
Metal-promoted Mo6S8 clusters: a platform for probing ensemble effects on the electrochemical conversion of CO2 and CO to methanol†
Joseph T.
Perryman
a,
Jessica C.
Ortiz-Rodríguez
ab,
Joshua W.
Jude
a,
Forrest P.
Hyler
a,
Ryan C.
Davis
c,
Apurva
Mehta
c,
Ambarish R.
Kulkarni
d,
Christopher J.
Patridge
e and
Jesús M.
Velázquez
 *a
*a
aDepartment of Chemistry, University of California, One Shields Avenue, Davis, California 95616, USA. E-mail: jevelazquez@ucdavis.edu
bDepartment of Chemistry, University of Puerto Rico, Cayey, Puerto Rico 00736, USA
cStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, California 94025, USA
dDepartment of Chemical Engineering, University of California, One Shields Avenue, Davis, California 95616, USA
eDepartment of Chemistry, D’Youville College, Buffalo, NY 14201, USA
First published on 21st August 2019
Abstract
Presented herein is an investigation of a promising ternary metal sulfide catalyst that is capable of electrochemically converting CO2 to liquid and gas fuels such as methanol and hydrogen. When promoted by copper, an extended structure of Chevrel-phase Mo6S8 clusters is capable of reducing CO2 and CO to methanol in aqueous conditions with an overpotential of −0.4 V vs. RHE. H2 gas is simultaneously and preferentially evolved during this process, contributing to total current densities as high as 35 mA cm−2. It has been observed that Cu2Mo6S8 displays unique catalytic activity in terms of product selectivity, and we attribute this activity to molybdenum sulfide cluster units based on the results of structural, electronic, and electroanalytical characterization. Also discussed is the formulation of an interesting electronic structure–function correlation founded on the basis of X-ray absorption spectroscopic analyses and corroborated by the results of electroanalytical evaluation, where it has been observed that introduction of metal promoting species into the Chevrel-phase framework encourages charge transfer into cluster chalcogen sites.
New conceptsSelective electrocatalyst materials comprised of Earth-abundant elements have been synthetic targets for years, although a lack of fundamental studies of catalyst electronic structure has hindered the progress of rational material design thus far. In this work, fundamental insights gleaned from electrochemistry and X-ray absorption spectroscopy are utilized in order to elucidate the role of metal promotion on active site geometry and electronic structure. As it relates to electrocatalytic CO2 reduction, the electronic structures of transition metal sulfide active sites have been tremendously under-investigated experimentally, hence the evidence presented in this work that demonstrates potential tunability of active site functionality may usher a new paradigm in experimental evaluation of energy conversion materials. |
Introduction
Growing interest in the fields of energy conversion and storage involves developing materials and methods that can facilitate production of liquid fuel products which can easily be incorporated into modern petroleum-based infrastructures.1,2 In certain difficult-to-decarbonize energy services such as long-distance shipping, aviation, and production of materials like steel and cement—which combined contribute to over 9.2 Gt of CO2 emissions per year—implementation of alternative energy systems based solely on hydrogen or solar energy is infeasible owing to incompatible gravimetric and volumetric energy densities.3 However, it is immediately apparent that continued production and subsequent combustion of fossil fuels may exacerbate detrimental anthropogenic effects on both public and environmental health. Hence it will be desirable for future methods of fuel production to utilize abundant feedstocks such as solar energy and water, as well as captured and/or atmospheric CO2.3 This will ensure that future energy cycles are effectively carbon-neutral and based on converted renewable energy.4 One potential route towards realizing carbon-neutrality is electrochemical reduction of captured CO2 to liquid fuels; this process will need to be facilitated by an earth-abundant, selective and efficient catalyst material that is stable under aqueous, near-neutral operating conditions.5 Unfortunately, highly selective CO2 reduction to energy-dense hydrocarbons and oxygenates in water remains a challenge for all known heterogeneous catalysts.6Transition metal chalcogenides are capable of withstanding reductive potentials over wide pH ranges,7 and are among the best earth-abundant hydrogen evolution catalysts under extreme pH conditions.8,9 Moreover, transition metal chalcogenide materials have been studied for decades, owing to their tunable electronic, structural, and catalytic properties.10–14 Chevrel-phase (CP) sulfides with formula MxMo6S8 (M = transition metal or alkali metal, x = 0–4) have been the subject of much study due to their high-temperature superconducting behavior, as well as their reputation as the first functional multivalent battery cathode materials.15–17 To the best of our knowledge, despite numerous studies evaluating their performance as hydrodesulfurization, hydrogen evolution, oxygen reduction, and oxygen evolution catalysts, no experimental work has elucidated the ability of Chevrel-phase materials to reduce CO2.8,14,18–20 The chemical composition of catalytically active sites for small-molecule electroreduction is known to have a direct effect on the adsorption strength of reaction intermediates, largely stemming from modulation of electronic structure and subtle alterations in the geometry of binding sites.21–24 In the context of heterogeneous catalyst surfaces, the former is known as the “ligand effect,” and often, changes to electronic structure through compositional modification lead to accompanying effects associated with unique active-site “ensembles,” wherein neighboring atoms at catalytically active binding sites afford unique coordination interactions for intermediate species with varying energetic favorability.25,26 In other words, the presence of multiple elements at an active binding site can afford a distribution of diverse and distinctive coordination environments for reaction intermediates—effectively allowing for independent variation of binding strength for preceding and succeeding adsorbates with relevance in a given catalytic reaction. These phenomena are illustrated graphically in Fig. 1 where intermediate binding over a metallic surface is compared to binding over a surface with a ternary active-site ensemble. Recent computational studies by Liu et al. suggest that CP sulfides may be promising candidates as selective catalysts for the formation of methanol, both from CO2 and from syngas (CO and H2).26,27 Specifics of a potential reaction mechanism are discussed later in this work, although it is noteworthy that key intermediate species in the CO2 reduction reaction (CO2RR) such as CO* and HCO* are thought to be stabilized at Mo binding sites by cationic promoting species, while adjacent S atoms facilitate sequential hydrogenation toward an energy-dense product.26,28 Binding strengths to these two intermediates in particular are often key factors in the rate of CO2 reduction to liquid fuels; hence it is critical that materials which promote favorable binding are investigated.25
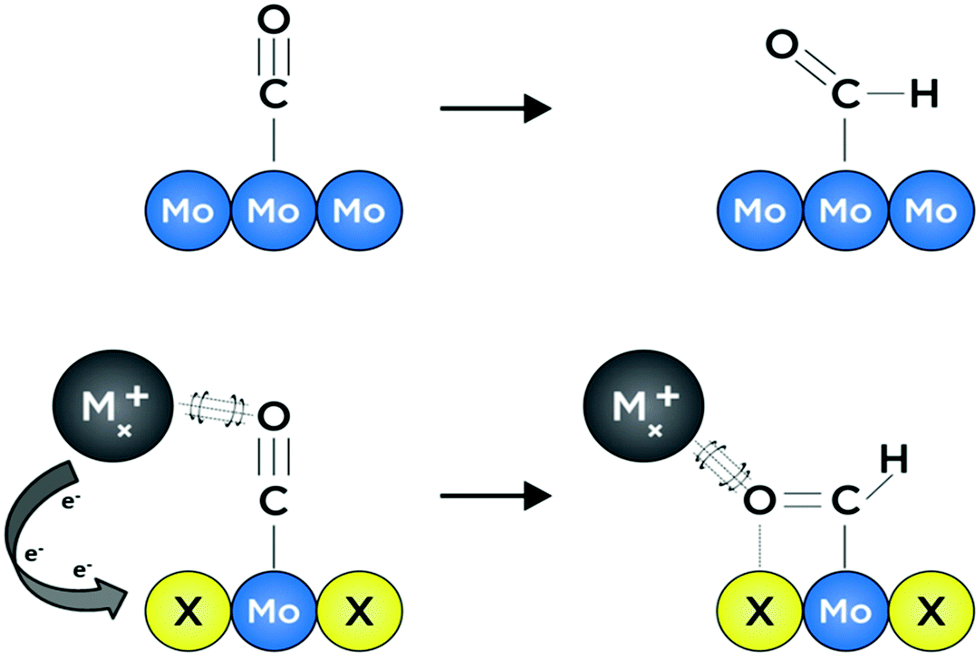 | ||
| Fig. 1 Schematic depiction of CO hydrogenation over a purely metallic surface (top), compared to a promoted metal chalcogenide surface (bottom) where synergistic chalcogen (yellow) inclusion at CO2RR active Mo sites (blue) encourages a unique intermediate binding geometry. Simplified Mx (grey) Coulombic stabilization of anionic intermediate domains is included for clarity. | ||
In this work, we report the unique catalytic ability of the Cu2Mo6S8 CP to reduce both CO2 and CO to methanol without also producing a myriad of other CO2RR products, and we provide a rationale for such behavior through interpretation of complimentary electronic and local structure analysis.
Experimental section
Chemicals and materials
MoS2 powder (99%, ∼325 mesh), Cu powder (99.995%, ∼100 mesh), Ni powder (99.8%, ∼325 mesh), Cr powder (99.85%, ∼100 mesh), and Pt mesh (99.99%) were used as purchased from Alfa Aesar. Mo powder (99.99%, ∼100 mesh), Na2CO3 (99.999% trace metals basis), and NaHCO3 (>99.5%) were used as purchased from Sigma Aldrich. Na2CO3 electrolyte solutions were prepared with 18.2 MΩ cm−1 water from a Thermo-Fisher Barnstead E-pure® purification system and did not require any pre-electrolysis treatment. Research-grade CO2 (99.999%) and CO (99.999%) were used as purchased from Matheson Gas. Ag/AgCl reference electrodes were purchased through ALS Japan. Selemion® anion exchange membrane was purchased from AGC Engineering and stored in ultra-pure deionized water prior to use in electrochemical experiments.Catalyst synthesis
CP catalysts were prepared by a direct microwave-assisted solid-state synthesis method adapted from literature methods.29 To obtain the desired CP, an appropriate metal powder (e.g. Cu, Cr, Ni), Mo powder, and MoS2 powder were mixed in stoichiometric ratios under N2 atmosphere, ball-milled for 24 h under N2 and pressed under 25 metric tons over a 20 mm surface area. Compressed powders were transferred to fused quartz tubes, and Al2O3 wool was tightly packed above the pellet under +5.0 mbar partial pressure of N2 as shown in Fig. S1 (ESI†). This reaction vessel was then transferred to a bath of ∼325 mesh graphite powder in a conventional microwave oven under Ar and irradiated with microwaves at a power of 1000 W for 10 minutes. In this process, graphite acts as a microwave susceptor and radiates heat to generate reaction temperatures >900 °C after less than one minute of irradiation. After allowing full phase-conversion over the course of a 10 minute reaction time, the quartz tube was removed from the graphite bath and immediately cooled in a room-temperature water bath. In order to obtain bare Mo6S8, Cu was chemically etched from a Cu2Mo6S8 pellet according to literature methods which involved bubbling O2 gas into a 6.0 M HCl solution overnight.30Structural and electronic characterization
Crystal structures and phase purity of as-synthesized CPs were analyzed via powder X-ray diffraction (PXRD) using a Bruker D8 Advance diffractometer with Cu K-alpha radiation (1.5406 Å) as well as at Stanford Synchrotron Radiation Lightsource (SSRL) beamline (BL) 2-1 (0.728068 Å). PXRD experiments at BL 2-1 were performed under He atmosphere with a 1.3 Tesla bend magnet and a Si(111) monochromator giving an energy resolution (ΔE/E) of 5 × 10−4. Refinement and powder pattern indexing shown in Fig. S2 (ESI†) was performed using the TOPAS analysis software by Bruker.Catalyst morphology and composition were analyzed before and after electrolysis using an FEI (Hillsboro, OR) 430 Nano Scanning Electron Microscope (SEM) and an FEI Scios DualBeam SEM with an Oxford Energy Dispersive X-ray (EDX) detector, respectively. Further elemental analysis was completed using a PHI Versaprobe 3 X-ray photoelectron spectrometer (XPS) to determine catalyst surface composition and oxidation state before and after electrolysis. Additional structural and electronic information was acquired through ex situ XAS at SSRL beam lines 4-1 and 4-3 using hard and soft/tender X-rays, respectively, in order to acquire X-ray Absorption Near-Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) information for Cu, Mo, and S.
X-ray absorption spectroscopy and data analysis
Near-edge (XANES) and fine structure (EXAFS) information was collected SSRL at BL 4-1 (molybdenum K-edge and copper K-edge) and BL 4-3 (sulfur K-edge). All samples were collected at room temperature (25 °C). BL 4-1 operates with a 20 pole 2.0 Tesla wiggler station and uses a liquid N2-cooled double-crystal Si(220) monochromator, giving an energy resolution (ΔE/E) of 1 × 10−4 and flux of 2 × 1012. BL 4-3 monochromator uses the Si(111) giving similar resolution and flux. Samples were collected in fluorescence and transmission with foil standards (Mo and Cu) collected simultaneously for calibration, while sodium thiosulfate was measured separately as a calibration standard for sulfur K-edge measurements. Data was calibrated for energy by comparing the first derivative of each sample spectrum with that of the respective elemental reference. Scans were performed in triplicate and merged to reduce noise at high k values. Processing was done using Athena and AUTOBK algorithm under the Demeter package.31 EXAFS modeling (Mo K-edge and Cu K-edge) was done using Artemis in the Demeter package. Theoretical EXAFS for Cu2Mo6S8 was calculated based on previous refinement for Cu2Mo6S8 from literature.31–33 Mo K-edge data was fit in R-space using Hanning window and multiple k-weight (1, 2, and 3) using a reciprocal space range of 2.0–12.5 Å−1 and a real space range of 1.0–3.2 Å. The scattering paths were calculated using FEFF 6.31 Dominant scattering paths for the Mo K-edge data, including Mo–S (4 paths), Mo–Mo (4 paths), and Mo–Cu (2 paths) were fit using a single energy parameter (E0), a passive reduction factor (S02) set to 0.83 for Mo K (found by fitting the Mo foil standard), bond length shift parameters (drS1, drS2, drMo1, drCu1), and Debye–Waller thermal parameter (ssS, ssMo, and ssCu) giving a total of 8 parameters in the fit. The un-promoted Mo6S8 CP was fit with the same scheme without Cu scattering paths. The Cu K-edge data was fit using the data range of 2.0–12.0 Å−1 in reciprocal space and 1.3–4.2 Å in real space. No Cu–Cu scattering paths appeared to contribute significantly to the EXAFS signal and therefore only Cu–S (2 paths), and Cu–Mo (4 paths) were given parameters of E0, drS1, drS2, drMo1, ssS, and ssMo. The passive reduction factor (S02) was set to 0.82, found by fitting the Cu foil standard. Theoretical XANES were calculated with FEFF 9.05 using full multiple scattering.34Detailed experimental methods related to electrochemical and computational analyses have been included for reference in the ESI.†
Results
Synthesis
The rapid, microwave-assisted solid-state synthetic method implemented in this work has proven capable of yielding highly phase-pure polycrystalline CP materials, including Cr1.73Mo6S8, Ni2Mo6S8, and Cu2Mo6S8. In order to observe changes to local and electronic structure with and without a ternary metal promoter atom, the un-promoted Mo6S8 CP was also synthesized according to a chemical etching process previously described in the Experimental section. Catalyst morphology can be seen for Cu2Mo6S8 in Fig. 2, while composition can be observed in the EDX scans shown in Fig. S3 (ESI†). Pawley refinement of synchrotron PXRD information has yielded lattice parameters of a = b = 9.6328 Å, c = 10.2229 Å with a unit cell volume of 821.502 Å3 for the Cu2Mo6S8 CP of interest in this study. These values are all in close agreement with literature values for the R-3H unit cell.16,35 Results of XRD analysis can be seen in Fig. S4 and Table S1 (ESI†).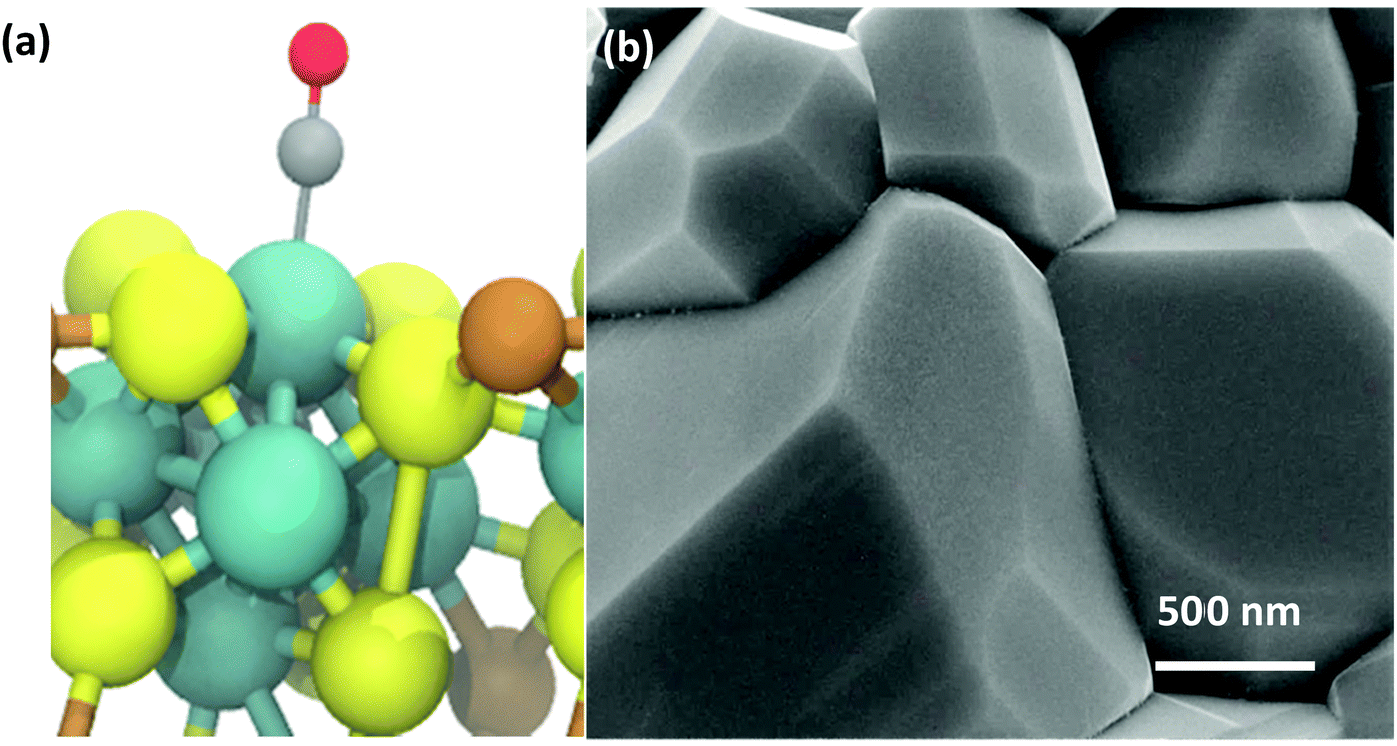 | ||
| Fig. 2 Computationally confirmed interaction between CO and a ternary CP active site ensemble (a). This illustrates that preferential binding of CO on a Cu2Mo6S8 CP surface occurs at Mo (blue) active sites, while the proximity of Cu (brown) to S sites (yellow) encourages promoter-to-chalcogen charge transfer. Scanning electron micrograph detailing the faceted morphology of polycrystalline Cu2Mo6S8 (b). | ||
Local and electronic structure
Cu2Mo6S8 and Mo6S8 CPs were analyzed at the Mo K-edge (20 keV), Cu K-edge (8.9 keV), and S K-edge (2.4 keV) XANES in order to elucidate spectral transitions that yield valuable information regarding frontier orbital population, oxidation state, coordination geometry, as well as charge transfer between species that constitute catalyst active site ensembles. Fig. 3 shows normalized μ(E) in the K-edge XANES regions for Mo, Cu, and S. Fig. 3a shows the Cu K-edge spectra for Cu2Mo6S8, with a Cu0 foil for reference. By measuring the position of the most intense peak in the 1st derivative (inflection point) of the spectra compared to the reference foil, we see that the Cu K-edge in Cu2Mo6S8 is shifted 2.5 eV higher than the reference. This indicates an oxidized Cu species in Cu2Mo6S8 as has been previously determined in literature as well as confirmed in this study by XPS analysis as shown in Fig. S5 (ESI†). Mo K-edge data is displayed in Fig. 3b for Mo6S8 and Cu2Mo6S8 along with a Mo0 foil for reference. The spectra show near overlap at the edge jump between Cu2Mo6S8 and Mo6S8 (∼Δ1 eV), indicating a negligible change in Mo oxidation state when Cu is introduced to the structure. Fig. 3c shows the S K-edge data for Cu2Mo6S8 and Mo6S8; a scan for the Na2S2O3 reference can be seen in Fig. S6 (ESI†). There are clear changes to the S electronic structure due to incorporation of the Cu promoter, as will be discussed further later.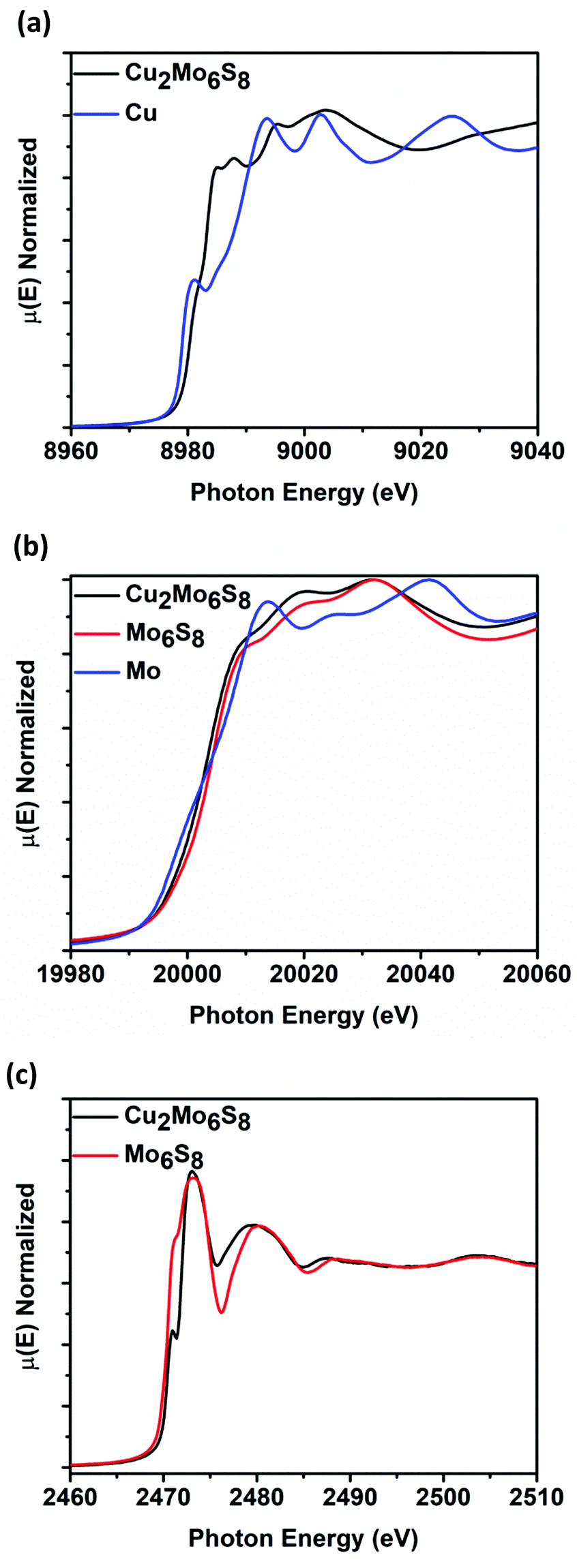 | ||
| Fig. 3 K-Edge XANES for Cu in Cu2Mo6S8 with a Cu0 foil for reference (a), K-edge XANES for Mo in Cu2Mo6S8 and Mo6S8, with a Mo0 foil for reference (b), and K-edge XANES for S in Cu2Mo6S8 and Mo6S8 (c). A reference scan using Na2S2O3 is shown in Fig. S6 of the ESI.† | ||
After multiple scans of the EXAFS region for all elemental components of Cu2Mo6S8 and un-promoted Mo6S8, it has been determined that all observed bond lengths are similar to expected values within the uncertainties of the fit. EXAFS information and theoretical fittings plotted in magnitude and real space for Mo and Cu are shown in Fig. 4a–d where we observe qualitatively different local Mo coordination when promoting species are present compared to un-promoted Mo6S8. Quantitative bond length information extracted from the EXAFS analysis shown in Fig. 4 is discussed in detail below.
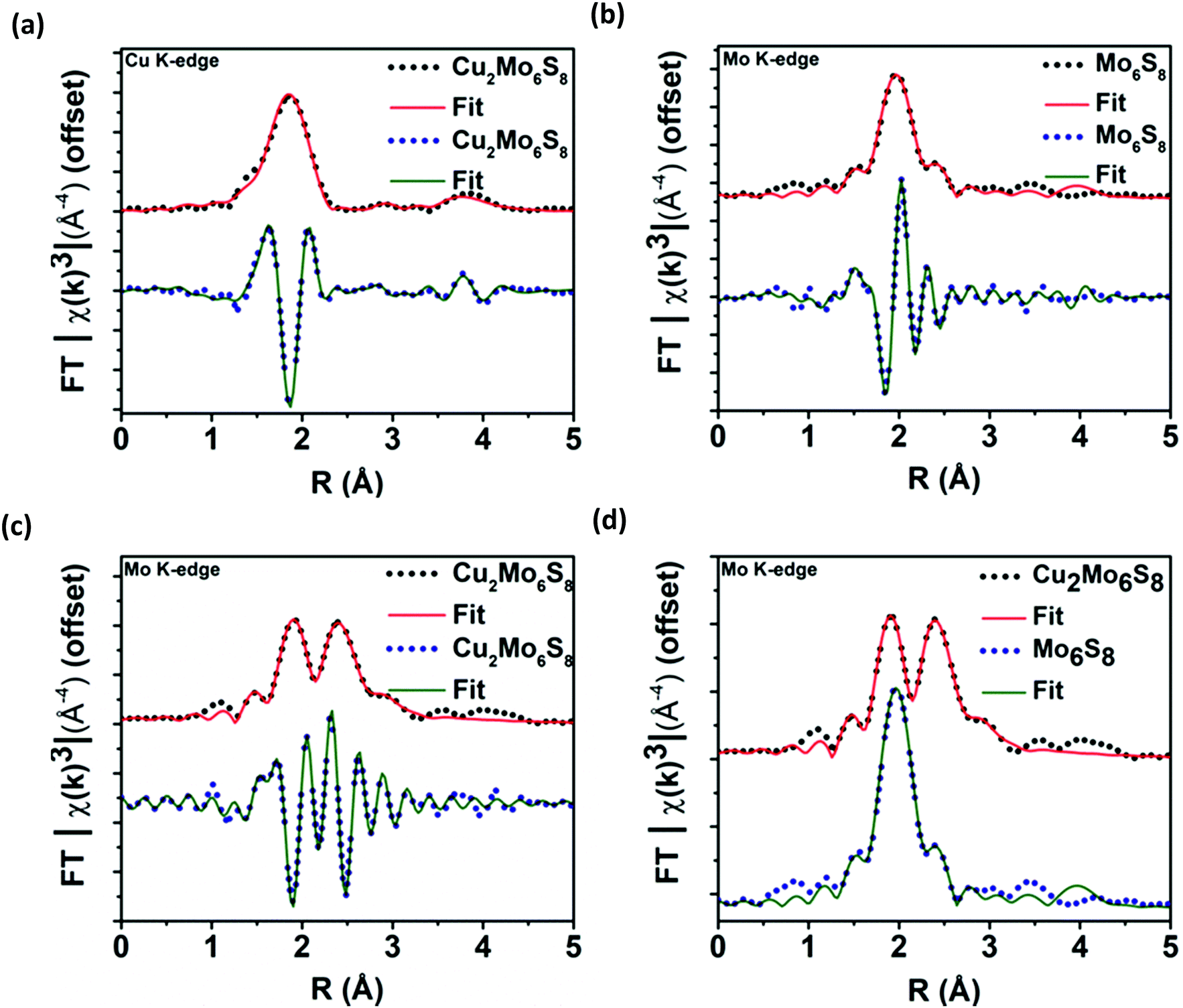 | ||
| Fig. 4 Processed EXAFS information plotted in magnitude and in real space for Cu in Cu2Mo6S8 (a), Mo in Mo6S8 (b), Mo in Cu2Mo6S8 (c), and for Mo in Cu2Mo6S8 overlaid with Mo in Mo6S8 to compare Mo–S and Mo–Mo signals (d). Qualitative differences in local Mo coordination as presented here are discussed quantitatively in the Results and Discussion sections. | ||
Catalyst evaluation
Cu2Mo6S8 was tested in neutral-pH controlled-potential electrolysis experiments with CO2 dissolved in solution under applied potentials ranging from −1.0 V vs. the reversible hydrogen electrode (RHE) to −0.4 V vs. RHE, as maximum CO2RR efficiency occurred within this potential window. We have confirmed that in this potential window, only two liquid-phase CO2 reduction products are formed after electrolysis; namely, formate and methanol. Nuclear magnetic resonance (NMR) spectra for these CO2RR products after electrolysis at −1.0 V vs. RHE are given in Fig. 5a and d.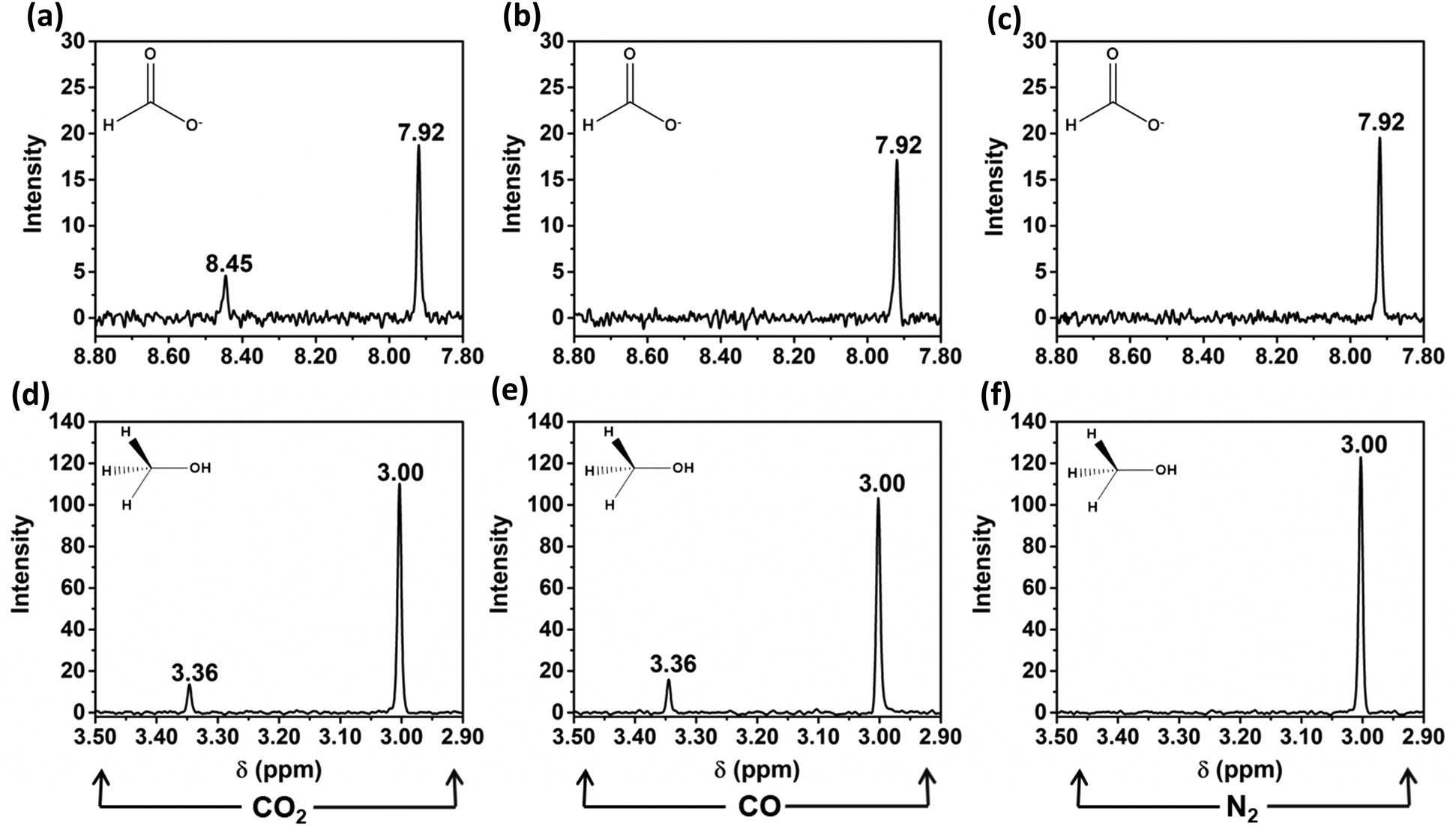 | ||
| Fig. 5 NMR signals for formate at 8.45 ppm (a–c) and for methanol at 3.36 ppm (d and e) detected after electrolysis under CO2 (a and d), CO (b and e), and N2 (c and f). Signals at 3.00 ppm and 7.92 ppm both correspond to the internal standard, N,N-dimethylformamide. All displayed NMR spectra are the result of electrolysis at −1.0 V vs. RHE in either 0.1 M Na2CO3 purged with CO2 or N2 or 0.1 M NaHCO3 purged with CO. | ||
When the target molecule was changed from CO2 to CO in a 0.1 M NaHCO3 solution, an NMR signal for formate was no longer observed, but the signal for methanol remained. This is evidence that the undesirable formate pathway was entirely suppressed upon removal of CO2 from the electrochemical cell. This observation is consistent with multiple accounts from literature that report a general lack of formate production when CO is implemented as a reduction feedstock, likely due to the impracticality of switching adsorbate-electrode coordination from C to O, as well as the required insertion of an additional oxygen atom.36Fig. 5b and e details this result by providing the insets required to visualize formate and methanol production or lack thereof when the electrochemical cell was purged with CO prior to electrolysis. To confirm that observed product formation in this study was the result of CO2 or CO reduction and not electrolyte interactions, N2 was purged into a near-neutral pH, 0.1 M NaHCO3 solution and controlled potential electrolysis was again performed at −1.0 V vs. RHE. Notably, during these N2 control experiments, neither formate nor methanol was observed, as shown in Fig. 5c and f.
As seen in Fig. S7 (ESI†), it was found that reductive current was dominated by hydrogen evolution. This is evidenced by large geometric current densities for hydrogen evolution compared to those for CO2RR products. It is worth noting however, that no systematic attempts were made to minimize hydrogen evolution on these CP catalysts through modifications to operating conditions (e.g. ionic liquid electrolyte,37 gas-diffusion methodologies38) or electrochemical cell constructs. In addition to inhibition of the parasitic formate pathway when CO2 is not present in solution, we see in Fig. S8 (ESI†) a general trend that faradaic efficiencies for methanol conversion from CO increase relative to those from CO2 at the same potentials. That is, despite lower solubility in water than CO2, CO is more efficiently converted to methanol on the Cu2Mo6S8 surface although geometric current densities are not entirely dissimilar between CO2 and CO reduction, indicative that hydrogen evolution remains the predominant reaction over these surfaces.
It was observed that Cu2Mo6S8 maintained its electrocatalytic performance over the course of multiple hours of electrolysis, even at the most negative potentials applied. This is evident in Fig. S9 (ESI†) by the stability of the reductive current over time. Further, catalyst stability is evidenced by high-resolution XPS spectra presented in Fig. S5 (ESI†) which indicate no significant changes to the catalyst surface aside from slight oxidation upon exposure to moisture and oxygen following chronoamperometry experiments, illustrated by the evolution of a Mo6+ signal in Fig. S10 (ESI†). Fig. S11 (ESI†) also shows survey scans of the catalyst surface before and after electrolysis which indicate no plating of foreign metals that could yield false positives of product formation. Lastly, to confirm product formation was not the result of chemical degradation, NMR and GC-TCD analyses were completed on a CO2-purged Na2CO3 electrolyte after Cu2Mo6S8 was left in solution at open circuit for 12 hours. Shown in Fig. S12 (ESI†), no products were detected after this experiment. Furthermore, H2S production which would indicate surface degradation during electrolysis was monitored via GC-MS. As shown in Fig. S13 (ESI†), no H2S production (m/z = 34) was observed. H2S was also un-detected qualitatively.
Discussion
Local coordination and electronic structure
CPs have a general structure that includes Mo6 octahedral clusters surrounded by S8 cubes, where S atoms lie at each face of the Mo6 octahedron as depicted in Fig. 6. These metal-sulfide units extend in a three-dimensional network to form a hexagonal lattice structure. Mo6S8 clusters in CPs are tilted in alignment along the ternary axis, with axial S atoms that connect Mo6S8 units together in offset chains through relatively short Mo–S bonds. This gives each Mo atom in the bulk network a square pyramidal pseudo-coordination, while Mo atoms exposed on the surface are relatively under-coordinated and exhibit square planar pseudo-coordination.19,39–41 Furthermore, as a result of this arrangement, large cavities exist between cluster units that are well-suited for occupation by various promoter cations of interest. | ||
| Fig. 6 Density of states for Mo6S8 (a) and Cu2Mo6S8 (b) calculated using the HSE06 functional. A noticeable DOS increase at more negative energies indicates charge transfer to the S atoms as evidenced by XAS. The grey region shows the d10 electronic configuration of Cu(I). Respective structures are represented above where Mo are blue spheres, S are yellow spheres, and Cu are red spheres. Further confirmation of charge transfer to S atoms can be seen in Fig. S18 (ESI†) which shows the result of Bader charge analysis. | ||
The absence of pre-edge features in the Mo K-edge XANES data shown in Fig. 3b, a signature of s → d-orbital transitions, suggested that the d-orbitals are fully occupied and/or there is little to no orbital mixing between p and d orbitals which indicates octahedral symmetry. The minimal shift in absorption onset and lack of alteration in the features of the near-edge spectra for Cu2Mo6S8 and Mo6S8 indicate insignificant change in the electronic structure of the Mo species.42 Mo K-edge EXAFS signals and modeling for Mo6S8 and Cu2Mo6S8 indicate a dramatic difference in local Mo coordination after Cu incorporation. The Mo6S8 has a lone peak (Fig. 4b) centered around 2.0 Å that originates from Mo–S nearest-neighbor scatterers with an additional shoulder feature near 2.5 Å. The diminished peak results from a shift in the Mo–S bond distances with axial Mo–S distances increasing to 2.57 Å. Upon Cu incorporation, Mo–Mo bond distances contract toward uniform distances (2.67 Å and 2.73 Å) and constructively interfere to form a new peak. Fitting parameters are included in Tables S2–S4 (ESI†), while aforementioned bond lengths for dominant/degenerate scatterers are shown in Table S5 (ESI†). The fitted bond lengths support small shifts in scattering distances that correlate between the Mo K-edge and Cu K-edge EXAFS modelling in Cu2Mo6S8. These results match closely with those corresponding to a series of magnesiated MgxMo6S8 compounds that was recently published.43 This previous work by Prendergast et al. showed that charge compensation during the introduction of an electron donating species actually proceeds through chalcogen atoms—this phenomenon is intriguing because charge compensation during intercalation usually results in monotonic oxidation of transition metal species, although in this case the delocalized electronic nature of the Mo6 cluster creates low-lying d-orbitals in CPs, hence such behavior was not observed for the CP sulfides studied here.44–46
The Mo6S8 cluster is deficient by four electrons and is metastable, requiring donation of at least two additional electrons in order for an extended structure to be directly synthesized.47 The oxidation state of Cu in Cu2Mo6S8 was verified as Cu(I) in earlier studies of the phase, and XPS results (Fig. S5, ESI†) closely match those of Cu2S.32,48 Hence, incorporation of Cu2 into Mo6S8 induces the donation of two electrons, serving the dual purpose of rendering the framework thermodynamically stable while simultaneously filling available S 3p orbitals and thereby raising the reactivity of the S p-band.23 This filling of the S p-orbitals can clearly be seen in the S K-edge comparison at 2471 eV between Cu2Mo6S8 and Mo6S8 in Fig. 3c, and is further exemplified graphically in the partial density of states (PDOS) calculations shown in Fig. 6b where a clear increase in S 3p PDOS at more negative energies indicates that charge is in fact transferred to S atoms in the structure upon ternary metal incorporation.
X-Ray absorption by S will cause core electrons to excite from 1s → 3p orbitals that mix to some degree with Mo 4d orbitals, giving rise to a small pre-edge shoulder that can be clearly seen in the S K-edge XANES spectra at approximately 2471 eV in Fig. 3c. As Cu donates electrons to the metal-sulfide cluster, these S orbitals are filled and the pre-edge feature diminishes in magnitude.41 The feature also appears to sharpen, as fewer transitions are left available for core S electrons. Interestingly, the aforementioned MgxMo6S8 study shows experimental data that indicates a completely masked pre-edge shoulder for Mg2Mo6S8 S K-edge XANES.43 This is because two Mg atoms would donate a total of four electrons, completing the ideal electron configuration of Mo6S8 and eliminating available pre-edge transitions.40,43 In contrast, Cu2Mo6S8 only yields two donated electrons (from two Cu+), hence the shoulder feature is still present and available S 3p orbitals are not completely filled.
To interpret the observed reactivity of CP sulfides in this work, we correlate potentially increased reactivity of the chalcogen species to this filling of 3p orbitals, as shown in the PDOS included in Fig. 6b. It is hypothesized that CO hydrogenation will be the rate-limiting step in the CO2RR to liquid fuels, hence being able to surmount the energy barrier associated with this step will rely strongly on the ability of the catalyst to facilitate hydrogenation. Thus, increasing the population of the S p-band should increase the reactivity of the chalcogen atoms which are thought to play a key role in hydrogenation.26,27
Effect of active site ensemble on reactivity
We observed that the Cu2Mo6S8 can reduce CO2 to methanol in aqueous electrolyte at low overpotentials and with only formate production as a competing CO2RR pathway. Such activity is unique to this particular sulfide catalyst, as no known metal chalcogenides produce methanol in aqueous electrolyte. Interestingly, while copper is one of few transition metals known to reduce CO2 to methanol (although methane is the preferred C1 fuel product over copper surfaces),49–51 it is not believed that its presence in Cu2Mo6S8 is the source of observed catalytic activity. Rather, it is thought that copper merely acts as an electron donor to the catalytically active Mo6S8 cluster, rendering the cluster electronically stable and allowing for a direct, high-temperature synthesis of a ternary active-site ensemble similar to the one illustrated in the bottom panel of Fig. 1.47 In fact, we were able to compute CO binding affinity at Cu sites in the extended structure as being −0.91 eV, which is more likely to result in dissociation than prolonged residence on Cu sites for further electroreduction. In contrast, binding energies for CO at Mo sites in Cu2Mo6S8 and Mo6S8 are much stronger at −1.61 eV and −1.50 eV, respectively. Additional support for this assertion that copper species do not contribute to observed product formation can be found in Fig. S14 (ESI†), where we provide NMR spectra that illustrate the production of formate and methanol during controlled potential electrolysis of Ni2Mo6S8 and Cr1.73Mo6S8 (see Fig. S15 (ESI†) for corresponding PXRD patterns for these CPs). Lastly, chronoamperometry was performed for un-promoted Mo6S8 to obtain a baseline for CO2 reduction activity, as shown in Fig. S16 (ESI†), where methanol is produced at −0.8 V vs. RHE. It has yet to be determined quantitatively whether the identity and stoichiometry of the metal promoter in a CP lattice has a significant effect on activity or selectivity, although such a phenomenon has been predicted by Liu et al.26Although faradaic yields for CO2RR products are low as a result of competing hydrogen evolution and mass transport limitations, methanol production efficiency as much as doubled at some potentials when CO was introduced as the target for reduction (Fig. S8, ESI†). This is a strong indicator that the pathway for methanol production on CP catalysts does proceed via CO hydrogenation as expected, and further indicates either that the interaction between CO2 and the Mo6S8 units is weaker than between CO and Mo6S8, or that non-polar and sparingly soluble CO2 does not readily diffuse to the polarized electrode/electrolyte interface during electrolysis—either (or both) situation may be the case.
The Cu2Mo6S8 catalyst investigated here displays among the lowest reported overpotentials for electrochemical methanol production in aqueous media.52 Based upon the results of this work, it is hypothesized that CP sulfides promote a reaction pathway that involves CO hydrogenation to methanol, and in order to reach the CO* intermediate of the CO2RR, it has been calculated that an associative mechanism occurs which involves the formation of an HOCO* intermediate as shown in Fig. S17 (ESI†), followed by an H2O*CO* intermediate. Formation of water through the reverse water-gas shift reaction may improve the energetics associated with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O cleavage that is inevitably required for methanol production from CO2. This hypothesis seems reasonable, as the Mo d-band center is relatively low-lying with respect to the Fermi level, and is therefore not prone to directly break a CO bond;23,27 the PDOS results presented here for Mo6S8 and Cu2Mo6S8 also support this Mo d-band positioning which can also be seen in Fig. 6. This hypothesis is supported by the production of methanol rather than methane by Cu2Mo6S8 in CO2 and CO saturated solutions.
O cleavage that is inevitably required for methanol production from CO2. This hypothesis seems reasonable, as the Mo d-band center is relatively low-lying with respect to the Fermi level, and is therefore not prone to directly break a CO bond;23,27 the PDOS results presented here for Mo6S8 and Cu2Mo6S8 also support this Mo d-band positioning which can also be seen in Fig. 6. This hypothesis is supported by the production of methanol rather than methane by Cu2Mo6S8 in CO2 and CO saturated solutions.
Kinetic limitations
It is widely acknowledged that competitive hydrogen evolution often contributes to low overall current efficiencies for the CO2RR in aqueous electrolytes. Hydrogen evolution in the electrolysis experiments presented here was vigorous enough that nucleation and de-nucleation of H2 gas may have induced mass-transport difficulties during electrolysis, as the CO2 target molecule—with intrinsically low solubility in water (∼35 mM for a saturated solution at 1 atm)—may have been unable to diffuse to the electrode–electrolyte interface against such intense gas evolution. To confirm such an assertion, further experimentation utilizing a rotating disk electrode or vapor-fed cell architecture may be required. It is also postulated that a Butler–Volmer trend for product formation was not observed in this study as a result of large currents for competitive hydrogen evolution at more negative applied potentials. This may explain the decrease in faradaic efficiency for both formate and methanol at more negative potentials.To further confound the electrochemical reduction of CO2 to methanol, production of methanol on a CP catalyst likely proceeds through the previously discussed CO hydrogenation pathway in multiple steps. The suspected rate-limiting CO hydrogenation step is likely to involve a large activation energy barrier owing to the high transition state energy required to yield the HCO* intermediate.26,27 While extensive intermediate studies have yet to be performed for Cu2Mo6S8, the relatively significant increase in FE towards methanol production when the target CO2 molecule was replaced by CO suggests that the reaction does proceed via CO hydrogenation, and that the reaction rate is inherently limited by weak adsorption interactions between catalyst active sites and CO2. Moreover, we believe that the simultaneous production of formate over CP surfaces when CO2 is the target molecule for reduction is the result of insufficient stabilization of the *HOCO intermediate (predicted by theory) relative to the HCOO* intermediate that follows proton-coupled electron-transfer to *CO2.26,27
To increase faradaic yields for CO2RR products compared with the HER, implementation of an electrolyte cation with a greater specific adsorptivity to the electrode surface may serve to tune the electric potential at the outer Helmholtz plane such that proton migration may be suppressed and partially negative atoms within CO2RR intermediate species may be further stabilized.53,54
Conclusions
Using local probe ex situ XAS, we have observed tuning of the S electronic structure of Mo6S8, potentially towards stronger activity for CO hydrogenation. We have also shown that metal promotion in CP frameworks leads directly to modular stabilization of reaction intermediates such as CO, ostensibly through synergistic interactions that include coulombic stabilization by ternary active-site ensembles. This idea has been emphasized in theoretical work by Liu et al. for CP systems, and we now have experimental evidence that these hypotheses are valid.We have demonstrated that Cu2Mo6S8, although previously unexplored as a CO2 reduction electrocatalyst, is capable of producing methanol—albeit at low partial current densities and low overall efficiency relative to hydrogen evolution. However, this reactivity was observed in aqueous electrolyte, at low overpotentials, and with a unique degree of selectivity relative to many existing electrocatalysts.49,52,55 Only two liquid-phase CO2 reduction products, formate and methanol, were formed during electrolysis on the investigated catalysts. Further, the competing formate pathway was successfully suppressed by the alternative implementation of a CO target molecule, which yielded methanol as the lone liquid-phase product.
It will be of significant fundamental value to incorporate advanced spectroscopic analyses that allow for identification of adsorbed reaction intermediates as a function of applied potential. This avenue of future exploration, in tandem with an operando investigation of catalyst surfaces, will allow for reliable determination and rationalization of CO2 and CO reduction mechanisms. Promising operando techniques such as grazing incidence XAS and XPS will yield necessary mechanistic information by elucidating changes to active-site local coordination and electronic configuration upon application of an applied potential and while in the presence of reduction target molecules dissolved in electrolyte.56–58
Future work is still required to establish the mechanism by which the investigated Chevrel-phase material reduces CO2 to methanol, as this knowledge will necessarily lead to an increased understanding of the role of active site multifunctionality on reaction efficiency. To develop a catalyst system whose tunable local and electronic structure can affect reactivity and selectivity could be a significant step forward in developing fundamental knowledge in energy conversion and storage research. The unique reactivity displayed by this metal-promoted Mo6S8 catalyst family warrants detailed investigation into the precise role of metal promotion in encouraging desirable reaction trajectories over promising energy-conversion catalysts. Hence, future work will elucidate the effect of tunable metal promotion on electronic structure of catalytically active sites such that control over reaction kinetics may be achieved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the University of California, Davis for start-up funding for this work. JTP thanks Chevron Corporation for funding through the UC Davis Institute of Transportation Chevron fellowship program. We also acknowledge funding support from the NSF through UC Davis ChemEnergy REU program, grant #1560479. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 (Project ID: 60905). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS of NIH.References
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- J. John-Paul, P. G. K. Surya Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- J. D. Wiensch, J. John, J. M. Velazquez, D. A. Torelli, A. P. Pieterick, M. T. McDowell, K. Sun, X. Zhao, B. S. Brunschwig and N. S. Lewis, ACS Energy Lett., 2017, 2, 2234–2238 CrossRef CAS.
- J. Jun, G. Minrui, S. Wenchao and Y. Yushan, Angew. Chem., Int. Ed., 2016, 55, 15240–15245 CrossRef PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- D. Lembke, S. Bertolazzi and A. Kis, Acc. Chem. Res., 2015, 48, 100–110 CrossRef CAS.
- Y. Yu, S.-Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS.
- T. Heine, Acc. Chem. Res., 2015, 48, 65–72 CrossRef CAS.
- S. A. Francis, J. M. Velazquez, I. M. Ferrer, D. A. Torelli, D. Guevarra, M. T. McDowell, K. Sun, X. Zhou, F. H. Saadi, J. John, M. H. Richter, F. P. Hyler, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Chem. Mater., 2018, 30(15), 4902–4908 CrossRef CAS.
- N. Alonso-Vante, Handbook of Fuel Cells, 2010 Search PubMed.
- Ø. Fischer, Appl. Phys., 1978, 16, 1–28 Search PubMed.
- R. Chevrel, M. Hirrien and M. Sergent, Polyhedron, 1986, 5, 87–94 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724 CrossRef CAS PubMed.
- S. J. Hilsenbeck, R. E. McCarley, A. I. Goldman and G. L. Schrader, Chem. Mater., 1998, 10, 125–134 CrossRef CAS.
- K. F. McCarty and G. L. Schrader, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 519–524 CrossRef CAS.
- N. Alonso-Vante, B. Schubert and H. Tributsch, Mater. Chem. Phys., 1989, 22, 281–307 CrossRef CAS.
- P. Liu and J. K. Norskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 RSC.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- A. Nilsson, L. Pettersson, B. Hammer, T. Bligaard, C. H. Christensen and J. K. Nørskov, Catal. Lett., 2005, 100, 111–114 CrossRef CAS.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- C. Liu and P. Liu, ACS Catal., 2015, 5, 1004–1012 CrossRef CAS.
- P. Liu, Y. Choi, Y. Yang and M. G. White, J. Phys. Chem. A, 2010, 114, 3888–3895 CrossRef CAS PubMed.
- K. Chan, C. Tsai, H. A. Hansen and J. K. Nørskov, ChemCatChem, 2014, 6, 1899–1905 CrossRef CAS.
- F. Murgia, P. Antitomaso, L. Stievano, L. Monconduit and R. Berthelot, J. Solid State Chem., 2016, 242, 151–154 CrossRef CAS.
- E. Lancry, E. Levi, Y. Gofer, M. Levi, G. Salitra and D. Aurbach, Chem. Mater., 2004, 16, 2832–2838 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- E. Levi, Y. Gofer, Y. Vestfreed, E. Lancry and D. Aurbach, Chem. Mater., 2002, 14, 2767–2773 CrossRef CAS.
- E. Levi, E. Lancry, A. Mitelman, D. Aurbach, G. Ceder, D. Morgan and O. Isnard, Chem. Mater., 2006, 18, 5492–5503 CrossRef CAS.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- A. M. Umarji, G. V. S. Rao, M. P. Janawadkar and T. S. Radhakrishnan, J. Phys. Chem. Solids, 1980, 41, 421–429 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- T. Hughbanks and R. Hoffmann, J. Am. Chem. Soc., 1983, 105, 1150–1162 CrossRef CAS.
- F. Thole, L. F. Wan and D. Prendergast, Phys. Chem. Chem. Phys., 2015, 17, 22548–22551 RSC.
- J. T. Perryman, F. P. Hyler, J. C. Ortiz-Rodriguez, A. Mehta, A. R. Kulkarni and J. M. Velazquez, J. Coord. Chem., 2019, 72, 1322–1335 CrossRef CAS.
- V. Kunzl, Collect. Czech. Chem. Commun., 1932, 4, 213–224 CrossRef CAS.
- L. F. Wan, J. Wright, B. R. Perdue, T. T. Fister, S. Kim, C. A. Apblett and D. Prendergast, Phys. Chem. Chem. Phys., 2016, 18, 17326–17329 RSC.
- C. J. Patridge, C. T. Love, K. E. Swider-Lyons, M. E. Twigg and D. E. Ramaker, J. Solid State Chem., 2013, 203, 134–144 CrossRef CAS.
- C. T. Love, A. Korovina, C. J. Patridge, K. E. Swider-Lyons, M. E. Twigg and D. E. Ramaker, J. Electrochem. Soc., 2013, 160, A3153–A3161 CrossRef CAS.
- C. J. Patridge, C. Jaye, T. A. Abtew, B. Ravel, D. A. Fischer, A. C. Marschilok, P. Zhang, K. J. Takeuchi, E. S. Takeuchi and S. Banerjee, J. Phys. Chem. C, 2011, 115, 14437–14447 CrossRef CAS.
- R. Chevrel and M. Sergent, Superconductivity in Ternary Compounds I, 1982 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, 1993 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Hori, Modern Aspects of Electrochemistry, 2008 Search PubMed.
- Y. Hori, Handbook of Fuel Cells, 2010 Search PubMed.
- G. Seshadri, C. Lin and A. B. Bocarsly, J. Electroanal. Chem., 1994, 372, 145–150 CrossRef CAS.
- B. N. Kabanov, I. I. Astakhov and I. G. Kiseleva, Russ. Chem. Rev., 1965, 34, 775 CrossRef.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS.
- A. D. Handoko, F. Wei, J. Lim, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- S. Yamamoto, H. Bluhm, K. Andersson, G. Ketteler, H. Ogasawara, M. Salmeron and A. Nilsson, J. Phys.: Condens. Matter, 2008, 20, 184025 CrossRef.
- B. Liu, E. N. Glass, R.-P. Wang, Y.-T. Cui, Y. Harada, D.-J. Huang, S. Schuppler, C. L. Hill and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2018, 20, 4554–4562 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh00745h |
| This journal is © The Royal Society of Chemistry 2020 |