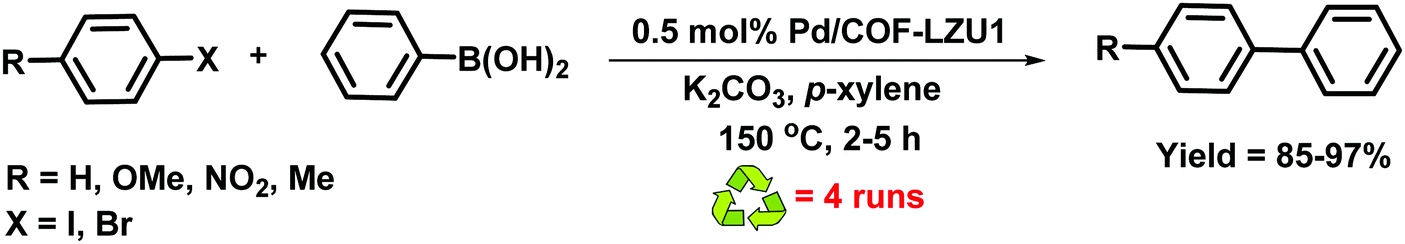DOI:
10.1039/C9MH00856J
(Review Article)
Mater. Horiz., 2020,
7, 411-454
Recent development of covalent organic frameworks (COFs): synthesis and catalytic (organic-electro-photo) applications
Received
3rd June 2019
, Accepted 16th September 2019
First published on 16th September 2019
Abstract
Covalent organic frameworks (COFs) are one of the most important and dynamic members of the porous organic materials and are constructed using reticular chemistry with the building blocks being connected via covalent bonds. Due to their fascinating properties such as large surface area; structural versatility; effortless surface modification; and high chemical stability, COFs are being widely deployed in catalytic; sensing; adsorption; gas storage; and many other valuable applications. This review summarizes selected and most recent espoused synthetic methodologies for their assembly including the formation of magnetic COFs. Potential insights in the field of catalysis; electrocatalysis; and photocatalysis, where COFs can serve as excellent platforms for supporting catalytic species, are also illustrated.
1 Introduction
Nature is a rich source of inspiration for new ideas. Over decades, chemists have been trying to mimic nature to synthesize complex systems that could perform advanced functions.1–5 In this regard, scientists have constructed numerous chemical architectures by assembling diverse building units in assorted manners.6–12 In particular, these new materials are continually developed and applied as catalysts in fine-chemical and petrochemical industries. However, the present scenario faces challenges in renewing catalytic species with greener, readily available, efficient materials, and advance assembly systems to achieve sustainable development. In this context, persistent effort goes into designing nano-porous materials with the domain ranging from conventional mesoporous zeolites to coordination polymers including metal–organic-frameworks (MOFs).13–17 Nevertheless, their linking chemistry purely relies on kinetically controlled irreversible coupling reactions which results in structural disorder and non-separable oligomers that restrain their catalytic applications. These efforts indicate the search for organic porous materials with long-range order structure and considerable crystallinity.18
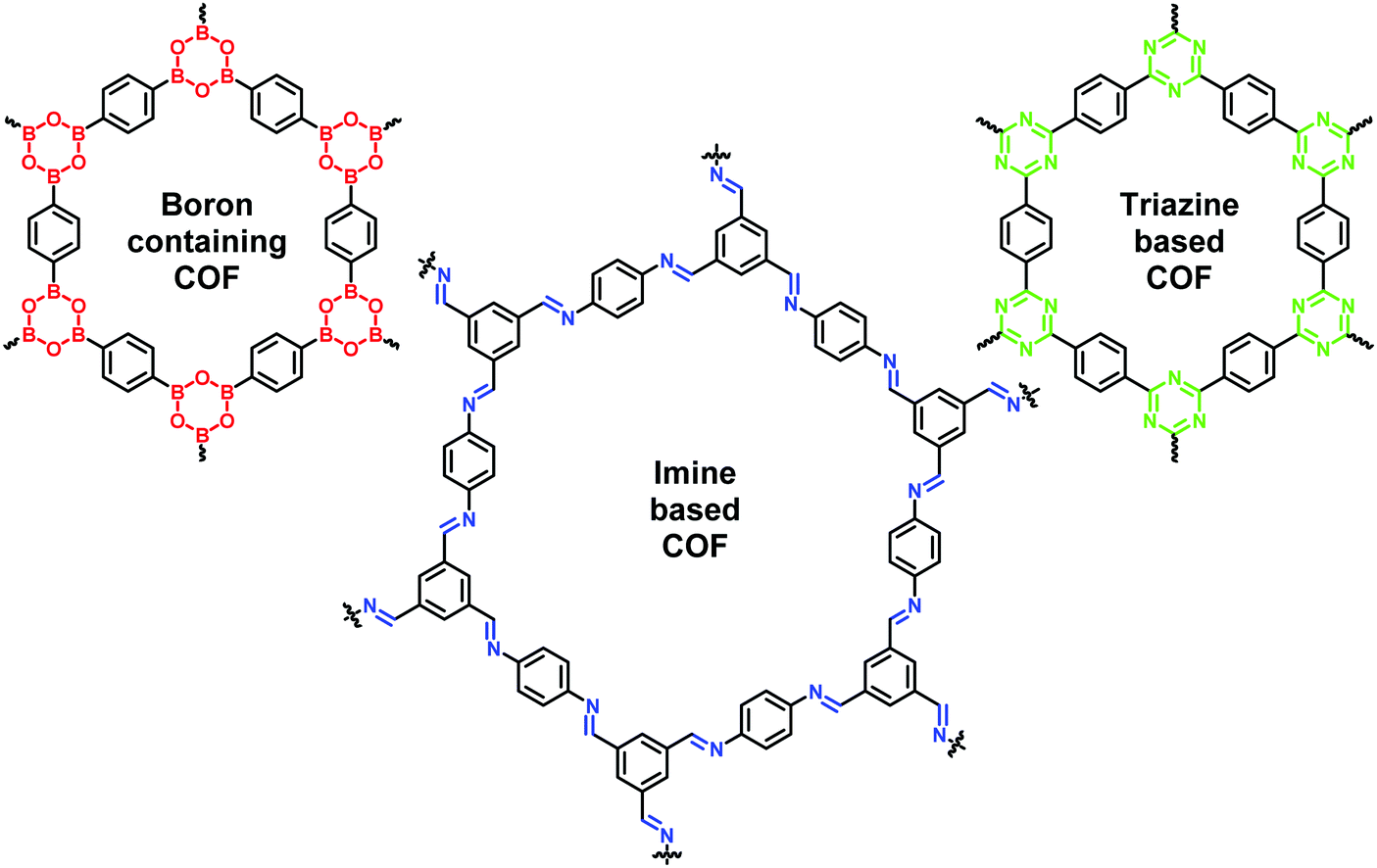
Yaghi and co-workers introduced COFs in 2005, for the first time, as crystalline porous organic polymers which are formed by reversible reactions such as condensation of two-dimensional (2D) and three-dimensional (3D) organic building precursors.6,19–21 They comprised light elements (such as H, B, C, N, O) and are connected by robust covalent bonds via reticular chemistry (Fig. 1). The noteworthy progress on COFs is mainly due to their self-healing ability and thermodynamically controlled dynamic covalent chemistry involved in their assembly which results in a long-range ordered crystalline structure. Unlike MOFs, the densities of COFs are usually lower and thus they display excellent stability in organic solvents, and even under varying acidic; basic; oxidative; and reductive conditions. Besides, COFs can withstand harsh conditions and can retain their ordered structure and crystallinity. This is attributed to the metal-free skeletal structure linked via strong covalent bonds rather than coordinate bonds that comprise MOFs. Moreover, the presence of hydrogen bonding and π–π stacking interaction in COFs further strengthens the skeletal and pore structure and protects them from solvation and hydrolysis. In comparison with inorganic zeolites and porous silica materials, COFs have higher porosity and bigger and tunable pore size which speed up the diffusion of reactants and desorption of products, thereby promoting higher yield and selectivity. Moreover, COFs exhibit ultrahigh theoretical specific surface area and high chemical stability which renders them as an ideal heterogeneous catalyst.19,22–27
 |
| | Fig. 1 Structures of diverse COFs. | |
In recent years, COFs have been uniquely designed and synthesized and are explored in numerous applications due to a number of very exclusive advantages22,28–50 such as low density, pre-designable and well-defined porous structures with uniform pore size ranging from angstroms to nanometers, large surface area, versatile covalent combination of rigid building units having multiconnectivities, structural regularity, tunable and effortless functionalization, sturdy nature that provides resistance to disintegration under adverse conditions and many more. These fascinating advantages have attracted many researchers working in the fields of gas and energy storage,16,51–60 sensing,61–66 adsorption of metal ions,67,68 catalysis,69–73 optoelectrics,74,75 and drug delivery,76–80 among others.81–83
Covalent triazine frameworks (CTFs) represent another class of porous polymers related to COFs encompassing aromatic triazine linkages (Fig. 1).84 They possess a high amount of nitrogen functionalities, high chemical and thermal stability and large structural diversity. However, due to the presence of strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, very few CTFs are crystalline. The energy band structure of CTFs can be modified by varying the building blocks that are connected to the electron-withdrawing triazine units, thus enabling their wider applications in photocatalytic processes. The progress of CTFs can be seen in a variety of other appliances such as gas adsorption85,86 and storage,87,88 energy storage,89–91 and heterogeneous catalysis.92,93
N bonds, very few CTFs are crystalline. The energy band structure of CTFs can be modified by varying the building blocks that are connected to the electron-withdrawing triazine units, thus enabling their wider applications in photocatalytic processes. The progress of CTFs can be seen in a variety of other appliances such as gas adsorption85,86 and storage,87,88 energy storage,89–91 and heterogeneous catalysis.92,93
The present review article is supported by selected and important synthetic methodologies for assembly of COFs and aims to provide perceptive information on their catalytic, electrocatalytic and photocatalytic applications.27,94,95 The literature accumulated in this review article reveals that COFs (1) can be synthesized under diverse experimental conditions, (2) can be exploited as potential catalytic support materials by entrapping metal salts/nanoparticles (NPs) offering activities and selectivities comparable to their homogeneous analogues, (3) can even serve as metal-free catalysts, (4) can be supported on various materials such as magnetic nanoparticles which enhances easy magnetic recoverability and (5) can be used to remove organic pollutants.
2 Synthetic routes for COFs
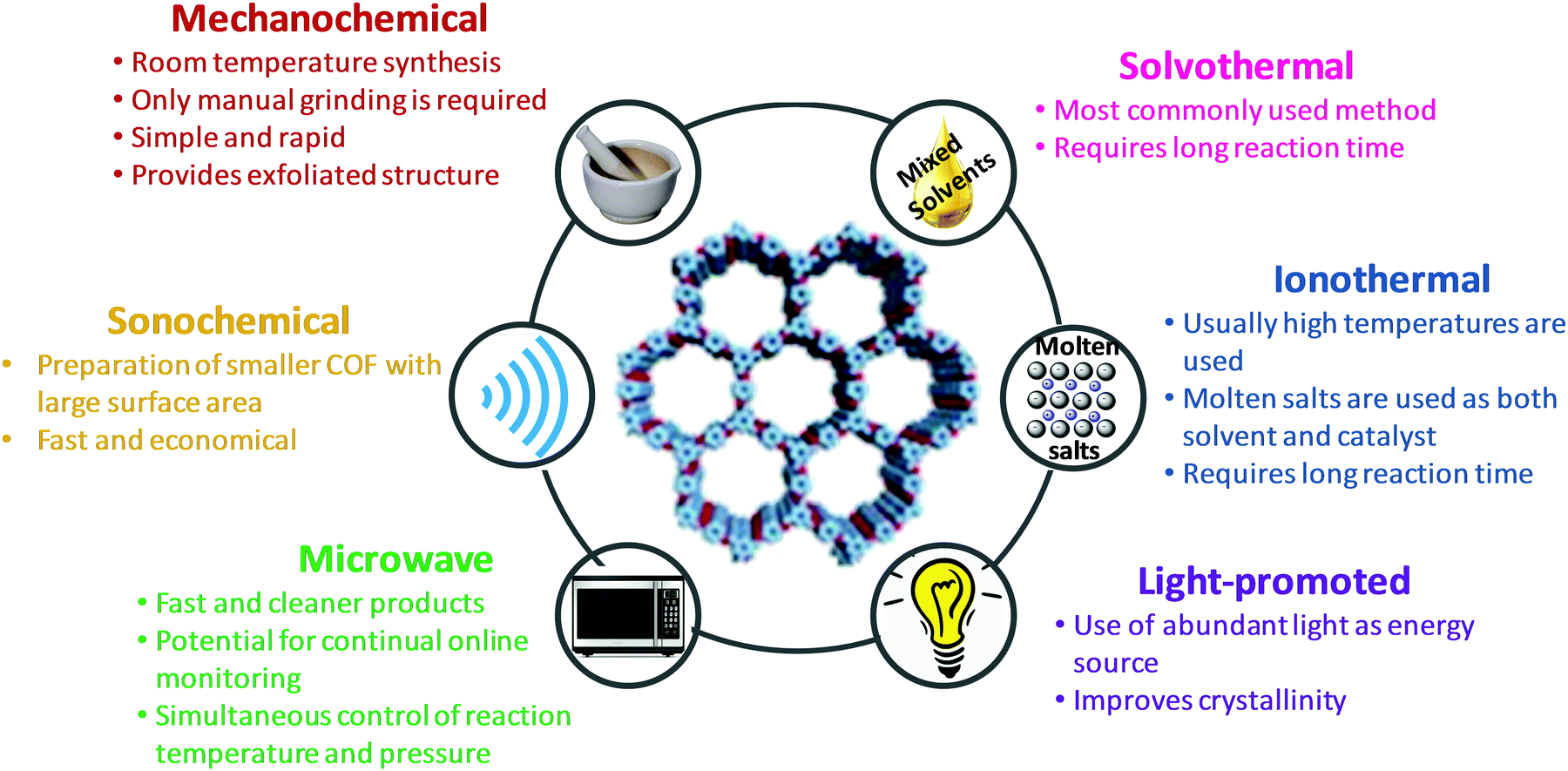
The key to forming highly ordered and crystalline COFs is the regulation of thermodynamic equilibrium during covalent bond formation including a pre-requisite knowledge of building units and synthetic routes (such as reaction media and conditions). Depending upon the desired porosity; surface functionalization; and structural regularity, COFs can be predesigned with careful understanding of the aforementioned parameters. Recently, attempts have been made to explore the synthetic prospects in a variety of manners; some of the common methods include solvothermal, ionothermal, microwave, sonochemical, mechanochemical and light-promoted methods for the successful formation of COFs.22,33,96–100
2.1 Solvothermal synthesis
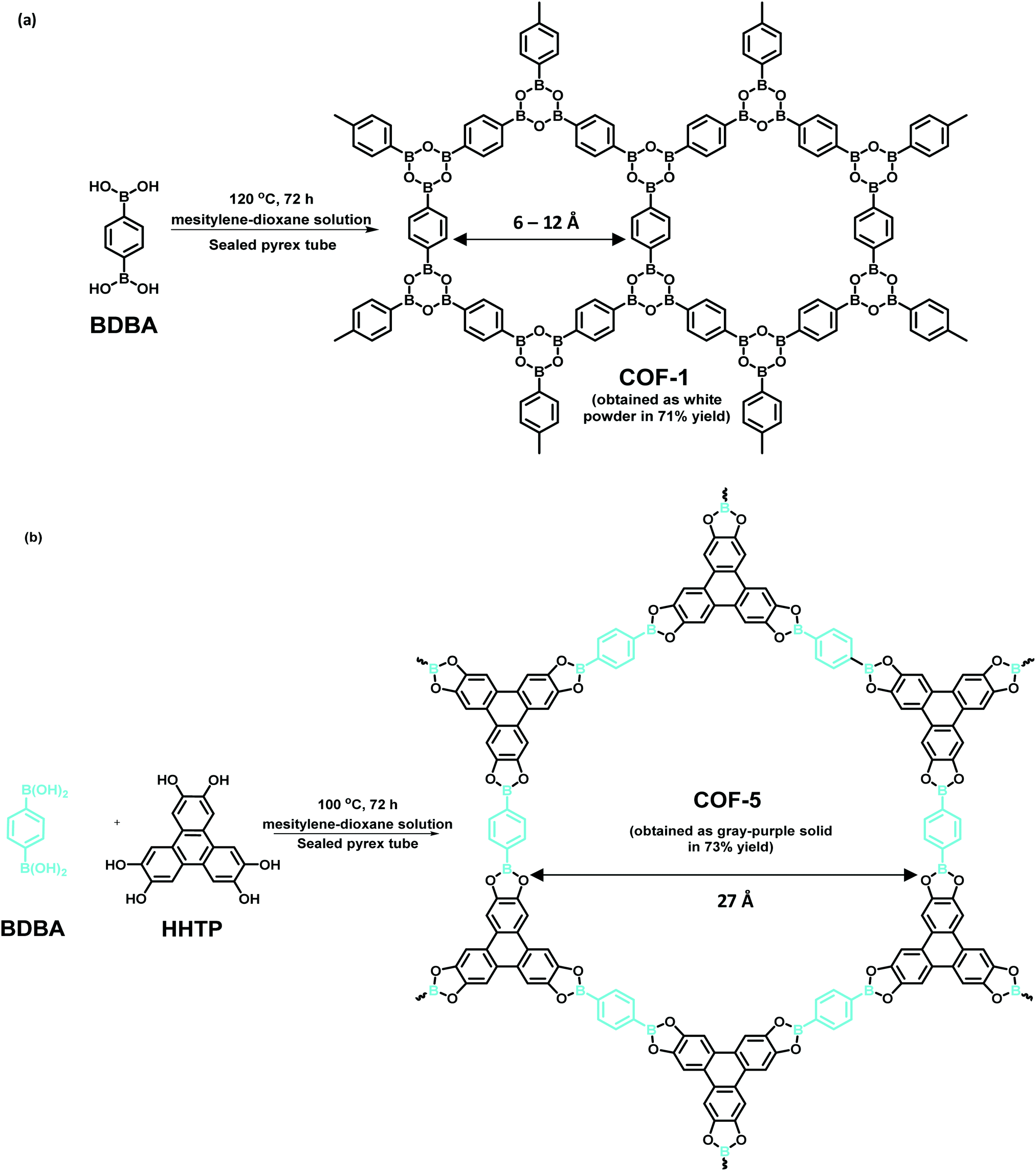
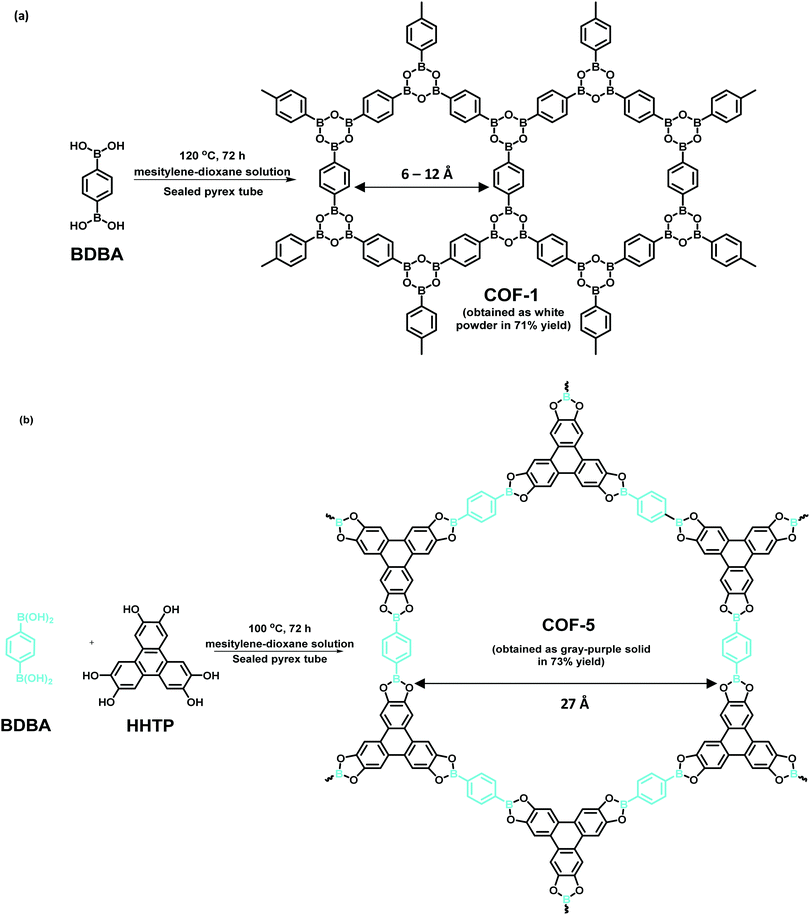
Usually, a variety of COFs are acquired via solvothermal methods, similar to typical protocols deployed for the synthesis of inorganic zeolites in autoclaves. This entails mixing of monomers in a combination of diverse solvents in a Pyrex tube followed by degassing, sealing and heating at a designated temperature for a given reaction time. Next, the precipitate is collected, washed and dried to produce solid powdered COFs. Historically, the first two 2D COFs were synthesized by Yaghi et al. using the solvothermal method (Fig. 2).6 COF-1 was assembled via self-molecular dehydration reaction of 1,4-benzenediboronic acid (BDBA), as a layered hexagonal framework. Analogously, COF-5 was synthesized using condensation of BDBA and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP), as a coplanar extended sheet structure. The presence of mesitylene-dioxane as a solvent mixture helps to control the diffusion of monomers into the solution, thereby expediting the nucleation of the COF. Besides, a sealed environment fulfils the availability of water for efficient crystalline growth of COFs.
 |
| | Fig. 2 Synthesis of the very first 2D COFs (a) COF-1 and (b) COF-5. | |
2.2 Ionothermal synthesis
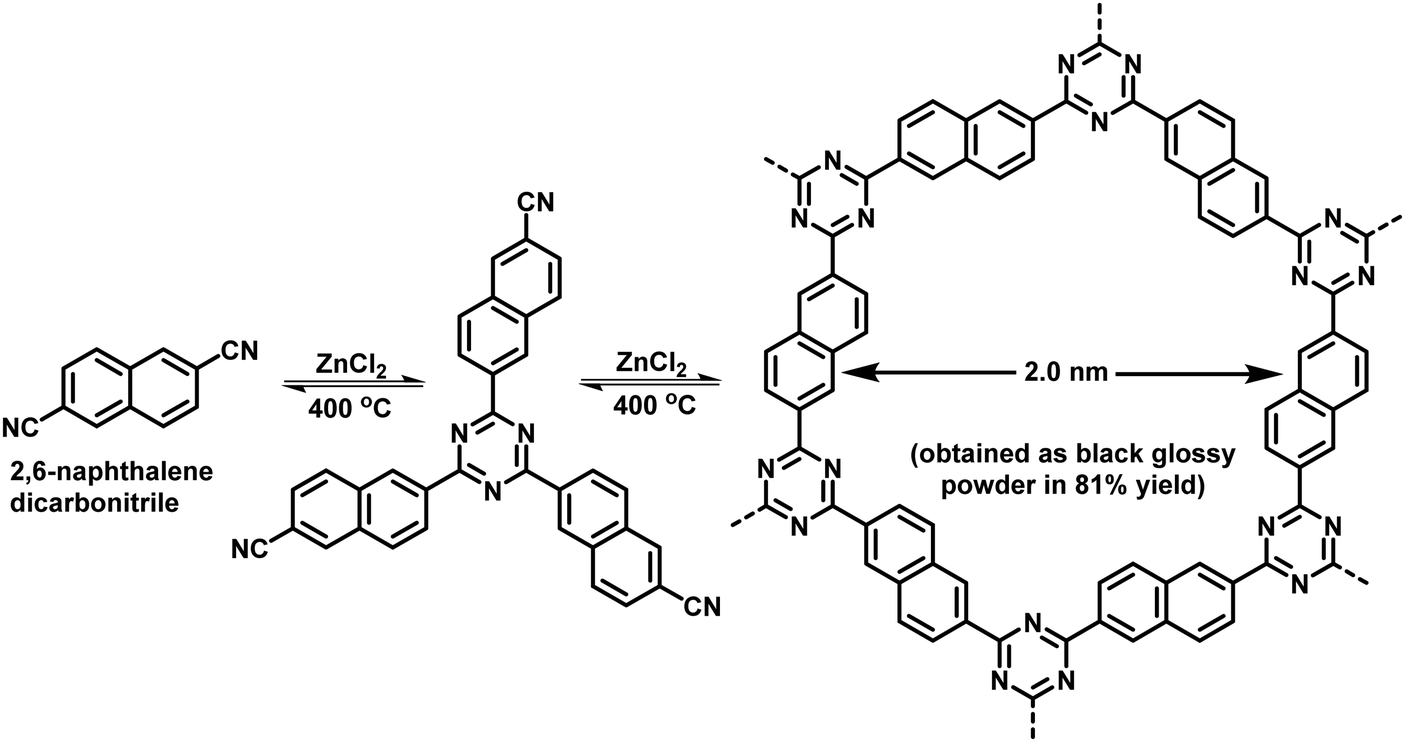
Ionothermal synthesis is a synthetic method wherein solids are formed in the presence of an ionic liquid/molten salt which serves as both solvent and a catalyst. The reaction is usually carried out at high temperatures (∼400 °C) and pressure in a thick-walled vessel such as a Pyrex tube.87 This method was used by Bojdys et al. for the synthesis of CTFs; CTF-2 was obtained by heating 2,6-naphthalenedicarbonitrile and ZnCl2 in a quartz glass ampoule at 400 °C for 40 h via a ionothermal condensation reaction (Fig. 3).101 This synthesis method involves very harsh reaction conditions such as high reaction temperature which not only limits the scope of building units but also invites unwanted decomposition and condensation side reactions. Moreover, the ensuing CTFs are amorphous in nature and lack long-range molecular order which may explain why this method did not receive much attention.
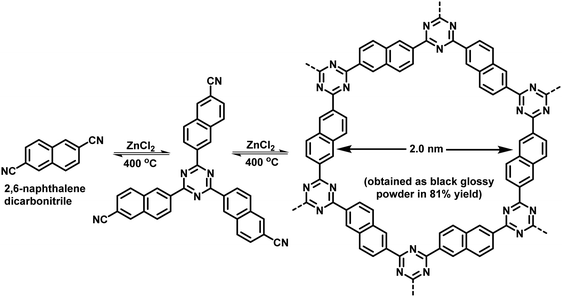 |
| | Fig. 3 Ionothermal synthesis of CTF-2. | |
It is believed that with the increase in crystallinity, charge transport within the framework intensifies, which further enhances the photoelectric properties, and therefore, methods are continuously being developed that result in the formation of crystalline CTFs.28 In this context, Liu et al. exploited the principle of slow nucleation and the production of low concentration of nuclei to improve crystallization, by performing in situ controlled oxidation of alcohol monomers in an open system polycondensation reaction (Fig. 4).102,103 This resulted in the formation of a series of highly crystalline CTFs, CTF-HUST-C1, CTF-HUST-C5 and CTF-HUST-C6. This strategy allowed a convenient synthesis of a series of crystalline CTFs with different monomers resulting in greater thermal stability and improved photocatalytic performance due to easier electronic transport and better absorption when compared to amorphous CTFs.
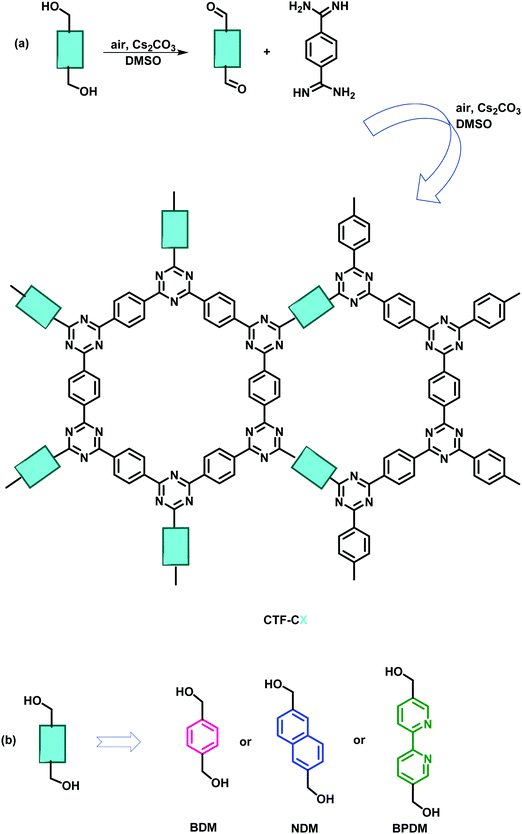 |
| | Fig. 4 (a) The general strategy of slowing down the nucleation process via in situ-controlled oxidation of alcohol monomers in an open system. (b) Monomers: BDM for CTF-HUST-C1, NDM for CTF-HUST-C5 and BPDM for CTF-HUST-C6. | |
2.3 Microwave (MW) synthesis
The use of microwaves as an alternate energy source in chemistry has been broadly explored during the past few years.104–106 It has numerous advantages over the solvothermal method such as considerable reduction of reaction times and the formation of cleaner products, which allows the possibility for expeditious COF synthesis.107–109 Other benefits include the potential for continual online monitoring, visual monitoring of the phase behavior in reactions and simultaneous control of reaction temperature and pressure. Recently, Cooper et al. synthesized 2D COF-5 and 3D COF-102 under MW irradiation at 100 °C while stirring them for 20 min which is 200 times faster than the previously reported solvothermal method.110 As can be seen from Fig. 5a, a gray precipitate was formed in the reaction vessel which resulted in the formation of COF-5. The unreacted starting materials and the trapped impurities in the porous structure were removed by washing with acetone (the purple color of supernatant is due to the formation of an oxidized form of HHTP) (Fig. 5b). Finally, the purified COF-5 was isolated as a gray solid in 68% yield (Fig. 5c). Thus, MW-assisted synthesis also allows enhancement in surface area of COF-5 (SBET(Microwave) = 2019 m2 g−1; SBET(Solvothermal) = 1590 m2 g−1) by removing impurities via a MW extraction process.
 |
| | Fig. 5 Images for the synthesis and purification of COF-5: (a) gray COF-5 with purple impurities, (b) removal of impurities by a MW extraction process and (c) purified gray COF-5. Reproduced with permission from ref. 110 Copyright 2009 American Chemical Society. | |
2.4 Sonochemical synthesis
Sonochemical synthetic methods are gaining great attention as alternative routes to conventional techniques since the process is fast and economical.111,112 Ahn et al. utilized this method to synthesize COF-1 and COF-5. COF-1 was prepared from BDBA using a mixture of mesitylene and 1,4-dioxane in a sonicating unit in just an hour;113 the sonication promoted the effective mixing of sparingly soluble BDBA in the mixed solvent system via a cavitation phenomenon. COF-1 prepared using this method was 400 times smaller than that of the conventionally prepared one, with nearly the same surface area. Similarly, COF-5 was prepared using BDBA and HHTP in the same solvent system.
2.5 Mechanochemical synthesis
A mechanochemical route is of considerable significance in the field of modern synthetic chemistry as it leads to simple, rapid, solvent-free and room temperature synthesis only by means of manual grinding.114,115 Recently, Banerjee et al. reported the formation of three COFs: TpPa-1, TpPa-2 and TpBD via Schiff base condensation reaction using a pestle and mortar (Tp = 1,3,5-triformylphloroglucinol, Pa-1 = p-phenylenediamine, Pa-2 = 2,5-dimethyl-p-phenylenediamine, BD = benzidine).116 The formation of these COFs can be seen by the naked eye as the color changes (Fig. 6). In addition, the obtained COFs had a graphene-like layered structure, unlike the COFs synthesized via a solvothermal method; this could be well attributed to the exfoliation of COF layers during the mechanochemical synthesis.
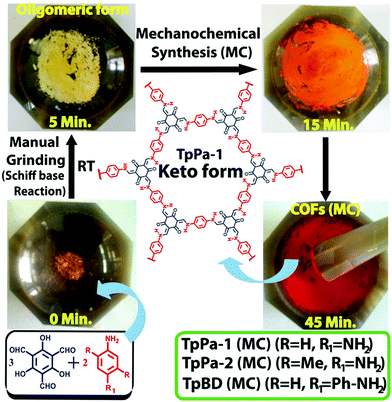 |
| | Fig. 6 Mechanochemical synthesis of TpPa-1, TpPa-2 and TpBD. Reproduced with permission from ref. 116 Copyright 2013 American Chemical Society. | |
2.6 Light-promoted synthesis
Recently, a light-promoted highly conjugated and crystalline COF (hcc-COF) has been synthesized under simulated sunlight irradiation of wavelength 200–2500 nm by mixing of 1,2,4,5-benzenetetramine tetrahydrochloride (BTA) and hexaketocyclohexane octahydrate (HCH) (in mixed solvents) in a quartz vial under an argon atmosphere in just 3 h (Fig. 7).117 A small amount of water and acetic acid was also added as a co-catalyst to induce efficient imine condensation. However, it was observed that under light-deficient conditions, an amorphous hcc-COF was obtained. Due to an extended conjugated structure, which facilitates charge transfer, the as-synthesized hcc-COF shows the highest electrical conductivity of 2.22 × 10−3 S m−1.
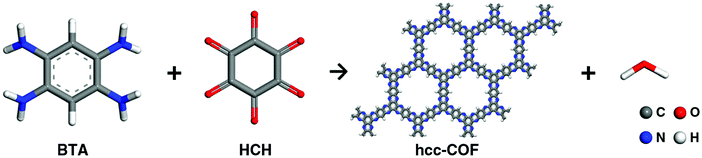 |
| | Fig. 7 Synthesis of hcc-COF. Reproduced with permission from ref. 117 Copyright 2019 Springer. Nature Publishing AG. | |
The properties and advantages of various synthetic methods of COFs are summarized in Fig. 8.
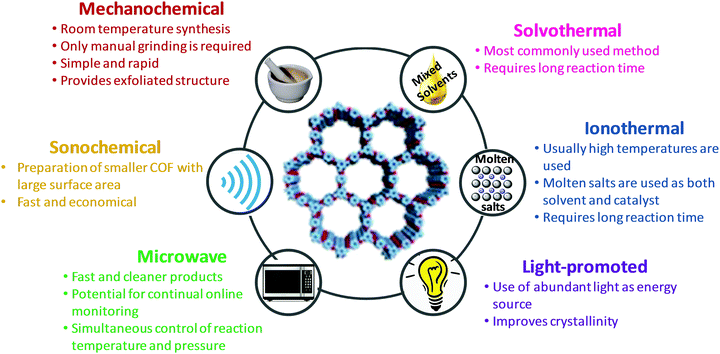 |
| | Fig. 8 Properties and advantages of various synthetic methods of COFs. | |
2.7 Synthesis of magnetic COFs
Inspired by the fascinating properties of magnetic nanoparticles (MNPs), which enable easy magnetic recovery, cost effectiveness and low toxicity, they can be exploited in a wide range of disciplines.118–130 It simplifies the procedures, enhances the efficiency, and allows recoverability and thus reusability. Therefore, considering the superiorities of MNPs and COFs, Cai et al. designed a unique gypsophila bouquet-shaped magnetic COF via a simple methodology (Fig. 9).
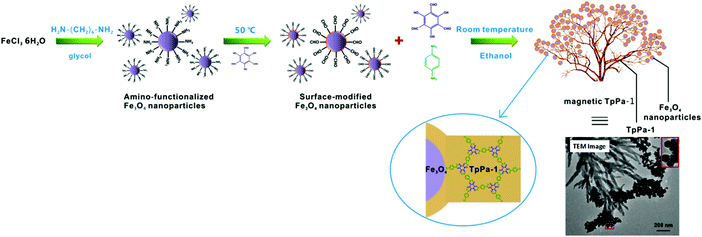 |
| | Fig. 9 Synthesis of gypsophila bouquet-shaped magnetic TpPa-1. Reproduced with permission from ref. 131 Copyright 2017 American Chemical Society. | |
Initially, surface-modified Fe3O4 NPs were synthesized by preferentially grafting Tp (COF monomer) onto amino-functionalized Fe3O4 NPs.131 This increased the local concentration of Tp on NPs and helped to trigger the binding of COF using Schiff base reaction. Furthermore, Tp and another monomer Pa-1, were added to consequently produce magnetic TpPa-1. The TEM image in Fig. 9 clearly shows that nanofibers of TpPa-1 sprout from clustered 30 nm amino-functionalized Fe3O4 NPs in order to acquire a structure like a gypsophila bouquet. Magnetic TpPa-1 possessed superparamagnetism besides a large surface area and high porosity.
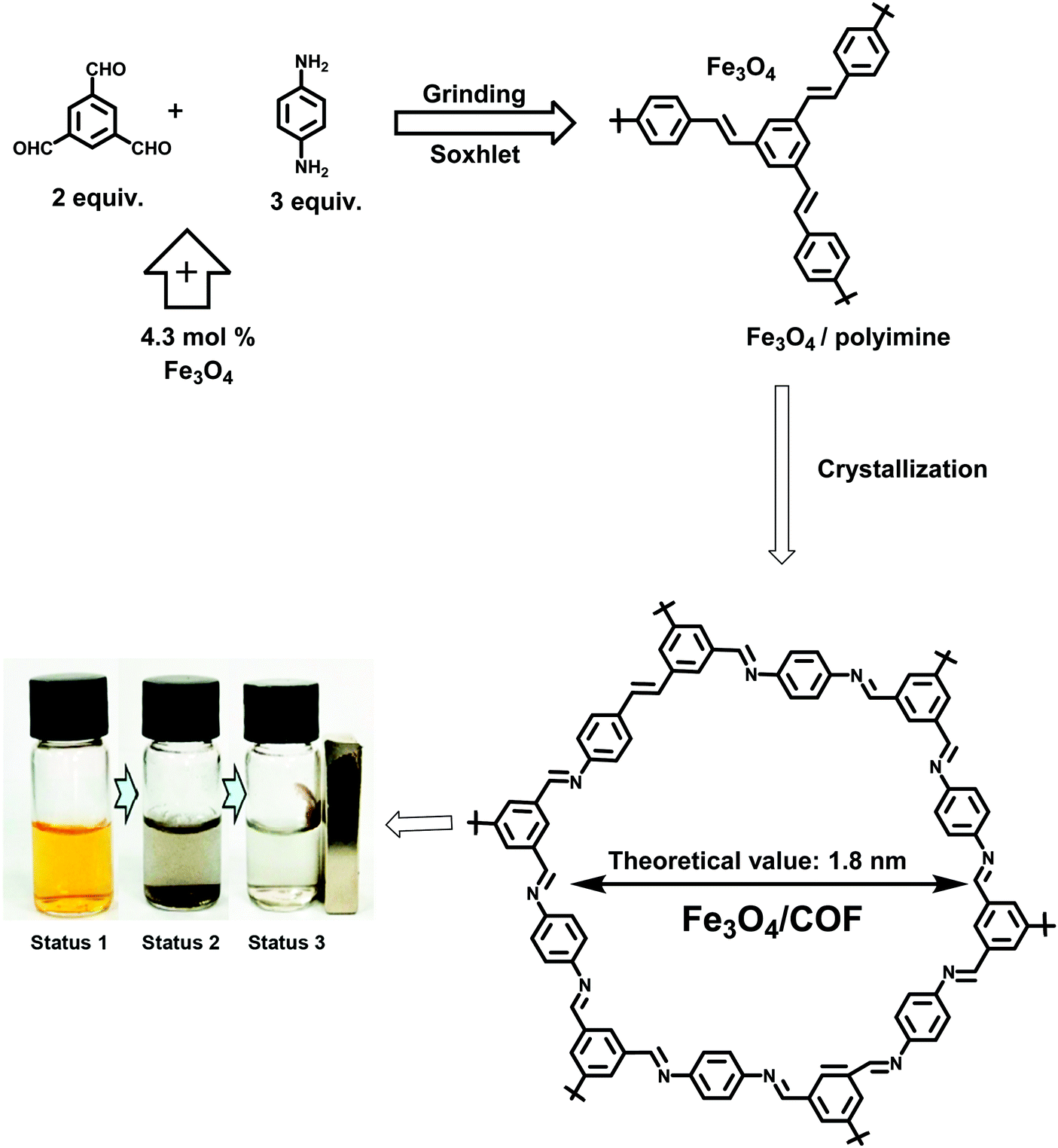
Similarly, a simple two-step synthesis involving, mechanochemical grinding along with crystallization, was deployed to obtain a combination of high quality COF and magnetic composite (Fig. 10).132
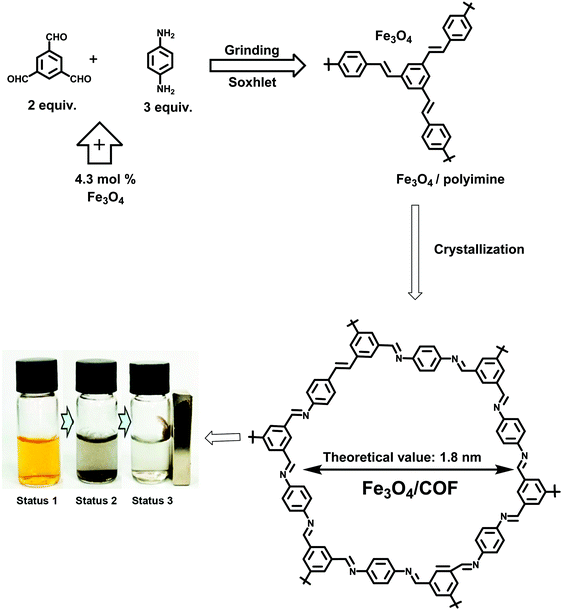 |
| | Fig. 10 Schematic illustration for the synthesis of Fe3O4/COF. Reproduced with permission from ref. 132 Copyright 2017 American Chemical Society. | |
For this, Fe3O4 (500–1200 nm), 1,3,5-triformylbenzene (TFB) and 1,4-diaminobenzene (DAB) were milled together with a few drops of 1,4-dioxane/acetic acid leading to the formation of an amorphous polymer network. Further crystallization was performed in a small amount of 1,4-dioxane/mesitylene/acetic acid at 70 °C for 5 days. Finally, the product was washed with toluene and methanol to yield Fe3O4/COF. The as-synthesized COF network forms a 15–35 nm thick organic shell on Fe3O4 particles. The COF prepared via this method was utilized for the adsorption of iodine from water and efficiently collected by using an external magnet (Fig. 10).132
Besides, many other groups have also synthesized magnetic COFs and have efficiently exploited them for various applications such as adsorption,133,134 and enrichment of peptides with simultaneous exclusion of proteins.135
3 Applications of COFs
COFs have enriched every aspect of catalytic transformations as potentially expedient organic-, electro- and photocatalysts owing to their interesting properties. In this section, various important reactions are discussed such as coupling reactions, oxidation reactions, oligomerization reactions, reduction reactions, asymmetric synthesis, tandem reactions, chemical fixation of CO2, cycloaddition reaction, condensation reaction, asymmetric Michael addition, water splitting, singlet oxygen generation, and some metal-free COF-catalyzed reactions. This review highlights the use of COFs that have proved their efficiency in these reactions.
3.1 Catalytic applications
COFs have diversified porosities, functional diversity, large surface area and excellent thermal stability.22,24 Therefore, in contrast to traditional amorphous solids and inorganic zeolites, COFs have been used as excellent candidates for catalytic applications.71,136 Besides, they offer numerous other features that are beneficial from the heterogeneous catalysis viewpoint: they can serve as hosts for catalytically active metal NPs; their well isolated structure minimizes aggregation of entrapped NPs; large spatial separation allows incorporation of multiple catalytic sites in the same COF, thereby imparting cooperative catalysis character; they have permeable channels that enable the substrates to gain a facile access to the catalytically active entities, thus allowing easy and efficient confined chemical reactions inside a nanoreactor and they are easily separable from reaction media, thus providing sustainability and economic viability. Several examples are presented in this section where COFs have been utilized in various metal-assisted and metal-free catalytic reactions.
3.1.1 Metal-containing COFs as catalysts.
3.1.1.1 C–C coupling reactions.
In recent years, numerous metal-supported catalysts have been established for cross-coupling reactions such as Pd-containing MOFs,137,138 Pd@silica,139–141 and Pd@MNPs,142,143 among others. However, examples on Pd@COFs are relatively very few.
In 2011, Wang et al. reported the imine-based 2D COF-LZU1 material for catalytic applications (Fig. 11).70 The imine-based 2D COF-LZU1 with an eclipsed layered sheet structure was easily prepared by a reaction between 1,3,5-triformylbenzene and 1,4-diaminobenzene, facilitating metal ion incorporation. By simple post-treatment, Pd(OAc)2 was added at room temperature to form Pd/COF-LZU1 which was then applied to catalyze Suzuki–Miyaura coupling reaction (Fig. 12). The superior prowess of Pd/COF-LZU1 was demonstrated by a wide substrate scope, producing excellent yields of the coupled products and easy recyclability of the catalyst which is attributed to its unique and supramolecular structure with easily accessible catalytic sites and fast diffusion of reactants and products from the COF surface.
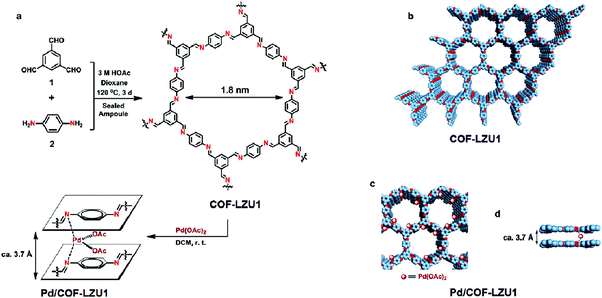 |
| | Fig. 11 (a and b) Proposed structure of COF-LZU1 and Pd/COF-LZU1 and (c and d) microporous channels and eclipsed layer sheet structure of Pd/COF-LZU1 (C: blue, N: red, Pd(OAc)2: brown spheres). Reproduced with permission from ref. 70 Copyright 2011 American Chemical Society. | |
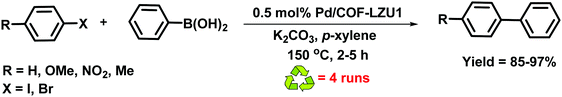 |
| | Fig. 12 Suzuki–Miyaura reaction catalyzed by Pd/COF-LZU1. | |
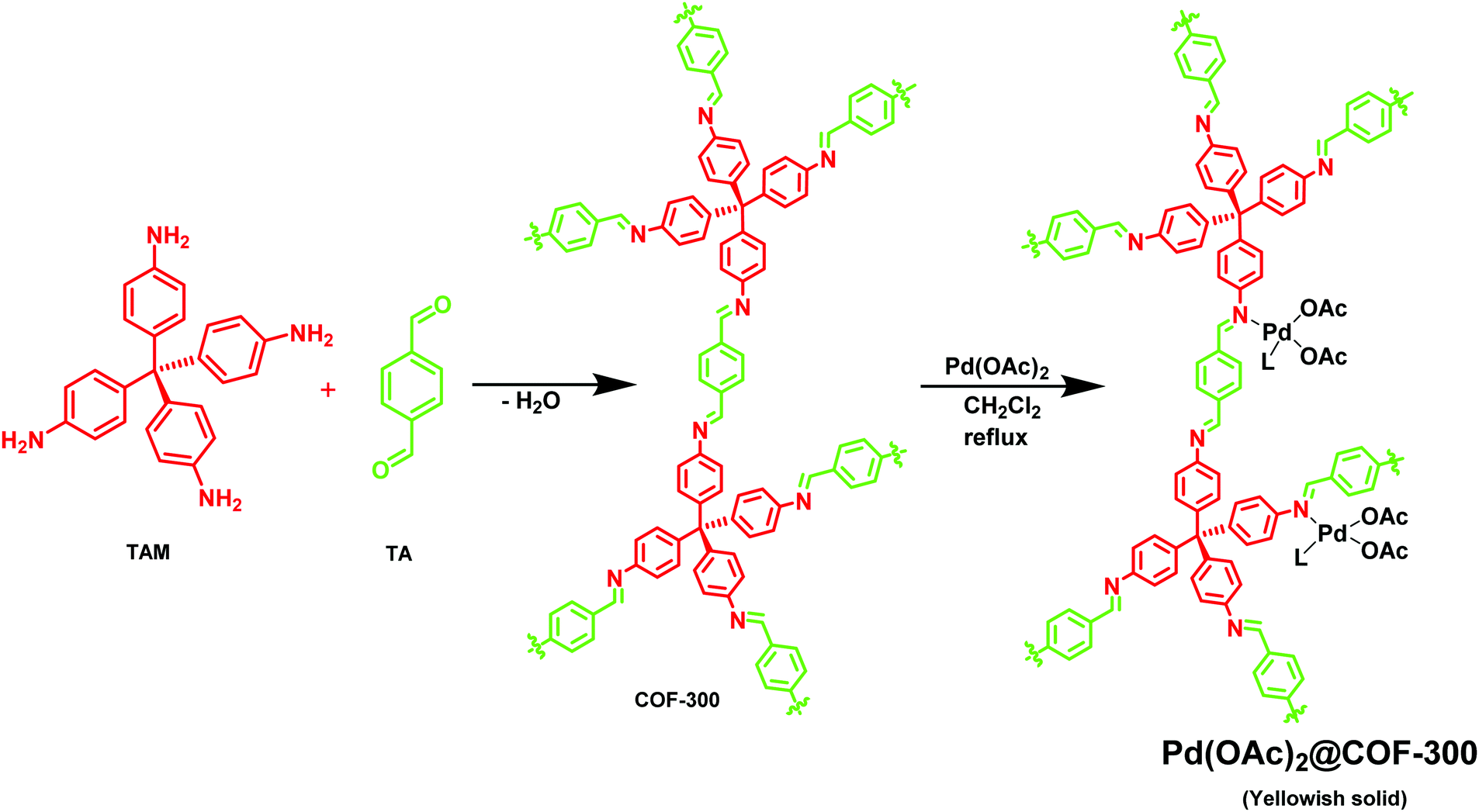
Esteves et al. synthesized an imine-linked COF-300 via a simple condensation of tetrakis-(4-aminobenzyl)methane (TAM) with terephthalaldehyde (TA) (Fig. 13);144 COF-300 has a high surface area of 1400–1500 m2 g−1 which can be used as a host material for Pd metal species. The catalytic activity of the Pd(OAc)2@COF-300 was evaluated in a Suzuki–Miyaura coupling reaction (Fig. 14); a series of boronic acids and aryl halides reacted under mild reaction conditions to generate coupled products in good to excellent yields. The applicability of the catalytic system was also tested in a continuous flow mode where Pd(OAc)2@COF-300 was filled in the glass column and all the other reactants and reagents were passed through the column for a defined residence time; 100% selectivity was observed during the reaction.
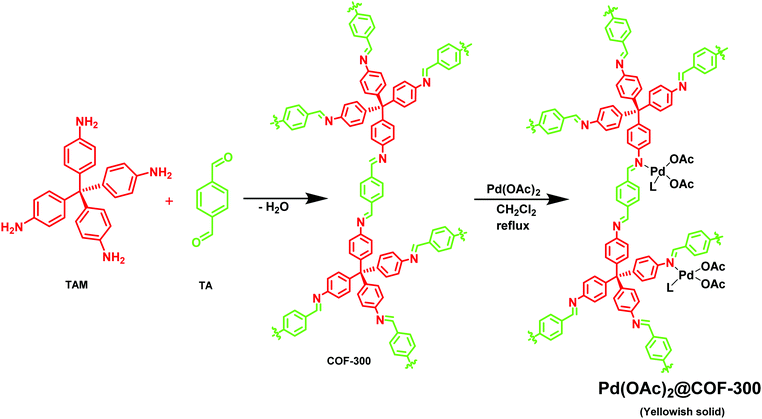 |
| | Fig. 13 Synthesis of Pd(OAc)2@COF-300. | |
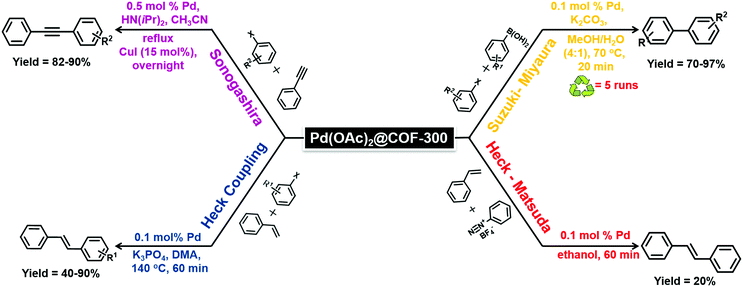 |
| | Fig. 14 Various C–C coupling reactions catalyzed by Pd(OAc)2@COF-300. | |
In addition, only a slight drop in the conversion was seen (from 56% to 44%) when the first aliquot was compared with the last. Encouraged by these results, the catalyst was also tested in other cross-coupling reactions: Heck, Sonogashira, and Heck-Matsuda (Fig. 14); isolated yields of 40–90% were attained in Heck and Sonogashira reactions. However, the desired product was barely observed in the case of Heck–Matsuda reaction which might be due to the highly polar diazonium salt having difficulty in accessing the hydrophobic channel of the COF or the inability of the salt to enter the pores of the COF owing to its large solvated size.
Similarly, a new and promising class of COF has been synthesized by Vaidhyanathan et al.145 where an amphiphilic triazine COF (trzn-COF) was used for loading of 5–20 nm sized Pd0 NPs (Fig. 15). Nitrogen-rich triazine mimics pyridyl groups that assist in the reduction of Pd2+ to Pd0 and interacts with Pd0 NPs. By contrast, ether bonds attached to the non-polar phenyl framework and Schiff bonds create an apolar matrix that mimics the environment present in homogeneous molecular catalysts. Pd-trzn-COF was then utilized for simultaneous or multi-fold Heck couplings, non-boronic acid C–C coupling and CO oxidation reactions (Fig. 16).
 |
| | Fig. 15 Schematic representation for the formation of triazine-based COF with Connolly representation showing stacking of hexagonal layers. Reproduced with permission from ref. 145 Copyright 2015 Springer Nature Publishing AG. | |
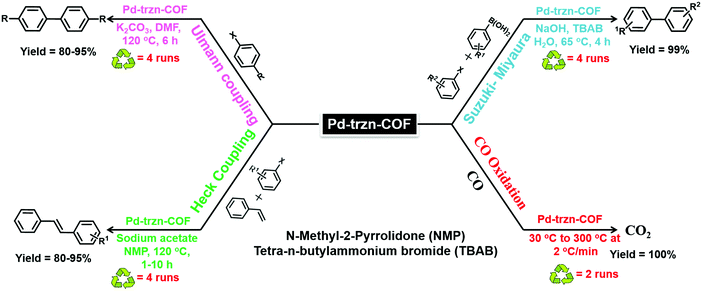 |
| | Fig. 16 Various C–C coupling reactions and CO oxidation reaction using Pd-trzn-COF as a catalyst. | |
Over the past few years, ultrafine and highly dispersed metal nanoparticles have been of interest to researchers because of their unique optical, mechanical and electronic properties.146,147 Amongst them, Pd and Pt noble metal nanoparticles have gained maximal attention in the field of catalysis due to their ultra-small size and high catalytic potential.148,149 However, these NPs aggregate on ordinary supports, and thus hamper the catalytic activities and reusability. In this context, Gu, Zhang et al. fabricated a thioether-based COF for the controlled growth of ultrafine metal NPs with narrow size distribution (1.7 ± 0.2 nm) (Fig. 17).150
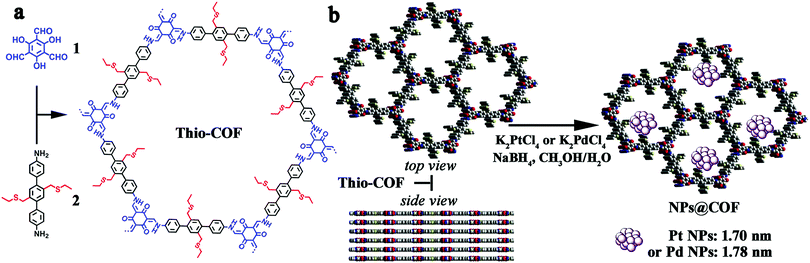 |
| | Fig. 17 (a) Synthesis of thioether-based COF, (b) schematic representation of PdNPs@COF and PtNPs@COF; top and side view of thio COF (S: yellow; N: blue; C: gray; O: red). Reproduced with permission from ref. 150 Copyright 2017 American Chemical Society. | |
The thioether groups provided suitable accommodation sites for the metal NPs via metal–sulfur interactions. The COF was established by the reaction of trialdehyde and thioether-substituted diamine via the solvothermal method. The ensuing PdNPs@COF and PtNPs@COF were then utilized in the Suzuki–Miyaura coupling reaction and for the reduction of 4-nitrophenol (Fig. 18); both catalysts showed superior catalytic activities in terms of mild reaction conditions, low catalyst loading, high product yields and easy recyclability and reusability without the loss of catalytic activity.
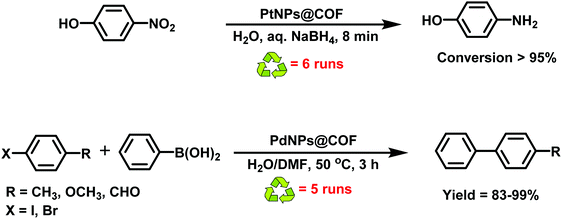 |
| | Fig. 18 Utilization of PtNPs@COF and PdNPs@COF as a catalyst in reduction of nitrophenol and Suzuki–Miyaura reaction, respectively. | |
 |
| | Fig. 19 Ionothermal synthesis of CTF. Reproduced with permission from ref. 151 Copyright 2010 American Chemical Society. | |
 |
| | Fig. 20 The oxidation of glycerol using Pd@CTF. | |
Similarly, Mu et al. synthesized hydroxyl groups bearing imine-based COFs by the condensation of 1,3,5-tris-(4-aminophenyl)triazine with 2,5-dihydroxyl-terephthalaldehyde under solvothermal (TAPT-DHTA-COFHX) (pore size = 2.30 nm) and reflux conditions (TAPT-DHTA-COFDMF) (pore size = 3.36 nm).152 Both COFs displayed high crystallinity as well as large surface area and showed an eclipsed stacking structure with active sites for the incorporation of Cu(OAc)2 which was coordinated via the interaction with hydroxyl groups and imine linkers (Fig. 21). The COFs were further utilized in the selective oxidation of styrene to benzaldehyde (Fig. 22). Besides showcasing remarkable catalytic performance, they also exhibited excellent recyclability without a significant loss of catalytic activity.
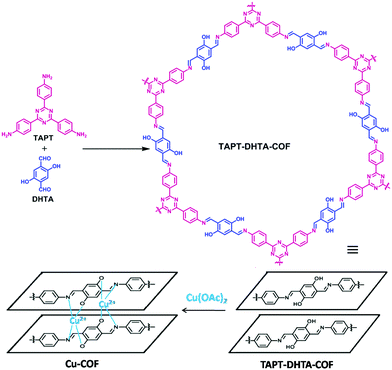 |
| | Fig. 21 Synthesis of TAPT-DHTA-COF and Cu-COF. | |
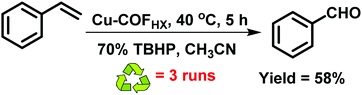 |
| | Fig. 22 Selective oxidation of styrene to benzaldehyde using Cu-COF as a catalyst. | |
 |
| | Fig. 23 Synthesis of (a) an imine-linked polymer network (IL-PON) through polyimine condensation, (b) a micro-CTF, and (c) a meso-CTF and expected coordination of Ni2+ to the nitrogen species in the frameworks. | |
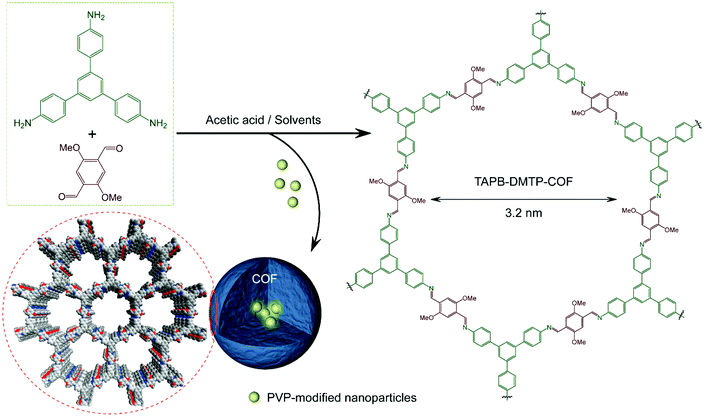 |
| | Fig. 24 Encapsulation of PVP-functionalized Au NPs into TAPB-DMTP-COF. Reproduced with permission from ref. 154 Copyright 2017 American Chemical Society. | |
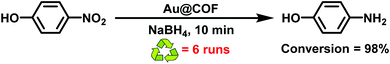 |
| | Fig. 25 Reduction of nitrophenol using Au@COF. | |
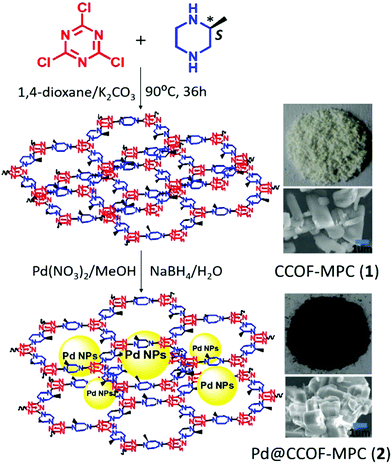 |
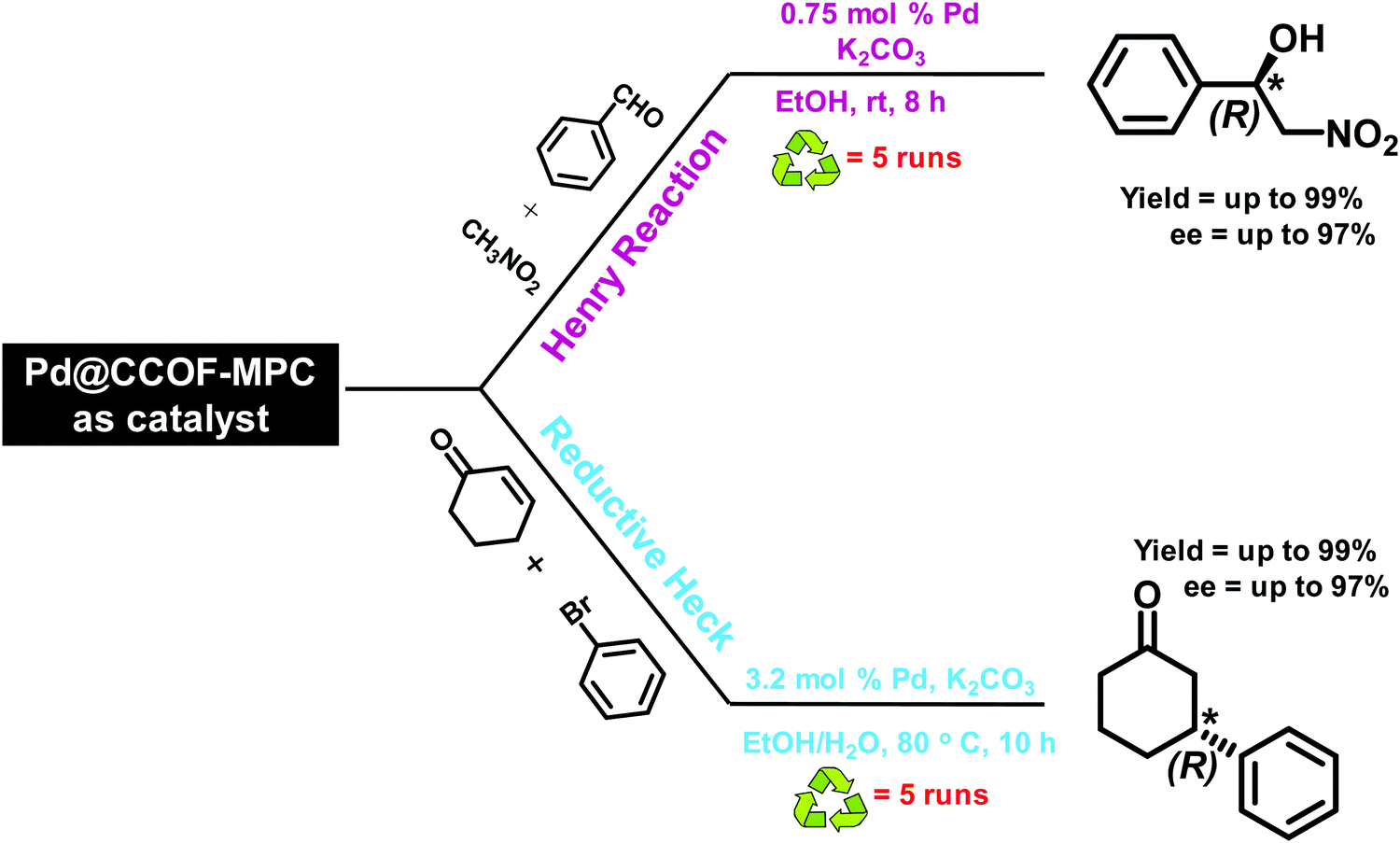
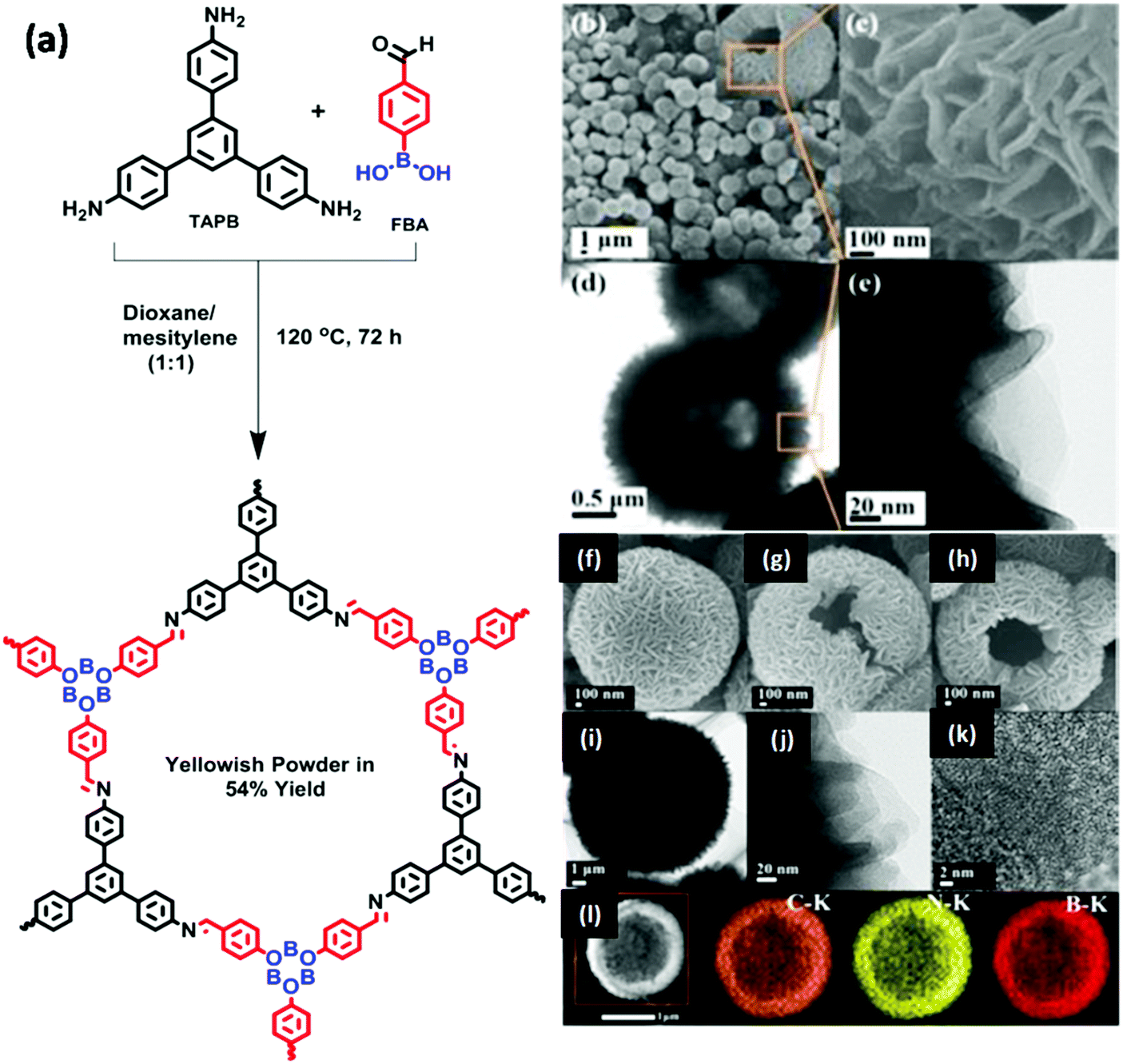
| | Fig. 26 Synthesis of CCOF-MPC and Pd@CCOF-MPC. Reproduced with permission from ref. 157 Copyright 2017 American Chemical Society. | |
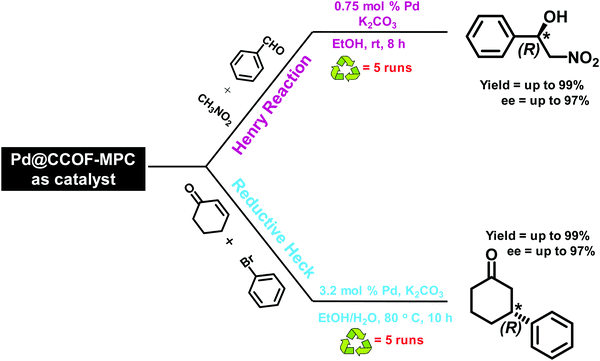 |
| | Fig. 27 Utilization of Pd@CCOF as a catalyst in asymmetric Henry and reductive Heck reaction. | |
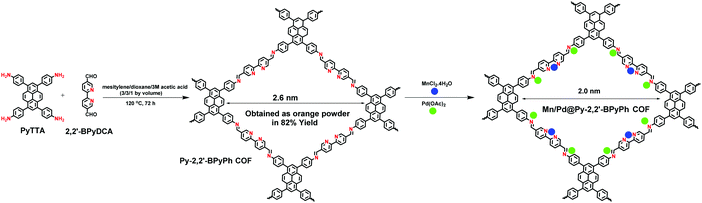 |
| | Fig. 28 Synthesis of Mn/Pd docked bimetallic COF, Mn/Pd@Py-2,2′-BPyPh. | |
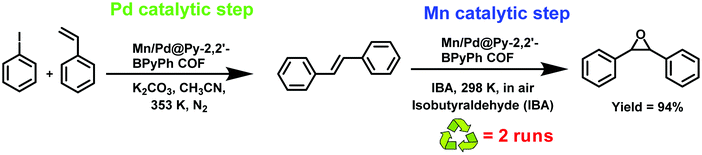 |
| | Fig. 29 Utilization of Mn/Pd@Py-2,2′-BPyPh in a tandem reaction. | |
3.1.2 COFs in metal-free catalysis.
3.1.2.1 Chemical fixation of CO2.
The emission of carbon dioxide is increasing as a result of anthropogenic activities and has caused severe health and environmental concerns.164,165 Not surprisingly, CO2 capture and its transformation into various value-added chemicals has gained much deserving attention.166,167 Numerous catalysts such as alkali metal salts,168 organometallic complexes169,170 and ionic liquids171,172 have been deployed but difficulty in separation due to their homogeneous nature has restricted their large-scale applications. Therefore, heterogenization of the above catalysts was next pursued using metal–organic frameworks,173,174 zeolites,175,176 silica177,178 and many other supports, however almost all are associated with the main drawback, which is metal toxicity and product contamination. To circumvent these shortcomings, Zhang, Liu et al. have utilized two imine-based COFs as metal-free and efficient organocatalysts for the conversion of CO2 to cyclic carbonates. COF-JLU6 (pore size 3.3 nm) was fabricated by the reaction of 1,3,5-tris-(4-aminophenyl)triazine and 2,5-dihydroxyterephthalaldehyde while COF-JLU7 (pore size 3.4 nm) utilized 1,3,5-tris-(4-aminophenoxy)benzene and 2,5-dihydroxyterephthalaldehyde, via solvothermal reactions (Fig. 30).179 When COFs were used as catalysts and tetra-n-butylammonium bromide (TBAB) as a co-catalyst, COF-JLU7 exhibited good catalytic performance which can be attributed to a large number of heteroatoms present in the walls of pores thus increasing the affinity and storage of CO2; COF-JLU7 can be reused for 5 consecutive runs without any appreciable loss in its catalytic activity.
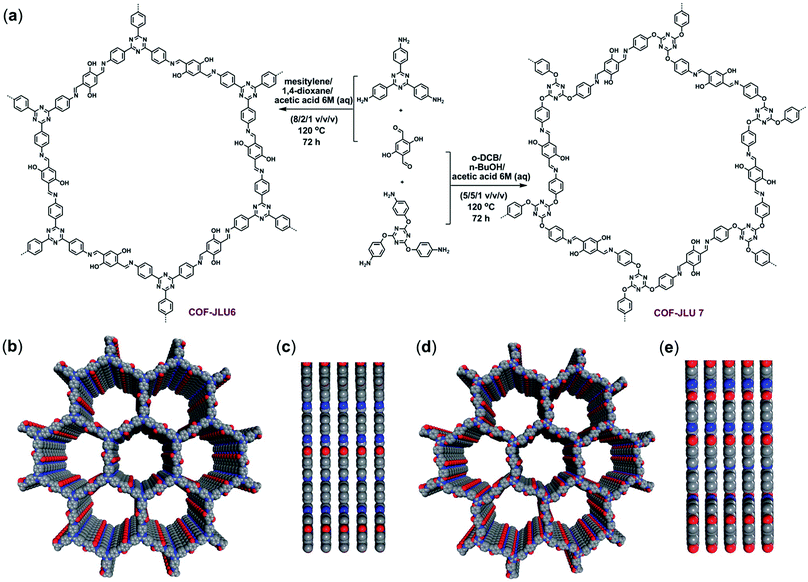 |
| | Fig. 30 (a) Schematic illustration for the formation of COF-JLU6 and COF-JLU7. (b) Top and (c) side view of COF-JLU6. (d) Top and (e) side view of COF-JLU7 (O: red; N: blue; C: gray). Reproduced with permission from ref. 179 Copyright 2018 Royal Society of Chemistry. | |
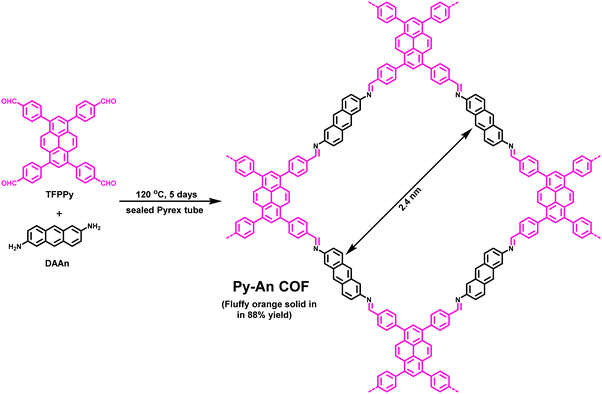 |
| | Fig. 31 Synthesis of Py-An COF. | |
The prepared heterogeneous catalyst was utilized in a Diels–Alder reaction of 9-hydroxymethylanthracene with N-substituted maleimide derivatives. With the aid of Py-An COF as a catalyst, all the reactions continued smoothly at room temperature and pressure. The ease of catalytic activity can be attributed to the enhanced accumulation of reactants within the pores of Py-An COF and the presence of C–H⋯π interactions between anthracene units of π-walls of COF and the 9-hydroxymethylanthracene reactant molecule (Fig. 32). Moreover, the prepared heterogeneous catalyst can be reused and recycled in up to four runs without loss of its catalytic activity after centrifugation.
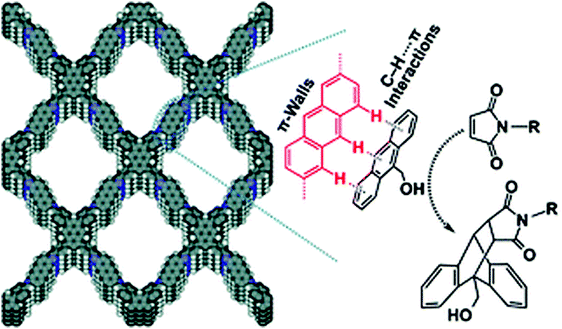 |
| | Fig. 32 C–H⋯π interactions between Py-An COF and the reactant molecule. Reproduced with permission from ref. 182 Copyright 2015 Royal Society of Chemistry. | |
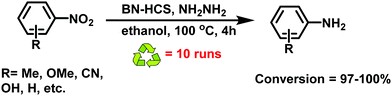 |
| | Fig. 33 Catalytic activity of BN-HCS in reduction of nitroarenes. | |
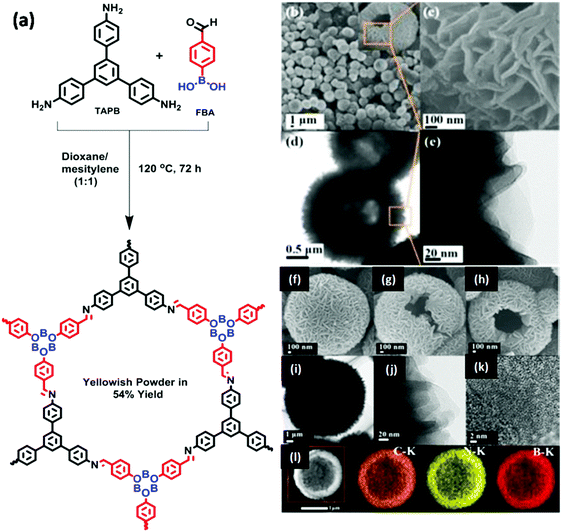 |
| | Fig. 34 (a) Synthesis of H-COF, SEM images of (b and c) H-COF, (f) BN-HCS-700, (g) BN-HCS-800, (h) BN-HCS-900, TEM images of (d and e) H-COF, (i and j) BN-HCS-800, (k) HR-TEM image of BN-HCS-800 and (l) EDX elemental mapping of BN-HCS-800. Reproduced with permission from ref. 185 Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
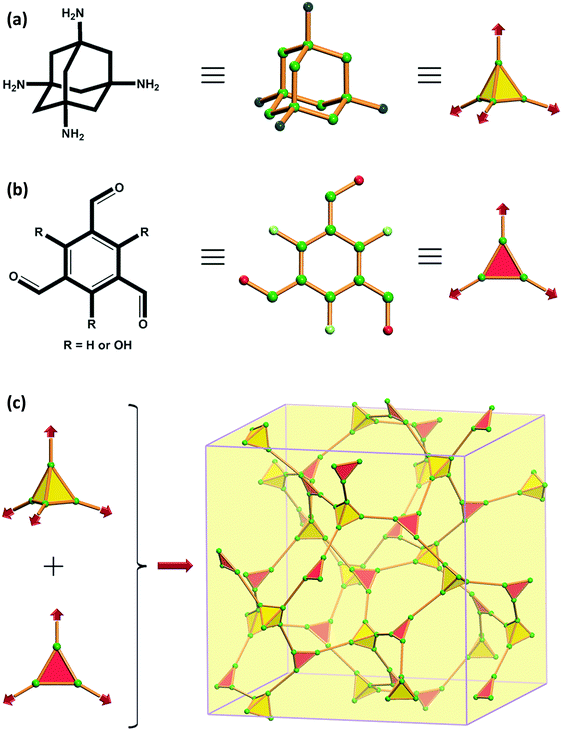 |
| | Fig. 35 Structure of (a) TAA, (b) TFB (RH) or TFP (ROH) and (c) condensation of TAA and TFB or TAA and TFP to give BF-COF-1 and BF-COF-2 respectively. Reproduced with permission from ref. 73 Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
 |
| | Fig. 36 Catalytic activity of BF-COFs in a Knoevenagel condensation reaction. | |
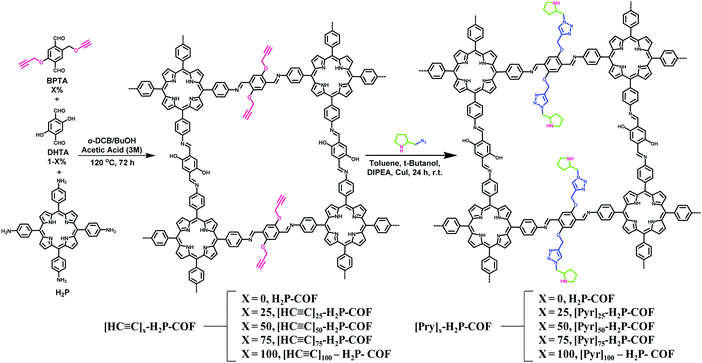 |
| | Fig. 37 Pore surface engineering of imine-based COF via condensation reaction and click chemistry. | |
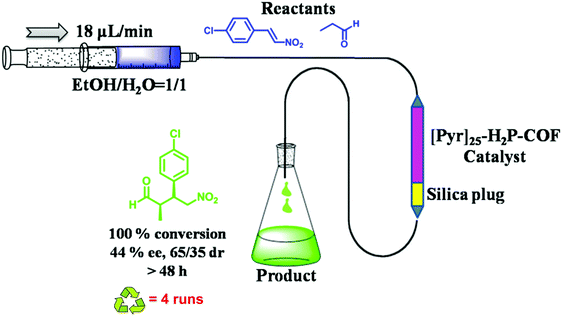 |
| | Fig. 38 Use of a continuous flow reactor for the Michael addition reaction using [Pyr]25-H2P-COF as a catalyst. | |
3.2 Electrocatalytic applications
Electrocatalytic reactions are regarded as one of the most direct and effective pathways for the interconversion of electrical and chemical energy.191–194 Since the beginning of the 1980s, the design and development of advanced electrocatalysts for various energy conversion processes, such as water splitting reactions and CO2 reduction, have garnered tremendous attention from the research community;195–198 these processes being often regarded as clean, green and renewable. Recently, researchers have found that doping of covalent structures with heteroatoms, such as N, can enhance electrochemical reactivity.199,200 In this regard, various COF-based electrocatalysts have been designed owing to their tunable structural and electronic properties, desired multi-element composition, large surface area and precisely controllable channels for the diffusion and transportation of ions;201 such deployment of COF-based electrocatalysts for various processes, is briefly mentioned below.
3.2.1 Oxygen reduction reactions (ORR) and oxygen evolution reactions (OER).
Fuel cell technology is considered to be one of the most attractive alternatives to current energy conversion processes because of its immense potential for high efficiency and environmental friendliness.202,203 To date, various electrochemical reactions have been carried out using Pt and other noble metal oxides,204–207 but limitations due to scarcity and high costs have hindered their large-scale use. Therefore, the commercialization of such clean energy technologies requires efficient, stable and affordable electrocatalysts with outstanding ORR and OER activity.
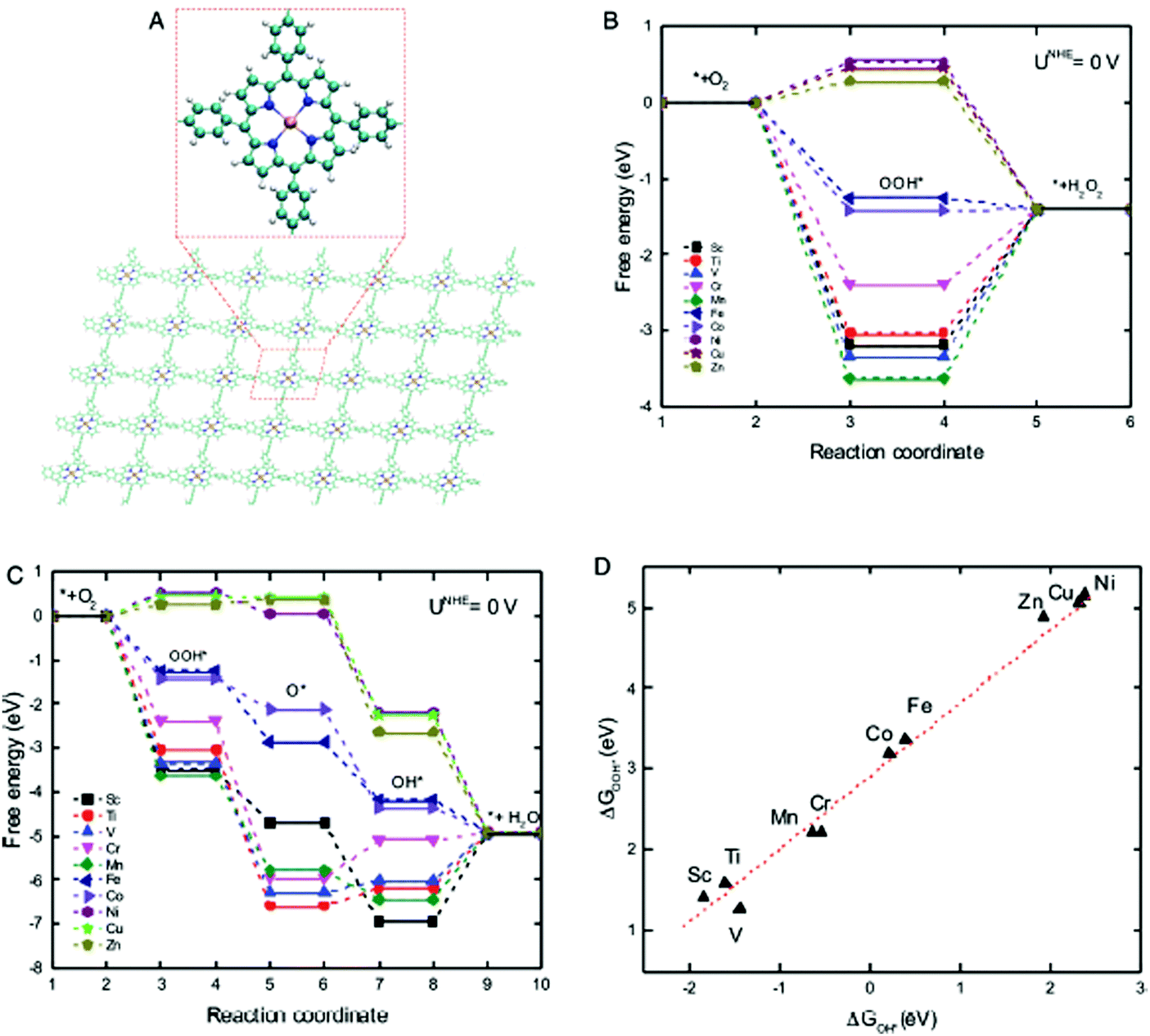
Lately, the use of biomimetic units, with inherently present active sites, has been explored in non-precious-metal ORR catalysis.208,209 However, their design requires understanding of intrinsic material characteristics, or descriptors, which can correlate their structural aspects with catalytic activities. A recent study by Xia et al. identified crystal field stabilization energy (CFSE) as an activity descriptor for predicting ORR/OER activities of transition metal (TM) containing COFs with porphyrin units (TM = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn) (Fig. 39A).95Fig. 39B and C show that among all the TMs, Fe- and Co-porphyrin-COF can spontaneously lead to both 2- and 4-electron transfer. Usually, overpotential (η), which represents the energy barrier needed to be overcome in ORR/OER, is considered as a measure of catalytic activity; the lower the η, the better the catalytic performance. Among all the 3d-TMs, Fe-porphyrin-COF presents the minimum overpotential; 0.381 V and 0.485 V for OER and ORR respectively, which is comparable to previously reported results.95 The study also predicted the ability to produce green oxidizer, H2O2, using COFs as electrocatalysts. H2O2 is currently synthesized industrially using a high-energy intensive anthraquinone process. CFSE descriptor predicts that among all the TM-COFs, Zn- and Cu-COFs could serve as the most efficient catalysts for H2O2 production due to lower overpotential values (1.15 V and 0.97 V for Cu and Zn respectively); however, the energy barriers are practically quite high. To design a better catalyst, the authors prepared certain COFs with alkaline earth metals (AM) (AM = Be, Mg, Ca, Sr, Ba). They introduced a new descriptor called orbital configuration energy (CE), making it possible to describe the catalytic activity of AM-COFs. Among all AM-COFs, Ca- and Sr-COFs showed quite low η of 0.27 and 0.29 V, respectively.
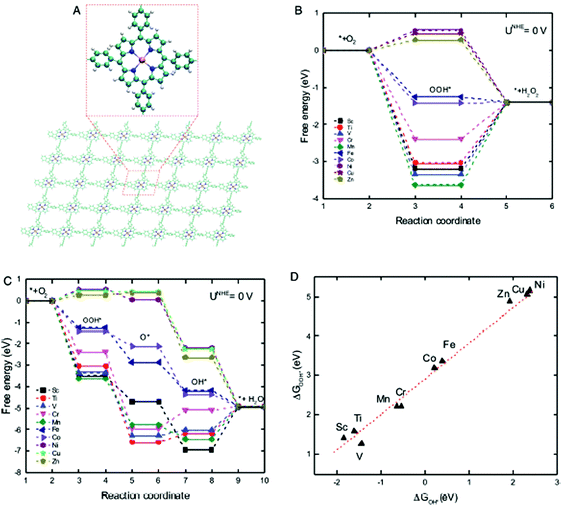 |
| | Fig. 39 (A) Schematic illustration of TM-COFs (TM = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn) (C: green, H: silver, N: blue, TM: deep gold spheres), free energy diagrams of TM-porphyrin-COFs for (B) 2-electron transfer and (C) 4-electron transfer ORR in an acid medium and (D) absorption energy of OOH* versus that of OH*. Reproduced with permission from ref. 95 Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
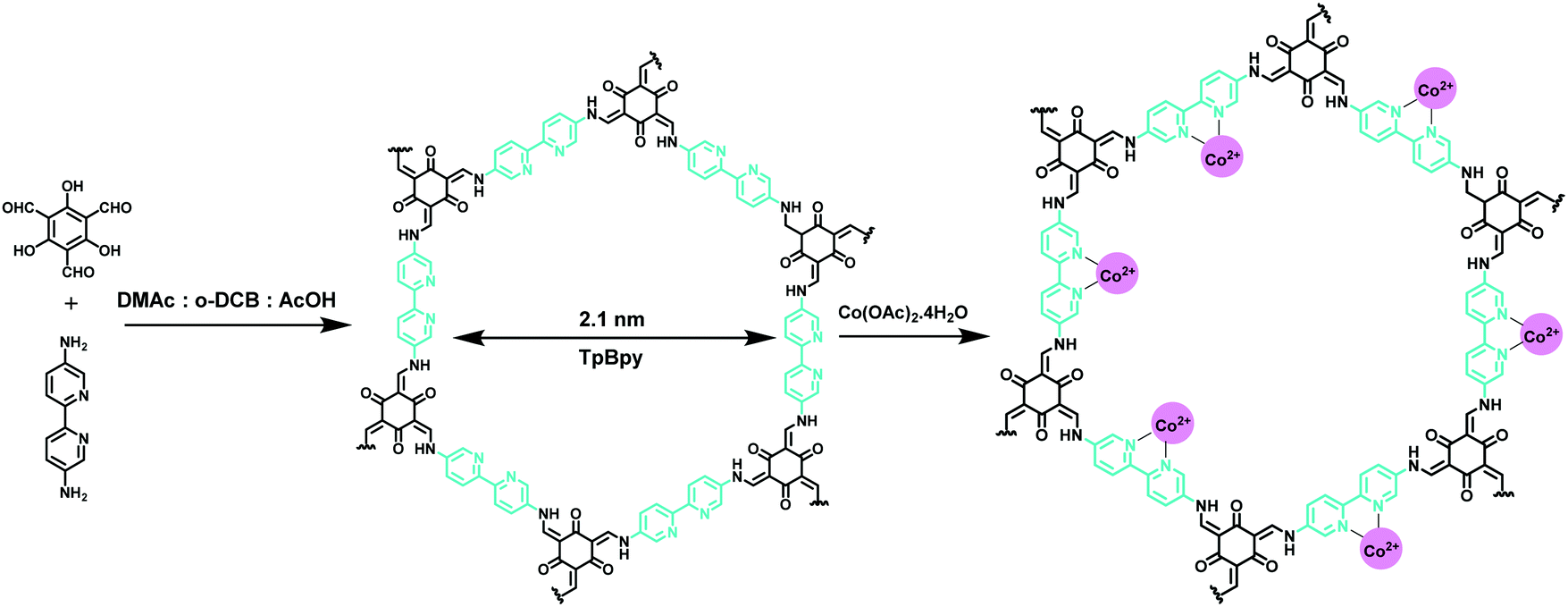
Similarly, Aiyappa et al. designed a cobalt-containing bipyridine-based COF (Co-TpBpy) as a heterogeneous electrocatalyst for the OER under neutral pH conditions (Fig. 40).210 The as-prepared COF retains a porous, robust and stable structure with a very high surface area of 450 m2 g−1 which allows easy passage of liberated O2, thereby minimizing leaching of the catalyst and improving the long-term stability during prolonged operation time and cycles. The fabricated COF showed high activity under neutral pH conditions in a phosphate buffer solution with an overpotential of 400 mV at a current density of 1 mA cm−2. Besides, the catalyst retained 94% of its activity even after 1000 cycles and 24 h of activity with a turnover frequency (TOF) of 0.23 s−1 and faradaic efficiency of 0.95.
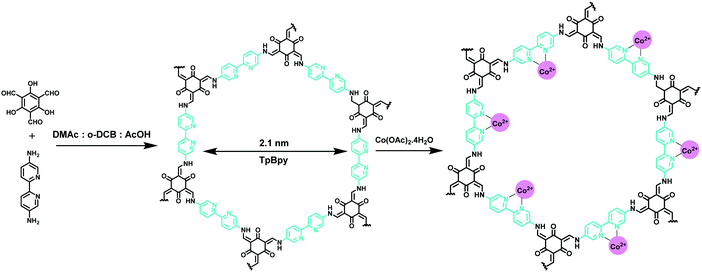 |
| | Fig. 40 Synthesis of Co-TpBpy. | |
3.2.2 Reduction of CO2.
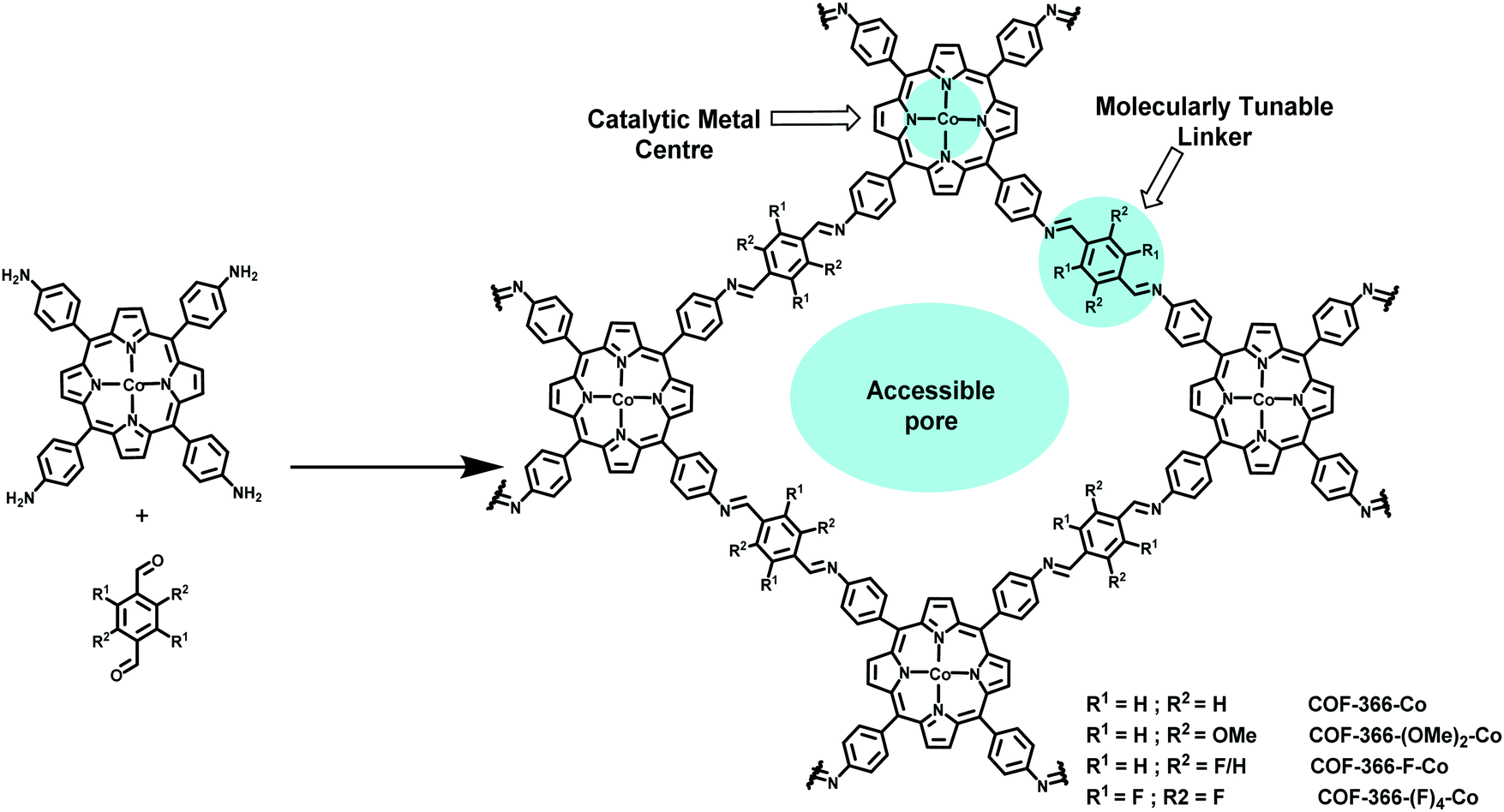
Concerning CO2 fixation, various techniques have been developed so far, among which electrochemical reduction of CO2 into value-added fuels appears to be a promising option.211–215 Zhang et al. have disclosed the successful reduction of CO2 in a value-added carbon product, carbon monoxide, by utilizing the tunable heterogeneous crystalline COF comprising cobalt porphyrins via electrochemical means using cheap, abundant, and environmentally benign reaction media, water;72 a highly active, stable, porous and selective catalyst was fabricated by the imine condensation of [5,10,15,20-tetrakis(4-aminophenyl)porphinato]cobalt [Co(TAP)] with 1,4-benzenedicarboxaldehyde (BDA) (Fig. 41). The SEM image of the fabricated COF-366-Co showed crystallites of rectangular rod-shaped morphology (∼50 nm in length) with a relatively narrow pore size distribution (10 to 18 Å). The COF-366-Co displayed high electrochemical performance at −0.67 V in CO2 saturated aqueous bicarbonate buffer having a faradaic efficiency for carbon monoxide (FECO) = 90%, TON of 1352 and TOF of 98 h−1 greater than the cobalt porphyrin unit alone. The catalyst has high stability (24 h) and could be reused up to five times without any significant loss of activity and can be grown over the surface of thin films on glassy carbon. Moreover, the expanded COF-367-Co analog was fabricated using biphenyl-4,4′-dicarboxaldehyde (BPDA) as the strut and was evaluated for the electrocatalytic generation of CO by varying the content of cobalt within the COF which manifests FECO = 91%, TON of 3901 and TOF of 165 h−1.
