
Parallelized identification of on- and off-target protein interactions†
Jiayi
Dou
ab,
Inna
Goreshnik
ab,
Cassie
Bryan
ab,
David
Baker
abc and
Eva-Maria
Strauch‡
 *abd
*abd
aDepartment of Biochemistry, University of Washington, Seattle, WA 98195, USA. E-mail: estrauch@uga.edu; Tel: +706 542 7725
bInstitute for Protein Design, University of Washington, Seattle, WA 98195, USA
cHoward Hughes Medical Institute, University of Washington, Seattle, WA 98195, USA
dDepartment of Microbiology, University of Washington, Seattle, WA 98195, USA
First published on 26th November 2019
Abstract
Genetic selection combined with next-generation sequencing enables the simultaneous interrogation of the functionality and stability of large numbers of naturally occurring, engineered, or computationally designed protein variants in parallel. We display hundreds of engineered proteins on the surface of yeast cells, select for binding to a set of target molecules by flow cytometry, and sequence the starting pool as well as selected pools to obtain enrichment values for each displayed protein with each target. We show that this high-throughput workflow of multiplex genetic selections followed by large-scale sequencing and comparative analysis allows not only the determination of relative affinities, but also the monitoring of specificity profiles for hundreds to thousands of protein–protein and protein–small molecule interactions in parallel. The approach not only identifies new interactions of designed proteins, but also detects unintended and undesirable off-target interactions. This provides a general framework for screening of engineered protein binders, which often have no negative selection or design step as part of their development pipelines. Hence, this method will be generally useful in the development of protein-based therapeutics.
 Eva-Maria Strauch | Eva-Maria Strauch is an Assistant Professor in the Department of Pharmaceutical and Biomedical Sciences at the University of Georgia, and holds an adjunct position at the Institute of Bioinformatics. Her laboratory uses and develops protein design and engineering methodologies for better vaccines, protein therapeutics, and delivery systems. Her research focuses on understanding, targeting, and repurposing the protein chemistry of viral surface proteins. She received her Ph.D. from the University of Texas at Austin under the direction of Prof. George Georgiou focusing on directed evolution. She was a postdoctoral fellow under the direction of Prof. David Baker at the University of Washington where she developed and applied both computational and experimental methods for the development of novel proteins and protein–protein interactions. More information can be found under http://strauchlab.com. |
Design, System, ApplicationProtein selections have been used for identifying and optimizing binding proteins, determining protease and enzyme specificity, and, more recently, identifying folded proteins or binders that were computationally designed. Proteins rarely act in isolation; they are part of pathways and only interact with specific molecules. Engineered proteins, such as antibodies or computationally designed proteins intended for therapeutic treatments, will be surrounded by many molecules in the human body. Most of the time these proteins have been developed in “isolation” and negative design or selection procedures to avoid off-target binding have not been applied. Sensitive methods to analyze interactions in a high-throughput manner to control for unexpected specific or “sticky” interactions should speed up lead discovery and facilitate drug development. |
Introduction
Protein display selections have been used to repeatedly select for a desired functionality from a diverse gene library until convergence on a small number of protein variants is achieved. With the advent of next-generation sequencing (NGS), the frequency of many variants from multiple genetic selections can be evaluated in parallel. The strength of selective pressure for the desired functionality determines the diversity of the gene pool after selection; depending on the selective pressure, weak or dysfunctional variants will be depleted or even disappear from the pools. The diversity that can be assessed in detail depends on the numbers of genes or gene fragments that can be sequenced. Currently, even a simple benchtop sequencer, such as the Miseq (Illumina), can obtain up to 35 million sequences in a single run, and the numbers are increasing due to constant improvements to the technology. Previous combination of selection experiments in conjunction with NGS have enabled the dissection of the specificities of laboratory-evolved PDZ domains with a set of peptide ligands.1 Relative affinities of a series of peptide–protein interactions can be extracted through variation in the positioning of the selection gates2 and the concentration of a target molecule.3 Further, selections and deep sequencing has been used to obtain detailed protein fitness landscapes with respect to their residue-level contributions to protein interactions, detection of binding epitopes,4,5 detailed binding and enzyme activity,6–13 and even protein stability using temperature variation as their selection criterion or protease resistance.14 These detailed landscapes have provided a deeper understanding of protein chemistry15 and have also revealed information about the usage of the genetic code;16 however thus far parallel screening of hundreds of unrelated proteins against a series of target molecules to address both affinity and specificity profiles at the same time has not been reported.Here, we describe an approach which queries hundreds of computationally re-designed proteins to not only quickly identify new protein–protein interactions (PPI) and small molecule–protein interactions (SMPI), but also comprehensively monitor promiscuous binding behavior and off-target interactions, which is crucial information for determination of lead candidates. We combine proteins to be queried into single pools and carry out selections against a series of target molecules. We then sequenced both the selected and the starting pools to obtain frequency changes in each pool which allows us to obtain a comprehensive binding profile addressing both relative affinities and specificities. Resulting profiles can provide crucial information for the determination of lead candidates in the development of protein-based therapeutics or will help to decipher protein interaction networks of naturally occurring proteins.
Experimental
Library construction
Gene fragments were synthesized (Gen9 Inc.) with 5′ and 3′ additions homologous to the pETCON plasmid17 (Fig. S1†) which has a size of about 6 kB, allowing recombination into the expression vector within the yeast cell. Genes encoding proteins designed to bind to protein targets (pool 1.A and pool 1.B, with partially overlapping genes), were additionally barcoded with an 18-base pair sequence after the stop codons. To clone these fragments into pETCON, we triple digested the vector DNA with NheI, XhoI and BglII to ensure linearization. After combining 2 μg of the pooled genes (average size of 450 bp) and 0.75 μg of the vector DNA, DNA was co-transformed into EBY100 yeast cell18 using electroporation.19 Transformed resulting library size was 5 × 106 transformed cells for the library of pool 1.A and 1.B. For the library generation of the potential small-molecule binding proteins (pool 2), we digested pETCON with NdeI and XhoI as the genes did not contain a gene-specific barcode. A total of 5 μg of the gene fragments and 1 μg digested pETCON vector were co-transformed in the same fashion as above. The transformation yielded 1 × 107 transformed cells.Gene pool selections
Yeast cells containing the synthetic gene libraries were grown overnight at 30 °C in 50 mL minimal medium containing 2% glucose, but lacking tryptophan and uracil. For induction, cell suspension was adjusted to an optical density (O.D.) of 1 at 600 nm, sub-cultured into SGCAA20 and grown at 22 °C for another 18 h. Before incubating with respective target molecules, yeast cells were washed once with phosphate buffered saline containing 0.1% BSA (PBSF) and normalized to an O.D. of 1 at 600 nm. The benchmark proteins, comprised of 5 variants of the translocated intimin receptor (TIR) protein, were tagged with individual 18 base-long barcodes and grown as 1 mL cultures independently. Cells were washed, normalized to an O.D. of 1, and combined equally. 50 μL of the mixed cells expressing the TIR variants were added to 1 mL of the normalized library pool.For PPI selections, we incubated 50 μL of the yeast pool in PBSF with 1 μM of biotinylated target protein for 3 h at 4 °C while rotating. To introduce avid conditions, 250 nM streptavidin–phycoerythrin (SAPE, Invitrogen) were added to the cell pool with the target protein and incubated for an additional hour at 4 °C. At this point, we also added 2 μg ml−1 anti-Myc FITC-labeled antibody. For 2.5 μM intimin, 625 nM SAPE was added. Before sorting, cells were washed once again with 1 mL ice-cold PBSF. For selections under non-avid conditions, cells were incubated with indicated concentrations for 3 h at 4 °C while rotating, washed once with ice-cold PBSF, and re-suspended in 100 μL PBSF before adding 35 nM SAPE and 2 μg mL−1 anti-Myc FITC-labeled antibody. Cells were incubated for an additional ∼40 min on ice, washed once more, and stored as a pellet on ice before sorting.
For SMPI selections, three different modifications of small molecules were utilized: 1). monovalent biotinylated ligands, 2). biotinylated-BSA conjugated ligands, and 3). biotinylated-70 K-dextran conjugated ligands. Fluorescent detection was enabled by incubation with SAPE and anti-Myc FITC conjugated antibody. For each ligand in category 1, 4 μM was pre-incubated with 1 μM SAPE to create additional avidity. For category 2, 2.5 μM biotinylated-BSA-ligand conjugates were mixed with 627 nM SAPE. For category 3, we combined 640 nM biotinylated-70 K-dextran ligand conjugates with 487 nM SAPE. To every 50 μL reaction, 1 μL anti-Myc-FITC was added. Cells (5 × 106) were labeled at room temperature for 2.5 hours while rotating and washed once with ice-cold PBSF before sorting.
For each target, at least 1 million cells were sorted using fluorescence-activated cell sorting (FACS) on a BD Influx sorter. Gates were drawn based on the signals observed (Fig. 3, 4 and S2†); cells that were only labelled with anti-cMyc-FITC conjugated antibody were used as a reference for background fluorescence at 580 nm and gates were drawn just above these populations.
DNA preparation and next-generation sequencing
Plasmids from cells of the starting and selected pools were extracted as previously described21 (see detailed procedure in ESI† Methods). Following a QIAgen PCR clean-up step producing a 30 μL DNA solution, 15 μL were subjected to PCR for the addition of selection-specific barcodes and flow cell adapters. For that, two PCR steps were performed. The first PCR used a set of “inner primers” to add the Illumina-specific primer annealing site that enables the use of primers included in the commercially available sequencing kit without the addition of custom sequencing primers. Additionally, we included a short 12 bp sequence, that can be used as a second barcode to label the selected gene pools. The 12mer sequences were designed to have maximal nucleotide diversity for 4 different sets of primers. This is important for the determination of cluster assignments by the Illumina machine and can be helpful when sequencing low sequence diversity libraries as it increases the apparent complexity detected by the machine. Primers were designed to have a lower annealing temperature for the first reaction (51 °C) (Table S3†). To add the Illumina flow-cell adapters and selection-specific barcodes, a second PCR step with a higher annealing temperature (64 °C) was performed using primers outer-F and a set of reverse primers containing various barcodes (Table S3†). Due to the significant difference between the two melting temperatures, a purification step for amplicon of the first PCR was not necessary and 2 μL of the first reaction served directly as template for the second reaction. All primers were PAGE purified. The first PCR step was performed for 14 cycles, whereas the second PCR was performed for 15 cycles. However, the cycles necessary for the first reaction depend on the efficiency of the DNA preparation from the yeast cells and may need more cycles, which can be monitored using qPCR. Resulting DNA fragments were gel purified and amounts were quantified by qPCR as instructed (Illumina qPCR manual).As all genes in the pool 1 libraries (PPI pools) were barcoded, we only sequenced the 18mer gene-specific barcodes (see ESI Methods and Fig. S1B, Table S3†), which allowed us to use a 50-cycle kit on a Miseq Sequencer (Illumina).21
As the pool 2 libraries (SMPI pools) do not contain gene-specific barcodes, we amplified and sequenced the whole gene. Plasmid-specific primers at the 5′ (upstream of the NheI site) and 3′ site (including the XhoI site) were used as amplification primers (Fig. S1, Table S3†). The final DNA prepared for sequencing was pooled using 5 times the amount of DNA for the reference pool. For sequencing whole genes (SMPI libraries, from pool 2 selection), a 300-cycle paired end sequencing run was performed.
Sequence analysis
Sequencing reads were split into the different populations based on their 12-bp and 6-bp selection-specific barcodes. Pools were treated identically for analysis and quality filtration. All sequences with an average quality score below 20 or if they contained any position with a score lower than 12 were rejected. Either barcodes or sequences between the restriction sites, depending on which library was analyzed, were extracted and counted. The frequency of each gene in each selected pool was normalized by its frequency in the reference pool and described as enrichment values. To provide an estimate of the reliability of the data, we incorporated a bootstrapping re-sampling step; we oversampled the library size by pulling 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sequences 50 times randomly from the raw sequencing data split into corresponding selection pools. This reduces artificially high appearing enrichment values caused by low sequencing coverage in the reference pool; spread of data can be monitored for any given input gene (see Fig. 3D and E and 4C and D). For each random draw of sequences (during our bootstrap analysis) frequencies from the reference pool (either starting library or cells selected for expression on the yeast surface) were then used to normalize all frequencies of each gene in any given selection.
000 sequences 50 times randomly from the raw sequencing data split into corresponding selection pools. This reduces artificially high appearing enrichment values caused by low sequencing coverage in the reference pool; spread of data can be monitored for any given input gene (see Fig. 3D and E and 4C and D). For each random draw of sequences (during our bootstrap analysis) frequencies from the reference pool (either starting library or cells selected for expression on the yeast surface) were then used to normalize all frequencies of each gene in any given selection.
For pool 1, the 18-bp gene specific barcodes were counted, and the frequencies obtained from the expression selection were used as the reference. Pool 2, which involved the identification of new small-molecule binders, was sequenced without gene-specific barcodes. Since several genes were too long to cover with the available sequencing cycles (300 × 2), we utilized only the forward sequences for counting. All obtained sequences appearing above 20 times in the reference pool were aligned to the input sequences using the basic local alignment search tool (BLAST) and the results were used to assign the sequenced fragments. The identified gene fragments were then used to count their occurrences in each selection pool. This analysis method can also be applied when input libraries contains unknown genes or open reading frames. For example, gene fragments occurring at a certain threshold in the input library can be quickly assigned by BLASTing against the examined organism's genome or any database.
Protein expression and purification
Intimins and designed proteins were expressed in E. coli. Details can be found in the ESI Methods.†Fluorescence polarization equilibrium binding assays
Fluorescence polarization-based affinity measurements of selected proteins were performed as noted previously22 using Alexa488-conjugated ligand. In a typical experiment, the concentration of the Alexa488-conjugated ligand was fixed below the dissociation constant (Kd) of the interaction being monitored and the effect of increasing concentrations of protein on the fluorescent anisotropy of Alexa488 was determined. Fluorescence anisotropy (r) was measured in 96-well plate format using a SpectraMax M5e microplate reader (Molecular Devices) with λex = 485 nm and λem = 538 nm and using a 515 nm emission cutoff filter. In all experiments, standard phosphate-buffer saline (PBS, pH 7.4) was used as the buffer system at room temperature.Results and discussion
Overview and workflow
The proteins of interest are displayed on the surface of yeast and evaluated for binding to biotinylated target molecules (Fig. 1). DNA fragments encoding the protein of interest with flanking regions containing a short sequence for homologous recombination into the surface expression vector were co-transformed into yeast cells with linearized plasmid DNA and evaluated in pools for display and binding to a set of target molecules. To compensate for possible overrepresentation of individual genes, the fraction of each clone in the starting pool was determined. To correct for distribution changes due to growth differences during induction or expression variation, selections were carried out for cells that expressed the protein on the surface of the yeast and used as the reference population. Plasmid DNA for the unsorted gene library and each selected pool was isolated and tagged via PCR with sequencing-chip tethering adapters and individual barcodes for each sorting experiment. After next-generation sequencing, sequences were counted, and gene frequencies were normalized to their corresponding reference pool. The enrichment values provide information on the affinity and specificity of each queried protein for each target and thereby portray specificity profiles.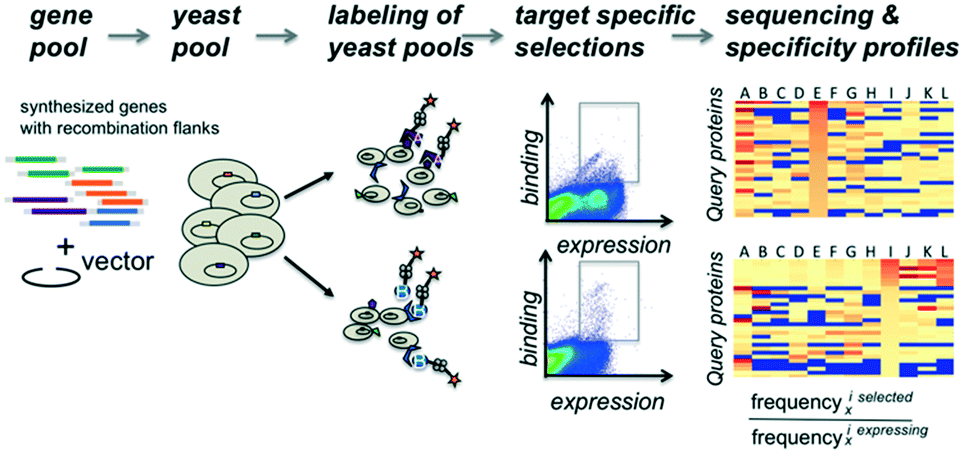 | ||
| Fig. 1 Workflow overview. Gene fragments with flanking sequences for homologous recombination into the surface expression vector are co-transformed into yeast cells with linearized plasmid DNA. Aliquots of the yeast cell pool expressing recombinant genes as a fusion to the Aga2 surface protein are incubated independently with various fluorescently labeled query molecules. After sorting of fluorescently labeled cells, each selected pool receives a selection-specific barcode. DNA fragments from each selected pool are sequenced and the occurrence of each gene within a given pool is counted. Counts are normalized by dividing their frequency in the selected pool by their frequency observed in the starting pool. | ||
We illustrate the usefulness of this approach in a variety of applications. First, we examined a control set of variants of the translocated intimin receptor (TIR) with point mutations spanning two orders of magnitude differences in affinity. We then used the method to assess binding affinity and specificity of designed binding proteins for a set of protein targets and small molecule targets respectively.
Tuning of sorting conditions
We reasoned that selections could be made sensitive to different affinity ranges by manipulating target concentrations, incubation conditions and sorting gate settings. As a model system, we chose the interaction between the cell surface adhesion protein intimin of the enteropathogenic Escherichia coli with its receptor protein TIR because a series of single point variants are known which tune the binding affinity over two orders of magnitude in the micromolar range (this is the range most relevant to initial binding screens). The interaction between wild type TIR and intimin has a dissociation constant of 577 nM, whereas the point mutations N300D, N300Q and N300A have dissociation constants of 5.5 μM, 20.2 μM and 69.9 μM respectively. Each TIR variant was tagged with a barcode and flanking regions for homologous recombination as described above. Duplicate constructs with different barcodes were generated for wild type TIR and the TIR variants N300D and N300Q to assess the variance in the enrichment values. In the sorting experiment we included 615 completely unrelated proteins (pool 1, see Methods and Table S1†) to mimic the practical of screening large numbers of unrelated clones in parallel. Pool 1 gene frequencies before and after selections, including all TIR variants, were obtained through sequencing and counting of the barcodes (see Methods ESI†).We performed selections at 500 nM intimin, which is close to the KD of the wild type TIR interaction, 1 μM and 2.5 μM with and without avidity effects (Fig. 2). Expression levels were detected by incubating with anti-cMyc-tag antibody conjugated to FITC, and binding of the biotinylated intimin was monitored through the addition of streptavidin conjugated to phycoerythrin (SAPE, see Methods ESI†). Gates were set to capture cells with red fluorescence (emission at 580 nm, Y-axis) greater than that of cells only labelled with anti-cMyc FITC-conjugated antibody (emission 530 nm, X-axis), unless noted otherwise (Fig. S2†). A distinct single population was observed at 500 nM intimin, and sequencing confirmed the enrichment of primarily wild type TIR. At 2.5 μM intimin, a second population appeared (Fig. 2A) corresponding to the enrichment of the N300D variant with a Kd of 5 μM (Fig. 2B). To boost apparent affinity through avidity effects, we incubated the biotinylated target protein with a 4:1 ratio to tetrameric streptavidin–PE (SAPE) to promote non-covalent oligomerization20 (see Methods ESI†). With 1 μM intimin under avid conditions, 4 distinct populations were observed (Fig. 2A), and the four TIR variants are enriched to different extents consistent with their dissociation constants. At 2.5 μM intimin conjugate, the populations start to merge as the signal saturates: a gate selecting cells above background captures all variants equally well, even the weak binder TIR N300A (Fig. 2). At 1 μM, the correlation of enrichment values with affinities depends on the positioning of the gates: more stringent gating separated variants according to their relative affinities, whereas more lenient gating resulted in little separation. Overall, as is evident in Fig. 2B, by varying the conditions the selection can be made responsive to different affinity ranges. The ratio of the unsorted frequencies of all TIR variants to the frequencies after selecting for expression was consistently around 1 (1.093 ± 0.3 when taking all 7 constructs into account) indicating that selection, preparation of the DNA of input and selected pools, and sequencing do not introduce significant biases (Fig. 2).
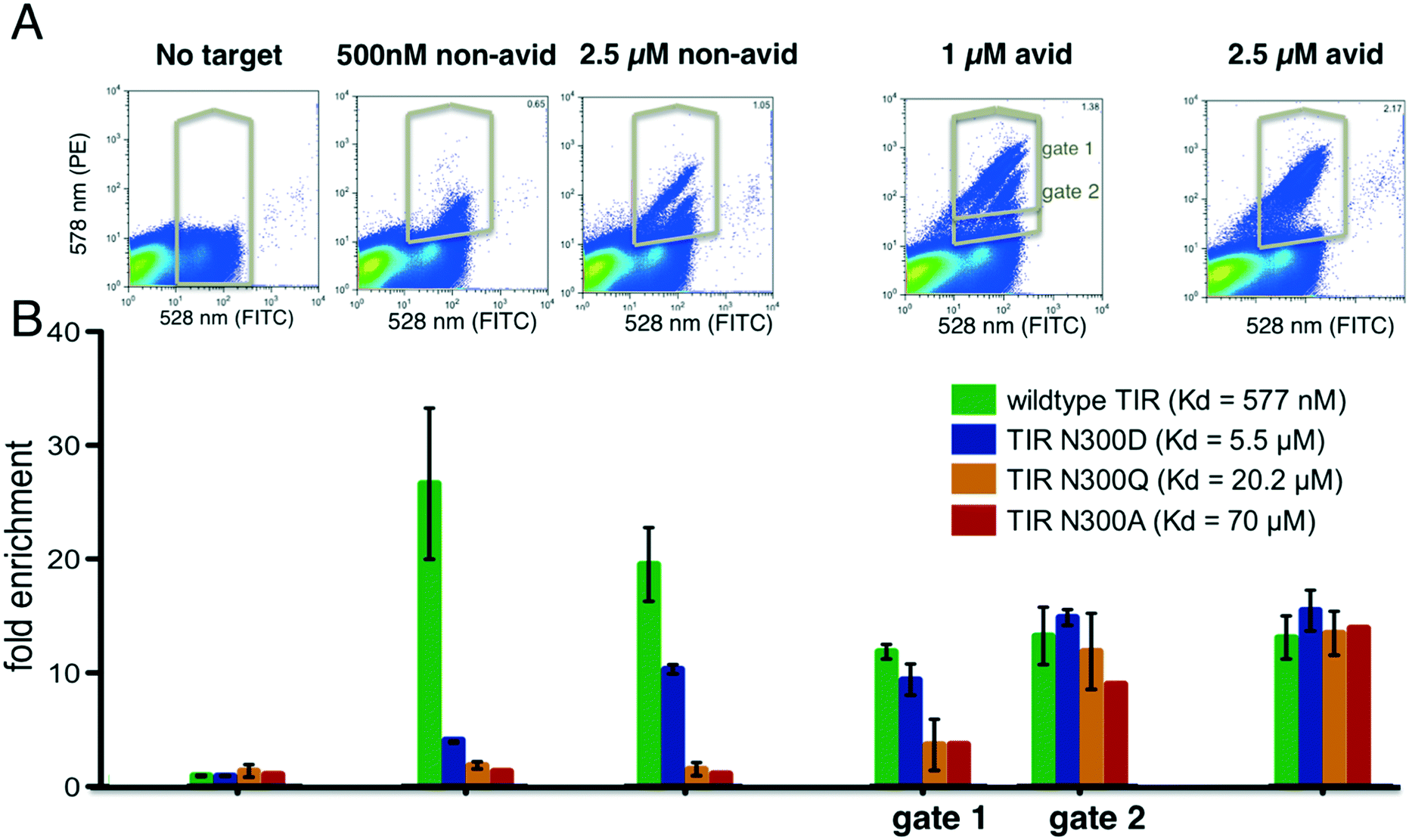 | ||
| Fig. 2 Tuning of enrichment ratio–affinity relationship. Various point mutants of the intimin receptor TIR from the enteropathogenic E. coli (variants span two orders of magnitude difference in their binding constants) expressed on the surface of yeast, were added to a pool of yeast cells expressing synthetic genes of re-designed proteins, incubated with indicated concentrations of biotinylated intimin (sorted via flow cytometry under indicated conditions). (A) FACS scatter plots showing binding (Y-axis) to intimin and expression (X-axis) of yeast pools containing 615 different genes and TIR variants. (B) Enrichment of different TIR variants after selection with biotinylated intimin under indicated conditions. Gene frequencies in selected pools were divided by the frequencies obtained after selecting for clones that displayed the C-terminal cMyc-tag (histogram on the very left side labelled “no target”). The first bar chart group shows the ratio of the frequencies from the unsorted library to the frequencies after selection for display. Error bars reflect the standard deviation of measurements obtained using two genetic constructs, each containing a different “gene-specific” barcode. Data without error bars reflects a single construct. Subsequent groups bars in the chart represent enrichments obtained after selections reflected in the histograms above each group. Under non-avid conditions and 500 nM of intimin, wild type is highly enriched, whereas under avid conditions and 2.5 μM intimin all variants are detected. | ||
Discovery of protein interactions via multiplex selections and evaluation of affinity and specificity profiles
We evaluated the binding properties of the first two partially overlapping libraries of designed proteins (pool 1.A and B) that were designed to binding to protein targets by incubating them with two panels of proteins. The first panel of seven target proteins have unrelated folds, no sequence similarity and are of viral or human origin, namely: H3 hemagglutinin (H3) of Influenza (A/Hong Kong/1/1968), Frizzled 7 (Fz), the surface protein L1 and a variant of L1 from smallpox, CTLA and the Ebola glycoprotein (GP). The second panel contained H3, Fz and PD1 as well as two hemagglutinin versions from H1 from A/South Carolina/1/18 (SC) and A/Solomon Islands/3/2006 (SI). The query proteins in the pooled library were computational re-designed proteins from a diverse set of existing natural proteins coming from 117 (for the PPI libraries) or 68 (for the SMPI library) different organisms (Table S2†). However, they all had in common that they could be expressed in E. coli as reported in their crystal structure deposit file in the protein database. To explore the use of the method for assessing protein–small molecule interactions, we interrogated the binding properties of a pool of 228 designed proteins of pool 2 to 7 conjugated small molecule ligands: cortisol (HCY), 17-hydroxyprogesterone (OHP), vitamin D (VitD), mycophenolic acid (MPA), apixaban (APN), artemisinin (ART), and biotin (BTN). With the exception of HCY and OHP, these small molecule targets are quite different in their molecular properties. Ligands were either conjugated directly to biotin, biotinylated BSA or dextran (BSA and dextran are used as a “carrier” molecule to increase avidity effects for detecting weak interactions). Successfully designed protein binders often have weak initial activity for their intended targets, as the design process still needs improvement. However, once active designs have been identified, only one or two mutations can improve affinity up to 100-fold.17 Hence, even designs with weak affinity can provide a valuable starting point for further optimization. On the other hand, designs that bind non-specifically to other proteins may be partially misfolded, which can cause a general “stickiness” as a result of possibly exposed hydrophobic core residues. These designs are less likely to be rescued by subsequent optimization as they likely have multiple, unpredictadable conformations for which several mutations are likely needed to establish a stable binding conformation. Although additional protease-based stability selection can eliminate some misfolded designs from the pool.14 Protease resistance assay cannot detect misfolding caused by domain swaps or aggregate formation which can still have cause nonspecific binding. Hence, for the initial test of designed proteins, the evaluation of binding specificity is as important as affinity, arguably even more important for hydrophobic interactions. Selection conditions that allow highly sensitive detection were chosen to ensure that a wide range of activity levels are monitored at once for evaluating binding specificity.To achieve the highest sensitivity, sorting gates for the identification of binding proteins were set so that any cell with signal above background was collected (Fig. S2,† for concentrations and incubation condition see Methods). To ensure complete coverage of the library, selections were oversampled by screening at least 200 times more yeast cells than the library size. To adjust for differences in the starting distributions, frequencies in selected populations were normalized by their corresponding frequencies in the reference population. For this purpose, a pool selected for expression and the unsorted pool populations were sequenced for PPI and SMPI libraries respectively.
A. Identification of protein–protein interactions
As expected, TIR variants bind specifically only to their cognate binding partner intimin (Fig. 3A) and not to any other target protein. Population distributions and gating conditions for selection against H3 HA and Fz are shown in Fig. 3B. The top 7 enriched designs from the selection for H3 HA showed specific binding to this target with one exception (Fig. 3C); the information that the protein design_II.60 also binds off-target proteins would have been missed if the design had been only screened against the target molecule. It is likely that several of the promiscuously binding designs do not fold into the expected conformation and would therefore be poor candidates for any further improvements or applications. Selection for Fz resulted in the identification of one highly enriched binding protein (Fig. 3D). We repeated the experiment by assembling a second pool (pool 1.B) of 230 designs with an overlap of 130 designs of pool 1.A (including identified binders from experiment 1). For the repeat, we re-started with transforming the linear gene fragments with linearized plasmid into yeast. For the repetition, 3-fold less H3 HA was used for the selections. The same binders were again identified (designs_II.23, 24, 52, 53, 55 in Fig. S6;† design_II.41 was not detected in any of the pools; likely the cloning of this gene fragment or its transformation failed). For Fz, we confirmed the significant enrichment for design_III.27 (Fig. 3E and S6†). To ensure the analysis was not biased by the sequencing counts, we ensured that there was no correlation between counts and enrichments (Fig. S4†).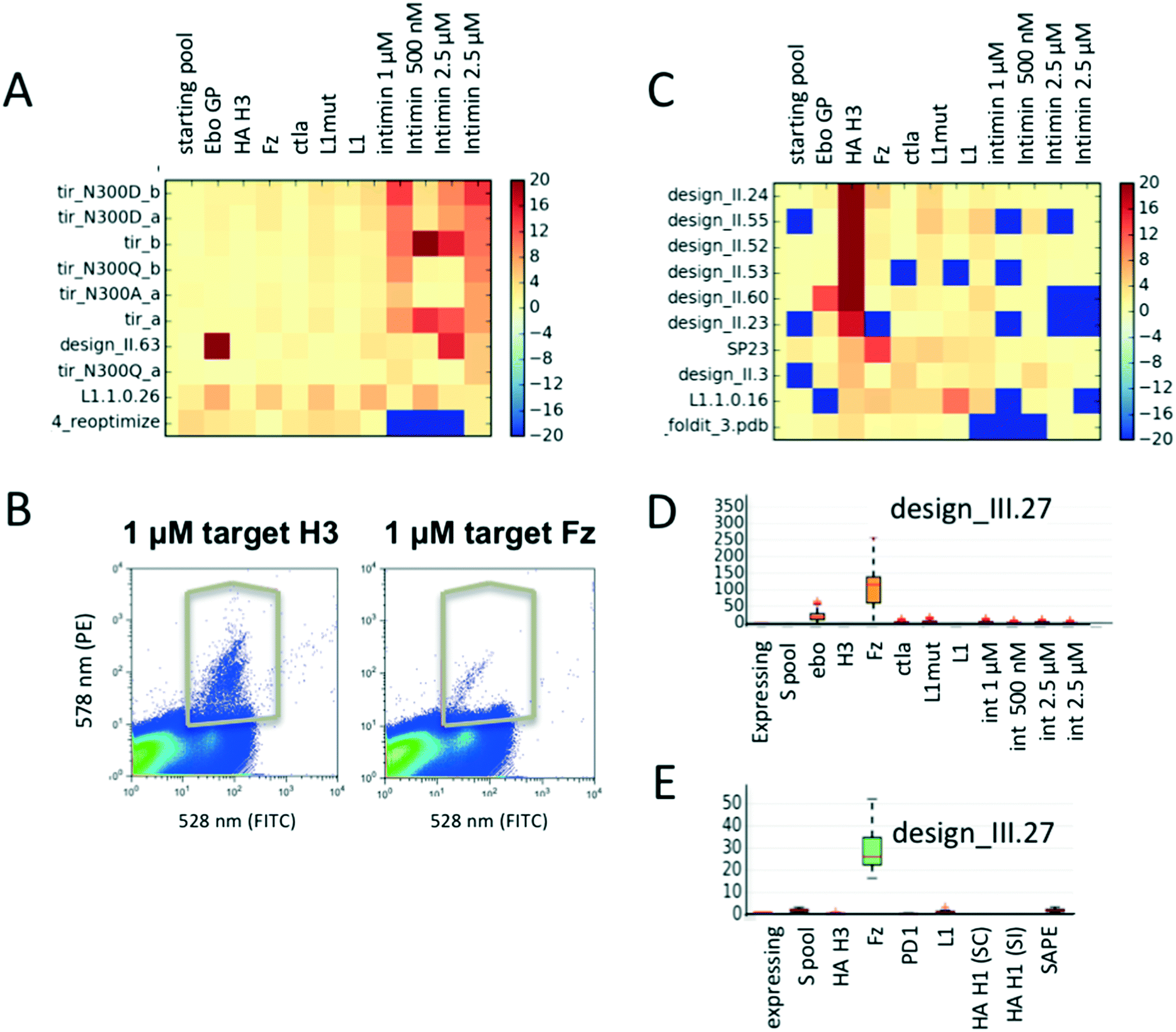 | ||
| Fig. 3 Parallel evaluation of protein binding activities and specificity. (A) Enrichment values of TIR variants selected for binding to various biotinylated intimin concentrations as well as several other biotinylated target proteins. As expected, the TIR variants are highly specific for their cognate binding partner intimin. Warm colors reflect enrichment, cold depletion. (B) Flow cytometer scatter plots demonstrating binding signal for influenza hemagglutinin of the H3 of the Hong Kong 1968 strain and Fz, as well as gates used for their selections. (C) Mean enrichment values of designed proteins sorted for proteins binding to target H3 hemagglutinin; missing data is set to −20, values with enrichment above 20 are capped at 20. Mean enrichment values are obtained through bootstrapping: a sample size of N = 20000 sequences were pulled from the raw sequencing data for R = 50 times. (D and E) Mean enrichments and specificity profiles for design_III.27 after sorting of two different library pools. Data spread for each sample can be seen in the boxplots. Larger spreads are a result of lower sequence counts for a given sequence. | ||
To investigate how many sequencing reads are necessary for a given library to obtain meaningful enrichments values, we simulated the effect of smaller numbers of reads. We calculated enrichment values for 3 newly identified new binders (design_II.52, 24 and design_III.27), and monitored them while decreasing the number of sequencing reads used for analysis (Fig. S3†). While the median enrichment values stabilize between 1.5–2 fold coverage of the library size, outliers occur less after 15–30 fold coverage of the library. Hence, the number of reads should be at least 20–30 fold greater than the library size to obtain reliable data. Both the number of yeast cells to be screened and sequencing reads depend on the distribution of the gene frequencies of the starting library. To get a rough estimate of the confidence for a given data-point, we applied a simple bootstrapping procedure (see Methods ESI†). Fluctuations in enrichment values occur when either the reference library or selected pool have low counts.
B. Selections of small molecule binders
Fluorescence activated cell sorting (FACS) was carried out for yeast cells containing pool 2 with each ligand conjugate. To increase the signal to noise ratio, we performed two rounds of sorting. Using the procedure described above (and in the Methods section ESI†), we identified several new binding proteins (Fig. 4A–D). We found that several failed designs for binding artemisinin (Fig. 4B: 186_Art, 286_ART, 259_ART and 365_ART) showed strong off-target binding to other hydrophobic ligands OHP, HCY and VitD. This observation indicates that featureless hydrophobic pockets (like the ones seen in those designs) cannot achieve desired affinity and specificity. Our analysis focused on two proteins designed for binding OHP, designs C9 and C10. While C9 does not show binding to the other ligands (Fig. 4B and C), C10 binds to a variety of other compounds (Fig. 4B and D). To verify that the enriched proteins indeed bind to these multiple compounds, we tested the designs as individual clones by flow cytometry (Fig. 4E). Design C10 showed a clear binding signal to 1 μM biotinylated OHP and 1 μM biotinylated VitD when displayed on yeast surface. Design C9 binds biotinylated OHP, but not biotinylated VitD or PE-FITC (Fig. 4E). We also tested binding of C10 to BSA-biotin, for which C10 showed a very low enrichment just above background; but binding was not observed indicating that these low values are within background noise levels. To determine whether these designs bind small molecules in solution and to obtain approximate binding affinities, genes were cloned into bacterial expression vectors and purified. Fluorescence anisotropy titrations determined that design C9 binds OHP with micromolar affinity and design C10 binds with a much weaker affinity (Fig. 4F). This biochemistry binding measurement using purified proteins confirmed the results from our cell-based selection assay. The fact that low-affinity binder C10 shows higher level of off-target binding towards VitD suggests that the less optimized hydrophobic pockets often leads to nonspecific interactions.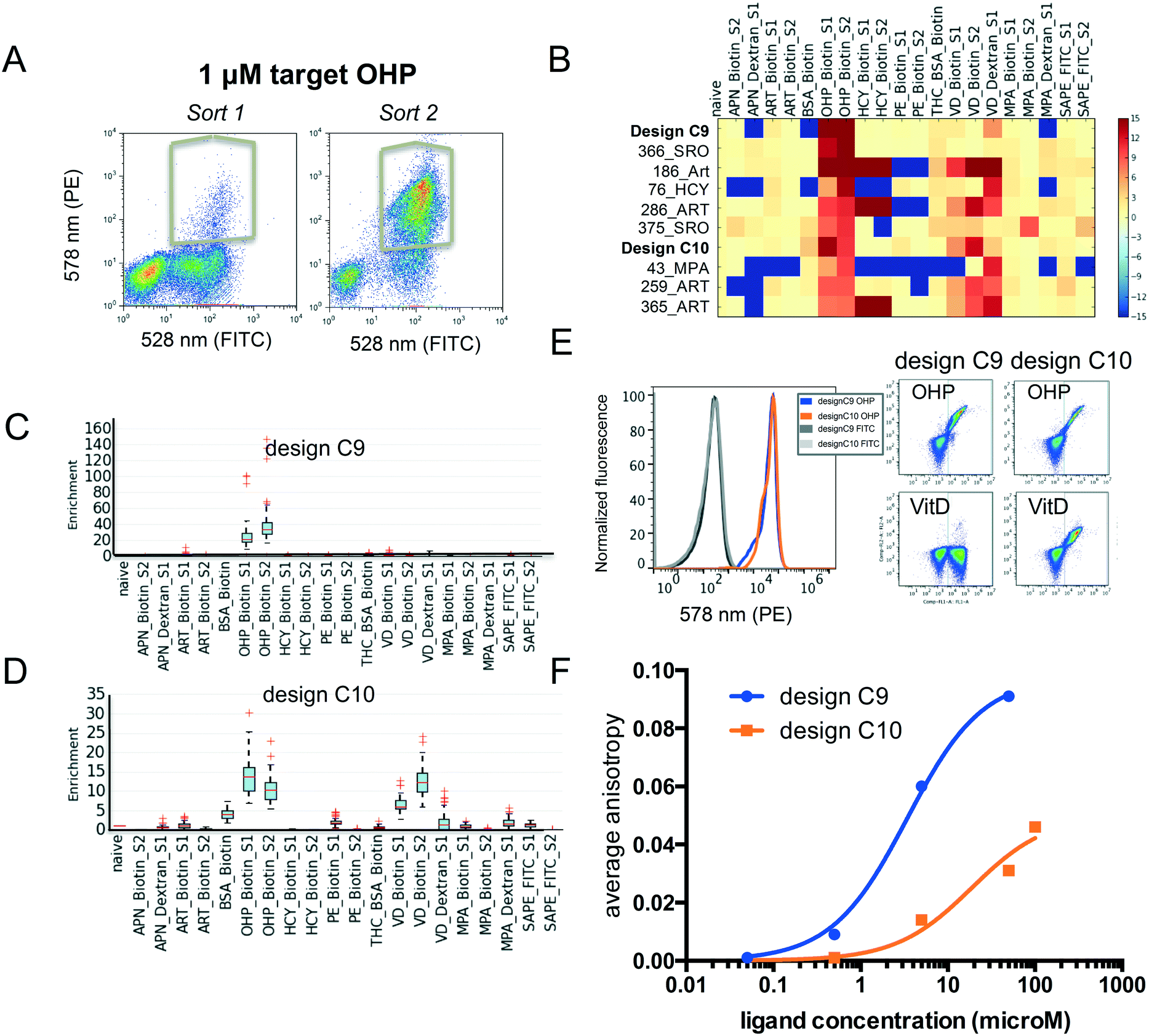 | ||
| Fig. 4 Evaluation of small molecule binding affinity and specificity. (A) Flow cytometer scatter plots demonstrating binding signal for OHP during sort 1 and 2, as well as gates used for selections. (B) Mean enrichment values sorted for binding to OHP-biotin. Values above 15 or below −15 were clipped and missing data set to −15. (C and D) Boxplots of the bootstrapped enrichments of the identified new binding proteins design C9 and C10. (E) Flow cytometry measurement of binding to indicated ligand–biotin conjugates. Scatter plots show binding of design C9 (left) and C10 (right) to 1 μM OHP-biotin. (F) Titrations of purified C9 and C10 measured via fluorescence anisotropy for binding to OHP conjugated to Alex488. | ||
Conclusions
We demonstrate that pooling a variety of unrelated genes and selecting for binding of their surface expressed proteins to multiple fluorescently labeled targets by flow cytometry enables a rapid assessment of relative affinities and specificity profiles. Such selections distinguish proteins that bind specifically to a desired molecule from those that bind non-specifically to multiple targets. In case of designed proteins, off-target binding can indicate problems with the structural integrity of the protein such as the exposure of hydrophobic core residues. For protein engineering in general, monitoring of off-target interactions is crucial for the development of novel protein-based therapeutics, diagnostics and synthetic sensors from engineered recombinant proteins or antibodies. Our method can be used in early discovery steps to facilitate decisions on lead candidates. The pooling strategy could likely be applied to other screening platforms such as phage display, ribosome display, and GFP reassembly assays as long as the proteins can be expressed by E. coli or in vitro as in case of ribosome display. As gene synthesis is becoming cheaper and genomic libraries are becoming readily available, high throughput analysis of protein interactions is becoming increasingly powerful. Highly parallel analyses as described here provide an effective way to extract maximum information content on binding affinity and specificity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Per Greisen and Daniel Adriano Silva Manzano for helpful discussions. We would also like to thank Chris Garcia for providing Frizzle 7 and Ian Wilson for providing the hemagglutinin variants. The work was supported by Defense Threat Reduction Agency Grant HDTRA1-16-C-0029. EMS was supported by 1R01AI140245 and 1R21AI143399.References
- A. Ernst, D. Gfeller, Z. Kan, S. Seshagiri, P. M. Kim, G. D. Bader and S. S. Sidhu, Mol. BioSyst., 2010, 6, 1782–1790 RSC.
- L. L. Reich, S. Dutta and A. E. Keating, J. Mol. Biol., 2015, 427(11), 2135–2150 CrossRef CAS PubMed.
- R. M. Adams, T. Mora, A. M. Walczak and J. B. Kinney, Elife, 2016, 5, e23156 CrossRef PubMed.
- C. A. Kowalsky, M. S. Faber, A. Nath, H. E. Dann, V. W. Kelly, L. Liu, P. Shanker, E. K. Wagner, J. A. Maynard, C. Chan and T. A. Whitehead, J. Biol. Chem., 2015, 290(44), 26457–26470 CrossRef CAS PubMed.
- T. Van Blarcom, A. Rossi, D. Foletti, P. Sundar, S. Pitts, C. Bee, J. Melton Witt, Z. Melton, A. Hasa-Moreno, L. Shaughnessy, D. Telman, L. Zhao, W. L. Cheung, J. Berka, W. Zhai, P. Strop, J. Chaparro-Riggers, D. L. Shelton, J. Pons and A. Rajpal, J. Mol. Biol., 2015, 427, 1513–1534 CrossRef CAS PubMed.
- B. V. Adkar, A. Tripathi, A. Sahoo, K. Bajaj, D. Goswami, P. Chakrabarti, M. K. Swarnkar, R. S. Gokhale and R. Varadarajan, Structure, 2012, 20, 371–381 CrossRef CAS PubMed.
- B. P. Roscoe, K. M. Thayer, K. B. Zeldovich, D. Fushman and D. N. Bolon, J. Mol. Biol., 2013, 425, 1363–1377 CrossRef CAS PubMed.
- Z. Deng, W. Huang, E. Bakkalbasi, N. G. Brown, C. J. Adamski, K. Rice, D. Muzny, R. A. Gibbs and T. Palzkill, J. Mol. Biol., 2012, 424, 150–167 CrossRef CAS PubMed.
- L. M. Starita, J. N. Pruneda, R. S. Lo, D. M. Fowler, H. J. Kim, J. B. Hiatt, J. Shendure, P. S. Brzovic, S. Fields and R. E. Klevit, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1263–E1272 CrossRef CAS PubMed.
- T. A. Whitehead, A. Chevalier, Y. Song, C. Dreyfus, S. J. Fleishman, C. De Mattos, C. A. Myers, H. Kamisetty, P. Blair, I. A. Wilson and D. Baker, Nat. Biotechnol., 2012, 30, 543–548 CrossRef CAS PubMed.
- C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Nature, 2013, 501, 212–216 CrossRef CAS PubMed.
- E. M. Strauch, S. J. Fleishman and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 675–680 CrossRef CAS PubMed.
- C. L. Araya, D. M. Fowler, W. Chen, I. Muniez, J. W. Kelly and S. Fields, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16858–16863 CrossRef CAS PubMed.
- G. J. Rocklin, T. M. Chidyausiku, I. Goreshnik, A. Ford, S. Houliston, A. Lemak, L. Carter, R. Ravichandran, V. K. Mulligan, A. Chevalier, C. H. Arrowsmith and D. Baker, Science, 2017, 357, 168–175 CrossRef CAS PubMed.
- D. M. Fowler and S. Fields, Nat. Methods, 2014, 11, 801–807 CrossRef CAS PubMed.
- E. Firnberg, J. W. Labonte, J. J. Gray and M. Ostermeier, Mol. Biol. Evol., 2014, 31, 1581–1592 CrossRef CAS PubMed.
- S. J. Fleishman, T. A. Whitehead, D. C. Ekiert, C. Dreyfus, J. E. Corn, E. M. Strauch, I. A. Wilson and D. Baker, Science, 2011, 332, 816–821 CrossRef CAS PubMed.
- L. Benatuil, J. M. Perez, J. Belk and C. M. Hsieh, Protein Eng., Des. Sel., 2010, 23, 155–159 CrossRef CAS PubMed.
- L. Benatuil, J. M. Perez, J. Belk and C. M. Hsieh, Protein Eng., Des. Sel., 2010, 23, 155–159 CrossRef CAS PubMed.
- G. Chao, W. L. Lau, B. J. Hackel, S. L. Sazinsky, S. M. Lippow and K. D. Wittrup, Nat. Protoc., 2006, 1, 755–768 CrossRef CAS PubMed.
- T. A. Whitehead, A. Chevalier, Y. Song, C. Dreyfus, S. J. Fleishman, C. De Mattos, C. A. Myers, H. Kamisetty, P. Blair, I. A. Wilson and D. Baker, Nat. Biotechnol., 2011, 30, 543–548 CrossRef PubMed.
- A. M. Rossi and C. W. Taylor, Nat. Protoc., 2011, 6, 365–387 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00118b |
| ‡ Current affiliation: Department of Pharmaceutical & Biomedical Sciences, University of Georgia, Athens, GA 30602, USA. |
| This journal is © The Royal Society of Chemistry 2020 |