
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperfast and controllable microfluidic inking of anti-inflammatory melanin-like nanoparticles inspired by cephalopods†
Shiqi
Wang‡
 a,
Saowanee
Wannasarit‡
ab,
Patrícia
Figueiredo
a,
Jiachen
Li
ac,
Alexandra
Correia
a,
Bing
Xia
c,
Ruedeekorn
Wiwattanapatapee
b,
Jouni
Hirvonen
a,
Dongfei
Liu
ad,
Wei
Li
*a and
Hélder A.
Santos
*ad
a,
Saowanee
Wannasarit‡
ab,
Patrícia
Figueiredo
a,
Jiachen
Li
ac,
Alexandra
Correia
a,
Bing
Xia
c,
Ruedeekorn
Wiwattanapatapee
b,
Jouni
Hirvonen
a,
Dongfei
Liu
ad,
Wei
Li
*a and
Hélder A.
Santos
*ad
aDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: wei.li@helsinki.fi; helder.santos@helsinki.fi
bDepartment of Pharmaceutical Technology, Faculty of Pharmaceutical Sciences, Prince of Songkla University, 90110 Hat Yai, Thailand
cKey Laboratory of Forest Genetics & Biotechnology (Ministry of Education of China), College of Science, Nanjing Forestry University, 210037 Nanjing, P. R. China
dHelsinki Institute of Life Science (HiLIFE), University of Helsinki, FI-00014 Helsinki, Finland
First published on 26th March 2020
Abstract
Here, a microfluidic approach for superfast melanin-like nanoparticle preparation with tunable size and monodispersity is reported. The particles formed have similar chemical composition to those prepared by the bulk method, and show reactive oxygen species scavenging behaviour and inflammatory macrophage phenotype switching capability, suggesting their potential for anti-inflammatory applications.
New conceptsWe propose a new concept utilizing the microfluidic technique for superfast, controllable and monodisperse melanin nanoparticle (MN) production, inspired by MN biosynthesis from cephalopods. With high biocompatibility, biodegradability and free radical scavenging abilities, melanin has been intensively used as an antioxidant, photothermal agent and surface coating material in the biomedical field. Although traditional MN preparation methods generate homogenous nanoparticles, the lengthy reaction times and unavoidable batch-to-batch variations limit the possibility to tailor particle production towards real-world applications. Thanks to the microfluidic technique, here we demonstrate that monodisperse MNs can be produced in a highly concentrated substrate solution within seconds for the first time. By simply adjusting the parameters of the microfluidic device, we can precisely manipulate the size and the morphology of the produced nanoparticles. These MNs made with the microfluidic method showed similar chemical composition to those prepared by the bulk method. The negligible toxicity towards macrophages, reactive oxygen species scavenging capability, and inflammation attenuating effects towards M1 phenotype macrophages suggest their potential for anti-inflammatory applications. |
Cephalopods, including octopuses, cuttlefish and squids, are well-known for their unique inking behavior when disturbed. The dark-colored ink comprises melanin nanoparticles (MNs) secreted from the ink sac, as well as chemicals and polymeric contents from the funnel gland.1 Cephalopod ink has been used in medicine since ancient times both in Western and Eastern culture, without knowing the real active components.2 Recent studies found that MNs from Sepia ink had superior antioxidant potency due to the reactive oxygen species (ROS) scavenging capability.3–5 Considering the prevalent oxidative stress closely associated with inflammation, especially chronic non-resolving inflammation associated diseases,6,7 MNs from cephalopod ink may show promising potential in anti-inflammatory applications.
The biosynthesis of melanin in cephalopods is a highly regulated process confined in intracellular vesicles called melanosomes (0.5–1 μm in diameter), where tyrosine and 3,4-dihydroxy-L-phenylalanine (DOPA) substrates undergo a series of enzyme-cascade reactions and finally form MNs.8 Unlike the ellipse-shaped melanin microparticles produced by vertebrates, MNs produced by cephalopods are uniform and spherical, with an average size of approximately 150 nm.9 By transmission electron microscopy (TEM) imaging, it was identified that one melanosome can accommodate up to 30 MNs.10 Such concentrated MNs made in the melanosomes are speculated to be synthesized from locally concentrated substrates in a highly catalytic environment, somehow uniquely developed in evolution.
Inspired by the efficient and well-controlled biosynthesis of MNs, we developed a glass capillary-based microfluidic device for producing monodisperse MNs from concentrated dopamine with fine-tuning of the size and morphology (Fig. 1a). Although some artificial MN preparation methods have been reported by bottom-up self-polymerization in alkaline solutions,11–13 only very dilute substrates were allowed at slow reaction rates (typically 1–3 day reaction time) to prepare homogenous MNs. At a higher substrate concentration, the reaction becomes too fast, leading to uncontrollable nanoparticle (NP) formation with heterogeneous properties. In contrast, microfluidic technology enables precise control of superfast liquid mixing and mass transfer in complex, miniaturized capillary networks.14–20 Within milliseconds, all the reagents efficiently mix, resulting in a homogenous environment throughout.21–23
 | ||
| Fig. 1 (a) The schematic illustration of MN preparation on a co-flow glass-capillary microfluidic device in parallel with a cuttlefish inking comparison. (b–d) TEM images of the MNs prepared by the microfluidic device with different parameters (D = 3, 4.5 and 12 cm, respectively). Scale bar = 500 nm in the main image and 100 nm in the inset. (e–g) DLS size distribution profiles of the MNs prepared by the microfluidic device with different parameters (D = 3, 4.5 and 12 cm, respectively). | ||
As shown in Fig. 1a, the co-flow microfluidic device for MN production is composed of three sequentially aligned glass capillaries. Dopamine hydrochloride was dissolved in Milli-Q water as the inner fluid and pumped into the inner tapered glass capillary, while concentrated sodium hydroxide (NaOH) solution, as the 1st outer fluid, was pumped between the inner and 1st outer cylindrical capillary. Once mixed with NaOH, dopamine started self-polymerization followed by oligomer aggregation to form MNs. Then, the 2nd outer fluid, concentrated hydrochloric acid (HCl), was pumped into the space between the 1st and the 2nd outer cylindrical capillaries. When the MNs in the 1st outer fluid encountered the HCl, the self-polymerization of dopamine, which is pH-dependent, was terminated due to the acidification of the solutions. Thus, by controlling the distance (marked as D in Fig. 1a) between the tip of the tapered capillary and the end of the 1st outer cylindrical capillary, the reaction time of dopamine polymerization was precisely controlled, which in turn tuned the size and morphology of the MNs. The variation of D shows the time resolution of MN particle formation by spatial resolution within the microfluidic chip, which provides unique insight into the reaction kinetics.
The size and morphology of the MNs produced by the microfluidic device with different D values (3 cm, 4.5 cm and 12 cm) were characterized by dynamic light scattering (DLS) and TEM. As shown in Fig. 1b–d, when D increased from 3 to 12 cm, the size of the MNs increased and the particles became denser with higher contrast in TEM images. The DLS results also confirm the trend, because the whole distribution profile shifted to larger sizes with an increase in D (Fig. 1e–g). Within 5.08 s (D = 3 cm), loosely assembled NPs with slightly irregular shape (Fig. 1b and inset) were already formed. The rough surface was possibly due to the random aggregation in such a short time. The particle size shown in TEM was smaller than that measured by DLS (Fig. 1e), suggesting that the NPs were possibly quite swollen and hydrated.13 After another 2.54 s (D = 4.5 cm), the hydrodynamic size of the NPs increased by 31 nm, with a smaller PDI. Notably, the MNs became rounder, solid and smooth (Fig. 1c and inset), indicating that the particle growth homogenized the particle surface, and the size distribution became narrower. On elongating the reaction time to 20.32 s (D = 12 cm), the hydrodynamic size of the MNs increased to 381 nm, but still with monodispersity (Fig. 1d and g). At this stage, the aggregation became more evident, and more merged particles could be identified in the TEM image. The overall results shown above suggest that monodisperse MNs of controllable sizes can be easily prepared within a few seconds by this novel microfluidic device, using 100 times more concentrated dopamine substrate (200 mg mL−1 dopamine) compared with the conventional bulk method.13
In comparison, we also used the same condition to produce MNs by the conventional bulk method. After rapidly mixing dopamine and NaOH solutions, HCl was immediately added to terminate the reaction. However, the MNs synthesized were polydisperse with micro-size aggregates (Fig. 2a and b). This was possibly because the reaction and subsequent particle formation were faster than the mixing. Therefore, the spatial and temporal heterogeneities led to uncontrollable particle formation and aggregation. A further increase of the reaction time to 20 h did not make the particles small or homogenous (Fig. S1, ESI†), suggesting that the aggregation right after the NaOH addition was irreversible.
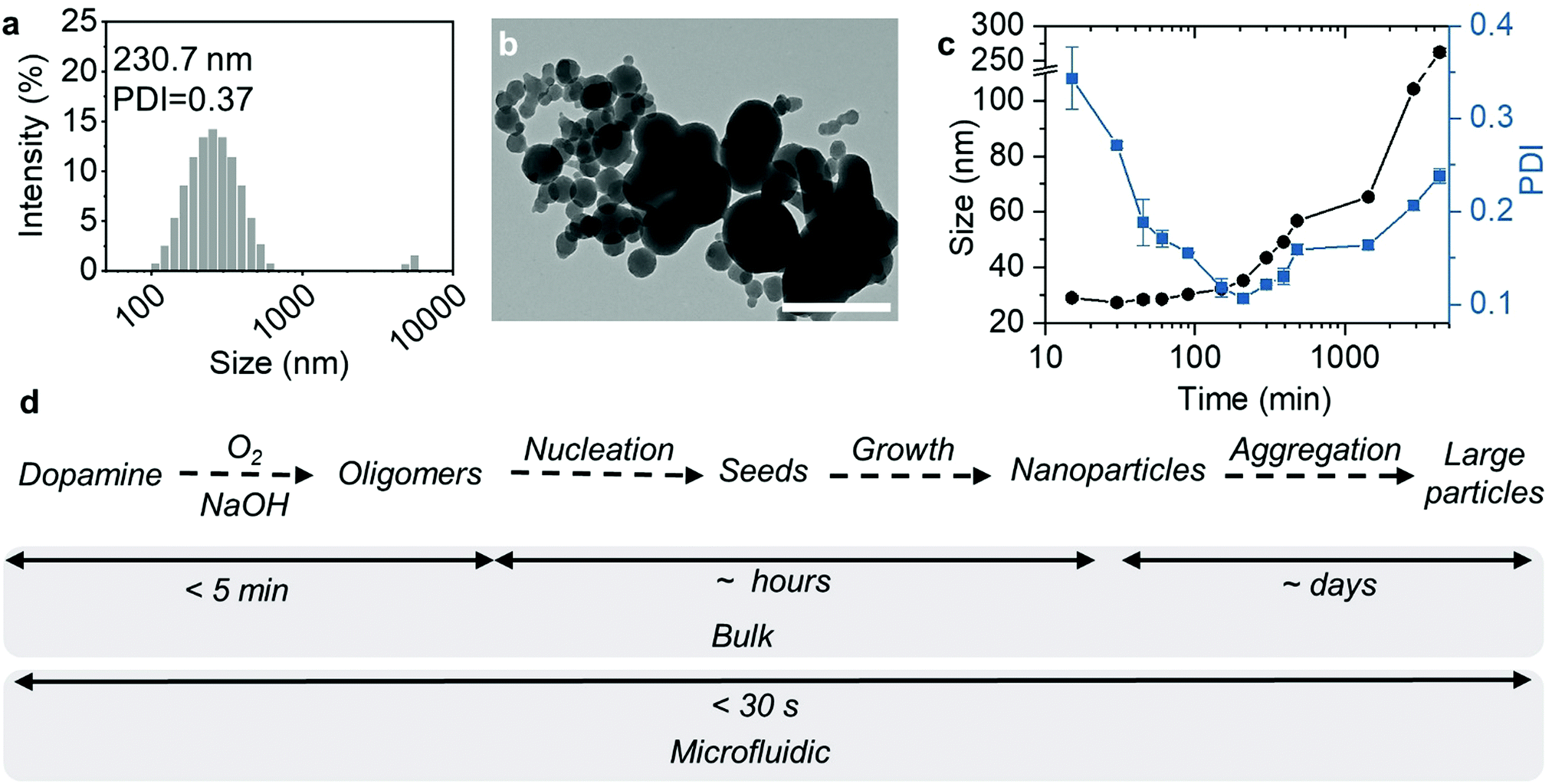 | ||
| Fig. 2 (a and b) DLS distribution profile and TEM image of the MNs prepared by the bulk method at 200 mg mL−1 dopamine. Scale bar = 500 nm. (c) The Z-average size and PDI of MNs prepared by the bulk method at 1.5 mg mL−1 dopamine. (d) The schematic showing MN formation kinetic steps in the conventional bulk method and microfluidic method. | ||
Then, we decreased the dopamine concentration to 1.5 mg mL−1, close to the concentration range reported in conventional bulk MN synthesis methods.13 As shown in Fig. 2c, MN formation was significantly slower at such a low concentration of dopamine. Although the colour of the reaction solution changed to dark yellow immediately after the reaction was initiated (Fig. S2, ESI†), the count rate in the first 5 min remained almost the same, without any identifiable particles (Fig. S3, ESI†). After 5 min, the count rate started to increase, indicating the formation of nanoparticles with light-scattering capabilities. During the first 1 h, the Z-average size of the particles remained almost the same, with the PDI greatly decreased. At the same time, the count rate increased. Since the count rate is proportional to c × r6 (c represents the particle concentration and r represents the particle size),24 this means that the number of MNs in the solution increased during the first hour, probably due to the particle nucleation. The supersaturation of soluble oligomers caused MNs of around 30 nm to nucleate continuously during this initial stage. Afterwards, the particle size grew steadily with time, with a slightly increased PDI, suggesting slow particle growth. After 24 h, the particles reached a size of 65.3 nm with a PDI of 0.16 (Fig. S4, ESI†). A further increase in the reaction time led to larger aggregates (104.2 nm at 48 h and 262.3 nm at 72 h) with larger size distribution.
Given the above results, we made a comparison of the MN formation kinetics with the bulk method using diluted dopamine and with the microfluidic method using concentrated dopamine. According to the literature, the generally accepted simplified MN formation mechanism involves 4 steps, including dopamine oxidation, oligomer formation, oligomer aggregation, and aggregate growth (Fig. 2d).25 First, dopamine was auto-oxidized by the dissolved oxygen in the alkaline solution to form quinone, and further converted to 5,6-dihydroxyindole (DHI) and indole-5,6-quinone (IDQ).26 Further oxidation of DHI and IDQ leads to the formation of oligomers (1.0–1.5 nm), which quickly aggregate and form “seeds”.27 The polymer grows steadily from these “seeds” by continuous addition of monomers or dimers by covalent or non-covalent bonding during the polymerization process,28,29 finally generating nano- or micro-sized particles.
At a low concentration of dopamine, the oxidation and oligomer formation were slow (>5 min). This allows for thorough mixing of all reactants in the solution before reaching the supersaturation of MN precursors. Therefore, the nucleation proceeded in a relatively homogenous environment, and the subsequent growth in such a dilute solution was possibly diffusion-controlled, resulting in monodisperse particles after hours or days. In the microfluidic system, all the steps were superfast, due to the high concentration of dopamine. The mixing was also fast and highly efficient, leading to a similarly homogenous environment for nanoparticle nucleation. Therefore, monodisperse particles, with precisely controlled initiation of self-polymerization of dopamine, aggregation of oligomers and termination of the reaction, can be produced in a microfluidic device within a few seconds.
As the MN formation kinetics on microfluidic chips differed from that of conventional bulk methods, we hypothesized that the structure and physiochemical properties were affected by the preparation methods. We first compared the size and morphology of the synthetic MNs with natural MNs from Sepia, which is considered as a standard of eumelanin.30 The Sepia MNs acquired from a commercial supplier after reconstitution in water showed a broad distribution profile with more than one peak (Fig. S5, ESI†). Even after the removal of aggregates using low speed centrifugation, there were still some micro-sized particles, as shown in the DLS size distribution profile (Fig. 3a). The morphology of these particles was compact and spherical (Fig. 3b), consistent with previous literature.13 The zeta-potential of all MNs was similar (around −30 mV) (Table S1, ESI†). Further stability studies at 37 °C in 50% human plasma showed no obvious size change over a 2 h incubation period (Fig. S6, ESI†). The PDI of the MNs in plasma was higher than that in water, probably due to the high protein content in the plasma, which caused a bimodal distribution.31,32
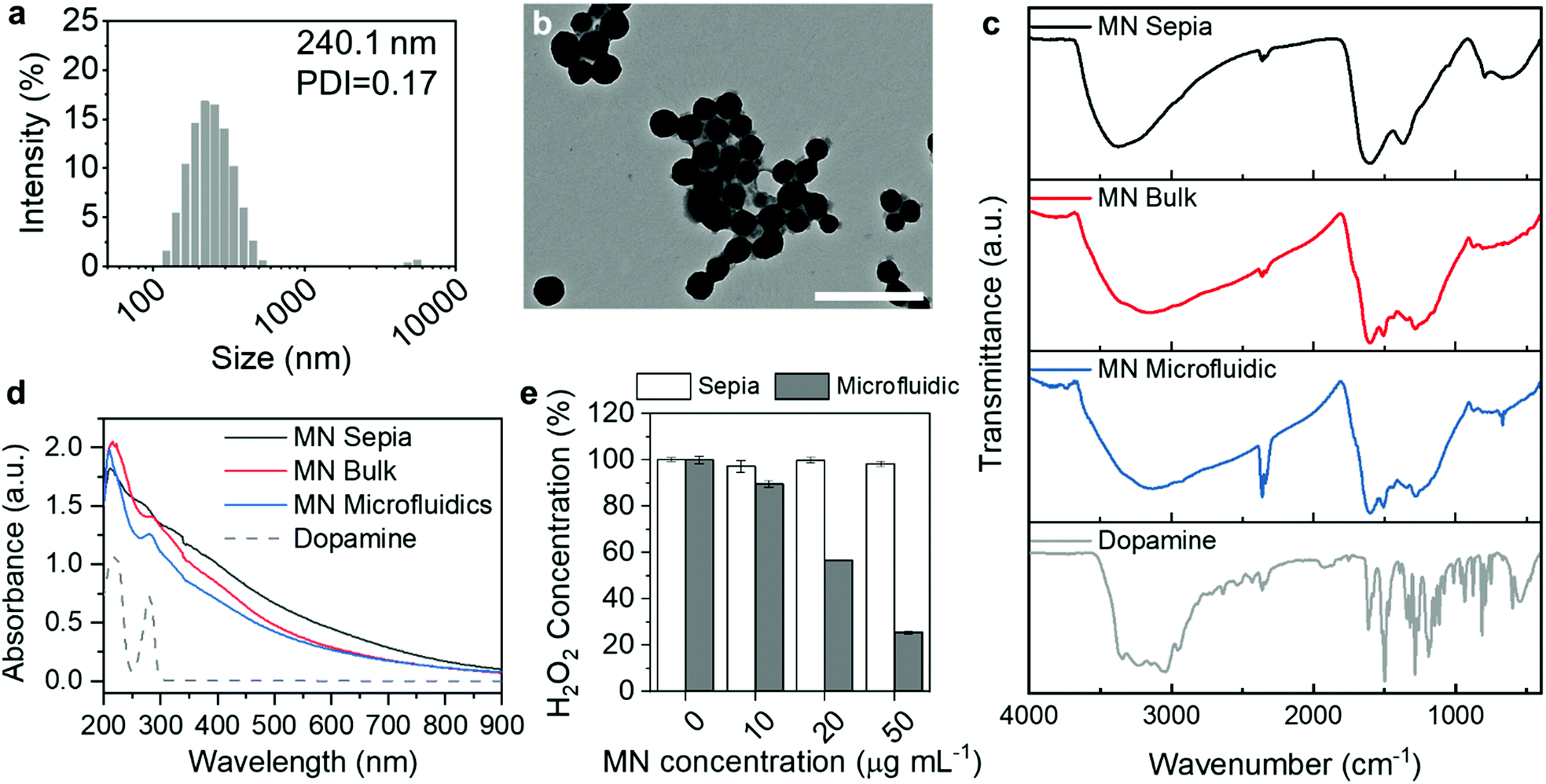 | ||
| Fig. 3 (a and b) DLS distribution profile and TEM image of the MNs from Sepia after low speed centrifugation. Scale bar = 500 nm. (c) The FTIR spectra of MNs from Sepia (black), MNs synthesized by the bulk method (red), MNs synthesized by the microfluidic method (blue) and dopamine (grey). (d) The UV-vis absorbance spectra of MNs from Sepia (black), MNs synthesized by the bulk method (red), MNs synthesized by the microfluidic method (blue) and dopamine (grey dash line). (e) The H2O2 scavenging capability of MNs at different concentrations (10, 20 and 50 μg mL−1). The results were normalized to the untreated controls. | ||
The chemical composition of MNs prepared by the microfluidic technique, prepared by the bulk method (slow reaction) and natural MNs from Sepia was examined by Fourier-transform infrared spectroscopy (FTIR). As shown in Fig. 3c, the MNs from Sepia had broad bands around 3380, 1590 and 1354 cm−1, corresponding to N–H stretching from secondary amine, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic ring stretching and C–N–C stretching from indole rings.33 The broad bands clearly differed from those sharp and split bands shown in the dopamine FTIR spectrum, indicating the heterogeneous polymeric nature of the MNs. The frequencies and the band shape are in good agreement with published frequencies of similar samples.33,34
C aromatic ring stretching and C–N–C stretching from indole rings.33 The broad bands clearly differed from those sharp and split bands shown in the dopamine FTIR spectrum, indicating the heterogeneous polymeric nature of the MNs. The frequencies and the band shape are in good agreement with published frequencies of similar samples.33,34
The synthetic MNs showed identical FTIR spectra, regardless of the preparation method (Fig. 3c). Notably, the synthetic MNs’ spectra showed several different bands between 1800 and 900 cm−1 compared with the natural MNs. First, there is an additional band at 1715 cm−1, corresponding to CO stretching (zoomed in FTIR spectra in Fig. S7, ESI†). This carbonyl group may originate from the ketonic carbonyl of quinone groups, or from the carboxylic acids on dihydroxyindole-2-carboxylic acid (DHICA) and other derivatives.35 Other differences include a series of bands emerging at 1610, 1510 and 1436 cm−1, compared with one broad and smooth band in the natural MNs’ spectrum. The multiple bands are all correlated to the CC aromatic ring stretching in different chemical environments, suggesting a heterogeneous structure. The strong bands at 1280 and 1224 cm−1 corresponding to the C–O vibration might originate from the hydroxyl groups from DHI.33,34 These differences in the FTIR spectra suggest that the chemical composition of synthetic MNs from the bulk and microfluidic methods is similar, but differs from the natural MNs.
The UV absorbance spectra of all MNs (Fig. 3d) showed typical broad absorbance from UV to near infra-red wavelengths, due to the chemical and structural disorder.36,37 The shapes of the absorbance spectra of MNs prepared by the microfluidic and MNs prepared by the bulk method are identical, with a small peak at 280 nm, suggesting residual dopamine or other low molecular weight precursors absorbed on the nanoparticles. MNs from Sepia had a smoother absorbance profile without any obvious peak, and the absorbance is slightly higher than that of the synthetic MNs in the infra-red region, which suggests that there might be some differences in the hierarchical assembly structure within the particles.38
Next, we evaluated the ROS scavenging capability of MNs synthesized by the microfluidic method. We first evaluated whether the MNs can scavenge hydrogen peroxide (H2O2), which is a main type of cellular ROS.7,39 MNs were incubated with H2O2 (10 μM, which is detrimental to cells and leads to necrosis40,41) for 1 h and the remaining H2O2 concentration was evaluated by Amplex™ UltraRed reagent. As shown in Fig. 3e, the H2O2 scavenging capability of the MNs is concentration-dependent. At 20 μg mL−1, the MNs decreased the H2O2 concentration by half, and further decreased it to 25% at a concentration of 50 μg mL−1. The H2O2 scavenging activity might be attributed to the DHICA moieties in the MNs, as shown in the FTIR spectrum.
Surprisingly, the Sepia MNs did not show H2O2 scavenging capability in the studied concentration range (Fig. 3e). It has long been known that Sepia MNs react with H2O2,42 and a high concentration of H2O2 bleaches natural MNs generated by various species.43 However, most previous studies regarding Sepia MNs used fresh MNs purified from Sepia ink, instead of lyophilized samples from a commercial supplier. There is evidence showing that the purification method affected the composition of the final MNs,44 as well as the morphology.9 It is likely that the commercial MNs, after purification, lyophilization and rehydration, lost the H2O2 scavenging capability.
Based on the H2O2 scavenging capability of the MNs prepared by the microfluidic technique, we decided to investigate the antioxidant and anti-inflammatory potential of the material in biological systems. As a proof-of-concept study, we decided to evaluate the MNs on macrophage cells, which are key players in inflammation development. Pro-inflammatory macrophages (M1 phenotype) secrete high levels of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, IL-12 and IL-23, and contribute to inflammation progression.45–47 In contrast, the M2 phenotype secretes anti-inflammatory cytokines (e.g., IL-10 and transforming growth factor-beta) and promotes tissue repair.48–50 Therefore, switching the M1 to the M2 phenotype using anti-inflammatory nanoparticles is a promising strategy for anti-inflammatory therapy.51
Before the evaluation of the anti-inflammatory potency, we first tested the cytotoxicity of the MNs on RAW 264.7 cells, which are typical murine macrophages. The cells were cultured with MNs for 6, 24 and 48 h, respectively, and the cell viability was assessed by the CellTiter-Glo® luminescence assay. As shown in Fig. 4a, the MNs had negligible cytotoxicity at concentrations lower than 100 μg mL−1. When the concentration increased to 500 μg mL−1, the cell viability was 75% at 6 h, 68% at 24 h and 65% at 48 h. The increased cytotoxicity was probably due to the cellular uptake of MNs, which was confirmed by flow cytometry and confocal microscopy (Fig. S8, ESI†). We further tested the cytotoxicity on THP-1 cells (human monocytes, which are commonly used for macrophage differentiation). The results were similar to those of RAW 264.7 (Fig. 4b), which showed decreased cell viability when the concentration was higher than 100 μg mL−1.
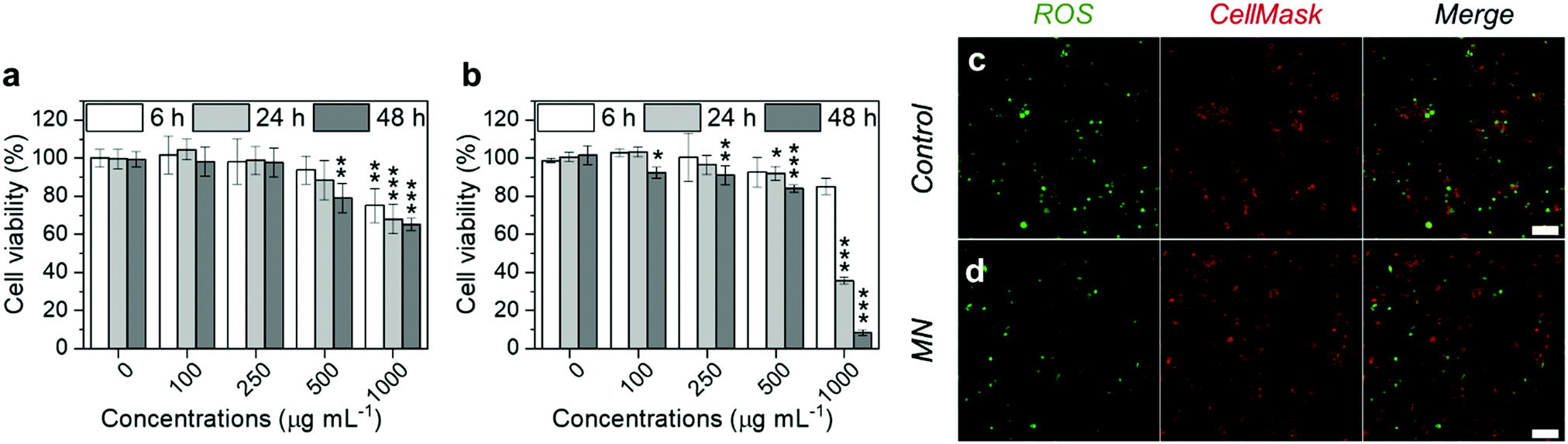 | ||
| Fig. 4 (a and b) The cytotoxicity of MNs prepared by the microfluidic method on RAW 264.7 cells (a) and THP-1 cells (b) after 6, 24 and 48 h incubation. The results were normalized to the untreated controls. Data are presented as the mean ± s.d. (n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001. (c and d) The confocal images showing the ROS level in RAW 264.7 cells stimulated by LPS and IFN-γ for 24 h, and then treated without particles (c) and with MNs at 20 μg mL−1 (d). The intracellular ROS level was indicated by the green fluorescence emitted by dichlorofluorescein. The plasma membranes were stained by CellMask™ Deep Red to outline the cells. | ||
The ROS scavenging capability of the MNs was evaluated on inflamed RAW 264.7 cells. Prior to the NP incubation, RAW 264.7 cells were stimulated by lipopolysaccharides (LPS) and interferon-gamma (IFN-γ), which are common reagents for inducing oxidative stress and inflammatory responses.46 Then the cells were treated with MNs for 5 h, before analyzing using the oxidant-sensing fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). Non-polar DCFH-DA can diffuse through cell membranes into the cytoplasm where it gets hydrolyzed by esterases and subsequently oxidized by ROS to become fluorescent.52 The fluorescence intensity of all cells was then analyzed by confocal microscopy. As shown in Fig. 4c and d, the prevalent green fluorescence emitted by oxidised DCFH in the positive control (cells stimulated by LPS and IFN-γ) confirmed the high ROS level. Cells treated with MNs showed slightly decreased green fluorescence compared with the positive control, although some cells were not fully recovered, possibly due to the short incubation time.
Further characterization of the ROS scavenging capability was performed on RAW 264.7 cells challenged by concentrated H2O2. Quantitative analysis using DCFH-DA suggested that the cells treated with MNs showed a lower ROS level compared with the positive control, and a higher concentration of MNs led to better scavenging effects (Fig. S9, ESI†). At 50 μg mL−1 of MNs, the ROS level became 10% lower after 45 min challenge, and 20% lower when the challenge time was elongated to 90 min. We also used a small molecule ROS scavenger, L-glutathione (GSH), as a control. The preloaded GSH caused a 14% decrease in ROS at 45 min, but almost no effects at 90 min, probably due to the fast metabolism.53
Next, we evaluated the anti-inflammatory therapeutic efficacy on RAW 264.7 macrophages. As shown in Fig. 5a, the macrophages in the resting state (M0) were first stimulated by LPS and IFN-γ for 1 day, and then treated with MNs. The cells stimulated by LPS and IFN-γ but without any treatment developed into the M1 phenotype (M1 control), while those M0 cells with anti-inflammatory cytokine stimulation (IL-4 and IL-13) for 3 consecutive days developed into the M2 phenotype (M2 control). The M1 and M2 phenotypes have distinguishable markers on the surface and cytokine secretion profiles.46 Therefore, we evaluated the expression of CD80 (M1 marker) and CD206 (M2 marker) by immunostaining and subsequent analysis by flow cytometry, as well as the concentration of TNF-α (M1 cytokines) and IL-10 (M2 cytokines) by enzyme-linked immunosorbent assay (ELISA), to identify the phenotypes after treatment.
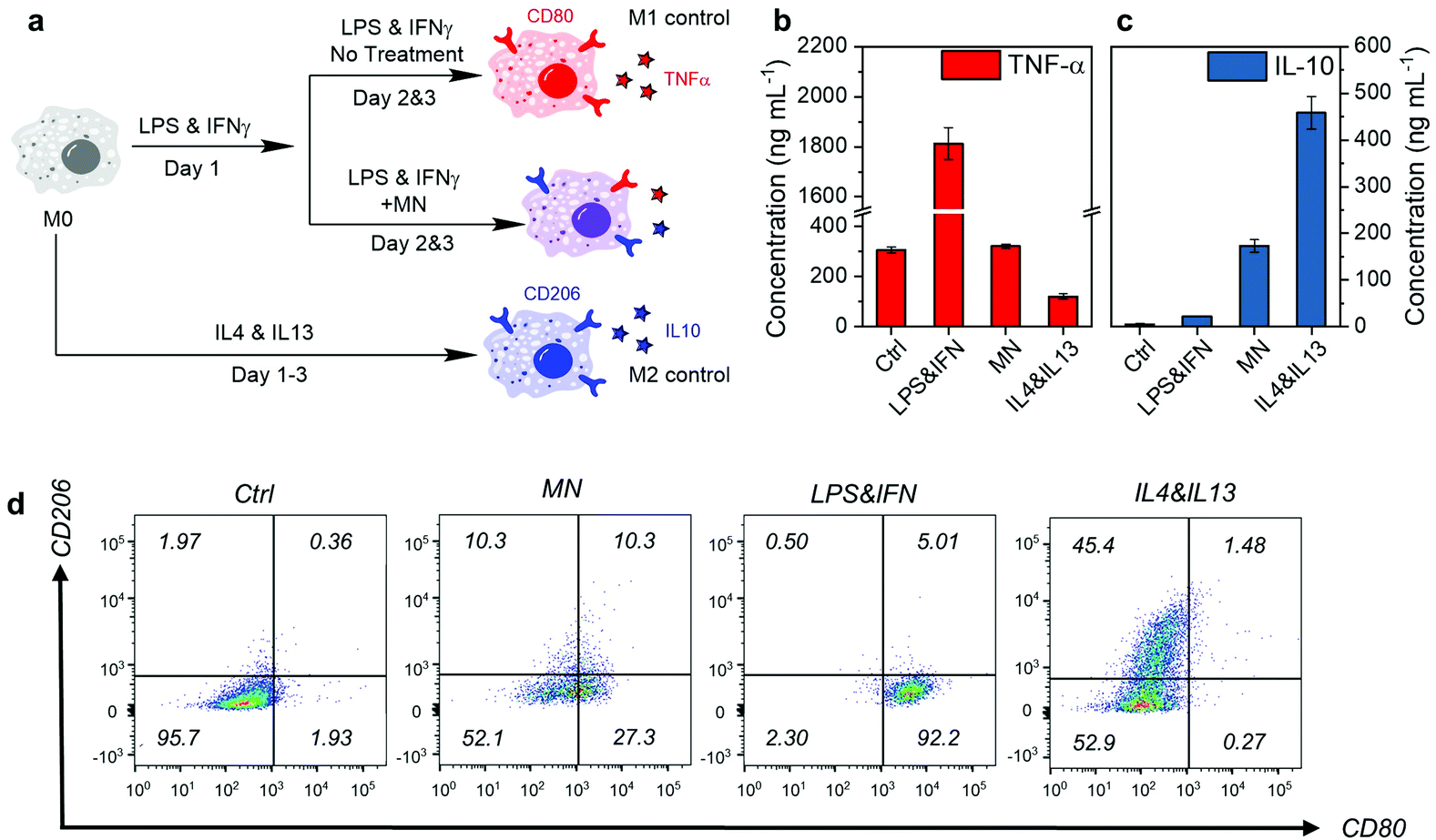 | ||
| Fig. 5 (a) Schematic representation of the macrophage stimulation and treatment. (b and c) The concentration of TNF-α and IL-10 in the macrophage culture medium after stimulation and treatment, quantified by ELISA. Data are presented as the mean ± s.d. (n = 3). (d) The flow cytometry analysis of macrophage marker CD80 and CD206 expression after immunostaining. Control (ctrl) represents cells without LPS and IFN-γ stimulation. MN represents cells stimulated by LPS and IFN-γ and treated with MNs at 20 μg mL−1 for 2 days. LPS&IFN represents the M1 control with continuous pro-inflammatory stimulation, while IL4&IL13 represents the M2 control with continuous anti-inflammatory stimulation. | ||
As shown in Fig. 5b and c, MNs reduced the pro-inflammatory cytokine TNF-α effectively to almost the M0 level. At the same time, the anti-inflammatory cytokine IL-10 secretion was elevated, suggesting potent inflammation-attenuating effects. Further investigation on the cell marker by flow cytometry showed that the M1 typical marker CD80 of MN treated cells decreased significantly compared with the M1 control (Fig. 5d). The M1 phenotype population (CD80+ CD206−) decreased from 92.2% to 27.3%, with an increase of 10% in the M2 phenotype (CD80− CD206+) population. Around half of the cells (52.1%) became double negative, suggesting the transformation into a resting M0 state. This means that the inflammation attenuation effect of the MNs was due to the macrophage phenotype conversion. The molecular mechanism behind the phenotype conversion by MNs is possibly related to the down-regulation of Toll-like receptor 4 expression (for sensing LPS) and NF-κB signaling pathway.54
Conclusions
In this study, we developed melanin-like nanoparticles by superfast and controlled preparation in microfluidic devices inspired by cephalopods. The MNs were generated from highly concentrated substrates within seconds, which is comparable to the biosynthesis. Characterized by DLS and TEM, the particles showed spherical morphology and monodisperse size, due to the superfast mixing and the precise control of initiation and termination of the reaction. Furthermore, the MNs prepared by the microfluidic method successfully reduced the pro-inflammatory cytokine level and induced the secretion of immune-suppressive cytokines IL-10 on macrophages, and altered the cell phenotype. The simple but advanced preparation of MNs by a microfluidic device, along with the inflammation attenuating property, makes the nanoparticles developed here a promising candidate for anti-inflammatory applications.Experimental
Details of the experimental procedures are provided in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Dr S. Wang acknowledges the financial support from the Jenny and Antti Wihuri Foundation. S. Wannasarit acknowledges financial support from the Thailand Research Fund under the Royal Golden Jubilee (RGJ) PhD program (PHD/0180/2556). Dr W. Li acknowledges financial support from the Finnish Cultural Foundation (grant no. 00190634) and the Academy of Finland (decision no. 322093). Prof. H. A. Santos acknowledges financial support from the HiLIFE Research Funds, the Sigrid Jusélius Foundation, the Norwegian Research Council/Nacamed AS, and the European Research Council Proof-of-Concept Research Grant (grant no. 825020). The authors also acknowledge the following core facilities funded by Biocenter Finland: Electron Microscopy Unit for TEM, the Light Microscopy Unit of the Institute of Biotechnology for the confocal microscope and the Flow Cytometry Unit for the flow cytometry analyser.Notes and references
- C. D. Derby, Biol. Bull., 2007, 213, 274–289 CrossRef CAS PubMed.
- C. Derby, Mar. Drugs, 2014, 12, 2700–2730 CrossRef CAS PubMed.
- B.-L. L. Seagle, E. M. Gasyna, W. F. Mieler and J. R. Norris, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16644–16648 CrossRef CAS PubMed.
- J.-P. Zhong, G. Wang, J.-H. Shang, J.-Q. Pan, K. Li, Y. Huang and H.-Z. Liu, Mar. Drugs, 2009, 7, 9–18 CrossRef PubMed.
- X. Guo, S. Chen, Y. Hu, G. Li, N. Liao, X. Ye, D. Liu and C. Xue, J. Food Sci. Technol., 2014, 51, 3680–3690 CrossRef CAS PubMed.
- S. K. Biswas, Oxid. Med. Cell. Longevity, 2016, 2016, 1–9 CrossRef PubMed.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Antioxid. Redox Signaling, 2014, 20, 1126–1167 CrossRef CAS PubMed.
- A. Palumbo, Pigm. Cell Res., 2003, 16, 517–522 CrossRef CAS PubMed.
- Y. Liu and J. D. Simon, Pigm. Cell Res., 2003, 16, 72–80 CrossRef CAS PubMed.
- U. Schraermeyer, Pigm. Cell Res., 1994, 7, 52–60 CrossRef CAS PubMed.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. M. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed.
- J. Yan, L. Yang, M.-F. Lin, J. Ma, X. Lu and P. S. Lee, Small, 2013, 9, 596–603 CrossRef CAS PubMed.
