
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of charge and hydrophilicity on the anti-fouling properties of kidney-inspired, polyester membranes†
Halan
Mohamed
 a,
Stephen
Hudziak
b,
Vishanigaa
Arumuganathan
a,
Zheyi
Meng
*ac and
Marc-Olivier
Coppens
*a
a,
Stephen
Hudziak
b,
Vishanigaa
Arumuganathan
a,
Zheyi
Meng
*ac and
Marc-Olivier
Coppens
*a
aEPSRC “Frontier Engineering” Centre for Nature Inspired Engineering and Department of Chemical Engineering, University College London, London, UK. E-mail: m.coppens@ucl.ac.uk
bDepartment of Electronic and Electrical Engineering, University College London, London, UK
cState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Material Science and Engineering, Donghua University, Shanghai, China. E-mail: mengzheyi@dhu.edu.cn
First published on 3rd July 2020
Abstract
Nature-inspired materials hold promise to tackle fouling experienced by membranes. Herein, a systematic approach is taken to explore the relative impact of charge and hydrophilicity on the anti-fouling properties of polyester membranes, both of which appear to play a role in the kidney's remarkable anti-fouling properties. Monofunctional and bifunctional polypropylene glycol (PPG) and polyethylene glycol (PEG) are used to modify polyester membranes. In both cases of hydrophilic modification and the addition of charge, a reduction in the adsorbed mass of lysozyme is experienced, as a rudimentary proxy for anti-fouling. Force spectroscopy measurements are used to quantify the change in adhesion force between the lysozyme functionalised tip and the modified PET surface. Adhesion forces decrease most between monofunctional and bifunctional PPG, which is supported by filtration results. This suggests that charge impacts the anti-fouling potential more significantly than hydrophilicity. This opens a new prospective avenue to modify membranes in order to improve their anti-fouling properties.
Design, System, ApplicationInspiration from nature presents an attractive approach to designing advanced materials. However, natural systems are often complicated with multiple functions carried out within the same structure. This renders it difficult to ascertain what exactly is responsible for a particular property. Consequently, mechanistic understanding of natural systems is necessary to discover which modifications are essential to adapt the inspiration to a new environment. In this work, the kidney's anti-fouling properties provide inspiration. Hydrophilic and negatively charged polysaccharide chains prevent fouling through a combination of a hydration layer and electrostatic repulsion. Hydrophilic modification of membranes to enhance anti-fouling properties is a relatively common technique. However, the interplay between charge and hydrophilicity has largely been unexplored in relation to their effect on anti-fouling. Understanding of this system can help guide the synthesis of more effective anti-fouling membranes. This work methodically explores this using polyester membranes modified with monofunctional and bifunctional polypropylene glycol (PPG) and polyethylene glycols (PEG). Monofunctional PPG and PEG are compared, as PPG is relatively more hydrophobic than PEG due to the extra alkyl group. Bifunctional PPG and PEG are positively charged at pH 7 due to their protonated amino group and, therefore, provide insight into the effect of charge. |
Introduction
Advancements in biotechnology over the past decades have heavily relied on the development and application of highly selective bio-separation processes. These include membrane processes, where permeation is typically pressure-driven and solutes are selectively rejected based on size. Under the umbrella of bio-separations, common applications include the recovery of bacteria and fungi from culture broths and the sterile filtration of nutrient media.1,2 Unfortunately, membranes used in bio-separations and other applications suffer from fouling, which leads to gradually declining productivity, with costs associated to regeneration, replacement and downtime. To operate at constant flux, increased transmembrane pressure is necessary, however, this can damage delicate biological materials due to higher shear stress.3One avenue of research into the synthesis of anti-fouling membranes has been the development of those inspired by nature. A common technique to improve the anti-fouling performance of membranes is by modification with hydrophilic materials. The aim of this scheme is to prevent fouling through the formation of a tightly bound hydration layer, which makes it thermodynamically unfavourable for foulants to displace the water molecules.4,5 This scheme is inspired by an example of fouling resistance observed in nature, namely, phosphocholine present on the surface of red blood cells. Phosphocholines contain zwitterionic groups, which create a condensed hydration layer.6 Other mechanisms learnt from nature have resulted in membrane designs that include tuneable or stretchable pores with liquid gating for anti-fouling transport of gases and liquids.7–9
The nature-inspired methodology used in this work advocates improved understanding of natural systems to identify what is truly the underlying cause of a desired property.10 This is especially important in cases where the environment in which the inspiration is used differs from that of the natural system, thus, requiring adaptations. Universality of structures observed in nature can also provide insight into common mechanisms that give rise to sought-after properties. For instance, the hydrophilic characteristic seen in phosphocholines is also observed in other biological systems. The glycocalyx present in the lumen of the kidney's glomerular blood vessels represents another structure that contains hydrophilic polysaccharide chains.11
The kidney exhibits superior anti-fouling properties in its function of filtering 140 L of urine from blood daily with no significant fouling.12 Glomerular filtration, also known as the ultrafiltration of blood, is carried out by the specialised blood vessel walls of the glomerulus. The overall charge of this membrane is negative, which aids the retention of plasma proteins through electrostatic repulsion as these proteins tend to be negatively charged.11–14 The glycocalyx, a negatively charged hydrophilic brush structure present in the lumen of the glomerulus, is credited as a selective layer for the retention of plasma proteins, and also as a hydrated layer to prevent fouling.15–19 Charge and hydrophilicity are regarded as the main factors attributed to the anti-fouling property of the kidney. Hydrophilic modification has already been introduced into artificial membrane systems to improve resistance against fouling.20–24 However, the combination of charge and hydrophilicity is not a common strategy to prevent fouling in membrane science.
This work methodically explores the combination of charge and hydrophilicity by comparing polyester membranes with monofunctional and bifunctional polypropylene glycol (PPG) and polyethylene glycols (PEG) (Fig. 1). Monofunctional PPG and PEG are compared to demonstrate the effect of hydrophilicity as PPG is relatively more hydrophobic than PEG due to the extra alkyl group. In addition to this difference in hydrophilicity, PPG has a branched structure unlike the linear structure of PEG. Bifunctional PPG and PEG are used to provide insight into the effect of charge. Both moieties and lysozyme are positively charged at the neutral pH used in this study as the pKa of the aliphatic amines is in the range of 10–11 and the pI of lysozyme is 11.35.25,26 The PPG and PEG moieties used are short-chain molecules containing around 6–8 repeating units to discount the consideration of complex chain conformations. The polyester substrates used are formed by track etching.27 Due to their regular straight pore structure, these membranes are used in sensing, nanogating and nanofluidics.28,29 Atomic force microscopy (AFM) and filtration tests are applied to compare the affinity and anti-fouling properties between the different PPG/PEG molecules and lysozyme.
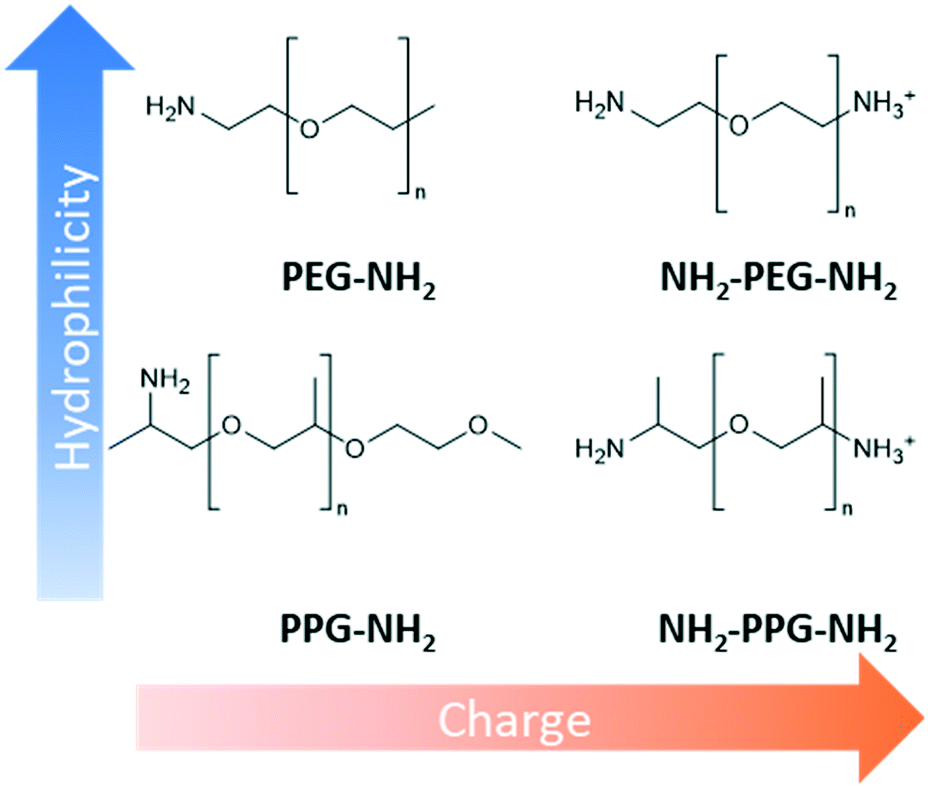 | ||
| Fig. 1 Comparison of short-chain moieties used with regards to hydrophilicity and charge at pH 7. | ||
Experimental
Materials
Polyethylene terephthalate (PET) track etched membrane discs (25 mm diameter, pore density 6 × 108 cm−2) with pore diameter of 100 nm were purchased from it4ip (Louvain-la-Neuve, Belgium). pentafluorophenol (PFP, >99%), n-heptane (>99%) and octyltrichlorosilane (97%) were purchased from Acros Organics. Hydrochloric acid (HCl, 37%), monofunctional poly(ethylene glycol) (PEG-NH2, n = 6), bifunctional poly(ethylene glycol) (NH2-PEG-NH2, n = 7), poly(propylene glycol) bis(2-aminopropyl ether) (NH2-PPG-NH2, average molecular weight Mn ∼ 400 g mol−1), O-(2-aminopropyl)-O′-(2-methoxyethyl)polypropylene glycol (PPG-NH2, Mn ∼ 600 g mol−1), lysozyme from chicken egg white (>90%), 3-aminopropyltriethoxysilane (APTES, 99%), glutaraldehyde (50% in water) and toluene (>99.5%) were procured from Sigma Aldrich. Ethanol (99.8%) and trifluoroacetic acid (TFA, 99%) were bought from Fisher Scientific. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, >98%) were purchased from Alfa Aesar. Potassium permanganate (KMnO4, >99%) and sulphuric acid (H2SO4, 1 M) were purchased from Honeywell Fluka Analytical. All materials were used as purchased with no further modification.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 vol/vol). The membranes were left to soak in the prepared solution for 2.5 h on a shaker at 100 rpm. The membranes were subsequently rinsed three times with ultrapure water, twice with HCl (6 N), ultrapure water, ethanol and then, dried finally. A solution of PFP (43 mM) and EDC (63 mM) in ethanol was prepared and the membranes were left to soak in the solution for 2 hours and rinsed twice with ethanol afterwards. The membranes were then immersed in an ethanol solution of the short chain moiety (10 mM) for 24 h before rinsing with ethanol and drying in ambient conditions overnight (Fig. 2).
:5 vol/vol). The membranes were left to soak in the prepared solution for 2.5 h on a shaker at 100 rpm. The membranes were subsequently rinsed three times with ultrapure water, twice with HCl (6 N), ultrapure water, ethanol and then, dried finally. A solution of PFP (43 mM) and EDC (63 mM) in ethanol was prepared and the membranes were left to soak in the solution for 2 hours and rinsed twice with ethanol afterwards. The membranes were then immersed in an ethanol solution of the short chain moiety (10 mM) for 24 h before rinsing with ethanol and drying in ambient conditions overnight (Fig. 2).
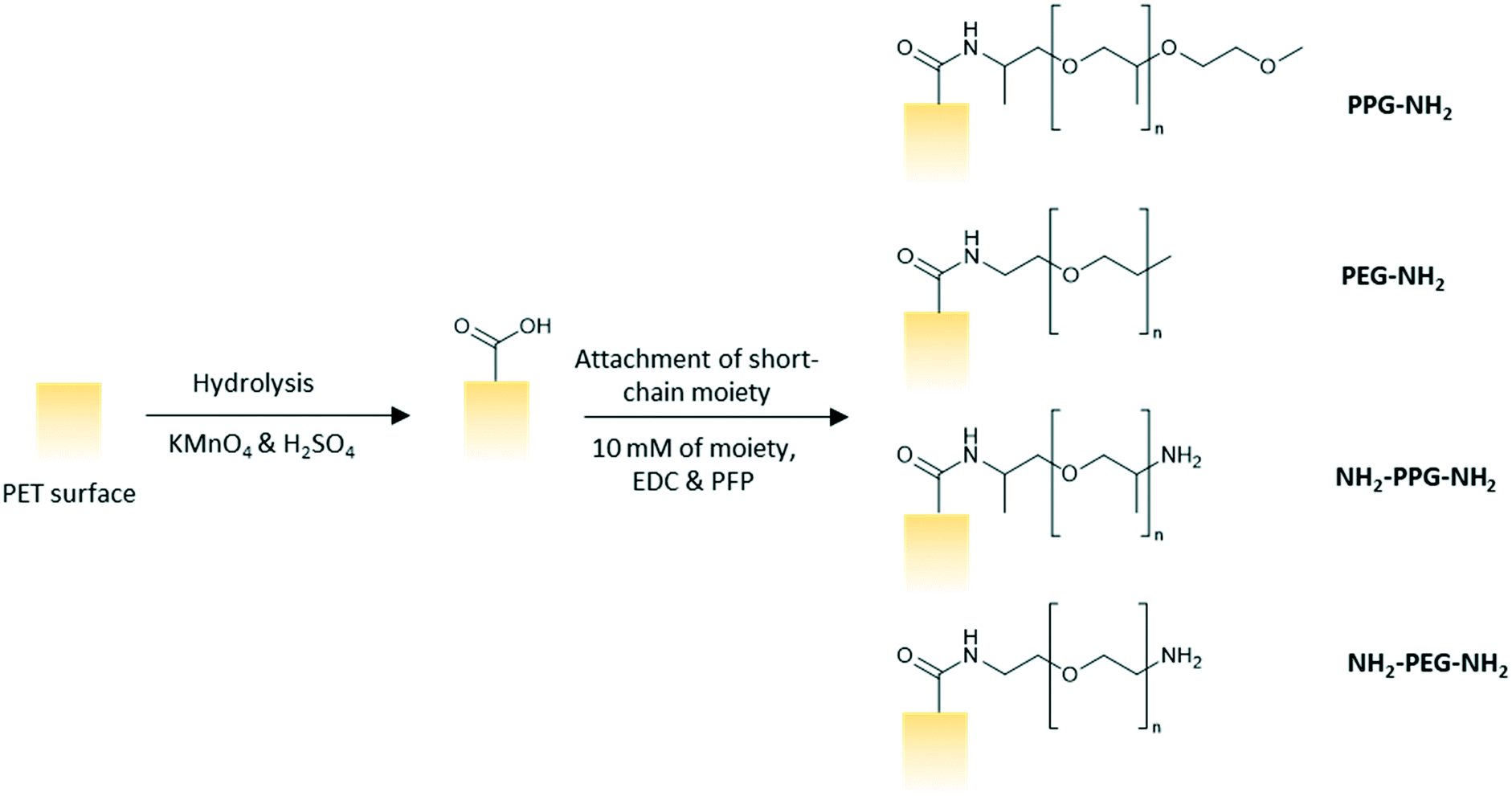 | ||
| Fig. 2 Schematic of modification process on either a PET coated silicon wafer or on a PET membrane surface (note: possible partial charges are not shown). | ||
Force spectroscopy experiments were conducted using a Flex-Axiom AFM in conjunction with a C3000 controller (Nanosurf) to measure the adhesion force between the lysozyme functionalised tip and the modified PET film. The spring constant and deflection sensitivity were measured using the thermal tuning method. The adhesion force was measured from the force vs. tip sample separation deflection curve. At least 64 approach-and-retraction cycles were carried out at five different locations on each sample.
Results
XPS
X-ray photoelectron spectroscopy was carried out to confirm successful grafting of the short chain moieties. Native PET does not contain any nitrogen, whereas monofunctional and bifunctional PEG and PPG do. Therefore, to confirm successful grafting, a N1s peak should be seen in the spectra for these modified membrane samples. Fig. 3(a) shows the spectrum for the unmodified PET, which is characteristic of polyethylene terephthalate. A sharp C1s peak can be seen at approximately 280 eV, along with its Auger peak at 1223 eV. Another sharp peak is seen at ∼530 eV, representative of O1s with a KLL Auger peak at 980 eV. These peaks are also observed in the spectra in Fig. 3(b)–(e). Individual scans for N1s peaks are necessary, as the proportion of nitrogen present in the samples is much lower than carbon and oxygen. Fig. 3(a) shows no peak for N1s, as expected, however, Fig. 3(b)–(e) all show a clear N1s peak at 400 eV, representative of the C–NH2 bond in the short-chain moieties. The absence of this peak in the unmodified PET proves that its presence in Fig. 3(b)–(e) is due to successful grafting of the monofunctional and bifunctional PEG and PPG onto the PET membranes.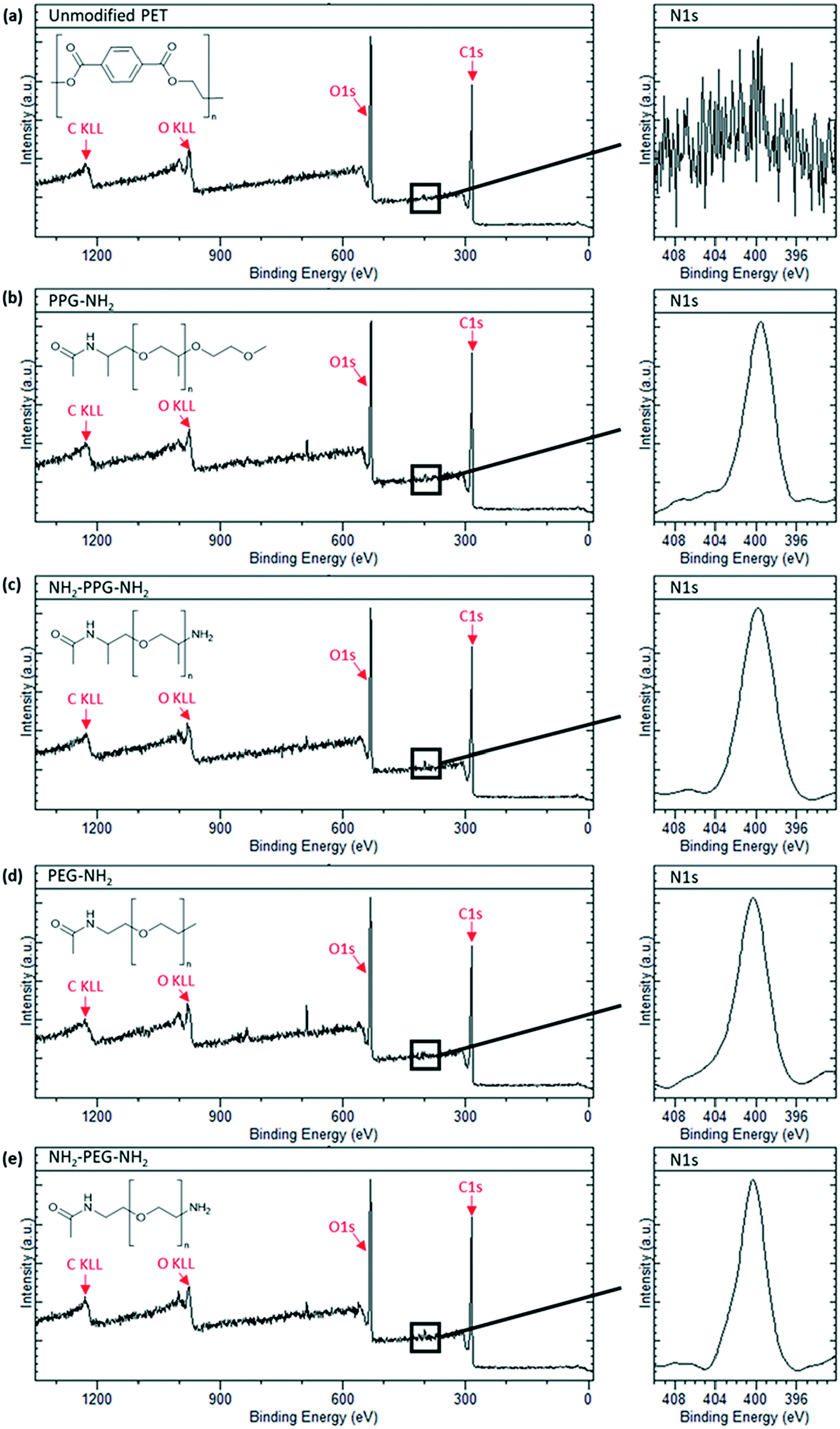 | ||
| Fig. 3 XPS spectra of sample types used with an insert of N1s peak scan. (a) Unmodified PET, (b) PPG-NH2, (c) NH2-PPG-NH2, (d) PEG-NH2 and (e) NH2-PEG-NH2 modified PET membranes. | ||
Surface topography
Scanning electron microscopy (SEM) was used to observe whether there was any obvious change in porosity and surface morphology caused by modification with the short-chain moieties. The membranes used in this work were formed through track etching;31 the pores seen in Fig. 4 are, indeed, characteristic of those formed using this technique. While there is no evident change in the morphology of the membranes at this scale, this is expected as the moieties used for modification have low degrees of polymerisation, thus any change is undetectable at this scale. The images suggest that there is no steric blockage of pores or change in morphology after the attachment of monofunctional and bifunctional PPG and PEG. In Fig. 4(b) and (c) some artefacts are seen in the micrographs; these are most likely areas that are topographically higher, resulting in charge accumulation. It is unlikely that these artefacts are due to aggregation of PEG-NH2 and NH2-PEG-NH2 chains, as they are roughly of the same scale as the pores (∼100 nm), whereas the chains have only 6 or 7 repeating units and are thus much smaller. Characterisation of surface roughness and water contact angle can be found in Table S1.† | ||
| Fig. 4 Scanning electron micrographs of (a) unmodified PET membrane, (b) PEG-NH2, (c) NH2-PEG-NH2, (d) PPG-NH2 and (e) NH2-PPG-NH2 modified PET membranes. The scalebar is 1 μm long. | ||
AFM
Force spectroscopy measurements were conducted with AFM to quantify the adhesion force between the lysozyme functionalised tip and the modified PET film on silicon wafers. When using a cantilever with a low spring constant and a sharp tip, this can prove to be a powerful tool sensitive to small interaction forces. As the cantilever approaches, the tip jumps to contact the surface through a combination of short-range forces, such as Van der Waals and capillary forces. Properties of the film, such as Young's modulus, can be measured once the tip contacts the surface by indenting the sample. Adhesion is measured during the retraction part of the cycle, as the tip pulls away from the surface due to a combination of non-specific interactions. Negative forces during retraction indicate attractive forces due to adhesion, whereas a positive force is due to repulsive interactions.These experiments replicated the conditions used for filtration tests; the samples were submerged in the prepared PBS solution (0.2 M, pH 7) with an additional drop added onto the cantilever to prevent air bubbles forming during the initial approach to the surface. At this pH, bifunctional PPG, PEG and lysozyme are positively charged as the pKa of the aliphatic amines is in the range of 10–11 and the pI of lysozyme is 11.35.25,26 XPS spectra to confirm the success of the lysozyme functionalisation process are shown in Fig. S2.†Fig. 5 shows a compilation of the adhesion forces measured; in most cases, the highest frequency of forces measured lies in the range of 0 to −0.2 nN.
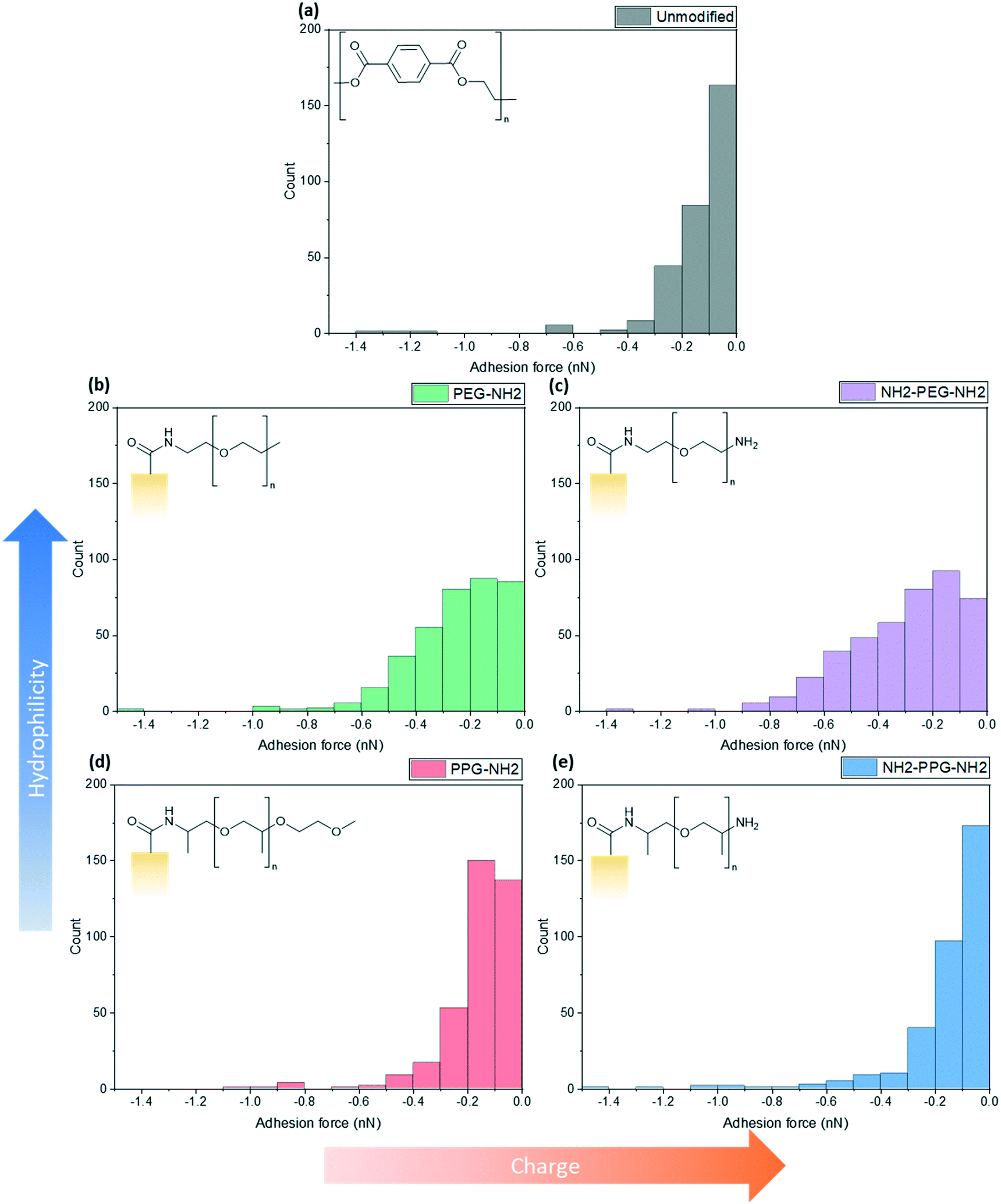 | ||
| Fig. 5 Calculated maximum adhesion forces for (a) unmodified PET, (b) PEG-NH2, (c) NH2-PEG-NH2, (d) PPG-NH2 and (e) NH2-PPG-NH2 modified PET membranes. | ||
Fig. 5(a) shows the adhesion force distribution for native PET; the observed range is expected, as the interactions that give rise to these forces will not be the same during each approach-and-retract cycle. Fig. 5(b) and (c) show a wider distribution of adhesion forces in comparison to Fig. 5(a), (d) and (e), which have sharper distributions. The change in adhesion force between samples is not significant, however, this is expected as the chain length of modifying groups is small in comparison to the thickness of the PET film, thus the dominant layer is still the PET.
Compared to the unmodified PET, PPG-NH2 shows a shift in the peak of the distribution from 0 to −0.1 nN to −0.1 to −0.2 nN. The change between these samples does not specifically correspond to an increase in hydrophobicity, as PPG is still relatively hydrophilic. However, the unmodified PET film will still have more exposed carboxylic acid groups after the hydrolysis step, in comparison to PPG-NH2, which has an alkyl group terminating the chain. This may increase the adhesion force experienced through hydrophobic interactions.
The charge due to the protonation of the additional amino group in NH2-PPG-NH2 has shifted the highest count of forces back to 0 to −0.1 nN, in comparison to PPG-NH2. The adhesion measurements between 0 and −0.1 nN for NH2-PPG-NH2 represent 50% of the total, whereas this range only accounted for 37% in the case of PPG-NH2. The higher positive charge on NH2-PPG-NH2 reduces the adhesion force, due to electrostatic repulsion between the positively charged lysozyme and the protonated amino group. The smaller adhesion force indicates fewer non-specific interactions that cause attractive forces.
Increasing relative hydrophilicity from PPG-NH2 to PEG-NH2 increases the probability of adhesion forces within the range −0.2 to −0.6 nN. This would suggest a higher fouling potential for PEG-NH2, which is discussed in the next section. A second, protonated amino group on PEG (NH2-PEG-NH2) does not lead to a significant change in measured adhesion forces (compare Fig. 5(b) and (c)). However, there is a clearer peak between −0.1 and −0.2 nN.
Fouling tests
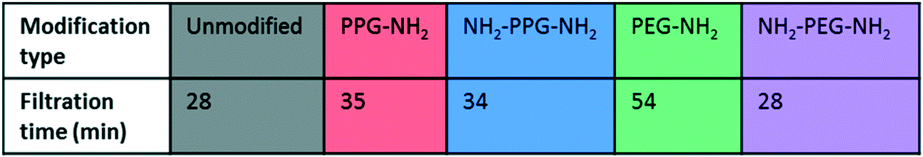
Filtration tests with lysozyme solutions were conducted to measure the flux decline through the membrane samples over time, due to fouling by lysozyme adsorption. In addition, the specific adsorbed mass of lysozyme was calculated for each sample type before and after forward flushing with PBS, to remove any loosely adsorbed protein. The flux was recorded over the entirety of the filtration time. As the time required to filter 10 mL of the feed solution differed for each sample type, Fig. 6(b) shows the flux only within the first 23 minutes. A summary of the total times required for filtration is given in Table 1. The drop in flux for samples past this time is due to the exhaustion of the feed, rather than a reflection of poor anti-fouling performance. Based on the trends seen in the AFM results presented in Fig. 5, it is expected that the best performing samples would be PPG-NH2 and NH2-PPG-NH2, as the adhesion forces between lysozyme molecules and the modified membrane surface are generally lower than for the other samples.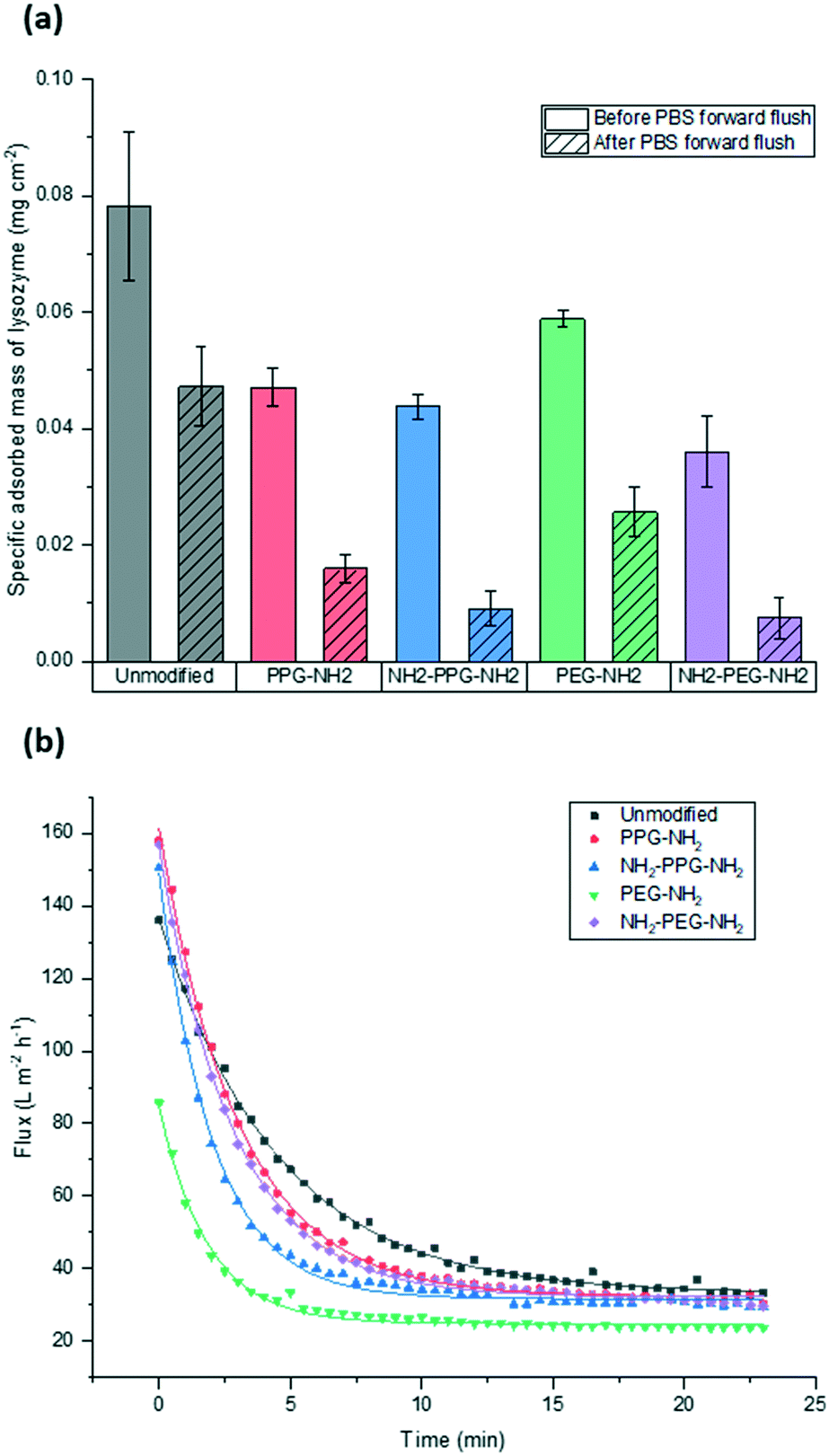 | ||
| Fig. 6 Comparison of (a) adsorbed mass of lysozyme before and after forward flushing with PBS and (b) recorded flux during fouling tests. Trendlines were added to guide the eye. | ||
|
The modified PET membranes show less propensity for irreversible fouling, compared to the bare, unmodified PET membrane, as seen in Fig. 6(a). NH2-PPG-NH2 and NH2-PEG-NH2 adsorb the smallest quantities of lysozyme post flushing with PBS, which would suggest that these moieties have a higher anti-fouling potential. NH2-PEG-NH2 has a slightly smaller, average adsorbed mass of 0.0075 mg cm−2 compared to NH2-PPG-NH2 samples, which adsorbed 0.0090 mg cm−2 on average; however, the overlapping error bars imply that the measured difference is insignificant. The decrease in adsorbed lysozyme after flushing is virtually identical at 79% for NH2-PEG-NH2 and NH2-PPG-NH2. Their electrically neutral counterparts, PEG-NH2 and PPG-NH2, adsorbed more lysozyme, which suggests that the addition of electrostatic repulsion has reduced fouling. Fig. 6(b) illustrates the flux decline experienced in the filtration of the lysozyme feed. The deviation in flux from the unmodified sample is low for the modified samples, which is expected as the chain length of the moieties used is small in comparison to the membrane pore size. PPG-NH2, NH2-PEG-NH2 and NH2-PPG-NH2 show the highest initial flux, however, the flux decline for these samples is larger than that of the unmodified membrane. This can be explained in conjunction with Fig. 6(a); the adsorption of lysozyme for the unmodified membrane decreases by 40% after forward flushing, whereas PPG-NH2, NH2-PEG-NH2 and NH2-PPG-NH2 decrease by 66%, 79% and 79%, respectively. This suggests that a greater proportion of the fouling experienced by these modified samples is reversible. Furthermore, although this initial decline is steeper for these samples, the value of the flux after stabilisation is roughly the same for the unmodified, PPG-NH2, NH2-PEG-NH2 and NH2-PPG-NH2 membranes. The filtration time for all modified samples but NH2-PEG-NH2 has increased in comparison to unmodified PET, as seen in Table 1. NH2-PPG-NH2 and PPG-NH2 have only slightly increased the required filtration time whilst maintaining a comparable flux and low lysozyme adsorption. The additional amino group in NH2-PPG-NH2 in comparison to PPG-NH2 has also decreased the adsorbed mass of lysozyme, without a significant change in flux. The addition of charge, via an additional amino group, has similarly decreased the mass of adsorbed lysozyme between NH2-PEG-NH2 and PEG-NH2, however, there is a more pronounced change in flux between these two, which suggests that PEG-NH2 is more susceptible to irreversible fouling without the protonated amino group. The samples functionalised with PEG-NH2 adsorb more lysozyme than those functionalised with PPG-NH2, and the flux is lower. Nevertheless, in both cases the anti-fouling potential has increased with reduced irreversible fouling, compared to the unmodified PET membrane.
Discussion
Effect of hydrophilicity
Monofunctional PPG and PEG were used to explore the effect of hydrophilicity on the propensity of fouling. PPG-NH2 was used as a relatively more hydrophobic treatment in comparison to PEG-NH2, due to the additional alkyl group. Hydrophilicity can induce anti-fouling properties, as it provides an energy barrier which makes adsorption of proteins unfavourable.4–7 Force spectroscopy was conducted to observe how the adhesion force changed through modification, using these short-chain moieties. It was seen in Fig. 5(b) and (d) that 40% of measured adhesion forces for PPG-NH2 occur between −0.1 to −0.2 nN, whereas the force distribution for PEG-NH2 does not have a distinct peak and has a wider distribution. 90% of adhesion forces occur between 0 and −0.3 nN for PPG-NH2, whereas that range only represents 68% of the data for PEG-NH2. Nevertheless, both show a change in adhesion force in comparison to unmodified PET, as seen in Fig. 5(a). The most distinct peak present in the native PET has shifted from 0 to −0.1 nN to −0.1 to −0.2 nN in PPG-NH2. However, in comparison to PET, PEG-NH2 exhibits a wider distribution of adhesion forces. 93% of the adhesion forces experienced by PEG-NH2 lie within the range of 0 to −0.5 nN, whereas 94% of adhesion forces are between 0 and −0.3 nN for the unmodified PET. The distribution of adhesion forces for PEG-NH2 was wider than for PPG-NH2, which suggests that there is a higher chance for more interactions between the modified surface and lysozyme.All this is consistent with the filtration results shown in Fig. 6(a) with an adsorbed mass of lysozyme of 0.026 mg cm−2 and 0.016 mg cm−2, respectively, for PEG-NH2 and PPG-NH2. PEG-NH2 also experienced a lower flux than PPG-NH2 in Fig. 6(b), which is likely due to reversible fouling, such as the formation of a cake layer, and, in the case of NH2-PEG, additional irreversible fouling, such as pore blockage. It would seem that monofunctional PEG experienced a greater proportion of irreversible fouling, as the mass of lysozyme adsorbed after flushing with PBS decreased only by 56%, as opposed to 66% for PPG-NH2. However, although monofunctional PPG performs better than monofunctional PEG, this does not necessarily mean that lower hydrophilicity due to the additional alkyl group is the cause. PPG also generates a hydration layer via hydrogen bonding between its ether groups and water molecules. More importantly, PPG has a lower glass transition temperature for polymers of the same molecular weight as PEG. As a result, the PPG chain is more flexible than PEG, which makes the formation of a hydration layer easier.32 Thus, in addition to the hydrophilicity of functional groups or moieties, the chain structure might also play an important role in the formation of the hydration layer.
Effect of charge
Bifunctional PPG and PEG were used to ascertain the impact of charge, in comparison to their neutrally charged, monofunctional counterparts. Charge provides an additional interaction, electrostatic repulsion, due to the overall positive charge of lysozyme and the positively charged, protonated amino group.7 Force spectroscopy revealed a reduction in absolute adhesion force between lysozyme and NH2-PPG-NH2, compared to PPG-NH2, as seen in Fig. 5(d) and (e). The shift in adhesion force peak from 0 to −0.1 nN to −0.1 to −0.2 nN between the unmodified PET and PPG-NH2 functionalised PET was reversed with the addition of the second amino group in bifunctional PPG. The measured adhesion forces between 0 and −0.1 nN for PPG-NH2 and NH2-PPG-NH2 represent 37% and 50% of the total, respectively, which suggests a lower fouling potential for NH2-PPG-NH2. Conversely, there was no significant change between PEG-NH2 and NH2-PEG-NH2, apart from a clearer peak between −0.1 to −0.2 nN. However, as the distribution of adhesion forces for both monofunctional and bifunctional PEG is large, it is hard to conclusively ascertain whether the addition of charge has changed adhesion significantly in this case.In Fig. 6(a) the addition of positive charges in both NH2-PPG-NH2 and NH2-PEG-NH2 has reduced the total adsorbed mass of lysozyme. The difference in flux is most prominent between monofunctional and bifunctional PEG, as illustrated in Fig. 6(b). PEG-NH2 has the lowest flux out of all short-chain moieties, whereas the flux for NH2-PEG-NH2 is similar to that of PPG-NH2 and NH2-PPG-NH2. It would seem that the hydrophilic nature of PEG-NH2 can resist irreversible fouling through the cake layer, which causes the reduced flux. However, monofunctional PEG still experiences a greater proportion of irreversible fouling than the other short chain moieties. The addition of a second amino group in NH2-PEG-NH2 results in a thinner cake layer, seen in the lower amount of adsorption of lysozyme before PBS flushing, due to the addition of electrostatic repulsion, which limits the adsorption of lysozyme. The reduction in adsorbed lysozyme after flushing with PBS has increased from 56% for monofunctional PEG to 79% for bifunctional PEG. This is also reflected in the time necessary to filter the same volume of lysozyme solution in Table 1; the sample functionalised by PEG-NH2 takes almost twice as long as NH2-PEG-NH2 to filter the same quantity of solution. Monofunctional and bifunctional PPG experience a similar trend, however, the fluxes for PPG-NH2 and NH2-PPG-NH2 are comparable. The addition of an amino group results in a reduced adsorbed mass of lysozyme, as illustrated in Fig. 6(a). Furthermore, the reduction in adsorbed lysozyme post PBS flushing has improved from 66% to 79% with the additional amino group. This is supported by the results in Fig. 5(d) and (e), which imply a reduced mass of lysozyme, as the adhesion force decreases with the extra amino group. Both bifunctional PPG and PEG show enhanced anti-fouling performance in comparison to their monofunctional counterparts.
Thus, charge exhibits a more pronounced effect on anti-fouling than hydrophilicity in this work. These findings are in agreement with computational studies of the adsorption process of biomacromolecules onto charged surfaces.33–35 In this work, we have simplified the nature of lysozyme to being positively charged, where, in reality, this is the overall net charge. Biomacromolecules have different domains that can be positively and negatively charged. It has been seen that where the surface is positively charged, such as in this work, the domains that are negatively charged can cause adsorption of the protein. As positively charged domains are not able to function as anchoring sites, this reduces the sites available for adsorption. During the approach to the membrane surface, if the orientation of the protein exposes a negatively charged domain, the side chains are able to penetrate both hydration layers and anchor onto the surface.33–35 However, if this orientation is only partially correct, the electrostatic interactions are weakened, and adsorption is not stable.33 Considering the low charge density used in this study, it is likely that anti-fouling properties can be further enhanced with higher charge densities.36,37
Conclusions
Fouling represents a major bottleneck in membrane separations. Nature can provide insights into how materials capable of tackling fouling could be designed. However, this is not a trivial task, as a deeper appreciation of the mechanisms used by natural systems is needed to make effective adaptations that translate this superior performance into the environment pertinent to applications. In this work, the kidney has been used as inspiration to guide the design of enhanced anti-fouling properties of polyester membranes. In kidneys, the combination of hydrophilic and negatively charged polysaccharide chains is understood to be an important factor in preventing fouling. Hydrophilic modification of membranes is a common method to increase the anti-fouling potential of surfaces, however, the addition of charge has remained largely unexplored.This work sought to explore the relative contributions of hydrophilicity and charge in the prevention of fouling. Modification of polyester membranes with monofunctional and bifunctional PPG and PEG were used to investigate this. Monofunctional PPG and PEG were used to determine the contribution of hydrophilicity, as PPG has an extra alkyl group, which makes it relatively more hydrophobic than PEG. Bifunctional PPG, PEG and lysozyme are positively charged at the neutral pH used in this study, as the pKa of the aliphatic amines is in the range of 10–11 and the pI of lysozyme is 11.35.25,26 The performance of materials with an additional amino group was compared to their neutrally charged counterparts.
XPS was used to attest to the successful grafting of the short chain moieties and SEM confirmed that there was no change in morphology. The adhesion force between the modified films and the lysozyme-immobilised tip was measured using force spectroscopy. This was compared to filtration tests to quantify the adsorbed mass of lysozyme on the membranes due to irreversible fouling. In terms of adsorbed mass of lysozyme, bifunctional PPG and PEG performed best. In comparison to the unmodified PET, NH2-PPG-NH2 adsorbed five times less lysozyme, whilst NH2-PEG-NH2 reduced the adsorbed lysozyme by 85%. Bifunctional PPG also exhibited the highest distribution of adhesion forces between 0 and −0.1 nN, which supports its lower propensity for fouling. The highest flux was measured for unmodified PET, however, this flux declines quickly as well. The next highest fluxes were measured for NH2-PPG-NH2, NH2-PEG-NH2 and PPG-NH2, which are approximately the same, however, the adsorbed mass of lysozyme for PPG-NH2 is higher than for the bifunctional PPG and PEG modified samples. Both bifunctional PPG and PEG show an increased resistance to irreversible fouling compared to their monofunctional counterparts, due to their protonated amino group at neutral pH. Monofunctional PEG exhibited a higher adsorbed mass of lysozyme and a lower flux than monofunctional PPG, although PEG-NH2 is relatively more hydrophilic.
The formation of a hydration layer is influenced not only by hydrophilic groups, but also by chain structures. This may explain why, once hydrophilicity was achieved in the samples investigated, the addition of charge was the most dominant factor to reduce the amount of adsorbed lysozyme. This enhanced anti-fouling property due to charge could be even further improved with higher charge densities to increase electrostatic repulsion between the foulant and the membrane surface.38–42
Conflicts of interest
The authors have no conflicts of interests to declare.Acknowledgements
The authors gratefully acknowledge financial support provided through an EPSRC Frontier Engineering Award (EP/K038656/1) and an EPSRC Doctoral Training Partnership (1910310). Xiao Xiao (School of Material Science and Engineering, Beihang University) and Yuheng Zhang (School of Chemistry, Beihang University), both funded by Beihang University's Yuanhang Global Study Summer Research Program, are also acknowledged for their contributions to obtaining preliminary data.References
- G. Subramanian, Bioseparation and Bioprocessing, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Z. F. Cui and H. S. Muralidhara, Membrane Technology: A Practical Guide to Membrane Technology and Applications in Food and Bioprocessing, Butterworth-Heinemann, Oxford, 2010 Search PubMed.
- A. C. Rayat, A. Chatel, M. Hoare and G. J. Lye, Curr. Opin. Chem. Eng., 2016, 14, 150–157 CrossRef.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS PubMed.
- G. Gunkel, M. Weinhart, T. Becherer, R. Haag and W. T. S. Huck, Biomacromolecules, 2011, 12, 4169–4172 CrossRef CAS PubMed.
- Y. X. Shen, P. O. Saboe, I. T. Sines, M. Erbakan and M. Kumar, J. Membr. Sci., 2014, 454, 359–381 CrossRef CAS.
- X. Hou, Natl. Sci. Rev., 2020, 7, 9–11 CrossRef.
- Z. Sheng, H. Wang, Y. Tang, M. Wang, L. Huang, L. Min, H. Meng, S. Chen, L. Jiang and X. Hou, Sci. Adv., 2018, 4(2), eaao6724 CrossRef PubMed.
- X. Hou, Y. Hu, A. Grinthal, M. Khan and J. Aizenberg, Nature, 2015, 519, 70–73 CrossRef CAS PubMed.
- M. O. Coppens, Curr. Opin. Chem. Eng., 2012, 1, 281–289 CrossRef CAS.
- B. Haraldsson, J. Nyström and W. M. Deen, Physiol. Rev., 2008, 88, 451–487 CrossRef CAS PubMed.
- R. Hausmann, M. Grepl, V. Knecht and M. J. Moeller, Curr. Opin. Nephrol. Hypertens., 2012, 21, 441–449 CrossRef CAS PubMed.
- L. Rügheimer, P. Hansell and M. Wolgast, Acta Physiol., 2008, 194, 335–339 CrossRef PubMed.
- M. Garsen, A. L. Rops, T. J. Rabelink, J. H. M. Berden and J. Van Der Vlag, Nephrol., Dial., Transplant., 2014, 29, 49–55 CrossRef CAS PubMed.
- M. J. C. Dane, B. M. Van Den Berg, M. C. Avramut, F. G. A. Faas, J. Van Der Vlag, A. L. Rops, R. B. G. Ravelli, B. J. Koster, A. J. Van Zonneveld, H. Vink and T. J. Rabelink, Am. J. Pathol., 2013, 182, 1532–1540 CrossRef CAS PubMed.
- S. Reitsma, D. W. Slaaf, H. Vink, M. A. M. J. Van Zandvoort and M. G. A. Oude Egbrink, Eur. J. Physiol., 2007, 454, 345–359 CrossRef CAS PubMed.
- C. Hjalmarsson, B. R. Johansson and B. Haraldsson, Microvasc. Res., 2004, 67, 9–17 CrossRef PubMed.
- M. J. C. Dane, B. M. van den Berg, D. H. Lee, M. G. S. Boels, G. L. Tiemeier, M. C. Avramut, A. J. van Zonneveld, J. van der Vlag, H. Vink and T. J. Rabelink, Am. J. Physiol.: Renal, Fluid Electrolyte Physiol., 2015, 308, F956–F966 CAS.
- M. Jeansson and B. Haraldsson, Am. J. Physiol.: Renal, Fluid Electrolyte Physiol., 2006, 290, 111–116 Search PubMed.
- J. S. Chen, Y. S. Ting, H. M. Tsou and T. Y. Liu, Surf. Coat. Technol., 2018, 344, 621–625 CrossRef CAS.
- D. M. Davenport, J. Lee and M. Elimelech, Sep. Purif. Technol., 2017, 189, 389–398 CrossRef CAS.
- C. Zhang, H. N. Li, Y. Du, M. Q. Ma and Z. K. Xu, Langmuir, 2017, 33, 1210–1216 CrossRef CAS PubMed.
- C. Liu, J. Lee, J. Ma and M. Elimelech, Environ. Sci. Technol., 2017, 51, 2161–2169 CrossRef CAS PubMed.
- X. Li, T. Cai and T. S. Chung, Environ. Sci. Technol., 2014, 48, 9898–9907 CrossRef CAS PubMed.
- S. Wanwimolruk, D. J. Birkett and P. M. Brooks, Mol. Pharmacol., 1983, 24, 458–463 CAS.
- L. R. Wetter and H. F. Deutsch, J. Biol. Chem., 1951, 192, 237–242 CAS.
- Z. Y. Meng and J. Zhai, Curr. Org. Chem., 2018, 22, 737–749 CrossRef CAS.
- X. Hou, Adv. Mater., 2016, 28, 7049–7064 CrossRef CAS PubMed.
- Z. Zhang, L. Wen and L. Jiang, Chem. Soc. Rev., 2018, 47, 322–356 RSC.
- E. C. Cho, D. H. Kim and K. Cho, Langmuir, 2008, 24, 9974–9978 CrossRef CAS PubMed.
- P. Apel, Radiat. Meas., 2001, 34, 559–566 CrossRef CAS.
- Z. Yi, L. Zhu, Y. Xu, J. Jiang and B. Zhu, Ind. Eng. Chem. Res., 2011, 50, 11297–11305 CrossRef CAS.
- K. Kubiak-Ossowska, B. Jachimska and P. A. Mulheran, J. Phys. Chem. B, 2006, 120, 10463–10468 CrossRef PubMed.
- K. Kubiak-Ossowska, M. Cwieka, A. Kaczynska, B. Jachimska and P. A. Mulheran, Phys. Chem. Chem. Phys., 2015, 17, 24070–24077 RSC.
- S. Tiwari, A. Gogoi and K. A. Reddy, Phys. Chem. Chem. Phys., 2019, 21, 24165–24176 RSC.
- A. Bratek-Skicki, V. Cristaudo, J. Savocco, S. Nootens, P. Morsomme, A. Delcorte and C. Dupont-Gillain, Biomacromolecules, 2019, 20, 778–789 CrossRef CAS PubMed.
- A. Bratek-Skicki, P. Eloy, M. Morga and C. Dupont-Gillain, Langmuir, 2018, 34, 3037–3048 CrossRef CAS PubMed.
- T. Chen, H. Yang, X. Wu, D. Yu, A. Ma, X. He, K. Sun and J. Wang, Langmuir, 2019, 35, 3031–3037 CrossRef CAS PubMed.
- M. J. Han, G. N. B. Baroña and B. Jung, Desalination, 2011, 270, 76–83 CrossRef CAS.
- M. Kumar and M. Ulbricht, Sep. Purif. Technol., 2014, 127, 181–191 CrossRef CAS.
- H. Guo, Y. Uehara, T. Matsuda, R. Kiyama, L. Li, J. Ahmed, Y. Katsuyama, T. Nonoyama and T. Kurokawa, Soft Matter, 2020, 16, 1897–1907 RSC.
- M. Vaselbehagh, H. Karkhanechi, S. Mulyati, R. Takagi and H. Matsuyama, Desalination, 2014, 332, 126–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0me00045k |
| This journal is © The Royal Society of Chemistry 2020 |