good contact between the surfaces to be repaired. The concept of autonomic self-healing, as we know it today, was introduced by Dry et al.31,32 in the early 1990s; mainly in cement and epoxy resins. However, it was not until the publication of White et al.13 that the definitive impulse for the development of self-healing polymeric materials began. This work is considered as the starting point of self-healing polymers (Fig. 3). According to White et al.,13 self-healing was given by the incorporation of a healing agent (dicyclopentadiene, DCPD) embedded in microcapsules and a platinum catalyst (Grubb's catalyst) dispersed in an epoxy resin. Upon the release of the agent and encountering the catalyst, polymerization of the DCPD would occur, sealing the crack. This method, in its initial stage, enabled efficiencies of up to 75% in the recovery of the maximum load in a fracture toughness test. The implementation of this methodology in elastomers, specifically in poly(dimethylsiloxane) (PDMS), was carried out by Keller et al.,33 who used a chemistry based on two types of microcapsules. They introduced a high molecular weight resin of PDMS functionalized with vinyl groups and a platinum catalyst in one, while in another they encapsulated an initiator and a PDMS copolymer with active sites that would serve to link the vinyl groups of the functionalized resin by the platinum catalyst action. This chemical reaction, also based on the polymerization of external agents, allowed efficiencies of up to 120% in the recovery of the tear strength. This first generation has been classified in several ways according to the arrangement of the healing agents and catalysts. The most widespread classification comprises five types: single-capsules, capsule/dispersed catalyst, phase-separated droplet/capsules, double-capsules, and all-in-one capsules. It is also possible to establish classifications according to encapsulation techniques (for example: in situ polymerization, sol–gel reaction, interface polymerization, and emulsion, among others).34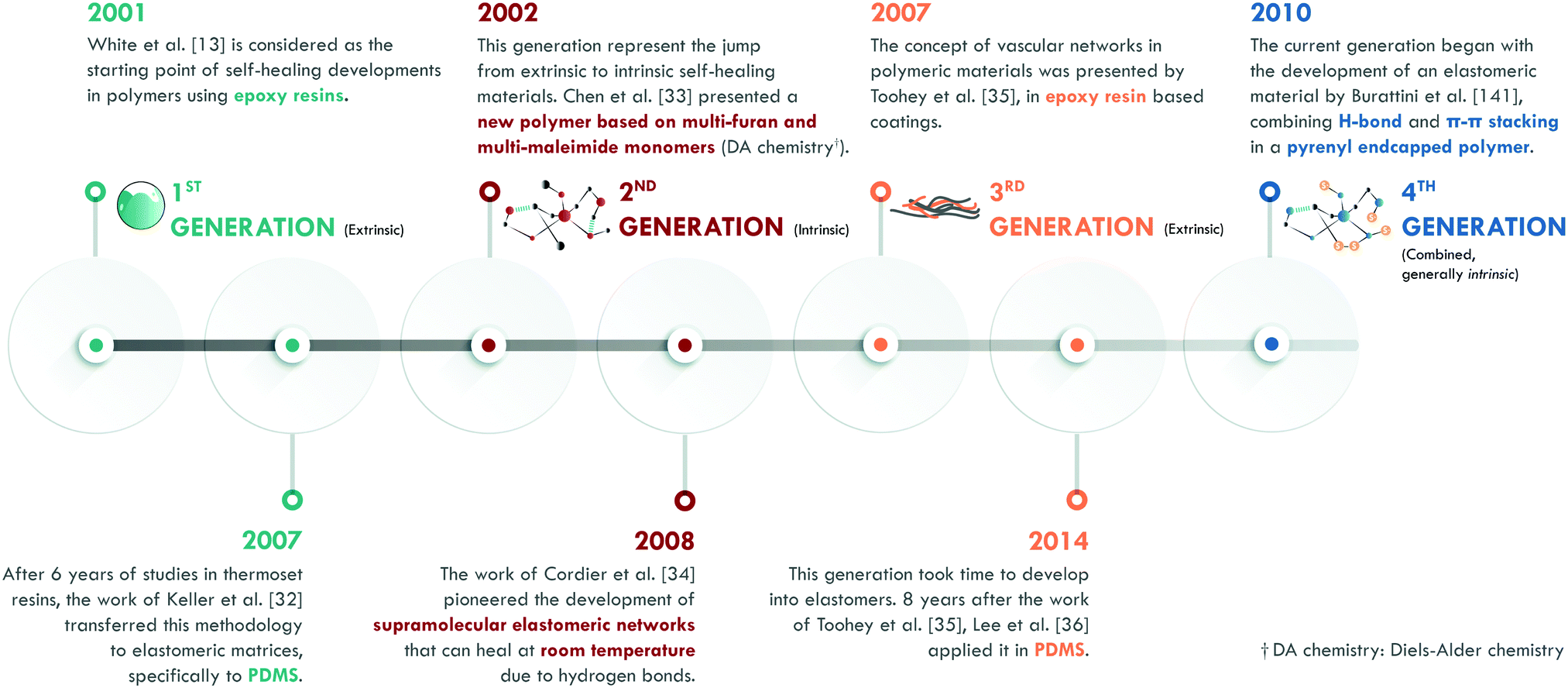 | ||
| Fig. 3 Timeline of generations in self-healing polymers. | ||
The second generation is based on the chemistry of the dynamic bonds. Any bond or interaction that is reversible under equilibrium conditions is considered dynamic and can be classified as: covalent and non-covalent. Although polymeric materials based on reversible chemistry had been developed in the past, Chen et al.35 specifically designed the first self-healing polymer based on multi-furan and multi-maleimide monomers (Diels–Alder chemistry). In this work, fracture toughness tests were carried out after healing at temperatures between 120 and 150 °C, achieving an efficiency of 57% in the recovery of the maximum load. At the same time, the effect of pressure on the repair process was evaluated, concluding that it had a minimal influence on the healing efficiency of this material. This work represented an important development in the field because it showed a recovery of 80% after subjecting the material to a third healing cycle, thus proving the occurrence of multiple healing cycles.
Years later, Cordier et al.36 first introduced the intrinsic self-healing methodology into an elastomer by designing and synthesizing molecules that could form chains and crosslinks through hydrogen bonds. Therefore, they had constructed a supramolecular network that was capable of restoring itself at room temperature. In this work, time dependence in the self-healing process was evidenced: longer times implied greater efficiencies. At the same time, they concluded that self-healing was not an instantaneous process.
The third generation of self-healing materials began with the work of Toohey et al.,37 although the concept had been explored almost twenty years earlier by Dry et al.31,32 The definitive stimulus for this generation took considerable time, due to the difficulties of incorporating vascular networks into a polymer matrix. This generation is usually classified according to the nature of the vascular network and its preparation technique. For example, electrospinning (coaxial electrospinning or emulsion spinning), solution blowing (coaxial solution blowing or emulsion blowing), and tubes and channel networks (micro/nano, such as hollow glass fibers and carbon nanotubes, among others).22 Toohey et al.37 used the previously described methodology by White et al.,13 but the DCPD healing agent was confined in a net embedded in the epoxy resin coating. The healing efficiency was also measured as the retention of properties in fracture toughness tests, with efficiencies of over 40% and supporting up to 7 healing cycles.
The difficulty of incorporating vascular networks in the matrix has hindered this generation in elastomers. Only one group has reported the incorporation of electrospun vascular networks in PDMS.38–41 Lee and coworkers38,39 prepared two co-axial electrospun networks with polyacrylonitrile (PAN) as the shell, and with either dimethylvinyl-terminated dimethylsiloxane (resin monomer) or methylhydrogen dimethylsiloxane (curing agent) as the core. Thus, the two core materials only interacted upon cutting the PAN shell. The self-healing efficiency was qualitatively evaluated as an anti-corrosive barrier, showing good performance. Thus, the design of resistant vascular networks that do not break during high shear, conventional processes remains a challenge.
A fourth generation is currently emerging within this field. However, this new generation does not mean that further developments using the previous approaches have stopped. On the contrary, these strategies are largely under development and are under study for both traditional and advanced applications. For example, the use of extrinsic mechanisms has shown a notable increase in coatings,39 instrument panels,42 and sponges,43 while intrinsic ones have turned out to be more versatile for general purpose elastomers in innovative applications such as nanogenerators,44 sensors,45 conductive elastomers,46 and even tires.20Table 1 summarizes some examples of the published work in the last five years in self-healing rubbers based on the first three generations.
| Healing mechanism | Rubber matrix | Ref. |
|---|---|---|
| a Water-soluble supramolecular polymer – WSP. b Polyacrylic amide (AAm)/choline chloride (ChCl)-co-maleic acid(MA)/ChCl – poly(AAm/ChCl-co-MA/ChCl). c Styrene butadiene styrene/ethylene-methacrylic acid copolymer elastomers – SBS/EMAA copolymer. | ||
| First generation: capsular self-healing systems | ||
| Single-capsule systems | ||
| Capsule based | Polyurethane – PU | 47 |
| Double-capsule systems | ||
| Capsule based | Polydimethylsiloxane – PDMS | 48 |
| Capsule based | Polyurethane – PU | 42 |
| Second generation: intrinsic self-healing systems | ||
| Non-covalent systems | ||
| Dipole–dipole interactions | Fluorinated elastomer | 49 |
| Hydrogen bonds | Epoxidized natural rubber – ENR | 45 and 50–53 |
| Hydrogen bonds | Butadiene rubber – BR | 19 |
| Hydrogen bonds | Polydimethylsiloxane – PDMS | 54–60 |
| Hydrogen bonds | Polyurea elastomer – PUE | 61 |
| Hydrogen bonds | Eucommia ulmoides ester elastomer | 62 |
| Hydrogen bonds | Lignin-based supramolecular elastomer | 63 |
| Hydrogen bonds | Supramolecular elastomer | 64–68 |
| Hydrogen bonds | Industrial acrylic elastomer | 69 |
| Hydrogen bonds | WSPa – photonic elastomer | 70 |
| Hydrogen bonds | Poly(AAm/ChCl-co-MA/ChCl)b | 46 |
| Hydrogen bonds | Hybrid polymer networks | 71 |
| Hydrogen bonds | Conductive elastomer | 72 |
| Ionic interactions | Natural rubber – NR | 18 and 73–75 |
| Ionic interactions | Epoxidized natural rubber – ENR | 76 |
| Ionic interactions | Brominated natural rubber – BNR | 77 |
| Ionic interactions | Brominated butyl rubber – BIIR | 78–80 |
| Ionic interactions | Ethylene propylene diene rubber – EPDM | 81 |
| Ionic interactions | Polydimethylsiloxane – PDMS | 82 and 83 |
| Ionic interactions | Polyampholyte-based elastomer | 84 |
| Ionic interactions | NR/BIIR | 85 |
| Ionic interactions | SBS/EMAA copolymerc | 86 |
| Ionic interactions | Poly(isobutylene-co-isoprene) | 87 and 88 |
| Metal–ligand coordination | Nitrile rubber – NBR | 89 |
| Metal–ligand coordination | Polydimethylsiloxane – PDMS | 16 and 90–98 |
| Metal–ligand coordination | Polyurethane – PU | 99 |
| Metal–ligand coordination | Aminopropyl methyl phenyl polysiloxanes – AMPS | 100 |
| Metal–ligand coordination | NBR/poly(vinyl chloride) | 101 |
| Host–guest interactions | Alkyl acrylate-based elastomer | 102 |
| Shape memory | ENR/poly(latic acid) | 103 |
| Covalent systems | ||
| Diels–Alder chemistry | Natural rubber – NR | 104 |
| Diels–Alder chemistry | Styrene butadiene rubber – SBR | 105 |
| Diels–Alder chemistry | Ethylene propylene diene rubber – EPDM | 106 |
| Diels–Alder chemistry | Polydimethylsiloxane – PDMS | 107–111 |
| Diels–Alder chemistry | Polyurethane – PU | 17 and 112–118 |
| Diels–Alder chemistry | Silicon-based elastomer | 119 and 120 |
| Diels–Alder chemistry | Styrene butadiene styrene rubber – SBS | 121–123 |
| Diels–Alder chemistry | Polymer/graphene-based material | 124 |
| Diels–Alder chemistry | Elastomer coatings | 125 |
| Diels–Alder chemistry | Soft robotics – pneumatic actuator elastomers | 126–130 |
| Disulfide bonds | Natural rubber – NR | 131–133 |
| Disulfide bonds | Styrene butadiene rubber – SBR | 20 and 134 |
| Disulfide bonds | Butadiene rubber – BR | 135 and 136 |
| Disulfide bonds | Chloroprene rubber – CR | 137 |
| Disulfide bonds | Polydimethylsiloxane – PDMS | 138 and 139 |
| Disulfide bonds | Polyurethane – PU | 140 and 141 |
| Boron-based bonds | Styrene butadiene rubber – SBR | 142 |
| Boron-based bonds | Silicon elastomer | 143 |
| Boron-based bonds | Poly(n-butyl acrylate) | 144 |
| Boron-based bonds | Boronic ester-based polymer | 145 |
| Imine bonds | NR latex-based elastomer | 44 |
| Imine bonds | Polydimethylsiloxane – PDMS | 146–148 |
| Transesterification reactions | Epoxidized natural rubber – ENR | 149–151 |
| Transesterification reactions | Thiol-epoxy elastomer | 152 |
| Alcoxyamine bonds | Poly(butyl methanol methacrylate) – PBMM | 153 |
| Oxime-carbamate bonds | Poly(oxime-urethane) – POU | 154 |
| Urea bonds | Polyurethane – PU | 155 |
| Third generation: vascular self-healing systems | ||
| Electrospinning (ES) based systems | ||
| Vascular based – coaxial ES | Polydimethylsiloxane – PDMS | 39–41, 43 and 156 |
| Channel network systems | ||
| Vascular based – micro | Polydimethylsiloxane – PDMS | 157 |
Despite all this work, the dichotomy between mechanical properties and self-healing efficiency is still an unresolved challenge. The combination of healing mechanisms as a way of positively balancing these two parameters is the core of the fourth generation.