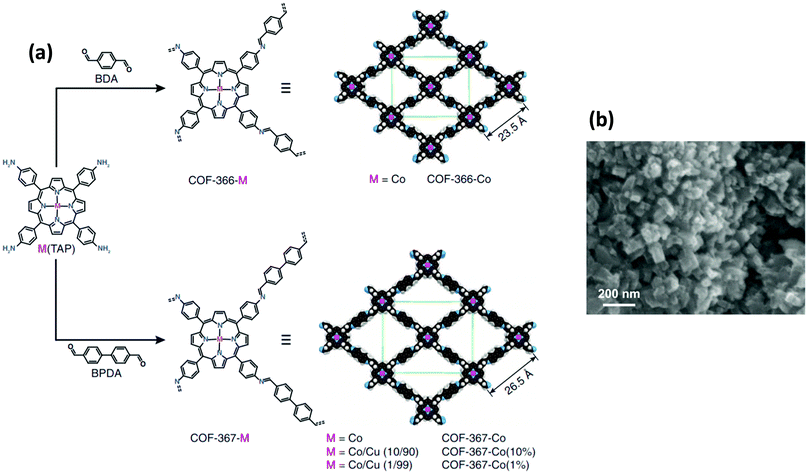 |
| | Fig. 41 (a) Design and synthesis of metalloporphyrin-derived 2D COFs and (b) SEM image of COF-366-Co. Reproduced with permission from ref. 72 Copyright 2015 AAAS. | |
Recently, Wen et al. reported the synthesis of a series of perfluorinated FN-CTFs-X (X = synthesis temperature) for the electrocatalytic reduction of CO2.216 CTFs were synthesized using the ionothermal method via one-step polymerization of tetrafluoroterepthalonitrile in the presence of molten ZnCl2 at various temperatures (Fig. 42); all of the materials exhibited a large surface area and displayed good CO2 adsorption capacities. Notably, FN-CTF-400 is a highly selective catalyst for the electrochemical reduction of CO2 into CH4 as its faradaic efficiency for CH4 production (FECH4) is extremely high (99.3%) in the potential range of −0.7 V to −0.9 V (Fig. 43). By contrast other FN-CTFs showed decreased selectivities for the production of CH4 (Fig. 43b–d). In addition, FN-CTF-400 demonstrated high catalytic stability as it maintained an FECH4 value of 91.7% even after 5 h of continuous electroreduction. The authors contend that FN-CTF-400 consisted mainly of a polymer and a small amount of pyrolyzed carbon due to synthesis at a lower temperature. The polymer assisted in CO2 capturing as well as in suppressing H2 evolution by creating a hydrophobic environment, while the N and F co-doped carbon framework provided suitable active sites for the production of CH4. However, in other FN-CTFs, the polymer got converted into graphite structures, which reduced CO2 into CO rather than converting it into CH4.
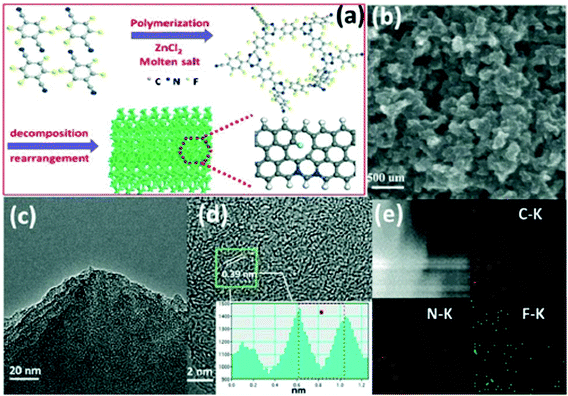 |
| | Fig. 42 (a) Ionothermal synthesis, (b) SEM image, (c) TEM image, (d) HRTEM image with line scan and (e) EDS elemental mapping of FN-CTF-400. Reproduced with permission from ref. 216 Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
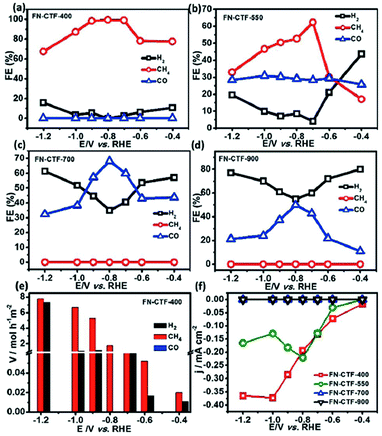 |
| | Fig. 43 Faradaic efficiencies for the production of CH4, H2 and CO versus electrode potential of (a) FN-CTF-400, (b) FN-CTF-550, (c) FN-CTF-700 and (d) FN-CTF-900, (e) rate of production of CH4, H2 and CO with FN-CTF-400 at different applied potentials and (f) current densities corresponding to CH4 production. Reproduced with permission from ref. 216 Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Another group also designed a cobalt-porphyrin-based COF for the electrocatalytic reduction of CO2 to CO.217 In the report, the authors grew a series of 2D oriented thin films of COFs (of ∼250 nm thickness), onto an electrode made of highly ordered pyrolytic graphite (HOPG), with a variety of electron-withdrawing functional groups on the linker (Fig. 44). The study revealed that the electronic character of the catalytically active cobalt center, which is embedded within the reticular superstructure of COFs, could be modulated by remote functionalization of the organic strut. This induces an electron-withdrawing effect on cobalt, which is proportional to the electronegativity and the amount of the installed functional groups. This also significantly improved the catalytic behavior of COFs by facilitating the first step of CO2 reduction, in which Co(II) was reduced to Co(I), by taking the electronic density away from Co, which rendered it more prone to reduction; the current density for CO formation is maximum for COF-366-F-Co, 65 mA mg−1 (Fig. 45). Furthermore, COFs showed remarkable reduction at low overpotential (550 mV) with high selectivity (faradaic efficiency of 87%) and stability (for more than 12 h without any significant loss in reactivity).
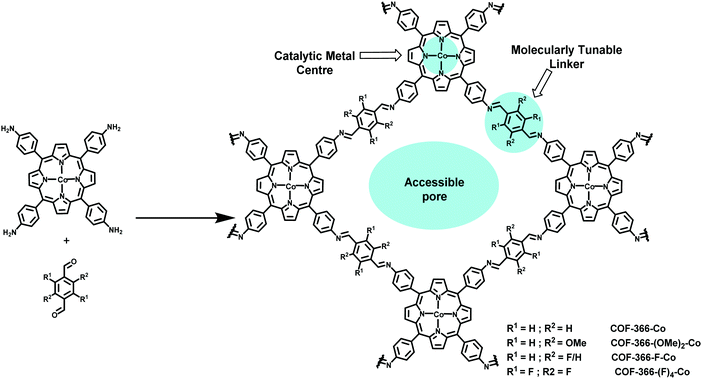 |
| | Fig. 44 Synthesis of cobalt-porphyrin-derived COFs. | |
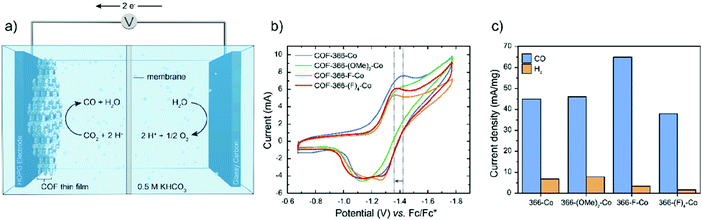 |
| | Fig. 45 Electrochemical characterization of COFs. (a) Illustration of the electrolysis cell, (b) cyclic voltammogramms of COFs in N,N-dimethylformamide with tetrabutylammoniumhexafluorophosphate as the electrolyte and (c) current densities per milligram of cobalt in different COF catalysts. Reproduced with permission from ref. 217 Copyright 2018 American Chemical Society. | |
3.3 Photocatalytic applications
Among various applications of 2D COFs, photocatalysis appears to be a green approach in converting solar energy to chemical energy in order to drive various organic transformations. Moreover, researchers have shown immense interest in exploring the full potential of this emerging class of porous, metal-free crystalline polymers aiming for photochemical reactivity; COFs provide a design-controllable platform with confined functional pore space having tunable electronic and photocatalytic properties. Due to these appealing features, they enable applications such as water splitting, photoredox reactions, selective CO2 reduction, singlet oxygen generation and photodegradation of organic pollutants.
3.3.1 Water splitting.
In recent years, COFs in their crystalline and porous forms combined with either metal clusters or organic ligands have been deployed in diverse areas including catalysis,218–220 sensing,221,222 and gas storage.223,224 This encouraged researchers to integrate the COFs with various other components, which subsequently led to achieving fascinating properties of the resulting hybrid materials such as large surface areas, long-range order structures, tunable band gaps, and superb visible light absorbance. Due to the aforementioned properties, the hybrid composites of COFs have received special attention in the field of photocatalysis.225 Moreover, researchers have also deployed the seamless sp2 carbon-conjugated 2D COFs to render hydrogen production that is facilitated by light harvesting, exciton migration, exciton splitting, and electron transfer.226
Recently, Zhang et al. synthesized a 2D COF with a stable MOF by covalently anchoring NH2-UiO-66 onto the surface of TpPa-1-COF (Fig. 46).227 This new material exhibited a highly porous framework with good crystallinity and large surface area and was exploited for photocatalytic H2 evolution under visible light irradiation. Amongst other MOF- and COF-based photocatalysts, this COF-MOF hybrid displayed ultra-high H2 evolution rates, which was attributed to the well-matched band gaps between NH2-UiO-66 and TpPa-1-COF, and the effective charge separation across the interface in the hybrid material.
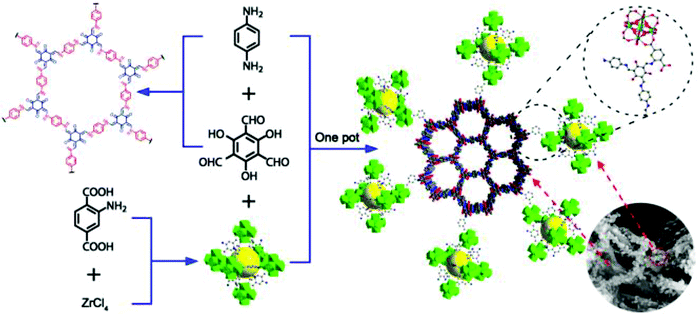 |
| | Fig. 46 Schematic illustration for the synthesis of NH2-UiO-66/TpPa-1-COF hybrid material. Reproduced with permission from ref. 227 Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Similarly, Lotsch et al. demonstrated the development of stable and tunable photocatalysts which were built on low molecular weight triazine-based compounds by circumventing the possibility of formation of amorphous materials by decreasing the reaction temperature to 300 °C and prolonging the reaction time to more than 150 h.228 This prevents incipient carbonization, unlike CTF-1 (Fig. 47). The group designed a series of phenyl-triazine oligomers (PTOs) by varying the parameters (such as a ratio of precursor to ZnCl2 salt and initial precursor dilution) for a multi-step electron transfer process to allow design of the band gap and modified band potential, dependent on various factors such as a nitrogen content of CTFs, number of ring systems and the extent of π–π interactions. In this regard, engineering the trimer (2,4,6-tris(p-cyanophenyl)-1,3,5-triazine) has been proven to be an effective approach to facilitating photocatalytic hydrogen evolution with a rate of 1076 (±278) mmol h−1 g−1 upon sunlight irradiation (400 (±20) nm) with an apparent quantum efficiency of 5.5 (±1.1)%. Furthermore, the presence of large excess of terminal nitrile moieties in the smaller oligomers synthesized via excess of ZnCl2 favors the dispersion of photocatalyst (by hydrogen bonding) and leads to high photocatalytic activity compared to the larger one.
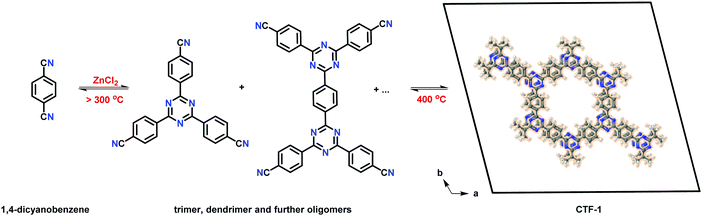 |
| | Fig. 47 Reaction mechanism for the formation of CTF-1 via trimerization and, ultimately, to an extended 2D CTF (right). The idealized crystal structure of fully condensed, crystalline CTF-1 is shown in an AA-type eclipsed configuration (viewed along the c-direction) (C: gray; N: blue; H: white). | |
Likewise, Cooper et al. have reported the fabrication and utilization of organic semiconductors for photocatalytic hydrogen production using ascorbic acid as a sacrificial electron donor and Pt as a co-catalyst.229 The crystalline, microporous and idealized eclipsed structure of FS-COF (where FS is fused sulfone) was fabricated by a Schiff-base condensation reaction of 2,4,6-triformylphloroglucinol (TFG) and 3,9-diamino-benzo[1,2-b:4,5-b′]bis[1]benzothiophene-5,5,11,11-tetraoxide where keto–enol tautomerization helps in enhancing the chemical stability (Fig. 48). Additionally, FS-COF shows high external quantum efficiency (3.2%) and high rate of hydrogen evolution (10.1 mmol g−1 h−1) which is 22 times higher than N3-COF (0.47 mmol g−1 h−1) with a steady photochemical reaction up to 50 h at 420 nm. Here, the high absorbance of photons, exfoliation and increasing the number of potential sites of FS-COF play a significant role in the photoreduction of water.
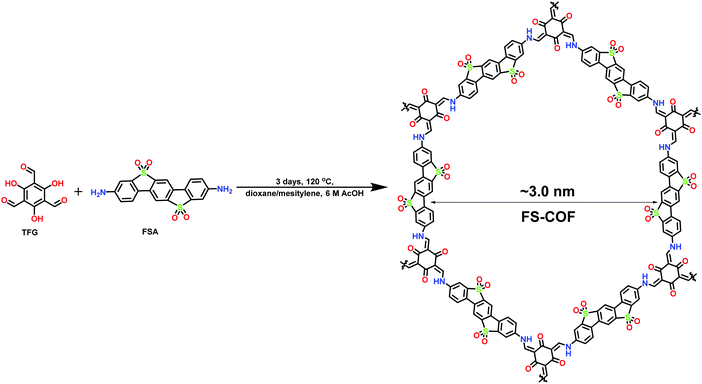 |
| | Fig. 48 Synthesis of FS-COF. | |
Furthermore, various 2D nanoporous, well-defined π-extended organic graphene analogs/semiconductors, produced through CC linkages with tunable band gap, were successfully exploited for water splitting under visible light. Zhang et al. utilized a solvothermal approach to synthesize reversible and self-healing 2D COFs namely g-C40N3-COF, g-C31N3-COF, and g-C37N3-COF having trans-disubstituted CC linkages by deploying 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) as the key building block with 4,4′′-diformyl-p-terphenyl (DFPTP), 4,4′-diformyl-1,1′-biphenyl (DFBP) and 1,3,5-tris(4-formylphenyl) benzene (TFPB), respectively via Knoevenagel condensation at the aryl methyl carbon atoms (Fig. 49).230 The honeycomb-like crystalline porous sp2 carbon-jointed pyridinyl structured COFs having high surface areas were deployed to carry out water splitting reaction at two different half reactions using 10 vol% of triethanolamine (TEOA) as a sacrificial reagent. The g-C40N3-COF shows high hydrogen evolution rate 206 μmol h−1 with the apparent quantum efficiency of 4.84% at λ = 420 nm over the other two COFs.
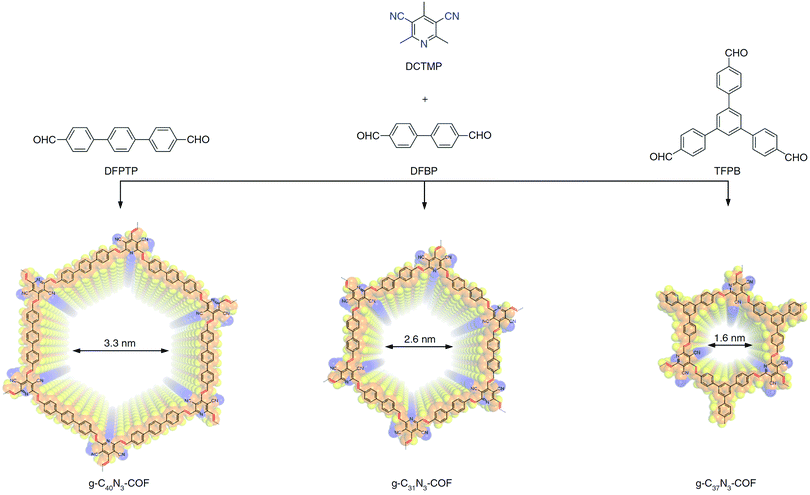 |
| | Fig. 49 Illustration of synthetic procedures of g-CxNy-COFs. Reproduced with permission from ref. 230 Copyright 2019 Springer Nature Publishing AG. | |
Recently, diacetylene-bridged COFs have received significant attention due to their high charge career mobility, conjugated framework, and potential of having active sites for facile migration of photogenerated excitons to the surface. In this respect, Pachfule et al. recently reported the synthesis of highly porous and chemically stable acetylene (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) and diacetylene (–CC–CC–) functionalized β-ketoenamine COFs and have applied them as photocatalysts for hydrogen generation from water.231 Here, two types of β-ketoenamine COFs bearing acetylene and diacetylene moieties, were synthesized via an acid-catalyzed solvothermal reaction. This was accomplished by reacting 1,3,5-triformylphloroglucinol (TP) with 4,4′-(ethyne-1,2-diyl)dianiline (EDDA) and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA) to form TP-EDDA and TP-BDDA, respectively, in the presence of acetic acid. The keto–enol tautomerism during ketoenamine formation of amines with TP, provided COFs their necessary chemical stability. It was observed that the diacetylene-based COF outperformed the acetylene-based COF in terms of photocatalytic activity which is attributed to its high porosity, easily accessible diacetylene functionalities, and considerable chemical stability. In order to analyze the effect of acetylene and diacetylene moieties on optical and catalytic properties, an isoreticular COF was synthesized with comparable pore apertures comprised from terphenylene edges (TP-DTP COF, DTP: 4,4′′-diamino-p-terphenyl). Diffuse reflectance ultraviolet-visible (UV-Vis) spectra displayed noticeable changes between the COFs (Fig. 50), where TP-DTP showed an absorbance edge near 500 nm, while red-shift of the absorbance edge to 520 and 525 nm was observed for TP-EDDA and TP-BDDA COFs, respectively. Besides, the absorbance tail got extended up to 675 nm for TP-BDDA, revealing specific red shift. In addition, optical band gaps were calculated from Tauc plots, which were found to be 2.42, 2.34, and 2.31 eV, for TP-DTP, TP-EDDA, and TP-BDDA, respectively, (Fig. 50a, inset), and are considered ideal for photocatalytic water splitting which needs a minimum band gap of ∼1.8 eV.
C–) and diacetylene (–CC–CC–) functionalized β-ketoenamine COFs and have applied them as photocatalysts for hydrogen generation from water.231 Here, two types of β-ketoenamine COFs bearing acetylene and diacetylene moieties, were synthesized via an acid-catalyzed solvothermal reaction. This was accomplished by reacting 1,3,5-triformylphloroglucinol (TP) with 4,4′-(ethyne-1,2-diyl)dianiline (EDDA) and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA) to form TP-EDDA and TP-BDDA, respectively, in the presence of acetic acid. The keto–enol tautomerism during ketoenamine formation of amines with TP, provided COFs their necessary chemical stability. It was observed that the diacetylene-based COF outperformed the acetylene-based COF in terms of photocatalytic activity which is attributed to its high porosity, easily accessible diacetylene functionalities, and considerable chemical stability. In order to analyze the effect of acetylene and diacetylene moieties on optical and catalytic properties, an isoreticular COF was synthesized with comparable pore apertures comprised from terphenylene edges (TP-DTP COF, DTP: 4,4′′-diamino-p-terphenyl). Diffuse reflectance ultraviolet-visible (UV-Vis) spectra displayed noticeable changes between the COFs (Fig. 50), where TP-DTP showed an absorbance edge near 500 nm, while red-shift of the absorbance edge to 520 and 525 nm was observed for TP-EDDA and TP-BDDA COFs, respectively. Besides, the absorbance tail got extended up to 675 nm for TP-BDDA, revealing specific red shift. In addition, optical band gaps were calculated from Tauc plots, which were found to be 2.42, 2.34, and 2.31 eV, for TP-DTP, TP-EDDA, and TP-BDDA, respectively, (Fig. 50a, inset), and are considered ideal for photocatalytic water splitting which needs a minimum band gap of ∼1.8 eV.
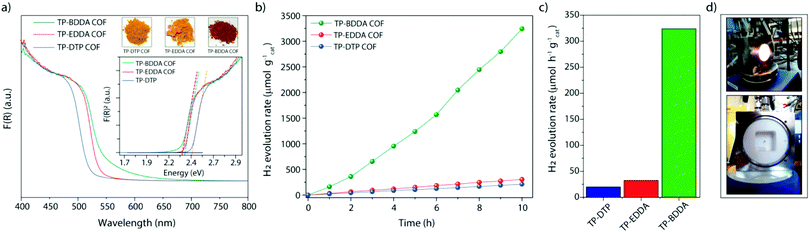 |
| | Fig. 50 (a) UV-Vis diffuse reflectance spectra of TP-BDDA, TP-EDDA, and TP-DTP. The inset shows Tauc plots and optical images of the COF powders. (b) Photocatalytic hydrogen evolution performance of TP-BDDA, TP-EDDA, and TP-DTP catalysts under visible light irradiation (≥395 nm) using TEOA as a sacrificial agent. (c) Comparison of photocatalytic hydrogen evolution rates. (d) Reactor setup for hydrogen evolution experiments. Reproduced with permission from ref. 231 Copyright 2017 American Chemical Society. | |
3.3.2 CO2 reduction.
Perceiving CO2 in the atmosphere as a raw material that may be used as a carbon source enables researchers to define a new chemistry along with addressing the issue of global warming.232 Hence, scientists have undertaken the design of a variety of active metal-based photocatalysts and semiconductors for the effective conversion of CO2 into valuable chemicals and fuel using solar energy.233–237 Utilization of MOF and TEOA (as reductant) for the CO2 reduction leads to the formation of HCOOH as a side product.237–243 COFs offer various advantages over MOF, namely high porosity, crystalline nature, large surface area, and ease of design and fine tuning, while using an organic moiety with high covalent interactions between them.20,56,244–249 Zhu et al. designed highly stable (chemically and thermally) metal-free azine-based COFs which exhibited high photocatalytic and electronic performance.250 Two crystalline azine-linked 2D COFs, namely ACOF-1 and N3-COF, were used as photocatalytic systems for the aqueous CO2 reduction into CH3OH, where water acted as an environmentally friendly and inexpensive reductant. Both of these COFs showed high surface area and π–π stacking between layers. The electrochemical and photocatalytic properties of ACOF-1 and N3-COF revealed a high absorbance in the region of 480–500 nm and an optical band gap of approximately 2.6 eV which suggest a better photocatalytic activity for CO2 reduction (Fig. 51). A stainless-steel reactor having a quartz window was utilized for the reduction under a CO2 pressure of 0.4 MPa at 80 °C. The importance of light as well as the photocatalyst for the reduction of CO2 with H2O was proven as no product formation took place in their absence. Decreasing the CO2 concentration was accompanied by a decrease in the yield of the reduced product.
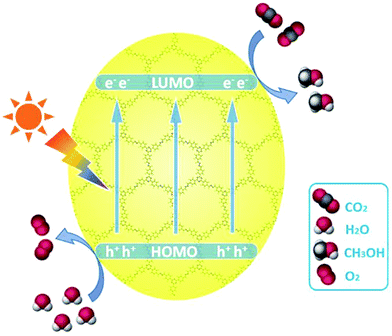 |
| | Fig. 51 Schematic diagram for the photocatalytic reduction of CO2 over azine-based COFs under visible light irradiation. Reproduced with permission from ref. 250 Copyright 2018 Elsevier B.V. | |
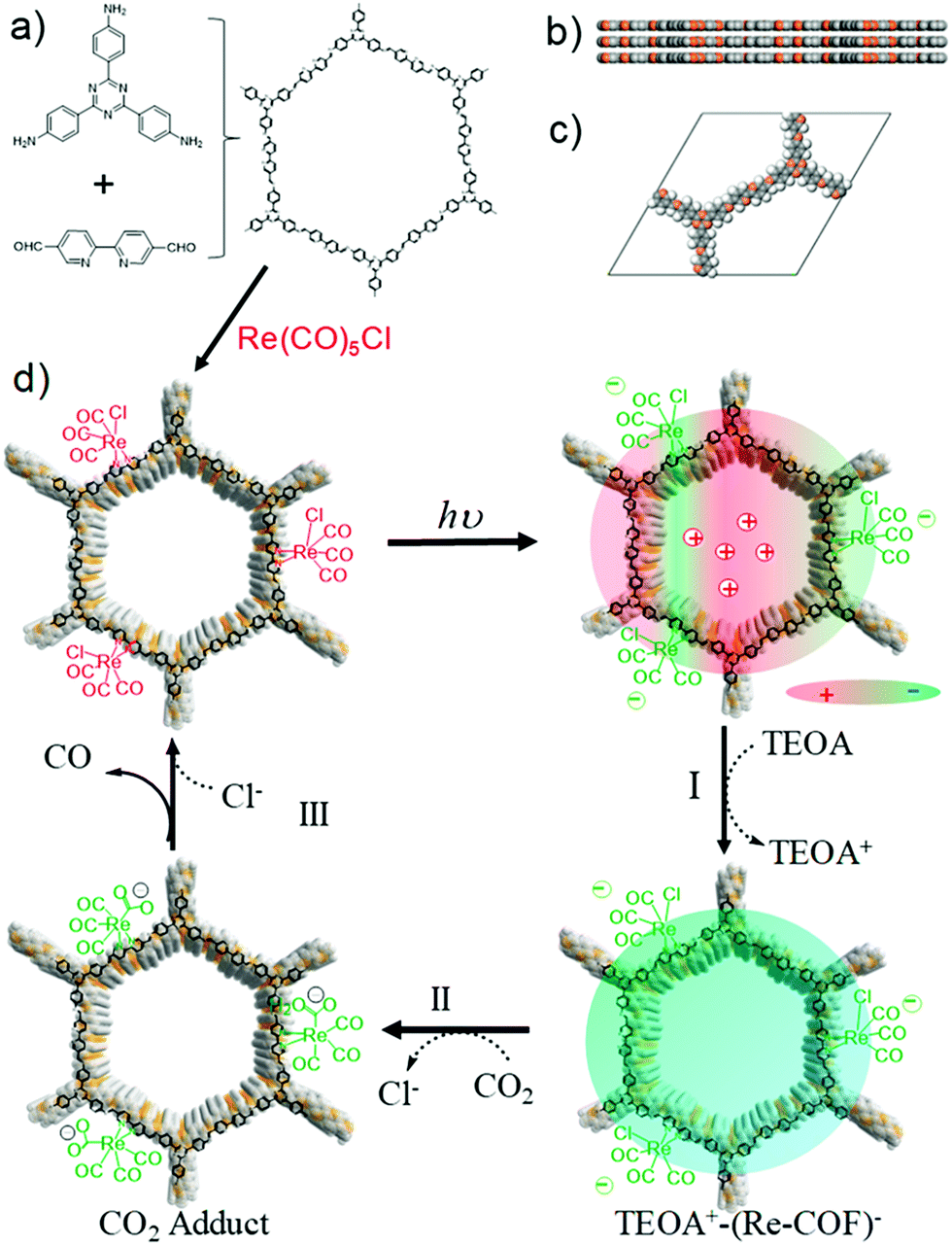
Zhang et al. recently reported the successful reduction of CO2 to CO by utilizing a hybrid 2D tricarbonylchloro(bipyridyl) Re integrated triazine COF (Re-COF) with high activity and selectivity (98%) upon irradiation with light.251 Solvothermal methodology was adopted for the stepwise fabrication of the crystalline Re-COF; triazine COF was achieved by using 2,2-bipyridyl-5,5-dialdehyde (BPDA) and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and a further Re moiety was introduced by post-modification with Re(CO)5Cl (Fig. 52). Here, the presence of Re over the COF helps in inhibiting the process of charge recombination and showed longer lifetime (τ = 171 ps). Moreover, transient optical and X-ray transient absorption (XTA) spectroscopy suggests that the system facilitates intramolecular charge transfer through electron transfer from photoexcited COF to Re(bpy)(CO)3Cl and clarify the lag in the charge recombination. The photocatalytically viable Re-COF was utilized for CO2 reduction to generate ∼15 mmol CO per g of catalyst constantly for >20 h after ∼15 min induction period in the presence of TEOA as a sacrificial donor upon irradiation with a Xe lamp; the system shows a high TON of 48 and can be reused and recycled up to 3 runs.
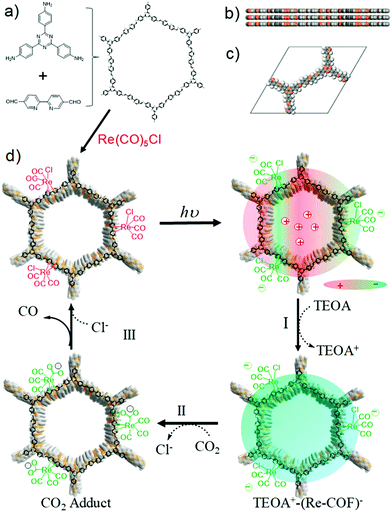 |
| | Fig. 52 (a) Synthesis of triazine COF and Re-COF; (b) side view and (c) unit cell of AA stacking COF; (d) proposed catalytic mechanism for CO2 reduction. Reproduced with permission from ref. 251 Copyright 2018 American Chemical Society. | |
In a similar manner, Zou et al. have fabricated and utilized a Ni bearing 3D COF for selective reduction of CO2 to CO (Fig. 53);252 effective use of 2,2′-bipyridine-based COF was made as a support to comprise single Ni sites (Ni-TpBpy) which is highly useful in confinement and interaction. Here, the synthesis of 2,2-bipyridine-based COF was carried out by the condensation of 1,3,5-triformylphloroglucinol with 5,5′-diamino 2,2′-bipyridine using solvothermal conditions and further treatment with Ni(ClO4)2 leading to Ni-TpBpy. The fabricated COF exhibited high activity for CO generation (4057 μmol g−1) under aqueous conditions within 5 h with an enhanced selectivity (96%) over H2 evolution (170 μmol g−1). The Ni-TpBpy-based COF showed a good turnover number (13.62) and quantum efficiency (0.3%). Moreover, the catalyst can be reused up to three runs with slight loss of catalytic activity; no major morphological and structural changes were observed.
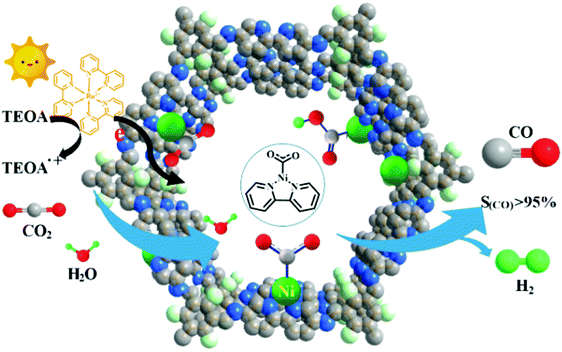 |
| | Fig. 53 Schematic diagram of photocatalytic selective reduction of CO2 over Ni-TpBpy. Reproduced with permission from ref. 252 Copyright 2019 American Chemical Society. | |
3.3.3 Photoredox organic transformations.
Owing to the considerable popularity of macromolecular organic semiconductor photocatalysts due to easy tuning of their optoelectronic properties by a suitable choice of donor and acceptor units along with their arrangement, they have been effectively deployed as a stable platform for visible-light photoredox reactions.253–266 Over the years, various transition metal complexes267–269 and organic dyes270,271 have been used as photocatalysts in many photoredox reactions, but their homogeneous nature, high cost, tedious recovery and poor recyclability often limit their large-scale applications. Considering the benefits of COFs, Liu et al. fabricated triazine-based COF-JLU5 by simple condensation of 1,3,5-tris-(4-aminophenyl)triazine with 2,5-dimethoxyterephthalaldehyde (Fig. 54).272 This COF fulfils the requirements of a promising photocatalytic system because of its periodic columnar π-array structure which arises from the layered stacking of 2D polymer sheets. This also allows, in principle, good exciton diffusion and migration. Additionally, it has several other advantages such as: (1) the –OMe group, adjacent to the Schiff base, increases the chemical stability by resonance effects; and (2) the electron donor–acceptor structure enhances the photocatalytic properties. With these features in hand, COF-JLU5 was utilized in the aerobic cross-dehydrogenative coupling (CDC) of N-aryltetrahydroisoquinolines (Fig. 55). All the reactions were carried out using a small amount of a photocatalyst, at room temperature using blue LEDs and under aerobic conditions; all products were obtained in good to excellent yields.
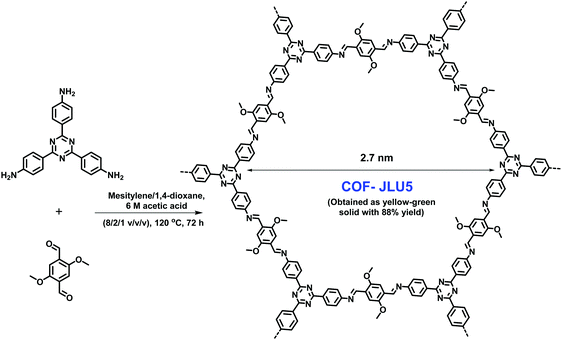 |
| | Fig. 54 Synthesis of COF-JLU5 by an imine condensation reaction. | |
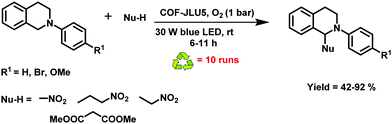 |
| | Fig. 55 Aerobic CDC using COF-JLU5. | |
Researchers have identified visible light as a green and sustainable source for promoting organic transformations.273–275 Recently, Huang et al. reported an asymmetric CTF as an efficient organic single-component semiconductor visible-light-active photocatalyst that exhibits a photogenerated energy transfer cascade.276 This asymmetric framework comprises four distinct molecular donor–acceptor domains, which enhances the intramolecular energy transfer and photoredox potential, and broadens the photogenerated absorption range. It was found to be superior over symmetric CTFs containing similar donor and acceptor moieties in terms of photocatalytic efficiency and this was demonstrated via a model visible light-driven dehydrogenation reaction of diphenylphosphine oxide and diphenylacetylene to form benzophosphole oxides (Fig. 56).
 |
| | Fig. 56 CTF-catalyzed visible-light-promoted formation of 1,2,3-triphenylphosphindole 1-oxide from diphenylphosphine oxide and diphenylacetylene. | |
Likewise, a Pd NP (2.2 nm) doped core–shell MOF@COF hybrid material was developed by Sun et al. in the form of Pd/TiATA@LZU1 that displayed excellent photocatalytic performance for tandem dehydrogenation and hydrogenation reactions in a continuous-flow microreactor and in a batch system.277 The material was well characterized by SEM, TEM and elemental mapping which revealed that TiATA@LZU1 core–shell particles with a size between 0.5 and 1.5 μm were formed with a shell thickness of ∼20 nm (Fig. 57). Synergy was found among the three units in the “donor–mediator–acceptor” system, where the MOF core behaved as an electron donor, the COF shell as a medium for electron transfer, and metal as an acceptor. Additionally, the photocatalytic tandem dehydrogenation of ammonia borane and hydrogenation of olefins was carried out in a newly fabricated dual-chamber microreactor by grafting Pd/TiATA@LZU1 on the inner wall. In situ evolved H2 gas instantaneously passed from the bottom chamber to the upper chamber via a gas-permeable membrane and was consumed in a hydrogenation reaction. This dual-chamber microreactor prevents contamination of products and refrains from mismatching of two reaction conditions that are usually encountered in bulk reactors.
 |
| | Fig. 57 The SEM images of (a) TiATA (b) LZU1, and (c) core–shell TiATA@LZU1, (d) TEM and (e) elemental mapping images of TiATA@LZU1. The scale bars in (a–e) are 500 nm, 5 μm, 200 nm, 0.1 μm, and 250 nm, respectively. Reproduced with permission from ref. 277 Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
To date, many groups have reported the use of precious metal-based photocatalytic systems for the selective oxidation of organic moieties but their costs and toxicity limit their practical utility.278–281 Recently, Zhang et al. have designed and synthesized thiophene-containing CTF-Th to carry out selective oxidation (Fig. 58).282 These metal-free frameworks offer highly selective visible-light-promoted oxidation of alcohols to aldehydes and ketones under environmentally benign and mild conditions. The framework imparts various desirable properties such as robustness, large structural variation and tunable optical and electronic properties. The group attained a very low highest occupied molecular orbital (HOMO) level at +1.75 V versus a saturated calomel electrode (SCE) which enhanced the photo-oxidation potential of the CTFs. The CTF precursor was successfully synthesized by using the cyclized trimer of 2,5-dicyanothiophene (DCT) and supporting it over mesoporous silica SBA-15 in the presence of TfOH vapors. Finally, a highly ordered catalyst was obtained as an insoluble yellow powder.
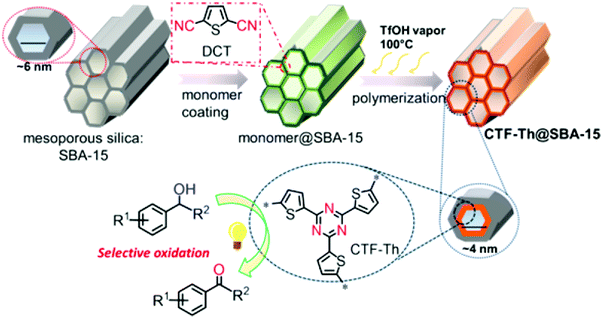 |
| | Fig. 58 Illustrated design and formation pathway for the CTF-Th on SBA-15 as a mesoporous nanoreactor. Reproduced with permission from ref. 282 Copyright 2017 American Chemical Society. | |
TEM images of CTF-Th@SBA-15 revealed an exquisite hexagonal cylindrical structure with 800 nm length, 500 nm diameter and a pore size of ca. 4 nm (Fig. 59). BET studies displayed the importance of SBA-15 in increasing the surface area of CTF-Th@SBA-15 via providing mesoporous channels.
 |
| | Fig. 59 (a) SEM image, and (b and c) HR-TEM of CTF-Th@SBA-15. Elemental mapping images of CTF-Th@SBA-15 in (d) carbon, (e) sulfur and (f) nitrogen. Reproduced with permission from ref. 282 Copyright 2017 American Chemical Society. | |
The catalytic efficiency was also verified by selective oxidation of alcohols using CTF-Th@SBA-15 as a photocatalyst under mild conditions; high conversion and selectivity was reported for eight substrates in the presence of oxygen as a clean oxidant (Fig. 60).
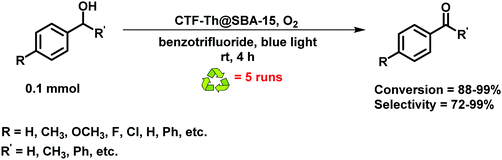 |
| | Fig. 60 Photocatalytic selective oxidation of benzyl alcohol under an oxygen atmosphere. | |
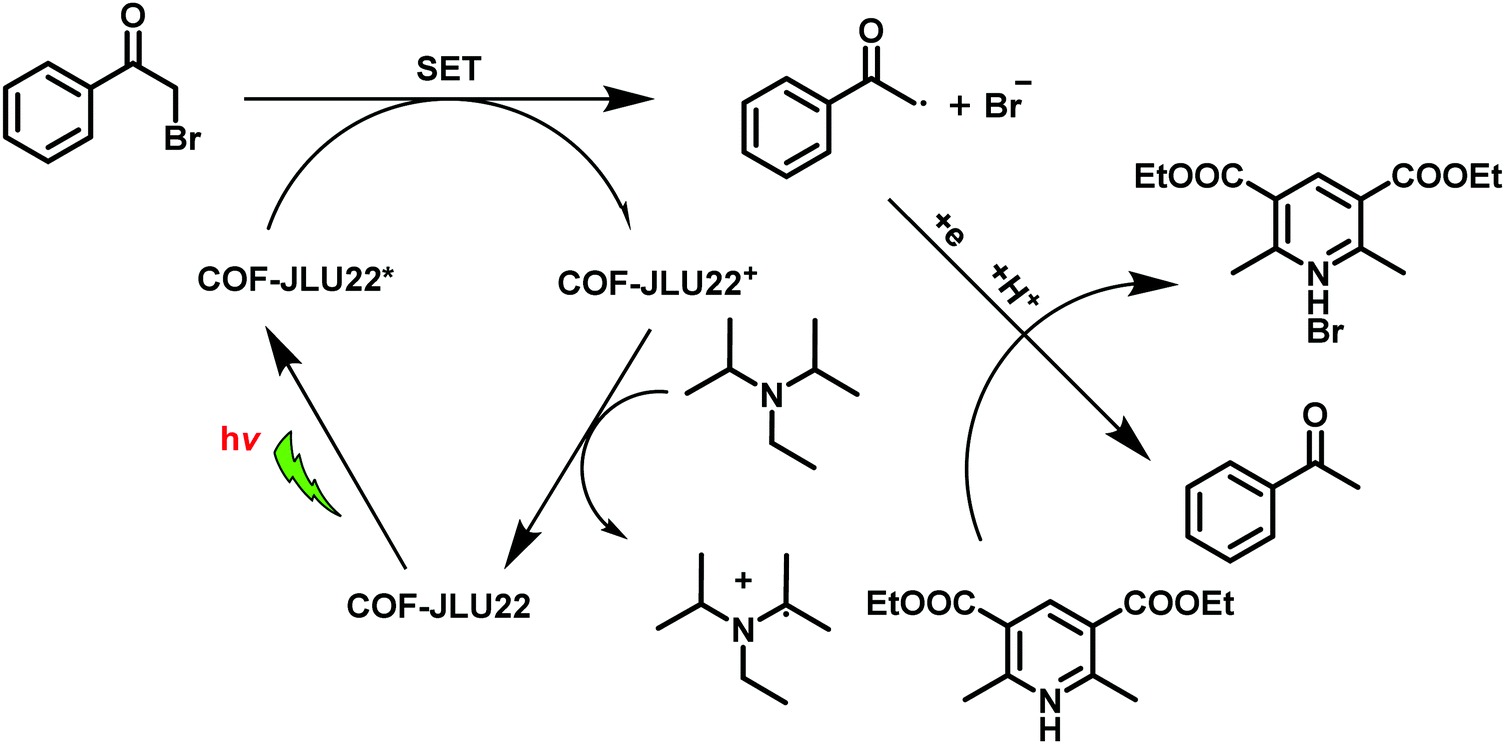
In view of the importance of visible-light active, heterogeneous photocatalysts, Li et al. designed an imine-based 2D COF with an electron donor and acceptor structure (COF-JLU22).283 For this, 1,3,6,8-tetrakis(4-aminophenyl)pyrene was utilized due to superb properties of pyrene-based compounds such as large surface area, high crystallinity, excellent stability and good photoelectric activity (Fig. 61). In addition, an electron-deficient linker 4,7-diphenylbenzo[c][1,2,5]thiadiazole was introduced into the catalyst's framework to form mesoporous channels that would improve the rate of reaction and transmission of matter. Moreover, the fabrication of such electron donor–acceptor COF can lead to red shift of the absorption wavelength and amplify the photocatalytic activity of the resulting COF. The final catalyst was found to possess large pore volume, high crystallinity and robust thermal, chemical and photochemical stability. Moreover, it was found to be highly efficient for photoreductive dehalogenation of phenacyl bromide derivatives and α-alkylation of aldehydes under visible-light irradiation (Fig. 62); the catalyst furnished good recyclability and was reused for five runs. The mechanism for both the reactions has been investigated and the probable mechanism for dehalogenation of phenacyl bromide was proposed in sequential steps: firstly, when visible-light irradiates, the excited state of COF-JLU22 generates photogenerated holes and electrons via charge separation. Secondly, this photogenerated hole takes electrons from sacrificial agent N,N-diisopropylethylamine (DIPEA), while the photogenerated electron transfers from the conduction band of COF-JLU22 (E1/2 = −0.86 V) to phenacyl bromide (E1/2 = −0.49 V) resulting in the C–Br cleavage which produces an α-carbonyl radical and bromide anion. Thirdly, this α-carbonyl radical abstracts a proton and an electron from the Hantzsch ester to furnish acetophenone as the final product (Fig. 63).
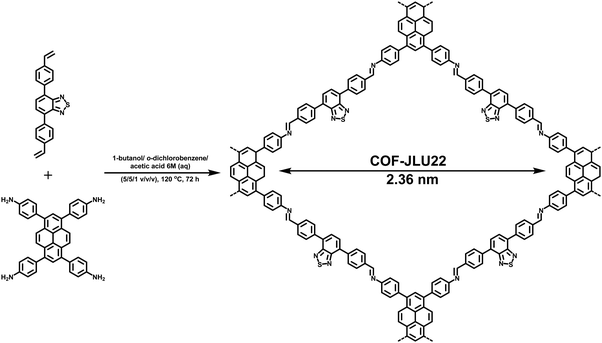 |
| | Fig. 61 Synthesis of COF-JLU22 by imine condensation reaction. | |
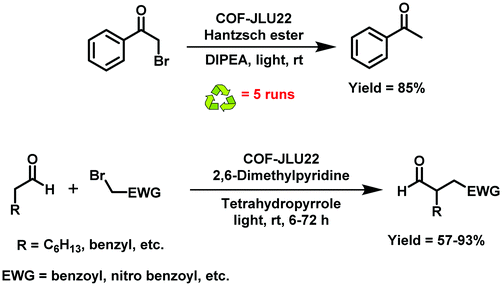 |
| | Fig. 62 Photoreductive dehalogenation reaction of phenacyl bromide and photocatalytic α-alkylation of aldehydes by COF-JLU22, respectively. | |
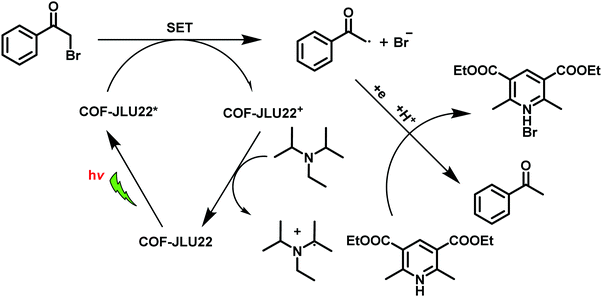 |
| | Fig. 63 Proposed reaction mechanism for the photoreductive dehalogenation reaction with COF-JLU22. | |
Likewise, hydrazone-based COFs have received tremendous attention as yet another example of a metal-free COF photocatalyst due to their possible structural variations, reversibility, and synthetic accessibility;284 synthesis under solvothermal conditions entails the condensation of 2,5-dimethoxyterephthalohydrazide (DMTH) with 1,3,5-triformylbenzene (TFB) (Fig. 64). In comparison to boronate ester linkage, hydrazone linkage provides improved hydrolytic and oxidative stability, in addition to providing powerful character through the reversibility of the imine bond and hydrogen bonding sites of an amide group. The synthesized TFB-COF photocatalyst exhibited excellent porosity, high crystallinity and good thermal and chemical stability. It displayed magnificent photocatalytic activity towards CDC reactions of a wide variety of substrates at room temperature which is attributed to the superb porosity features with high BET surface area as well as the light-harvesting efficacy of the catalyst (Fig. 65). Moreover, this metal-free catalytic system provides advantages like operational simplicity, broad substrate scope, ambient reaction conditions, easy recoverability, and good recyclability.
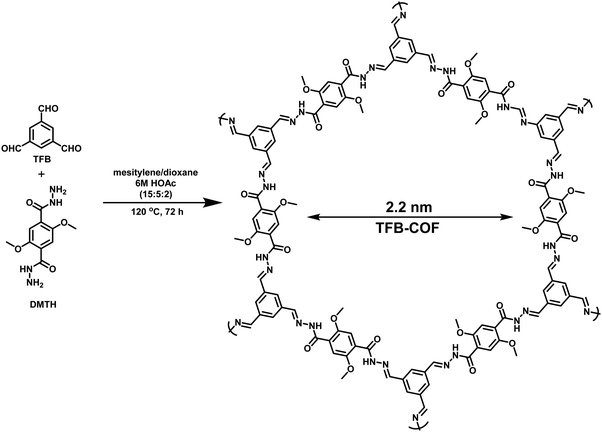 |
| | Fig. 64 Schematic representation for the synthesis of TFB-COF. | |
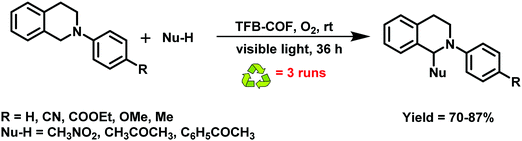 |
| | Fig. 65 Reaction substrate extension of photocatalytic coupling reactions of N-aryltetrahydroisoquinolines using TFB-COF as a catalyst. | |
3.3.4 Environment remediation.
Wastewater treatment has been of concern for a long time as it contains numerous impurities originating from a variety of anthropogenic activities.285,286 Visible-light-responsive photocatalysts offer a sustainable route to alleviate environmental issues by directly harvesting energy from sunlight. Earlier, many groups have outlined various TiO2-based inorganic semiconductors for water treatment.287,288 Recently, an upsurge in the development of organic photocatalysts has been observed for the degradation of organic pollutants in comparison to inorganic photocatalysts. This led to the investigation of assorted organic photocatalytic materials, and among them, COFs garnered tremendous attention due to their periodic architectures which offer a strategy to integrate photocatalytic units into the extended structures in a highly controlled and predictable fashion. Moreover, their optoelectronic properties and band gaps can be tailored effectively by selecting appropriate building units, linking mode and stacking pattern.
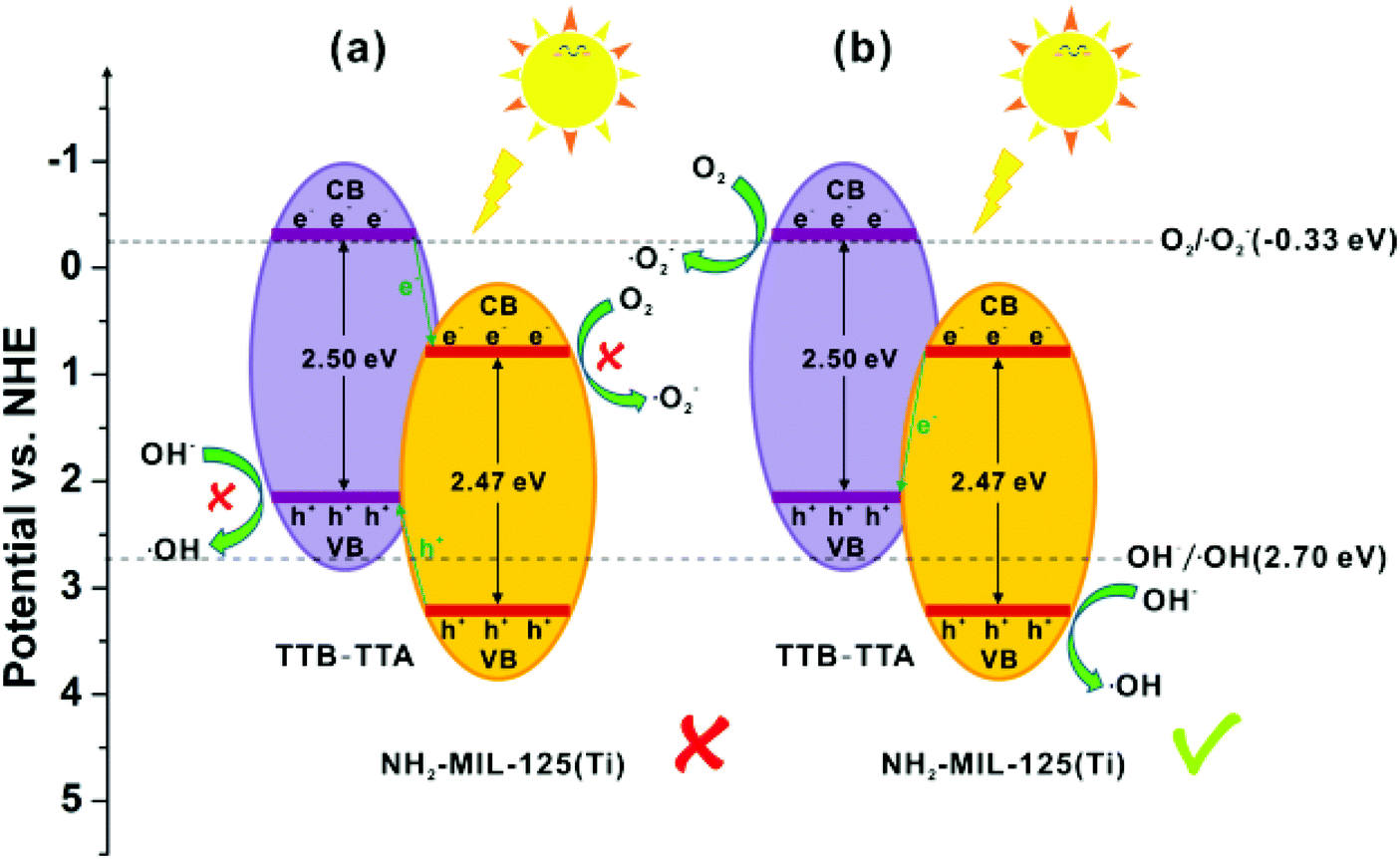
Inspired by the structural diversity of COFs, various photocatalytic materials have been developed. However, identical COF systems have limited building units due to structural periodicity which makes it difficult for them to simultaneously fulfil all the requirements for enhancing photocatalytic activity that include a wide absorption range, large surface area, high charge-separation efficiency, and long-term stability. To circumvent these problems, recently, He et al. developed an effective strategy to synthesize a series of multicomponent heterojunction materials by covalently integrating MOFs with COFs.289 Three common amine-functionalized MOFs, NH2-MIL-53(Al), NH2-MIL-125(Ti) and NH2-UiO-66(Zr), were used as dopants, with highly photocatalytically active TTB-TTA (a COF synthesized from 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (TTB) and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA)) to form heterogeneous photocatalysts (Fig. 66); they were investigated and compared for their photoelectric and photocatalytic performance. Amongst them, the band gaps matched well for NH2-MIL-125(Ti) and TTB-TTA, which resulted in largely accessible surface area, porous network, high crystallinity and facilitated effective charge separation across the covalent heterojunction interface. Consequently, this MOF-COF hybrid system exhibited superior photocatalytic activity for the degradation of organic pollutants under visible light irradiation in comparison to pure NH2-MIL-125(Ti) and TTB-TTA materials (Fig. 67). On account of detected active oxidant species, a direct Z-scheme mechanism was generated between NH2-MIL-125(Ti) and TTB-TTA, where photoinduced electrons in NH2-MIL-125(Ti) combined with holes in TTB-TTA, thus enhancing the charge carrier transfer. Furthermore, this photocatalyst was found to be stable due to COF encapsulation and covalent linkages.
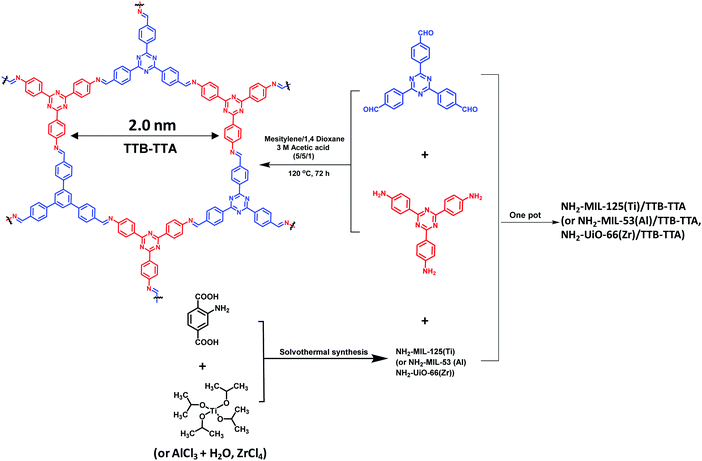 |
| | Fig. 66 Schematic illustration of the synthesis of three MOF/COF hybrid materials. | |
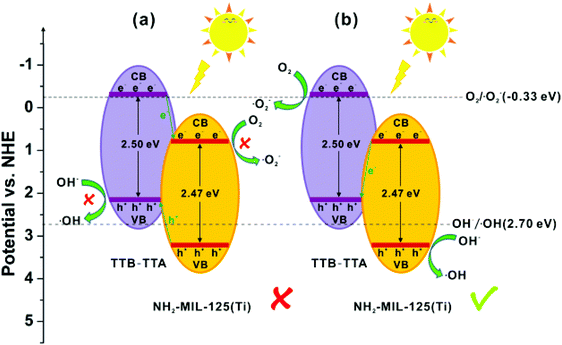 |
| | Fig. 67 (a) Type-II heterojunction and (b) Z-scheme charge-transfer mechanisms for the NH2-MIL-125(Ti)/TTB-TTA system. Reproduced with permission from ref. 289 Copyright 2019 Elsevier B.V. | |
The same group investigated the structural variation and interpretation of chemical units in the precursors of a series of imine-based COFs to their photoelectric properties, and finally with the subsequent effect on photocatalytic degradation of dyes.290 For this, three different COFs bearing visible-light catalytically active substituents of a triazine ring were fabricated from the same aldehyde but different monomers of nitrogen-containing functional groups (Fig. 68). By precisely adjusting the building blocks of COFs, the relationship between structure, physicochemical properties and photocatalytic performance of a synthesized catalyst was analyzed. It was found that photocatalytic performance was enhanced with increasing density of active centers and degree of conjugation in the frameworks. This suggested that they are directly correlated with the charge generation and recombination kinetics, which greatly influences the photocatalytic capacity. Amongst these, the COFA+C had a highly delocalized π-system and available active sites, and hence, it exhibited magnificent photocatalytic activity in degrading organic pollutants under visible light irradiation. In fact, COFA+C was found to possess the highest degradation efficiency when compared to its analogues COFA+B and COFA+D, as it was able to completely degrade methyl orange (MO) after 30 minutes of visible light irradiation, while COFA+B only removed 29.6% MO, and almost no change was observed with COFA+D.
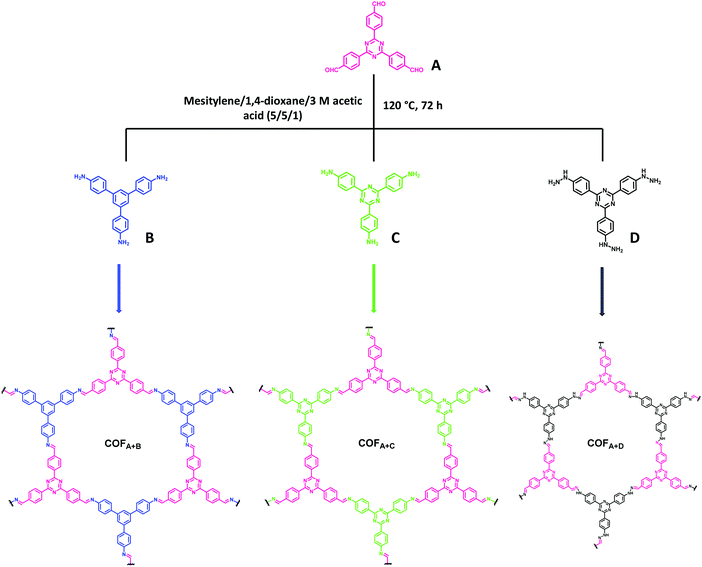 |
| | Fig. 68 Synthesis of COF via Schiff base chemistry of different nitrogen-containing building blocks with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde. | |
3.3.5 Singlet oxygen generation.
Photosensitized generation of singlet oxygen has gained special attention all over the world for its utility in various applications such as wastewater treatment,291 fine chemical synthesis,292,293 photodynamic applications such as blood sterilization,294,295 sunlight-activated herbicides and insecticides, photo-oxidation, DNA damage, photodynamic therapy (PDT) of cancer and polymer science.296 Recently, researchers have focused their attention on synthesizing a light-activated COF which can be further utilized for the generation of single oxygen. Lately, Jiang et al. have synthesized a zig-zag, flat and twisted squaraines-linked 2D COF having high stability, extended π-conjugation and lower band gap energy with enhanced absorbance.297 Here, the synthesis of CuP-SQ COF was carried out under solvothermal conditions between squaric acid (SA) and copper(II) 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAP-CuP) (Fig. 69).
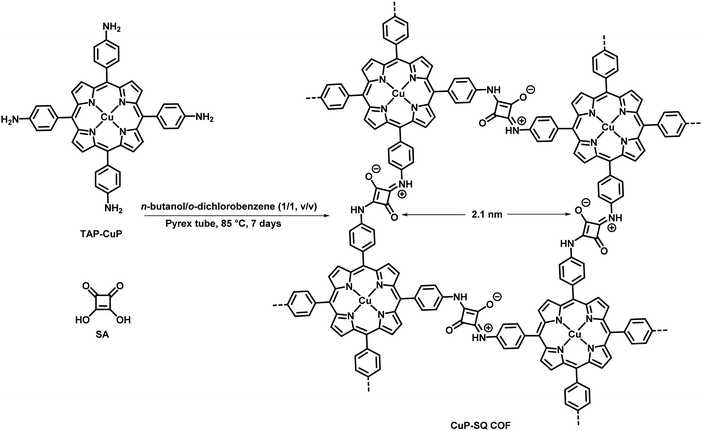 |
| | Fig. 69 Fabrication of CuP-SQ COF through the condensation of SA with TAP-CuP. | |
The high photocatalytic activity of CuP-SQ COF was exploited for the production of singlet oxygen via the activation of molecular oxygen in the presence of 1,3-diphenylisobenzofuran (DPBF) as a label. It was believed that the well-defined ordered structure of CuP-SQ COF undergoes the triplet state upon photoirradiation and is responsible for the effective generation of singlet oxygen.
Similarly, Wang et al. have obtained singlet oxygen from novel 3D porphyrin-based COFs (3D-Por-COF and 3D-CuPor-COF) incorporated with photoelectric units.298 The high surface area, microporous, crystalline and heterogeneous framework was fabricated by the [4+4] imine condensation reaction (Fig. 70). The pore size distribution curves of 3D-Por-COF reveal a major peak at 0.60 and 1.07 nm, while in the case of 3D-CuPor-COF peaks were present at 0.63 and 1.18 nm. The photoexcited triplet state of porphyrin-derived COFs was availed to activate the molecular oxygen by energy transfer to generate the singlet oxygen using 9,10-dimethylanthracene (DMA) as a label (Fig. 71). The best results were obtained with 3D-Por-COF in comparison to 3D-CuPor-COF which showed high photocatalytic degradation of DMA (99%) in oxygen-saturated CH3CN solution within the time period of 90 min upon irradiation with 500 nm. Moreover, this heterogeneous framework was found to be highly efficient and retained its structural and catalytic properties even after three cycles. Furthermore, the photophysical and photoredox property can be easily tuned and modified by the insertion of any particular metal in the porphyrin moiety.
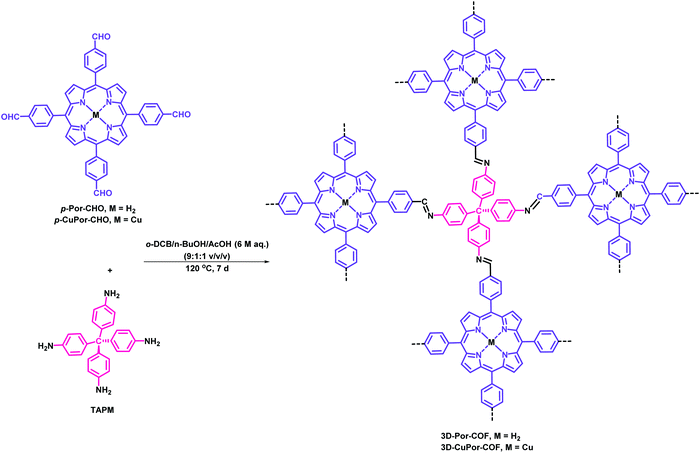 |
| | Fig. 70 Fabrication of 3D-Por-COF and 3D-CuPor-COF. | |
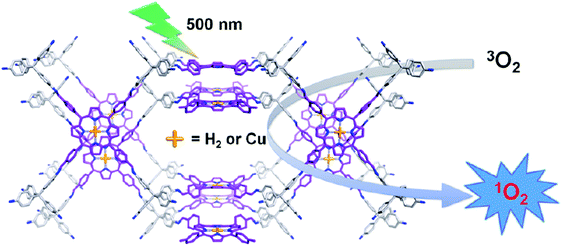 |
| | Fig. 71 Schematic diagram of 3D-Por-COF and 3D-CuPor-COF generating singlet oxygen. Reproduced with permission from ref. 298 Copyright 2017 American Chemical Society. | |
Jiang et al. have reported the synthesis of a series of 2D COFs having interlayer H-bonding interaction to lock the sheets into planar conformation via repressing the torsional strain of edge units thus leading to the extension of π-conjugation.299 The highly photocatalytically active, stable, crystalline and porous COFs for the generation of singlet oxygen were fabricated by employing different molar ratios of a mixture of dihydroxyterephthalaldehyde (DHTA) and terephthaladehyde (TA) as a monomer to react with 5,10,15,20-tetrakis(4′-tetraphenylamino)porphyrin derivatives (MP; M = Cu, H2, and Ni) (Fig. 72). Notably, H-bonding plays a significant role in the photocatalytic singlet oxygen generation by using 1,3-diphenylisobenzofuran (DPBF) as a label. Isosbestic points reveal the successful conversion of molecular oxygen into singlet oxygen upon irradiation of light over the oxygen-saturated DMF solution of DPBF using CuP-DHPh COF.
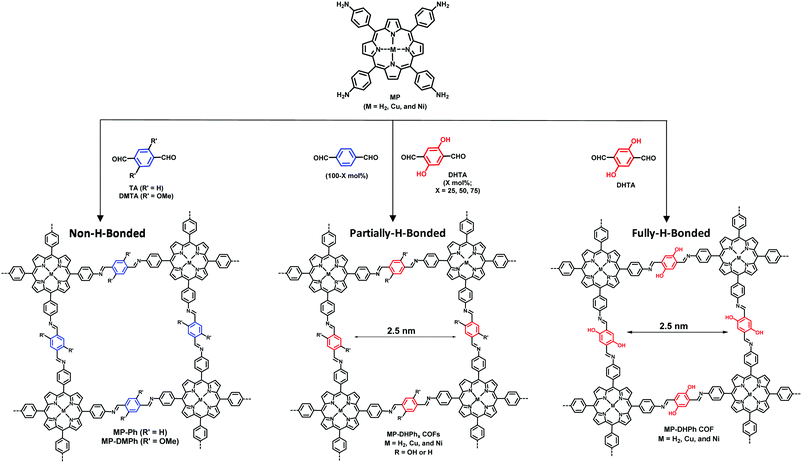 |
| | Fig. 72 Schematic of the fabrication of 2D porphyrin COFs and tunable content of hydrogen-bonding structures. | |
3.4 Miscellaneous applications
We have overviewed so far, the catalytic, electrocatalytic and photocatalytic applications of COFs in organic and energy conversion reactions. However, there is another potentially wider domain for the applications of COFs. Recently, several researchers have begun exploring the ability of COFs as chemosensory materials due to their exceptional structural tunability and properties.63,64,131,300–302 There is a growing need for sensitive and selective detection of inorganic and organic substances for a range of applications such as environmental monitoring, medical diagnostics, food quality control, chemical threat detection, and laboratory/industrial productions. Consequently, a series of COF-based chemical sensors have been fabricated for the selective and efficient detection of toxic metal ions, nitro explosives and even moisture. Besides, their porosity, high thermal stability and lightweight nature have captured attention for various gas adsorption and storage applications such as CO2, H2, CH4, and NH3.30,51,303–308 When functionalized with photoelectric moieties, COFs exhibit unique optical and electrical properties, which prompts their use in semiconduction and photoconduction applications.309,310 Furthermore, they can also serve as potential candidates for conductive membranes and as drug delivery agents.76,311–313
4 Conclusions and future scope
In summary, COFs have emerged as porous and partly crystalline organic materials that allow sufficiently precise atomic integration of organic molecules into ordered structures. They possess unique characteristics such as tunable molecular design, high porosity with controllable pore size, adjustable electronic/physical properties, and excellent stability. These properties have provided COF materials with superior potential in the field of gas and energy storage, adsorption, and catalysis applications. In this context, a variety of synthetic methods have been developed so far including solvothermal, ionothermal, mechanochemical, sonochemical, microwave and light-promoted synthesis.
In terms of catalysis, many researchers have exploited the porous nature of COFs in order to support metal species and other catalytic entities, which enhances the efficiency of their use as heterogeneous catalysts. COFs allow large availability of functional groups onto their surface, which can strongly coordinate with metals. This diminishes the possibility of metal leaching and subsequent contamination of the product stream. Moreover, this type of catalyst can be easily recovered from the reaction media via simple centrifugation or filtration, thereby allowing recycling without a significant loss of catalytic activity. Offering these advantages, COFs have been widely used in a variety of organic transformations including C–C coupling, oxidation, reduction, asymmetric synthesis, and many others. Even, tandem reactions are performed by developing bimetallic docked COFs. Besides being used as metal supports, metal-free COFs have been designed as heterogeneous organocatalysts to carry out efficient CO2 fixation, cycloaddition, condensation, and even photocatalytic and electrocatalytic reactions because of their collective π-array structure. We believe that a properly designed combination of COFs and photoactive materials such as carbon nitrides will certainly open the door for advanced electrocatalytic and photocatalytic applications.
The progress made over the years has improved long-term prospects of this new type of porous material. However, continuous effort aimed at improving design, and large-scale synthesis under standard conditions, is still required. One aspect, which is of utmost importance, is the relatively poor moisture tolerance of COFs, therefore, newer methods with moisture tolerant linkers and reactions need to be developed to impart stability under ambient conditions. Additionally, the generation of multifunctional COFs with designing of both pores and skeletons, must be addressed. Besides, more structural details such as slipped distance and determination of defects in the framework of the COF family are yet to be fully explored. More investigations into the kinetic and thermodynamic parameters are also required to enable preparation of high-quality COF materials. Moreover, earnest efforts are required in the field of computational modelling to study the specific interactions between COF materials and guest species. Although, some promising results have already been accomplished, the progress of COFs is still in its infant stages. It is believed that in the not too distant future, further developments in COFs will make significant advancements towards authentic applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Priya Yadav, Radhika Gupta and Pooja Rana gratefully acknowledge UGC and CSIR, Delhi, India for awarding research fellowships. The authors gratefully acknowledge the support by the Operational Program Research, Development and Education – European Regional Development Fund, project no. CZ.02.1.01/0.0/0.0/16_019/0000754, CZ.02.1.01/0.0/0.0/15_003/0000416 and LO1305 of the Ministry of Education, Youth and Sports of the Czech Republic.
References
- M. C. Feiters, A. E. Rowan and R. J. Nolte, Chem. Soc. Rev., 2000, 29, 375–384 RSC.
- R. Trask, H. Williams and I. Bond, Bioinspiration Biomimetics, 2007, 2, P1–P9 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS.
- J. M. Zayed, N. Nouvel, U. Rauwald and O. A. Scherman, Chem. Soc. Rev., 2010, 39, 2806–2816 RSC.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- P. J. Stang, Chem. – Eur. J., 1998, 4, 19–27 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS PubMed.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2013, 26, 310–322 CrossRef.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- A. I. Cooper, CrystEngComm, 2013, 15, 1483 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef CAS.
- N. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- A. Nimibofa, E. A. Newton, E. A. Tobin and W. Donbebe, Am. J. Mater. Sci., 2017, 7, 185–195 Search PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS.
- M. Mastalerz, Angew. Chem., Int. Ed., 2008, 47, 445–447 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS.
- G. Q. Lin, H. M. Ding, D. Q. Yuan, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- C. Wang, Y. Wang, R. L. Ge, X. D. Song, X. Q. Xing, Q. K. Jiang, H. Lu, C. Hao, X. W. Guo, Y. A. Gao and D. L. Jiang, Chem. – Eur. J., 2018, 24, 585–589 CrossRef CAS PubMed.
- K. K. Wang, H. L. Huang, D. H. Liu, C. Wang, J. P. Li and C. L. Zhong, Environ. Sci. Technol., 2016, 50, 4869–4876 CrossRef CAS.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- W. Ji, L. L. Xiao, Y. H. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS.
- A. Yassin, M. Trunk, F. Czerny, P. Fayon, A. Trewin, J. Schmidt and A. Thomas, Adv. Funct. Mater., 2017, 27, 1700233 CrossRef.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Chem. Sci., 2019, 10, 3796–3801 RSC.
- Z. Miao, G. Liu, Y. Cui, Z. Liu, J. Li, F. Han, Y. Liu, X. Sun, X. Gong, Y. Zhai, Y. Zhao and Y. Zeng, Angew. Chem., Int. Ed., 2019, 58, 4906–4910 CrossRef CAS.
- X. W. Wu, X. Han, Y. H. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS.
- X. L. Li, C. L. Zhang, S. L. Cai, X. H. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef.
- J. N. Guo, Y. Li, Y. H. Cheng, L. M. Dai and Z. H. Xiang, ACS Nano, 2017, 11, 8379–8386 CrossRef CAS.
- X. Han, Q. C. Xia, J. J. Huang, Y. Liu, C. X. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS.
- W. Cao, W. D. Wang, H.-S. Xu, I. V. Sergeyev, J. Struppe, X. Wang, F. Mentink-Vigier, Z. Gan, M.-X. Xiao, L.-Y. Wang, G.-P. Chen, S.-Y. Ding, S. Bai and W. Wang, J. Am. Chem. Soc., 2018, 140, 6969–6977 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. c. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS.
- M. G. Rabbani, A. K. Sekizkardes, Z. Kahveci, T. E. Reich, R. Ding and H. M. El-Kaderi, Chem. – Eur. J., 2013, 19, 3324–3328 CrossRef CAS.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. GoddardIii, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS.
- Z. Li, X. Feng, Y. Zou, Y. Zhang, H. Xia, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825–13828 RSC.
- J.-T. Yu, Z. Chen, J. Sun, Z.-T. Huang and Q.-Y. Zheng, J. Mater. Chem., 2012, 22, 5369–5373 RSC.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS.
- R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC.
- Y. Meng, G. Q. Lin, H. M. Ding, H. P. Liao and C. Wang, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., 2008, 120, 8958–8962 CrossRef.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613–6616 RSC.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159–7171 RSC.
- W. Zhang, L.-G. Qiu, Y.-P. Yuan, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2012, 221, 147–154 CrossRef.
- N. Huang, L. Zhai, H. Xu and D. Jiang, J. Am. Chem. Soc., 2017, 139, 2428–2434 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS.
- D. Ma, Y. Wang, A. Liu, S. Li, C. Lu and C. Chen, Catalysts, 2018, 8, 404 CrossRef.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., 2014, 126, 2922–2926 CrossRef.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., 2013, 125, 2992–2996 CrossRef.
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS.
- L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC.
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef CAS.
- H. Zhao, Z. Jin, H. Su, X. Jing, F. Sun and G. Zhu, Chem. Commun., 2011, 47, 6389–6391 RSC.
- M.-X. Wu and Y.-W. Yang, Chin. Chem. Lett., 2017, 28, 1135–1143 CrossRef CAS.
- C. T. Stoppiello, H. Isla, M. Martínez-Abadía, M. W. Fay, C. D. J. Parmenter, M. J. Roe, B. Lerma-Berlanga, C. Martí-Gastaldo, A. Mateo-Alonso and A. N. Khlobystov, Nanoscale, 2019, 11, 2848–2854 RSC.
- X. H. Guo, Y. Tian, M. C. Zhang, Y. Li, R. Wen, X. Li, X. F. Li, Y. Xue, L. J. Ma, C. Q. Xia and S. J. Li, Chem. Mater., 2018, 30, 2299–2308 CrossRef CAS.
- D. Jiang, X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao and Q. Jiang, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201904291.
- X. Ma and T. F. Scott, Commun. Chem., 2018, 1, 98 CrossRef.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS.
- J. Liu, W. Zan, K. Li, Y. Yang, F. Bu and Y. Xu, J. Am. Chem. Soc., 2017, 139, 11666–11669 CrossRef CAS.
- F. Xu, S. Yang, G. Jiang, Q. Ye, B. Wei and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 37731–37738 CrossRef CAS.
- S. N. Talapaneni, T. H. Hwang, S. H. Je, O. Buyukcakir, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2016, 55, 3106–3111 CrossRef CAS.
- G. Zhu, S. Shi, M. Liu, L. Zhao, M. Wang, X. Zheng, J. Gao and J. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 12612–12617 CrossRef CAS.
- H. S. Jena, C. Krishnaraj, G. Wang, K. Leus, J. Schmidt, N. Chaoui and P. Van Der Voort, Chem. Mater., 2018, 30, 4102–4111 CrossRef CAS.
- W. B. Chen, Z. F. Yang, Z. Xie, Y. S. Li, X. Yu, F. L. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998–1004 RSC.
- C. Y. Lin, L. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2017, 29, 1606635 CrossRef PubMed.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 RSC.
- F. Zhao, H. Liu, S. Mathe, A. Dong and J. Zhang, Nanomaterials, 2017, 8, 15 CrossRef.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS.
- V. Nguyen and M. Grünwald, J. Am. Chem. Soc., 2018, 140, 3306–3311 CrossRef CAS.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS PubMed.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS.
- Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS.
- V. Polshettiwar, M. N. Nadagouda and R. S. Varma, Aust. J. Chem., 2009, 62, 16–26 CrossRef CAS.
- D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1–47 RSC.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- H. Wei, S. Chai, N. Hu, Z. Yang, L. Wei and L. Wang, Chem. Commun., 2015, 51, 12178–12181 RSC.
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS.
- P. Cintas and J.-L. Luche, Green Chem., 1999, 1, 115–125 RSC.
- S.-T. Yang, J. Kim, H.-Y. Cho, S. Kim and W.-S. Ahn, RSC Adv., 2012, 2, 10179–10181 RSC.
- T. Friščić, J. Mater. Chem., 2010, 20, 7599–7605 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett and W. Jones, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS.
- S. Kim and H. C. Choi, Commun. Chem., 2019, 2, 60 CrossRef.
- M. B. Gawande, R. Luque and R. Zboril, ChemCatChem, 2014, 6, 3312–3313 CrossRef CAS.
- M. B. Gawande, Y. Monga, R. Zboril and R. K. Sharma, Coord. Chem. Rev., 2015, 288, 118–143 CrossRef CAS.
- R. K. Sharma, S. Dutta, S. Sharma, R. Zboril, R. S. Varma and M. B. Gawande, Green Chem., 2016, 18, 3184–3209 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., 2008, 120, 2319–2321 CrossRef.
- R. Gupta, M. Yadav, R. Gaur, G. Arora and R. K. Sharma, Green Chem., 2017, 19, 3801–3812 RSC.
- G. Arora, M. Yadav, R. Gaur, R. Gupta and R. K. Sharma, ChemistrySelect, 2017, 2, 10871–10879 CrossRef CAS.
- R. Gaur, M. Yadav, R. Gupta, G. Arora, P. Rana and R. K. Sharma, ChemistrySelect, 2018, 3, 2502–2508 CrossRef CAS.
- R. K. Sharma, R. Gaur, M. Yadav, A. K. Rathi, J. Pechousek, M. Petr, R. Zboril and M. B. Gawande, ChemCatChem, 2015, 7, 3495–3502 CrossRef CAS.
-
R. K. Sharma, M. Yadav and M. B. Gawande, Ferrites and Ferrates: Chemistry and Applications in Sustainable Energy and Environmental Remediation, ACS Publications, 2016, pp. 1–38 Search PubMed.
- R. K. Sharma, M. Yadav, R. Gaur, Y. Monga and A. Adholeya, Catal. Sci. Technol., 2015, 5, 2728–2740 RSC.
- R. K. Sharma, M. Yadav, Y. Monga, R. Gaur, A. Adholeya, R. Zboril, R. S. Varma and M. B. Gawande, ACS Sustainable Chem. Eng., 2016, 4, 1123–1130 CrossRef CAS.
- S. He, T. Zeng, S. Wang, H. Niu and Y. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 2959–2965 CrossRef CAS.
- Y. Liao, J. Li and A. Thomas, ACS Macro Lett., 2017, 6, 1444–1450 CrossRef CAS.
- Y. Li, C.-X. Yang and X.-P. Yan, Chem. Commun., 2017, 53, 2511–2514 RSC.
- R. Wang and Z. Chen, Microchim. Acta, 2017, 184, 3867–3874 CrossRef CAS.
- G. Lin, C. Gao, Q. Zheng, Z. Lei, H. Geng, Z. Lin, H. Yang and Z. Cai, Chem. Commun., 2017, 53, 3649–3652 RSC.
- H. Xu, X. Chen, J. Gao, J. Lin, M. Addicoat, S. Irle and D. Jiang, Chem. Commun., 2014, 50, 1292–1294 RSC.
- F. X. L. i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- Y. Huang, Z. Lin and R. Cao, Chem. – Eur. J., 2011, 17, 12706–12712 CrossRef CAS.
- V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS.
- Y. Wan, H. Wang, Q. Zhao, M. Klingstedt, O. Terasaki and D. Zhao, J. Am. Chem. Soc., 2009, 131, 4541–4550 CrossRef CAS.
- R. K. Sharma, M. Yadav, R. Gaur, R. Gupta, A. Adholeya and M. B. Gawande, ChemPlusChem, 2016, 81, 1312–1319 CrossRef CAS.
- P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435–4437 RSC.
- Z. Yinghuai, S. C. Peng, A. Emi, S. Zhenshun and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917–1922 CrossRef.
- R. S. Gonçalves, A. B. de Oliveira, H. C. Sindra, B. S. Archanjo, M. E. Mendoza, L. S. Carneiro, C. D. Buarque and P. M. Esteves, ChemCatChem, 2016, 8, 743–750 CrossRef.
- D. Mullangi, S. Nandi, S. Shalini, S. Sreedhala, C.
P. Vinod and R. Vaidhyanathan, Sci. Rep., 2015, 5, 10876 CrossRef.
- T. Torimoto, K.-i. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka and S. Kuwabata, Appl. Phys. Lett., 2006, 89, 243117 CrossRef.
- T. Hayashi, T. Ohno, S. Yatsuya and R. Uyeda, Jpn. J. Appl. Phys., 1977, 16, 705–717 CrossRef CAS.
- X. Feng, J. D. Sloppy, T. J. LaTempa, M. Paulose, S. Komarneni, N. Bao and C. A. Grimes, J. Mater. Chem., 2011, 21, 13429–13433 RSC.
- K. Y. Cho, Y. S. Yeom, H. Y. Seo, P. Kumar, A. S. Lee, K.-Y. Baek and H. G. Yoon, J. Mater. Chem. A, 2015, 3, 20471–20476 RSC.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed.
- C. E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537–541 CrossRef CAS.
- M. Mu, Y. Wang, Y. Qin, X. Yan, Y. Li and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 22856–22863 CrossRef CAS.
- E. Rozhko, A. Bavykina, D. Osadchii, M. Makkee and J. Gascon, J. Catal., 2017, 345, 270–280 CrossRef CAS.
- X. Shi, Y. Yao, Y. Xu, K. Liu, G. Zhu, L. Chi and G. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 7481–7488 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2006, 10, 2143–2173 CrossRef.
- C. Bolm, T. Rantanen, I. Schiffers and L. Zani, Angew. Chem., Int. Ed., 2005, 44, 1758–1763 CrossRef CAS.
- H.-C. Ma, J.-L. Kan, G.-J. Chen, C.-X. Chen and Y.-B. Dong, Chem. Mater., 2017, 29, 6518–6524 CrossRef CAS.
- K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551–564 RSC.
- R. A. Bunce, Tetrahedron, 1995, 51, 13103–13159 CrossRef CAS.
- J. Zhou, Chem. – Asian J., 2010, 5, 422–434 CrossRef CAS PubMed.
- F. X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718–724 CrossRef CAS PubMed.
- Y.-Z. Chen, Y.-X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS.
- W. Leng, R. Ge, B. Dong, C. Wang and Y. Gao, RSC Adv., 2016, 6, 37403–37406 RSC.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS.
- R. S. Tol, Energy Policy, 2005, 33, 2064–2074 CrossRef.
- S. M. Benson and F. M. Orr, MRS Bull., 2008, 33, 303–305 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef.
- S. Wang, S. Yan, X. Ma and J. Gong, Energy Environ. Sci., 2011, 4, 3805–3819 RSC.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuehn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS.
- K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2011, 2, 180–183 CrossRef.
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS.
- R. K. Sharma, R. Gaur, M. Yadav, A. Goswami, R. Zbořil and M. B. Gawande, Sci. Rep., 2018, 8, 1901 CrossRef.
- Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS.
- Y. Zhi, P. Shao, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2018, 6, 374–382 RSC.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- R. R. Schmidt, Acc. Chem. Res., 1986, 19, 250–259 CrossRef CAS.
- Y. Wu, H. Xu, X. Chen, J. Gao and D. Jiang, Chem. Commun., 2015, 51, 10096–10098 RSC.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- L. Li, L. Li, C. Cui, H. Fan and R. Wang, ChemSusChem, 2017, 10, 4921–4926 CrossRef CAS.
- U. P. Tran, K. K. Le and N. T. Phan, ACS Catal., 2011, 1, 120–127 CrossRef CAS.
- Y. Yang, H.-F. Yao, F.-G. Xi and E.-Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS.
- M. Opanasenko, A. Dhakshinamoorthy, M. Shamzhy, P. Nachtigall, M. Horáček, H. Garcia and J. Čejka, Catal. Sci. Technol., 2013, 3, 500–507 RSC.
- T. I. Reddy and R. S. Varma, Tetrahedron Lett., 1997, 38, 1721–1724 CrossRef CAS.
- E. Angeletti, C. Canepa, G. Martinetti and P. Venturello, J. Chem. Soc., Perkin Trans. 1, 1989, 105–107 RSC.
-
Nat. Rev. Chem., 2018, 2, 0125.
- I. S. Pieta, A. Rathi, P. Pieta, R. Nowakowski, M. Hołdynski, M. Pisarek, A. Kaminska, M. B. Gawande and R. Zboril, Appl. Catal., B, 2019, 244, 272–283 CrossRef CAS.
- Y. H. Hou, Y. P. Liu, R. Q. Gao, Q. J. Li, H. Z. Guo, A. Goswami, R. Zboril, M. B. Gawande and X. X. Zou, ACS Catal., 2017, 7, 7038–7042 CrossRef CAS.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. X. Huang, R. Silva, X. X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS.
- C. F. Zinola, M. E. Martins, E. P. Tejera and N. P. Neves Jr., Int. J. Electrochem., 2012, 874687 CAS.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS.
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- C. Y. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef.
-
B. C. Steele and A. Heinzel, Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 224–231 Search PubMed.
- H. Chang, S. H. Joo and C. Pak, J. Mater. Chem., 2007, 17, 3078–3088 RSC.
- S. Dai, J.-P. Chou, K.-W. Wang, Y.-Y. Hsu, A. Hu, X. Pan and T.-Y. Chen, Nat. Commun., 2019, 10, 440 CrossRef.
- M. Chisaka and H. Morioka, Catal. Sci. Technol., 2019, 9, 611–619 RSC.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., 2005, 117, 2170–2173 CrossRef.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- X. Guo, C. Qian, R. Shi, W. Zhang, F. Xu, S. Qian, J. Zhang, H. Yang, A. Yuan and T. Fan, Small, 2019, 1804855 CrossRef.
- A. Marianov and Y. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 3838–3848 CrossRef CAS.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- G. A. Olah, A. Goeppert and G. S. Prakash, J. Org. Chem., 2008, 74, 487–498 CrossRef PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288–10293 CrossRef CAS PubMed.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- Y. Wang, J. Chen, G. Wang, Y. Li and Z. Wen, Angew. Chem., 2018, 130, 13304–13308 CrossRef.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- H. Li, Q. Pan, Y. Ma, X. Guan, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 14783–14788 CrossRef CAS PubMed.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519–15522 CrossRef CAS PubMed.
- W. Xia, C. Qu, Z. Liang, B. Zhao, S. Dai, B. Qiu, Y. Jiao, Q. Zhang, X. Huang, W. Guo, D. Dang, R. Zou, D. Xia, Q. Xu and M. Liu, Nano Lett., 2017, 17, 2788–2795 CrossRef CAS PubMed.
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249–267 CrossRef CAS PubMed.
- Y. Zhao, Chem. Mater., 2016, 28, 8079–8081 CrossRef CAS.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- F.-M. Zhang, J.-L. Sheng, Z.-D. Yang, X.-J. Sun, H.-L. Tang, M. Lu, H. Dong, F.-C. Shen, J. Liu and Y.-Q. Lan, Angew. Chem., 2018, 130, 12282–12286 CrossRef.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- P. Pachfule, A. Acharjya, J. r. m. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- M. E. El-Khouly, E. El-Mohsnawy and S. Fukuzumi, J. Photochem. Photobiol., C, 2017, 31, 36–83 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Grätzel, Nature, 1987, 327, 506–508 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- S. Wang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308–2320 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- K. S. Lakhi, D.-H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J.-H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- R. K. Yadav, A. Kumar, N.-J. Park, K.-J. Kong and J.-O. Baeg, J. Mater. Chem. A, 2016, 4, 9413–9418 RSC.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- Y. Fang, Y. Ma, M. Zheng, P. Yang, A. M. Asiri and X. Wang, Coord. Chem. Rev., 2018, 373, 83–115 CrossRef CAS.
- Y. Su, L. Zhang and N. Jiao, Org. Lett., 2011, 13, 2168–2171 CrossRef CAS.
- C. C. Nguyen, N. N. Vu and T.-O. Do, J. Mater. Chem. A, 2015, 3, 18345–18359 RSC.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- P. Rana, R. Gaur, R. Gupta, G. Arora, J. Anireddy and R. K. Sharma, Chem. Commun., 2019, 55, 7402–7405 RSC.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682–2685 RSC.
- Y. Zhi, Z. Li, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2017, 5, 22933–22938 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- W. Huang, J. Byun, I. Rörich, C. Ramanan, P. W. M. Blom, H. Lu, D. Wang, L. Caire da Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8316–8320 CrossRef CAS.
- D. Sun, S. Jang, S. J. Yim, L. Ye and D. P. Kim, Adv. Funct. Mater., 2018, 28, 1707110 CrossRef.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Nature, 1999, 398, 227–230 CrossRef CAS.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334–342 CrossRef CAS.
- W. Liu, Q. Su, P. Ju, B. Guo, H. Zhou, G. Li and Q. Wu, ChemSusChem, 2017, 10, 664–669 CrossRef CAS PubMed.
-
M. Yadav, R. Gupta and R. K. Sharma, Advances in Water Purification Techniques, Elsevier, 2019, pp. 355–383 Search PubMed.
-
R. K. Sharma, M. Yadav and R. Gupta, Chemistry and Water, Elsevier, 2017, pp. 183–205 Search PubMed.
- D. Friedmann, C. Mendive and D. Bahnemann, Appl. Catal., B, 2010, 99, 398–406 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS.
- S. He, Q. Rong, H. Niu and Y. Cai, Appl. Catal., B, 2019, 247, 49–56 CrossRef CAS.
- S. He, B. Yin, H. Niu and Y. Cai, Appl. Catal., B, 2018, 239, 147–153 CrossRef CAS.
- M. E. Jiménez-Hernández, F. Manjón, D. García-Fresnadillo and G. Orellana, Sol. Energy, 2006, 80, 1382–1387 CrossRef.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- R. Ciriminna, R. Delisi, Y.-J. Xu and M. Pagliaro, Org. Process Res. Dev., 2016, 20, 403–408 CrossRef CAS.
- W. M. Sharman, C. M. Allen and J. E. Van Lier, Drug Discovery Today, 1999, 4, 507–517 CrossRef CAS PubMed.
- N. L. Oleinick, A. R. Antunez, M. E. Clay, B. D. Rihter and M. E. Kenney, Photochem. Photobiol., 1993, 57, 242–247 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS PubMed.
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS PubMed.
- M. Rong, L. Lin, X. Song, T. Zhao, Y. Zhong, J. Yan, Y. Wang and X. Chen, Anal. Chem., 2014, 87, 1288–1296 CrossRef.
- C. Zhang, S. Zhang, Y. Yan, F. Xia, A. Huang and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- M. Margreth, R. Schlink and A. Steinbach, Pharm. Sci. Encycl. Drug Discov. Dev. Manuf., 2010, 1–34 Search PubMed.
- S. B. Kalidindi, K. Yusenko and R. A. Fischer, Chem. Commun., 2011, 47, 8506–8508 RSC.
- J. Lan, D. Cao and W. Wang, Langmuir, 2009, 26, 220–226 CrossRef PubMed.
- R. W. Tilford, S. J. Mugavero III, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS.
- E. Klontzas, E. Tylianakis and G. E. Froudakis, Nano Lett., 2010, 10, 452–454 CrossRef CAS PubMed.
- J. Lan, D. Cao, W. Wang and B. Smit, ACS Nano, 2010, 4, 4225–4237 CrossRef CAS PubMed.
- B. Nikolic, IEEE Trans. Electron Devices, 2008, 55, 71–83 Search PubMed.
- X. Ding, L. Chen, Y. Honsho, X. Feng, O. Saengsawang, J. Guo, A. Saeki, S. Seki, S. Irle, S. Nagase, V. Parasuk and D. Jiang, J. Am. Chem. Soc., 2011, 133, 14510–14513 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- H. Hashemzadeh and H. Raissi, J. Phys. D: Appl. Phys., 2018, 51, 345401 CrossRef.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Priya
Yadav
*a,
Priya
Yadav