
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular assembly by time-programmed acid autocatalysis†
Guido
Panzarasa
 *,
Tianqi
Sai
,
Alexandre L.
Torzynski
,
Katrina
Smith-Mannschott
and
Eric R.
Dufresne
*,
Tianqi
Sai
,
Alexandre L.
Torzynski
,
Katrina
Smith-Mannschott
and
Eric R.
Dufresne
Laboratory of Soft and Living Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: guido.panzarasa@mat.ethz.ch
First published on 20th November 2019
Abstract
Autocatalytic pH clocks can be useful to control self-assembly in the time domain. Their applications are, however, limited by the currently available toolbox. We describe here an approach for the design of a dynamic pH switch that generates intense alkali-to-acid changes after a tailorable lagtime (from minutes to hours), and we demonstrate its application for the time-controlled supramolecular self-assembly of nanofibers.
Design, System, ApplicationControlling self-assembly in time with small-molecule networks is a fundamental step for the development of dynamic materials. Chemical clocks are especially promising as in situ switches, but the available toolbox of such small-molecule networks is limited. To help solve this problem, we describe a practical approach for programming sudden and intense (up to 7 units) alkali-to-acid pH changes after a tailorable lagtime. Our approach is based on coupling the bromate-sulfite acid-autocatalyzed reaction with cyclic esters (δ-gluconolactone and 1,3-propanesultone) as slow generators of hydrogen ions. The initial delay is thus dictated by the nature and concentration of the cyclic ester and can be extended from minutes to hours. We demonstrate the usefulness of our method for controlling pH-driven supramolecular self-assembly. |
Chemical feedback is defined as the influence of a species on the rate of its own production.1 Autocatalytic reactions, in which a product catalyzes its own formation, are prototypical examples of positive chemical feedback. As such, they play key roles in complex (bio)chemical dynamics,2,3 for both living systems and artificial materials. In closed systems, autocatalysis often manifests as an exponential product increase after a controllable time lag, a behavior which is usually referred to as a “chemical clock”4 and can be accompanied by a sudden change of pH and/or redox potential. Such dynamic switches5 are useful for the design of time-controlled self-assembly, which, in a broader perspective, could translate in the development of new materials.6–8 Examples of previously reported applications include the self-assembly of colloidal particles,9–11 micelles,12 and the triggering of thiol-acrylate polymerization.13,14 Nevertheless, the vast majority of these systems are based on acid-to-alkali systems, such as the urease-urea14 and the methylene glycol-sulfite clock reaction.10
We describe here an easy method to program the autonomous activation of acid-autocatalyzed reactions, based on the hydrolysis of cyclic esters. Our approach allows to tailor the lagtime and dramatically increases the magnitude of the pH change (ΔpH ca. 7). Finally, we demonstrate its application for time-domain programming of supramolecular self-assembly of a pH-responsive perylene diimide, as sketched in Fig. 1.
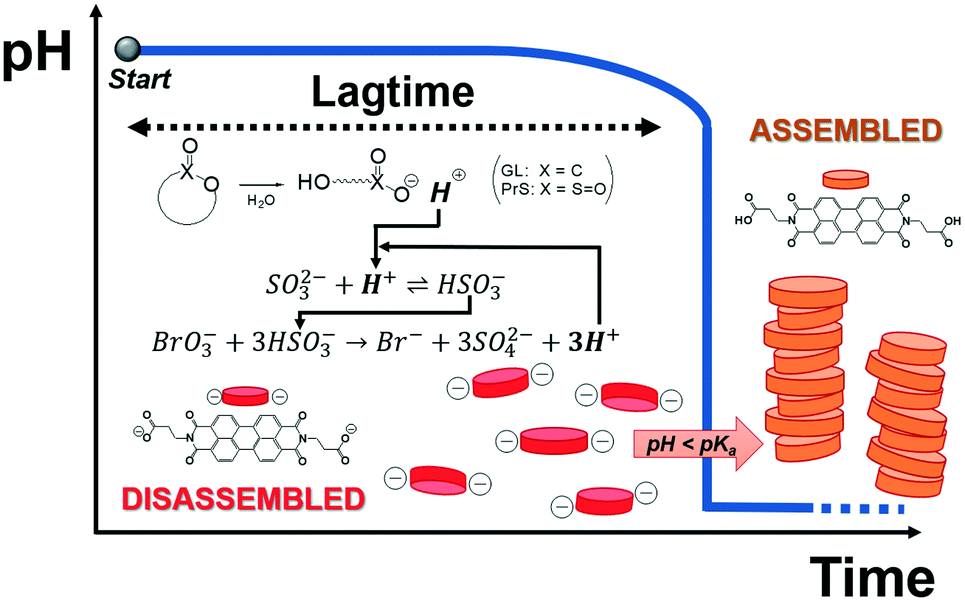 | ||
| Fig. 1 Supramolecular assembly of a pH-responsive perylene diimide triggered by programmed acid autocatalysis. | ||
Acid-autocatalyzed reactions proceed slowly in neutral or basic conditions (pH ≥ 7), while for pH < 6 their rapid acceleration manifests in a sudden drop of pH, which can be up to 2–3 units. Reactions of this kind have been described for dynamic particle self-assembly,15 chemomechanical hydrogel actuation,16 chemical morphogenesis,17 and for the design of blood-clotting models.18 Since the delay time, or lagtime, cannot be flexibly tuned by changing the initial reactants concentrations, these reactions are usually triggered by the direct addition of acid to lower the pH. The necessity of external control is an obvious limitation for the use of acid-autocatalyzed reactions as switches. Furthermore, having to start at a neutral or slightly acidic pH narrows the applicability range.
It has long been known that the hydrolysis of esters is associated with complex dynamics. In 1937, R. Skrabal studied the saponification of di- and trichloroacetic esters using a clock reaction approach.19 In 1992, P. L. Luisi et al. showed that micelles could be autocatalytically generated from the alkaline hydrolysis of a fatty acid ester.20 Lactones, cyclic esters of carboxylic acids, have been recently demonstrated for controlling the behavior of chemical clocks, such as the methylene glycol–sulfite9 and the chlorite-iodide21 reactions, through their hydrolysis. Compared to linear esters, cyclic esters have an enhanced tendency towards hydrolysis, which is driven by the release of ring strain.22 δ-Gluconolactone (GL) is one of the most popular: it is non-toxic, readily soluble in water (0.59 g mL−1) and it rapidly hydrolyzes to gluconic acid (pKa 3.86) (eqn (1)). The application of GL as an in situ acid generator for the controlled gelation of dipeptides and other low molecular weight hydrogelators,23–26 demonstrated by D. J. Adams et al., is another example of the importance of this approach for soft matter research.
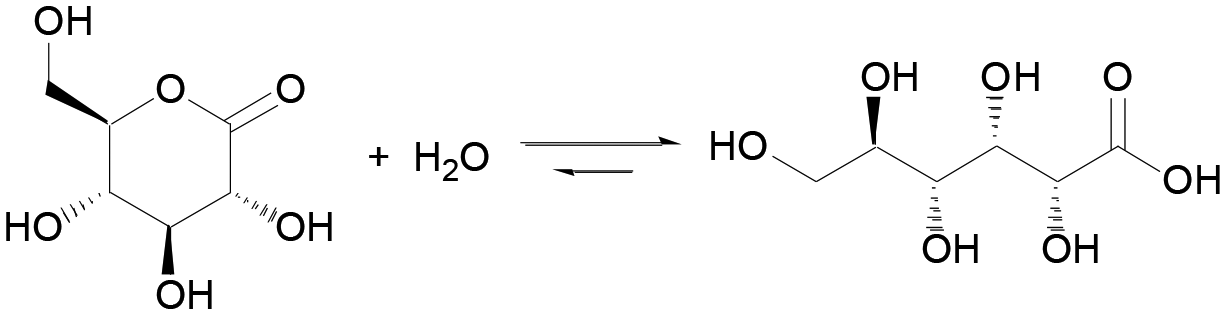 | (1) |
Here we employ also sultones as in situ acid generators for the programming of acid autocatalysis, and we demonstrate their advantages compared to lactones. Sultones are the cyclic esters of sulfonic acids. 1,3-Propanesultone (PrS) is one of the simplest sultones, and is a useful sulfoalkylating agent in organic synthesis, medicinal chemistry, and surfactant production. It has good solubility in water (0.1 g mL−1), with which it slowly reacts yielding 1-hydroxypropanesulfonic acid (pKa 1.53) (eqn (2)).
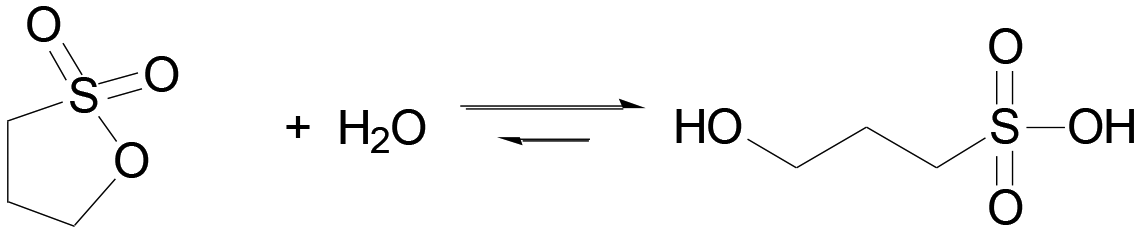 | (2) |
δ-Gluconolactone and 1,3-propanesultone have very different hydrolysis behavior. At room temperature (20 °C), the hydrolysis constant of GL is27kGL = 1 × 10−4 s−1 and is much lower for PrS: kPrS = 1 × 10−5 s−1 (corresponding to a 14.8 h half-life).28,29 At alkaline pH, this difference is even more evident: GL is hydrolyzed in minutes27 (kGL OH = 4 × 103 M−1 s−1), while the half-life of PrS is of the order of hours.30 These differences enable to efficiently tune the activation time of acid autocatalysis.
To demonstrate the programming of acid autocatalysis with cyclic esters we chose the bromate-sulfite (BS) reaction, thanks to its well-understood behavior. The thermodynamic equations31 for the BS reaction can be formulated as follows (eqn (3) and (4)):
| SO32− + H+ ⇌ HSO3− | (3) |
| BrO3− + 3HSO3− → Br− + 3SO42− + 3H+ | (4) |
The BS reaction produces three equivalents of hydrogen ions for each equivalent used, resulting in an autocatalytic pH decrease. The following concentrations were used:32 sodium bromate [NaBrO3] = 220 mM and sodium sulfite [Na2SO3] = 60 mM. In these conditions, the self-activated reaction required more than 2.5 h to display a pH change ΔpH of ∼3.7 units, starting at pH 9.5 (the alkalinity is due to sodium sulfite) (Fig. 2A and B).
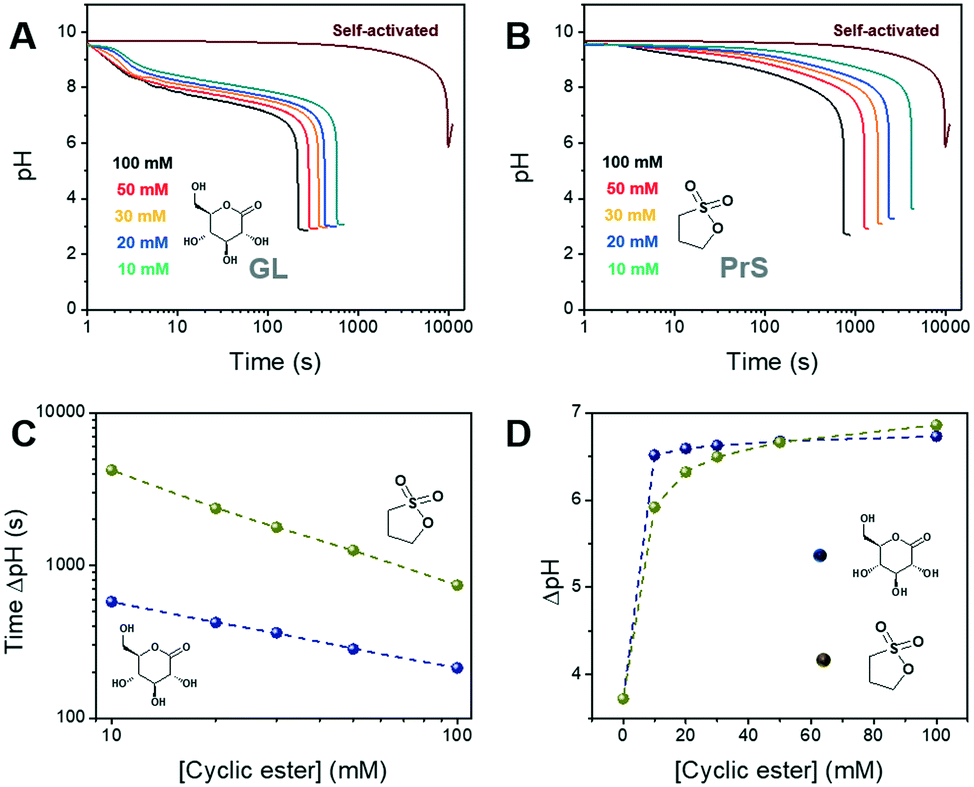 | ||
| Fig. 2 pH-Time profiles for the activation of the bromate-sulfite (BS) clock reaction using different concentrations of (A) δ-gluconolactone and (B) 1,3-propanesultone. Relationship between the concentration of cyclic ester and (C) the time of maximum pH change, (D) the magnitude of the pH change achieved. | ||
In our approach, the hydrogen ions necessary for the BS reaction to start are produced by the hydrolysis of δ-gluconolactone or 1,3-propanesultone. As shown in Fig. 2A and B, both cyclic esters are able to trigger the bromate-sulfite reaction. GL produced a decrease of about 1.5 pH units in the reaction mixture immediately after its addition, while with PrS the pH decrease was much more gradual, as expected, given its slower hydrolysis rate. Independent of the cyclic ester used, the initial pH remained fairly constant until the drop produced by the onset of acid autocatalysis. This is because the generated hydrogen ions are quickly scavenged by sulfite, transforming into bisulfite (eqn (3); k3 = 5 × 1010 M−1 s−1),27 which then reacts with bromate (eqn (4)). This pH-buffering ability of sulfite was confirmed by dedicated control experiments (Fig. S1†). With acid autocatalysis, the pH drop is sharper than the one produced by cyclic esters' hydrolysis alone.
The time of the onset of acid autocatalysis was determined by the nature and the concentration of the cyclic ester used, and could be flexibly tuned from minutes to hours (Fig. 2C). In the concentration range we tested (between 100 mM and 10 mM), the use of δ-gluconolactone corresponded to activation times between 3 and 15 min, while with 1,3-propanesultone lagtimes between 15 min and 1 h were obtained; even longer times (ca. 2 h) were obtained with 1,4-butanesultone (Fig. S2†). Increasing or reducing further the concentration of the chosen cyclic ester might allow a wider range of activation times. In addition, both GL and PrS can generate dramatic pH changes (ΔpH) between the initial and final state, from ∼3.7 for the self-activated reaction to a maximum of ∼6.8 with 100 mM PrS (Fig. 2D). The advantages of in situ acid generation compared to direct, external acid addition can be appreciated from Fig. S3,† which shows the effect of model acids on the bromate-sulfite reaction. Unsurprisingly, the direct addition of acid resulted in the drastic lowering of the initial pH and in dramatically faster (≤2 min) activation times.
We then used our approach for the time-domain control of pH-driven supramolecular self-assembly. Supramolecular building blocks offer enticing possibilities for the development of time-responsive materials,33,34 especially because intermolecular interactions, such as π–π stacking and hydrogen-bonding, can be used to facilitate self-assembly.35
Perylene diimides (PDI), thanks to the π–π attractive interactions between the perylene cores, are prone to assemble into ordered 1D nanostructures such as rods and fibers.36,37 This and other remarkable qualities, such as high physico-chemical stability and good optoelectronic properties, make them useful supramolecular building blocks.38–40
We chose the pH-responsive perylene diimide derivative (3-aminopropanoic acid)-perylene-3,4:9,10-tetracarboxylic acid diimide (“PDIacid”). With an apparent pKa of 4.9 (determined by titration), PDIacid is soluble in water at alkaline pH (pH > pKa), because the negative charges generated by the deprotonation of the carboxylic acid groups can overcome the π–π attractive interaction between the perylene cores, keeping them separated. On the other hand, when pH < pKa, protonation results first in a color change from pink to yellow, and then in the formation of supramolecular aggregates (Fig. 3A and C).
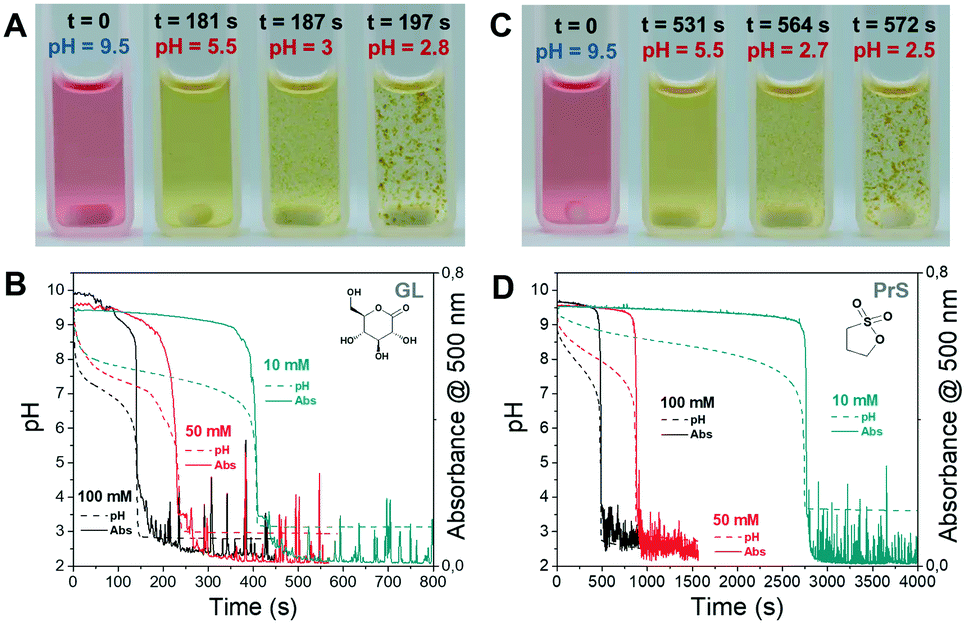 | ||
| Fig. 3 Self-assembly of PDIacid triggered by the BS reaction in presence of cyclic esters. Experimental pictures: (A) 100 mM GL, (C) 100 mM PrS. (B and D) Evolution of pH and absorbance at 500 nm as a function of time. Noise in absorbance curves is due to the formation of large scattering aggregates. | ||
In the deprotonated state, PDIacid has a strong absorption peak centered at 500 nm, which disappears when it is protonated (Fig. S4†). Coupled pH and UV-vis measurements showed that the rapid pH decrease brought by acid autocatalysis resulted in the supramolecular self-assembly of PDIacid (Fig. 3B and D). Stirring promoted further aggregation, resulting in the production of macroscopic structures (Fig. 4B and D, Movie S1 and S2†).
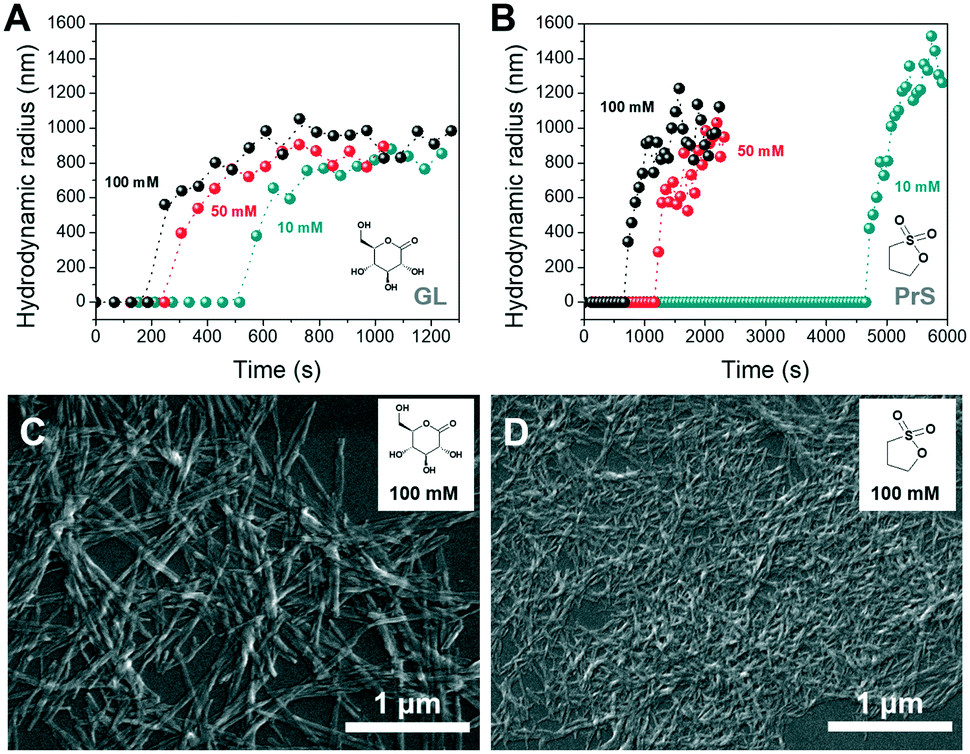 | ||
| Fig. 4 The sudden assembly of PDIacid, driven by the BS reaction in presence of different concentrations of GL (A) and PrS (B), as revealed by dynamic light scattering (DLS). (C and D) Representative scanning electron microscopy (SEM) images of the supramolecular assemblies obtained with both cyclic esters. | ||
Dynamic light scattering (DLS) was used to obtain further insight on the self-assembly process of PDIacid. As shown in Fig. 4A and C, particles with hundreds of nanometers in size appeared abruptly after a lagtime, which was dependent of the concentration and nature of cyclic ester (Fig. 4A and B). The results also suggested that the assemblies stopped growing soon after their formation.
Scanning electron microscopy (SEM) was used to investigate the morphology of the PDIacid assemblies obtained with both cyclic esters. From Fig. 4C and D it is clear that PDIacid assembled into fibers with hundred of nanometers to micrometers in length. This morphology is typical for perylene bisimide assemblies and originates from the π–π stacking between the perylene cores. Hydrogen bonding between the lateral carboxylic acid groups could also be involved.41 However, the fibers obtained with the two cyclic esters have significantly different diameters. This difference is not yet explained, and it deserves further investigation.
Conclusions
We show how to program the activation time of an acid-autocatalyzed reaction, using cyclic esters as in situ slow generators of hydrogen ions. The lagtime can be tuned from minutes to hours, depending on the cyclic ester and its concentration. Applying our approach to the time-programming of supramolecular self-assembly of a pH-responsive perylene diimide, we demonstrate that the activation of acid autocatalysis results in the sudden formation of ordered structures. Our findings are not limited to the bromate-sulfite clock, and can be extended to other acid-autocatalytic systems (such as the chlorite–tetrathionate reaction) as well as to other building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the SNF National Centre of Competence in Research ‘Bio-Inspired Materials’ for partial funding, and the SNSF Grant 200021-172827.Notes and references
- I. R. Epstein, J. A. Pojman and Q. Tran-Cong-Miyata, in Nonlinear Dynamics with Polymers: Fundamentals, Methods and Applications, ed. J. A. Pojman and Q. Tran-Cong-Miyata, Wiley-VCH Verlag GmbH & Co., 2010, pp. 5–19 Search PubMed.
- A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 CrossRef CAS PubMed.
- Z. Dadon, N. Wagner and G. Ashkenasy, Angew. Chem., Int. Ed., 2008, 47, 6128–6136 CrossRef CAS PubMed.
- G. Lente, G. Bazsa and I. Fábián, New J. Chem., 2007, 31, 1707 RSC.
- T. Bánsági and A. F. Taylor, Tetrahedron, 2017, 73, 5018–5022 CrossRef.
- L. Heinen and A. Walther, Soft Matter, 2015, 11, 7857–7866 RSC.
- H. C. Wang, Y. Zhang, C. M. Possanza, S. C. Zimmerman, J. Cheng, J. S. Moore, K. Harris and J. S. Katz, ACS Appl. Mater. Interfaces, 2015, 7, 6369–6382 CrossRef CAS PubMed.
- H. Frisch and P. Besenius, Macromol. Rapid Commun., 2015, 36, 346–363 CrossRef CAS PubMed.
- E. Tóth-Szeles, J. Horváth, G. Holló, R. Szcs, H. Nakanishi and I. Lagzi, Mol. Syst. Des. Eng., 2017, 2, 274–282 RSC.
- G. Panzarasa, A. Osypova, A. Sicher, A. Bruinink and E. R. Dufresne, Soft Matter, 2018, 14, 6415–6418 RSC.
- G. Panzarasa, A. L. Torzynski, T. Sai, K. Smith-Mannschott and E. R. Dufresne, Transient Supramolecular Assembly by Programmable pH Cycles, ChemRxiv 2019, DOI:10.26434/chemrxiv.9938105.v1.
- H. E. Cingil, N. C. H. Meertens and I. K. Voets, Small, 2018, 14, 1802089 CrossRef PubMed.
- G. Hu, C. Bounds, J. A. Pojman and A. F. Taylor, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2955–2959 CrossRef CAS.
- E. Jee, T. Bánsági, A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS PubMed.
- B. Bohner, G. Schuszter, H. Nakanishi, D. Zámbó, A. Deák, D. Horváth, A. Toth and I. Lagzi, Langmuir, 2015, 31, 12019–12024 CrossRef CAS PubMed.
- J. Horváth, I. Szalai, J. Boissonade and P. De Kepper, Soft Matter, 2011, 7, 8462–8472 RSC.
- F. Gauffre, V. Labrot, J. Boissonade, P. De Kepper and E. Dulos, J. Phys. Chem. A, 2003, 107, 4452–4456 CrossRef CAS.
- R. R. Pompano, H. W. Li and R. F. Ismagilov, Biophys. J., 2008, 95, 1531–1543 CrossRef CAS PubMed.
- R. Skrabal, Monatsh. Chem., 1937, 71, 298–308 CrossRef.
- P. A. Bachmann, P. L. Luisi and J. Lang, Nature, 1992, 357, 57–59 CrossRef CAS.
- G. Panzarasa and E. R. Dufresne, Chaos, 2019, 29, 071102 CrossRef PubMed.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- A. Z. Cardoso, A. E. Alvarez Alvarez, B. N. Cattoz, P. C. Griffiths, S. M. King, W. J. Frith and D. J. Adams, Faraday Discuss., 2013, 166, 101–116 RSC.
- E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1864–1867 RSC.
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CAS.
- K. Kovacs, R. E. McIlwaine, S. K. Scott and A. F. Taylor, Phys. Chem. Chem. Phys., 2007, 9, 3711–3716 RSC.
- F. G. Bordwell, C. Edward Osborne and R. D. Chapman, J. Am. Chem. Soc., 1959, 81, 2698–2705 CrossRef CAS.
- R. F. Fischer, Ind. Eng. Chem., 1964, 56, 41–45 CrossRef CAS.
- A. Mori, M. Nagayama and H. Mandai, Bull. Chem. Soc. Jpn., 1971, 44, 1669–1672 CrossRef CAS.
- T. G. Szántó and G. Rábai, J. Phys. Chem. A, 2005, 109, 5398–5402 CrossRef PubMed.
- Z. Viranyi, I. Szalai, J. Boissonade and P. De Kepper, J. Phys. Chem. A, 2007, 111, 8090–8094 CrossRef CAS PubMed.
- O. K. Aramballi J. Savyasachi, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Maile and G. Thorfinnur, Chem, 2017, 764–811 Search PubMed.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- F. Biedermann and Hans-Jörg Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513–1528 RSC.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- B. Wang and C. Yu, Angew. Chem., Int. Ed., 2010, 49, 1485–1488 CrossRef CAS PubMed.
- A. Datar, K. Balakrishnan and L. Zang, Chem. Commun., 2013, 49, 6894–6896 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, additional figures and experimental movies. See DOI: 10.1039/c9me00139e |
| This journal is © The Royal Society of Chemistry 2020 |