3. Fourth generation of self-healing rubbers
Since the work of Burattini et al.,158 the literature on combined self-healing mechanisms is steadily growing (Fig. 4), and has focussed on intrinsic self-healing mechanisms, always searching for an optimal combination of dynamic bonds, either covalent (those that require a stimulus for reversibility) or non-covalent (those that are intrinsically reversible due to their lability).159 Such a strategy has also been considered for different, but not distant, purposes in elastomers, such as recyclability;160–163 however, this section concentrates on self-healing articles. Table 2 summarizes the publications of the fourth generation of self-healing rubbers, where σR refers to the tensile strength prior to healing, η is the healing efficiency, and T is the temperature at which the healing was performed. The discussion of this review is focused on these characteristics.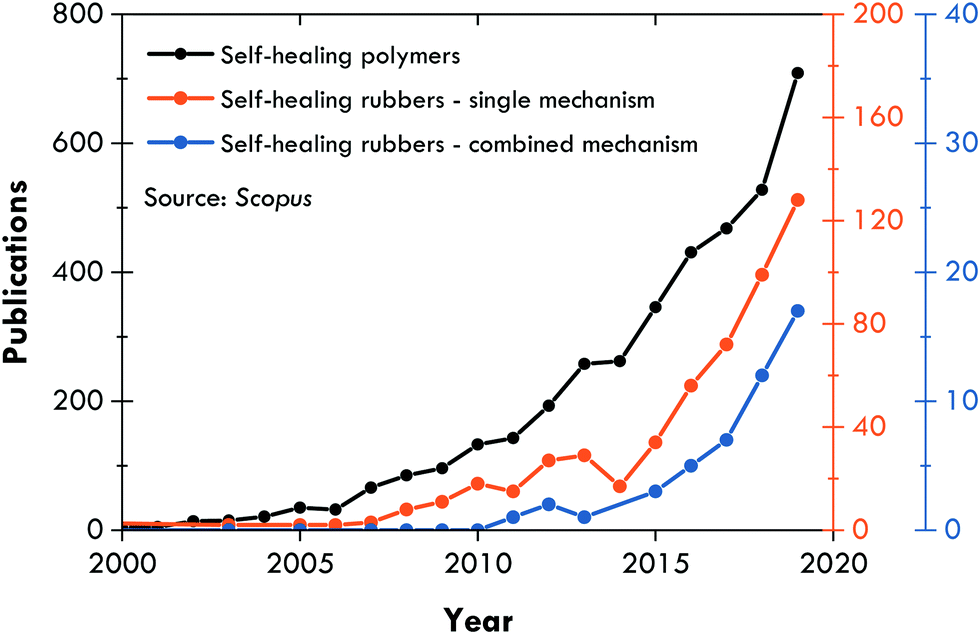 | ||
| Fig. 4 Evolution in the number of publications on self-healing rubbers. | ||
| First healing mechanism | Second healing mechanism | Rubber matrix | σ R (MPa) | η (%) | T (°C) | Ref. |
|---|---|---|---|---|---|---|
| a Curcumin polymer block – P-Cur. b The self-healing process was only qualitatively evaluated by profilometry. c Poly(butylene succinate) – PBS. d The self-healing process was evaluated by optical microscopy under different external stimuli. e Hyperbranched polyazomethine – hbPAM. f The self-healing efficiency was calculated for the elongation at break. | ||||||
| Combined non-covalent systems | ||||||
| Hydrogen bonds | π–π stacking | Pyrenyl end-capped polymer | 0.20 | 80 | 100 | 158 |
| Hydrogen bonds | π–π stacking | Polysiloxane-based polymer | 3.28 | 60 | 120 | 164 |
| Hydrogen bonds | Ionic interactions | Carboxylated SBR – XSBR | 1.30 | 92 | RT | 165 |
| Hydrogen bonds | Ionic interactions | NR | 18.50 | 79 | 50 | 166 |
| Hydrogen bonds | Ionic interactions | Polymer complexes | 27.00 | 92 | RT | 167 |
| Hydrogen bonds | Ionic interactions | PDMS | 0.08 | 115 | 80 | 168 |
| Hydrogen bonds | Ionic interactions | PDMS | — | 90 | RT | 169 |
| Hydrogen bonds | Ionic interactions | BIIR | 5.60 | 39 | 70 | 170 |
| Hydrogen bonds | Ionic interactions | Self-healing elastomer | 13.50 | 82 | RT | 171 |
| Hydrogen bonds | Metal–ligand coordination | IR | 21.00 | 73 | 80 | 172 |
| Hydrogen bonds | Metal–ligand coordination | P-Cura | 1.80 | 98 | RT | 173 |
| Hydrogen bonds | Metal–ligand coordination | PDMS | 2.60 | 90 | RT | 174 |
| Hydrogen bonds | Metal–ligand coordination | PU | 14.80 | 92 | RT | 175 |
| Hydrogen bonds | Metal–ligand coordination | Acrylic copolymer | 3.85 | 89 | 80 | 176 |
| Hydrogen bonds | Metal–ligand coordination | PDMS copolymer | — | 80 | 177 | |
| Hydrogen bonds | Metal–ligand coordination | PU | 7.13 | 71 | RT | 178 |
| Hydrogen bonds | Host–guest interactions | Acrylic copolymer | 8.60 | 75 | RT | 179 |
| Hydrogen bonds | Host–guest interactions | PU | 11.07 | 93 | 100 | 180 |
| Hydrogen bonds | van der Waals forces | PU | 0.35 | 100 | 50 | 181 |
| Hydrogen bonds | Dative bonds B–O | PBSc/PDMS | 0.18 | 86 | RT | 182 |
| Hydrogen bonds | Dipole–dipole interactions | SBS | 3.50 | 28 | 80 | 183 |
| Metal–ligand coordination | π–π stacking | PDMS | 0.30 | 100 | RT | 184 |
| Ionic interactions | Ionic Interactions | NR | 2.60 | 75 | 80 | 185 |
| Combined covalent systems | ||||||
| Disulfide bonds | Imine bonds | PDMS | 0.15 | 95 | RT | 186 |
| Disulfide bonds | Imine bonds | PU | 34.60 | — | 187 | |
| Disulfide bonds | Imine bonds | hb-PAMe | 4.00 | 91 | RT | 188 |
| Combined non-covalent/covalent systems | ||||||
| Hydrogen bonds | Disulfide bonds | Poly(urea-urethane) | 0.84 | 97 | RT | 189 |
| Hydrogen bonds | Disulfide bonds | PU | 9.50 | 96 | Sunlight | 190 |
| Hydrogen bonds | Disulfide bonds | Poly(urea-urethane) | 7.70 | 97 | 60 | 191 |
| Hydrogen bonds | Disulfide bonds | PU | 5.01 | 100 | 60 | 192 |
| Hydrogen bonds | Disulfide bonds | Polyurea | — | 150 | 193 | |
| Hydrogen bonds | Disulfide bonds | ENR | 9.30 | 98 | 120 | 194 |
| Hydrogen bonds | Disulfide bonds | PU | 25.00 | 90 | 100 | 195 |
| Hydrogen bonds | Disulfide bonds | PU | 3.39 | 95 | 80 | 196 |
| Hydrogen bonds | Disulfide bonds | PU | 0.34 | 88 | RT | 197 |
| Hydrogen bonds | Disulfide bonds | PU | 20.00 | 94 | 90 | 198 |
| Hydrogen bonds | Disulfide bonds | PDMS | — | Various | 199 | |
| Hydrogen bonds | Diels–Alder chemistry | PDMS/PU | 1.10 | 99 | 140, 80 | 200 |
| Hydrogen bonds | Diels–Alder chemistry | PU | 37.11 | 92 | 120, 60 | 201 |
| Hydrogen bonds | Boron-based bonds | ENR | 1.86 | 99 | RT | 202 |
| Hydrogen bonds | Imine bonds | PDMS | 0.16 | 93 | RT | 203 |
| Hydrogen bonds | Imine bonds | BR | 1.52 | >100 | 80 | 204 |
| Hydrogen bonds | Imine bonds | PDMS | 0.40 | 95 | RT | 205 |
| Hydrogen bonds | Ditelluride bonds | WSP | 19.00 | 86 | RT | 206 |
| Hydrogen bonds | Diselenide bonds | WSP | 15.34 | 84 | RT | 207 |
| Shape memory | Disulfide bonds | PU | 23.00 | 41 | 80 | 208 |
| Shape memory | Disulfide bonds | PU | 5.40 | 94 | 80 | 209 |
| Shape memory | Disulfide bonds | PU | 26.00 | 98 | 70 | 210 |
| Ionic interactions | Diels–Alder chemistry | Acrylic copolymer | 13.00 | 86 | 60 | 211 |
| Metal–ligand coordination | Boron-based bonds | ENR | 9.00 | 85 | 80 | 212 |
| Metal–ligand coordination | Imine bonds | PDMS | 0.05 | 88 | RT | 213 |
| Metal–ligand coordination | Diels–Alder chemistry | PU | 3.25 | 100 | RT | 214 |
| Metal–ligand coordination | Disulfide bonds | Poly(urea-urethane) | 9.40 | 100 | 80 | 215 |
3.1. Combined non-covalent systems
Intrinsic self-healing mechanisms of non-covalent nature comprise all those weak interactions that can occur between different families of atoms, such as van der Waals interactions, π–π stacking, dipole–dipole interactions, hydrogen bonding, ionic interactions, metal–ligand coordination and host–guest interactions. Some authors208,209 attribute self-healing capability to the existence of the shape memory effect (SM). It is not clear if SM can be considered as a self-healing mechanism itself. We have included it in this section because, without a doubt, it assists the self-healing process, especially in its initial stages, when the best possible contact between the surfaces is required, contributing to achieving high healing efficiencies. Fig. 5 summarizes all non-covalent interactions and their basic definition.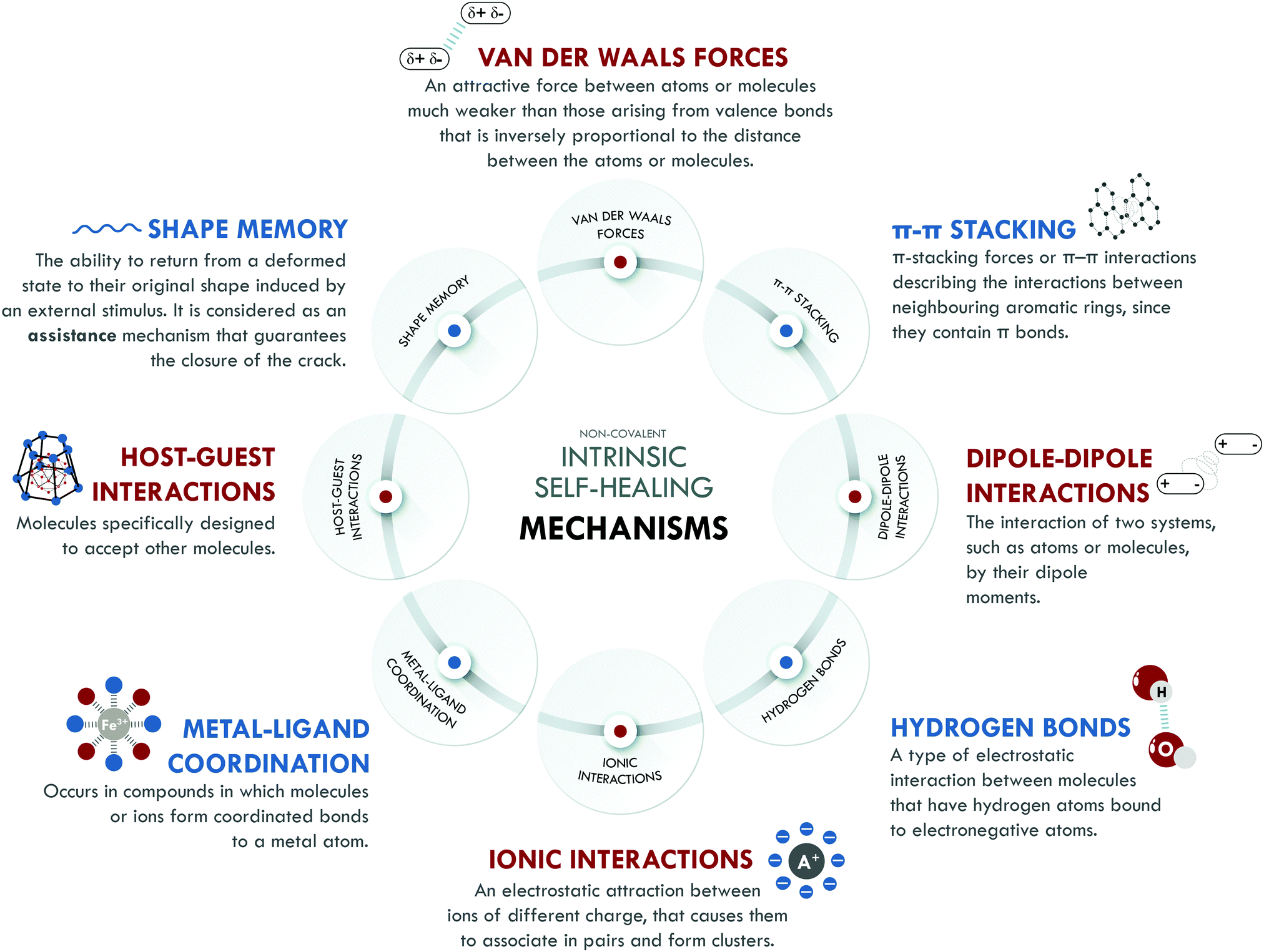 | ||
| Fig. 5 Non-covalent intrinsic self-healing mechanisms. | ||
Non-covalent systems are characterized by having low bonding energy compared to pure covalent systems,216 so they usually have higher healing efficiencies since this facilitates the restoration of the broken bonds even at room temperature. The non-covalent interaction most used in elastomers is based on hydrogen bonds, which have been combined with other interactions of the same nature to design self-healing materials with diverse performances. Burattini et al.158 reported a combination of healing moieties (π–π stacking and hydrogen bonds) in an elastomeric network based on polyimide and polyurethane with pyrenyl end groups. The π–π stacking was due to the π–electron-deficient diimide groups and the π-electron-rich pyrenyl units. Meanwhile, the formation of hydrogen bonds occurred at the intermolecular level, between the terminal residues of the pyrenyl groups in polyurethane. This material reached a tensile strength of 0.2 MPa, with a healing efficiency higher than 80% with respect to the tensile strength. Following the same combination of intrinsic mechanisms, Zuo et al.164 designed, for the first time, a fluorescent polysiloxane-based elastomer with 60% healing efficiency at room temperature and a tensile strength of 3.28 MPa. In this study, supramolecular interactions were confirmed through molecular simulation. The prepared materials presented interesting potential for bioimaging purposes.
The second most commonly used non-covalent mechanism employs ionic interactions. Xu et al.165 successfully combined them with hydrogen bonds in a carboxylated styrene-butadiene rubber (XSBR) filled with chitosan nanoparticles. The formation of ionic clusters enabled the configuration of a crosslinked supramolecular network with reversible bonds at room temperature that reached healing efficiencies of up to 92% with a tensile strength of 1.3 MPa. Sattar et al.166 used this same combination for the first time to prepare silica filled (SiO2) natural rubber (NR) compounds. Their strategy involved the ionization of the natural proteins and lipids of the elastomer to produce the ionic crosslinks. The dynamic supramolecular network was formed by adding magnesium sulfate (MgSO4), which provided Mg2+ ions, and formed electrostatic pairs with the negative charged lipids, arising from the acidic ionization. This procedure resulted in healing efficiencies of 79% at 50 °C with an excellent tensile strength of 18.5 MPa. The formation of the ionic network was confirmed by Fourier-transform infrared spectroscopy (FTIR) and by comparison with deproteinized NR, in which the repair efficiency was only 52%.
Combined hydrogen bonds and ionic bonds were also considered by Guo et al.,167 giving rise to a non-covalent network with the highest tensile strength and the best resistance/efficiency ratio reported. They prepared ternary polymer complexes of branched poly(ethyleneimine) (bPEI), poly(acrylic acid) (PAA) and poly(ethylene oxide) (PEO) (bPEI/PAA/PEO). These ternary complexes facilitated the formation of electrostatic interactions, between the bPEI and PAA, and of hydrogen bonds, between PAA and PEO (Fig. 6). Thanks to a positive effect between both interactions, an elastomeric material was obtained with a tensile strength of 27.4 MPa and a healing efficiency of 92% at room temperature in a high humidity atmosphere. More recently, the same joint mechanisms were explored in a special purpose rubber, brominated butyl rubber (BIIR), with opposite results. Stein et al.170 designed an elastomeric network of BIIR modified with one uracil and one imidazole moiety. The latter provided the ionic groups that associate in so-called ionic clusters, while the former, with a bifunctional structure containing two diamidopyridyl moieties, was responsible for the formation of hydrogen bonds. The incorporation of hydrogen bonds worsened the healing efficiency achieved with only ionic interactions, since they decreased the tensile strength to 5.7 MPa and the healing efficiency to 39% from a tensile strength of 10.7 MPa and a healing efficiency of 74% at 70 °C.
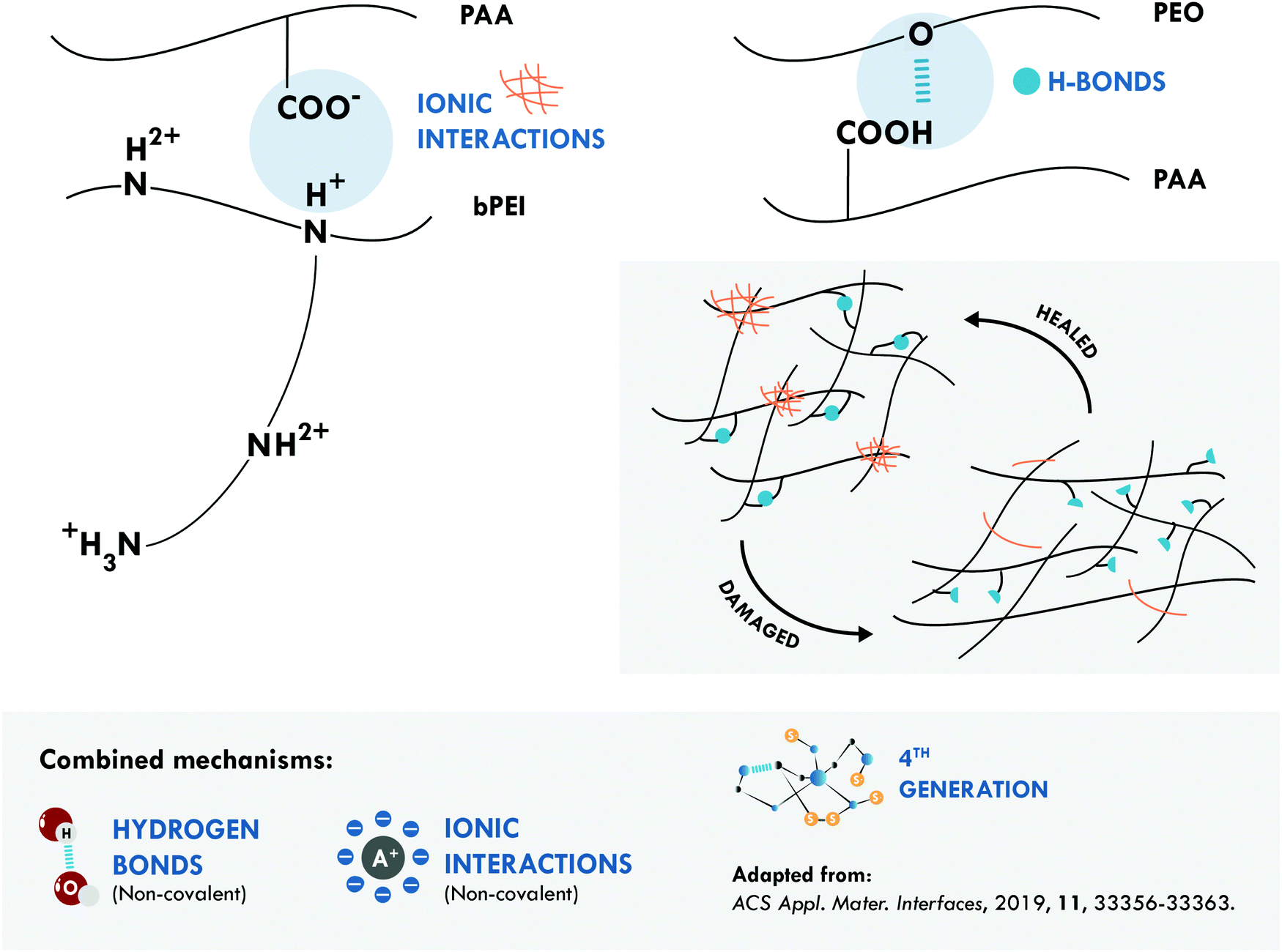 | ||
| Fig. 6 Combination between hydrogen bonds and ionic interactions in bPEI/PAA/PEO.167 ©2019. Adapted with permission from the American Chemical Society. | ||
The metal–ligand coordination bond is another non-covalent option widely combined with hydrogen bonds. One of the most relevant studies was performed by Liu et al.172 They reported poly(isoprene) (IR) with unprecedented mechanical properties, 21 MPa, and good healing efficiency, 72% at 80 °C. These results were attributed to three factors: first, to the dynamic nature of both mechanisms; second, to the mobility of the IR chains, which facilitated self-healing at the selected temperature and time; and third, to a positive effect thanks to the combined mechanisms (Fig. 7).
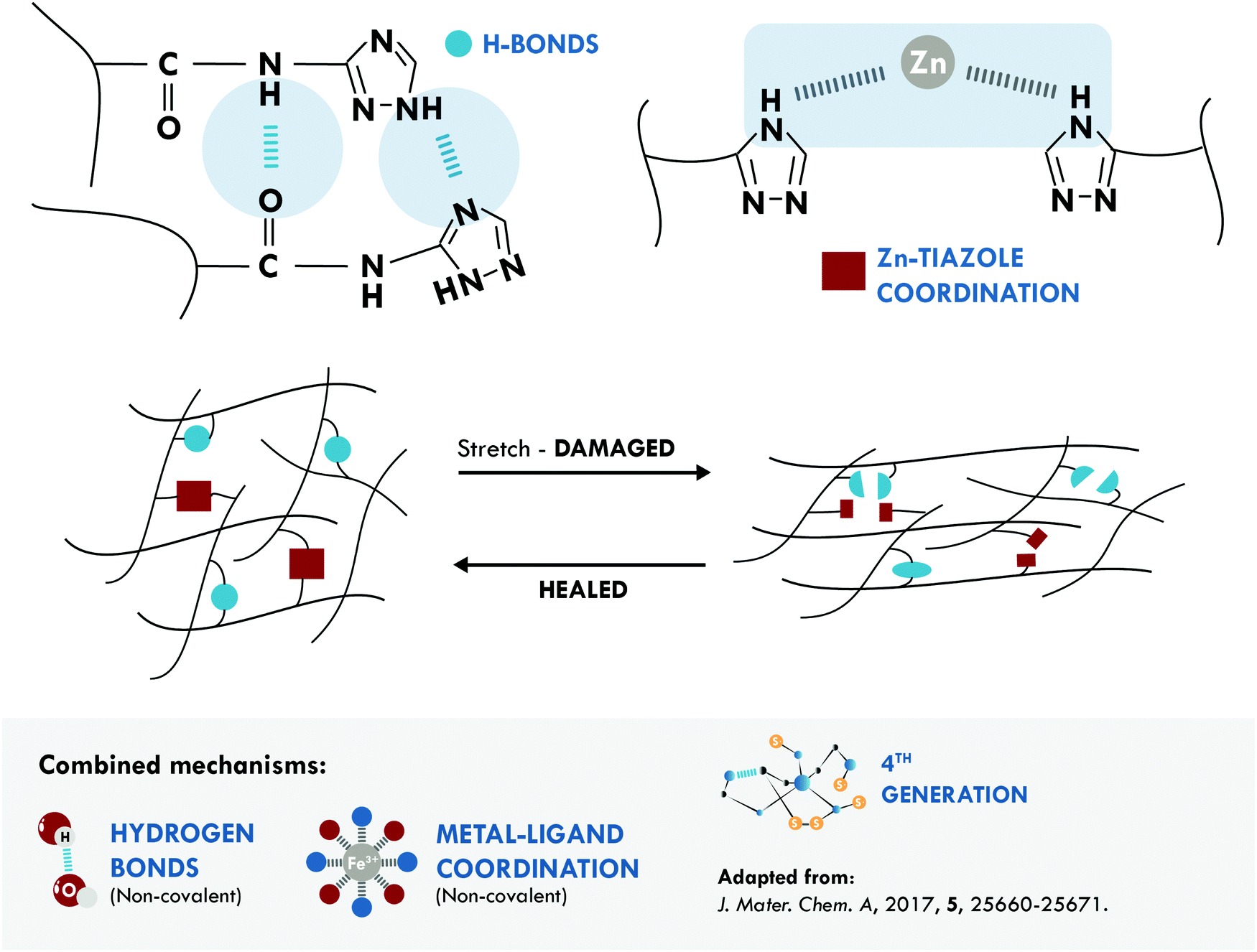 | ||
| Fig. 7 Combination between hydrogen bonds and metal–ligand coordination in IR.172 ©2017. Adapted with permission from the Royal Society of Chemistry. | ||
These dual mechanisms were also reported in polyurethane (PU). PU and its variants are the most studied materials from the point of view of intrinsic mechanisms, so it is not surprising that it is one of the first materials in which a network with three types of dynamic bonds has been reported: one covalent and two non-covalent, the latter being responsible for self-healing at room temperature. Zhang et al.175 prepared a Cu(II)-dimethylglyoxime-urethane-complex-based polyurethane elastomer. Here, the metal–ligand coordination was given by the copper ions and the hydrogen bonds were between amino groups and esters of the main chain. A tensile strength of 14.8 MPa and an efficiency of 92% were achieved, with potential applications in wires.
In PU, two other mechanisms together with hydrogen bonds have also been reported: host–guest interactions and van der Waals forces, with very different results. Xiao et al.180 designed a network based on the combination of host–guest interactions and multiple hydrogen bonds in waterborne PU (WBPU), an environmentally friendly option that decreases the effect of the release of volatile organic components into the atmosphere. They reported a tensile strength of 11.07 MPa and efficiency of 93%, close to the work described above with metal–ligand coordination, but at a temperature of 100 °C. On the other hand, Chen et al.181 combined hydrogen bonds with van der Waals forces in poly(urethane urea) (PUU) in which a branched poly(propylene carbonate) (PPC) diol was used as the soft segment, obtaining a tensile strength of 0.35 MPa, with 100% healing efficiency at 50 °C. The origin of these results was ascribed to the bond energy of the van der Waals force, which is considerably low compared to other non-covalent mechanisms.
In summary, there is no clear tendency in the effect of combined non-covalent systems on healing and mechanical performance. Both properties seem to be strongly influenced by the energy of the selected bonds, the chemical structure of the matrix and the effect generated when combining different mechanisms. Moreover, not all the combinations are a priori useful; the positive combination effect clearly depends on the matrix and the competition between the interactions.
3.2. Combined covalent systems
Intrinsic covalent mechanisms are related to all those chemical bonds that can be formed between different atoms and can be dynamic under an external stimulus. Fig. 8 schematically summarizes some of these bonds and their basic definition. Clear examples are disulfides, which can undergo metathesis reactions, or Diels–Alder chemistry, where Diels–Alder and retro-Diels–Alder reactions occur at different temperatures. These bonds are of higher energy compared to non-covalent bonds,216 so their contribution is usually associated with the mechanical performance of the material. However, their reversibility is a key factor in achieving high healing efficiencies.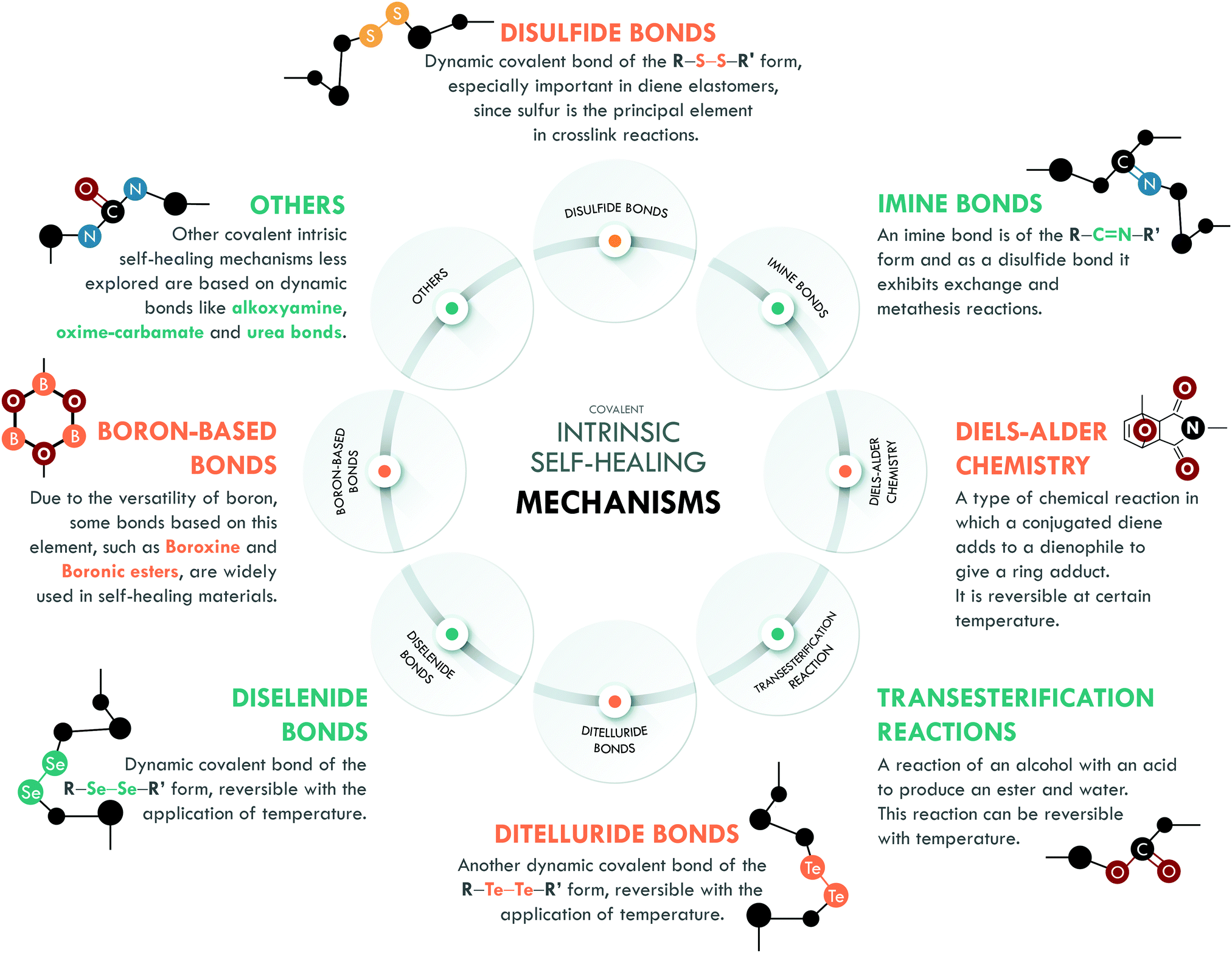 | ||
| Fig. 8 Covalent intrinsic self-healing mechanisms. | ||
The combination of covalent systems has been mildly reported with only two articles in recent years, using the same dual system with disulfide and imine bonds. Lv et al.186 applied aromatic disulfides, whose metathesis can occur at room temperature, contrary to aliphatic disulfides, which require an external stimulus. They obtained PDMS that could be repaired in just 4 h; however, its mechanical properties, typical of siloxane-based systems, were merely 0.15 MPa. Self-healing is mainly ascribed to the disulfide bond, which acted as a sacrificial bond, while the imine bond would mostly act as semi-permanent crosslink points for the elasticity and maintaining the original shape. Although the mechanical strength is low, this work serves as a proof of concept for certain applications (e.g. adhesives), in which high mechanical performance is not necessary. Lee et al.187 used the same approach in PU, studying the metathesis of both dynamic bonds (Fig. 9). The elastomer preparation was carried out in two stages; first, they prepared a Schiff base from biomaterials such as cystine and vanillin, responsible for providing disulfide and imine bonds. Subsequently, the base was mixed with 1,4-butanediol (to ensure miscibility) and added to the precursors of PU (isophorone diisocyanate, IPDI, and poly(propylene glycol)-based thiol, PPG). They monitored the healing process through optical microscopy and reported complete healing of cracks in 120 min with the use of heat (65 °C) and UV irradiation. The material also exhibited a recyclability efficiency of 97% but the healing efficiency was not reported as a measure of the retention of tensile properties.
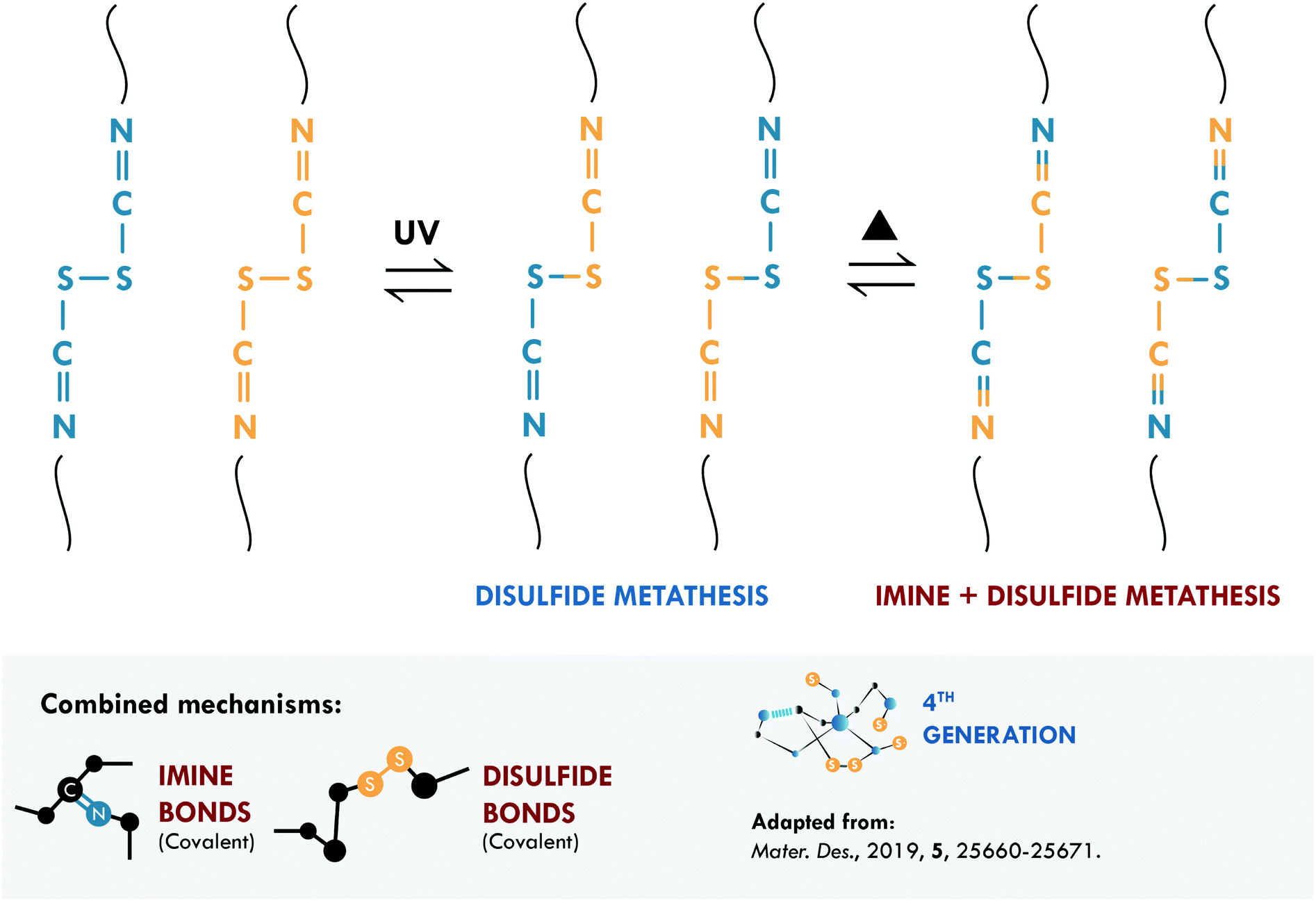 | ||
| Fig. 9 Imine and disulfide metathesis in PU.187 ©2019. Adapted with permission from Elsevier. | ||
The limited literature of combined covalent-based mechanisms is a clear indication that these dual systems are not the preferred option in elastomeric matrices, nor in any other type of polymer. We can speculate and attribute such low development to two facts. On one side, the imperative need for an external stimulus (in most cases) for assuring the reversibility of the covalent bonds. On the other side, the high bond energy that limits molecular dynamics and, thus, partially restricts reversibility and healing.
3.3. Combined non-covalent/covalent systems
The combination of covalent and non-covalent bonds is one of the most explored systems among the fourth generation, obtaining materials with good mechanical performance and high healing efficiencies. For these dual systems, we can observe a prevalence of PU as a material, and hydrogen bonds/disulfide bonds as a designated combination. Rekondo et al.189 were the first to apply this strategy and used aromatic disulfide metathesis to design a self-healing network of PUU. They reached 97% healing efficiency thanks to the aromatic disulfides, which are in constant exchange at room temperature, and to the urea groups, which can form quadruple hydrogen bonds. However, the mechanical properties were limited for high-performance applications. Xu et al.190 solved this limitation by developing an interesting system where healing was promoted by sunlight, with potential application in the manufacture of smart photosensitive polymers with high tensile strength. Although pure UV light was known to generate disulfide exchange, it was unknown if the low UV content in sunlight (between 3% and 5%) would be able to generate the same effect. They demonstrated, by mass spectroscopy and liquid chromatography (HPLC), that the UV component of sunlight was capable of generating a photochemical exchange reaction of disulfides in small molecules. Here, this exchange was favored by the formation of hydrogen bonds between amino groups adjacent to the disulfide groups of the main chain. They reported healing efficiencies of up to 96% with a tensile strength of 9.5 MPa. The development of PU with enough mechanical robustness continued to grow. Hu et al.195 introduced 2-ureido-4[1H]-pyrimidione (UPy)-functionalized side groups in the hard segment of thermoplastic polyurethane (TPU). The introduction of these functional groups formed quadruple hydrogen bonds and generated a supramolecular network that increased the crosslink density of the hard domains. The result of this combination was a material with a tensile strength of 25 MPa and a healing efficiency of 90% at a temperature of 100 °C.All these advances in PU have enabled its use in innovative applications, such as 3D printing, which seems quite distant for other elastomeric materials. Li et al.196 reported one of the first examples of self-healing PU manufactured using digital light 3D printing (DLP), with potential application in the manufacture of sensors and flexible electronics. Their elastomer reached a tensile strength of 3.39 MPa with a healing efficiency of 95% after 12 h at 80 °C. At the same time, they proved that the material supports multiple healing cycles. The healing protocol was applied directly on a piece produced with a honeycomb structure, which corroborated the applicability and success of the designed system, being able to bend freely and withstand high deformation without failing.
More recently, Liu et al.198 continued exploring different options on PU under this combination of healing mechanisms. They incorporated poly(vinyl alcohol)-graft-(ε-caprolactone) (PVA-PCL) into isocyanate terminated PU with disulfide bonds. The incorporation of PVA-PCL increased the formation of hydrogen bonds, which acted as physical crosslink points. They reported an improved tensile strength, of 20 MPa, and a positive effect of the hydrogen and disulfide bonds on the healing efficiency, reaching 94% at 90 °C. All the work done with PU highlights the good balance that can be reached between mechanical performance and healing capability. However, in most cases the healing procedure applies high temperatures (>60 °C). Thus, room temperature self-healing is still a milestone to overcome in the development of high performance PU.
The second most studied system is a general purpose elastomer, in particular epoxidized natural rubber (ENR). Cheng et al.194 considered two known principles: the vulcanization of the double bond of the main chain of cis-1,4 poly(isoprene) and the opening reaction of the epoxy ring. They performed a typical vulcanization reaction, to create multiple sulfur bonds in the main chain, together with the addition of aromatic disulfides, to couple to the radical generated during the ring-opening reaction (Fig. 10). Simultaneously, the ring-opening reaction generated multiple hydroxyl groups forming hydrogen bonds that assist the self-healing process. This combination resulted in a tensile strength of 9.30 MPa and 98% healing efficiency at 120 °C, compared to only 22% of an equivalent disulfide-free sample.