- K.-Y. Ju, Y. Lee, S. Lee, S. B. Park and J.-K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Adv. Drug Delivery Rev., 2018, 128, 54–83 CrossRef CAS PubMed.
- D. Liu, S. Cito, Y. Zhang, C.-F. Wang, T. M. Sikanen and H. A. Santos, Adv. Mater., 2015, 27, 2298–2304 CrossRef CAS PubMed.
- D. Liu, H. Zhang, S. Cito, J. Fan, E. Mäkilä, J. Salonen, J. Hirvonen, T. M. Sikanen, D. A. Weitz and H. A. Santos, Nano Lett., 2017, 17, 606–614 CrossRef CAS PubMed.
- W. Li, D. Liu, H. Zhang, A. Correia, E. Mäkilä, J. Salonen, J. Hirvonen and H. A. Santos, Acta Biomater., 2017, 48, 238–246 CrossRef CAS PubMed.
- F. Araújo, N. Shrestha, M.-A. Shahbazi, D. Liu, B. Herranz-Blanco, E. M. Mäkilä, J. J. Salonen, J. T. Hirvonen, P. L. Granja, B. Sarmento and H. A. Santos, ACS Nano, 2015, 9, 8291–8302 CrossRef PubMed.
- H. Zhang, W. Cui, X. Qu, H. Wu, L. Qu, X. Zhang, E. Mäkilä, J. Salonen, Y. Zhu, Z. Yang, D. Chen, H. A. Santos, M. Hai and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7744–7749 CrossRef CAS PubMed.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Lab Chip, 2017, 17, 1856–1883 RSC.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698–711 CrossRef CAS PubMed.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS PubMed.
- J. Shang and X. Gao, Chem. Soc. Rev., 2014, 43, 7267–7278 RSC.
- M. Arzillo, G. Mangiapia, A. Pezzella, R. K. Heenan, A. Radulescu, L. Paduano and M. D’Ischia, Biomacromolecules, 2012, 13, 2379–2390 CrossRef CAS PubMed.
- N. F. Della Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. D’Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS.
- N. F. Della Vecchia, A. Luchini, A. Napolitano, G. D’Errico, G. Vitiello, N. Szekely, M. D’Ischia and L. Paduano, Langmuir, 2014, 30, 9811–9818 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- M. d’Ischia, K. Wakamatsu, A. Napolitano, S. Briganti, J.-C. Garcia-Borron, D. Kovacs, P. Meredith, A. Pezzella, M. Picardo, T. Sarna, J. D. Simon and S. Ito, Pigm. Cell Melanoma Res., 2013, 26, 616–633 CrossRef PubMed.
- L. L. Chaikov, M. N. Kirichenko, S. V. Krivokhizha and A. R. Zaritskiy, J. Biomed. Opt., 2015, 20, 057003 CrossRef PubMed.
- B. Okutucu, A. Dınçer, Ö. Habib and F. Zıhnıoglu, J. Biochem. Biophys. Methods, 2007, 70, 709–711 CrossRef CAS PubMed.
- S. A. Centeno and J. Shamir, J. Mol. Struct., 2008, 873, 149–159 CrossRef CAS.
- M. L. Roldán, S. A. Centeno and A. Rizzo, J. Raman Spectrosc., 2014, 45, 1160–1171 CrossRef.
- M. L. Alfieri, R. Micillo, L. Panzella, O. Crescenzi, S. L. Oscurato, P. Maddalena, A. Napolitano, V. Ball and M. D’Ischia, ACS Appl. Mater. Interfaces, 2018, 10, 7670–7680 CrossRef CAS PubMed.
- R. Micillo, L. Panzella, M. Iacomino, G. Prampolini, I. Cacelli, A. Ferretti, O. Crescenzi, K. Koike, A. Napolitano and M. D’Ischia, Sci. Rep., 2017, 7, 41532 CrossRef CAS PubMed.
- C.-T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 3859 CrossRef CAS PubMed.
- K.-Y. Ju, M. C. Fischer and W. S. Warren, ACS Nano, 2018, 12, 12050–12061 CrossRef CAS PubMed.
- J. R. Hoidal, Am. J. Respir. Cell Mol. Biol., 2001, 25, 661–663 CrossRef CAS PubMed.
- F. Antunes and E. Cadenas, Free Radicals Biol. Med., 2001, 30, 1008–1018 CrossRef CAS.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed.
- J. M. Menter, A. M. Patta, T. D. Hollins, C. L. Moore and I. Willis, Photochem. Photobiol., 1998, 68, 532–537 CrossRef CAS PubMed.
- R. A. W. Smith, B. Garrett, K. R. Naqvi, A. Fülöp, S. P. Godfrey, J. M. Marsh and V. Chechik, Free Radicals Biol. Med., 2017, 108, 110–117 CrossRef CAS PubMed.
- M. Magarelli, P. Passamonti and R. Carlo, Rev. CES Med. Vet. Zootec., 2010, 5, 18–28 Search PubMed.
- P. J. Murray, Annu. Rev. Physiol., 2017, 79, 541–566 CrossRef CAS PubMed.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J.-L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS PubMed.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS PubMed.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS PubMed.
- A. Ortega-Gómez, M. Perretti and O. Soehnlein, EMBO Mol. Med., 2013, 5, 661–674 CrossRef PubMed.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS PubMed.
- K. L. Spiller and T. J. Koh, Adv. Drug Delivery Rev., 2017, 122, 74–83 CrossRef CAS PubMed.
- E. J. Bland, T. Keshavarz and C. Bucke, Mol. Biotechnol., 2001, 19, 125–132 CrossRef CAS PubMed.
- A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711–760 CrossRef CAS PubMed.
- L. Jin, F. Yuan, C. Chen, J. Wu, R. Gong, G. Yuan, H. Zeng, J. Pei and T. Chen, Inflammation, 2019, 42, 658–671 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental details and additional data. See DOI: 10.1039/d0mh00014k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |