
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst decade of π-electronic ion-pairing assemblies
Yohei
Haketa
,
Kazuki
Urakawa
and
Hiromitsu
Maeda
 *
*
Department of Applied Chemistry, College of Life Sciences, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: maedahir@ph.ritsumei.ac.jp
First published on 30th March 2020
Abstract
Ion-pairing assemblies consisting of appropriately designed charged π-electronic species afford various functional supramolecular assemblies, including crystals and soft materials; they are formed by the anisotropic arrangement of charged π-electronic species through electrostatic and other weak noncovalent interactions. The design, synthesis and combination of charged π-electronic species (ion-pair formation) are crucial for the preparation of functional dimension-controlled assemblies. This article includes the prologue and progress of ion-pairing assemblies comprising π-electronic ions. Synthesis of π-electronic ions, preparation of ion pairs, fabrication of assemblies as crystals, gels and liquid crystals and their characteristic properties are summarized.
 Yohei Haketa | Yohei Haketa received his Ph.D. degree in 2011 from Ritsumeikan University, under the guidance of Prof. Hiromitsu Maeda, focusing on the assemblies of π-conjugated anion receptors. He was selected as a Research Fellow of the Japan Society for the Promotion of Science (JSPS) in 2009–2012. After working in Asahi Kasei Corporation as a researcher (2012–2015), he joined the group of Prof. Hiromitsu Maeda at Ritsumeikan University as a postdoctoral fellow in 2015 and started an academic career as an assistant professor in 2017, and, in 2019, he became a lecturer. |
 Kazuki Urakawa | Kazuki Urakawa received his Ph.D. degree in 2018 from Kumamoto University, under the guidance of Prof. Hayato Ishikawa, focusing on the synthesis of π-conjugated systems. He was selected as a Research Fellow of the Japan Society for the Promotion of Science (JSPS) in 2017–2018. After working in Adeka Corporation as a researcher for half a year, he joined the group of Prof. Hiromitsu Maeda as a postdoctoral fellow in October 2018 and will start an academic career as an assistant professor in April 2020. |
 Hiromitsu Maeda | Hiromitsu Maeda received his Ph.D. degree in 2004 from Kyoto University, under the guidance of Prof. Hiroyuki Furuta (Kyushu University) and Prof. Atsuhiro Osuka, after spending three months in the Sessler group, the University of Texas at Austin, in 2001. In 2004, he started an academic career in the Department of Bioscience and Biotechnology, Faculty of Science and Engineering, Ritsumeikan University. In 2008, he was transferred to the College of Pharmaceutical Sciences, wherein he was promoted to a professor in 2014. In 2016, he moved to the Department of Applied Chemistry, College of Life Sciences. He has been awarded several prizes, including ChemComm Emerging Investigator Lectureship (2012) and Fellow of the RSC (2015). |
Design, System, ApplicationOrdered arrangements of π-electronic species have been achieved by designing and synthesizing charged π-electronic species as the building blocks of assemblies. Stacking alignments of π-electronic ions are constructed by the incorporation of π-electronic ions which have suitable geometries and electronic states. It is noteworthy that combinations of π-electronic cations and anions are crucial for adjusting the assembling modes as well as resultant bulk-state properties. π-Electronic ion-pairing assemblies can exhibit versatile electronic properties including charge-carrier transport properties. Further modifications of π-electronic systems enable the preparation of fascinating π-electronic ion pairs and functional ion-pairing assemblies. |
1. Introduction
1-1. Background on ion-pairing assemblies
Over the past several decades, π-electronic molecular assemblies have been extensively investigated for implementation in functional electronic and optical devices such as photovoltaic cells, light-emitting diodes and field-effect transistors.1 The assembling modes of π-electronic molecules determine their assembling states and properties. In particular, a crucial factor for controlling the properties of organic electronics is the ordered arrangement of π-electronic systems (overlap of orbitals), which enables charge-carrier mobility.2 Thus far, various noncovalent interactions such as hydrogen-bonding, van der Waals, π–π and dipole–dipole interactions have been combined to assemble π-electronic molecules in desired arrangements. Importantly, the incorporation of electrostatic interactions into appropriately designed π-electronic species provides anisotropically ordered molecular arrangements as dimension-controlled assemblies, including low-dimensional crystals (fibres, sheets, etc.), supramolecular gels and liquid crystals.3,4 Such highly organized assemblies based on interactions between charged species have recently become known as ionic self-assemblies (ISAs).5 The components of ISAs are ion pairs associated through a combination of geometrically and electronically distinct charged species by noncovalent interactions. Therefore, these assemblies of ion pairs possess tuneable properties depending on the constituent cations and anions. To achieve the desired properties, it is essential to prepare appropriately designed π-electronic ion pairs with suitable geometries and electronic states that result in ordered arrangements and the formation of dimension-controlled ion-pairing assemblies with diverse modes.1-2. Concepts
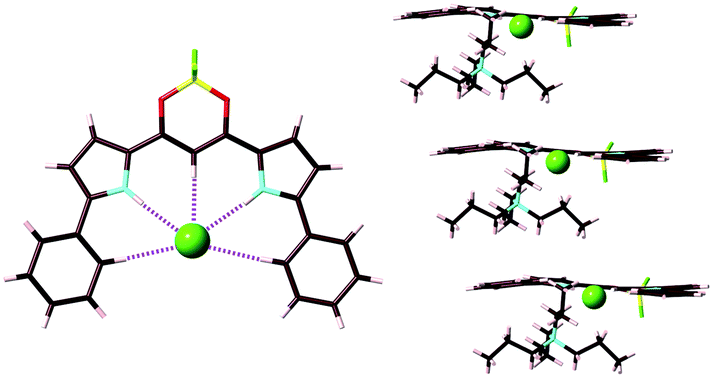
In 1828, Magnus' green salt (Fig. 1a), a pair of planar positively and negatively charged PtII complexes, [Pt(NH3)4]2+ and [PtCl4]2−, was reported.6 A single-crystal X-ray structure of Magnus' green salt reported in 1957 exhibited oppositely charged PtII complexes aligned by electrostatic interactions, forming an anisotropic quasi one-dimensional linear array of PtII ions (Fig. 1b).7 This fascinating ion-pairing structure of Magnus' green salt has stimulated numerous studies regarding the use of 1D arrays as wires for electrically conductive materials. Such an alignment of oppositely charged planar species results in a highly stable structure due to the electrostatic interactions. The remarkable structure of Magnus' green salt clearly illustrated that the geometries of constituent ions affect the packing structure and the resultant properties. Further modifications of Magnus' green salt with organic ligands resulted in the improvement of various functions, such as solubility and processability.8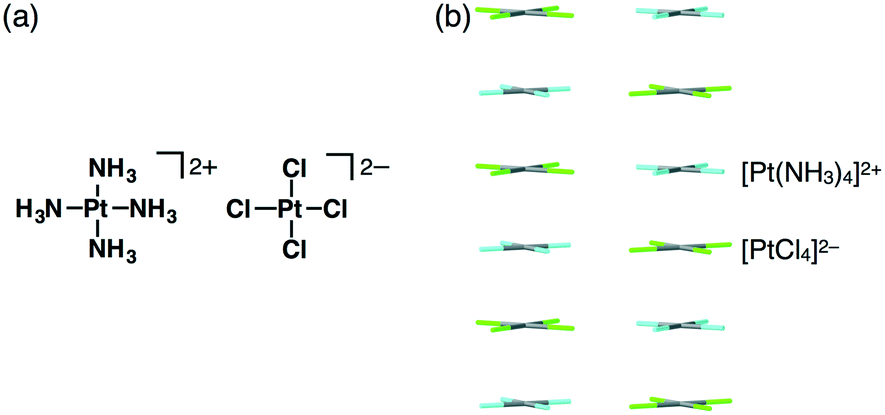 | ||
| Fig. 1 Magnus' green salt: (a) ion-pair and (b) packing structures. Atom colour code in (b): grey, light blue and green refer to platinum, nitrogen and chlorine, respectively. | ||
Essentially, a planar geometry is suitable for components of stacking-based supramolecular assemblies. A variety of planar charged species can be rationally designed on the basis of π-electronic charged species. With regard to the key concept of ion-pairing assemblies based on π-electronic charged species, a charge-by-charge assembly (Fig. 2 left) is defined as an assembling mode comprising alternately stacked positively and negatively charged species, whereas a charge-segregated assembly (Fig. 2 right) results from the stacking of identically charged species, attained by overcoming electrostatic repulsion.3 In addition, partial contributions by charge-by-charge and charge-segregated assemblies provide intermediate assembling modes (Fig. 2 centre). A charge-by-charge mode is suitable for constructing columnar assemblies because of the attractive force between oppositely charged π-electronic species on the basis of π–π stacking interactions.
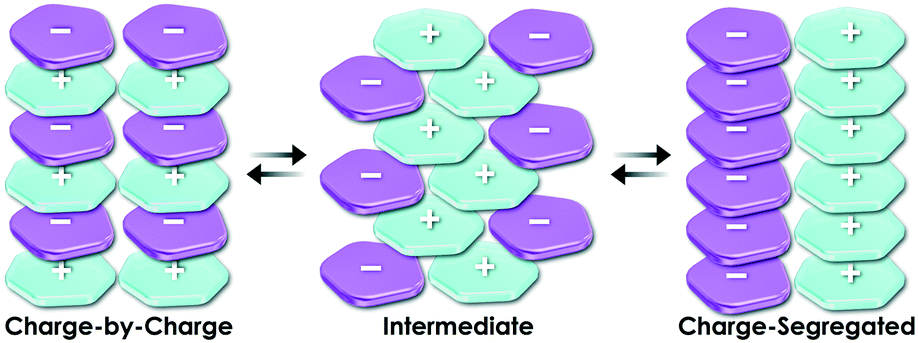 | ||
| Fig. 2 Conceptual diagram of assembling modes comprising π-electronic ions. | ||
π-Electronic ion pairs can essentially be classified as follows: (i) pairs of non-π-electronic cations and π-electronic anions (section 2), (ii) pairs of π-electronic cations and non-π-electronic anions (section 3) and (iii) pairs of π-electronic cations and π-electronic anions (genuine π-electronic ion pairs) (section 4). Practically, desired ion pairs are prepared by ion metathesis: a π-electronic cation with an inorganic counteranion and a π-electronic anion with an inorganic countercation are mixed, affording a π-electronic ion pair after purification including the removal of an inorganic salt (Fig. 3a). The design and synthesis of π-electronic ions that form ion-pairing assemblies, especially in the case of π-electronic anions, are more challenging compared to those of electronically neutral molecules. Although positive charges in cations can be stabilized by delocalization within π-electronic systems, stabilization of anions is challenging due to their excess electrons, which induce oxidation and electrophilic reactions. Thus far, complexes composed of electronically neutral host anion-responsive π-electronic molecules (receptors) and guest inorganic anions can be used as pseudo π-electronic anions (Fig. 3b). In particular, anion binding by π-electronic systems induces the transformation of hard guest anions to soft anions as anion complexes, and the properties of the anion complexes can be controlled by π-electronic receptor molecules. Therefore, the synthesis of π-electronic receptors is also another important factor in the production of various ion-pairing assemblies. The combination of electronically neutral receptors, guest anions and countercations can provide diverse ion pairs. The chemistry of artificial receptors has been primarily developed for sensors, transporters, etc.,9 but there is significant potential for receptor–anion complexes to be used as components of ion-pairing dimension-controlled assemblies.
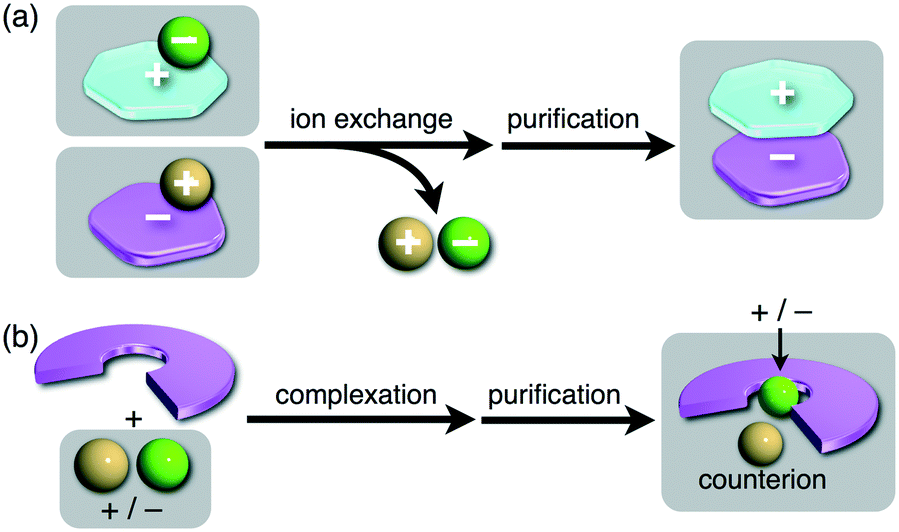 | ||
| Fig. 3 Preparation of π-electronic ion pairs: (a) ion metathesis providing π-electronic ion pairs and (b) ion complexation providing ion pairs of π-electronic receptor–ion complexes and counterions. | ||
1-3. History of ion-pairing dimension-controlled assemblies
Among various π-electronic anion receptors, since 2005, we have investigated a series of dipyrrolyldiketone BF2 complexes (e.g., 1a–c, Fig. 4), which exhibit effective [1 + 1]-type anion-binding properties through the inversion of two pyrrole rings.10 The anion-binding constant of the parent 1a for Cl− is 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 in CH2Cl2 at r.t., suggesting the possibility of using anion complexes as building blocks for assemblies. Thus far, various modifications of dipyrrolyldiketone BF2 complexes have been investigated, and their anion complexes were used as components of various ion-pairing assemblies in the forms of crystals, supramolecular gels and liquid crystals.3b,c In solution, anion-driven helical structures of covalently linked oligomers undergo chirality induction by ion-pairing with chiral countercations, displaying circularly polarized luminescence.11 As the details of a series of anion receptors are summarized in other review articles,3 a brief history and several topics regarding ion-pairing assemblies based on receptor–anion complexes are discussed in this section.
000 M−1 in CH2Cl2 at r.t., suggesting the possibility of using anion complexes as building blocks for assemblies. Thus far, various modifications of dipyrrolyldiketone BF2 complexes have been investigated, and their anion complexes were used as components of various ion-pairing assemblies in the forms of crystals, supramolecular gels and liquid crystals.3b,c In solution, anion-driven helical structures of covalently linked oligomers undergo chirality induction by ion-pairing with chiral countercations, displaying circularly polarized luminescence.11 As the details of a series of anion receptors are summarized in other review articles,3 a brief history and several topics regarding ion-pairing assemblies based on receptor–anion complexes are discussed in this section.
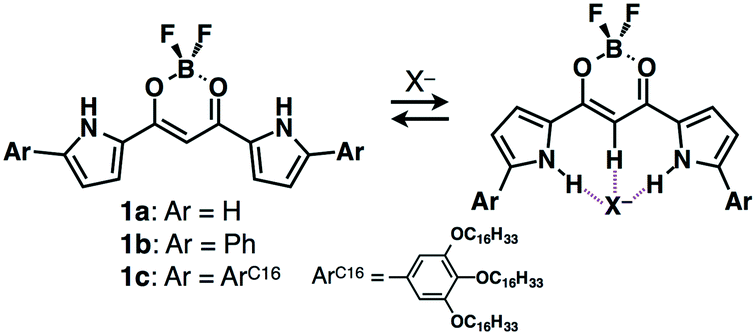 | ||
| Fig. 4 Dipyrrolyldiketone BF2 complexes 1a–c and [1 + 1]-type anion-binding behaviour. | ||
In 2007, the solid-state structure of a receptor–anion complex 1b·Cl− with a tetrapropylammonium cation (TPA+) was revealed by single-crystal X-ray analysis; the ion pair TPA+–1b·Cl− formed charge-by-charge stacking of 1b·Cl− and TPA+ (Fig. 5).12 [1 + 1]-Type receptor–anion complex 1b·Cl− formed a planar geometry through hydrogen bonding of the pyrrole NH, bridged CH and phenyl-o-CH. Electrostatic interactions between 1b·Cl− and TPA+ stabilized the columnar structure. It is noteworthy that this initial finding was crucial for further investigations of ion-pairing assemblies because introduction of planar cations instead of bulky TPA+ allows for contribution of π–π stacking interactions. Among planar π-electronic cations, the trioxatriangulenium cation (TOTA+) as a Cl− salt was initially attempted for combination with 1b for the crystallization (Fig. 6a).13 In fact, the precursor of the TOTA+ ion pair was prepared as a Cl− salt, and the final ring closure reaction was conducted with LiI, followed by anion exchange with aqueous KCl. However, Cl− was partially exchanged with I− during the ring closing reaction. Therefore, the single-crystal X-ray analysis of the obtained anion complex of 1b revealed the existence of both 1b·Cl− and 1b·I−, in the disordered state, with countercation TOTA+, forming a charge-by-charge stacking columnar structure comprising alternately stacked 1b·X− (X− = Cl− and I−) and TOTA+ with distances of 3.38 and 3.50 Å (Fig. 6b(i)). This observation indicated that anion exchange from softer I− to harder Cl− based on the soft countercation (TOTA+) is not straightforward, as it follows the hard and soft acid and base (HSAB) theory. Therefore, π-electronic cations that do not require any reagents including anions except for Cl− were necessary.
 | ||
| Fig. 5 Single-crystal X-ray structure of TPA+–1b·Cl−. | ||
 | ||
| Fig. 6 (a) TOTA+ and TATA+ and (b) single-crystal X-ray structures of (i) TOTA+–1b·X− represented as a Cl− complex and (ii) TATA+–1b·Cl−. | ||
Following a literature survey, the π-electronic triazatriangulenium (TATA+) cation, whose structure is similar to TOTA+, was selected as an alternative (Fig. 6a).14 The synthetic process of TATA+ allows for much easier control of counteranion purity. Cl− in the intermediate species is not excluded because the final ring closure reaction does not require reagents that induce anion exchange. The combination of 1b and TATA+ afforded the ion pair TATA+–1b·Cl−, forming a charge-by-charge assembly, similar to TOTA+–1b·X− (X− = Cl− and I−), with a shorter distance of 6.85 Å between the 1b·Cl− units than the ion pair with TPA+ (7.29 Å) (Fig. 6b(ii)).15 Crystallization with planar TATA+ also allowed for the possibility of controlling the geometries (stoichiometries) and packing structures of receptor–anion complexes, as seen in the Cl− complexes of 1a: a Cl−-bridged 1D structure was formed in the TBA+ ion pair,10 whereas a [2 + 1]-type planar receptor–Cl− complex was constructed in the TATA+ ion pair.16 Importantly, the peripheral receptor substituents are readily introduced to form dimension-controlled assemblies based on the ordered arrangement of constituent ionic species.
Aliphatic π-electronic receptor 1c formed a supramolecular gel from hydrocarbon solvents such as octane (10 mg mL−1).12 Interestingly, the addition of Cl− as a solid TBA+ salt to the gel of 1c resulted in a gradual transition to the solution state, which was attributed to the formation of a soluble ion pair consisting of 1c·Cl− and aliphatic TBA+. In sharp contrast to gel decomposition by the addition of the bulky TBA salt, the addition of the π-electronic cation salt TATA·Cl afforded dimension-controlled assemblies based on charge-by-charge stacking structures.15 The ion pair TATA+–1c·Cl− afforded an opaque octane gel with a gel-to-solution transition temperature of 35 °C (Fig. 7a). Furthermore, X-ray diffraction (XRD) analysis, polarized optical microscopy (POM) and differential scanning calorimetry (DSC) of the ion pair TATA+–1c·Cl− as the mesophase revealed the formation of a hexagonal columnar (Colh) structure based on charge-by-charge assembly. These significant findings regarding the formation of ion-pairing dimension-controlled assemblies afforded valuable insights into the design of π-electronic ions and their combinations.
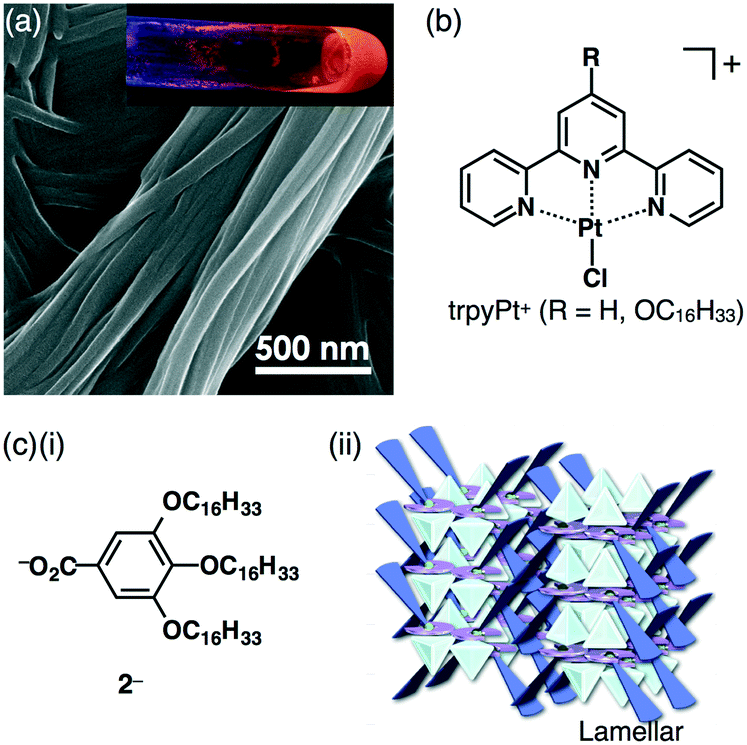 | ||
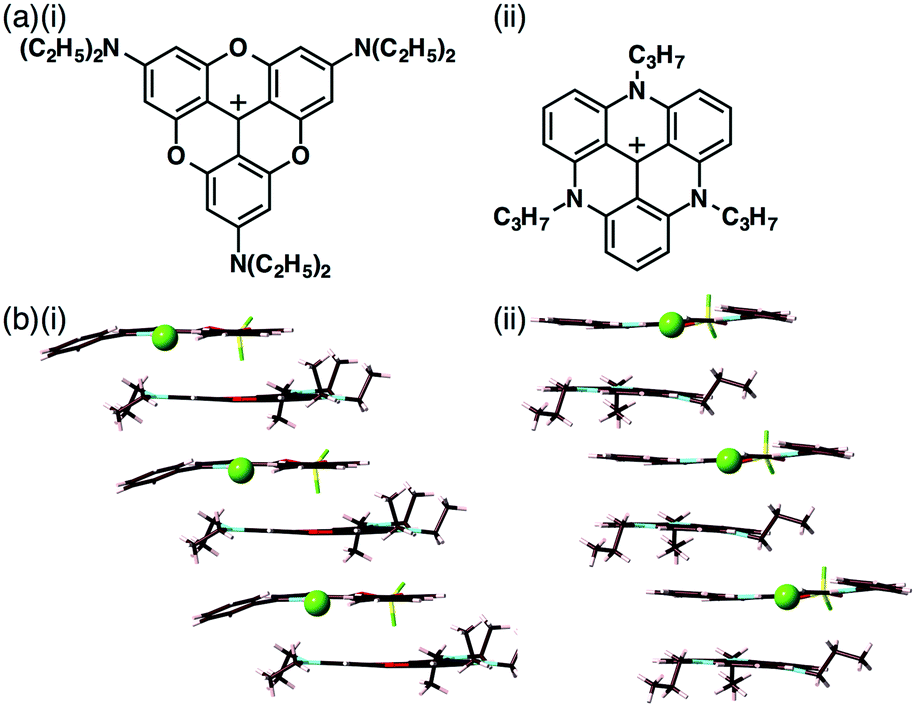
| Fig. 7 (a) SEM image of TATA+–2c·Cl− at 20 °C (inset: photograph of the gel under UV365 nm light), (b) PtII complexes as π-electronic trpyPt+ cations and (c) (i) aliphatic benzoate 2− obtained as a TBA+ ion pair and (ii) packing structure of TBA+–1b·2−. | ||
An advantage of forming dimension-control assemblies from π-electronic receptors, bound anions and coexisting cations is the variety of possible combinations, which in turn allows for tuning of the functionality of the materials. In the solution state, the receptor molecules bind anions through hydrogen bonding in equilibrium. Thus, it is important to investigate solid-state binding modes by single-crystal X-ray analysis. Since the first report of ion-pairing dimension-controlled assemblies in 2010,15 various ion pairs based on receptor–anion complexes have been prepared. Modified anions with π-electronic receptors provide tuneable π-electronic anionic species as the building components of ion-pairing assemblies.17 Control of the geometries and electronic states of counterion species for π-electronic anions (receptor–anion complexes) is also essential for achieving functional ion-pairing assemblies.18 Based on the modifications of diverse cationic species, the combination of Cl− complexes of dipyrrolyldiketone BF2 complexes and the positively charged π-ligand–metal complex trpyPt+ afforded fascinating ion-pairing assemblies (Fig. 7b).19 The charge-segregated assembly of trpyPt+–1c·Cl− exhibited hole and electron mobilities of 0.7 and 0.6 cm2 V−1 s−1, respectively, suggesting that ion-pairing dimension-controlled assemblies have characteristic properties such as electric conductivity. Furthermore, the complexation of 1b with 3,4,5-trihexadecyloxyphenyl-substituted benzoate 2− as a TBA+ salt formed lamellar mesophases derived from charge-by-charge assemblies (Fig. 7c).20 The ion-pairing material showed a moderately efficient charge-carrier transport ability due to the highly organized assembling state, as demonstrated in the value of 0.05 cm2 V−1 s−1 for TBA+–1b·2−.
Focusing on the components of ion-pairing assemblies, the introduction of a negatively charged site in anion receptors constructs self-assembled dimers with countercations.21 Carboxylate-appended derivatives of dipyrrolyldiketone BF2 complexes exhibited narcissistic self-sorting dimerization behaviours.21a Such effective self-associating dimeric structures can also be formed by the introduction of a cationic site onto anion-appended anion receptors. In fact, zwitterionic π-electronic systems 3a–c formed self-associating dimeric structures in the solution state (Fig. 8).21b Such self-assembled dimerized forms are stabilized by hydrogen bonding between the anionic moieties and interaction sites as well as by charge delocalization.
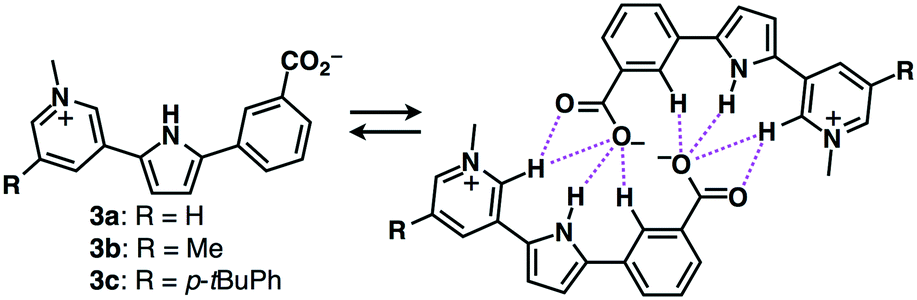 | ||
| Fig. 8 Zwitterionic π-electronic systems 3a–c. | ||
These examples demonstrate the possibility of diverse combinations of anion-responsive π-electronic molecules,22 modified anions and coexisting cations, which produce highly organized dimension-controlled ion-pairing assemblies. The introduction of functional units to the component(s) results in tuneable functionalities. On the basis of the concepts of ion-pairing dimension-controlled assemblies established by the above-mentioned examples, a wide range of ion-pairing materials have been prepared using π-electronic ions for potential applications such as ferroelectricity and electric conductivity.
2. Ion-pairing assemblies based on π-electronic anions with non-π-electronic cations
2-1. Ion-pairing assemblies of deprotonated species
Ion pairs consisting of receptor–ion complexes and counterions are remarkable as they lend themselves to precise tuning of their properties by modification of the building components as electronically neutral π-electronic receptors, guest ions and counterions. Because π-electronic receptors and ions are noncovalently bonded, receptor–ion complexes can possibly dissociate. Therefore, the fabrication of stable non-complexing ion pairs with ideal geometries and electronic states is desired for the formation of various assembling structures that exhibit tuneable properties and robustness.The deprotonation of an acid unit introduced into an appropriate π-electronic molecule affords anion-appended π-electronic species. In fact, this strategy has been widely applied for the formation of ionic liquid crystal materials.5 Sulfonates,23 carboxylates24 and hydroxides25 are often used as anionic components, whose countercations are exchangeable by ion metathesis. For example, BODIPY-sulfonate–ammonium ion pairs23 exhibited Colh mesophases. Notably, the important point is that the combination of appropriate anions and cations can provide characteristic properties. The precisely designed BODIPY-sulfonate–ammonium ion pair 4 (Fig. 9) showed an efficient interionic energy transfer from the BODIPY sulfonate dye to the ammonium-appended BODIPY dye both in solution and in the Colh mesophases.23b
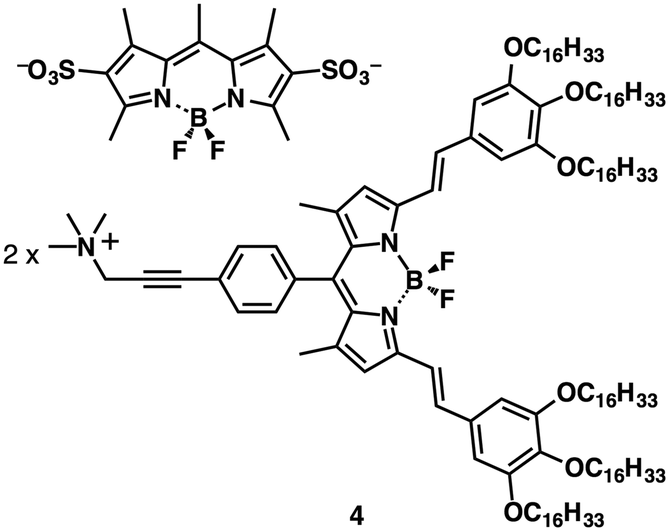 | ||
| Fig. 9 BODIPY-sulfonate–ammonium ion pair 4. | ||
Introduction of stimuli-responsive units to ionic components can produce stimuli-responsive ion-pairing assemblies (Fig. 10).26 For example, azobenzene carboxylates bearing aliphatic chains with TBA+, TBA+–5−, exhibited photo-induced crystal–crystal mesophase transitions via trans–cis isomerization (Fig. 10a).26a On the other hand, trans–cis isomerization of anion-complex ion pair TBA+–1a·5− was inhibited by energy transfer from the azobenzene unit to the receptor–anion complex as well as by the close packing around the azobenzene unit that reduced the free volume, providing a photo-stable assembly.26b Meanwhile, the ion pair of an azobenzene anion bearing alkyl chains on both sides, TBA+–6−, exhibited comparably improved photo-responsive properties in the anion-binding form TBA+–1a·6− in the bulk state (Fig. 10b and c).26c Interestingly, TBA+–1c·6− afforded a photo-responsive supramolecular octane gel, suggesting that these fascinating properties were realized by an appropriate combination of ion pairs.
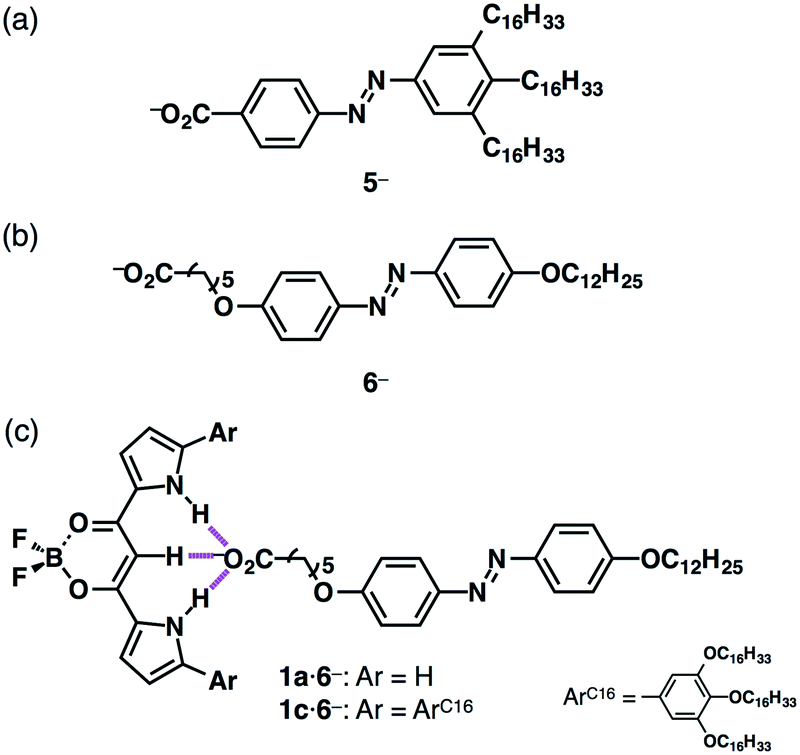 | ||
| Fig. 10 Azobenzene carboxylates with aliphatic chains: (a) 5− obtained as a TBA+ ion pair, (b) 6− obtained as a TBA+ ion pair and (c) receptor–carboxylate complexes 1a,c·6− obtained as TBA+ ion pairs. | ||
2-2. Deprotonation of dipyrrolyl π-systems: π-electronic anions stabilized by intramolecular hydrogen bonding
The design of the anion shapes is essential in order to achieve an anisotropic alignment of π-electronic ions. Maintaining the stabilities of deprotonated species is likewise an important factor. A strategy for stabilizing deprotonated anions is the use of intramolecular hydrogen bonding between the anionic components and proximally located hydrogen-bonding donors. Partial delocalization of negative charges also enhances the stability of π-electronic anions. On the basis of the above strategies, dipyrrolylnitrophenols 7a–c were designed as precursors of π-electronic anions (Fig. 11a).27 Their deprotonation by tetrabutylammonium hydroxide (TBAOH) delivered π-electronic anions 7a−–c− as TBA+ ion pairs via the inversion of pyrrole rings and intramolecular hydrogen bonding between the pyrrole NH and the anionic phenoxide oxygen. Density functional theory (DFT) calculations also revealed the delocalization of negative charges into the core phenoxide units with the electron-withdrawing nitro group. Various countercations can be introduced to form π-electronic ion pairs, which give rise to charge-by-charge assemblies in the crystal state.27a The single-crystal X-ray analysis of deprotonated species of π-extended TBA+–7b− revealed the formation of charge-by-charge assemblies with a superior anisotropic columnar orientation due to more efficient stacking (Fig. 11b). Meanwhile, deprotonated species bearing aliphatic chains 7c−–TBA+ formed a liquid crystal mesophase of Colh structure, as observed in the POM image (Fig. 11c).27b The deprotonation of a pyrrole NH of appropriately designed pyrrole-based π-electronic systems can provide corresponding anionic species that are accompanied by countercations.28,29 For example, deprotonation of a single NH of dipyrrolylquinoxalines (DPQs) 8a,b by TBAOH resulted in the formation of ion pairs of anionic DPQ− and TBA+ (Fig. 12a).29 TBA+–8a− exhibited a solid-state charge-by-charge columnar assembly (Fig. 12b), whereas the ion pair of aliphatic TBA+–8b− formed an octane gel (10 mg mL−1) based on fibril morphologies (Fig. 12c).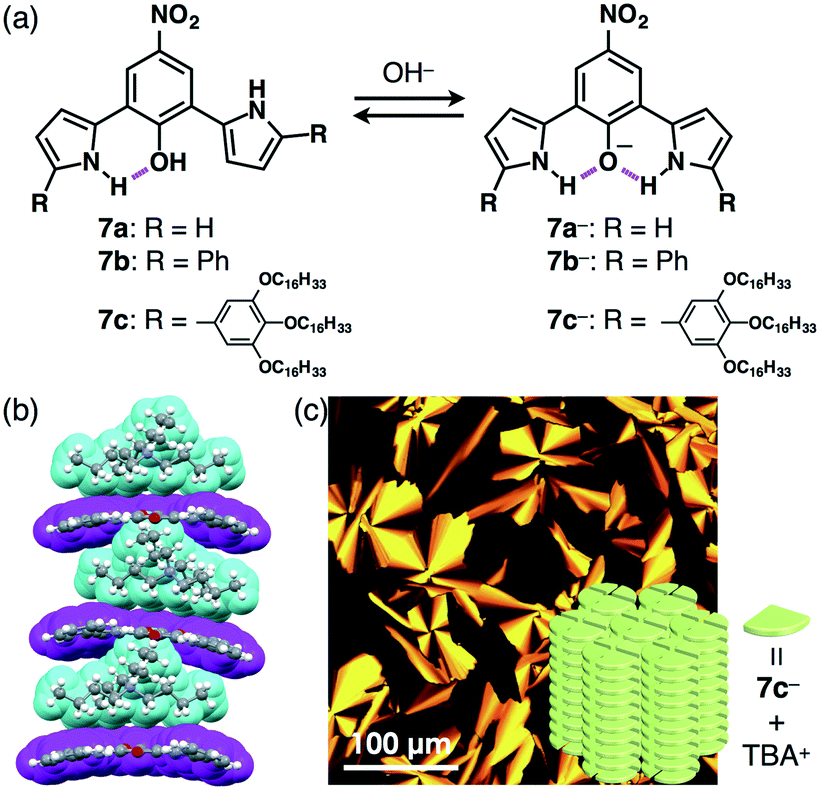 | ||
| Fig. 11 (a) Dipyrrolylnitrophenols 7a–c and formation of deprotonated species 7a−–c− obtained as TBA+ ion pairs, (b) single-crystal X-ray structure of TBA+–7b− (cyan: TBA+, magenta: 7b−) and (c) POM image of the mesophase of TBA+–7c− and the packing model. | ||
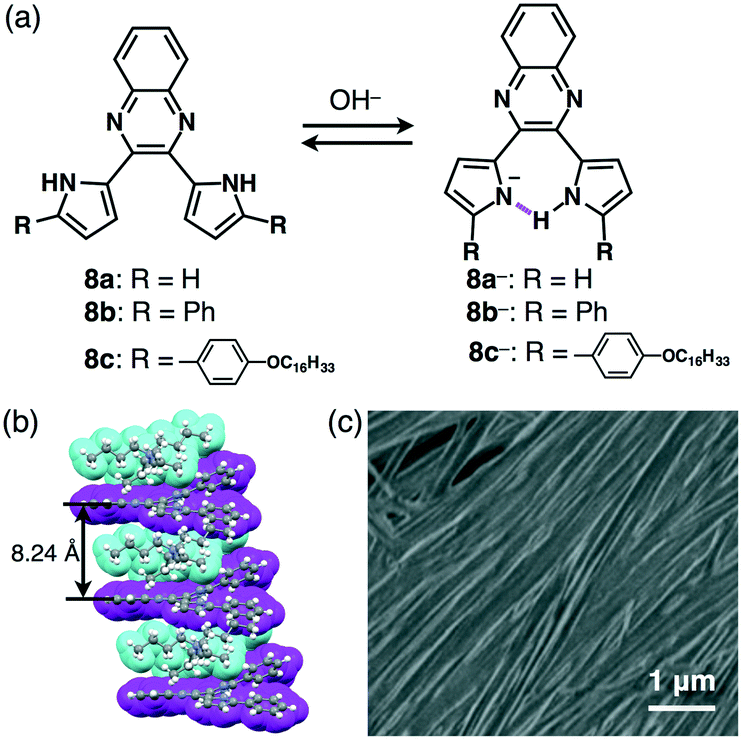 | ||
| Fig. 12 (a) Dipyrrolylquinoxalines 8a–c and formation of deprotonated species 8a−–c− obtained as TBA+ ion pairs, (b) single-crystal X-ray structure of TBA+–8b− (cyan: TBA+, magenta: 8b−) and (c) SEM image of TBA+–8c− as the octane xerogel (10 mg mL−1). | ||
2-3. Deprotonation of hydroxy-substituted porphyrins for π-electronic anions
Incorporation of π-electronic anions with an extensive π-conjugated system is meaningful for electronic materials. Larger π-electronic molecules with an acid unit can provide stable anionic species owing to more effective delocalization of negative charges within the core π-electronic unit.30 The deprotonation of meso-hydroxy-substituted porphyrin 9 by treatment with TBAOH provided the anionic species 9− (Fig. 13) as indicated by UV/vis absorption and 1H NMR spectral changes.30a The single-crystal X-ray analysis of TBA+–9− revealed the packing structure consisting of the alternately stacked column of 9− and TBA+. Interestingly, the PdII complex 9′ coordinated at meso-oxygen and β-carbon was obtained from 9 by treatment with Pd(OAc)2, rac-BINAP and Cs2CO3 (Fig. 14a).30b The coupling reaction of 9′ with PhMgBr afforded β-phenyl-substituted 10a. Subsequently, according to similar procedures, 10a was converted to β-diphenyl-substituted 10bvia the PdII complex 10a′. Deprotonation of the OH unit of 10a,b by TBAOH provided the π-electronic anions 10a−,b− (Fig. 14b(i)). The ion pairs TBA+–10a−,b− formed charge-by-charge assemblies in the solid state, as elucidated by single-crystal X-ray analysis (Fig. 14b(ii)). The introduction of aryl moieties capable of more efficient hydrogen bonding would provide more stabilized anionic π-electronic species.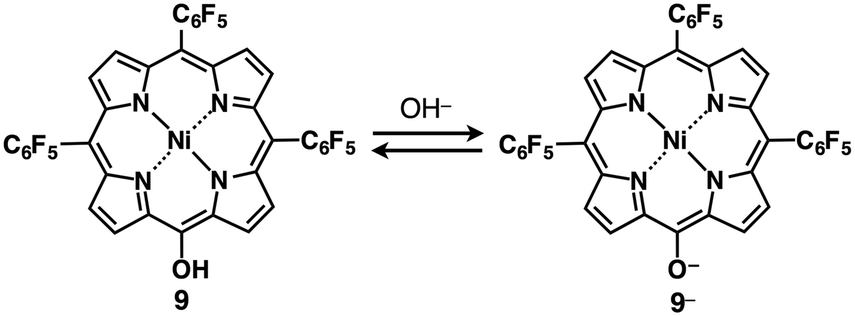 | ||
| Fig. 13 meso-Hydroxy-substituted porphyrin 9 and formation of deprotonated species 9− obtained as a TBA+ ion pair. | ||
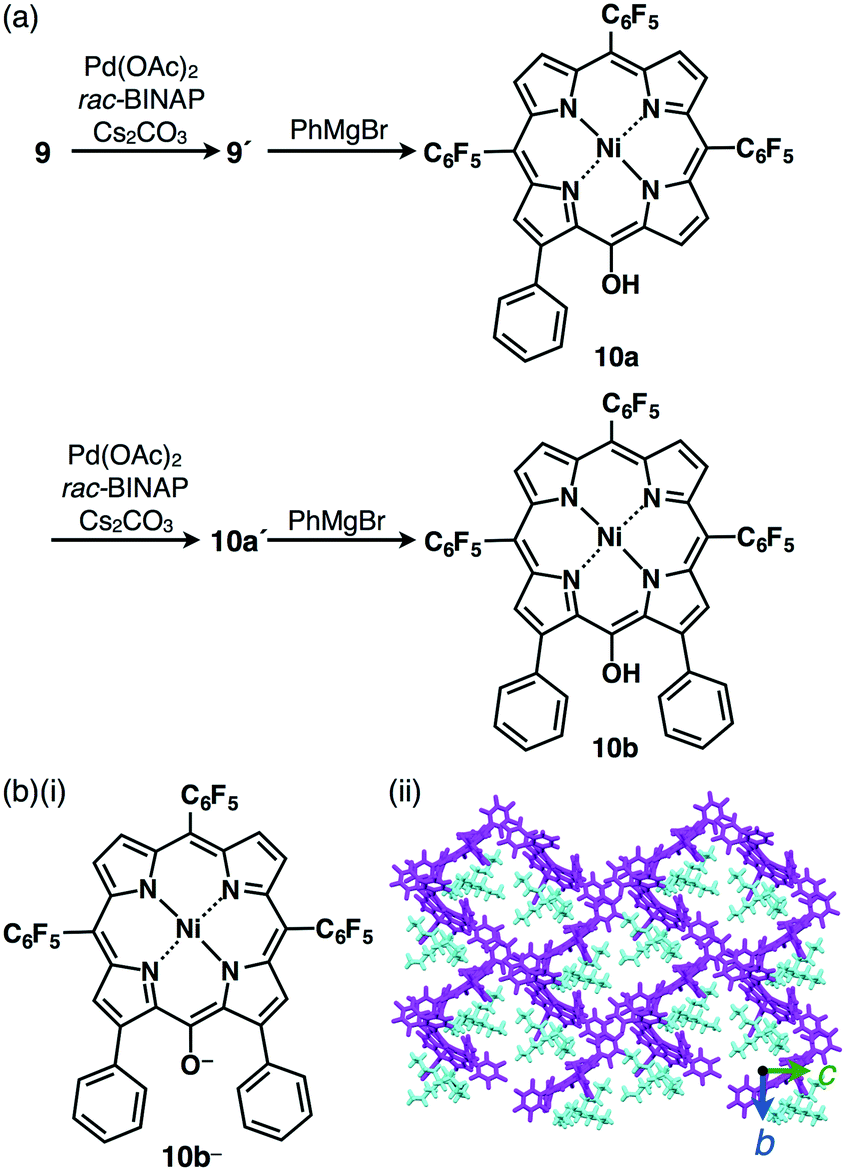 | ||
| Fig. 14 (a) Synthesis of β-phenyl-substituted porphyrins 10a,b and (b) (i) deprotonated species 10b− obtained as a TBA+ ion pair and (ii) single-crystal X-ray structure of TBA+–10b− (cyan: TBA+, magenta: 10b−). | ||
2-4. Ion-pairing assemblies based on genuine π-electronic anions
π-Electronic anions are more difficult to prepare than cations, because anions suffer from oxidation and electrophilic attacks. Although π-electronic anions have problematic stability, there are some examples of genuine π-electronic anions. For example, Kuhn's anion, reported by Kuhn and Rewicki in 1967, has a large π-conjugated core unit, which enables the delocalization of negative charges.31 Thus far, several ion pairs of Kuhn's anion have been prepared,32 but no dimension-controlled assemblies have been reported. On the other hand, π-electronic systems, such as cyclopentadienides (Cp−), with electron-withdrawing moieties stabilize π-electronic anions by their aromaticity and can thus be used as building units of ion-pairing assemblies. White et al. prepared an ion pair consisting of penta(methoxycarbonyl)-substituted Cp− (PMCp−) (Fig. 15a) and a tetramethylammonium cation, forming a charge-by-charge assembly in the solid state.33 Similarly, cyano substituents decrease the electron density of π-electronic anions, as seen in pentacyanocyclopentadienide (PCCp−) (Fig. 15a).34,35 Based on the ion-exchange strategy, a Na+ salt of PCCp− and Cl− salts of desired cations were mixed under appropriate conditions, affording the corresponding ion pairs of PCCp−.36 Cations with bulky geometries afforded charge-segregated assemblies. For example, the ion pairs of PCCp− with a triethylammonium cation by Wood and Wright et al.37 and with butyltrimethylammonium (C4H9(CH3)3N+) and TPA+ provided charge-segregated assemblies based on the stacking columnar structures of PCCp− in the crystal state (Fig. 15b).36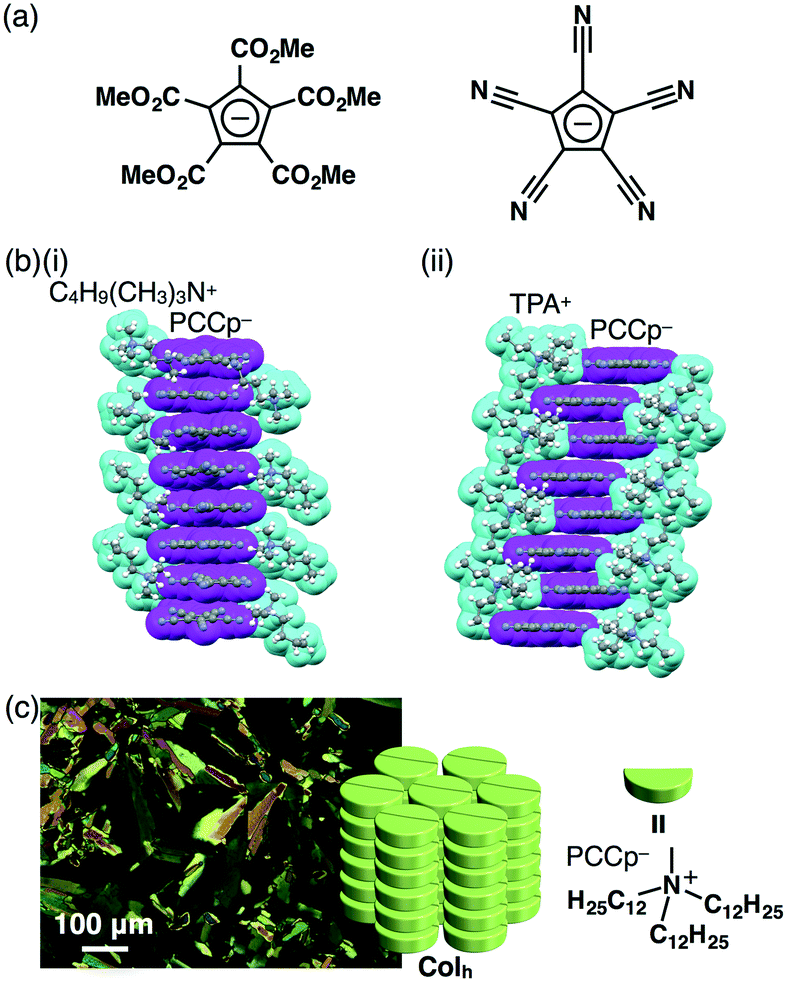 | ||
| Fig. 15 (a) PMCp− (left) and PCCp− (right), (b) single-crystal X-ray structures of (i) C4H9(CH3)3N+–PCCp− and (ii) TPA+–PCCp− (cyan: cation, magenta: PCCp−) and (c) POM and packing model of (C12H25)3CH3N+–PCCp− at 70 °C upon cooling. | ||
Charge-segregated dimension-controlled assemblies are fascinating, because the alignment of identically charged π-electronic ions shows effective charge-carrier transport properties. Accordingly, ion pairs consisting of PCCp− with aliphatic cations such as dimethyldioctadecylammonium ((C18H37)2(CH3)2N+) and tridodecylmethylammonium ((C12H25)3CH3N+) (Fig. 15c) have been prepared.36 The ion pairs (C18H37)2(CH3)2N+–PCCp− and (C12H25)3CH3N+–PCCp− formed smectic B (SmB) and Colh phases, respectively, in their mesophases. The characteristic diffraction peaks at ∼0.60 and ∼0.36 nm observed in the XRD corresponded to the repeating distances of the ammonium cations and PCCp−, respectively (Fig. 15c). These observations strongly suggested the formation of charge-segregated assemblies in the mesophases. Furthermore, the stacking columnar assembly of (C12H25)3CH3N+–PCCp− showed hole-transporting properties (0.4 cm2 V−1 s−1) in the film prepared by drop-casting of a CHCl3 solution, as revealed by the FI-TRMC method.
3. Ion-pairing assemblies based on π-electronic cations with non-π-electronic anions
3-1. π-Electronic cations for ion-pairing assemblies
Due to the stability of π-electronic cations compared to π-electronic anions, a myriad of methods exist for the preparation of cationic systems. The fascinating electronic and optical properties of dyes and fluorescence materials based on intriguing geometrical features of π-electronic cations have motivated broad research fields. Here, π-electronic cations are classified as genuine positively charged π-conjugated systems and cationic metal complexes of π-electronic ligands which do not compensate the whole positive charge of the complexed metal cations. It is imperative to design the geometries of π-electronic cations such that they enable the delocalization of electrons and the induction of π–π stacking with identically or oppositely charged π-electronic ions. To date, several types of π-electronic cationic species are in use as building units of ion-pairing assemblies.3-2. Genuine π-electronic cations
Stable genuine π-electronic cations have been reported involving fascinating synthetic routes. One of the original carbocations is the cycloheptatrienyl (Ch+; tropylium) cation, a six-π-electron aromatic system (Fig. 16a). Ch+ has been investigated as a component of bioactive species and an electron-accepting unit. In terms of extended π-systems useful for further fine tuning of electronic states and rigid planar geometries, Müllen et al. have investigated π-electronic cationic systems by incorporating heteroatoms into polycyclic aromatic hydrocarbons (PAHs).38 In 2009, they reported on a positively charged 9-phenylbenzo-[1,2]quinolizino[3,4,5,6-fed]phenanthridinylium (PQP+) cation (Fig. 16b) that exhibited π–π stacking structures.38b Although clear mesophases were not observed, PQP+-based ion pairs exhibited the fascinating anisotropic arrangement of PQP+ depending on the counteranions.38a Moreover, alkylsulfonate counteranions induced morphological control of sub-micrometer-scale aggregates.38c Furthermore, in 2000, Laursen et al. reported the synthesis of TATA+ salts, which formed cation-stacking columnar structures.14,39 The introduction of alkyl chains on the edges of TATA+ showed anion-dependent morphological changes caused by the specific packing structures of TATA+.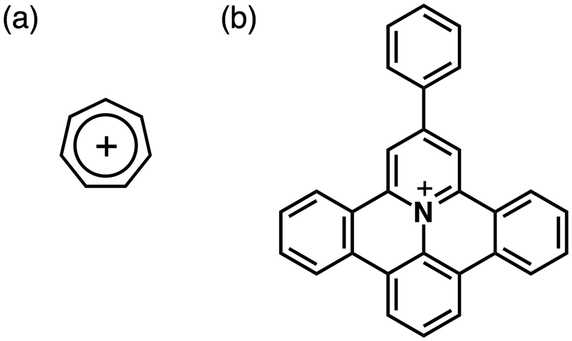 | ||
| Fig. 16 π-Electronic cations: (a) Ch+ and (b) PQP+. | ||
3-3. Metal complexes of π-electronic ligands for π-electronic cations
An effective way to prepare planar π-electronic cationic species is to make use of the complexation of appropriate π-electronic ligands and metal cations.40–43 Square-planar geometries of d8 metal complexes are crucial for the formation of stacking-based assemblies. Thus far, various metal-coordinated π-electronic cations have been investigated such as the complexes of PtII and AuIII (d8 states for both) with acyclic π-electronic ligands.40,41 PtII complexes of terpyridines (Fig. 17a) and AuIII complexes of phenylbipyridines (Fig. 17b) afford planar monovalent π-electronic systems, which can be used as the building units of ion-pairing assemblies. In fact, Yam et al. reported on luminescent terpyridyl–PtII complexes bearing a diynyl moiety as OTf− ion pairs.44 The terpyridine and diynyl ligands coordinated to PtII in a square-planar geometry, which exhibited linear-chain packing and dimeric structures. Furthermore, Che et al. have investigated phenylbipyridine–AuIII complexes possessing an alkynyl unit as PF6− ion pairs.45 In the solid state, electrostatic interactions as well as metal–metal and π–π interactions are important for controlling the assembling modes of the ion-pairing assemblies.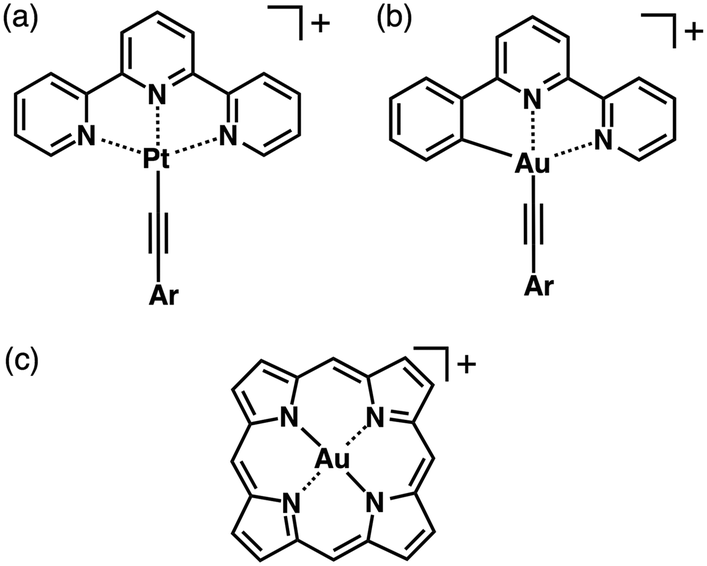 | ||
| Fig. 17 (a) Terpyridine–PtII, (b) phenylbipyridine–AuIII and (c) porphyrin–AuIII complexes as π-electronic cations. | ||
3-4. Porphyrin–AuIII complexes as planar π-electronic cations
Regarding assemblies based on stacking of π-electronic units, an extended rigid planarity is crucial. An effective way to achieve this is to use the metal complexes of large π-conjugated systems. Porphyrin is a dianionic tetradentate π-electronic ligand, which forms complexes with various metal cations. Its complexation with trivalent metal cations afforded positively charged complexes, which resulted in the formation of planar cationic species. An important strategy is the use of AuIII, which provides planar porphyrin–AuIII complexes (Fig. 17c) as stable π-electronic cations that require no axial ligand, resulting in the formation of ion pairs with various anions. The counteranions of porphyrin–AuIII complexes can be exchanged by ion metathesis with metal salts of target anions.The electron deficiency of porphyrin–AuIII complexes as cations has been useful in many research fields. The preparation of a porphyrin–AuIII complex was first reported in 1969.46 Since then, porphyrin–AuIII complexes have been used as anticancer agents,47 electron-accepting units in donor–acceptor systems48 and also precursors for porphyrin–AuII complexes.49 Although there are a few number of studies on single-crystal structures and irregularly shaped nanoscale aggregates of porphyrin–AuIII ion pairs,50 their dimension-controlled assemblies as soft materials have not been reported. Assemblies of electronically neutral porphyrins have been investigated extensively, whereas dimension-controlled assemblies of charged porphyrin–AuIII ion pairs have not been developed. Through peripheral modifications of porphyrins, the complexation with AuIII can afford a variety of π-electronic ion pairs. Therefore, there are significant advantages in using porphyrin–AuIII complexes as components of stacking assemblies especially in the case of ion pairs with π-electronic counteranions because of the potential formation of assemblies based on collaborative π–π stacking and electrostatic interactions.
Based on the aforementioned background, we prepared porphyrin–AuIII complex ion pairs with appropriate peripheral substituents to control the electronic states as well as the bulk-state ion-pairing assemblies.51 In 1977, Tulinsky et al. disclosed the crystal structure of a tetraphenylporphyrin (TPP) AuIII complex as a Cl− ion pair 11+–Cl− (Fig. 18a).52 In their initial report, the X-ray structure of the Cl− ion pair was represented by a line depicting the coordination of Cl− to the porphyrin–AuIII centre. In order to prove the nature of the Cl− ion pair, we also prepared 11+–Cl− in order to examine whether Cl− acts as an anion coordinated to AuIII or as a proximally located anion without coordination bonding. The single-crystal X-ray analysis of 11+–Cl− elucidated the formation of two crystal pseudo-polymorphs (type A and B). The type A polymorph showed a partially charge-segregated assembly with a single co-crystallized CHCl3 molecule (Fig. 18b). On the other hand, the type B polymorph exhibited a columnar structure based on a charge-by-charge assembly of 11+ and Cl− associated with four co-crystallized CHCl3 molecules. It is noteworthy that the proximal Au⋯Cl− distances in the type A and B polymorphs are 3.00 and 3.12 Å, respectively, being comparable to the sum of the ionic radii of Au3+ and Cl− (3.18 Å), suggesting the formation of contact ion pairs. Importantly, the line passing through both Au and Cl has angles of 76.9° and 80.2° to the porphyrin plane for type A and B structures, respectively. It should be noted that these angles are not 90°, indicating that the Cl− is not coordinated to the core AuIII but is located proximally around the porphyrin–AuIII complex, as a result of electrostatic interaction.51
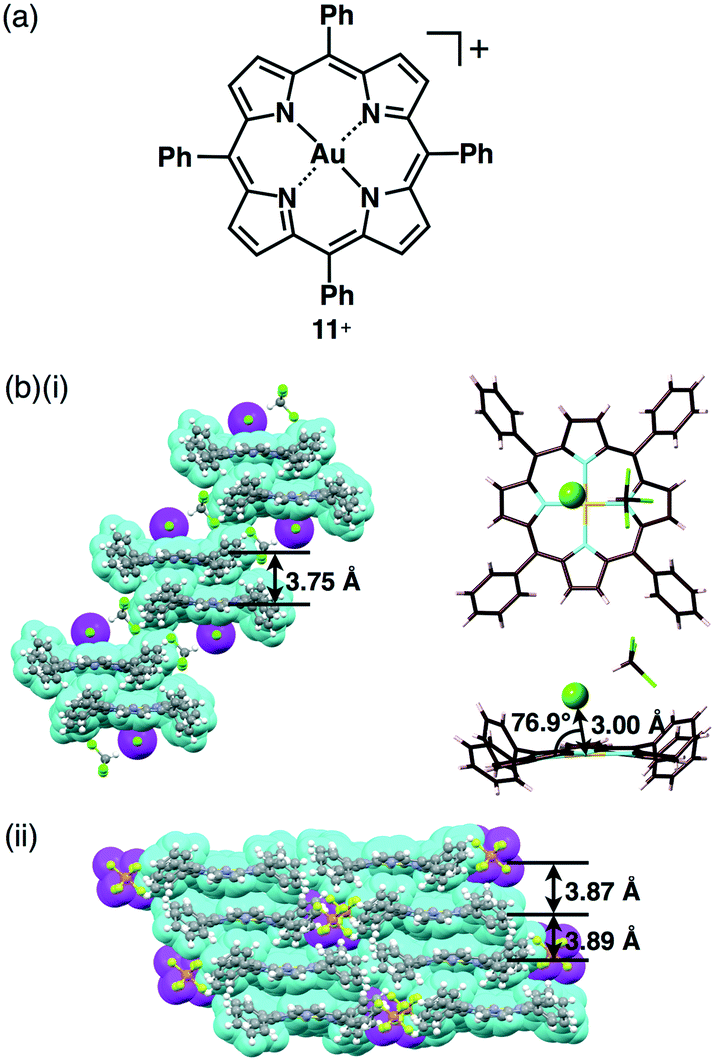 | ||
| Fig. 18 (a) Porphyrin–AuIII complex 11+ obtained as Cl−, BF4− and PF6− ion pairs and (b) single-crystal X-ray structures of (i) 11+–Cl− (type A) (packing (left) and ion pair structure (right)) and (ii) 11+–BF4− (cyan: 11+, magenta: anion). | ||
The counteranion of porphyrin–AuIII can be exchanged by ion metathesis with the metal salts of target anions. In some cases, starting from the Cl− ion pair, silver (Ag+) salts of the anions were used for anion exchange by removing AgCl as an insoluble salt. Ion metathesis also provided 11+–X− (X− = BF4− and PF6−) (Fig. 18a). In contrast to 11+–Cl−, ion pairs with bulky anions, 11+–BF4− and 11+–PF6−, formed columnar structures of charge-segregated assemblies with separately stacked 11+, with distances of 3.7–3.9 Å (Fig. 18c).51
3-5. Effect of peripheral modifications for electron-deficient π-electronic cations
An advantage for using porphyrins as scaffolds is their potential for modulation of electronic states by the introduction of peripheral electron-donating and withdrawing substituents. The pentafluorophenyl (C6F5) unit is considered an electron-withdrawing group in comparison to phenyl units and is often used to stabilize porphyrin core π-electronic systems. AuIII complexation of meso-C6F5-substituted porphyrins has been successfully achieved under reaction conditions that utilize HAuCl4 and AgOTf, as reported by Zhang and Zhang et al.53 Recently, we prepared ion pairs of porphyrin–AuIII complexes 12a+–c+, which were partially or totally substituted with C6F5 groups at the meso positions (Fig. 19a), in order to control the electronic states and resulting ion-pairing assemblies.54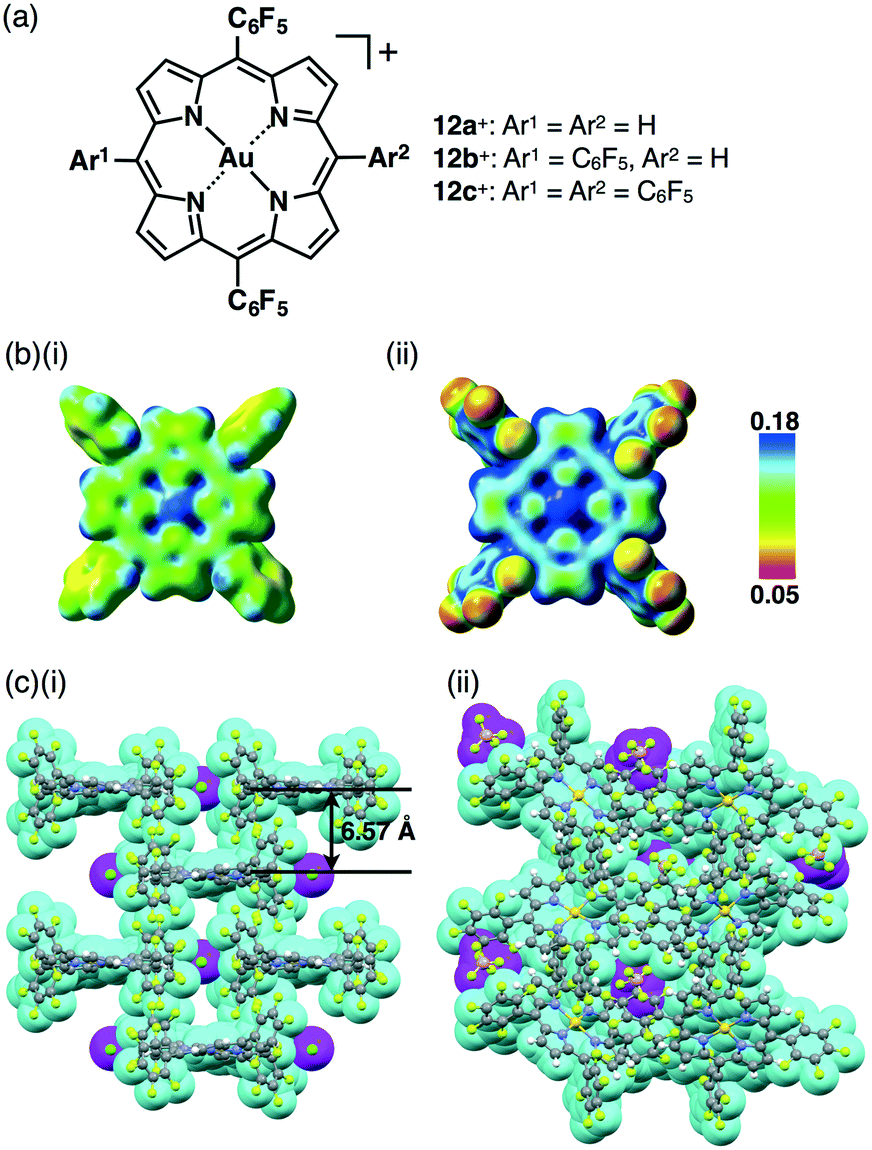 | ||
| Fig. 19 (a) Porphyrin–AuIII complexes 12a+–c+ obtained as OTf−, Cl−, BF4− and PF6− ion pairs, (b) electrostatic potential (ESP) mapped on the electron density isosurface (δ = 0.01) for (i) 11+ and (ii) 12c+ and (c) single-crystal X-ray structures of (i) 12c+–Cl− and (ii) 12c+–BF4− (cyan: 12c+, magenta: anion). | ||
The electron-deficient character of C6F5-substituted porphyrin–AuIII complexes was confirmed by electrostatic potential (ESP) diagrams, suggesting that the electron deficiency of the porphyrin core increases with the introduction of C6F5 units (Fig. 19b). The single-crystal X-ray analysis of the ion pairs revealed valuable insight into the ion-pairing assemblies with substituent dependencies. For example, tetra-C6F5-substituted 12c+–Cl− formed a columnar structure based on a charge-by-charge assembly for 12c+ and Cl− associated with four co-crystalized CHCl3 molecules with a distance of 6.57 Å between two 12c+ planes (Fig. 19c(i)). On the other hand, the ion pair with a bulky anion, 12c+–BF4−, formed a columnar structure based on a charge-segregated assembly (Fig. 19c(ii)), which was stabilized by hydrogen-bonding and F–π55 interactions. On the other hand, in the case of 12b+–PF6−, stacking dimers of 12c+ were formed in the assembly in order to cancel their dipole moments. This type of electron-withdrawing character of peripheral substituents is crucial for tuning interionic interactions.54
3-6. Aliphatic porphyrin–AuIII complexes for dimension-controlled assemblies
Introduction of aliphatic long alkyl chains into an appropriate π-electronic unit affords soft materials comprising the stacking core π-units supported by van der Waals interactions of alkyl chains. This can be applied to electronically neutral porphyrin systems; meso and pyrrole-β positions can be modified. The core modification with AuIII complexation affords cationic species, which can exist as an ordered arrangement of core units resulting from electrostatic interactions.As modified derivatives, meso-trialkoxyphenyl-substituted porphyrins as free bases were reported as liquid states, indicating a less ordered molecular arrangement.56 The AuIII complexes of alkoxy-substituted porphyrins 13n+–X− (n = 8, 12, 16 and 20; X− = Cl−, BF4− and PF6−) were successfully prepared with KAuCl4 and NaOAc in refluxing AcOH/CH2ClCH2Cl and further counteranion exchanges for BF4− and PF6− ion pairs (Fig. 20a).51 DSC and POM revealed mesophase information, for 13n+–Cl− (n = 12, 16 and 20), 13n+–BF4− (n = 16 and 20) and 13n+–PF6− (n = 12) that formed mesophases, in contrast to other ion pairs showing liquid or crystal states. Synchrotron XRD analysis revealed characteristic anion-dependent ordered assemblies. Interestingly, 13n+–Cl− (n = 12, 16 and 20) had Colh structures in the mesophases; for example, 1316+–Cl− showed a Colh structure at 100 °C (cooling) with a = 3.73, c = 0.36 nm and Z = 1 (ρ = 1.44) (Fig. 20b). The peak at 0.36 nm indicated the π–π stacking distance of porphyrin–AuIII complex cations based on a charge-segregated assembly. The formation of a charge-segregated assembly was also observed for ion pairs of bulky anions 1516+–BF4− and 1520+–BF4−, which showed Colh structures at 45 °C (heating) and 52 °C (cooling), respectively (Fig. 20c). The broad peaks at 0.42 nm, which were observed in both 15n+–BF4− (n = 16 and 20), arose from the less ordered stacking arrangement of porphyrin–AuIII complex cations. This can be explained by HSAB theory: soft anions tend to interact with the porphyrin–AuIII complexes as soft cations. Consequently, bulky counteranions hinder the ordered arrangement of ion pairs.
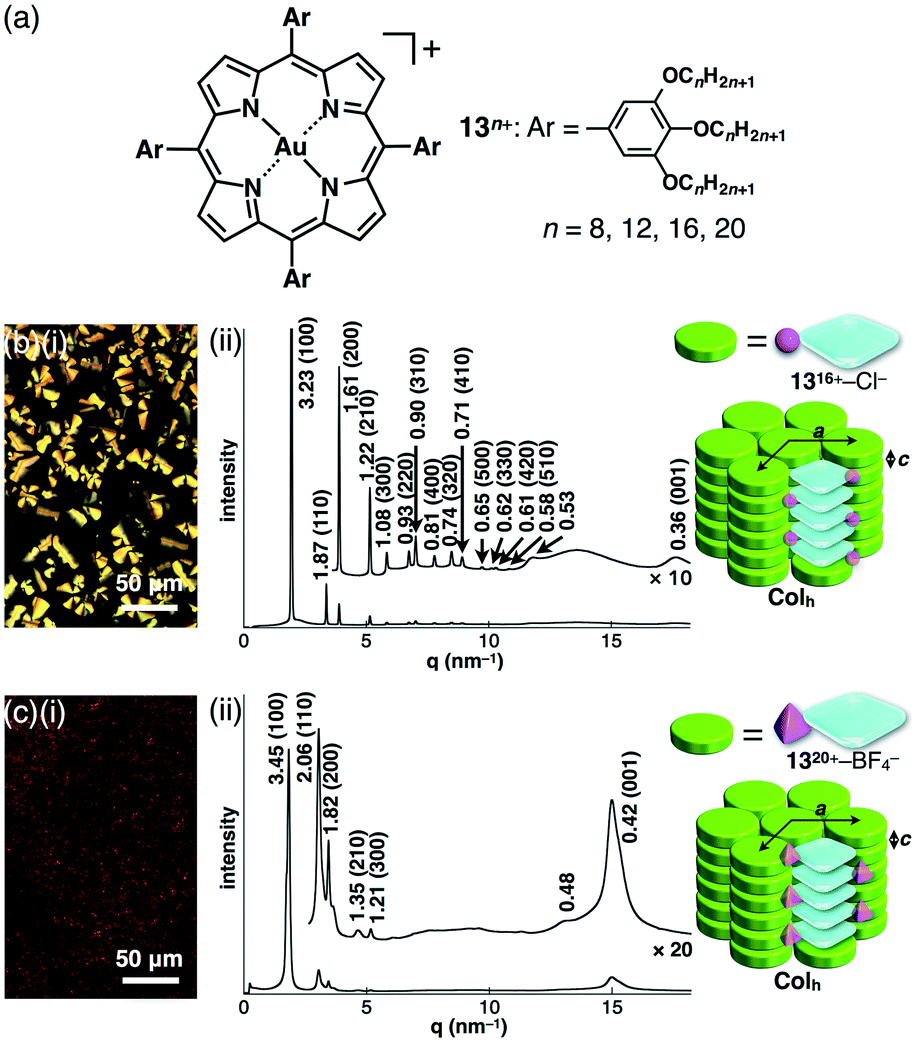 | ||
| Fig. 20 (a) meso-Trialkoxyphenyl-substituted porphyrin–AuIII complexes 13n+ (n = 8, 12, 16 and 20) obtained as Cl−, BF4− and PF6− ion pairs and mesophase properties: (i) POM images and (ii) XRD patterns and proposed assembled models of (b) 1316+–Cl− at (i and ii) 100 °C and (c) 1320+–BF4− at (i) 35 °C and (ii) 52 °C, upon cooling from Iso. | ||
4. Ion-pairing assemblies based on π-electronic cations and π-electronic anions (genuine π-electronic ion pairs)
4-1. π-Electronic ion pairs comprising genuine π-electronic ions
Ion pairs comprising π-electronic ions (cations and anions) can possess fascinating properties due to their geometrical features and characteristic electronic and optical properties. For example, various donor–acceptor systems have been investigated using π-electronic ion pairs consisting of PMCp−, as seen in the solution state as well as in the solid state.33c Ion pairs comprising PMCp− and π-electronic cations, such as Ch+, 2,4,6-trimethylpyrylium and N-methylpyridinium, give rise to charge-transfer complexes.33aPCCp− is also an effective building unit for ion-pairing assemblies in combination with various cations for the formation of a variety of ordered ion-pairing assemblies.34,35 We prepared a series of ion pairs of PCCp− (see also section 2) (Fig. 21a) via an ion-exchange method starting from Na+–PCCp−. For example, the single-crystal X-ray analysis of Ch+–PCCp− (Fig. 21a(i)) revealed a charge-by-charge stacking assembly mode, fabricated through π–π and electrostatic interactions, with a distance of ca. 3.4 Å (Fig. 21b(i)). We additionally reported on solid-state assembled structures of PCCp− ion pairs with TATA+ (Fig. 21a(ii)) and crystal violet cations.36 In the solid state, π-electronic cations and anions in TATA+–PCCp− alternately stack, providing a two-by-two charge-by-charge assembly (Fig. 21b(ii)). The charge-by-charge columnar structure is stabilized by electrostatic interactions as well as the stacking of π-electronic ions.
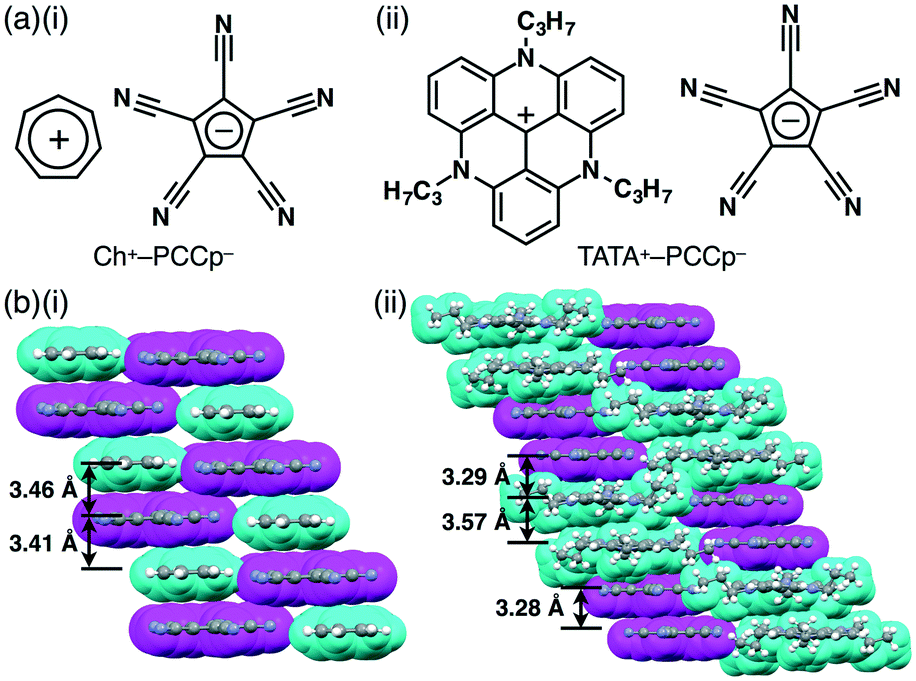 | ||
| Fig. 21 π-Electronic ion pairs (a) (i) Ch+–PCCp− and (ii) TATA+–PCCp− and (b) representative packing modes (represented by space-filling models) of the single-crystal X-ray structures: (i) Ch+–PCCp− and (ii) TATA+–PCCp− (cyan: cation, magenta: PCCp−). | ||
4-2. π-Electronic ion pairs comprising porphyrin–AuIII complexes
Ion metathesis using appropriate conditions (solvents, temperatures, counterions, etc.) affords a range of ion pairs. It is important to purify ion pairs prior to their use in materials for further applications. Purification processes, such as silica gel chromatography, ion-exchange chromatography and precipitation (crystallization), are indispensable for obtaining pure and stoichiometrically appropriate ion pairs. In particular, solvent conditions, the choice of which depends on the solubilities of the starting ion pairs and formed π-electronic ion pairs, are essential for purification. By employing ion metathesis, we have prepared various π-electronic ion pairs.Treating a Cl− salt of 11+ with Na+–PCCp− afforded exchanged ion pairs 11+–PCCp− and NaCl with small amounts of unexchanged starting ion pairs (Fig. 22a).51 These starting materials tend to remain and need to be carefully removed. In this case, silica gel column chromatography was used to purify the target ion pairs. Interestingly, ion pairs possessing small (hard) counteranions of 11+ are more polar in character as evidenced by smaller Rf values on TLC. 11+–PCCp− formed a solid-state charge-by-charge assembly (Fig. 22a) in contrast to 11+–BF4− and 11+–PF6−, exhibiting charge-segregated assemblies (Fig. 22b(i)). A columnar structure comprising alternately stacked 11+ and PCCp− was observed with stacking distances of 3.37 and 3.40 Å between the porphyrin core plane and PCCp−. The charge-by-charge column is stabilized by π–π stacking and electrostatic interactions of oppositely charged species.51,57
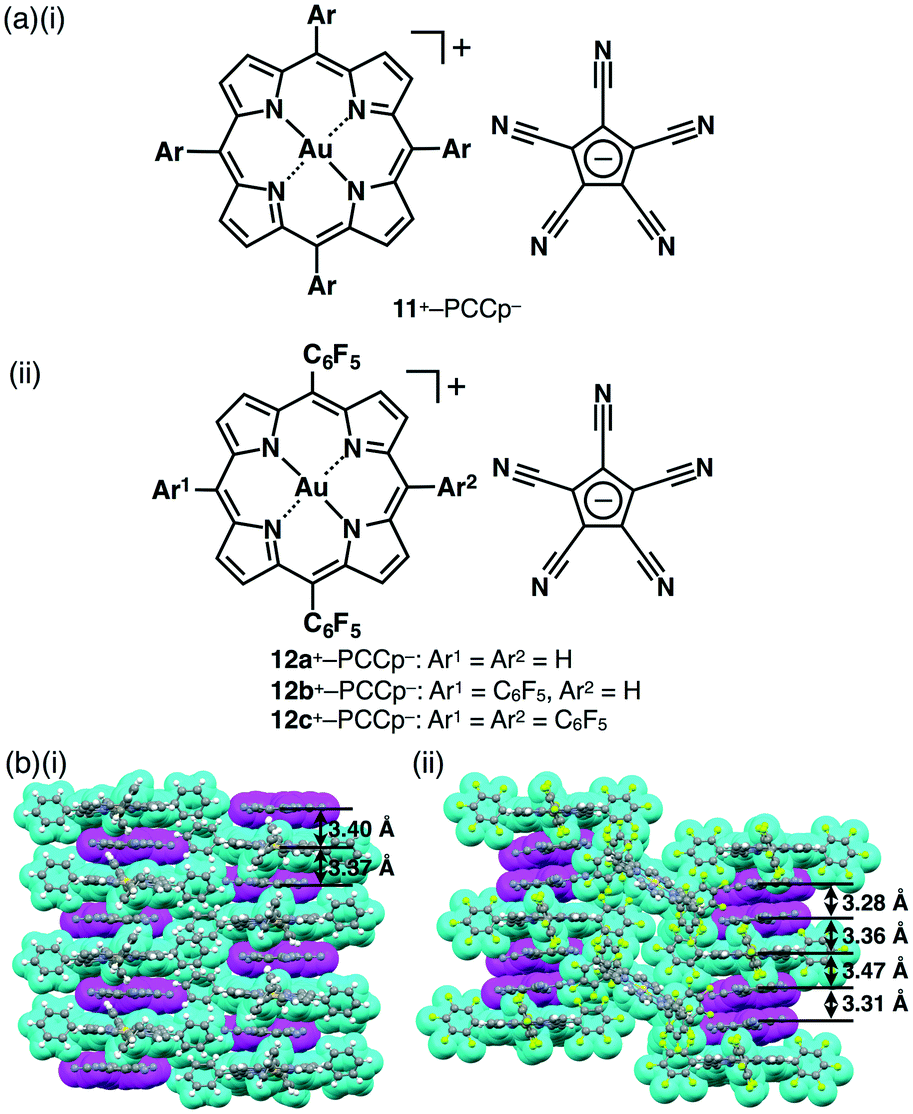 | ||
| Fig. 22 π-Electronic ion pairs (a)(i) 11+–PCCp− and (ii) 12a–c+–PCCp− and (b) representative packing modes (represented by space-filling models) of the single-crystal X-ray structures: (i) 11+–PCCp− and (ii) 12c+–PCCp− (cyan: cation, magenta: PCCp−). | ||
As shown in section 3, porphyrin–AuIII complexes with partially or totally substituted electron-withdrawing groups exhibited tuneable electronic states at the core π-electronic units. Combination with appropriate π-electronic anions promotes electron-transfer behaviour and the development of further materials. Therefore, we extended our research to the combination of a variety of ion pairs comprising modified porphyrin–AuIII complexes and π-electronic anions. For example, π-electronic ion pair 12c+–PCCp− (Fig. 22a(ii)) underwent the formation of a charge-by-charge assembly of 12c+ and PCCp− (1:2) along with a columnar assembly of 12c+. The PCCp− dimers with stacking distances of 3.28 and 3.31 Å were stabilized by hydrogen-bonding and π–π interactions (Fig. 22b(ii)).54
4-3. π-Electronic ion pair comprising positively and negatively charged porphyrin–metal complexes
Counteranions of π-electronic porphyrin–AuIII cations can be exchanged by the introduction of deprotonated species of extended π-electronic units with an appropriate acid moiety, such as hydroxy and carboxy units (section 2). Deprotonation of NiII porphyrin 9 by NaOH affords Na+–9−. π-Electronic ion pair 11+–9− (Fig. 23a) can be prepared from the ion metathesis of 11+–Cl− and Na+–9− (Fig. 13).30a The ion-pairing formation of 11+ and 9− was revealed by the 1H NMR signal shifts, based on the effects of (i) the aromatic ring current and (ii) proximally located charges. Consequently, the 1H NMR signals of 11+–9− (1.0 × 10−3 M) were shifted upfield compared to those of 11+–Cl− and TBA+–9−. This observation suggested the influence of the aromatic ring current derived from the interaction between these cations and anions in the solution state.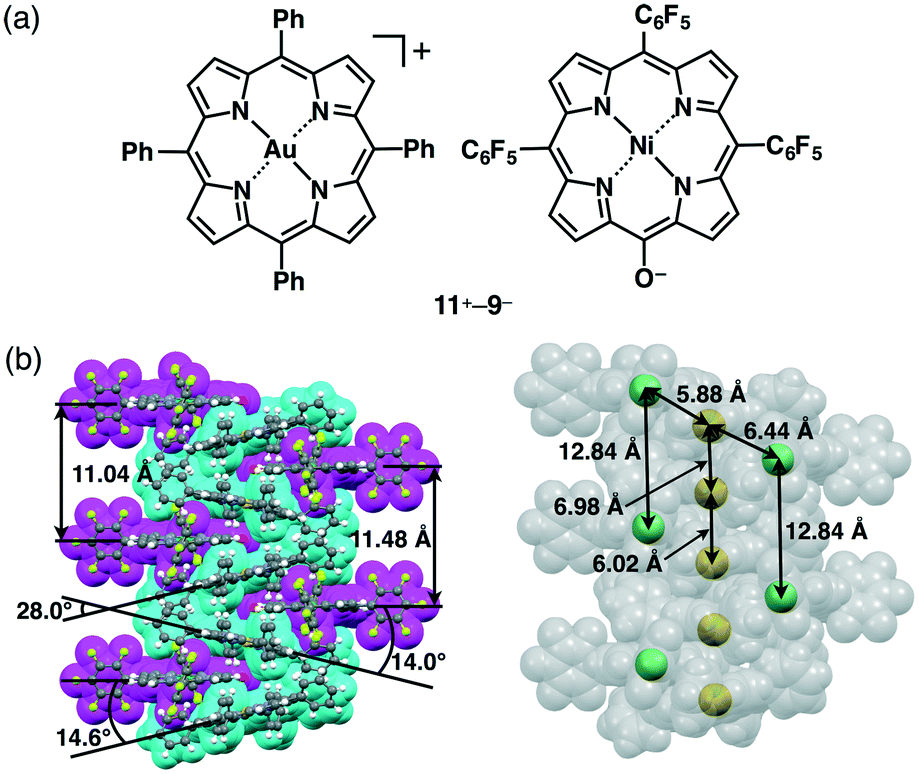 | ||
| Fig. 23 (a) π-Electronic ion pair 11+–9− and (b) single-crystal X-ray structure of 11+–9− (cyan: 11+, magenta: 9−). | ||
The single-crystal X-ray analysis of 11+–9− revealed a charge-by-charge columnar assembly with (C–)O⋯Au distances of 3.03 and 3.55 Å and the dihedral angles between the mean planes of 11+ and 9− were 14.0° and 14.6° (Fig. 23b), suggesting closely located oppositely charged ions. Interestingly, the metal ions, Au3+ and Ni2+, were arranged in a zigzag fashion with Au⋯Ni distances of 5.88 and 6.44 Å. The distances for identical metal ions Au3+ and Ni2+ in the columnar direction were 6.02/6.98 and 12.84 Å, respectively. The heterometals were arranged through the formation of ion-pairing assemblies comprising appropriately designed positively and negatively charged porphyrin–metal complexes.51 This observation demonstrates an advantage of ion pairs, which can consist of more than two types of ionic species.
4-4. Dimension-controlled assemblies based on π-electronic ion pairs
As seen in the crystal structure of 11+–PCCp−, which formed a charge-by-charge columnar assembly, the alternate stacking of oppositely charged π-electronic ions was critical for constructing stable materials. π-Electronic cations and anions can afford fascinating ion-pairing dimension-controlled assemblies. We disclosed the first examples of dimension-controlled assemblies comprising π-electronic ion pairs.51 π-Electronic ion pairs 13n+–PCCp− (n = 8, 12, 16 and 20) (Fig. 24a) were prepared from aliphatic porphyrin–AuIII complex ion pairs 13n+–Cl− (n = 8, 12, 16 and 20) and Na+–PCCp− in CH3CN/CH2Cl2, followed by purification with silica gel chromatography. The ion-pairing dimension-controlled assembly of 1316+–PCCp− fabricated a supramolecular octane gel (10 mg mL−1) (Fig. 24b(i)), as a result of the entanglement of fibrous morphologies with diameters of 1–3 μm and lengths of >100 μm (Fig. 24b(ii and iii)). Other anion combinations did not afford such a fibrous morphology, and thus, it can be concluded that charge-by-charge assembly of π-electronic ions is crucial for stabilizing the columnar assembly. DSC and POM revealed a remarkably wide-range mesophase temperature region (36 to 292 °C for the cooling process of 1316+–PCCp−) with dendritic textures (Fig. 24c(i)). The synchrotron XRD analysis of the ion pairs revealed well-defined Colh structures with perfect charge-by-charge assemblies in mesophases. For example, in the mesophase at 280 °C (cooling), 1316+–PCCp− exhibited a Colh structure with a = 3.46, c = 0.71 nm and Z = 1 (ρ = 0.88) (Fig. 24c(ii)). The diffraction peak at 0.71 nm is nearly twice that of a π–π stacking distance, indicating the distance between identical π-electronic charged species arising from the formation of a charge-by-charge columnar assembly, as observed in the crystal structure of 11+–PCCp− (Fig. 22b(ii)). The geometry of π-electronic PCCp− fits well with the core planes of porphyrin–AuIII complexes and thus enables the formation of highly anisotropic charge-by-charge stacking assemblies. Such assembling behaviour was confirmed in the XRD patterns of sheared mesophase samples.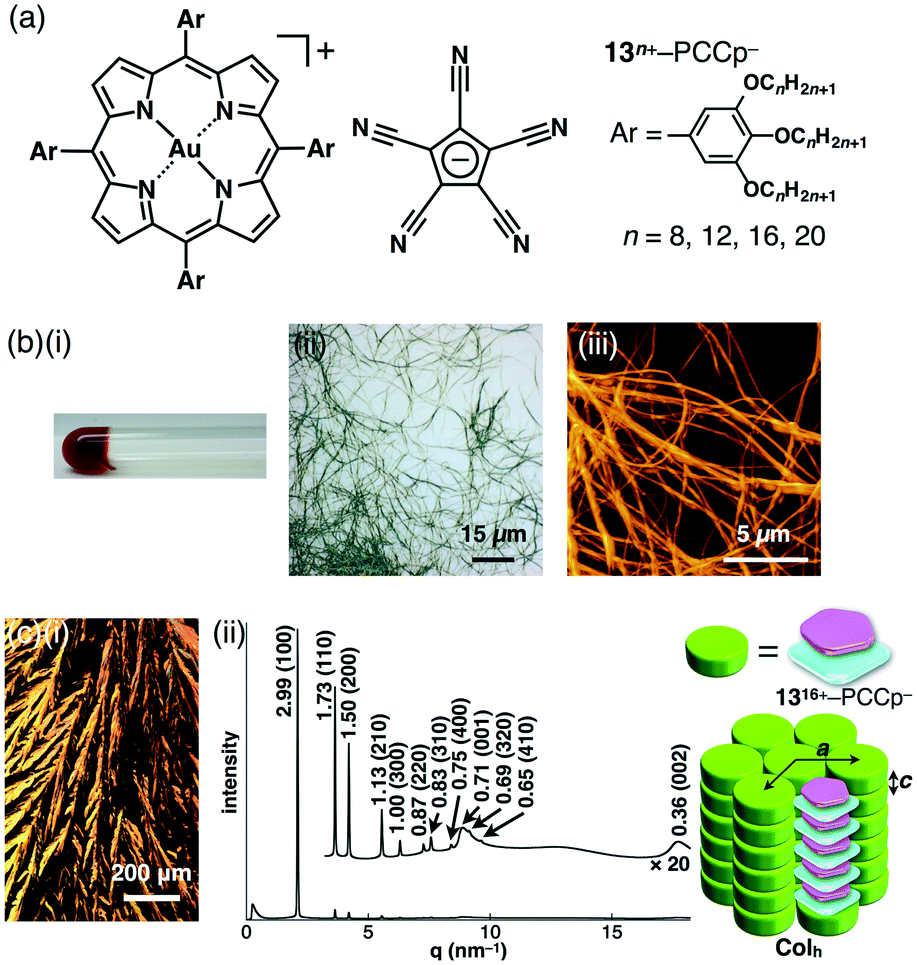 | ||
| Fig. 24 (a) π-Electronic ion pair 13n+–9− (n = 8, 12, 16 and 20), (b) octane gel (10 mg mL−1) of 1316+–PCCp−: (i) photograph of the gel, (ii) OM and (iii) AFM images and (c) mesophase properties of 1316+–PCCp−: (i) POM image and (ii) XRD pattern and a proposed assembled model at 280 °C upon cooling from Iso. | ||
Conclusions
Inspired by the structures of ion pairs comprising receptor–anion complexes and countercations, the chemistry of ion-pairing assemblies that contain genuine π-electronic ions was explored. Ordered arrangements of π-electronic species were achieved by designing and synthesizing charged π-electronic species as the building blocks. Stacking alignments of π-electronic ions, charge-by-charge and charge-segregated assemblies, were constructed by the incorporation of π-electronic ions with suitable geometries and electronic states. Importantly, combinations of π-electronic cations and anions are crucial for adjusting the assembling modes as well as resultant bulk-state properties. Ion-pairing dimension-controlled assemblies based on π-electronic ions exhibit versatile electronic properties, such as charge-carrier transporting properties. Further modifications of π-electronic systems enable the preparation of fascinating π-electronic ion pairs and their associated functional ion-pairing assemblies. As seen in inorganic materials, the ion-pairing strategies demonstrated in this article provide versatile approaches for the fabrication of functional materials based on π-electronic systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The recent contributions of our group reported herein were mainly supported by JSPS KAKENHI Grant Numbers JP26288042 and JP18H01968 for Scientific Research (B), JP19K05444 for Scientific Research (C), JP26107007 for Scientific Research on Innovative Areas “Photosynergetics” and the Ritsumeikan Global Innovation Research Organization (R-GIRO) project, 2017–2022.References
- Books on supramolecular assemblies for electronic materials: (a) Supramolecular Soft Matter, ed. T. Nakanishi, Wiley, 2011 Search PubMed; (b) Functional Supramolecular Architectures, ed. P. Samorì and F. Cacialli, Wiley, 2011 Search PubMed; (c) Unimolecular and Supramolecular Electronics I: Chemistry and Physics Meet at Metal-Molecule Interfaces, Topics in Current Chemistry, ed. R. M. Metzger, Springer, 2012 Search PubMed; (d) Supramolecular Materials for Opto-Electronics, ed. N. Koch, RSC, 2015 Search PubMed.
- (a) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed; (b) S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC; (c) T. Kato, T. Yasuda, Y. Kamikawa and M. Yoshio, Chem. Commun., 2009, 729–739 RSC; (d) T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- (a) H. Maeda, Bull. Chem. Soc. Jpn., 2013, 86, 1359–1399 CrossRef CAS; (b) Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894–2909 RSC; (c) Y. Haketa and H. Maeda, Bull. Chem. Soc. Jpn., 2018, 91, 420–436 CrossRef CAS.
- Selected reviews on ionic liquid crystals: (a) K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed; (b) K. V. Axenov and S. Laschat, Materials, 2011, 4, 206–259 CrossRef CAS PubMed; (c) K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- (a) C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS; (b) C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed.
- (a) G. Magnus, Poggendorff's Ann. Phys., 1828, 90, 239–242 CrossRef; (b) G. Magnus, Ann. Chim. Phys., 1829, 40, 110–111 Search PubMed.
- M. Atoji, J. W. Richardson and R. E. Rundle, J. Am. Chem. Soc., 1957, 79, 3017–3020 CrossRef CAS.
- W. Caseri, Platinum Met. Rev., 2004, 48, 91–100 CrossRef CAS.
- (a) Fundamentals and Applications of Anion Separations, ed. R. P. Singh and B. A. Moyer, Kluwer Academic/Plenum Publishers, 2004 Search PubMed; (b) Anion Sensing, Topics in Current Chemistry, ed. I. Stibor, Springer, 2005, vol. 255, pp. 1–238 Search PubMed; (c) J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, RSC, 2006 Search PubMed; (d) Recognition of Anions, Structure and Bonding, ed. R. Vilar, Springer, 2008, vol. 129, pp. 1–252 Search PubMed; (e) Anion Recognition in Supramolecular Chemistry, Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer, 2010, vol. 24, pp. 1–413 Search PubMed; (f) Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. García-España, Wiley, 2012 Search PubMed.
- H. Maeda and Y. Kusunose, Chem. – Eur. J., 2005, 11, 5661–5666 CrossRef CAS PubMed.
- (a) Y. Haketa and H. Maeda, Chem. – Eur. J., 2011, 17, 1485–1492 CrossRef CAS PubMed; (b) Y. Haketa, Y. Bando, K. Takaishi, M. Uchiyama, A. Muranaka, M. Naito, H. Shibaguchi, T. Kawai and H. Maeda, Angew. Chem., Int. Ed., 2012, 51, 7967–7971 CrossRef CAS PubMed.
- H. Maeda, Y. Haketa and T. Nakanishi, J. Am. Chem. Soc., 2007, 129, 13661–13674 CrossRef CAS PubMed.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255–12263 CrossRef CAS.
- (a) B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS; (b) B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773–1783 CrossRef CAS PubMed . See also ref. 15 for the preparation of TATA+–Cl–.
- Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079–10083 CrossRef CAS PubMed.
- Y. Haketa, M. Takayama and H. Maeda, Org. Biomol. Chem., 2012, 10, 2603–2606 RSC.
- Y. Haketa, Y. Honsho, S. Seki and H. Maeda, Chem. – Eur. J., 2012, 18, 7016–7020 CrossRef CAS PubMed.
- (a) B. Dong, Y. Terashima, Y. Haketa and H. Maeda, Chem. – Eur. J., 2012, 18, 3460–3463 CrossRef CAS PubMed; (b) B. Dong, T. Sakurai, Y. Honsho, S. Seki and H. Maeda, J. Am. Chem. Soc., 2013, 135, 1284–1287 CrossRef CAS PubMed; (c) B. Dong, T. Sakurai, Y. Bando, S. Seki, K. Takaishi, M. Uchiyama, A. Muranaka and H. Maeda, J. Am. Chem. Soc., 2013, 135, 14797–14805 CrossRef CAS PubMed.
- R. Sekiya, Y. Tsutsui, W. Choi, T. Sakurai, S. Seki, Y. Bando and H. Maeda, Chem. Commun., 2014, 50, 10615–10618 RSC.
- H. Maeda, K. Naritani, Y. Honsho and S. Seki, J. Am. Chem. Soc., 2011, 133, 8896–8899 CrossRef CAS PubMed.
- (a) H. Maeda, K. Kinoshita, K. Naritani and Y. Bando, Chem. Commun., 2011, 47, 8241–8243 RSC; (b) H. Maeda, T. Okubo, Y. Haketa and N. Yasuda, Chem. – Eur. J., 2018, 24, 16176–16182 CrossRef CAS PubMed.
- Peripheral modifications of anion-responsive π-electronic molecules: (a) R. Yamakado, T. Sakurai, W. Matsuda, S. Seki, N. Yasuda, S. Akine and H. Maeda, Chem. – Eur. J., 2016, 22, 626–638 CrossRef CAS PubMed; (b) V. Lakshmi, Y. Haketa, R. Yamakado, N. Yasuda and H. Maeda, Chem. Commun., 2017, 53, 3834–3837 RSC; (c) S. Sugiura, W. Matsuda, W. Zhang, S. Seki, N. Yasuda and H. Maeda, J. Org. Chem., 2019, 84, 8886–8898 CrossRef CAS PubMed; (d) Y. Watanabe, Y. Haketa, K. Nakamura, S. Kaname, N. Yasuda and H. Maeda, Chem. – Eur. J., 2020, 26 DOI:10.1002/chem.202000634 , in press.
- (a) F. Camerel, G. Ulrich, J. Barberá and R. Ziessel, Chem. – Eur. J., 2007, 13, 2189–2200 CrossRef CAS PubMed; (b) J. H. Olivier, J. Barberá, E. Bahaidarah, A. Harriman and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 6100–6103 CrossRef CAS PubMed.
- Selected examples of carboxylate-based ion pairs: Y. Zakrevskyy, B. Smarsly, J. Stumpe and C. F. J. Faul, Phys. Rev. E, 2005, 71, 021701 CrossRef CAS PubMed.
- Selected examples of hydroxide-based ion pairs: J. Kadam, C. F. J. Faul and U. Scherf, Chem. Mater., 2004, 16, 3867–3871 CrossRef CAS.
- (a) R. Yamakado, M. Hara, S. Nagano, T. Seki and H. Maeda, Chem. – Eur. J., 2017, 23, 9244–9248 CrossRef CAS PubMed; (b) R. Yamakado, M. Hara, S. Nagano, T. Seki and H. Maeda, Chem. Lett., 2018, 47, 404–407 CrossRef CAS; (c) R. Yamakado, Y. Haketa, M. Hara, S. Nagano, T. Seki and H. Maeda, Chem. Commun., 2019, 55, 10269–10272 RSC.
- (a) H. Maeda, A. Fukui, R. Yamakado and N. Yasuda, Chem. Commun., 2015, 51, 17572–17575 RSC; (b) H. Maeda, Y. Takeda, Y. Haketa, Y. Morimoto and N. Yasuda, Chem. – Eur. J., 2018, 24, 8910–8916 CrossRef CAS PubMed.
- (a) I. Heldt and U. Behrens, Z. Anorg. Allg. Chem., 2005, 631, 749–758 CrossRef CAS; (b) M. Becker, J. Harloff, T. Jantz, A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2012, 5658–5667 CrossRef CAS.
- R. Yamakado, H. Ishibashi, Y. Motoyoshi, N. Yasuda and H. Maeda, Chem. Commun., 2019, 55, 326–329 RSC.
- (a) Y. Sasano, N. Yasuda and H. Maeda, Dalton Trans., 2017, 46, 8924–8928 RSC; (b) Y. Sasano, Y. Haketa, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, Chem. – Eur. J., 2019, 25, 6712–6717 CrossRef CAS PubMed.
- R. Kuhn and D. Rewicki, Angew. Chem., Int. Ed. Engl., 1967, 6, 635–636 CrossRef CAS.
- K. Okamoto, T. Kitagawa, K. Takeuchi, K. Komatsu, T. Kinoshita, S. Aonuma, M. Nagai and A. Miyabo, J. Org. Chem., 1990, 55, 996–1002 CrossRef CAS.
- (a) E. Le Goff and R. B. LaCount, J. Am. Chem. Soc., 1963, 85, 1354–1355 CrossRef CAS; (b) M. I. Bruce, P. A. Humphrey, B. W. Skelton and A. H. White, Aust. J. Chem., 1984, 37, 2441–2446 CrossRef CAS; (c) M. I. Bruce, P. A. Humphrey, B. W. Skelton and A. H. White, Aust. J. Chem., 1986, 39, 165–169 CrossRef CAS.
- O. W. Webster, J. Am. Chem. Soc., 1965, 87, 1820–1821 CrossRef CAS.
- T. Sakai, S. Seo, J. Matsuoka and Y. Mori, J. Org. Chem., 2013, 78, 10978–10985 CrossRef CAS PubMed.
- Y. Bando, Y. Haketa, T. Sakurai, W. Matsuda, S. Seki, H. Takaya and H. Maeda, Chem. – Eur. J., 2016, 22, 7843–7850 CrossRef CAS PubMed.
- R. J. Less, M. McPartlin, J. M. Rawson, P. T. Wood and D. S. Wright, Chem. – Eur. J., 2010, 16, 13723–13728 CrossRef CAS PubMed.
- (a) D. Wu, L. Zhi, G. J. Bodwell, G. Cui, N. Tsao and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5417–5420 CrossRef CAS PubMed; (b) D. Wu, W. Pisula, V. Enkelmann, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 9620–9621 CrossRef CAS PubMed; (c) D. Wu, R. Liu, W. Pisula, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2791–2794 CrossRef CAS PubMed.
- D. Shi, C. Schwall, G. Sfintes, E. Thyrhaug, P. Hammershøj, M. Cárdenas, J. B. Simonsen and B. W. Laursen, Chem. – Eur. J., 2014, 20, 6853–6856 CrossRef CAS PubMed.
- A review of cationic PtII complexes: V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- A review of cationic AuIII complexes: R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed.
- A book including nonplanar cationic metal complexes: Advances in Organometallic Chemistry, ed. P. J. Pérez, Elsevier, 2018 Search PubMed.
- Selected examples of nonplanar cationic metal complexes: (a) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef PubMed; (b) Y. Ma, H. Liang, Y. Zeng, H. Yang, C.-L. Ho, W. Xu, Q. Zhao, W. Huang and W.-Y. Wong, Chem. Sci., 2016, 7, 3338–3346 RSC; (c) Y. Ma, S. Liu, H. Yang, Y. Zeng, P. She, N. Zhu, C.-L. Ho, Q. Zhao, W. Huang and W.-Y. Wong, Inorg. Chem., 2017, 56, 2409–2416 CrossRef CAS PubMed; (d) Y. Ma, S. Zhang, H. Wei, Y. Dong, L. Shen, S. Liu, Q. Zhao, L. Liu and W.-Y. Wong, Dalton Trans., 2018, 47, 5582–5588 RSC.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed.
- W. Lu, K. T. Chan, S.-X. Wu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752–755 RSC.
- E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373–376 CrossRef CAS.
- Selected reports on porphyrin–AuIII complexes as anticancer agents: (a) C.-M. Che, R. W.-Y. Sun, W.-Y. Yu, C.-B. Ko, N. Zhu and H. Sun, Chem. Commun., 2003, 1718–1719 RSC; (b) Y. Wang, Q.-Y. He, R. W.-Y. Sun, C.-M. Che and J.-F. Chiu, Eur. J. Pharmacol., 2007, 554, 113–122 CrossRef CAS PubMed; (c) R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem. – Eur. J., 2010, 16, 3097–3113 CrossRef CAS PubMed; (d) L. He, T. Chen, Y. You, H. Hu, W. Zheng, W.-L. Kwong, T. Zou and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 12532–12536 CAS.
- Selected reports on porphyrin–AuIII complexes as electron-accepting units: (a) A. M. Brun, A. Harriman, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8657–8663 CrossRef CAS; (b) K. Kilså, J. Kajanus, A. N. Macpherson, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2001, 123, 3069–3080 CrossRef PubMed; (c) M. Andersson, M. Linke, J.-C. Chambron, J. Davidsson, V. Heitz, L. Hammarström and J.-P. Sauvage, J. Am. Chem. Soc., 2002, 124, 4347–4362 CrossRef CAS PubMed; (d) M. P. Eng, T. Ljungdahl, J. Andréasson, J. Mårtensson and B. Albinsson, J. Phys. Chem. A, 2005, 109, 1776–1784 CrossRef CAS PubMed; (e) J. Fortage, J. Boixel, E. Blart, L. Hammarstöm, H. C. Becker and F. Odobel, Chem. – Eur. J., 2008, 14, 3467–3480 CrossRef CAS PubMed.
- (a) Z. Ou, W. Zhu, Y. Fang, P. J. Sintic, T. Khoury, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2011, 50, 12802–12809 CrossRef CAS PubMed; (b) Z. Ou, T. Khoury, Y. Fang, W. Zhu, P. J. Sintic, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2013, 52, 2474–2483 CrossRef CAS PubMed; (c) S. Preiß, J. Melomedov, A. Wünsche von Leupoldt and K. Heinze, Chem. Sci., 2016, 7, 596–610 RSC; (d) S. Preiß, C. Förster, S. Otto, M. Bauer, P. Müller, D. Hinderberger, H. H. Haeri, L. Carella and K. Heinze, Nat. Chem., 2017, 9, 1249–1255 CrossRef PubMed.
- R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem. – Eur. J., 2010, 16, 3097–3113 CrossRef CAS PubMed.
- Y. Haketa, Y. Bando, Y. Sasano, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, iScience, 2019, 14, 241–256 CrossRef CAS PubMed.
- R. Timkovich and A. Tulinsky, Inorg. Chem., 1977, 16, 962–963 CrossRef CAS.
- H. Lv, B. Yang, J. Jing, Y. Yu, J. Zhang and J.-L. Zhang, Dalton Trans., 2012, 41, 3116–3118 RSC.
- H. Tanaka, Y. Haketa, N. Yasuda and H. Maeda, Chem. – Asian J., 2019, 14, 2129–2137 CrossRef CAS PubMed.
- (a) S. Kawahara, S. Tsuzuki and T. Uchimaru, J. Phys. Chem. A, 2004, 108, 6744–6749 CrossRef CAS; (b) P. Li, J. M. Maier, E. C. Vik, C. J. Yehl, B. E. Dial, A. E. Rickher, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2017, 56, 7209–7212 CrossRef CAS PubMed.
- (a) A. Nowak-Król, D. Gryko and D. T. Gryko, Chem. – Asian J., 2010, 5, 904–909 CrossRef PubMed; (b) S. Maruyama, K. Sato and H. Iwahashi, Chem. Lett., 2010, 39, 714–716 CrossRef CAS.
- Solid-state ion-pairing assemblies consisting of porphyrin–AuIII complexes and receptor–anion complexes have been investigated: H. Tanaka, Y. Haketa, Y. Bando, R. Yamakado, N. Yasuda and H. Maeda, Chem. – Asian J., 2020, 15, 494–498 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |