Crystal structure and thermoresponsive luminescence of a 9,10-bis(phenylethynyl)anthracene-based cyclophane†
Received
13th August 2019
, Accepted 2nd September 2019
First published on 2nd September 2019
Abstract
Introducing a cyclic structure to luminophores is a promising approach for achieving external stimuli-responsive luminescence. This is because luminescent cyclophanes containing flexible linkers tend to form several molecular assembled states. However, previously reported cyclophanes exhibiting thermoresponsive and/or mechanoresponsive luminescence have not given crystals suitable for single crystal X-ray structure analysis because of the flexible cyclic molecular structures. Such analysis is important because solved crystal structures can show unambiguous correlation between the arrangement of luminophores and photophysical properties. Here, we report the crystal structure of a cyclophane featuring a 9,10-bis(phenylethynyl)anthracene group and the thermoresponsive luminescence. The cyclophane was designed with shorter flexible oligo(ethyleneglycol) chains used as linkers bridging the luminophore and another aromatic group. The two different π-conjugated groups were orthogonally arranged in the individual molecule, and the luminophores partially overlapped between adjacent molecules. The cyclophane showed a supercooled nematic phase at room temperature upon cooling. Thermal treatment for the kinetically trapped state led to a transition to another crystalline state and, consequently, a change in photoluminescence colour. Emission spectroscopic studies and emission lifetime measurements revealed that the luminophores formed excimers in the supercooled nematic phase, whereas no excimer formation was observed for the crystalline phases.
 Yoshimitsu Sagara | Dr. Yoshimitsu Sagara is currently an Assistant Professor at the Research Institute for Electronic Science in Hokkaido University, Japan. He received his Ph.D. from the University of Tokyo in 2009. After working as a JSPS postdoctoral fellow in Prof. Takashi Kato's group for one year, he joined Prof. Tetsuo Nagano's group as a JSPS postdoctoral fellow in the University of Tokyo. He then joined Prof. Christoph Weder's group as a JSPS Postdoctoral Fellow for Research Abroad. He was appointed to his current position in 2015, and was appointed as a PRESTO Researcher of the Japan Science and Technology Agency (JST) in 2017. His research interests include various external stimuli-responsive luminescent materials and their applications. |
Design, System, Application
Luminescent cyclophanes featuring flexible linkers that bridge aromatic groups are promising candidates for external stimuli-responsive luminescent materials. However, the flexible molecular structures prevent the formation of crystals suitable for single crystal X-ray structure analysis. Here, we demonstrate that appropriate molecular design can overcome this issue while maintaining external stimuli-responsive luminescence character. Our design strategies can be applied to other luminescent cyclophanes, which furthers understanding on the correlation between molecular packing and stimuli-responsive luminescent properties. This may promote the development of more sophisticated photofunctional materials based on cyclophanes.
|
Introduction
Cyclophanes are cyclic compounds containing aromatic groups bridged by aliphatic linkers. Cyclophanes have been investigated in synthetic chemistry because of their highly symmetric structures and fascinating three-dimensional molecular shapes.1–12 The cavities of cyclophanes can intercalate guest molecules or ions, thus cyclophanes have also been well studied in supramolecular chemistry.7,13–15 When luminophores are used as the aromatic groups of cyclophanes, the photophysical properties can change upon intercalating guest molecules or ions.16–23 Therefore, luminescent cyclophanes are attractive candidates for molecular probes that quantitatively evaluate the concentrations of guests in solution. In such cases, the cyclophane effectively exhibits photofunctions as an individual molecule.
Cyclophanes in condensed states have recently attracted much attention. Various cyclophanes show a liquid-crystalline phase, and their phase transition behaviours have been well studied and compared to those of linear analogues.24–32 Furthermore, thermal and/or mechanical stimuli could induce changes in the photophysical properties of luminescent cyclophanes. Changes in luminophore arrangements, which result from thermal and/or mechanical stimuli-induced phase transitions, lead to external stimuli-responsive luminescence.33–41
Since the first report of a thermo- and mechanochromic luminescent cyclophane having two 9,10-bis(phenylethynyl)anthracene moieties in 2016,33 several luminescent cyclophanes have been prepared by our group.34–39,41 Their stimuli-responsive luminescence properties and phase transition behaviour have been investigated. Cyclophanes containing only one luminophore featuring hexaethyleneglycol or pentaethyleneglycol linkers show supercooled nematic (SC-N) liquid-crystalline phases, when the photoluminescent cyclophanes in the nematic phase are rapidly cooled to room temperature.35,38,39 Thermal treatment to the thermodynamically metastable states induces phase transitions to thermodynamically stable crystalline phases, which results in changes in photoluminescence colours.35,38,39 Transition from the SC-N to crystalline phases could be used for the reversible emission colour switching of triplet–triplet annihilation-based photon upconversion38 and changes in linearly-polarized photoluminescence properties.39 However, though the flexible molecular structures allow us to access thermoresponsive luminescence, obtaining crystals suitable for single crystal X-ray structure analysis is difficult. Consequently, no crystal structures of luminescent cyclophanes have been solved in our group so far. Molecular arrangements clarified by such analysis should give significant information on the relationship between the molecular assembled structure and photophysical properties.
Here we demonstrate that the crystal structure of a luminescent cyclophane featuring long and flexible linkers can be solved by adopting an appropriate molecular design. The obtained cyclophane 1 (Fig. 1) can access a SC-N phase and exhibit an emission colour change from yellow to green upon a phase transition to another crystalline phase on heating.
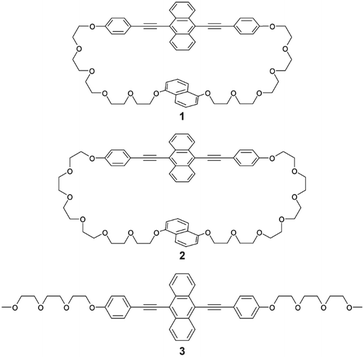 |
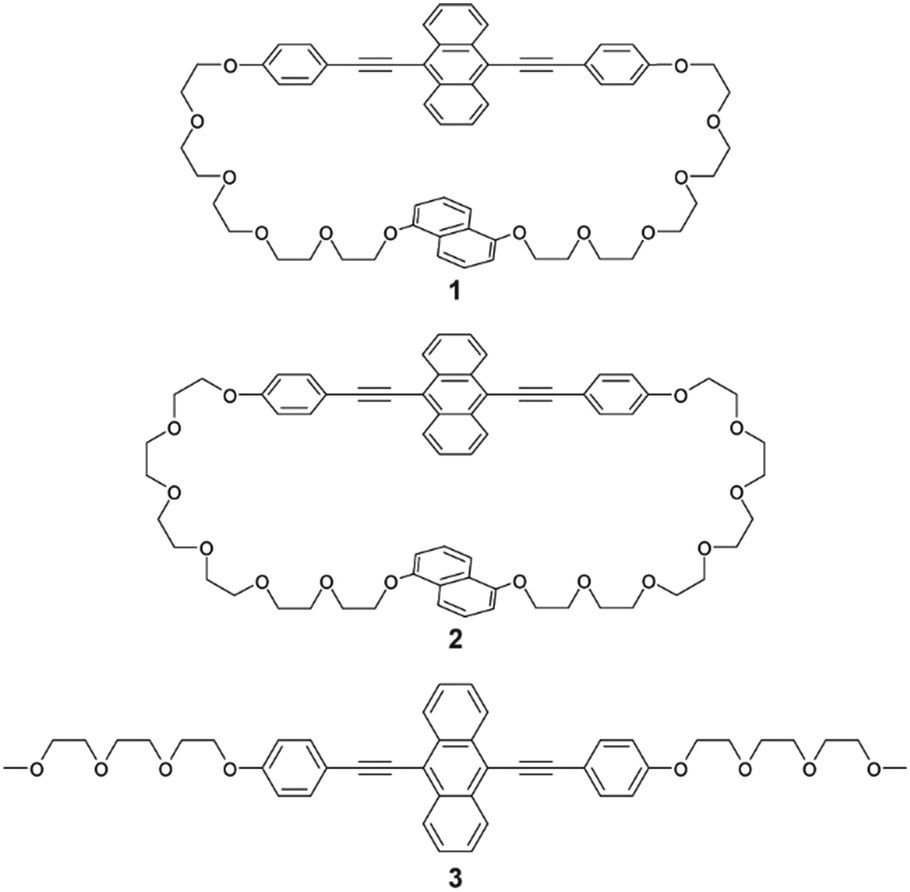
| | Fig. 1 Molecular structures of 9,10-bis(phenylethynyl)anthracene-based cyclophane 1, previously reported cyclophane 2, and linear analogue 3. | |
Results and discussion
Molecular design
Cyclophane 1 was designed based on the phase transition behaviour and thermoresponsive luminescence exhibited by previously reported cyclophane 2 (Fig. 1).38 The 9,10-bis(phenylethynyl)anthracene-based cyclophane 2 with hexaethyleneglycol linkers exhibited yellow emission in the nematic phase under excitation at 365 nm. When cyclophane 2 in the nematic phase on the glass substrate was put on a heat sink, a SC-N phase was obtained which maintained yellow emission. Annealing at 80 °C resulted in a phase transition from the SC-N phase to a crystalline phase, and the emission colour changed from yellow to green. To retain the thermoresponsive luminescence character and obtain suitable single crystals, slightly shorter oligo(ethyleneglycol) linkers and the same luminophore were used for cyclophane 1. The newly-designed cyclophane was prepared through Williamson ether reaction from 9,10-bis(4-hydroxyphenylethynyl)anthracene and naphthalene with two bromo-terminated pentaethyleneglycol groups.
Crystal structure
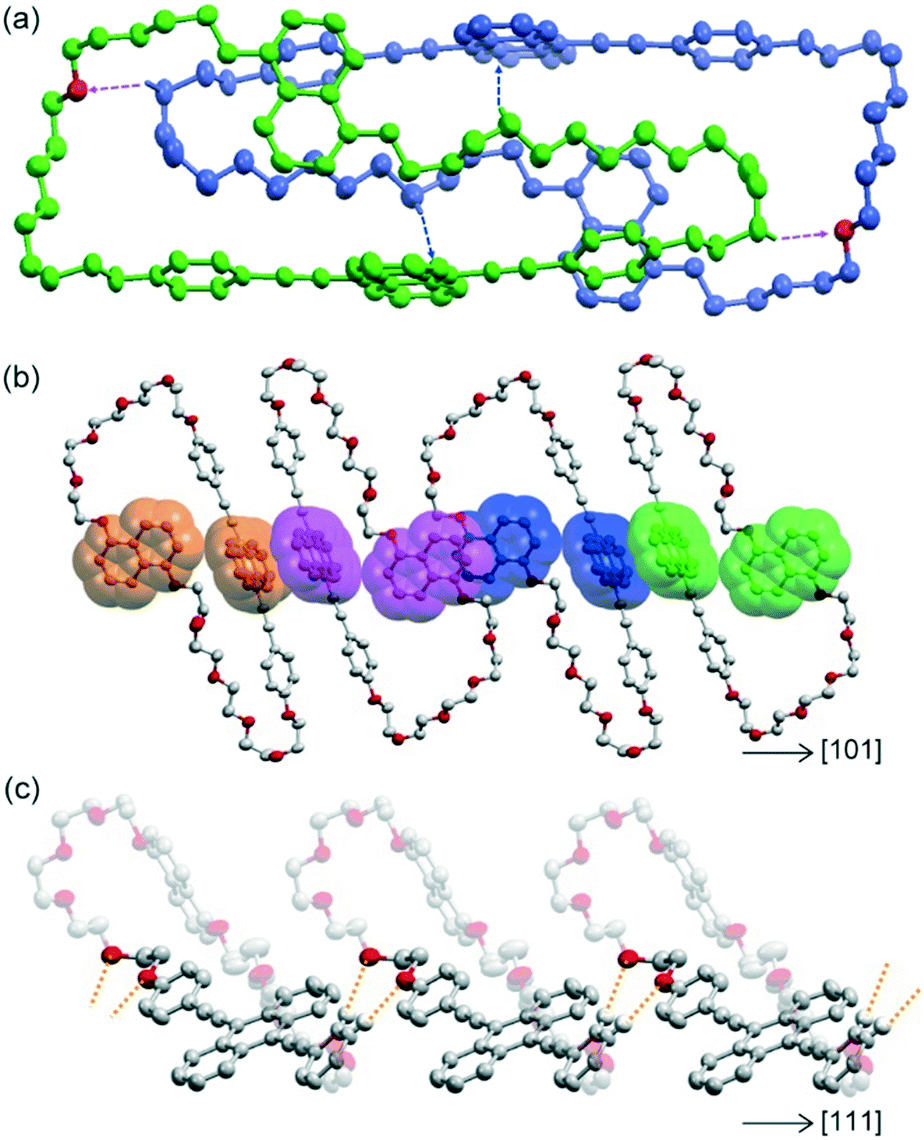
Yellow-emissive crystals suitable for single crystal X-ray structure analysis (Cr1 phase) were obtained from tetrahydrofuran solution by slow evaporation. Cyclophane 1 in the Cr1 phase was crystallized with the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , where one molecule of cyclophane 1 was crystallographically independent. Fig. 2a shows the dimer structure of cyclophane 1 in the Cr1 phase. The naphthalene and anthracene rings were orthogonally arranged with a torsion angle of 86.20° in an individual molecule. Cyclophane 1 was dimerized by two C–H⋯π interactions between the pentaethyleneglycol chain and anthracene ring with a shortest C⋯C distance of 3.551 Å. Two intra-dimer C–H⋯O interactions were also observed between pentaethyleneglycol chains, with a C⋯O distance of 3.504 Å and a C–H⋯O angle of 145.74°. Dimer assemblies interacted with each other by π⋯π interactions, as shown in Fig. 2b. Adjacent naphthalene rings were parallel and partially overlapped with an inter-planer distance of 3.545 Å, forming offset face-to-face π⋯π interactions.42 The same type of π⋯π stacking was also observed between interdimer anthracene rings with an inter-planer distance of 3.403 Å. These π⋯π interactions were alternately arranged in the [01] and [101] directions, forming a two-dimensional (2D) layered assembly parallel to the (111) plane. The 2D assemblies interacted via C–H⋯O interactions between one of the two phenylene rings of the luminophore and the pentaethyleneglycol chain, with C⋯O distances of 3.375 and 3.500 Å and C–H⋯O angles of 128.27 and 175.45° (Fig. 2c). These significant intermolecular interactions formed the three-dimensional packing structure.
, where one molecule of cyclophane 1 was crystallographically independent. Fig. 2a shows the dimer structure of cyclophane 1 in the Cr1 phase. The naphthalene and anthracene rings were orthogonally arranged with a torsion angle of 86.20° in an individual molecule. Cyclophane 1 was dimerized by two C–H⋯π interactions between the pentaethyleneglycol chain and anthracene ring with a shortest C⋯C distance of 3.551 Å. Two intra-dimer C–H⋯O interactions were also observed between pentaethyleneglycol chains, with a C⋯O distance of 3.504 Å and a C–H⋯O angle of 145.74°. Dimer assemblies interacted with each other by π⋯π interactions, as shown in Fig. 2b. Adjacent naphthalene rings were parallel and partially overlapped with an inter-planer distance of 3.545 Å, forming offset face-to-face π⋯π interactions.42 The same type of π⋯π stacking was also observed between interdimer anthracene rings with an inter-planer distance of 3.403 Å. These π⋯π interactions were alternately arranged in the [01] and [101] directions, forming a two-dimensional (2D) layered assembly parallel to the (111) plane. The 2D assemblies interacted via C–H⋯O interactions between one of the two phenylene rings of the luminophore and the pentaethyleneglycol chain, with C⋯O distances of 3.375 and 3.500 Å and C–H⋯O angles of 128.27 and 175.45° (Fig. 2c). These significant intermolecular interactions formed the three-dimensional packing structure.
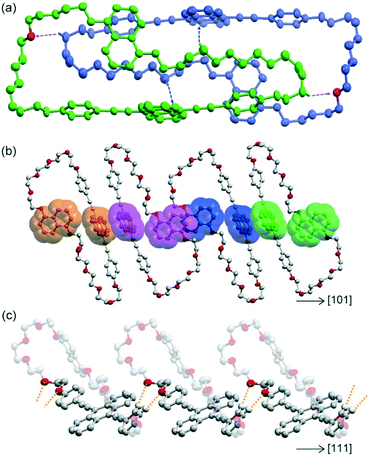 |
| | Fig. 2 Crystal structure of cyclophane 1 in the Cr1 phase. Hydrogen atoms are omitted for clarity except for those involved in C–H⋯O and C–H⋯π interactions. (a) Dimer structure of cyclophane 1. Blue and purple arrows correspond to C–H⋯π and C–H⋯O interactions, respectively. Each molecule is coloured green or blue except for oxygen atoms which interact with C–H bonds. (b) Adjacent luminophores form partially π-stacked structures in the plane parallel to (111). The π⋯π-stacking between naphthalene rings and between anthracene rings is alternately arranged in the [−101] and [101] directions. Luminophores in the same molecules are depicted in the same colour. (c) Intermolecular C–H⋯O interactions along the [111] direction are depicted as orange-dotted lines, which occur between 2D assemblies of dimers parallel to the (111) plane. | |
Phase transition behaviour
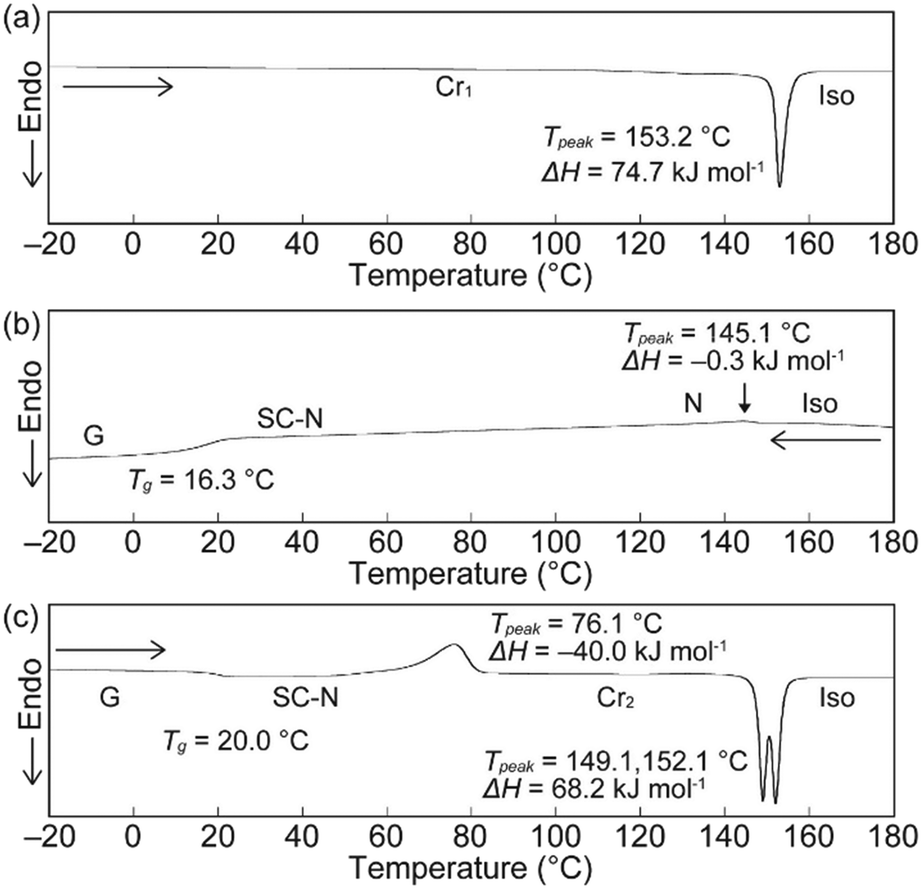
The phase transition behaviour of cyclophane 1 was investigated, as well as changes in its emission colour upon phase transition. A differential scanning calorimetry (DSC) curve obtained for the crystals described above (Cr1 phase) showed one endothermic peak at 153.2 °C on heating, which is ascribed to a transition to the isotropic state (Fig. 3a). Upon cooling from the isotropic state at a rate of 10 °C min−1, a small exothermic peak appeared at 145.1 °C (Fig. 3b). The fluidic phase just below 145.1 °C was characterized as a nematic (N) phase based on the small enthalpy change of −0.3 kJ mol−1, low viscosity, and schlieren texture observed in polarized optical microscopic (POM) images (Fig. 4a). The N phase exhibited yellow emission under excitation at 365 nm. On further cooling, no clear peaks were observed in the DSC trace and the sample showed a glass transition at 16.3 °C (Fig. 3b). The viscosity of the N phase gradually increased on cooling, suggesting that the nematic phase became a SC-N phase while retaining the schlieren texture (Fig. 4b) and yellow emission. In the case of cyclophane 2, a peak ascribed to a transition from the nematic to crystalline phase appeared in the DSC trace measured with the same cooling rate.38 Further rapid cooling was required to obtain the SC-N phase of 2. The DSC trace recorded for 1 on the 2nd heating from the glass (G) phase (−20 °C) displayed a clear exothermic peak, which corresponded to a transition from the SC-N to another crystalline (Cr2) phase at 76.1 °C. The photoluminescence colour changed from yellow to green upon this phase transition. There was also an obvious change in the POM image (Fig. 4c). Because the peak was exothermic, the SC-N phase was thermodynamically metastable whereas the Cr2 phase was thermodynamically stable. The Cr2 phase became an isotropic state after the two peaks at 149.1 and 152.1 °C. This suggested that the Cr2 phase differed from the Cr1 phase.
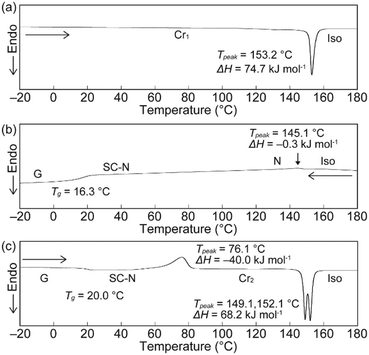 |
| | Fig. 3 DSC curves of cyclophane 1 on the (a) 1st heating, (b) 1st cooling, and (c) 2nd heating. Scanning rate was 10 °C min−1. | |
 |
| | Fig. 4 POM images of cyclophane 1 in the (a) nematic phase at 120 °C upon cooling, (b) supercooled nematic phase at r.t. upon cooling, and (c) Cr2 phase at r.t. after heating. All POM images were recorded in the presence of a coverslip. Scale bars: 100 μm. | |
X-ray diffraction measurements
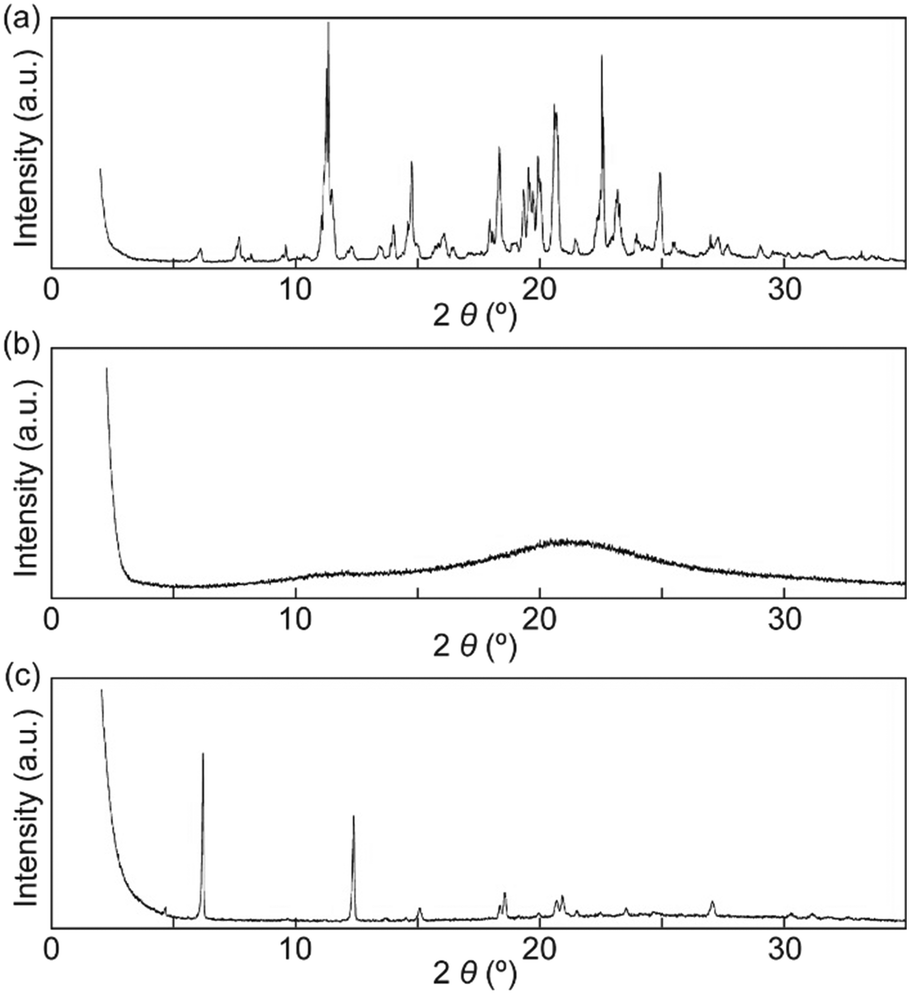
X-ray diffraction (XRD) patterns were recorded to confirm that the SC-N phase had nematic molecular order and that the Cr2 phase differed from the Cr1 phase. Many peaks were observed in the XRD pattern for the Cr1 phase, which indicated a well-ordered molecular packed structure (Fig. 5a). The XRD pattern for the Cr1 phase corresponded well with a simulated pattern obtained from the single crystal X-ray analysis of 1 (Fig. S3†). In contrast to the XRD pattern obtained for the Cr1 phase, the pattern of the SC-N phase showed no clear peaks (Fig. 5b). Therefore, the nematic molecular order was well retained on cooling from the nematic phase to room temperature (r.t.), which was consistent with the POM observations (Fig. 4a and b). After annealing the SC-N phase at 100 °C for 5 min, several sharp peaks appeared in the XRD pattern (Fig. 5c), and the pattern differed to that of the Cr1 phase. These results clarified that the nematic molecular order in the SC-N phase was converted to crystalline order that differed from the initial crystalline molecular assembly.
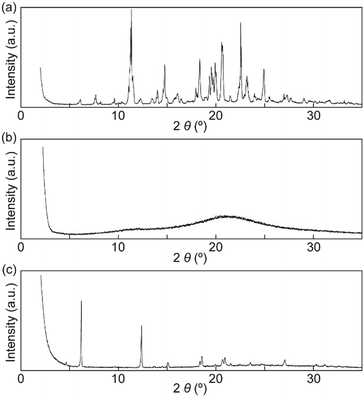 |
| | Fig. 5 XRD patterns of cyclophane 1 in the (a) Cr1 phase, (b) SC-N phase, and (c) Cr2 phase. All XRD patterns were recorded at r.t. | |
Photophysical properties
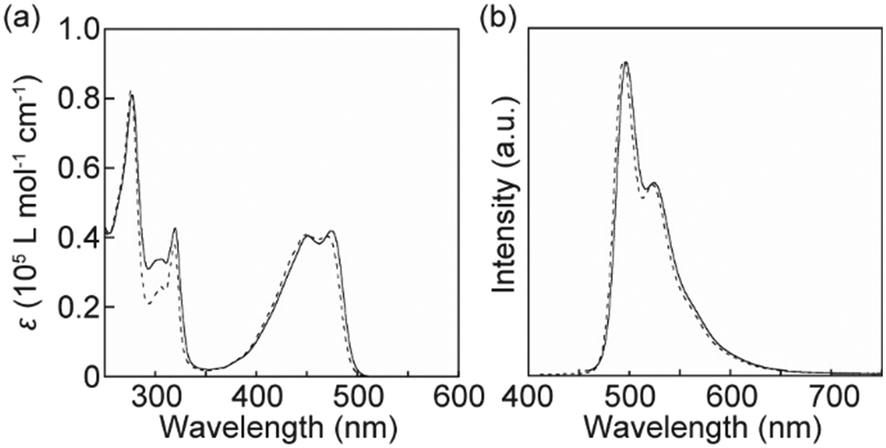
The photophysical properties of cyclophane 1 in solution and in the condensed states were investigated to obtain more insight into the thermoresponsive luminescence behaviour. Fig. 6 shows absorption and photoluminescence spectra for 1 and the linear analogue 3 (ref. 34) in CHCl3. The absorption of cyclophane 1 between 400 and 500 nm differed slightly to that of the CHCl3 solution of 3 (Fig. 6a). This indicated that feeble ground state electronic interactions between the luminophore and naphthalene moieties occurred in solution. The naphthalene group of 1 caused an increase in absorption around 300 nm. The photoluminescence spectrum of cyclophane 1 in CHCl3 displayed two peaks at 496 and 524 nm (Fig. 6b). Though tiny changes were also observed between the emission spectra of 1 and 3 in CHCl3 solution, the monomer emission feature of 9,10-bis(4-phenylethynyl)anthracene was retained after cyclization. A THF solution of 1 displays quite similar absorption and photoluminescence spectra to those of the CHCl3 solution of 1 (Fig. S4†).
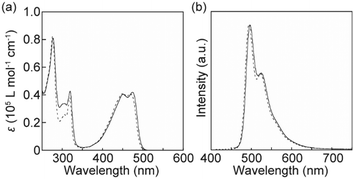 |
| | Fig. 6 (a) Absorption and (b) photoluminescence spectra of cyclophane 1 (solid line) and the linear analogue 3 (dashed line) in CHCl3 solution (c = 1.0 × 10−5 M). All spectra were recorded at r.t. The excitation wavelength for the photoluminescence spectra was 400 nm. | |
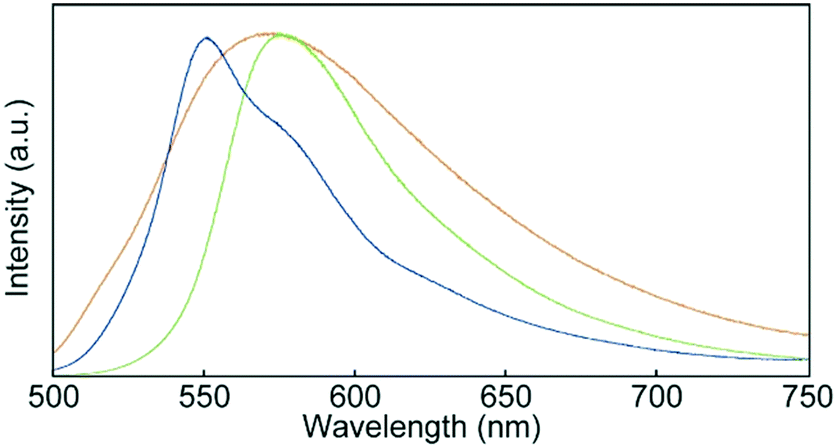
Fig. 7 shows photoluminescence spectra recorded for 1 in the condensed states. One peak was observed at 575 nm in the emission spectrum of the Cr1 phase. As the luminophores partially overlapped in the Cr1 phase (Fig. 2), the emission band was red-shifted from that of 1 in CHCl3 solution (Fig. 6b). Self-absorption also contributed to the observed red-shift. The SC-N phase displayed a broad emission band with a maximum at 571 nm. The profile of the broad emission was ascribed to excimers of the luminophores, because some luminophores could fully overlap each other in the SC-N phase with low molecular order. Energy transfer from monomer-like emission species to excimer sites resulted in the yellow photoluminescence. After the thermal stimuli-induced phase transition from the SC-N phase to Cr2 phase, the emission maximum blue-shifted to 550 nm and a shoulder appeared around 580 nm. This feature also differed from the spectrum of the initial Cr1 phase, confirming the difference in luminophore arrangements between the two crystalline phases.
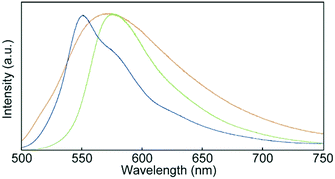 |
| | Fig. 7 Photoluminescence spectra of cyclophane 1 in the Cr1 (green), SC-N (yellow), and Cr2 (blue) phases. Spectra were recorded at r.t. with an excitation wavelength of 400 nm. | |
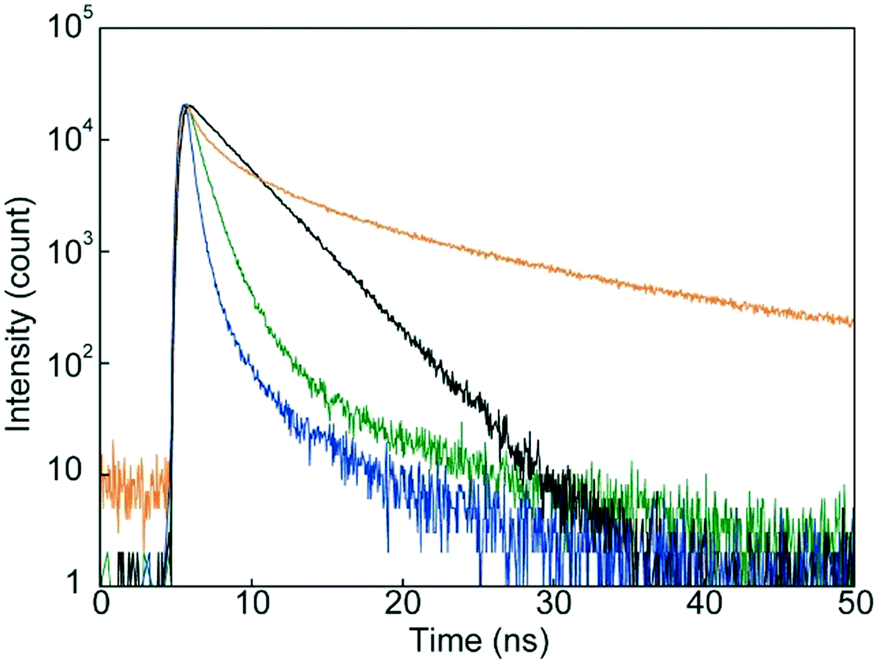
We conducted emission lifetime measurements for 1 in CHCl3 and in the condensed states (Fig. 8). An emission decay profile collected for 1 in CHCl3 solution could be fitted to a single exponential decay function to afford an emission lifetime of 3.0 ns, which is characteristic of monomer emission of the luminophore.33,34 The decay profiles of 1 in the condensed states could be fitted with multi-exponential decay functions. The fact that a relatively longer emission lifetime of 17 ns was obtained for the SC-N phase confirmed excimer formation in the SC-N phase. Such long emission lifetimes have been reported for other compounds having 9,10-bis(phenylethynyl)anthracene moieties groups.43 In contrast, emission species with shorter lifetimes were dominant in the two crystalline phases. As clarified by XRD and DSC measurements, the difference in molecular assembled structures between the Cr1 and Cr2 phases resulted in the change in the emission decay profiles.
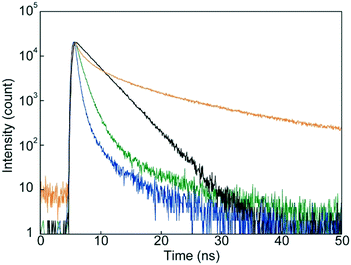 |
| | Fig. 8 Emission decay profiles of cyclophane 1 in dilute CHCl3 solution (black), in the Cr1 phase (green), in the SC-N phase (yellow), and in the Cr2 phase (blue). All data were collected at r.t. with an excitation wavelength of 405 nm. | |
Conclusions
We have demonstrated that careful molecular design makes it possible to obtain single crystals and solve the crystal structure of luminescent cyclophanes retaining thermoresponsive luminescent character. In the crystal structure of the Cr1 phase, 9,10-bis(phenylethynyl)anthracene and naphthalene groups were orthogonally arranged in individual cyclophane molecules. Two luminophores partially overlapped each other from adjacent cyclophanes. After cooling from the nematic phase, cyclophane 1 exhibited the yellow-emitting SC-N phase where excimer emission was dominant. Thermal treatment induced a phase transition to another crystalline phase showing green emission. The molecular design adopted here can be applied to other flexible cyclophanes retaining various photofunctions. Trials to prepare luminescent cyclophanes with shorter linkers are ongoing in our group.
Experimental
General methods
All reagents and solvents were purchased from Tokyo Kasei, Kanto and Fujifilm Wako. Unless otherwise noted, all reactions were carried out under nitrogen atmosphere. Flash silica gel column chromatography was performed with a Biotage Isolera flash system equipped with SHOKO-scientific Purif-Pack-EX cartridges. Recycling preparative gel permeation chromatography (GPC) was conducted with a Japan Analytical Industry LaboACE. 1H NMR spectra were recorded using a JEOL JNM-ECX 400 spectrometer, and all chemical shifts are quoted on the δ-scale in ppm relative to the signal of tetramethylsilane (at 0.00). 13C NMR spectra were recorded using a JEOL JNM-ECX 400 spectrometer, and all chemical shifts (δ) are reported in ppm using solvents as the internal standard (CDCl3 at 77.16). Coupling constants (J) are reported in Hz and relative intensities are also shown. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were recorded on an AB SCIEX TOF/TOF 5800. POM observations were carried out with an Olympus BX-60 optical microscope equipped with a Shimadzu Moticam 1080. DSC measurements were performed using a Hitachi DSC7020 with a scanning rate of 10 °C min−1 under nitrogen atmosphere. Powder XRD patterns were recorded with a Rigaku SmartLab diffractometer. UV-vis absorption spectra were measured using a JASCO V-550 spectrophotometer. Steady-state fluorescence spectra were measured using a JASCO FP-6500 spectrophotometer. Time-resolved fluorescence measurements were carried out using a Hamamatsu Photonics Quantaurus-Tau instrument.
Single crystal X-ray structure analysis
Single crystals suitable for X-ray crystallography were obtained from tetrahydrofuran solution by slow evaporation. A single crystal was mounted on a glass capillary with epoxy resin. Crystallographic data was collected using a Rigaku R-AXIS RAPID diffractometer with Mo Kα radiation (λ = 0.71075 Å) from a graphite monochromator at 273 K. The initial structure was solved using SHELXT software44 and expanded using Fourier techniques and refined on F2 by the full-matrix least-squares method SHELXL-2018/1 (ref. 45) compiled into Yadokari-XG.46,47 The parameters were refined using anisotropic temperature factors, except for the hydrogen atoms, which were refined using the riding model with a fixed C–H bond distance. The crystallographic data are summarized in Table S1 in the ESI.† CCDC 1946087 (cyclophane 1) contains the supplementary crystallographic data for this paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Ms. Ai Tokumitsu, Global Facility Center of Hokkaido University, for elemental analysis. This work was supported by JSPS KAKENHI (grant no. JP18H02024, JP18H01949, and JP19K15517) and the Takahashi Industrial and Economic Research Foundation. This work was also supported by the Japan Science Technology Agency (JST), PRESTO (grant no. JPMJPR17P6). We thank Aidan G. Young, PhD, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Notes and references
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef CAS.
- D. J. Cram and H. Steinberg, J. Am. Chem. Soc., 1951, 73, 5691–5704 CrossRef CAS.
- D. J. Cram and J. M. Cram, Acc. Chem. Res., 1971, 4, 204–213 CrossRef CAS.
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- T. Umemoto, S. Satani, Y. Sakata and S. Misumi, Tetrahedron Lett., 1975, 16, 3159–3162 CrossRef.
- T. Kawashima, T. Otsubo, Y. Sakata and S. Misumi, Tetrahedron Lett., 1978, 19, 5115–5118 CrossRef.
- C. Seel and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1992, 31, 528–549 CrossRef.
- J. O. Jeppesen, M. B. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115–5132 CrossRef CAS PubMed.
- P. Chen and F. Jakle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed.
- R. Reuter and H. A. Wegner, Chem. Commun., 2011, 47, 12267–12276 RSC.
- H. Hopf, Isr. J. Chem., 2012, 52, 18–19 CrossRef CAS.
- I. Roy, S. Bobbala, J. Zhou, M. T. Nguyen, S. K. M. Nalluri, Y. Wu, D. P. Ferris, E. A. Scott, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 7206–7212 CrossRef CAS PubMed.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1995, 95, 2529–2586 CrossRef CAS.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1996, 2169–2175 RSC.
- O. Baudoin, F. Gonnet, M.-P. Teulade-Fichou, J.-P. Vigneron, J.-C. Tabet and J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2762–2771 CrossRef CAS.
- M. Inouye, K. Fujimoto, M. Furusyo and H. Nakazumi, J. Am. Chem. Soc., 1999, 121, 1452–1458 CrossRef CAS.
- H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495–504 CrossRef CAS PubMed.
- P. P. Neelakandan and D. Ramaiah, Angew. Chem., Int. Ed., 2008, 47, 8407–8411 CrossRef CAS PubMed.
- D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158–4168 RSC.
- L. Qiu, C. Zhu, H. Chen, M. Hu, W. He and Z. Guo, Chem. Commun., 2014, 50, 4631–4634 RSC.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- V. Percec, A. D. Asandei and G. Ungar, Chem. Mater., 1996, 8, 1550–1557 CrossRef CAS.
- V. Percec, A. D. Asandei and P. Chu, Macromolecules, 1996, 29, 3736–3750 CrossRef CAS.
- B. Neumann, D. Joachimi and C. Tschierske, Adv. Mater., 1997, 9, 241–244 CrossRef CAS.
- V. Percec, P. J. Turkaly and A. D. Asandei, Macromolecules, 1997, 30, 943–952 CrossRef CAS.
- B. Neumann, T. Hegmann, R. Wolf and C. Tschierske, Chem. Commun., 1998, 105–106 RSC.
- B. Neumann, T. Hegmann, C. Wagner, P. R. Ashton, R. Wolf and C. Tschierske, J. Mater. Chem., 2003, 13, 778–784 RSC.
- T. Hegmann, B. Neumann, R. Wolf and C. Tschierske, J. Mater. Chem., 2005, 15, 1025–1034 RSC.
- P. R. Ashton, D. Joachimi, N. Spencer, J. F. Stoddart, C. Tschierske, A. J. P. White, D. J. Williams and K. Zab, Angew. Chem., Int. Ed. Engl., 1994, 33, 1503–1506 CrossRef.
- V. Percec, M. Kawasumi, P. L. Rinaldi and V. E. Litman, Macromolecules, 1992, 25, 3851–3861 CrossRef CAS.
- Y. Sagara, Y. C. Simon, N. Tamaoki and C. Weder, Chem. Commun., 2016, 52, 5694–5697 RSC.
- Y. Sagara, C. Weder and N. Tamaoki, RSC Adv., 2016, 6, 80408–80414 RSC.
- Y. Sagara, C. Weder and N. Tamaoki, Chem. Mater., 2017, 29, 6145–6152 CrossRef CAS.
- Y. Sagara and N. Tamaoki, RSC Adv., 2017, 7, 47056–47062 RSC.
- Y. Sagara, N. Tamaoki and G. Fukuhara, ChemPhotoChem, 2018, 2, 959–963 CrossRef CAS.
- K. Mase, Y. Sasaki, Y. Sagara, N. Tamaoki, C. Weder, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2018, 57, 2806–2810 CrossRef CAS PubMed.
- Y. Sagara, A. Seki, Y. Kim and N. Tamaoki, J. Mater. Chem. C, 2018, 6, 8453–8459 RSC.
- K. Nabeya, T. Muraoka, N. Hoshino, M. Aizawa, T. Kajitani, T. Akutagawa, A. Shishido, T. Fukushima and K. Kinbara, Mater. Chem. Front., 2018, 2, 969–974 RSC.
- Y. Sagara, T. Muramatsu and N. Tamaoki, Crystals, 2019, 9, 92 CrossRef.
-
L. Leigh and J. B. Leonard, in The Importance of π-Interactions in Crystal Engineering: Frontiers in Crystal Engineering, ed. R. T. T. Edward and Z. S. Julio, Wiley, Chichester, 2012, pp. 109–124 Search PubMed.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
K. Wakita, Yadokari-XG, Software for Crystal Structure Analyses, 2001 Search PubMed.
- C. Kabuto, S. Akine, T. Nemoto and E. Kwon, Nippon Kessho Gakkaishi, 2009, 51, 218–224 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of compound 1, crystallographic data of 1, and Hirshfield surface analysis for 1. CCDC 1946087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9me00105k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Kiyonori
Takahashi
a,
Takayoshi
Nakamura
a and
Nobuyuki
Tamaoki
*ab,
Kiyonori
Takahashi
a,
Takayoshi
Nakamura
a and
Nobuyuki
Tamaoki