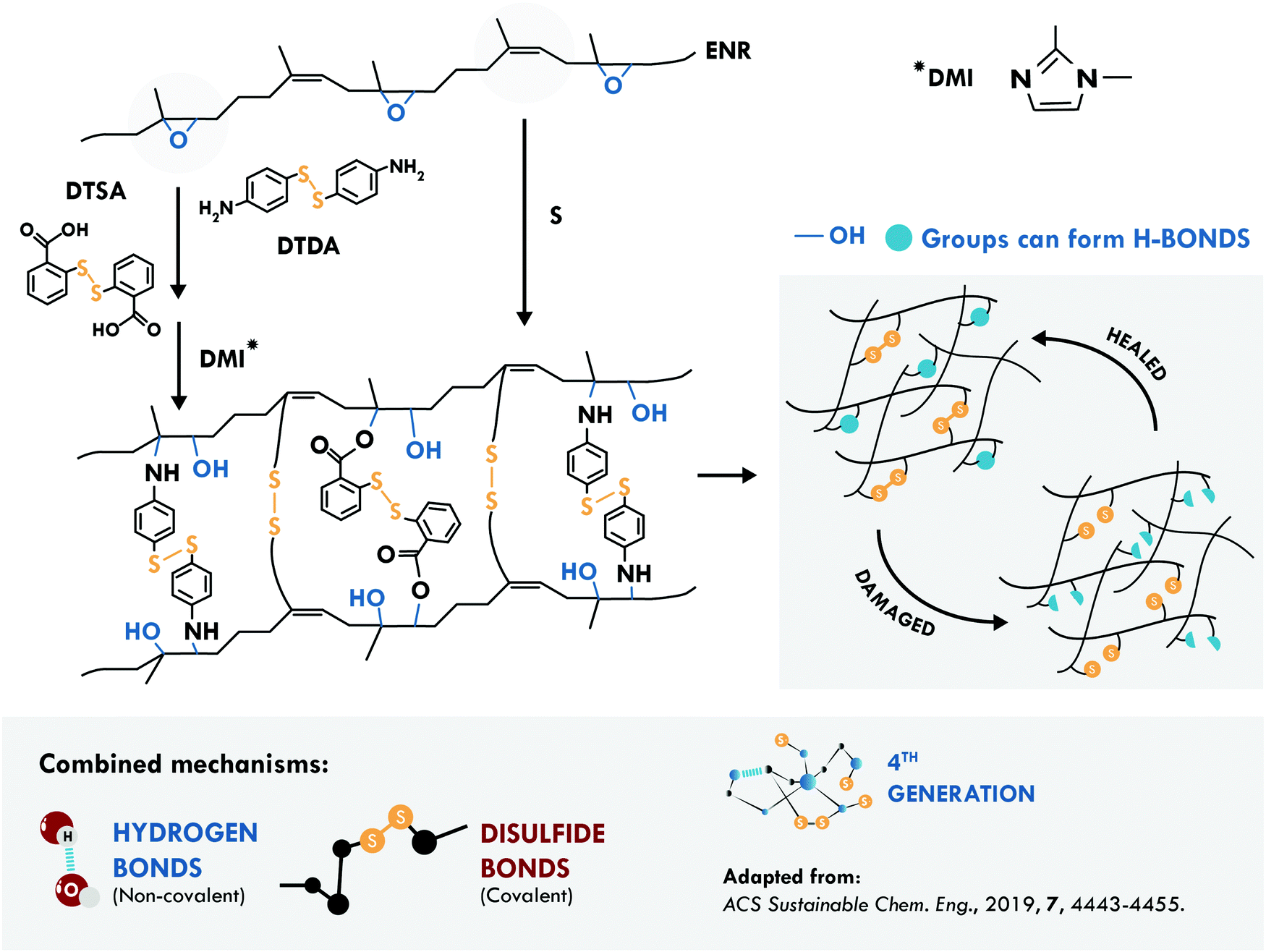 | ||
| Fig. 10 Combination between disulfide and hydrogen bonds in ENR.194 ©2019. Adapted with permission from the American Chemical Society. | ||
The metathesis reaction can also occur in other bonds formed by chalcogens (elements of group 16 of the periodic table, like sulfur). Fan et al. studied the exchange of ditelluride206 and diselenide.207 In both cases, water-dispersed supramolecular polymers (WSP) were prepared with aliphatic molecules with exchangeable bonds and pendant UPy groups. The latter, as shown in other studies, are capable of forming quadruple hydrogen bonds, with an important contribution to the mechanical properties and healing efficiency. This system resembled that of hydrogen and disulfide bonds, but with an interesting contribution: the exchange reaction occurred thanks to visible light. Both studies reported tensile strengths between 15 and 19 MPa with healing efficiencies around 85% at room temperature.
In 1950, Otto Diels and Kurt Alder received the Nobel Prize in Chemistry for their work on diene reactions.217 More than half a century later, Diels–Alder (DA) chemistry has widely been used in the design of dynamic networks to overcome the irreversibility of most crosslink reactions.218 Hence, it is not surprising that this chemistry represents one of the main mechanisms used in the design of self-healing materials. The DA reaction has been combined with hydrogen bonds for the design of two self-healing materials. Zhao et al.200 applied it in poly(siloxane-urethane). In one case, they used isocyanate terminated PDMS combined with a DA diol (PDMS-DA-PU), while in a second case, they employed mixtures of the isocyanate terminated PDMS with a PCL diol and a DA diol (PDMS/PCL-DA-PU) (Fig. 11). Following the evolution of a crack, they concluded that the PCL system hindered the repair; however, it increased the stiffness of the soft segments. Meanwhile, the sample without PCL allowed total healing of the crack, recovering 96% of their initial tensile stress (1.1 MPa) compared to 83% efficiency of the PDMS/PCL-DA-PU samples (tensile strength of 3.25 MPa). The low mobility of PCL hindered the approach of the furan and maleimide groups to reform the DA adduct, but the hydrogen bonds could compensate and maintain a good healing efficiency with a considerable increase in the mechanical properties. Besides, the biocompatibility of the material was proved by cytotoxicity evaluation and animal wound healing experiments, making it a good candidate for applications of smart self-healing artificial skin.
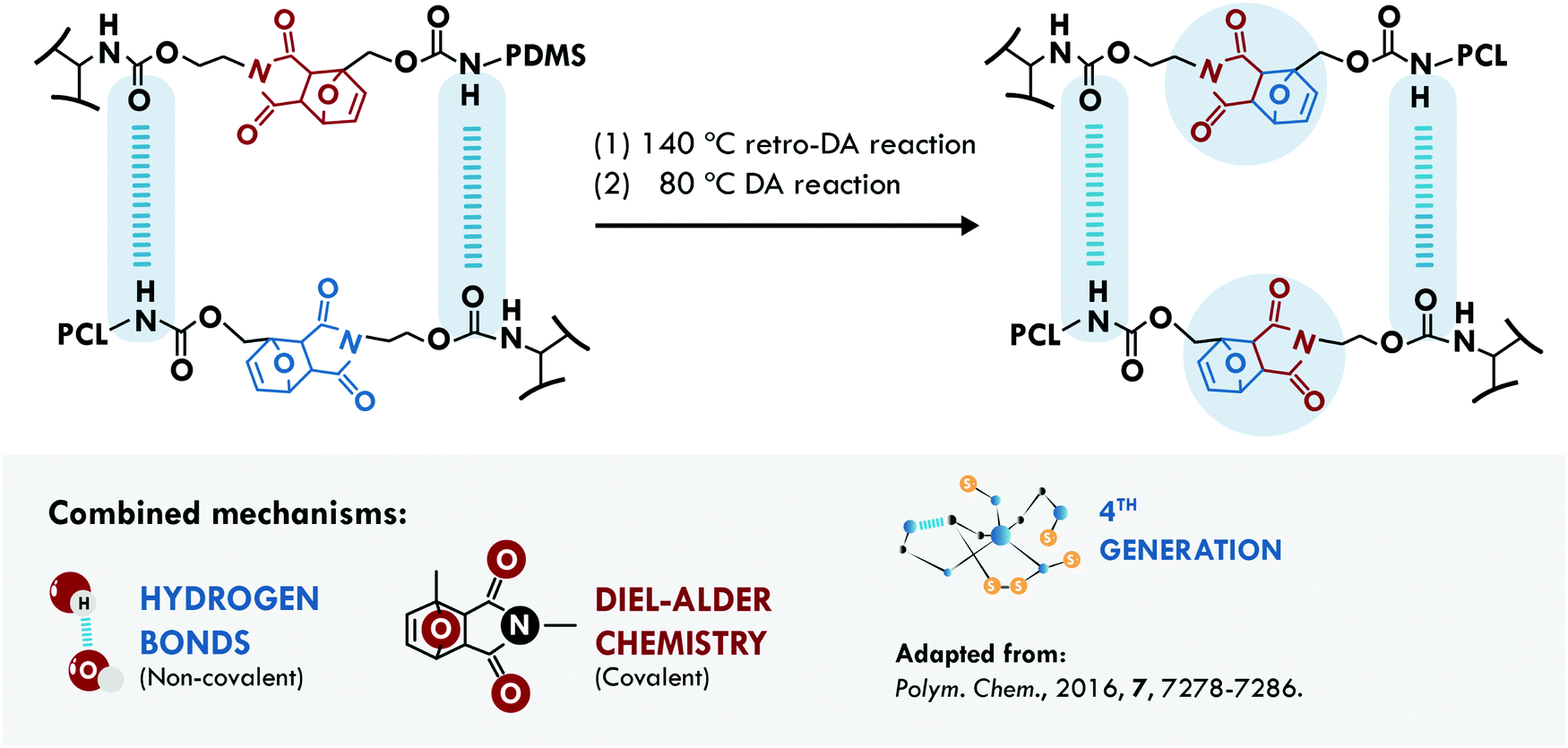 | ||
| Fig. 11 Combination between DA chemistry and hydrogen bonds in PDMS-DA-PU.200 ©2016. Adapted with permission from the Royal Society of Chemistry. | ||
Yang et al.201 also explored the combination of DA chemistry with hydrogen bonds. They prepared a reversible dual network by combining tri(2-furyl) phosphoramide (TFP) with a maleimide end-capped PU elastomer (MPU). With a precise stoichiometric relationship, these systems enabled the crosslinking of PU with TFP via DA chemistry. The healing mechanism comprised three stages: the rupture of the DA adduct through the rDA reaction at high temperature; then, the diffusion of the TFP and PU chains along the damaged surface; and, finally, the reformation of the DA bonds and hydrogen bonds between the hard segments of MPU and between MPU and TFP at 60 °C. This results in a self-healing elastomer with unprecedented mechanical properties (37.11 MPa) and a healing efficiency of 92%.
Another dynamic covalent link combined with hydrogen bonds is the boroxine bond. Guo et al.202 modified an ENR matrix with 3-aminophenylboronic acid (APB). This introduced a functionality with pendant secondary amines and boron-hydroxyl groups thanks to the ring-opening reaction of the epoxy ring. The formation of boroxine bonds between the boron and hydroxyl groups, through the dehydration reaction, and the formation of hydrogen bonds between the secondary amines, gave the material a reversible character. This rubber exhibited a healing efficiency of 91% at room temperature (based on toughness) and, even after 10 healing cycles, continued to reach high efficiencies (71%), this reduction being attributed to the irreversible rupture of covalent bonds after each test. This material not only exhibited great self-healing performance, but also generated a fluorescent response under deformation and showed sensitivity to external stimuli such as strain and humidity. They also tested the material in the throat of a volunteer and observed the strain-dependent sensing of different sounds and expressions, proving its potential application to identify human actions.
The imine bond has also been used repeatedly for the development of dual elastomeric networks with hydrogen bonds, mainly in PDMS. Yan et al.203 developed a network with high elongation and healing efficiency, 93% at room temperature, capable of being repaired at low temperatures, such as −20 °C. This study is one of the few cases that considered freezing temperatures in self-healing. Meanwhile Yang et al.205 reported the creation of a PDMS sensor with a healing efficiency of more than 70% at room temperature and in different media such as air, water and artificial sweat. These sensors were capable of detecting human movements. Hence, the use of self-healing silicones as soft electronics and sensors and in other applications that do not require high mechanical properties is a very promising field.
All the studies described above involved the use of hydrogen bonds as the non-covalent contribution; however, they are not the only system used. Shape memory has also been shown to facilitate self-healing. Xu et al.208 and Chang et al.209 used it in combination with disulfide bonds to develop PU. In both cases, shape memory eliminated the gap between the two areas of the damaged surface, guaranteeing good contact and ensuring high healing efficiency. Other non-covalent systems were used as ionic interactions combined with DA chemistry in an acrylic copolymer.211 In the last year, metal–ligand coordination has also been combined with different covalent systems and matrices such as ENR (with boron-based bonds212), PDMS (with imine bonds213), PU (with DA chemistry214) and PUU (with disulfide bonds215), but with very different results that limit establishing yet a clear trend for these combinations.
4. Outlook and perspectives
This minireview has defined what we dubbed “generations of self-healing materials”, providing a short overview of their evolution and focusing on the latest advances in the field. Thus, it has been shown that over the last twenty years self-healing materials have evolved from simple systems that only supported a single healing cycle to systems capable of: supporting multiple cycles; being activated through sunlight, UV light or temperature; being repaired in different media than air; or being healed at room temperature within a few minutes. Remarkably, all these systems can be processed using conventional techniques and with more innovative methods such as 3D printing.These improvements have mostly arisen over a short period of time with the development of the fourth generation of self-healing materials. These systems have evolved from the knowledge gained over previous generations, which are still under study. They are based on the combination of different healing mechanisms and could represent the path for making the definitive jump to commercial applications. One plausible explanation for the success of this approach is that covalent and non-covalent mechanisms complement each other: dynamic covalent bonds bring considerable improvements in mechanical properties and partially contribute to the self-healing capability, while non-covalent interactions act as the main sacrificial bonds providing substantial progress to the healing efficiency. In the near future, combinations will surely not be limited to two mechanisms, but it is likely that multiple combinations will be explored (three or more), seeking further positive effects.219 The use of fillers or other additives as carriers of additional healing mechanisms could also be a promising option,220,221 while molecular dynamics studies seem mandatory for simulating interactions between combined mechanisms and for predicting their effect on the self-healing capability.181,222–224
Fig. 12 shows the evolution between generations with a general and not-material specific trend towards self-healing elastomers with excellent mechanical performance (tensile strength higher than 10 MPa) and healing efficiencies higher than 80% in the absence of external stimuli. Special attention should be paid to general-purpose elastomers, such as natural rubber, or styrene-butadiene rubber, since most of the developments reported in the fourth generation concern rubbers for specific applications, such as polyurethanes or silicones. However, and despite great efforts, self-healing rubbers, and self-healing materials in general, continue to present serious limitations that should be resolved in the near future.
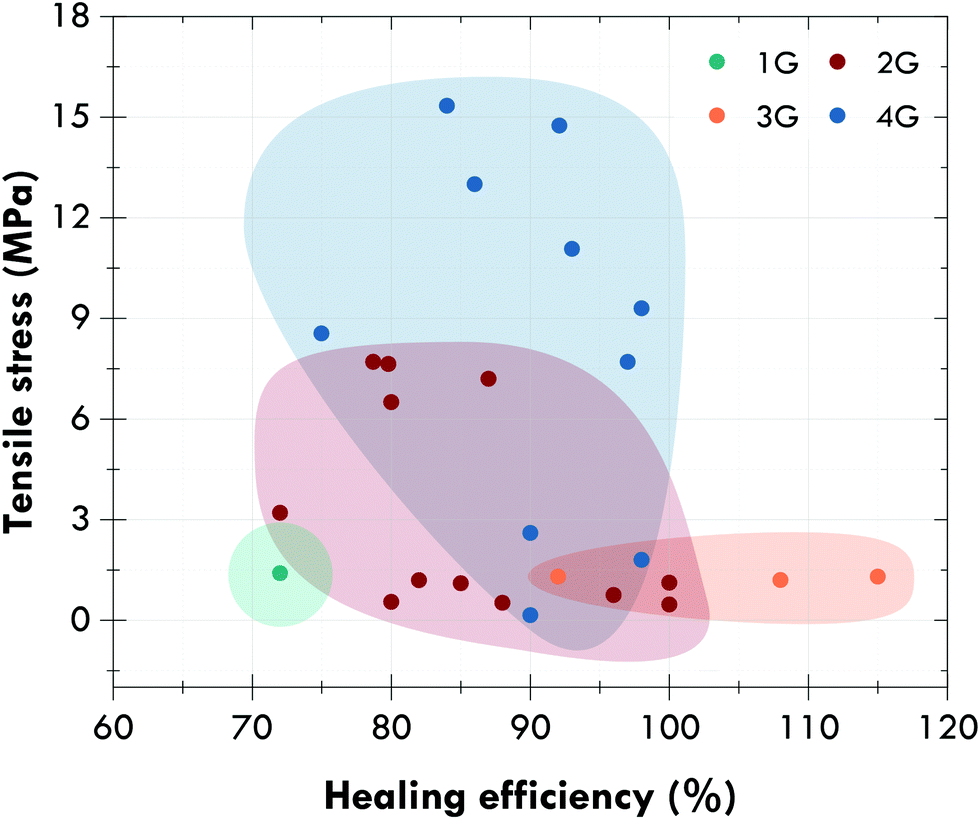 | ||
| Fig. 12 Ashby diagram of the four generations of self-healing elastomers. | ||
One of the main limitations is the absence of a unified protocol that permits quantifying the healing capability and, thus, establishing comparisons between systems or families of materials. This limitation becomes more important when trying to compare generations, where measuring protocols are different. As an illustrative example, healing within the first generation (encapsulated extrinsic systems) is usually measured through fracture toughness (KIC) tests, while in the second and fourth (essentially intrinsic systems), healing is predominantly measured by tensile tests. This difference in criteria, even within the same generation, makes it difficult to establish fair comparisons between materials. The second limitation is related to the type of damage; it is urgent to think about their real scalability. Evaluating the tensile behavior and fracture toughness or monitoring superficial cracks is insufficient to ensure the behavior in service of self-healing materials. This scalability also refers to the evaluation of self-healing not only from the point of view of structural properties, but also regarding key limitations and properties defined for specific applications. Fatigue resistance, thermal and electrical conductivity, aging and the dichotomy itself between the dynamic nature of the reversible bonds and the stability of the materials (for example, chemical and thermal), among others, should be considered as soon as possible, to make the practical applicability of these strategies a reality.
Finally, according to World Bank estimates, global waste will grow 70% by 2050, increasing from 2.1 billion tons registered in 2016 to 3.4 billion tons.225 More than 12% of this waste will be of polymeric origin. This review has presented the latest advances related to an effective alternative for reducing waste, extending the lifetime, and contributing to the evolution of smart materials. We hope that it will serve as an inspiration to researchers around the world to join efforts and make progress towards a more sustainable society. The world needs it.
List of abbreviations
| AAm | Acrylic amide |
| AMPS | Aminopropyl methyl phenyl polysiloxanes |
| APB | 3-Aminophenylboronic acid |
| BIIR | Brominated butyl rubber |
| BNR | Brominated natural rubber |
| bPEI | Branched poly(ethyleneimine) |
| BR | Butadiene rubber |
| ChCl | Choline chloride |
| CR | Chloroprene rubber |
| DA | Diels–Alder |
| DCPD | Dicyclopentadiene |
| DTDA | 4,4-Dithiodianiline |
| DTSA | 2,2-Dithiodibenzoic acid |
| EMAA | Ethylene-methacrylic acid |
| ENR | Epoxidized natural rubber |
| EPDM | Ethylene propylene diene rubber |
| ES | Electrospinning |
| FTIR | Fourier-transform infrared spectroscopy |
| hbPAM | Hyperbranched polyazomethine |
| HPLC | High-performance liquid chromatography |
| HTPB | Hydroxy-terminated polybutadiene |
| IPDI | Isophorone diisocyanate |
| IR | Poly(isoprene) |
| IIR | Butyl rubber |
| MA | Maleic acid |
| MPU | Maleimide end-capped polyurethane elastomer |
| NBR | Nitrile rubber |
| NR | Natural rubber |
| PAA | Poly(acrylic acid) |
| PAN | Polyacrylonitrile |
| PBMM | Poly(butyl methanol methacrylate) |
| PBS | Poly(butylene succinate) |
| PCL | ε-Caprolactone |
| P-Cur | Curcumin polymer block |
| PDMS | Poly(dimethylsiloxane) |
| PEO | Poly(ethylene oxide) |
| PMMA | Poly(methyl methacrylate) |
| POU | Poly(oxime-urethane) |
| PPC | Poly(propylene carbonate) |
| PPG | Poly(propylene glycol)-based thiol |
| PS | Polystyrene |
| PU | Polyurethane |
| PUE | Polyurea elastomer |
| PUU | Poly(urea-urethane) |
| PVA | Poly(vinyl alcohol) |
| PVAc | Poly(vinyl acetate) |
| SBR | Styrene butadiene rubber |
| SBS | Styrene butadiene styrene rubber |
| SEM | Scanning electron microscopy |
| SM | Shape memory |
| TFP | Tri(2-furyl) phosphoramide |
| TPU | Thermoplastic polyurethane |
| UPy | 2-Ureido-4[1H]-pyrimidione |
| UV | Ultraviolet |
| WBPU | Waterborne polyurethane |
| WSP | Water-soluble supramolecular polymer |
| XSBR | Carboxylated styrene butadiene rubber |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Ministry of Science and Innovation of Spain for a research contracts (MAT2015-73392-JIN and PID2019-107501RB-I00) and M. Hernández Santana for a Ramón y Cajal contract (RYC-2017-22837).References
- J. Korhonen, A. Honkasalo and J. Seppälä, Ecol. Econ., 2018, 143, 37–46 CrossRef.
- M. A. Reuter, A. van Schaik, J. Gutzmer, N. Bartie and A. Abadías-Llamas, Annu. Rev. Mater. Res., 2019, 49, 253–274 CrossRef CAS.
- S. Ramarad, M. Khalid, C. T. Ratnam, A. L. Chuah and W. Rashmi, Prog. Mater. Sci., 2015, 72, 100–140 CrossRef CAS.
- K. Aoudia, S. Azem, N. Ait Hocine, M. Gratton, V. Pettarin and S. Seghar, Waste Manage., 2017, 60, 471–481 CrossRef CAS PubMed.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179–211 CrossRef CAS.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- J. Dahlke, S. Zechel, M. D. Hager and U. S. Schubert, Adv. Mater. Interfaces, 2018, 5, 1800051 CrossRef.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- M. Hernández Santana, M. den Brabander, S. García and S. van der Zwaag, Polym. Rev., 2018, 58, 585–609 CrossRef.
- Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. B, 2019, 7, 4876–4926 RSC.
- R. V. S. P. Sanka, B. Krishnakumar, Y. Leterrier, S. Pandey, S. Rana and V. Michaud, Front. Mater., 2019, 6 Search PubMed.
- S. Van der Zwaag and E. Brinkman, in Self Healing Materials: Pioneering Research in the Netherlands, ed. S. Van der Zwaag and E. Brinkman, IOS Press, The Netherlands, 2015 Search PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- B. J. Blaiszik, N. R. Sottos and S. R. White, Compos. Sci. Technol., 2008, 68, 978–986 CrossRef CAS.
- X.-F. Wu, A. Rahman, Z. Zhou, D. D. Pelot, S. Sinha-Ray, B. Chen, S. Payne and A. L. Yarin, J. Appl. Polym. Sci., 2013, 129, 1383–1393 CrossRef CAS.
- X. Y. Jia, J. F. Mei, J. C. Lai, C. H. Li and X. Z. You, Macromol. Rapid Commun., 2016, 37, 952–956 CrossRef CAS PubMed.
- L. Feng, Z. Yu, Y. Bian, J. Lu, X. Shi and C. Chai, Polymer, 2017, 124, 48–59 CrossRef CAS.
- C. Xu, L. Cao, B. Lin, X. Liang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17728–17737 CrossRef CAS PubMed.
- D. Wang, J. Guo, H. Zhang, B. Cheng, H. Shen, N. Zhao and J. Xu, J. Mater. Chem. A, 2015, 3, 12864–12872 RSC.
- J. Araujo-Morera, M. Hernández Santana, R. Verdejo and M. A. López-Manchado, Polymers, 2019, 11, 2122 CrossRef CAS PubMed.
- B. Willocq, J. Odent, P. Dubois and J.-M. Raquez, RSC Adv., 2020, 10, 13766–13782 RSC.
- M. W. Lee, S. An, S. S. Yoon and A. L. Yarin, Adv. Colloid Interface Sci., 2018, 252, 21–37 CrossRef CAS PubMed.
- Y. M. Malinskii, V. V. Prokopenko, N. A. Ivanova and V. A. Kargin, Polym. Mech., 1970, 6, 240–244 CrossRef.
- Y. M. Malinskii, V. V. Prokopenko, N. A. Ivanova and V. S. Kargin, Polym. Mech., 1970, 6, 382–384 CrossRef.
- Y. M. Malinskii, V. V. Prokopenko and V. A. Kargin, Polym. Mech., 1970, 6, 969–972 CrossRef.
- K. Jud, H. H. Kausch and J. G. Williams, J. Mater. Sci., 1981, 16, 204–210 CrossRef CAS.
- R. P. Wool and K. M. O’Connor, J. Appl. Phys., 1981, 52, 5953–5963 CrossRef CAS.
- R. P. Wool and K. M. O’Connor, J. Polym. Sci., Polym. Lett. Ed., 1982, 20, 7–16 CrossRef CAS.
- L. Zhai, A. Narkar and K. Ahn, Nano Today, 2020, 30, 100826, DOI:10.1016/j.nantod.2019.100826.
- M. D. Ellul and A. N. Gent, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 1953–1968 CrossRef CAS.
- C. Dry, Int. J. Mod. Phys. B, 1992, 06, 2763–2771 CrossRef CAS.
- C. M. Dry and N. R. Sottos, Proc. SPIE 1916, Smart Structures and Materials 1993: Smart Materials, 1993, 1916.
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399–2404 CrossRef CAS.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175–220 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS PubMed.
- M. W. Lee, S. An, C. Lee, M. Liou, A. L. Yarin and S. S. Yoon, J. Mater. Chem. A, 2014, 2 Search PubMed.
- S. An, M. Liou, K. Y. Song, H. S. Jo, M. W. Lee, S. S. Al-Deyab, A. L. Yarin and S. S. Yoon, Nanoscale, 2015, 7, 17778–17785 RSC.
- M. W. Lee, S. An, H. S. Jo, S. S. Yoon and A. L. Yarin, ACS Appl. Mater. Interfaces, 2015, 7, 19546–19554 CrossRef CAS PubMed.
- M. W. Lee, S. An, H. S. Jo, S. S. Yoon and A. L. Yarin, ACS Appl. Mater. Interfaces, 2015, 7, 19555–19561 CrossRef CAS PubMed.
- U. S. Chung, J. H. Min, P.-C. Lee and W.-G. Koh, Colloids Surf., A, 2017, 518, 173–180 CrossRef CAS.
- M. W. Lee, S. An, Y.-I. Kim, S. S. Yoon and A. L. Yarin, Chem. Eng. J., 2018, 334, 1093–1100 CrossRef CAS.
- Y. Zhu, Q. Shen, L. Wei, X. Fu, C. Huang, Y. Zhu, L. Zhao, G. Huang and J. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 29373–29381 CrossRef CAS PubMed.
- Q. Guo, X. Zhang, F. Zhao, Q. Song, G. Su, Y. Tan, Q. Tao, T. Zhou, Y. Yu, Z. Zhou and C. Lu, ACS Nano, 2020, 14(3), 2788–2797, DOI:10.1021/acsnano.9b09802.
- R. Li, T. Fan, G. Chen, K. Zhang, B. Su, J. Tian and M. He, Chem. Mater., 2020, 32(2), 874–881, DOI:10.1021/acs.chemmater.9b04592.
- Z. He, S. Jiang, N. An, X. Li, Q. Li, J. Wang, Y. Zhao and M. Kang, J. Mater. Sci., 2019, 54, 8262–8275 CrossRef CAS.
- Z. Yin, J. Guo, J. Qiao and X. Chen, Colloid Polym. Sci., 2020, 298, 67–77, DOI:10.1007/s00396-019-04587-2.
- Y. Cao, H. Wu, S. I. Allec, B. M. Wong, D. S. Nguyen and C. Wang, Adv. Mater., 2018, 30, 1804602 CrossRef PubMed.
- J. Nie, W. Mou, J. Ding and Y. Chen, Composites, Part B, 2019, 172, 152–160 CrossRef CAS.
- L. Cao, D. Yuan, C. Xu and Y. Chen, Nanoscale, 2017, 9, 15696–15706 RSC.
- S. Utrera-Barrios, M. Hernández Santana, R. Verdejo and M. A. López-Manchado, ACS Omega, 2020, 5, 1902–1910 CrossRef CAS PubMed.
- C. Xu, J. Nie, W. Wu, Z. Zheng and Y. Chen, ACS Sustainable Chem. Eng., 2019, 7, 15778–15789 CrossRef CAS.
- D.-D. Zhang, Y.-B. Ruan, B.-Q. Zhang, X. Qiao, G. Deng, Y. Chen and C.-Y. Liu, Polymer, 2017, 120, 189–196 CrossRef CAS.
- P.-F. Cao, B. Li, T. Hong, J. Townsend, Z. Qiang, K. Xing, K. D. Vogiatzis, Y. Wang, J. W. Mays, A. P. Sokolov and T. Saito, Adv. Funct. Mater., 2018, 28 Search PubMed.
- J. Kang, D. Son, G. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. Tok and Z. Bao, Adv. Mater., 2018, 30, e1706846 CrossRef.
- E. Ogliani, L. Yu, I. Javakhishvili and A. L. Skov, RSC Adv., 2018, 8, 8285–8291 RSC.
- L. Chen, H. Peng, Y. Wei, X. Wang, Y. Jin, H. Liu and Y. Jiang, Macromol. Chem. Phys., 2019, 220 Search PubMed.
- Y. Liu, K. Zhang, J. Sun, J. Yuan, Z. Yang, C. Gao and Y. Wu, Ind. Eng. Chem. Res., 2019, 58(47), 21452–21458, DOI:10.1021/acs.iecr.9b03953.
- R. Du, Z. Xu, C. Zhu, Y. Jiang, H. Yan, H. C. Wu, O. Vardoulis, Y. Cai, X. Zhu, Z. Bao, Q. Zhang and X. Jia, Adv. Funct. Mater., 2019, 30 Search PubMed.
- L. Zhang, D. Wang, L. Xu, X. Zhang, A. Zhang and Y. Xu, J. Mater. Chem. C, 2020, 8, 2043–2053, 10.1039/c9tc05612b.
- X. Qi, J. Zhang, L. Zhang and D. Yue, J. Mater. Sci., 2020, 55, 4940–4951, DOI:10.1007/s10853-019-04272-3.
- L. Liu, L. Zhu and L. Zhang, Macromol. Chem. Phys., 2018, 219 Search PubMed.
- A. M. Grande, S. J. Garcia and S. van der Zwaag, Polymer, 2015, 56, 435–442 CrossRef CAS.
- L. Liu, W. Zhang, N. Ning and L. Zhang, Chem. Eng. J., 2019, 375 Search PubMed.
- F. Sordo, S.-J. Mougnier, N. Loureiro, F. Tournilhac and V. Michaud, Macromolecules, 2015, 48, 4394–4402 CrossRef CAS.
- B. Wu, Z. Liu, Y. Lei, Y. Wang, Q. Liu, A. Yuan, Y. Zhao, X. Zhang and J. Lei, Polymer, 2020, 186, 122003, DOI:10.1016/j.polymer.2019.122003.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- F. Fan and J. Szpunar, J. Appl. Polym. Sci., 2015, 132, 42135 Search PubMed.
- M. Li, B. Zhou, Q. Lyu, L. Jia, H. Tan, Z. Xie, B. Xiong, Z. Xue, L. Zhang and J. Zhu, Mater. Chem. Front., 2019, 3, 2707–2715 RSC.
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 CrossRef.
- R. Li, T. Fan, G. Chen, H. Xie, B. Su and M. Hui He, Chem. Eng. J., 2020, 393, 124685, DOI:10.1016/j.cej.2020.124685.
- C. Xu, L. Cao, X. Huang, Y. Chen, B. Lin and L. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 29363–29373 CrossRef CAS.
- S. R. Khimi, S. N. Syamsinar and T. N. L. Najwa, Mater. Today: Proc., 2019, 17, 1064–1071 CAS.
- N. L. N. Thajudin, N. S. Sardi, M. H. Zainol and R. K. Shuib, J. Rubber Res., 2019, 22, 203–211, DOI:10.1007/s42464-019-00025-8.
- Y. Liu, Z. Li, R. Liu, Z. Liang, J. Yang, R. Zhang, Z. Zhou and Y. Nie, Ind. Eng. Chem. Res., 2019, 58, 14848–14858 CrossRef CAS.
- X. Yang, J. Liu, D. Fan, J. Cao, X. Huang, Z. Zheng and X. Zhang, Chem. Eng. J., 2020, 389, 124448, DOI:10.1016/j.cej.2020.124448.
- A. Das, A. Sallat, F. Bohme, M. Suckow, D. Basu, S. Wiessner, K. W. Stockelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS PubMed.
- H. H. Le, F. Böhme, A. Sallat, S. Wießner, M. auf der Landwehr, U. Reuter, K.-W. Stöckelhuber, G. Heinrich, H.-J. Radusch and A. Das, Macromol. Mater. Eng., 2017, 302, 1600385 CrossRef.
- A. Das, A. Sallat, F. Bohme, E. Sarlin, J. Vuorinen, N. Vennemann, G. Heinrich and K. W. Stockelhuber, Polymers, 2018, 10, 94 CrossRef PubMed.
- Z.-F. Zhang, K. Yang, S.-G. Zhao and L.-N. Guo, Chin. J. Polym. Sci., 2019, 37, 700–707 CrossRef CAS.
- F. B. Madsen, L. Yu and A. L. Skov, ACS Macro Lett., 2016, 5, 1196–1200 CrossRef CAS.
- Z. Liu, P. Hong, Z. Huang, T. Zhang, R. Xu, L. Chen, H. Xiang and X. Liu, Chem. Eng. J., 2020, 387 Search PubMed.
- Y. Peng, L. Zhao, C. Yang, Y. Yang, C. Song, Q. Wu, G. Huang and J. Wu, J. Mater. Chem. A, 2018, 6, 19066–19074 RSC.
- H. H. Le, S. Hait, A. Das, S. Wiessner, K. W. Stoeckelhuber, F. Boehme, R. Uta, K. Naskar, G. Heinrich and H. J. Radusch, eXPRESS Polym. Lett., 2017, 11, 230–242 CrossRef CAS.
- W. Wu, Y. Zhou, J. Li and C. Wan, J. Appl. Polym. Sci., 2020, 48666, DOI:10.1002/app.48666.
- M. Suckow, A. Mordvinkin, M. Roy, N. K. Singha, G. Heinrich, B. Voit, K. Saalwächter and F. Böhme, Macromolecules, 2017, 51, 468–479 CrossRef.
- A. Sallat, A. Das, J. Schaber, U. Scheler, E. S. Bhagavatheswaran, K. W. Stöckelhuber, G. Heinrich, B. Voit and F. Böhme, RSC Adv., 2018, 8, 26793–26803 RSC.
- Z. F. Zhang, X. T. Liu, K. Yang and S. G. Zhao, Macromol. Res., 2019, 27, 803–810 CrossRef CAS.
- X. Y. Jia, J. F. Mei, J. C. Lai, C. H. Li and X. Z. You, Chem. Commun., 2015, 51, 8928–8930 RSC.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- Y. L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y. C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- Y. Lei, Q. Huang, S. Shan, Y. Lin and A. Zhang, New J. Chem., 2019, 43, 17441–17445, 10.1039/c9nj03909k.
- I. Oh, S. I. Jeon, I. J. Chung and C.-H. Ahn, Macromol. Res., 2019, 27, 435–443 CrossRef CAS.
- Y. Lei, W. Huang, Q. Huang and A. Zhang, New J. Chem., 2019, 43, 261–268 RSC.
- H. Tan, Q. Lyu, Z. Xie, M. Li, K. Wang, K. Wang, B. Xiong, L. Zhang and J. Zhu, Adv. Mater., 2019, 31, e1805496 Search PubMed.
- Z. Li, Y. Shan, X. Wang, H. Li, K. Yang and Y. Cui, Chem. Eng. J., 2020, 394 Search PubMed.
- B. Peng, H. Li, Y. Li, Z. Lv, M. Wu and C. Zhao, Chem. Eng. J., 2020, 395 Search PubMed.
- Z. Wang, C. Xie, C. Yu, G. Fei, Z. Wang and H. Xia, Macromol. Rapid Commun., 2018, 39, e1700678 CrossRef PubMed.
- D. Yu, X. Zhao, C. Zhou, C. Zhang and S. Zhao, Macromol. Chem. Phys., 2017, 218 Search PubMed.
- H. Wang, W. Liu, Z. Tu, J. Huang and X. Qiu, Ind. Eng. Chem. Res., 2019, 58, 23114–23123 CrossRef CAS.
- S. Nomimura, M. Osaki, J. Park, R. Ikura, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2019, 52, 2659–2668 CrossRef CAS.
- J. Huang, L. Cao, D. Yuan and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 40996–41002 CrossRef CAS PubMed.
- P. Tanasi, M. Hernández Santana, J. Carretero-González, R. Verdejo and M. A. López-Manchado, Polymer, 2019, 175, 15–24 CrossRef CAS.
- X. Kuang, G. Liu, X. Dong and D. Wang, Macromol. Mater. Eng., 2016, 301, 535–541 CrossRef CAS.
- Z. Jia, S. Zhu, Y. Chen, W. Zhang, B. Zhong and D. Jia, Composites, Part A, 2020, 129, 105709 CrossRef.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, J. Mater. Chem. B, 2016, 4, 982–989 RSC.
- A. Nasresfahani and P. M. Zelisko, Polym. Chem., 2017, 8, 2942–2952 RSC.
- J. Wang, C. Lv, Z. Li and J. Zheng, Macromol. Mater. Eng., 2018, 303, 1800089 CrossRef.
- Q. Yan, L. Zhao, Q. Cheng, T. Zhang, B. Jiang, Y. Song and Y. Huang, Ind. Eng. Chem. Res., 2019, 58(47), 21504–21512, DOI:10.1021/acs.iecr.9b04355.
- L. Zhao, X. Shi, Y. Yin, B. Jiang and Y. Huang, Compos. Sci. Technol., 2020, 186, 107919, DOI:10.1016/j.compscitech.2019.107919.
- Y. Fang, X. Du, S. Yang, H. Wang, X. Cheng and Z. Du, Polym. Chem., 2019, 10, 4142–4153 RSC.
- L. Feng, Y. Bian, C. Chai and X. Qiang, J. Polym. Environ., 2020, 28, 647–656, DOI:10.1007/s10924-019-01633-6.
- S. Yang, S. Wang, X. Du, X. Cheng, H. Wang and Z. Du, Polym. Chem., 2020, 11, 1161–1170, 10.1039/c9py01531k.
- T. T. Truong, S. H. Thai, H. T. Nguyen, D. T. T. Phung, L. T. Nguyen, H. Q. Pham and L.-T. T. Nguyen, Chem. Mater., 2019, 31, 2347–2357 CrossRef CAS.
- Y. Fang, J. Li, X. Du, Z. Du, X. Cheng and H. Wang, Polymer, 2018, 158, 166–175 CrossRef CAS.
- G. Fu, L. Yuan, G. Liang and A. Gu, J. Mater. Chem. A, 2016, 4, 4232–4241 RSC.
- T. Wang, W.-C. Yu, C.-G. Zhou, W.-J. Sun, Y.-P. Zhang, L.-C. Jia, J.-F. Gao, K. Dai, D.-X. Yan and Z.-M. Li, Composites, Part B, 2020, 193 CAS.
- S. Schäfer and G. Kickelbick, Polymer, 2015, 69, 357–368 CrossRef.
- Z. Gou, Y. Zuo and S. Feng, RSC Adv., 2016, 6, 73140–73147 RSC.
- J. Bai, H. Li, Z. Shi, M. Tian and J. Yin, RSC Adv., 2015, 5, 45376–45383 RSC.
- J. Bai, Q. He, Z. Shi, M. Tian, H. Xu, X. Ma and J. Yin, Polymer, 2017, 116, 268–277 CrossRef CAS.
- J. Bai and Z. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 27213–27222 CrossRef CAS PubMed.
- G. Li, P. Xiao, S. Hou and Y. Huang, Carbon, 2019, 147, 398–407 CrossRef CAS.
- J. Brancart, R. Verhelle, J. Mangialetto and G. V. Assche, Coatings, 2018, 9 Search PubMed.
- S. Terryn, G. Mathijssen, J. Brancart, D. Lefeber, G. V. Assche and B. Vanderborght, Bioinspiration Biomimetics, 2015, 10, 046007 CrossRef PubMed.
- S. Terryn, J. Brancart, D. Lefeber, G. Van Assche and B. Vanderborght, Sci. Robot., 2017, 2, eaan4268 CrossRef.
- S. Terryn, J. Brancart, D. Lefeber, G. Van Assche and B. Vanderborght, IEEE Robot. Autom. Lett., 2018, 3, 16–21 Search PubMed.
- E. Roels, S. Terryn, J. Brancart, R. Verhelle, G. Van Assche and B. Vanderborght, Soft Robot., 2020 DOI:10.1089/soro.2019.0081.
- S. Terryn, E. Roels, J. Brancart, G. V. Assche and B. Vanderborght, Actuators, 2020, 9 Search PubMed.
- M. Hernández, A. M. Grande, W. Dierkes, J. Bijleveld, S. van der Zwaag and S. J. García, ACS Sustainable Chem. Eng., 2016, 4, 5776–5784 CrossRef.
- M. Hernández, A. M. Grande, S. van der Zwaag and S. J. Garcia, ACS Appl. Mater. Interfaces, 2016, 8, 10647–10656 CrossRef PubMed.
- M. Hernández, M. M. Bernal, A. M. Grande, N. Zhong, S. van der Zwaag and S. J. García, Smart Mater. Struct., 2017, 26, 085010 CrossRef.
- M. Hernández Santana, M. Huete, P. Lameda, J. Araujo, R. Verdejo and M. A. López-Manchado, Eur. Polym. J., 2018, 106, 273–283 CrossRef.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- H. Xiang, J. Yin, G. Lin, X. Liu, M. Rong and M. Zhang, Chem. Eng. J., 2019, 358, 878–890 CrossRef CAS.
- H. P. Xiang, M. Z. Rong and M. Q. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 2715–2724 CrossRef CAS.
- H. P. Xiang, M. Z. Rong and M. Q. Zhang, Polymer, 2017, 108, 339–347 CrossRef CAS.
- L. Zhao, Y. Yin, B. Jiang, Z. Guo, C. Qu and Y. Huang, J. Colloid Interface Sci., 2020, 573, 105–114 CrossRef CAS PubMed.
- Y. Xu and D. Chen, Macromol. Chem. Phys., 2016, 217, 1191–1196 CrossRef CAS.
- L. Zhang, T. Qiu, X. Sun, L. Guo, L. He, J. Ye and X. Li, Polymers, 2020, 12 Search PubMed.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 24224–24231 CrossRef CAS PubMed.
- J. Cao, D. Han, H. Lu, P. Zhang and S. Feng, New J. Chem., 2018, 42, 18517–18520 RSC.
- C. Zhang, Z. Yang, N. T. Duong, X. Li, Y. Nishiyama, Q. Wu, R. Zhang and P. Sun, Macromolecules, 2019, 52, 5014–5025 CrossRef CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- J. Sun, X. Pu, M. Liu, A. Yu, C. Du, J. Zhai, W. Hu and Z. L. Wang, ACS Nano, 2018, 12, 6147–6155 CrossRef CAS PubMed.
- P. Wang, L. Yang, B. Dai, Z. Yang, S. Guo, G. Gao, L. Xu, M. Sun, K. Yao and J. Zhu, Eur. Polym. J., 2020, 123, 109382 CrossRef.
- X. Lei, Y. Huang, S. Liang, X. Zhao and L. Liu, Mater. Lett., 2020, 268, 127598, DOI:10.1016/j.matlet.2020.127598.
- C. Xu, R. Cui, L. Fu and B. Lin, Compos. Sci. Technol., 2018, 167, 421–430 CrossRef CAS.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- Z. Feng, J. Hu, H. Zuo, N. Ning, L. Zhang, B. Yu and M. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 1469–1479 CrossRef CAS PubMed.
- X. Yang, Y. Guo, X. Luo, N. Zheng, T. Ma, J. Tan, C. Li, Q. Zhang and J. Gu, Compos. Sci. Technol., 2018, 164, 59–64 CrossRef CAS.
- K.-H. Wu, L.-F. Feng, X.-P. Gu, C.-L. Zhang and S. Shen, Ind. Eng. Chem. Res., 2018, 57, 946–953 CrossRef CAS.
- D. Fu, W. Pu, Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. A, 2018, 6, 18154–18164 RSC.
- B. Zhao, H. Ding, S. Xu and S. Zheng, ACS Appl. Polym. Mater., 2019, 1(11), 3174–3184, DOI:10.1021/acsapm.9b00830.
- M. W. Lee, S. Sett, S. An, S. S. Yoon and A. L. Yarin, ACS Appl. Mater. Interfaces, 2017, 9, 27223–27231 CrossRef CAS PubMed.
- M. W. Lee, S. S. Yoon and A. L. Yarin, ACS Appl. Mater. Interfaces, 2017, 9, 17449–17455 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- Z. Jiang, A. Bhaskaran, H. M. Aitken, I. C. G. Shackleford and L. A. Connal, Macromol. Rapid Commun., 2019, 40, 1900038 CrossRef PubMed.
- L. Li and J. K. Kim, Rubber Chem. Technol., 2014, 87, 459–470 CrossRef CAS.
- J. Liu, S. Wang, Z. Tang, J. Huang, B. Guo and G. Huang, Macromolecules, 2016, 49, 8593–8604 CrossRef CAS.
- C. Li, Y. Wang, Z. Yuan and L. Ye, Appl. Surf. Sci., 2019, 484, 616–627 CrossRef CAS.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193–199 CrossRef CAS.
- Y. Zuo, Y. Zhang, T. Yang, Z. Gou and W. Lin, New J. Chem., 2018, 42, 14281–14289 RSC.
- C. Xu, J. Nie, W. Wu, L. Fu and B. Lin, Carbohydr. Polym., 2019, 205, 410–419 CrossRef CAS PubMed.
- M. A. Sattar, S. Gangadharan and A. Patnaik, ACS Omega, 2019, 4, 10939–10949 CrossRef PubMed.
- H. Guo, X. Fang, L. Zhang and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 33356–33363 CrossRef CAS PubMed.
- H. Sun, X. Liu, S. Liu, B. Yu, N. Ning, M. Tian and L. Zhang, Chem. Eng. J., 2020, 384, 123242 CrossRef CAS.
- G. Ye, Z. Song, T. Yu, Q. Tan, Y. Zhang, T. Chen, C. He, L. Jin and N. Liu, ACS Appl. Mater. Interfaces, 2020, 12(1), 1486–1494, DOI:10.1021/acsami.9b17354.
- S. Stein, A. Mordvinkin, B. Voit, H. Komber, K. Saalwächter and F. Böhme, Polym. Chem., 2020, 11, 1188–1197, 10.1039/c9py01630a.
- X. Wang, D. Liang and B. Cheng, Compos. Sci. Technol., 2020, 193 CAS.
- J. Liu, J. Liu, S. Wang, J. Huang, S. Wu, Z. Tang, B. Guo and L. Zhang, J. Mater. Chem. A, 2017, 5, 25660–25671 RSC.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J. C. Lai, L. Liu, C. H. Li, X. Yan, C. Liu, J. B. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, e1801435, DOI:10.1002/adma.201801435.
- X. Wu, J. Wang, J. Huang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 7387–7396 CrossRef CAS PubMed.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- X. Cui, Y. Song, J.-P. Wang, J.-K. Wang, Q. Zhou, T. Qi and G. L. Li, Polymer, 2019, 174, 143–149 CrossRef CAS.
- B. Yi, P. Liu, C. Hou, C. Cao, J. Zhang, H. Sun and X. Yao, ACS Appl. Mater. Interfaces, 2019, 11, 47382–47389 CrossRef CAS PubMed.
- S. Xu, D. Sheng, Y. Zhou, H. Wu, H. Xie, X. Tian, Y. Sun, X. Liu and Y. Yang, New J. Chem., 2020, 44, 7395–7400 RSC.
- J. B. Hou, X. Q. Zhang, D. Wu, J. F. Feng, D. Ke, B. J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12105–12113 CrossRef CAS PubMed.
- L. Xiao, J. Shi, K. Wu and M. Lu, React. Funct. Polym., 2020, 148, 104482, DOI:10.1016/j.reactfunctpolym.2020.104482.
- J. Chen, F. Li, Y. Luo, Y. Shi, X. Ma, M. Zhang, D. W. Boukhvalov and Z. Luo, J. Mater. Chem. A, 2019, 7, 15207–15214 RSC.
- M. Tang, P. Zheng, K. Wang, Y. Qin, Y. Jiang, Y. Cheng, Z. Li and L. Wu, J. Mater. Chem. A, 2019, 7, 27278–27288 RSC.
- C. Ellingford, A. M. Wemyss, R. Zhang, I. Prokes, T. Pickford, C. Bowen, V. A. Coveney and C. Wan, J. Mater. Chem. C, 2020, 8, 5426–5436 RSC.
- J. F. Mei, X. Y. Jia, J. C. Lai, Y. Sun, C. H. Li, J. H. Wu, Y. Cao, X. Z. You and Z. Bao, Macromol. Rapid Commun., 2016, 37, 1667–1675 CrossRef CAS PubMed.
- J. Liu, C. Xiao, J. Tang, Y. Liu and J. Hua, Ind. Eng. Chem. Res., 2020, 59(28), 12755–12765, DOI:10.1021/acs.iecr.0c01538.
- C. Lv, K. Zhao and J. Zheng, Macromol. Rapid Commun., 2018, 39, 1700686 CrossRef PubMed.
- S.-H. Lee, S.-R. Shin and D.-S. Lee, Mater. Des., 2019, 172, 107774 CrossRef CAS.
- X. Dai, Y. Du, Y. Wang, Y. Liu, N. Xu, Y. Li, D. Shan, B. B. Xu and J. Kong, ACS Appl. Polym. Mater., 2020, 2, 1065–1072 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 RSC.
- Y. Yang, X. Lu and W. Wang, Mater. Des., 2017, 127, 30–36 CrossRef CAS.
- X. Jian, Y. Hu, W. Zhou and L. Xiao, Polym. Adv. Technol., 2018, 29, 463–469 CrossRef CAS.
- T. Li, Z. Xie, J. Xu, Y. Weng and B.-H. Guo, Eur. Polym. J., 2018, 107, 249–257 CrossRef CAS.
- B. Cheng, X. Lu, J. Zhou, R. Qin and Y. Yang, ACS Sustainable Chem. Eng., 2019, 7, 4443–4455 CrossRef CAS.
- J. Hu, R. Mo, X. Jiang, X. Sheng and X. Zhang, Polymer, 2019, 183, 121912 CrossRef CAS.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef CAS.
- W. Yao, X. Chen, Q. Tian, C. Luo, X. Zhang, H. Peng and W. Wu, Chem. Eng. J., 2020, 384, 123375, DOI:10.1016/j.cej.2019.123375.
- M. Liu, J. Zhong, Z. Li, J. Rong, K. Yang, J. Zhou, L. Shen, F. Gao, X. Huang and H. He, Eur. Polym. J., 2020, 124, 109475 CrossRef CAS.
- H. Guo, Y. Han, W. Zhao, J. Yang and L. Zhang, Nat. Commun., 2020, 11, 2037 CrossRef CAS PubMed.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, Polym. Chem., 2016, 7, 7278–7286 RSC.
- S. Yang, S. Wang, X. Du, Z. Du, X. Cheng and H. Wang, Chem. Eng. J., 2020, 391, 123544, DOI:10.1016/j.cej.2019.123544.
- Q. Guo, B. Huang, C. Lu, T. Zhou, G. Su, L. Jia and X. Zhang, Mater. Horiz., 2019, 6, 996–1004 RSC.
- H. Yan, S. Dai, Y. Chen, J. Ding and N. Yuan, ChemistrySelect, 2019, 4, 10719–10725 CrossRef CAS.
- H. Zhang, D. Wang, N. Wu, C. Li, C. Zhu, N. Zhao and J. Xu, ACS Appl. Mater. Interfaces, 2020, 12(8), 9833–9841, DOI:10.1021/acsami.9b22613.
- Z. Yang, H. Li, L. Zhang, X. Lai and X. Zeng, J. Colloid Interface Sci., 2020, 570, 1–10 CrossRef CAS PubMed.
- W. Fan, Y. Jin, L. Shi, W. Du, R. Zhou, S. Lai, Y. Shen and Y. Li, ACS Appl. Mater. Interfaces, 2020, 12(5), 6383–6395, DOI:10.1021/acsami.9b18985.
- W. Fan, Y. Jin, L. Shi, W. Du and R. Zhou, Polymer, 2020, 190, 122199 CrossRef.
- Y. Xu and D. Chen, Mater. Chem. Phys., 2017, 195, 40–48 CrossRef CAS.
- K. Chang, H. Jia and S.-Y. Gu, Eur. Polym. J., 2019, 112, 822–831 CrossRef CAS.
- Y.-m. Ha, Y.-O. Kim, S. Ahn, S.-k. Lee, J.-s. Lee, M. Park, J. W. Chung and Y. C. Jung, Eur. Polym. J., 2019, 118, 36–44 CrossRef CAS.
- Y. Peng, Y. Yang, Q. Wu, S. Wang, G. Huang and J. Wu, Polymer, 2018, 157, 172–179 CrossRef CAS.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
- J. Pignanelli, B. Billet, M. Straeten, M. Prado, K. Schlingman, M. J. Ahamed and S. Rondeau-Gagne, Soft Matter, 2019, 15, 7654–7662 RSC.
- C. Lin, D. Sheng, X. Liu, S. Xu, F. Ji, L. Dong, Y. Zhou and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2228–2234 CrossRef CAS.
- Y.-H. Li, W.-J. Guo, W.-J. Li, X. Liu, H. Zhu, J.-P. Zhang, X.-J. Liu, L.-H. Wei and A.-L. Sun, Chem. Eng. J., 2020, 393, 124583, DOI:10.1016/j.cej.2020.124583.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- Nature, 1950, 166(4230), 889, DOI:10.1038/166889a0.
- B. Zhang, Z. A. Digby, J. A. Flum, E. M. Foster, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2015, 6, 7368–7372 RSC.
- X. Kuang, K. Chen, C. K. Dunn, J. Wu, V. C. F. Li and H. J. Qi, ACS Appl. Mater. Interfaces, 2018, 10, 7381–7388 CrossRef CAS PubMed.
- C. Lv, J. Wang, Z. Li, K. Zhao and J. Zheng, Composites, Part B, 2019, 177, 107270 CrossRef CAS.
- W.-J. Lee and S.-H. Cha, Polymers, 2020, 12 Search PubMed.
- A. C. Balazs, Mater. Today, 2007, 10, 18–23 CrossRef CAS.
- T. Zhang, B. L. Mbanga, V. V. Yashin and A. C. Balazs, Mol. Syst. Des. Eng., 2017, 2, 490–499 RSC.
- Z. Zhang, J. Liu, S. Li, K. Gao, V. Ganesan and L. Zhang, Macromolecules, 2019, 52, 4154–4168 CrossRef CAS.
- S. Kaza, L. Yao, P. Bhada-Tata and F. Van Woerden, What a waste 2.0: a global snapshot of solid waste management to 2050, The World Bank, 2018 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |