
Macromolecular design strategies toward tailoring free volume in glassy polymers for high performance gas separation membranes
Tanner
Corrado
and
Ruilan
Guo
 *
*
Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: rguo@nd.edu
First published on 27th September 2019
Abstract
The gas separation membrane field has seen exceptional growth since its inception, with extensive research being performed to find novel, high performance polymer membrane materials for application in industrial separations. In this review, we examine recent advances of polymer design strategies to overcome the permeability/selectivity tradeoff relationship and the diminished performance caused by physical aging. We explore fundamental structure–property relationships within current state-of-the-art polymer materials to discern promising routes for continued membrane materials research and development, highlighting promising macromolecular architectures and design strategies providing tailorable polymer free volume for high performance gas separation membranes. Finally, we provide our perspective on encouraging design routes moving forward.
 Tanner Corrado | Tanner Corrado is a graduate student at the University of Notre Dame. He joined the Chemical and Biomolecular Engineering Department at Notre Dame in the fall of 2016, following completion of his bachelor's degree in chemistry from Linfield College. In spring 2018, he was awarded a National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) Fellowship. With the guidance of Professor Ruilan Guo, his doctoral research has focused on the design of iptycene-based ladder polymers for membrane gas separation applications. |
 Ruilan Guo | Ruilan Guo is an Associate Professor of Chemical and Biomolecular Engineering at the University of Notre Dame. She earned her B.E. and M.E. degrees at Beijing University of Chemical Technology, her Ph.D. at Georgia Institute of Technology, and was a postdoctoral fellow working with the late Professor James E. McGrath at Virginia Tech before she joined Notre Dame. Guo's research focuses on developing functional polymer membrane materials for gas separation, fuel cell, and water treatment. She is a recipient of the Department of Energy Early Career Research Award, ACS Petroleum Research Fund for Doctoral New Investigators, and “2017 Class of Influential Researchers” by I&EC Research. She also serves on the Editorial Advisory Board of ACS Applied Energy Materials. |
Design, System, ApplicationThis review article provides an overview of most recent advances of polymer design strategies that enable fine tuning of free volume microporosity to overcome the two major materials challenges in the field of gas separation membranes, i.e., the permeability/selectivity tradeoff and the diminished performance caused by physical aging. The discussion has been emphasized on examining polymer design motifs and fundamental structure/property relationships to highlight the effectiveness of these strategies. Limitations and constraints of the discussed strategies are also reviewed to discern promising routes for continued membrane materials research and development. While this review focuses on gas separation membranes, the design strategies examined in this paper have much wider applicability in many other related fields such as water purification membranes and electrochemical devices where polymer functionality design and microstructure control plays a critical role for their performance under various conditions. |
1. Introduction
Membrane separation technology shows promise as an alternative to conventional separation systems such as distillation and sorption methods due to its smaller footprint and spatial requirements, mechanical simplicity in ease of operation, and, most importantly, the freedom from phase changes or a thermal driving force that helps reduce nearly 90% of energy consumption compared to traditional thermal separations.1–7 Since their initial industrial success in the late 1970s for the separation of hydrogen from the purge gas streams (nitrogen, argon and methane) in ammonia synthesis plants, gas separation membrane applications have grown into a sizable market, with the four largest commercial operations having sales between $1.0–$1.5 billion per year.2,4 Compared to other existing membrane-mediated chemical separations such as ultrafiltration (UF), nanofiltration (NF), and reverse osmosis (RO), gas separations are much more difficult to achieve due to much smaller size differences (typically several tenths of an Ångstrom difference) between gas molecules. For example, air separations involve the separation of O2 and N2 that have a size difference of only 0.18 Å. From a materials development standpoint, such a narrow size separation requires precise control of polymer membrane structure.Optimal polymer membranes will work with high combinations of permeability (gas throughput) and selectivity (separation efficiency) to minimize the necessary membrane surface area to ensure sufficient product purity and high separation capacity. Existing commercial polymer membranes follow the solution-diffusion mechanism for the separation of gases,4,8 whereby gas transport and separation occurs based on the diffusivity of gas molecules through the membrane (kinetic contribution) and the solubility of gas molecules in the polymer (i.e., gas-polymer interaction).2,9 However, inherent to most dense, pure polymer membrane systems following this transport mechanism are several challenges that have limited new membranes ability to replace the fewer than ten membrane materials that have been in commercial use for decades.4
First is the empirical tradeoff relationship between permeability and selectivity, which has limited polymer membranes in achieving significant gains in overall separation performance.10 The primary difficulty involved in overcoming the tradeoff between permeability and selectivity is a lack of precise control over the gas transport pathways, i.e., free volume architecture, within polymer membranes. Ideally, perfect gas separation occurs when the membrane contains free volume elements that have narrowly distributed “pore” sizes in the size range between the gases attempting to be separated. However, most conventional polymer membranes have a broad and random size distribution of microvoids that are not able to provide high permeability (large free volume size) and high selectivity (narrow size distribution) simultaneously. Significant research effort is underway focusing on innovative macromolecular designs and development of polymers with finely tuned microporosity. Though specific structure varies among these microporous polymers, a common structural feature is that they utilize a highly rigid, ladder-like backbone to limit the ability for the polymer chain to pack efficiently (for high permeability) and reduce rotational freedom (for selective micropores). These polymers have helped redefine 2015 upper bounds for several gas pairs due to their interconnected free volume elements in the size-region of gas separation, with what is imagined as a combination of selective ultramicropores connected with larger, more permeable regions, leading to significantly enhanced size-sieving and simultaneously improved permeability relative to typical glassy polymers.11 This enormous range of separation performance in polymers illustrates the extraordinary sensitivity of gas permeability/selectivity to polymer material structure. While initial separation performance (mostly examined using single gas permeation tests) is exceptional within these high free volume polymers, they all suffer from significant permeability reductions over time due to another major issue with polymer gas separation membranes – physical aging.
Physical aging occurs in glassy polymers generally being in a non-equilibrium state with excess free volume relative to the densely-packed equilibrium state of the polymer. Over time, polymer chains undergo relaxation via local, segmental motion toward a pseudo-equilibrium state of chain packing, which causes a reduction in free volume, and therefore, permeability. While this aging does tend to follow the tradeoff relationship, with a selectivity increase as the permeability decreases, the drop-off in permeability in high-free-volume polymers is significantly higher than the gain in selectivity. This issue arises due to the non-permanent nature of polymer free volume due to its basis being the instantaneous chain conformations, which while in great excess initially, will undergo serious densification over time. This temporary or instantaneous free volume is also a major contributing factor to another significant challenge to gas separation membranes – plasticization. Plasticization is a phenomenon that occurs from condensable gases, such as carbon dioxide, sorbing into the polymer membrane. As the concentration of the condensable gas inside the membrane increases, the polymer swells, causing chain reorganization that leads to increases in free volume and loss of size-sieving capability. This reorganization typically promotes the permeation of large gases more than small ones, leading to significant decreases in selectivity in mixed-gas or commercial testing that is not observed in pure-gas permeation analysis.
This review presents an overview of macromolecular strategies and motifs that are able to exquisitely tailor the polymer free volume architecture (total fraction, size, and size distribution) to address the major challenges associated with polymeric membranes for gas separations as described above. The discussion focuses on many recent advances occurring within the last 15 years in the field of polymer materials for gas separation membranes, with an emphasis on elucidating the fundamental structure–property relationships while highlighting the impact of structure manipulation on free volume architecture and consequent gas transport properties. To stay focused, we limit our discussion to pure, glassy polymer membrane materials (excluding perfluoropolymers); thus, mixed-matrix membranes, facilitated transport membranes, and polymer-derived carbon molecular sieve (CMS) membranes are not covered. In this review, the fundamental scientific principles that govern gas transport in dense polymers, polymer free volume concept and its characterization methods, and the correlation between free volume and gas transport in polymers are described. The discussion of macromolecular design on free volume manipulation is focused on two themes: 1) strategies to tailor free volume to achieve high permeability-selectivity combinations; and 2) strategies to reinforce free volume architecture for improved physical aging resistance, with an aim to identify effective approaches in achieving fast and selective gas transport and mitigating physical aging in polymer gas separation membranes. Finally, some perspectives and future research directions are provided for polymer gas separation membranes.
2. Gas transport in glassy polymers and free volume
2.1. Gas transport in polymer membranes
The performance of a gas separation membrane is often gauged in terms of permeability and selectivity coefficients which describe gas throughput and separation efficiency, respectively. Permeability, P (unit: Barrer), of a gas in a membrane is the steady-state gas flux normalized by the membrane thickness and the trans-membrane pressure difference.12 Each gas has a different permeability coefficient in each polymer, and the use of polymers to separate gas mixtures is achieved based on the different permeation rate of different gases. The solution-diffusion model describes transport of gases in dense polymeric membranes, where permeation is controlled by the thermal motion of polymer chains, providing a means for molecular diffusion through the membrane.8,9 Within this model, gases first sorb into the upstream face of the membrane, diffuse through the membrane down a concentration gradient, and desorb from the downstream side of the membrane. Using the solution-diffusion model, the permeability coefficient (PA) for each gas can be expressed as the product of individual solubility (SA) and diffusion (DA) coefficients that arise from the gases' ability to dissolve in the membrane and diffuse through it as seen in eqn (1).| PA = DA × SA | (1) |
The diffusion coefficient, DA (unit: cm2 s−1), is a measure of the speed (i.e., the mean squared displacement per unit time) a gas moves through the polymer due to random thermal motions. It can vary by several orders of magnitude due to modifications to the polymer structure as detailed in later sections. Solubility or sorption coefficient, SA (unit: cm3 (STP) per cm3 polymer atm), reflects the gas's solubility in the polymer, which strongly depends on gas molecule condensability (e.g., gas critical temperature) but has also been shown to be sensitive to polymer structure, as certain functionalities exhibit significant effects on polymer-gas interactions, providing a route toward tunability of solubility and solubility selectivity through polymer backbone manipulation.13 A membranes ability to separate a set of gases is commonly and conveniently defined as the ideal selectivity (αA/B), which is simply the ratio of the individual gas permeabilities (gas A being the more permeable one) through the membrane as given by eqn (2). Following the solution diffusion model, it can be further broken down as a product of the diffusivity and solubility selectivities.
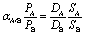 | (2) |
It should be noted that most membrane research reports values in ideal selectivities obtained from single-gas permeation tests (i.e., using pure gas as the feed). For non-interacting gases, ideal selectivities serves as a useful indication of the membrane's true performance. However, when interacting gases such as CO2 are involved, mixed gas permeation data are much more representative of actual membrane performance since they account for the competitive adsorption and possibly diffusional effects between the polymer and gases.
A very recent analysis of upper-bound redefining polymers of intrinsic microporosity (PIMs) has indicated that not all gases in these super-rigid, high free volume polymers follow the solution-diffusion mechanism. Correlations of diffusion coefficients with effective gas diameters has suggested that smaller gases like helium and hydrogen may experience the large fractional free volumes of PIMs as interconnected, and therefore undergo permeation primarily through the pore diffusion mechanism.14
2.2. Polymer free volume and its correlation with gas transport in membranes
One of the most important factors contributing to gases ability to permeate through a membrane is the polymer's free volume. While different definitions have been used for free volume in different areas, free volume in polymers generally refers to the amount of void space not occupied by the molecules of the polymer chains.15 Typically the free volume is reported as the fractional free volume (FFV), shown in eqn (3), which is a reduced form of the free volume which reports a percentage of free volume within the polymer. It is calculated as the difference between the measured specific volume, Vsp, (i.e., reciprocal of polymer density), and the occupied volume, Vo, calculated based on theory (e.g., related to the van der Waal's volume using a group contribution method).16–18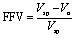 | (3) |
Free volume arises in polymers mostly due to inefficient packing between the polymer chains which leads to openings in the polymer large enough for gases to permeate through from local segmental motion. In this regard, free volume is largely a non-equilibrium quantity, which, over time, will decrease due to polymer chain relaxation towards their equilibrium packing. This causes the major problem of physical aging for polymer gas separation membranes that leads to great reduction in gas permeability over time.
While fractional free volume analysis provides a relative amount of unoccupied space within a polymer membrane, it does not deliver any specific information on the microstructure of the polymer, such as its average pore sizes or size distributions. To obtain a full picture of the free volume architecture (i.e., fractional free volume, free volume size and size distribution) within polymers, a variety of techniques have been developed. A summary of the working principle, attainable free volume information and the limitations for each of the techniques is given in Table 1 along with relevant references. A multitude of these techniques are often performed on the same polymer to help provide the clearest overall picture on the free volume architecture of the polymer membrane.
| Technique | Working principle | Attainable information | Limitations | Ref. |
|---|---|---|---|---|
| Group contribution method | • Coupled with density measurement• Free volume fraction is defined by the difference between polymer specific volume (calculated from density) and the theoretical occupied volume (i.e., the sum of the van der Waal's volume of groups comprising the polymer chain) | • Polymer film density• Total fractional free volume in the polymer, i.e., the percent of unoccupied volume in the polymer | • No information on size or size distributions of the free volume elements• Results sensitively depend how structural groups are defined. Generally decomposing the polymer chain into fewer large groups is preferred | 16–18 |
| Positron annihilation lifetime spectroscopy (PALS) | • Probe: triplet-state positronium (o-Ps, 1.06 Å)• Measuring lifetimes of o-Ps trapped in the free volume microcavities • o-Ps trapped in larger free volume voids has longer lifetimes due to reduced local electron density for delayed annihilation • Mean size of free volume elements determined via the Tao-Eldrup formula |
• The average size of free volume microcavities• Free volume size distribution can be determined using computer programs such as PATFIT (for finite-term analysis), CONTIN (for continuous analysis), or maximum entropy lifetime method (MELT; continuous analysis) | • Assumption that the free volume microcavities are spherical• Have to be performed in an inert atmosphere to eliminate the contribution of additional o-Ps decay due to the reaction with sorbed oxygen • Hard to determine the total free volume |
9, 19–24 |
| Physical adsorption (Physisorption) | • Probe: commonly N2 (3.64 Å)• Measuring N2 adsorption isotherm (77 K) • Pore size distribution determined by analyzing isotherm with nonlocal density functional theory (NLDFT) or other variations of DFT |
• Average pore size and size distribution via data fitting | • Not suitable for probing the microporosity due to large probe size• Inaccuracy due to polymer swelling • Results dependent on a chosen regularization parameter that can instill artifacts into pore size/size distribution data |
25–27 |
| Inverse gas chromatography | • Probe: organic vapors (typically > 5 Å)• Measuring the retention times of solute probe in a column containing the polymer of interest to determine the partial molar enthalpy of mixing (ΔHm) • The mean size of free volume elements in the polymer is determined by the coordinates of ΔHm at minimum versus solute critical volume |
• Temperature averaged size of free volume elements | • Only applicable for high free volume polymers due to large probe size• Inaccuracy in overestimating of large pore size due to polymer swelling during analysis • Insensitive to possible temperature-based changes in microstructure |
9, 28 |
| 129Xe NMR spectroscopy | • Probe: 129Xe (4.4 Å)• Measuring the chemical shift (δ) of 129Xe nuclei sorbed in a polymer which is sensitive to the local environment (e.g., the microcavity size hosting it) • The microcavity size is quantified by an empirical relationship between δ (zero pressure) and the mean free path of a Xe atom in a microcavity |
• Size of free volume elements• Able to probe various domains and phases in highly heterogeneous systems (e.g., block copolymers, polymer blends, composites) • With additional measurements of chemical shifts at various pressures, can get size distributions |
• Assumes spherical or cylindrical pore geometry• Precise temperature control during experiment is required to obtain accurate chemical shift data • Van der Waals size of Xe atom relatively uncertain (listed between 3.2–4.9 Å) leads to minor shifts in radii of free volume elements depending on chosen size for Xe |
9, 29–31 |
| X-ray scattering/diffraction | Small-angle X-ray scattering (SAXS):• Measuring the scattering pattern • Scattering of X-rays at small angles arises from the presence of spatial electron density fluctuation caused by the irregular placement of atoms such as micro voids |
• Free volume size distribution• Able to analyze the free volume with respect to temperature and pressure | • Less straightforward to use and interpret for amorphous polymers | 32–35 |
| Wide-angle X-ray diffraction (WAXD):• Measuring the diffraction pattern • d-Spacing (calculated from Bragg's law) can be related to either inter-chain or inter-segmental distances, which define the pore dimensions |
• A rough estimation of pore dimensions based on d-spacing• Able to analyze the pore dimensions with respect to temperature and pressure | • Indirect correlation between d-spacing and free volume size• Inaccuracy in peak assignment due to the very broad halos exhibited by amorphous polymers • No size distribution information |
||
| Molecular dynamic (MD) simulations | • Force field based methods• Integrating the equation of motion of polymer chains contained in a cubic simulation box with periodic boundaries • Free volume information is extracted from the configuration output of MD simulations via geometrical methods or energetic methods |
• Free volume size, size distribution, and connectivity• Visualization of polymer chain packing | • Long computation time for reliable results• Size constraints of the simulation box • Results dependent on specific analysis model |
36–40 |
Frequently the fractional free volume is related to the diffusion coefficient and permeability following an Arrhenius-type dependence. The free volume generated from inefficient packing due to rigid backbones or bulky side groups allows increased diffusion of penetrant gases through the polymer membrane due to an increase in the diffusion coefficient which is directly proportional to the free volume in the membrane, as shown by, eqn (4) where A and B are constants dependent on the specific gas and polymer.9
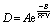 | (4) |
Following eqn (4), plots of the diffusion coefficient (D) versus 1/FFV provide a strong correlation between a polymer's fractional free volume and its diffusion coefficient, where larger fractional free volumes lead to increased diffusivity through the membrane.
For typical glassy polymer membranes, solubility has a relatively weak dependence on free volume, providing direct analysis of fractional free volumes effect on permeability by substituting eqn (4) into eqn (1) to get eqn (5), where Ap = SA × A.9
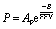 | (5) |
Strong correlations are frequently observed between fractional free volume and permeability by plotting permeability as a function of the reciprocal of FFV within similar polymer structural families, further highlighting the importance of targeting increased fractional free volume in new polymer designs, while also working to control free volume sizes to maintain size-sieving capabilities. However, for high-free-volume polymers such as PIMs, it has been shown that high solubility coefficients are a significant factor contributing to their performance above the upper bounds. While their solubility selectivity generally falls in the same range as typical glassy polymers, their solubility coefficients are usually much higher. This indicates that their exceptionally high free volume may play an important role in increasing solubility in addition to diffusivity, and therefore increasing overall permeability, while still having sieving controlled by diffusivity selectivity.41 In addition to diffusivity selectivity, solubility or sorption selectivity also plays an important role in gas separation. It has been shown that glassy polymers may have comparable to even higher solubility selectivity values, dependent on gas pair, relative to rubbery polymers, providing an alternative route to tuning performance through manipulation of polymer structure.42
3. Design strategies of tailoring free volume to overcome the permeability/selectivity tradeoff
3.1. Permeability/selectivity tradeoff and the upper bound limits
One of the most prominent challenges plaguing gas separation membranes is a natural tradeoff between a polymer's permeability and its selectivity, i.e., highly permeable polymers normally show poor size sieving properties while highly selective polymers typically have low permeabilities. An original analysis of this tradeoff relationship emerged in 1991, when Robeson plotted pure gas separation performance data for hundreds of polymers, encompassing many common gas pairs, on log–log plots of permeability of the more permeable gas versus selectivity.43 What was observed was an apparent, empirical upper bound line above which little to no data was present, with the upper right hand corner consisting of the most desirable combinations of permeability and selectivity. This early investigation noted the slope of the upper bound correlated with the difference in diameters of the different gases in each tested pair, with the suggestion that the upper bound-defining polymers contained relatively narrow free volume distribution in appropriate size ranges.43 Further corroborating the empirical findings of Robeson, Freeman followed with a theoretical justification for the form of the upper bound lines shown in eqn (6), by deriving equations for model parameters λA/B and βA/B provided below (eqn (7) and (8), respectively), where dB and dA are the kinetic diameters for the larger (dB) and smaller (dA) gases in the pair, SA and SB define the solubility coefficients for the more (SA) and less (SB) permeable gases, and a and b are fixed parameters for glassy polymers from the linear free energy model.9,44 This leaves only f, which is an adjustable constant depicting the product of the equilibrium chain spacing and the energy required to open a free volume hole for penetrant gas diffusion, set to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 cal mol−1 which provides the best fit to the reported upper bound data.9,44
600 cal mol−1 which provides the best fit to the reported upper bound data.9,44| αA/B = βA/B/PAλA/B | (6) |
| λA/B = (dB/dA)2 − 1 | (7) |
 | (8) |
Based on this theoretical explanation, Freeman proposed a series of polymer design strategies to advance the location of the upper bound (while noting the slope will remain unchanged as it is solely dependent on kinetic diameters for the specific gas molecules in the pair). To advance separation performance, βA/B should be increased through two potential routes: 1) stiffening of the polymer chain and/or 2) improving the solubility selectivity of the polymer, both simultaneously accompanied by heightened fractional free volume from increased interchain spacing. This ideally leads to simultaneous achievement of both gas permeability and selectivity, and advancement in polymer performance above the upper bound. However, this strategy is only effective when interchain distances do not become so large that diffusion in the polymer is no longer governed by thermal motion of polymer segments.44 After years of extensive research efforts following the proposed design strategies, the upper bounds were reevaluated in 2008, upon which only minor shifts in their location were observed for most gas pairs.45 However, more recent advances in polymer performance have seen larger shifts in the upper bounds for H2/CH4, H2/N2, and O2/N2 in 2015 and for CO2/CH4 and CO2/N2 in 2019.11,46 In the following sections (section 3.2 through section 3.7), we review and analyze various polymer design techniques in an attempt to overcome the tradeoff relationship between permeability and selectivity. Upper bound plots are frequently employed to compare the performance of various polymer structures in order to highlight the effectiveness of different design strategies in terms of performance enhancement. It should be noted that the separation performance reported in the literature as well as in this review is largely based on pure-gas permeation data.
3.2. Bulky bridging structures in the backbone
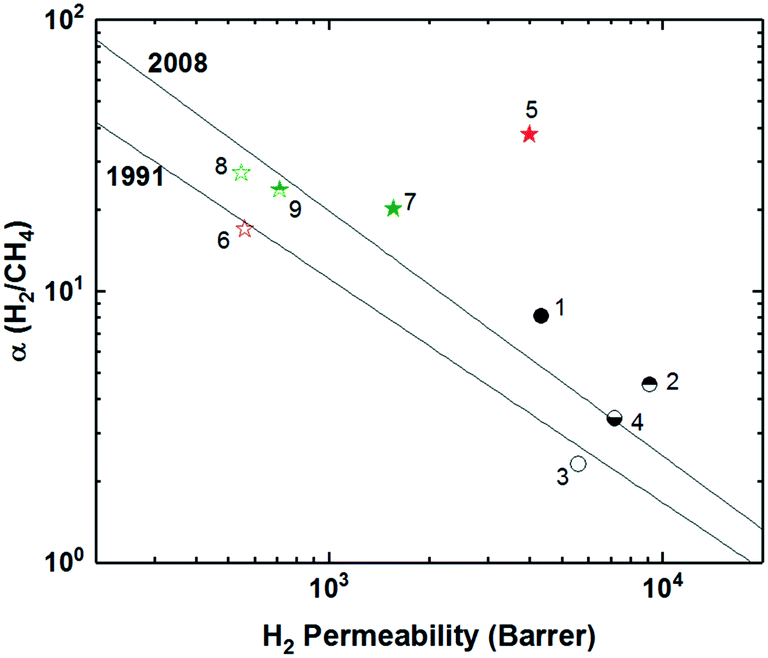
To increase interchain spacing to help overcome the upper bound tradeoff, a common method that has been explored in attempts to increase free volume and overall performance involves the inclusion of bulky bridging units in the repeating unit along the polymer backbone. Following this design strategy, extensive research has been done within the realm of polyimides, which have long been employed in commercial separation membranes due to their excellent physical properties (thermal, chemical, and mechanical stability) while being solution-processable and providing sufficient selectivities, albeit at relatively lower permeabilities.47 The prototypical commercial polymer membrane material is that of Matrimid® (Fig. 1), an aromatic polyimide comprised of the bridging units diaminophenylindane (DAPI) and 3,3′-4,4′-benzophenone tetracarboxylic dianhydride (BTDA) which has strong selectivity for CO2/CH4 (αCO2/CH4 = 36) with a CO2 permeability of ∼10 Barrer, one of the best combinations among commercial membranes (point 1, Fig. 2).3,48 With its location well below the 1991 upper bound, wide-ranging research has been undertaken in attempts to create new aromatic polyimides with greater separation performance while preserving the robust physical properties within the polymer family through exploration of bulky monomers to help disrupt polymer chain packing for improved free volume and consequent permeabilities. | ||
| Fig. 1 Structures of various glassy polymers utilizing bulky, bridging structures with tunable side groups for improved interchain spacing or free volume. | ||
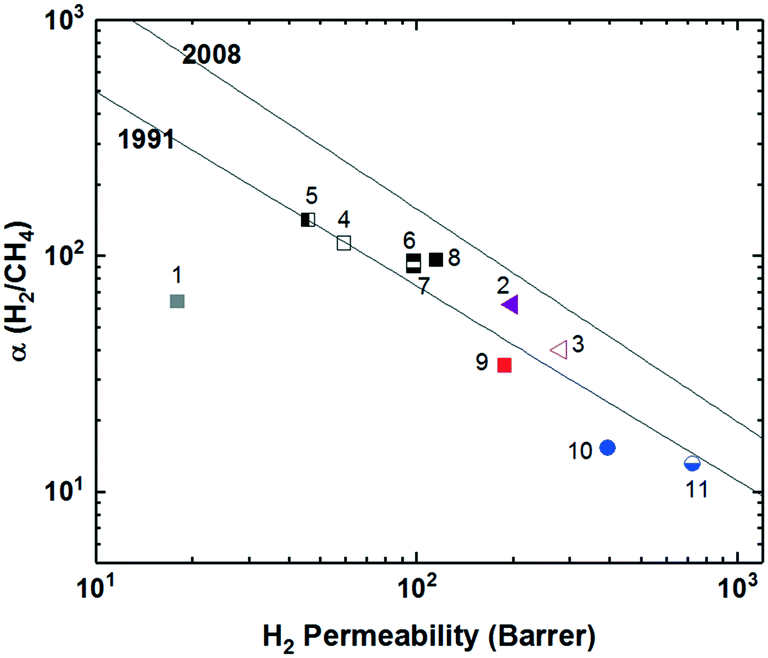 | ||
| Fig. 2 Representative polymers utilizing various bulky, bridging structures plotted against the Robeson upper bound for H2/CH4. Polymers labeled as follows: 1) Matrimid®; 2) 6FDA-DAT1; 3) 6FDA-DAT2; 4) 6FDA-1,4-trip_CF3; 5) 6FDA-1,4-trip_CF3:D 2:1; 6) 6FDA-1,4-trip_CF3:D 1:1; 7) 6FDA-1,4-trip_CF3:D 1:2; 8) 6FDA-1,4-trip_CF3:D 1:3; 9) 6FDA-PPDA(CF3); 10) PI-3B; 11) PI-3F. | ||
Polyimides provide two primary components for monomer modification – the dianhydride and diamine units – which can independently tune the final polymer properties by changing the bulkiness of the moiety and polymer backbone rigidity (presence of flexible or rotatable groups). Most of the new polyimide designs have been focused on varying the structure of diamine monomers due to their relatively easier synthesis and broader structure varieties compared to dianhydride monomers. A noticeable new series of polyimides with bulky moieties in recent literature is represented by iptycene-containing polyimides. Iptycenes are a family of hierarchical, three-dimensional molecules with rigid, fused ring structures, and the simplest as well as the most studied iptycene structure in polymers is triptycene.1,49–55 The motivation behind this design is that iptycenes' shape-persistent, bulky structures are highly effective in disrupting chain packing, leading to high fractional free volume (high permeability), and they are capable of instilling microporosity into the polymers enabling size sieving. An early report on incorporating the triptycene moiety into polyimides is 6FDA-DATRI (Fig. 1) which was based on 2,6-diaminotriptycene linked directly with 6FDA dianhydride in an attempt to create a highly rigid backbone.56 This design led to a fractional free volume of 0.226 and strong permeability/selectivity combinations, with separation performance surpassing the upper bound for several gas pairs (point 2, Fig. 2). Following this result, an analogous polyimide was reported utilizing an extended triptycene unit with greater internal free volume to examine polymer properties after addition of the benzene ring (6FDA-DAT2, Fig. 1).57,58 This resulted in increased proportions of larger pores while preserving the smaller size-sieving pores seen in 6FDA-DATRI (called 6FDA-DAT1 in this paper), leading to approximately 75% increases in permeability with only minor selectivity decreases (point 3, Fig. 2).57,58
More systematic studies on iptycene-based polyimides for gas separation membranes were reported by the Guo group to elucidate the fundamental structure–property relationship for these innovative iptycene polymer series, with an emphasis on exploration of structure manipulations for maximized separation performance.59–63 To systematically study the contributions of a disruptive bridging unit and side group modifications, a series of triptycene-incorporated diamines containing pendant groups of varying size were polymerized with 6FDA (6FDA-trip-1,4 series in Fig. 1). Overall, similar to the 6FDA-DATRI series, the addition of the triptycene moiety with adjacent substituent groups of varying sizes provided a route to simultaneously improved permeability and selectivity over commercial glassy membranes, leading to their separation performance approaching the 1991 upper bounds (point 4, Fig. 2).59 While the exchange of the triptycene unit led to higher selectivities for most gas pairs, the presence of flexible ether bonds in the backbone resulted in less desirable permeabilities than the aforementioned 6FDA-DATRI series without flexible linkages, stressing the importance of restricted backbone mobility in combination with the bulky pendant groups for optimum separation performance. In attempts to enhance the backbone rigidity of the initially reported triptycene-based polyimide series, the Guo group modified their initial triptycene-based diamine to remove the flexible ether bond such that the triptycene unit could be directly connected to the imide rings at the 1,4 positions. To study the synergetic effect of the ether bond and bulky trifluoromethyl groups, a copolymer series was synthesized with systematically varied content of the “direct” triptycene repeat unit (6FDA-1,4-trip_CF3:D copolymer series, Fig. 1). Compared to 6FDA-1,4-trip_CF3 with flexible ether bonds, copolymers containing 50% or greater of the triptycene-direct bridging group showed a large jump in free volume (e.g., FFV increased from 16.7% to 18.6% from 6FDA-1,4-trip_CF3 to 6FDA-1,4-trip_CF3:D 1:3) and permeabilities with mostly maintained selectivities, approaching the 2008 upper bound (points 5–8, Fig. 2). This design strategy further highlights the importance of the inflexible polymer backbone, as free volume and permeability improved with increasing backbone rigidity even sacrificing the incorporation of the bulky trifluoromethyl group, which was the most promising substituent in the previous study.62
Stemming from the results of the triptycene-based polyimide series, study of the larger iptycene unit, pentiptycene, was a logical next step.60,61 A bulkier, inflexible H-shaped scaffold containing five hierarchically connected arene rings, pentiptycene provides even more intrinsic, internal free volume than triptycene and further frustration of chain packing. An identical series to the initial triptycene-based diamines incorporating substituent groups on adjacent aromatic rings was synthesized with the only modification consisting of the substitution of the pentiptycene unit in for the triptycene (6FDA-PPDA series, Fig. 1). Similar to observations in the comparison between 6FDA-DAT1 and 6FDA-DAT2, replacement of the triptycene with bulkier pentiptycene (point 9, Fig. 2) significantly improved permeability of all gases with well-maintained selectivities. In general, design tactics utilizing amplified amounts of internal free volume elements within the iptycene family tend to lead to increased permeabilities with little selectivity tradeoff, improving overall separation performance.
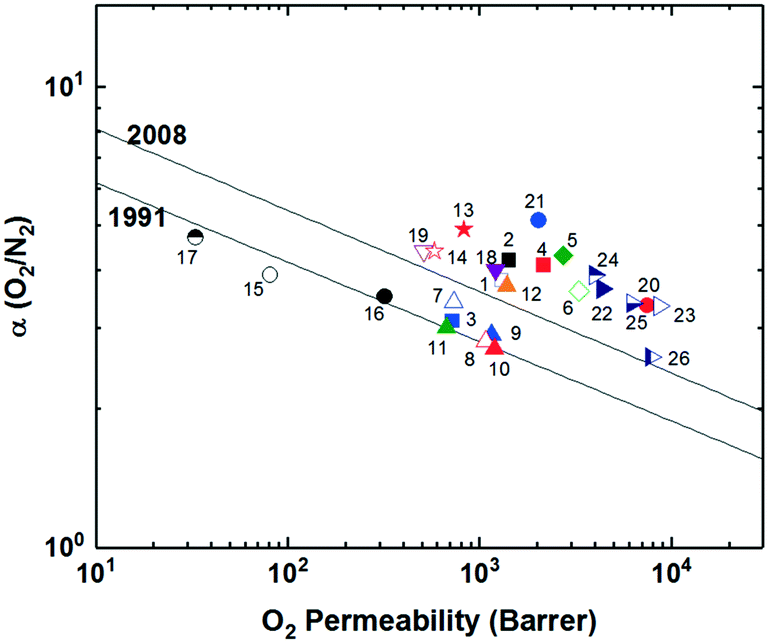
Recently, a series of sterically restricted polyimides (PI-3B and PI-3F, Fig. 1) were synthesized by combining commercially available dianhydrides with a new diamine coined BAN-3, containing a cumbersome pendant naphthalene group along with four isopropyl groups ortho to the amine positions and examined on their free volume and transport properties.64 As expected, incorporating BAN-3 led to high fractional free volume values of over 0.19 and fairly high permeabilities. Combining BAN-3 with the oft-studied bulky, trifluoromethyl containing 6FDA unit (PI-3F, point 11 in Fig. 2) led to the highest permeabilities of the series, albeit with moderate selectivities. When the smaller, more rigid BPDA was joined with BAN-3 (PI-3B, point 10 in Fig. 2), a drop in permeability was observed, accompanied by a sharp increase in selectivity for the separation of O2/N2, approaching the 2008 upper bound with an O2 permeability of 81.3 Barrer and O2/N2 selectivity of 5.0.64,65 Important in this polymer design is the individual contributions of each incorporated monomer, and how the pieces fit together to provide optimal microstructures for the greatest combinations of permeability and selectivity. Overall, increased backbone rigidity and the presence of disruptive pendant groups leads to promising enhancements in separation performance.
3.3. Substituent group and side chain manipulation
Incorporation of projecting bulky side groups or side chains is another common way to improve polymer free volume for better gas separation performance. This strategy was initially done quite extensively on commercial polysulfones, through analysis of various unwieldly groups located between the sulfone bonds to explore their effects on free volume and separation performance.66–72 In general, bulky side groups tend to frustrate chain packing leading to increases in free volume and permeability while the selectivity change varies depending on the nature of the side groups. However, an unusual trend of the effect of substituent groups on gas transport properties was observed for the abovementioned iptycene-based polyimides, where the internal free volume (IFV) uniquely associated with iptycene units may accommodate substituent groups of certain size. Within the 6FDA-1,4-trip series (Fig. 1), the bulky triptycene units with adjacent aromatic rings contained either no substituent (6FDA-1,4-trip_para), a methyl group (6FDA-1,4-trip_CH3), or a trifluoromethyl unit (6FDA-1,4-trip_CF3). It was observed that free volume and permeabilities increased from the methyl, to the para, to the trifluoromethyl substituted polymers, (points 1–3 in Fig. 3). What is unusual here is that the incorporation of a methyl substituent led to reductions in both the free volume and permeabilities. This result was attributed to the possibility of the methyl group, with an estimated occupied volume of 29.5 Å3, partially occupying the internal free volume elements of the triptycene unit between the clefts, which have an approximate volume of 31.0 Å3 per cleft, leading to reduced free volume and permeability relative to the unsubstituted counterpart.73 However, incorporation of the CF3 substituent led to expectedly simultaneous increases in free volume and permeability due to its larger size than that of the internal free volume elements. Similar observations were reported in pentiptycene-based 6FDA-PPDA series (Fig. 1), a –CH3 side group led to the lowest free volume and permeabilities within the series, while the –CF3 substituent again led to the largest (points 4–6 in Fig. 3), further underlining the ability of the smaller pendant group to “partially fill” the free volume elements intrinsic to the iptycene unit.60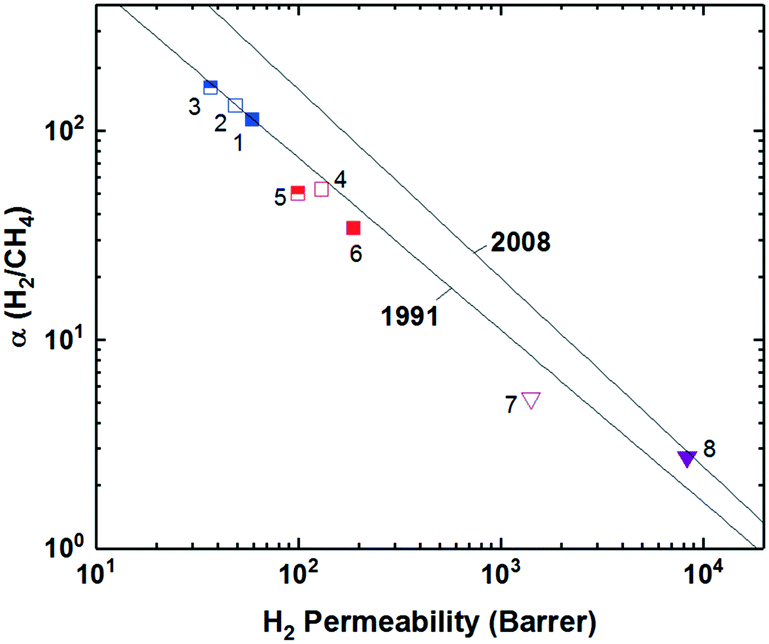 | ||
| Fig. 3 Representative polymer series utilizing various substituent groups and/or side chains plotted against the Robeson upper bound for H2/CH4. Polymers labeled as follows: 1) 6FDA-1,4-trip_para; 2) 6FDA-1,4-trip_CH3; 3) 6-FDA-1,4-trip_CF3; 4) 6FDA-PPDA(H); 5) 6FDA-PPDA(CH3); 6) 6FDA-PPDA(CF3); 7) OMe-ROMP; 8) CF3-ROMP. | ||
Recently, a few innovative polymer designs utilizing unique approaches for side chain manipulation have also advanced toward the 2008 upper bounds for certain gas pairs. Smith and coworkers explored the previously untapped realm of bottlebrush style polymers with super-rigid, microporosity generating side chains attached to flexible polymer backbones.74 This new design strategy has a few potential advantages relative to the traditional super-rigid ladder-type polymers in that high molecular weight can be easily achieved by circumventing condensation polymerization with ring-opening metathesis polymerization (ROMP) and various functionalities can be incorporated pre-polymerization to possibly control the morphology of the generated micropores. Two rigid monomers were created as norbornyl–anthracenyl moieties containing either a methoxy (OMe-ROMP, Fig. 1) or trifluoromethyl substituent (CF3-ROMP, Fig. 1) which underwent a solvent-free iterative Diels-Alder reaction to give ladder-type, super-rigid oligomers containing 2–9 repeat units with terminal norbornene groups. From here, high molecular weight polymers could be obtained via ROMP polymerization. In general, even with the flexible backbone, the incorporation of ultra-rigid ladder-type side chains produced highly permeable polymers. Specifically, CF3-ROMP (point 7 in Fig. 3) showed extraordinary permeability (e.g., H2 permeabilities of ≈8300 Barrer), along with sufficient selectivity to approach the 2008 upper bound for H2/CH4 separations. OMe-ROMP (point 8 in Fig. 3), while still quite permeable and with improved selectivity, performed significantly worse than CF3-ROMP. The replacement of the trifluoromethyl unit with the methoxy group led to a significant performance drop from near the 2008 upper bound to below the 1991 upper bound. These results were consistent with the much higher BET surface areas of 700 m2 g−1 for CF3-ROMP than 146 m2 g−1 for OMe-ROMP, indicating the incorporation of a bulkier, stiffer trifluoromethyl group protruding off the rigid side-chain further frustrated chain packing.74,75
Also utilizing norbornyl-based monomers, albeit in a different manner, were Long and coworkers, who recently synthesized a series of polynorbornenes containing various CO2-philic moieties for targeted carbon-dioxide based separations.13 Of the five polynorbornenes in the series (P1–P5, Fig. 1), P1 contained a triethoxysilyl substituent, P2–P4 contained increasing amounts of an ethylene glycol-like tris(2-methoxyethoxy)silyl substituent, ending in P5, a homopolymer consisting of all ethylene-glycol like substituents off the pendant silyl group. The goal of their fundamental structure–property analysis was to investigate the effect of incorporating the more CO2-philic ethylene glycol-like unit in the polymer in order to enhance CO2-based separation performance. As content of the CO2-philic group increased from 0–100% from P1–P5, a minor decrease in CO2 permeability from 936.6 to 754.8 Barrer was observed. However, this was accompanied by an increase of CO2/N2 selectivity of 124% due to greatly reduced nitrogen permeability, putting P5 about at the 2008 upper bound, a near vertical increase in performance on the upper bound plot relative to P1. This remarkable jump in performance was ascribed to greatly enhanced CO2 solubility selectivity in P5 relative to P1, with little contribution from diffusivity selectivity. Noticeably, the superior solubility selectivity didn't arise from increasing CO2 solubility with greater content of the CO2-philic group, but was actually attributed to large declines in N2 solubility.13
Within polymers of intrinsic microporosity (PIMs) which utilize a more rigid, ladder-type polymer backbone (detailed discussion on backbone manipulation is provided in section 3.4 and section 3.5), pendant substituent effects have been explored as well. Investigation of the spirobifluorene unit used in PIM-SBF through molecular simulation suggested the inclusion of methyl substituents would further increase microporosity.76 This motivated study of a series of solution-processable spirobifluorene-based PIMs (PIM-SBF-2 to PIM-SBF-5, Fig. 4) that contained either methyl (PIM-SBF-2 to PIM-SBF-4) or tert-butyl (PIM-SBF-5) side groups extending off the spirobifluorene unit, and their performance was compared to non-substituted PIM-SBF-1 (Fig. 4).77 Two other PIM-SBFs (PIM-SBF-6 and 7, Fig. 4) containing fused-benzo substituents were unable to dissolve in chloroform, and therefore unable to be cast into films and characterized for their separation performance. PIM-SBF-2, which contained four methyl substituents off the SBF unit, had much higher permeability than all other tested PIM-SBF polymers. It was followed in permeability by PIM-SBF-5 with two bulky tert-butyl groups present on the SBF unit, then by PIM-SBF-3 and 4, which contained 2 methyl substituents apiece in varied locations, but saw only minor changes in performance. The presence of four methyl groups versus two tert-butyl groups on the SBF unit provided starkly different free volume and overall performance. While modeling predicted both substituents would lead to increased microporosity, PIM-SBF-2 had a much higher concentration of ultramicropores (<7 Å) relative to PIM-SBF-1 and PIM-SBF-5, while PIM-SBF-5 had a larger presence of micropores greater than 10 Å. This difference in free volume distribution led to PIM-SBF-2 having exceptional combinations of permeability and selectivity, much more selective than the also super-permeable PIM-SBF-5. Additionally, the presence of the tert-butyl group was proposed to act as a molecular turnstile due to its free rotation, which could further promote transport of the larger gases.77 A later exploration of the PIM-SBF series involved the attachment of a fused tetramethyltetrahydronaphthalene (TMN) to the spirobifluorene unit (PIM-TMN-SBI, Fig. 4), which yielded permeabilities second only to PIM-SBF-2, albeit at slightly lower selectivities as well.78 The substantial difference in gas separation performance between the various substituted spiro units can be observed in Fig. 6. These results highlight the free volume control attributed to modifications of backbone rigidity as well as effects of side groups on free volume size and distribution. Minor modifications such as methyl groups versus tert-butyl, or fused benzene versus fused TMN substituents, yield vastly different effects on polymer solubility, free volume, and gas transport, providing a valuable route for tunability of gas separation performance.
 | ||
| Fig. 4 Structures of spirobisindane-based PIMs utilizing various pendant groups. | ||
A systematic fundamental study was performed on a series of semi-ladder polyimides utilizing a triptycene-dianhydride unit (TPDA) with either a diisopropyl, dipropyl, or diethyl bridgehead substituent. These substituted-TPDA units were polymerized with a series of diamines to give KAUST-PIs-1 to 7 (Fig. 5), containing the diisopropyl substituent, KAUST-PI-1′ and KAUST-PI-5′ with the dipropyl bridgehead group, and lastly KAUST-PI-5′′ containing the diethyl unit.79 This permitted analysis of the effect of different substituent groups within the same stiff polyimide backbone. Exchanging the bridged diisopropyl bridgehead group for the linear dipropyl in KAUST-PI-1 (point 5 in Fig. 6) caused a drastic drop in separation performance, moving KAUST-PI-1′ (point 6 in Fig. 6) below the 2008 upper bound with simultaneous decreases in permeability and selectivity. This performance drop off can be attributed to the lower BET surface area of KAUST-PI-1′ (610 m2 g−1) relative to KAUST-PI-1 (750 m2 g−1), along with a decreased fraction of both micropores and ultramicropores when the linear substituent is used, leading to fewer and less selective free volume elements. For the KAUST-PI-5 series, which looked at diisopropyl, dipropyl, and diethyl bridgehead substituents, a similar decrease in permeability and reduction in BET surface area was observed after replacing the diisopropyl group (KAUST-PI-5, point 7 in Fig. 6) with the dipropyl (KAUST-PI-5′, point 8 in Fig. 6). However, unlike the KAUST-PI-1 series, this substitution had increased selectivity in tandem with its reduced permeability, showing that the performance modification of the bridgehead substituent is also dependent on the diamine used in the polymer backbone. The 6FpDA-based polymers saw a lesser increase in permeability for the smaller gases (3–4 fold for H2 and O2) relative to the TMPD-incorporated polyimides which observed 5–7 fold improvements for the same gases when going from the propyl to the isopropyl substituent. The larger gases followed the opposite trend, with the TMPD-based polymers seeing smaller increases in permeability (3 fold) for N2 and CH4 while the 6FpDA-polyimides experienced 4–5 fold increases. These performance trends, seen in Fig. 4, along with the sorption data and microstructure analysis, suggest the branched substituents create more size-discriminatory free volume elements relative to the more flexible, linear propyl groups. The effect of linear substituents of different length was investigated by comparing KAUST-PI-5′ and KAUST-PI-5′′, containing propyl and ethyl substituents, respectively. Shortening of the linear substituent length actually led to a 50% increase in the permeabilities of KAUST-PI-5′′ due to an increase in the solubility coefficients, although this permeability increase was accompanied by reduced selectivities, yet KAUST-PI-5′′ still moved closer to the 2008 upper bound upon shortening of the linear chain (point 9, Fig. 6). As evidenced by the ladder-like polymers discussed above, adjustment of pendant groups off the polymer backbone or modification of already present bridging units with further bulky side chains provides wide-ranging opportunities for performance tunability.
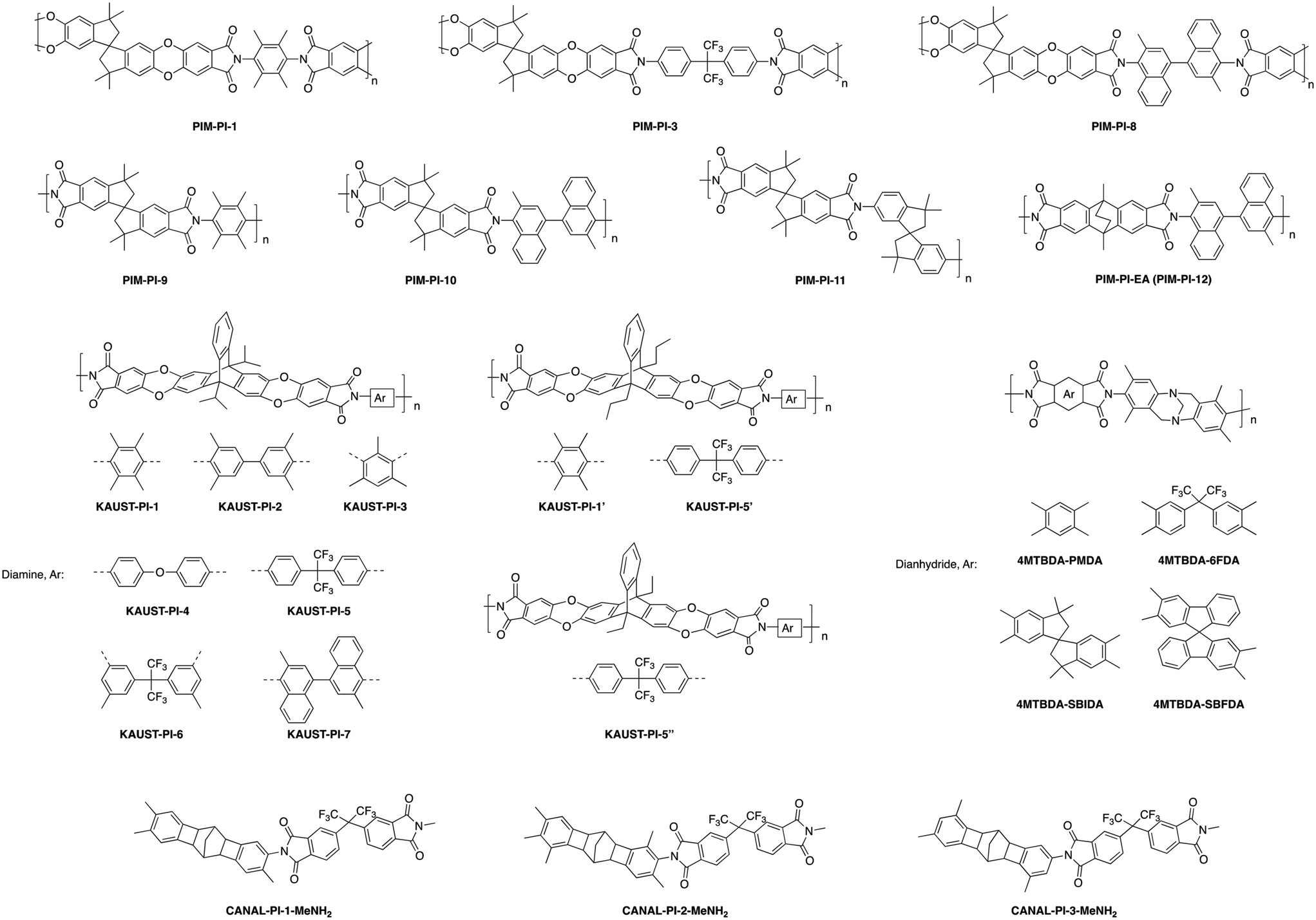 | ||
| Fig. 5 Polymer structures for inflexible, semi-ladder-type polymers containing various substituent groups. | ||
 | ||
| Fig. 6 Semi-ladder and ladder-type polymers utilizing various pendant structures within their respective series plotted against the Robeson upper bound for H2/CH4. Polymers labeled as follows: 1) PIM-SBF; 2) PIM-SBF-2; 3) PIM-SBF-5; 4) PIM-TMN-SBI; 5) KAUST-PI-1; 6) KAUST-PI-1′; 7) KAUST-PI-5; 8) KAUST-PI-5′; 9) KAUST-PI-5′′. | ||
3.4. Rigid, contorted ladder-type backbones
The arguably most effective strategy to stiffen the polymer backbone is to construct ladder-like structures where single-bond connections are replaced with two-bond connections with restricted rotation ability. The archetype for the ladder-type polymer of intrinsic microporosity for gas separation membranes, is PIM-1 (Fig. 4), which has a rigid, contorted backbone, and a spiro-center affording the contortion site.80,81 The ladder structure is achieved by combining 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane and tetrafluoroterepthalonitrile through a double aromatic nucleophilic reaction, providing a bifunctional reactive site that links the monomers together at two points, leading to significantly limited rotation relative to conventional non-ladder type glassy polymers. In addition, the contorted spirobisindane unit generates the twisted, ladder-type chains that are too inflexible to pack closely. Since the initial reporting of PIM-1 and its upper bound redefining performance, the study of novel polymers of intrinsic microporosity has grown significantly, with the publication of many new, high free volume, high performance polymers.In efforts to continue improving performance following this groundbreaking macromolecular design, the McKeown group made modifications to the backbone of the PIM-1 unit in an attempt to further rigidify the polymer chains.76 In this study, the spirobisindane unit of PIM-1 was replaced with a spirobifluorene unit (PIM-SBF, Fig. 4), which molecular modeling indicated would further reduce flexibility around the spirocenter due to the addition of the fused benzene rings. This substitution resulted in an excellent increase in performance of PIM-SBF (point 2, Fig. 8) relative to PIM-1 (point 1, Fig. 8), with the rigidified SBF backbone leading to similar permeabilities but greater selectivities, arising from enhanced diffusivity selectivity from better molecular sieving.
Even with the upgraded rigidification from incorporation of the SBF unit, the combination of relatively flexible dioxane rings and the conformational flexibility of the spirocenter still provided design variables with room for improvement. This motivated study of potential alternatives, with a goal of incorporating greater shape-persistence into the backbone to further enhance gas selectivities in these ultra-permeable polymers. Molecular modeling indicated that bridged, bicyclic units could provide a less flexible polymer backbone than the spirocenters and dioxane rings used in PIM-1 and PIM-SBF.82 Following their simulation results, the McKeown group synthesized two new polymers, integrating new inflexible units into the polymer backbones – ethanoanthracene (EA) and Tröger's base (TB), i.e., PIM-SBI-TB and PIM-EA-TB (Fig. 7).82,83 In PIM-SBI-TB, the substitution of the dioxane rings for the Tröger's base unit provided an opportunity for direct comparison of the performance of Tröger's base relative to the dioxane rings in PIM-1, while also providing a bridge for evaluation of the wholly new PIM-EA-TB, which was a significant departure from the conventional PIM-1 chain construction. Unexpectedly, PIM-SBI-TB (point 3, Fig. 8) showed relatively modest performance compared to other highly permeable PIM polymers, falling at or below the 1991 upper bound for most gas pairs, demonstrating that the Tröger's base unit alone is not enough to instill strong performance. However, when Tröger's base was combined with the inflexible bridged, bicyclic EA structure (PIM-EA-TB, point 4 in Fig. 8), outstanding combinations of permeability and selectivity were achieved for hydrogen-related separations and air (O2/N2) separation. This remarkable performance was credited to the rigidity instilled from combination of the EA and TB units, leading to superior diffusivity selectivity and greater overall size sieving. While classified as a PIM, PIM-EA-TB showed an unordinary property relative to most PIMs in that hydrogen permeated more than carbon dioxide, further reinforcing its promotion of small gas transport and superior size sieving that is not often observed in ultra-permeable membranes.82
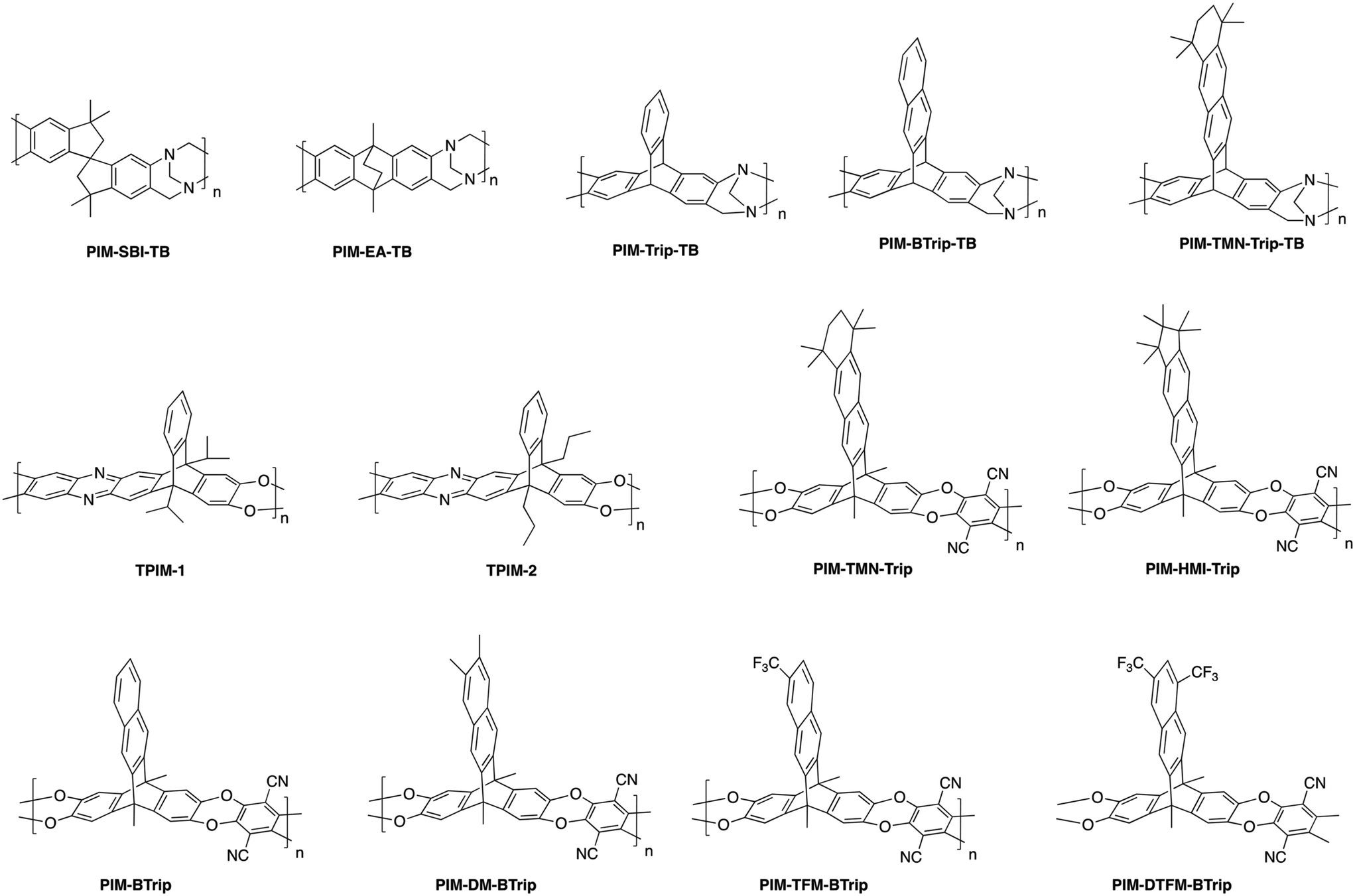 | ||
| Fig. 7 Structures for representative rigid, ladder-type polymers. | ||
 | ||
| Fig. 8 Upper bound plots for representative semi-ladder (blue region) and ladder-type (green region) PIMs. Polymers labeled as follows: 1) PIM-1; 2) PIM-SBF; 3) PIM-SBI-TB; 4) PIM-EA-TB; 5) PIM-Trip-TB; 6) PIM-BTrip-TB; 7) PIM-PI-1; 8) PIM-PI-8; 9) PIM-PI-9; 10) PIM-PI-10; 11) PIM-PI-11; 12) PIM-PI-EA; 13) KAUST-PI-1; 14) KAUST-PI-2; 15) CANAL-PI-1-MeNH2; 16) CANAL-PI-2-Me2NH2; 17) CANAL-PI-3-MeNH2; 18) TPIM-1; 19) TPIM-2; 20) PIM-TMN-Trip; 21) PIM-TMN-Trip-TB; 22) PIM-BTrip; 23) PIM-HMI-Trip; 24) PIM-DM-BTrip; 25) PIM-TFM-BTrip; 26) PIM-DTFM-BTrip. | ||
With the new polymerization methods available utilizing the formation of Tröger's base, further modifications were sought to explore even greater separation performance in high free volume polymers. A new ladder polymer utilizing the triptycene unit in place of the ethanoanthracene was synthesized utilizing the Tröger's base polymerization, PIM-Trip-TB (Fig. 7), with the hopes of increased selectivity.84 This was based on the hypothesis that the rotary motion of the methyl substituents could promote transport of the larger gases and reduce selectivity, therefore their removal would help enhance size-sieving ability. As a result, PIM-Trip-TB (point 5, Fig. 8) showed further enhancement of gas separation performance relative to PIM-EA-TB, with increases in permeability for all gases, and selectivity for most gas pairs. This rise in performance was achieved through further heightening of the diffusivity selectivity, arising from the higher chain stiffness of PIM-Trip-TB along with a growth in the amount of smaller free volume elements relative to PIM-EA-TB. This transitioned performance even further above the 2008 upper bound for O2/N2 separation as seen in Fig. 8, helping emphasize the profound impact on performance from installation of iptycene units and their intrinsic, size-sieving microporosity within the polymer.
Following the observations of Pinnau's triptycene-based polyimide study, where the integration of the benzotriptycene moiety (BTrip, an extended triptycene structure) ascribed more free volume into a polyimide compared to triptycene, leading to increased permeability, a benzotriptycene unit was substituted for triptycene in a ladder polymer with Tröger's base dubbed PIM-BTrip-TB (Fig. 7).85,86 The installation of the benzotriptycene unit did lead to greater permeabilities for all gases (point 6, Fig. 8), however, it was accompanied by slightly lower selectivities than PIM-TRIP-TB and PIM-EA-TB for all gas pairs, although performance for each gas pair was still exceptional, being at or above the 2008 upper bound.86 These results tracked well with the similar triptycene versus benzotriptycene polyimide analysis (6FDA-DAT1 versus 6FDA-DAT2, Fig. 1), where the benzotriptycene-incorporated polymer had a higher proportion of micropores >10 Å, but fewer in the selective ultramicroporous range (<7 Å).85,86
After the initial success of standard PIMs, exploration into a new subset of polyimides incorporating the design ideas of typical ladder PIMs was performed. Substantial advancement within polyimides has been observed with the creation of polyimides of intrinsic microporosity (PIM-PIs), which incorporate rigid, ladder-like units with the promising features of ladder-PIMs into semi-ladder polyimides through customization of the diamine and dianhydride units to optimize gas separation features.87 The first successful PIM-PIs (PIM-PIs 1,3 and 8, Fig. 5) incorporated a spirobisindane-based dianhydride with three aromatic diamines that had previously created highly permeable polyimides when incorporated into the polymer backbone. PIM-PI-8 (point 8, Fig. 8) combined the spirobisindane-based dianhydride with 3,3′-dimethylnaphthidine to provide the highest observed permeabilities for a polyimide at the time, accompanied by moderate selectivity. Following the initial PIM-PI series, McKeown and coworkers developed a new series of PIM-PIs by designing a spirobisindane-based dianhydride that eliminated the dioxane rings, creating a dianhydride unit directly off the aromatic ends of the spirobisindane structure, leading to an increase in rigidity. This upgraded dianhydride was then polymerized with various diamines to give PIM-PI-9 to PIM-PI-11 (Fig. 5). PIM-PI-9 (point 9, Fig. 8) and 10 (point 10, Fig. 8) showed enhancements in permeability relative to the original, less flexible PIM-PIs containing the same diamine but the extended spirobisindane dianhydride (PIM-PI-1 and 8). The increase in permeability did, however, come at the cost of some selectivity. Additionally, the presence of methyl groups next to the imide linker in PIM-PI-9 and 10 provided significant restriction of rotation, leading to a rigidified backbone relative to the highly contorted but freely rotating PIM-PI-11 (point 11, Fig. 8), providing increases in permeability for both polymers respectively.88
In an attempt to rigidify the polymer backbone of PIM-PIs further, the same group replaced the spirobisindane-based dianhydride with a more rigid ethanoanthracene-based dianhydride and prepared PIM-PI-EA (also termed PIM-PI-12, Fig. 5),82,89 which provided a direct comparison of the performance of the innovative EA group with that of the previous SBI unit used in PIM-PI-10. Though BET surface area was slightly lower for PIM-PI-EA (616 m2 g−1) compared to PIM-PI-10 (699 m2 g−1), PIM-PI-EA (point 12, Fig. 8) showed increased permeabilities for the smaller gases with similar or slightly lower values for the larger gases relative to PIM-PI-10. This led to a combination of greater permeability with enhanced selectivities for the tested gas pairs, moving PIM-PI-EA above the 2008 upper bound. The exceptional increases in selectivity appeared to be due to simultaneous enhancement of both diffusivity selectivity and solubility selectivity, similar to the observation in Tröger's base polymers PIM-EA-TB versus PIM-SBI-TB.
Further exploration of Tröger's base incorporated PIM-PI's was performed by McKeown and coworkers through analysis of dianhydride units of varying rigidity combined with 4MTBDA, which is a monomer containing a Tröger's base group endcapped with arene units containing a diamine linkage point with ortho methyl substituents to provide steric hindrance to rotation upon polymerization (Fig. 5).90 This monomer was polymerized with a series of dianhydrides: the oft-studied 6FDA, pyrometallic dianhydride (PMDA), a spirobisindane-based dianhydride (SBIDA), and a spirobifluorene-based dianhydride (SBFDA), all which were able to be cast into robust films from chloroform. Gas separation performance for the series correlated well with the rigidity of the dianhydride unit (PMDA > SBFDA > SBIDA > 6FDA), with the most rigid PMDA slightly outperforming SBFDA, and both providing performance near or above the 2008 upper bound for most gas pairs. While the SBIDA-incorporated polymer had similar or even slightly greater permeabilities for most gases, it saw a modest drop off in selectivity relative to its more rigid counterparts. Lastly, the most flexible 6FDA-based PIM-PI yielded the lowest overall performance, with significant drop-offs in permeability with similar to large decreases in selectivity as well, further highlighting the extensive effects backbone rigidity has on separation performance.
Pinnau and coworkers further explored the realm of PIM-PIs with the incorporation of a series of bridgehead substituted triptycene units polymerized with two different diamines containing methyl-groups ortho to the amine, further restricting rotation around the final imide ring. These polymers, KAUST-PI-1 and KAUST-PI-2 (Fig. 5), utilized a similar triptycene unit to the high-performing TPIM-1, each containing branched isopropyl groups, which help act as spacers to frustrate polymer chain packing.91 Both KAUST-PIs (point 13 and 14, Fig. 8) surpassed the 2008 upper bound for all tested gas pairs, further highlighting the substantial impact of the incorporation of super-rigid polymer backbones incorporated with triptycene units. KAUST-PI-1, the greater performing of the two due to its less flexible diamine unit, was found to have a bimodal pore size distribution, with large proportions of both size-selective ultramicropores as well as permeability promoting micropores, leading to high diffusivity selectivity and molecular sieving behavior. Unlike most PIMs, the KAUST-PIs exhibited higher H2 permeability relative to CO2, which is only also observed in PIM-EA-TB, accentuating the exceptional size-sieving of these polymers where in typical PIMs, the enhanced solubility of CO2 in the high free volume environment typically leads to greater permeabilities.91 KAUST-PI-1's exceptional rigidity, its incorporation of an intrinsically microporous triptycene unit, and the presence of a bulky, branched isopropyl group combined to provide some of the best combinations of permeability and selectivity observed in PIM-PIs, as seen in Fig. 8.
Following the initial success of the KAUST-PI series, a fundamental study on structure–property relationships was performed with a new series of KAUST-PIs utilizing the triptycene-dianhydride unit (TPDA) with either a diisopropyl, dipropyl, or diethyl bridgehead substituent, which were polymerized with a series of diamines to study effects of varying rigidity and bulkiness within the polymer backbone (Fig. 5).79 KAUST-PI-3, which incorporated a 2,4,6-trimethyl-1,3-phenylenediamine (TrMPD), showed mostly similar selectivities, although at the cost of permeability relative to KAUST-PI-1. KAUST-PI-4 and 6, containing 4,4′-oxidianiline (ODA) and 5,5′-(hexafluoroisopropylidene)-di-o-toluidine (ATAF) respectively gave much lower permeabilities with moderate selectivities, due to their more flexible diamine component. KAUST-PI-5, which utilized the 4,4′-(hexafluoroisopropyl)dianiline (6FpDA) diamine, saw an increase in permeability relative to KAUST-PI-4 and 6 coinciding with a drop off in permeability. With the exception of KAUST-PI-7, all KAUST-PIs had greater H2 permeation than CO2. KAUST-PI-7 showed the opposite trend, with a much larger CO2 permeability than KAUST-PI-1 and a lower H2 permeability, in addition to greatly reduced selectivities for all gas pairs. This unique result was attributed to the bulky 3,3′-dimethyl-naphtidine (DMN) diamine structure, which may have created larger, non-selective free volume elements, permitting increased permeability but greatly reduced selectivity due to reduced diffusivity selectivity. This study discerns the prominence of the effect of backbone rigidity on final gas separation performance, as incorporation of more flexible diamines as bridging structures led to drastic reductions in performance, with many sliding well below the 2008 upper bound.79
A new series of rigid, semi-ladder polyimides has recently been reported with the combination of 6FDA and a sequence of substituted ladder diamines synthesized via catalytic arene-norbornene annulation (CANAL).92–95 Initial pure-ladder CANAL-based polymers were created with desirable features for gas separation, however they were mechanically unfit to form robust thin films able to be tested for gas permeation properties.92–95 In this polymer series, CANAL-PI-1-MeNH2, CANAL-PI-2-Me2NH2, and CANAL-PI-3-MeNH2 (Fig. 5) utilize a series of CANAL diamines with varied locations and numbers of methyl substituents near the amine connections. The rigid, ladder-based diamine with its tunable methyl substituent location enabled customizable microporosity, providing a range of performance results due to the unique microporous structure observed in each polymer. CANAL-PI-3-MeNH2 (point 17, Fig. 8), with its methyl substituent meta to the imide linkage, experienced the highest selectivity and lowest permeability of the group, while moving the methyl group ortho to the amine led to an increase in permeability at the cost of some selectivity as observed in CANAL-PI-1-MeNH2 (point 15, Fig. 8). CANAL-PI-2-Me2NH2 (point 16, Fig. 8), with methyl groups located on both sides of the amine group, led to a significant jump in free volume and permeability, with a CO2 permeability of 1691 Barrer relative to PCO2 = 419 and PCO2 = 157 for CANAL-PI-1-MeNH2 and CANAL-PI-3-MeNH2, respectively. However, this increase did come with a large tradeoff in selectivity, maintaining similar distance from the 2008 upper bound as the other two polymers in the series.92 This new polymer series provides an intriguing design strategy as well as a new synthesis route toward further rigidified polymer backbones with customizable microporosity if the full ladder CANAL polymers can be successfully formed into free-standing films.
3.5. Full ladder polymers with iptycene unit
While most PIMs discussed to this point have featured moderate to significant contortion in the polymer backbone to disrupt chain packing for high free volume and permeability, a few ladder polymers have been synthesized with super-rigid, ribbon-like, iptycene containing backbones, but with minimal contortion relative to aforementioned PIMs. The two-dimensional, ribbon-like geometry was first observed by the Pinnau group with the synthesis of TPIM-1 and TPIM-2 (Fig. 7).96 These AB-type ladder polymers were synthesized via reaction between dihydroxyl units on one end of the monomer with difluoro-functional groups on the opposite end of the monomer in a self-polymerization, which allows achievement of high molecular weight polymers without strict stoichiometric control as is typical in most polycondensation reactions. TPIM-1 and TPIM-2 differ only in the substitution located at the triptycene bridgehead, with TPIM-1 containing a branched, isopropyl group while TPIM-2 has a linear, propyl unit. Though the attached side chain had a fairly significant effect on each polymer's microstructure and separation performance, both polymers showed extraordinary upper bound performance for hydrogen separations due to their ultra-rigid polymer backbones. TPIM-1 (point 18, Fig. 8), with the branched isopropyl substituent, saw simultaneously narrower ultramicropores and a larger amount of >10 Å micropores, which led to considerably larger permeabilities and selectivities relative to TPIM-2 (point 19, Fig. 8) with the more flexible, linear propyl substituent. While not as permeable as other previously mentioned ladder polymers containing Tröger's base units such as PIM-EA-TB, PIM-Trip-TB, and PIM-BTrip-TB, TPIM-1 showed much higher selectivities at high permeabilities, leading to the noteworthy performance relative to the upper bound, one of the best amongst all PIMs to date.96Following the success of the previously reported triptycene-based ladder PIMs, especially with the extraordinary permeability/selectivity combination observed in TPIM-1, another triptycene containing, two-dimensional, ribbon-like polymer was targeted by McKeown and coworkers.78 PIM-TMN-Trip (Fig. 7) combined a triptycene unit that was extended with a fused tetramethyltetrahydronaphthalene (TMN) structure and methyl substituted at the bridgehead with the common tetrafluoroterephthalonitrile monomer used in the synthesis of PIM-1. This created a 2D, ribbon-like polymer soluble in organic solvents and thus able to be formed into a film and characterized. PIM-TMN-Trip showed an exceptional BET surface of 1050 m2 g−1 as well as a larger contribution of ultramicropores relative to comparable iptycene-based ladder polymers (e.g., PIM-TMN-SBI, PIM-Trip-TB and PIM-BTrip-TB). High fractional free volume of 0.309 ± 0.005 was calculated via simulations for PIM-TMN-Trip, which agreed well with experimental results of 0.314 ± 0.005. This momentous free volume translated to ultrahigh permeabilities in PIM-TMN-Trip (point 20, Fig. 8), with hydrogen and carbon dioxide permeabilities of 16900 and 33300 Barrer respectively for a methanol-treated 195 μm thick film, with sufficient selectivity to surpass the 2008 upper bound. To attempt to differentiate between the performance of a 2D and 3D polymer containing the TMN-extended triptycene unit, a three-dimensional polymer, PIM-TMN-Trip-TB (Fig. 7), which contained a nearly identical triptycene unit (only lacking the methyl substituent on the bridgehead) was polymerized via the Tröger's base method to create a contorted structure.78 However, the same level of ultrapermeability was not obtained for the three-dimensional PIM-TMN-Trip-TB (point 21, Fig. 7) a methanol-treated 125 μm thick film, albeit with minor increases in selectivity for most gas pairs. Nonetheless, the PIM-TMN-Trip-TB did show exceptional O2/N2 separation, with an O2 permeability of 2030 Barrer accompanied by an O2/N2 selectivity of 5.13.78 Comparisons were done with the well-studied super-permeable polymer PTMSP, which emphasized the enhanced selectivity and superior size-sieving of the TMN-extended triptycene-based polymer due to the greater proportion of ultramicropores relative to PTMSP.78,81 However, both PIM-TMN-Trip and PIM-TMN-Trip-TB experienced exceptional separation properties, especially for the O2/N2 gas pair, where they make up two of the greatest outliers on the upper bound plot shown in Fig. 8. The drop-off in permeability from PIM-TMN-Trip to the three-dimensional PIM-TMN-Trip-TB highlights the effects of the disrupted chain packing experienced by the ribbon-like, two-dimensional polymer. Intriguingly, PIM-TMN-Trip and TPIM-1 provide an interesting contrast in terms of gas separation performance outcome for two-dimensional ladder polymers relative to other high-performance PIMs - both fall on the near-opposite ends of the performance spectrum, with PIM-TMN-Trip being one of the most permeable PIMs to date, while TPIM-1 holds some of the highest selectivities, at more moderate permeabilities, within the scope of PIMs.
Extending off the exceptional performance of PIM-TMN-Trip, a series of similar 2D ladder polymers (whose structures can be seen in Fig. 7) were synthesized exchanging the TMN unit for either no substituent (PIM-BTrip, point 22, Fig. 8), a solubilizing hexamethylindane (HMI) group (PIM-HMI-Trip, point 23, Fig. 8), a dimethyl-substituted benzotriptycene (PIM-DM-BTrip, point 24, Fig. 8), a single trifluoromethyl (TFM) unit (PIM-TFM-BTrip, point 25, Fig. 8), or two trifluoromethyl pendant groups (PIM-DTFM-BTrip, point 26, Fig. 8).46 All polymers provided exceptional separation performance, surpassing the 2008 upper bounds for all gas pairs. The unsubstituted PIM-BTrip specifically showed tremendous selectivity performance, approaching and surpassing the 2015 upper bound for H2/N2 and O2/N2 separations due to its excellent size-sieving capability. Upon attachment of substituent groups, increases in permeability with tradeoffs in selectivity were observed for most gas pairs. These ultra-rigid 2D ladder polymers that utilize no substituent group (PIM-BTrip) or smaller substituent groups (PIM-DM-BTrip, PIM-TFM-BTrip, and PIM-DTFM-BTrip) experienced a substantial enough jump in performance to define a new proposed 2019 upper bound for CO2/CH4 and CO2/N2 separations. The ladder polymers containing the larger TMN and HMI solubilizing groups, while ultrapermeable, were slightly less selective, placing them just below the proposed 2019 upper bound. The design principles utilized in these 2D ladder polymers provide a promising strategy toward further exploration of advancement of separation performance, evidenced by their redefinition of current upper bounds.
The resulting improvements in overall gas separation performance for the aforementioned ladder-type polymers arises from a series of design strategies: 1) rigidification of the polymer backbone, 2) incorporation of intrinsic free volume elements (e.g., iptycene units) into the polymer backbone, 3) control of polymer “dimensionality”, and 4) fine tailoring of free volume size distribution through adjustment of backbone substituent groups. The integration and optimization of these elements provides excellent opportunities for advancements in separation performance in polymer membranes.
3.6. Backbone linkage geometry
The modification of backbone linkage geometry within similar polymers represents another design strategy that can lead to differences in chain packing and thus separation performance. The Guo group studied the effects of linkage geometry in a series of triptycene-based diamines that incorporated the amine groups para (6FDA-1,4-trip_para, Fig. 9) or ortho (6FDA-1,4-trip_ortho, Fig. 9) for 1,4 connections off the triptycene unit, while also examining the properties of an analogous polyimide utilizing a 2,6 connection off the triptycene moiety (6FDA-2,6-trip_para, Fig. 9) similar to 6FDA-DATRI.97 The ortho connection led to reduced free volume and permeabilities relative to its para analog, while the 2,6 connection off the triptycene backbone saw slightly lower permeability with similar selectivity than its 1,4 equivalent. This is likely due to the greater steric hindrance within 6FDA-1,4-trip_para, as it contains two blades protruding off the polymer backbone relative to the one in 6FDA-2,6-trip_para, leading to greater packing disruption and likely better resistance to partial filling from neighboring polymer chains.97 The stark differences in performances observed between the assorted polymer linkages can be seen in Fig. 10, highlighting effects of simple changes to the connection sites used on the polymer backbone.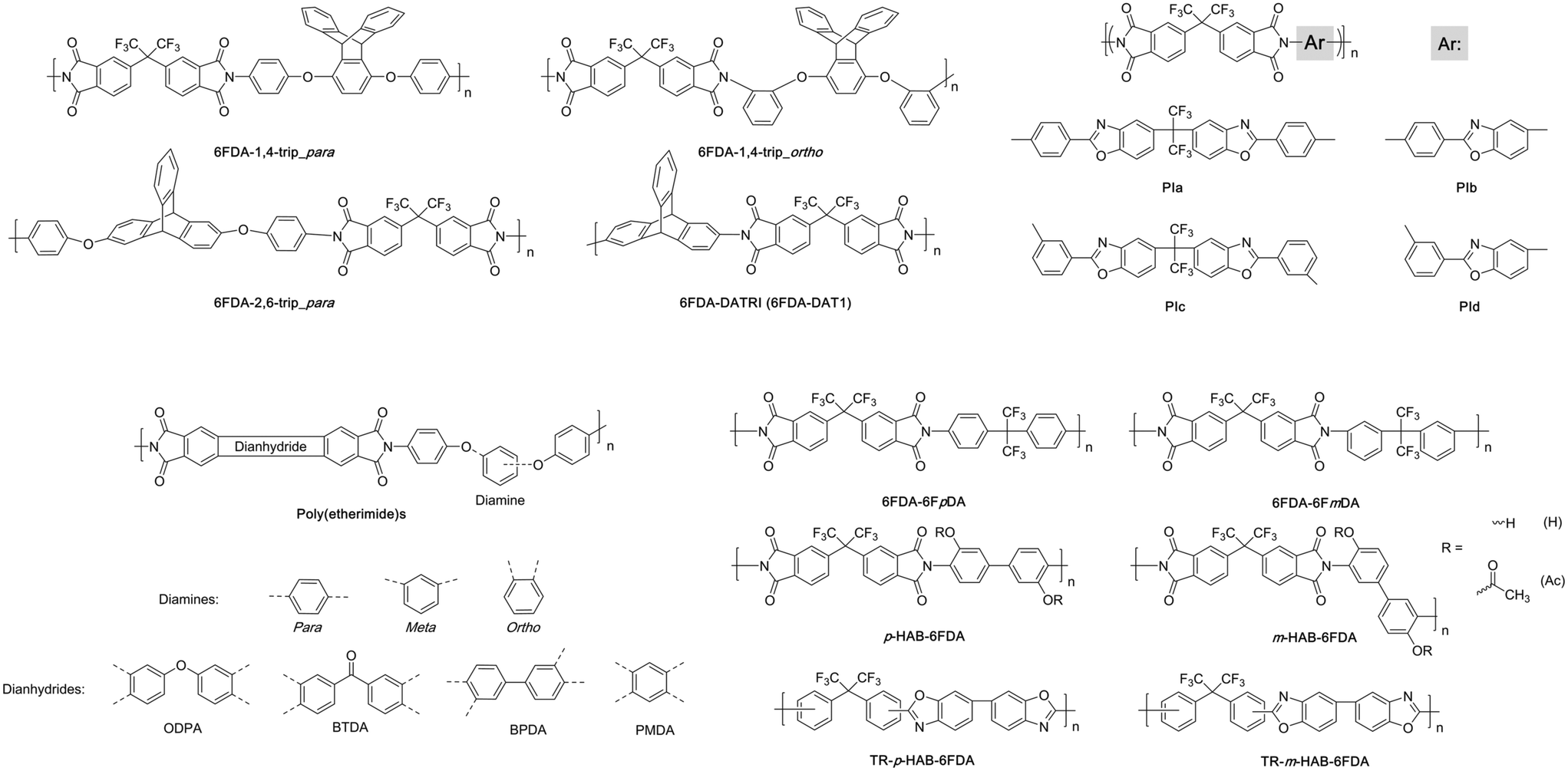 | ||
| Fig. 9 Polymer structures featuring assorted backbone linkage geometries. | ||
 | ||
| Fig. 10 Representative polymers consisting of varied linkage geometries plotted against the Robeson upper bound for CO2/CH4. Polymers labeled as follows: 1) 6FDA-1,4-trip_para; 2) 6FDA-1,4-trip_ortho; 3) 6FDA-2,6-trip_para; 4) 6FDA-6FmDA; 5) 6FDA-6FpDA; 6) m-HAB-6FDA; 7) p-HAB-6FDA; 8) m-HAB-6FDA-Ac; 9) p-HAB-6FDA-Ac; 10) m-HAB-6FDA-TR; 11) p-HAB-6FDA-TR; 12) m-HAB-6FDA-Ac-TR; 13) p-HAB-6FDA-Ac-TR; 14) PIa; 15) PIb; 16) PIc; 17) PId. | ||
Recently, a systematic study was also performed on a series of poly(etherimide)s comprised of flexible diamines utilizing either para, ortho, or meta linkages with a sequence of dianhydrides of varying rigidity (Fig. 9). While all polymers fell in the range of lower permeabilities with high selectivities, within the two series that saw results for all linkage types for different dianhydrides, permeabilities were observed in the order of para > meta > ortho.98 This outcome was also observed in a pair of 6FDA-based polyimides (6FDA-6FmDA and 6FDA-6FpDA, Fig. 9) analyzing the difference between meta and para linkages on separation performance.99 Blatant differences were seen in permeability between the two, with the para-connection providing a CO2 permeability of 63.9 Barrer, more than 12 times greater than the meta-connections (5.1 Barrer), stemming from more dense packing observed in 6FDA-6FmDA relative to 6FDA-6FpDA (FFVs of 0.1748 and 0.1897 respectively). This more efficient packing did lead to higher selectivities in the meta-based polyimide, with a CO2/CH4 selectivity of 63.8 compared to 39.9 for 6FDA-6FpDA (points 6 and 5 respectively, Fig. 10). A modification as simple as the location of the connection site can lead to drastic transformations in performance and provide a design route to tunable performance through these adjustments.
A multi-pronged study of thermally rearrangeable (TR) polyimides was performed which analyzed the effect of isomeric para and meta connected polyimides on both their precursor and post-thermal rearrangement (TR) performance (m- and p-HAB-6FDA and m- and p-HAB-6FDA-Ac, Fig. 9).100 For the two sets of isomeric polyimide precursors, which have either hydroxyl or acetyl groups ortho to the imide ring, insignificant differences in performance were observed between the permeabilities of the polymers, quite dissimilar than previously reported traditional polyimides. This is attributed to the presence of the ortho polar functional groups in the polyimide backbones, whose influence might outweigh the effects of isomeric differences. Similarly, after thermal treatment, only minor increases in permeability were observed for the para connections compared to the meta linkages, regardless of the substituent group, as seen in Fig. 10. This similarity in performance can possibly be attributed to the significant structural rearrangements that occur during the TR process, overshadowing any distinctions that might have been observed between the isomeric backbones. As observed in the isomeric TR-precursor polyimides as well as in their final thermally rearranged state, competitive effects of other functional groups or post-polymerization modification can outweigh effects in linkage geometry. However, linkage geometry can be a useful tool for tailoring polymer microstructure and separation performance as is often observed in polymers using isomeric ortho, meta, or para connections.
A series of partially thermally rearranged poly(benzoxazole-co-imide)s were explored consisting of two different diamines – one with and one without trifluoromethyl units on the polymer backbone – and both the para (p-BFO and p-BOA) and meta (m-BFO and m-BOA) isomers of each diamine were examined to analyze the effect of isomeric structure on the polymer series.101 The combination of trifluoromethyl groups and varied isomeric backbones led to interesting differences in molecular packing, with the two para polymers having the highest (PIa, Fig. 9) and lowest (PIc, Fig. 9) FFVs, while the two meta polymers (PIb and PId, Fig. 9) saw nearly identical free volumes. This resulted in PIa having the highest permeabilities for all gases, and the para-based polymers providing the best performance for CO2-based separations. Similar to previous polymers, the para-based trifluoromethyl containing polymer (PIa, point 14 in Fig. 10) showed higher permeabilities than its meta isomer (PIb, point 15 in Fig. 10). However, within the BOA series, a unique trend was observed in that the meta isomer was more permeable for hydrogen, while the para isomer had greater CO2 permeability. This led to greater separation performance from the meta isomer for hydrogen-based separations, but carbon dioxide-based separations saw enhanced performance from the para isomer (Fig. 10). Highlighted in this result is the strong effect linkage geometry can have on polymer microstructure and gas separation performance, where simply changing the connection architecture can provide different separation profiles for each gas pair.
3.7. Thermally rearrangeable (TR) polymer systems
Gaining attraction as a promising strategy for producing high performance membranes are thermally rearranged (TR) polymers, primarily based on polybenzoxazoles (PBOs). TR-PBOs are created from a thermal conversion process of functionalized polyimides containing functional groups (typically hydroxyl groups) ortho to the imide ring.10 The thermal rearrangement from the polyimide to the polybenzoxazole provides a route to typically enhanced free volume dependent on the structure of the original polymer precursor as well as the size of the thermally labile functional groups next to the imide ring. Reviews by Kim et al.102 and Galizia et al.2 both provide sections covering thermally rearranged polymers in greater detail. Since publication of these reviews, promising, innovative iptycene-incorporated TR polymers have been reported, as highlighted below.103,104A new series of TR polymers was systematically studied using similar design strategies to the aforementioned methods, incorporating the three-dimensional triptycene unit in increasing quantities into a series of polyimides with either a hydroxyl or acetyl group ortho to the imide ring (Fig. 11).103 This permitted study of both the effect of rising triptycene integration into the backbone along with consequences of removal of a larger substituent group during the TR process. As the amount of triptycene in the polymer increased, drastic enhancements in selectivity were observed with only minor sacrifices in permeability, leading to large selectivity increases on the upper bound plots for both the triptycene-based PBOs (TPBOs, points 1–4 in Fig. 12), placing their performance well above the 2008 upper bound for both hydrogen and carbon dioxide-based separations. The initial ortho functional group also played a prominent role in overall performance, with removal of the acetyl group (TPBO-Ac-1.0 versus TPBO-1.0) during the TR process providing membranes with greater permeabilities and slightly lower selectivities for the TPBO homopolymers, but with comparable selectivities at higher permeabilities for the rest of the series (TPBO-Ac-0.25 to 0.75 versus TPBO-0.25 to 0.75, points 1–3 and 5–7 respectively in Fig. 12). These results highlight the considerable enhancement in performance from inclusion of the triptycene unit due to its size-sieving internal free volume elements and rigid, bulky structure, in addition to the customizability in microstructure and separation performance arising from the tunable, thermally labile functional groups ortho to the imide ring.103 Fully incorporating triptycene into the backbone of the two TR polymers shown above provides some of the highest combinations of permeability and selectivity observed in O2/N2 separation, with drastic enhancements in selectivity relative to other high performing TR polymers and PIM-type polymers.
 | ||
| Fig. 11 Structures for various triptycene-based thermally rearranged (TR) polymers. | ||
 | ||
| Fig. 12 Upper bound plot for triptycene-based TR polymers for CO2/CH4 separation: polymers labeled as follows: 1) TPBO-0.25; 2) TPBO-0.5; 3) TPBO-0.75; 4) TPBO-1; 5) TPBO-Ac-0.25; 6) TPBO-Ac-0.5; 7) TPBO-Ac-0.75; 8) TPBO-Ac-1; 9) TPI-PBO-0.25; 10) TPI-PBO-0.5; 11) TPI-PBO-0.75. | ||
A major prohibitive challenge observed in TR polymers is the compromised mechanical properties that arise from the thermal treatment process, potentially due to thermal degradation at the high temperatures involved. To address this issue, a series of triptycene-containing poly(benzoxazole-co-imide) membranes were synthesized in an attempt to simultaneously enhance mechanical properties and separation performance through the incorporation of a rigid, non-TR-able triptycene unit.104 These copolymers were achieved by combining increasing amounts of the non-TR-able triptycene-based TPI segments with the TR-able o-hydroxyl PHI unit (TPI-PHI-Precursor and TPI-PBO, Fig. 11). Upon thermal rearrangement of all polymers within the series, strong separation performance around the 2008 upper bound was observed for both H2/CH4 and CO2/CH4 separations (Fig. 12), greater than the performance observed for a pure 6FDA-6FAP homopolymer.104,105 Overall, the incorporation of the non-TR-able triptycene-based segment resulted in good mechanical properties and enhanced separation performance, due to potential presence of pi-pi stacking between the benzene blades and the disruption of chain packing and instillation of intrinsic microporosity from the iptycene unit.61,104
4. Physical aging and polymer design strategies to overcome
4.1. The physical aging phenomenon
Physical aging is one of the major materials-related challenges gas separation membranes must overcome to achieve implementation in commercial applications. This is particularly pertinent considering the current trend in new polymer membrane material design largely focuses on high free volume or microporous polymers. Polymers below their glass transition temperature are typically not in an equilibrium state. Physical aging in these glassy polymers refers to the observed changes in polymer properties as a function of only storage time, free of any outside influence, driven by a gradual approach toward equilibrium.9,106–108 A simple understanding of excess free volume and its reduction due to physical aging can be achieved by analyzing a plot of specific volume or enthalpy versus temperature for a polymer (Fig. 13). This phenomenon provides a unique challenge for glassy gas separation membranes, in that their initial state is out-of-equilibrium, with excess free volume and a driving force toward a denser equilibrium state. While glassy polymers don't experience long-range chain motion as rubbery polymers do under ambient conditions, they still undergo local motions (e.g., single bond rotation, substituent group motion), and over time this leads to more dense chain packing and reductions in free volume, and therefore, loss of gas permeability, as the polymer attempts to approach its unattainable state of equilibrium, densely-packed chain packing. Physical aging in polymer gas separation membranes has been well studied, with extensive analysis on the effects of film thickness and polymer free volume on the aging process.107,109–113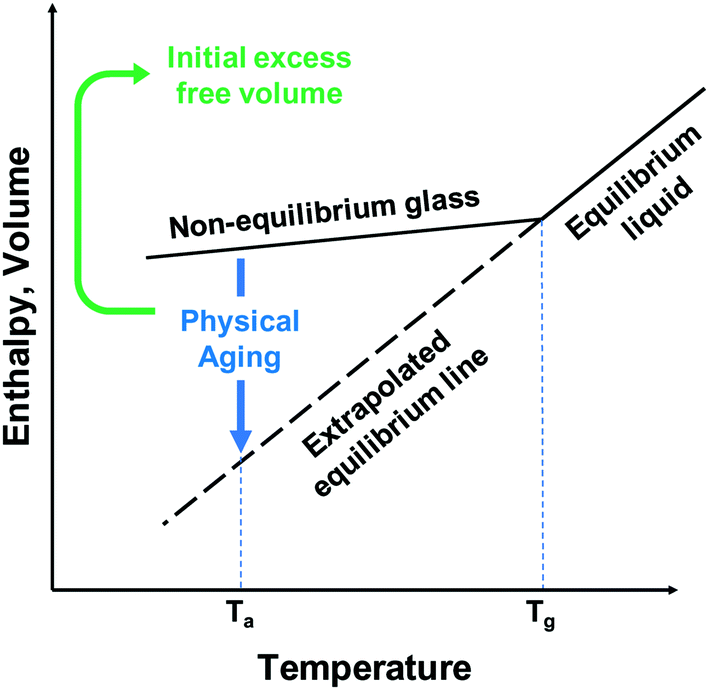 | ||
| Fig. 13 Schematic of typical effects of physical aging on enthalpy or volume for glassy polymers as a function of temperature, where Tg is the glass transition temperature and Ta is the constant temperature of the environment in which the polymer experiences aging. | ||
The relationship between free volume and physical aging generates a series of challenges that have been observed in high-free-volume polymers. While exceptional initial performance is observed utilizing super-rigid polymer backbones in attempts to maximize free volume and permeability, this often causes these high-performance polymers to experience larger non-equilibrium free volume relative to more flexible polymers. For example, a well-studied ultra-permeable polymer, poly(1-trimethylsilyl-1-propyne) (PTMSP), showed a decrease in N2 permeability from around 34000 Barrer to 11000 Barrer in only 27 days.114,115 Additionally, as previously mentioned, the first polymer of intrinsic microporosity, PIM-1 experienced an 80% decrease in nitrogen permeability after 1380 days of aging.116 While continued reductions in free volume are generally observed over extended periods of time, an “aging knee” has been observed in some PIM-type polymers, where the majority of permeability decline occurs within approximately the first 15 days, after which permeability reductions (and often accompanying selectivity increase) are quite modest even after extensive aging periods.117 However, a subsequent phase of aging is proposed to occur in high-free-volume polymers as well, where gradual reorganization of the chains over long time frames due to the greatly restricted mobility of the super-rigid backbones produces considerable redistribution of free volume and further declines in permeability.116
With the nontrivial changes in gas transport properties of initially high-performance polymers over time due to physical aging, a myriad of approaches have been explored to mitigate or overcome the issue. Low et al. provides an excellent review on substantial efforts that have been undertaken focusing on copolymer and polymer blending, chemical crosslinking, and inorganic–organic composite membranes.5 In this article, we focus on pure polymer design strategies that have showed promising aging resistance and/or graceful aging profiles where the selectivity enhancement outweighs the permeability losses. It should be mentioned that there is a lack of standard approach for physical aging tests across the reported studies in the literature. Direct comparison between different polymer strategies is frequently not available due to different aging time and/or aging conditions were applied. However, plots of selectivity gain vs. permeability loss are analyzed and provided regardless of their aging history, which should shed some light on the effectiveness of various design strategies in combatting physical aging.
4.2. Rigid backbones with limited rotational freedom and side-chain manipulation
One of the most common approaches to combatting physical aging (as well as overcoming the tradeoff relationship) through polymer design has been the rigidification of the backbone chain. The theory behind rigid backbones mitigating physical aging proposes that more rigid polymer chains will lead to reduced flexibility and further prohibit chain motion, reducing aging over time.Physical aging performance was analyzed for a set of bulky, iptycene-containing polymers – 6FDA-DAT1 and 6FDA-DAT2.58 After 150 days of aging, both membranes experienced declines in permeability of approximately 15–20%, with no meaningful changes in selectivity (Fig. 15 and 16). The design of these membranes is meant to take advantage of the iptycenes ability to imbue natural free volume into the polymer, while also disrupting chain packing. The authors noted that the two polymers have somewhat flexible imide linkages due to the lack of any groups ortho to the imide bond that could provide steric hindrance toward rotation. This flexibility likely causes the polymers to be fairly close to equilibrium quickly after casting, hence the only minor decreases in permeability after extensive aging. Therefore, the polymer microstructure and ensuing stable, strong performance can be attributed to the more permanent nature of the free volume elements ascribed by the iptycene units.58
The previously discussed rigid, semi-ladder polyimides KAUST-PI-1 and 2 underwent aging studies to probe their aging behaviors relative to reported pure ladder-PIMs. KAUST-PI-1, which only has 150-day aging data, saw some of the lowest decreases in permeability relative to other PIM-PIs and ladder PIMs with less than 20% loss in H2 permeability and a reduction in O2 permeability lower than 40% (Fig. 14 and 16). These were accompanied by >50% and >20% increases in αH2/N2 and αO2/N2 respectively, leading to performance translation steeper than the 2008 upper bound with aging. Conversely, KAUST-PI-2 saw quite comparable increases in selectivity albeit with >40% permeability loss for H2 and >60% loss for O2, with performance tracking relatively parallel to the upper bound with aging, although at a significantly larger aging time of 870 days (Fig. 14 and 16).116 It appears aging behavior in the KAUST-PIs is dependent on the backbone flexibility, as the more rigid KASUT-PI-1 performs better with initial performance while also enjoying an enhanced aging profile.91,116
 | ||
| Fig. 14 Physical aging effects on O2 permeability and O2/N2 selectivity for a variety of rigid polymers. Polymers labeled as follows: 1) KAUST-PI-1; 2) KAUST-PI-2; 3) PIM-PI-EA; 4) CANAL-PI-1-MeNH2; 5) CANAL-PI-2-Me2NH2; 6) CF3-ROMP; 7) OMe-ROMP; 8) PIM-1; 9) PIM-SBF; 10) PIM-SBF-2; 11) PIM-SBF-5; 12) PIM-EA-TB; 13) PIM-Trip-TB; 14) PIM-BTrip-TB; 15) TPIM-1; 16) TPIM-2; 17) PIM-TMN-Trip; 18) PIM-BTrip. | ||
Another semi-ladder PIM polyimide, PIM-PI-EA, experienced promising aging behavior as well.89 PIM-PI-EAs aging performance after 273 days rivalled that of KAUST-PI-2, moving further above the 2008 upper bound for both H2/N2 and O2/N2 separations (Fig. 14 and 16). Greater selectivity improvements relative to permeability losses during aging occurred due to preferential reduction of N2 permeability relative to that of H2 and O2, with PIM-EA-TB experiencing decreases in permeability of 32% and 52% for the smaller H2 and O2 gases, but a 65% decrease in permeability of the larger N2 gas. This corresponded to a 90% increase in αH2/N2 along with a 35% increase in αO2/N2 as seen in Fig. 14 and 16. Although the ladder polymer PIM-EA-TB experiences better overall separation performance after aging for H2/N2 and O2/N2 separations, with larger permeabilities at similar or slightly improved selectivities, PIM-PI-EA actually enjoys a better aging profile, seeing higher gains in selectivity along with smaller losses in permeability.89
Similar resistance to major permeability loss in triptycene-based polyimides was observed for CANAL-PIs.92 The more initially permeable CANAL-PI-2-Me2NH2 experienced only a 24% decrease in oxygen permeability after aging 101 days, a similar aging rate to the less permeable CANAL-PI-1-MeNH2 (Fig. 14). Hydrogen permeability was affected much less than the other gases as well for both tested CANAL-PIs, with each experiencing a 10% reduction in permeability. Compared to the 6FDA-DAT-1, while CANAL-PI-2-Me2NH2 saw a fractionally greater percentage decrease in O2 permeability (24% to 15%), it still maintained an O2 permeability nearly 6 times greater, owing to its more rigid backbone with restricted rotation around the imide linkage. However, the CANAL-PIs do not experience as strong of a size-sieving effect, conceivably due to having fewer natural free volume elements in the size range of gas diameters as is observed in iptycenes, and therefore do not experience as graceful of an aging profile as observed in Fig. 14.92
The innovative bottlebrush polymer, CF3-ROMP, with its flexible backbone but super-rigid side chains, exhibited ultrapermeability upon initial gas permeation testing. As physical aging has been observed to have more dramatic effects on polymers with greater initial permeabilities, an aging study was warranted. While long-term aging data is not available for this recently reported polymer, aging behavior was tracked at a series of time points up to 1000 hours (just under 42 days), and a linear trend was observed in a double-log plot of permeability versus aging time, indicating the polymer had not approached its quasi-steady state equilibrium.74 Additionally, this linear change in permeability over 42 days was counter to what was observed in the KAUST-PI series as well as in PIM-1, in which the largest decrease in permeability was observed during the first 15 days or so.117,118 Permeability decreases seen in CF3-ROMP were of lower magnitudes for the smaller kinetic diameter gases relative to the larger ones, leading to increased selectivity with aging as is typically observed with the tradeoff relationship (Fig. 14–16). However, the level of selectivity increase relative to permeability loss for all reported gas pairs led to a promising aging profile that caused the polymer to cross above the 2008 upper bound for H2/CH4 separation. This improved performance over time was attributed to enhanced diffusivity selectivities with time, due to reduction of the larger micropores into smaller ultramicropores in the size-sieving regime. In attempts to analyze the changes in free volume microstructure with time, wide-angle X-ray scattering (WAXS) experiments were performed in tandem with the permeation experiments at identical time stamps, with the same sample being used throughout the measurements to analyze distinct changes from aging within the same polymer, independent of the film history. This was done for both CF3-ROMP and OMe-ROMP, in addition to PIM-1, for microstructure comparison with aging. Consistent with the permeation results, the CF3-ROMP WAXS patterns showed minor decreases in intensity only for larger d-spacing values, with negligible reductions in intensity for d-spacing below 3.14 Å. Conversely, OMe-ROMP and PIM-1 saw intensity reductions across all ranges of d-spacing, although slightly more pronounced in the larger regime. This was consistent with the aging performances, where CF3-ROMP saw smaller reductions in overall permeability while achieving greater improvements in selectivity relative to the tradeoff.74 Overall, this innovative polymer design led to ultrapermeability combined with slow aging and a decent aging profile, although extended aging data is necessary to have a clearer picture of final aging performance as the polymer approaches the pseudo-steady state equilibrium.
4.3. Rigid, contorted ladder-type backbones
While initial performance for the prototypical polymer of intrinsic microporosity, PIM-1, created a new design strategy for exceptional performance, its aging properties highlighted a substantial challenge within this innovative polymer type – major permeability decreases over time. For example, PIM-1's oxygen permeability decreased from 2270 to 317 Barrer over an extensive 1380 day aging period, underlining the potential drawbacks of the new design scheme.5 With these obstacles, design elements have been explored not only to push initial separation performance above the upper bound, but to also mitigate potential aging or provide promising enhancement of separation properties after aging.For the aforementioned PIM-SBF series, extensive aging studies (>1200 days) were performed to analyze the aging effect on gas separation performance for these new high-performance polymers.77 After these long aging periods, all the polymers observed much greater losses in permeability for CO2 relative to H2, with H2 actually permeating faster in the aged polymers than CO2, similar to more conventional glassy polymers (Fig. 14–16). This is attributed to the large losses in free volume, which correlates with the carbon dioxide solubility, and therefore its permeability, while the smaller H2 gas has little solubility dependence and is less effected. During the aging process, while substantial permeability is lost, notable improvements in selectivity are observed for H2/N2 and O2/N2 separations, leading to increased overall gas separation performance relative to the upper bounds. As these high free volume polymers are aging, there appears to be preferential compaction of the larger micropores, effectively reducing permeability of the larger gases more than the smaller ones and increasing the amount of size-discriminatory free volume elements within the polymers. Most notably, after aging almost 1300 days, PIM-SBF-2 exhibited an O2 permeability of 910 Barrer accompanied by an O2/N2 selectivity of 5.5, propelling it from a bit above the 2008 upper bound during fresh performance close to the 2015 upper bound after aging.77 From Fig. 14–16, we can see a strong aging profile for SBF-2, with exceptional aging performance for H2/N2 stemming from a >200% increase in selectivity after aging. This indicates the addition of the methyl substituents to the SBF units provides a route to size-selective ultramicropores upon aging, which is not as evident in the unsubstituted PIM-SBF backbone.77
A redesign of the initial PIM-type polymer replacing the spirobisindane unit with ethanoanthracene or triptycene, and the tetrafluoroterephthalonitrile moiety with Tröger's base, led to ladder-PIMs (PIM-EA-TB and PIM-Trip-TB) with exceptional separation performance. Both polymers showed attractive performance even after aging for hydrogen (Fig. 16) and air (Fig. 14) separations. However, a significant difference was observed in their gas separation capabilities for CO2-related applications, in which PIM-EA-TB saw large losses of permeability and only minor increases in selectivity, while PIM-Trip-TB observed steeper gains in selectivity relative to permeability reduction, keeping performance at the 2008 upper bound for CO2/N2 while surpassing it for CO2/CH4 (point 10, Fig. 15). While the EA-based polymer had greater initial H2/N2 performance, PIM-Trip-TB showed better combinations of both permeability and selectivity post-aging. This heightened performance can be attributed to higher diffusivity selectivities for PIM-Trip-TB, with the greater size-sieving ascribed to the higher proportion of smaller free volume elements observed from sorption experiments. The 100-day aged PIM-Trip-TB film displayed an H2 permeability of 4740 Barrer and an H2/N2 selectivity of 25.1, greatly exceeding the upper bound. Its air separation performance was especially outstanding, with a selectivity of 5.7 for the aged film and an O2 permeability of 1073 Barrer, highlighting the exceptional size-sieving capability of the material for the similarly sized gas pair.84
 | ||
| Fig. 15 Physical aging effects on CO2 permeability and CO2/CH4 selectivity for a variety of rigid, bulky polymers. Polymers labeled as follows: 1) 6FDA-DAT1; 2) 6FDA-DAT2; 3) PIM-PI-EA; 4) CF3-ROMP; 5) OMe-ROMP; 6) PIM-SBF; 7) PIM-SBF-2; 8) PIM-SBF-5; 9) PIM-EA-TB; 10) PIM-Trip-TB; 11) PIM-BTrip-TB; 12) PIM-TMN-Trip; 13) PIM-BTrip; 14) 6FDA-1,4-trip_para; 15) 6FDA-1,4-trip_CH3; 16) 6FDA-1,4-trip_CF3; 17) 6FDA-PPDA(H); 18) 6FDA-PPDA(CH3); 19) 6FDA-PPDA(CF3) (points 17 and 18 overlapping). | ||
Aging of the subsequent, more permeable PIM-BTrip-TB also exhibited a graceful aging profile, with movement parallel to the upper bound for most gas pairs, but a steeper movement above the 2008 upper bound for the separation of oxygen and nitrogen (Fig. 14). Although momentous decreases in permeability were observed after 166 days (PO2 dropped from 3290 to 1170 Barrer), PIM-BTrip-TB exhibited quite similar performance to PIM-Trip-TB, with slightly greater O2 permeability (1170 to 1073 Barrer) and marginally lower selectivity (5.4 to 5.7), also placing it well above the 2008 upper bound.86
For ladder-type polymers with inflexible contorted backbones, further restriction of the polymer backbone led to exceptional initial performance, followed by substantial losses in permeability after extended aging times. However, for polymers with unbending backbones containing iptycene units (PIM-Trip-TB, PIM-BTrip-TB), accompanying selectivity increases often outweighed or at least countered the loss in permeation, moving parallel or even advancing further above the upper bounds. Similar results, but not quite as high performing or consistent across all gas pairs, were observed for the PIM-SBF series and PIM-EA-TB. These results emphasize the importance of instilling groups within the polymer backbone that can extend off and provide intrinsic free volume, which has been well reported for iptycenes.85
4.4. Full ladder polymers with iptycene units
Ribbon-like, two-dimensional ladder polymers like TPIM-1 and TPIM-2 initially showed separation performance in the same regime as ladder polymers with more contorted backbones such as PIM-EA-TB, PIM-Trip-TB, and PIM-BTrip-TB.96,116 However, the aging behavior of the TPIM series behaved drastically different, seeing much greater reductions in permeability with relatively extreme increases in selectivity. For example, both ladder polymers experienced similar, quite large reductions in O2 permeability of around 95% with >50% and >110% increases in O2/N2 selectivity (Fig. 14). For most high-performing PIM polymers, gains in H2/N2 selectivity typically outweigh lessened H2 permeability due to its small kinetic diameter relative to other gases, as seen in Fig. 16.116 TPIM-1 and TPIM-2, albeit being toward the higher end of H2 permeability loss (74% and 65% respectively), experienced H2/N2 selectivity increases an order of magnitude large than other PIMs after 780 days of aging. It seems that the two-dimensional ribbon-like polymers, while initially containing sizable excess free volume, may approach more efficient-packing over extended times than more contorted polymers, leading to reduction of more micropores into size-selective ultramicropores than in comparable highly permeable three-dimensional polymers, generating loftier increases in selectivity.116 | ||
| Fig. 16 Physical aging effects on H2 permeability and H2/N2 selectivity for a variety of rigid, bulky polymers. Polymers labeled as follows: 1) 6FDA-DAT1; 2) 6FDA-DAT2; 3) KAUST-PI-1; 4) KAUST-PI-2; 5) PIM-PI-EA; 6) CF3-ROMP; 7) OMe-ROMP; 8) PIM-SBF; 9) PIM-SBF-2; 10) PIM-SBF-5; 11) PIM-EA-TB; 12) PIM-Trip-TB; 13) PIM-BTrip-TB; 14) TPIM-1; 15) TPIM-2; 16) PIM-TMN-Trip; 17) PIM-BTrip; 18) 6FDA-PPDA(H); 19) 6FDA-PPDA(CH3); 20) 6FDA-PPDA(CF3). | ||
Within the TPIM-series, interesting differences in aging were observed which showed strong dependence on the presence of different substituent groups extending off each triptycene unit. Analysis of O2/N2 separation elucidated a notable effect of the triptycene bridgehead substituent group on aging-enhanced gas separation performance, which can most obviously be observed in Fig. 14. Striking differences were observed in O2/N2 selectivity after aging albeit similar ∼95% reductions in O2 permeability. TPIM-2, containing the linear propyl substituent, saw an increase in αO2/N2 around 55%, which was slightly higher, but mostly in the same range as other reported PIMs. Comparatively, TPIM-1 saw an increase of 115% for αO2/N2, up to 8.6 at a permeability of 61 Barrer (Fig. 14).116 This superior aging performance pushed TPIM-1 further above the 2008 upper bound for both H2/N2 and O2/N2, helping to redefine the 2015 upper bound.11 These results provided an interesting insight into backbone substituent effects on aged-film gas separation performance, as the choice of substituent led to tunability of the size of free volume elements, with the branched isopropyl group generating more microcavities between the kinetic diameters of O2 (3.46 Å) and N2 (3.64 Å) compared to the linear propyl group, spawning boosted aging performance for O2/N2 separation (and greater performance overall).116
Another 2D polymer, the ultrapermeable PIM-TMN-Trip, also displayed an attractive aging trend, but one that was a bit different than the aforementioned TPIM series.78 Over an aging period of 365 days the ultrapermeable PIM-TMN-Trip, with an initial PH2 = 16900 Barrer, saw only a 42% decrease in H2 permeability with a 78% increase in αH2/N2, extending its performance even further above the 2008 upper bound. For air separation, PIM-TMN-Trip experienced a decrease in O2 permeability of 58% accompanied by a 30% increase in selectivity, moving its separation performance up to the 2015 upper bound. These values align well with what has been observed in the majority of high-performing PIMs, with TPIM-1 and 2 appearing to be outliers.78
A series of 2D-ladder polymers similar to PIM-TMN-Trip utilized varied substituent groups off the outermost benzene ring to further explore the effects of pendant group presence on separation performance and aging trends.46 All reported polymers tended to experience greater increases in selectivity than reductions in permeability, resulting in promising aging profiles. Most notably, after 718 days of aging PIM-BTrip experienced selectivity increases of 164%, 324%, and 106% for the gas pairs CO2/CH4 (point 13, Fig. 15), H2/N2 (point 17, Fig. 16), and O2/N2 (point 18, Fig. 14) respectively, with permeability losses of only around 80% for CO2 and O2 and 60% for H2. This led to performance for the aged polymer above the 2015 upper bounds and defining the 2019 upper bounds. These results indicate that the 2D, ribbon-like backbone structure alone is not enough to achieve much larger selectivity enhancements during the aging process, but that control of free volume elements through the substituent groups extending off the rigid polymer backbone play an essential role in initial and aged ultramicropore size and polymer size sieving capability.
4.5. Configurational free volume elements
Highlighted in the section are some iptycene-containing polymers that showed unusual aging enhanced gas transport behavior where the aged polymers experienced not only expectedly increased selectivity but also unexpectedly improved or well-maintained permeability. This is represented by two series of iptycene-based polyimides (6FDA-1,4-trip series and 6FDA-PPDA series) with relatively flexible backbones containing ether linkages, when a bulky substituent group was instilled on adjacent aromatic rings, permeabilities were actually observed to increase over time (Fig. 15 and 16).59–61,63 Within the triptycene-based series, incorporation of the substituent groups surprisingly led to enhancements in permeability over time, with approximately 25% and 40% increases in permeability for 6FDA-1,4-trip_CH3 and 6FDA-1,4-trip_CF3 after aging for 9 months, as seen in Fig. 15, while the unsubstituted 6FDA-1,4-trip_para experienced typical aging behavior with more permeability loss than selectivity gain. This was attributed to the partial filling mechanism, where the bulky groups that initially (partially) occupy the internal free volume (IFV) of the triptycene unit may potentially unblock the IFV microcavities due to local motion with aging, leading to improved free volume and thus increased permeability.63 Additionally, due to the size-sieving nature of the iptycene's IFV microporosity, selectivity was still maintained or increased with aging, as was observed for CO2/CH4 separation in 6FDA-1,4-trip_CH3 and 6FDA-1,4-trip_CF3 (Fig. 15) and for H2/N2 separation as seen in the methyl and trifluoromethyl substituted 6FDA-PPDA series (Fig. 16).61,63 Similar results were observed within a pentiptycene-based polyimide series utilizing the same set of substituent groups. In this case, the unsubstituted backbone chain saw little effect from aging with insignificant changes in overall permeability. However, the incorporation of the methyl and trifluoromethyl groups did similarly lead to simultaneous increases in H2 permeability and selectivities for hydrogen-based separations, as seen in Fig. 16.61 This study corroborates the partial filling mechanism proposed for the triptycene series, where the intriguing interaction between the IFV microcavities and the adjacent substituent groups is first observed. Such unique free volume architecture in these iptycene-based polyimides was clearly evidenced by PALS analysis showing a bimodal size distribution of free volume elements with small-sized microcavities featuring the IFV and large-sized population for the regular free volume.59–61,63While termed “internal free volume” in these studies, this fraction of free volume microcavities in the iptycene-containing polymers originates from intrinsic molecular configuration (defined by chemical bonds) of iptycene units rather than transient, morphable chain conformation (e.g., inefficient chain packing, spiro-center with limited rotational freedom) as seen in common glassy polymers (Fig. 17). Distinct from conformational free volume which collapses rather rapidly over time as observed in high-free-volume polymers, configuration free volume is permanent in nature, as such, it is non-collapsible, similar to the case of inorganic molecular sieves. Recent studies by Galizia et al. on extensive experimental and theoretical examination of sorption behavior of a triptycene-containing thermally rearranged (TR) polymer (TPBO-0.25 as mentioned earlier in section 3.7)119,120 further highlighted triptycene's ability to both rigidify the polymer backbone as well as provide configurational free volume elements that could house condensing gas/vapor molecules such as CO2 while upholding its dimensional stability. Moreover, it has been discovered that thermal annealing of the membrane at 220 °C for 10 days does not produce any detectable physical aging as evidenced by almost identical CO2 sorption behavior before and after extensive thermal annealing, which is rarely observed in glassy polymers.121 These results, combined with the physical aging study in abovementioned studies, confirmed that configuration-based free volume provides not only exceptional size sieving ability, but also a fundamentally new strategy and a promising solution to address the undesired physical aging and plasticization issues inevitably encountered by existing microporous polymer membranes.
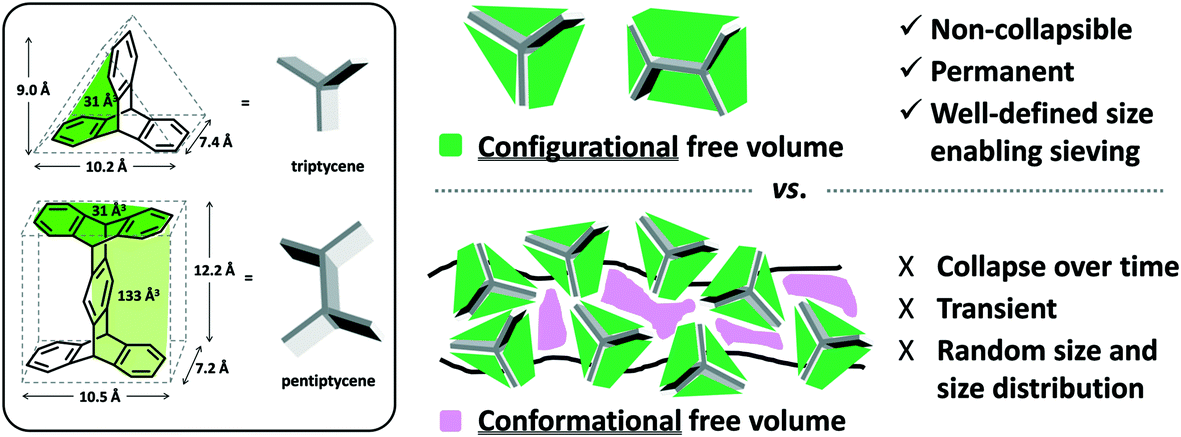 | ||
| Fig. 17 A schematic contrasting configuration-based and conformation-based free volume in iptycene-containing polymers. | ||
5. Summary and outlook
Developing polymer materials with specifically designed structures and functionality for gas separation membranes is and will continue to be a vibrant and productive field of research. In this article, we have reviewed most recent advances of innovative polymer design strategies that are directed to address the major materials challenges for gas separation membranes: permeability-selectivity tradeoff and physical aging. These topics are also identified as two major research themes in a report of “A Research Agenda for a New Era in Separation Science (2019)” recently released by a scientific committee organized by The National Academies of Sciences, Engineering, and Medicine.122The prevailing idea for improving the performance of glassy polymers is to inhibit intersegmental chain packing while simultaneously hindering backbone mobility. Since the initial report of PIM-1 in 2004, and the birth of polymers of intrinsic microporosity, extensive research efforts have been undertaken with the goal of greatly increasing polymer chain rigidity, while also incorporating architectures into the polymer backbone that further frustrate chain packing, instill high fractional free volume, and create intrinsic ultramicropores both initially and after aging. A major concern that must be considered here is the complexity and difficulties that are generally associated with the synthesis of rather exotic monomers and the polymerization to afford high molecular weight polymers. While the super-rigid backbones often lead to initial ultrapermeability followed by permeability declines with aging, analysis of the aforementioned high-performance polymers suggests the incorporation of more permanent, configuration-based free volume elements through introduction of bulky, shape-persistent, hierarchical structures, such as what is present in the iptycene-based moiety, into rigid ladder-like polymers is one of the most promising routes to superior gas separation performance with attractive aging behavior. We expect that new materials following this approach will be developed. Additionally, design strategies involving the incorporation of bulky side groups provide another route for disrupting chain packing and creating size-sieving microcavities. As observed in the polymers mentioned in this review, modification in substituent group size (e.g. ethyl versus propyl) and shape (i.e. linear propyl versus branched isopropyl) exhibited substantial differences in final polymer performance and aging properties, providing effective and possibly relatively simple routes toward fine tunability of microporosity.
Based on the design strategies utilized to achieve the many exceptional polymers highlighted here, it is apparent that continued efforts should be made toward targeting the incorporation of molecular architectures into ultra-rigid polymer backbones that impart “permanent” free volume elements, instilling microcavities in the size-range of the relevant gas pairs and continuing to tune these micro- and ultramicropores through the attachment of favorable side groups. At this point, there is still significant opportunity to explore novel structures of super-rigid polymers and to discover innovative structural architectures that can instill exceptional separation properties into polymer membranes. Based on the analysis of aforementioned novel glassy polymers for gas separation membranes in this review, some guidelines for future polymer designs might be considered: incorporation of hierarchical structures containing configurational free volume elements that are non-collapsible and in the size-range of targeted gas pairs, such as the internal free volume in iptycene units, can promote separation performance and enhance physical aging resistance; ultra-rigid backbones such as in semi-ladder and full-ladder type polymers commonly show exceptionally high permeability, and appropriate incorporation of size sieving molecular moieties is necessary to yield sufficient selectivities to redefine upper bounds; manipulating the locations for substituent group and/or side chain provides effective routes to finely tune free volume architecture as well as promote polymer-gas interactions for enhanced sorption coefficient, which sometimes may be underappreciated in glassy polymer membranes. Additionally, considerations should be given to the synthesis and processability challenges that novel polymer membrane materials must overcome to achieve commercialization, where in addition to exceptional separation performance, new polymers must withstand the necessary thermal, mechanical, and chemical stability requirements of the targeted processes, while also being sufficiently economical to produce. Finally, there seems to be significant potential to integrate new experimental techniques with molecular modeling and simulation towards the development of rational design rules and structure–property relationships for gas separation membranes, which will ultimately lead to great advances in the design and development of revolutionary new materials for energy efficient membrane separation systems capable of enhancing and ultimately replacing existing inefficient separation processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Division of Chemical Sciences, Biosciences, and Geosciences, Office of Basic Energy Sciences of the U.S. Department of Energy under award no. DE-SC0019024, and the National Science Foundation under award no. CBET-1603414. We also gratefully acknowledge the National Science Foundation Graduate Research Fellowship Program for the award (grant no. DGE-1841556) to T.C. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation and the Department of Energy.References
- J. R. Weidman and R. Guo, Ind. Eng. Chem. Res., 2017, 56, 4220–4236 CrossRef CAS.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- R. W. Baker and B. T. Low, Macromolecules, 2014, 47, 6999–7013 CrossRef CAS.
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- Y. Wang, X. Ma, B. S. Ghanem, F. Alghunaimi, I. Pinnau and Y. Han, Mater. Today Nano, 2018, 3, 69–95 CrossRef.
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS.
- Y. Yampolskii, I. Pinnau and B. D. Freeman, Materials Science of Membranes for Gas and Vapor Separation, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 CrossRef.
- R. Swaidan, B. Ghanem and I. Pinnau, ACS Macro Lett., 2015, 4, 947–951 CrossRef CAS.
- K. Ghosal and B. D. Freeman, Polym. Adv. Technol., 1994, 5, 673–697 CrossRef CAS.
- C. R. Maroon, J. Townsend, K. R. Gmernicki, D. J. Harrigan, B. J. Sundell, J. A. Lawrence, S. M. Mahurin, K. D. Vogiatzis and B. K. Long, Macromolecules, 2019, 52, 1589–1600 CrossRef CAS.
- A. Fuoco, C. Rizzuto, E. Tocci, M. Monteleone, E. Esposito, P. M. Budd, M. Carta, B. Comesaña-Gándara, N. B. McKeown and J. C. Jansen, J. Mater. Chem. A, 2019, 7, 20121–20126 RSC.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63 RSC.
- D. W. Van Krevelen and K. Te Nijenhuis, Properties of Polymers, Elsevier, 2009 Search PubMed.
- J. Park and D. R. Paul, J. Membr. Sci., 1997, 125, 23–39 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. J. Tao, J. Chem. Phys., 1972, 56, 5499–5510 CrossRef CAS.
- V. P. Shantarovich, Z. K. Azamatova, Y. A. Novikov and Y. P. Yampolskii, Macromolecules, 1998, 31, 3963–3966 CrossRef CAS.
- V. P. Shantarovich, I. B. Kevdina, Y. P. Yampolskii and A. Y. Alentiev, Macromolecules, 2000, 33, 7453–7466 CrossRef CAS.
- M. Eldrup, D. Lightbody and J. N. Sherwood, Chem. Phys., 1981, 63, 51–58 CrossRef CAS.
- Y. Kobayashi, J. Chem. Soc., Faraday Trans., 1991, 87, 3641 RSC.
- Y. Kobayashi, K. Haraya, Y. Kamiya and S. Hattori, Bull. Chem. Soc. Jpn., 1992, 65, 160–163 CrossRef CAS.
- G. Kupgan, T. P. Liyana-Arachchi and C. M. Colina, Langmuir, 2017, 33, 11138–11145 CrossRef CAS.
- C. Lastoskie, K. E. Gubbins and N. Quirke, J. Phys. Chem., 1993, 97, 4786–4796 CrossRef CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- Y. Yampolskii and N. Belov, Macromolecules, 2015, 48, 6751–6767 CrossRef CAS.
- B. Nagasaka, T. Eguchi, H. Nakayama, N. Nakamura and Y. Ito, Radiat. Phys. Chem., 2000, 58, 581–585 CrossRef CAS.
- T. Emmler, K. Heinrich, D. Fritsch, P. M. Budd, N. Chaukura, D. Ehlers, K. Rätzke and F. Faupel, Macromolecules, 2010, 43, 6075–6084 CrossRef CAS.
- T. Suzuki, M. Miyauchi, H. Yoshimizu and Y. Tsujita, Polym. J., 2001, 33, 934–938 CrossRef CAS.
- F. Y. Li, Y. Xiao, Y. K. Ong and T.-S. Chung, Adv. Energy Mater., 2012, 2, 1456–1466 CrossRef CAS.
- A. G. McDermott, P. M. Budd, N. B. McKeown, C. M. Colina and J. Runt, J. Mater. Chem. A, 2014, 2, 11742–11752 RSC.
- A. G. McDermott, G. S. Larsen, P. M. Budd, C. M. Colina and J. Runt, Macromolecules, 2011, 44, 14–16 CrossRef CAS.
- N. Ritter, I. Senkovska, S. Kaskel and J. Weber, Macromolecules, 2011, 44, 2025–2033 CrossRef CAS.
- D. Hofmann, M. Entrialgo-Castano, A. Lerbret, M. Heuchel and Y. Yampolskii, Macromolecules, 2003, 36, 8528–8538 CrossRef CAS.
- D. Hofmann, M. Heuchel, Y. Yampolskii, V. Khotimskii and V. Shantarovich, Macromolecules, 2002, 35, 2129–2140 CrossRef CAS.
- D. Hofmann, L. Fritz, J. Ulbrich, C. Schepers and M. Böhning, Macromol. Theory Simul., 2000, 9, 293–327 CrossRef CAS.
- D. Hofmann, L. Fritz, J. Ulbrich and D. Paul, Comput. Theor. Polym. Sci., 2000, 10, 419–436 CrossRef CAS.
- S. Misra and W. L. Mattice, Macromolecules, 1993, 26, 7274–7281 CrossRef CAS.
- L. M. Robeson, M. E. Dose, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2017, 525, 18–24 CrossRef CAS.
- L. M. Robeson, Q. Liu, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2015, 476, 421–431 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- H. Sanaeepur, A. Ebadi Amooghin, S. Bandehali, A. Moghadassi, T. Matsuura and B. Van der Bruggen, Prog. Polym. Sci., 2019, 91, 80–125 CrossRef CAS.
- Y. Huang and D. R. Paul, Ind. Eng. Chem. Res., 2007, 46, 2342–2347 CrossRef CAS.
- A. Bashir-Hashemi, H. Hart and D. L. Ward, J. Am. Chem. Soc., 1986, 108, 6675–6679 CrossRef CAS.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS.
- Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 2011, 6377–6403 CrossRef CAS.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- E. Hoffmeister, J. E. Kropp, T. L. McDowell, R. H. Michel and W. L. Rippie, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 55–72 CrossRef CAS.
- S. Luo, K. A. Stevens, J. S. Park, J. D. Moon, Q. Liu, B. D. Freeman and R. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 2306–2317 CrossRef CAS.
- T. M. Long and T. M. Swager, J. Am. Chem. Soc., 2002, 124, 3826–3827 CrossRef CAS PubMed.
- Y. J. Cho and H. B. Park, Macromol. Rapid Commun., 2011, 32, 579–586 CrossRef CAS PubMed.
- S. A. Sydlik, Z. Chen and T. M. Swager, Macromolecules, 2011, 44, 976–980 CrossRef CAS.
- F. Alghunaimi, B. Ghanem, N. Alaslai, R. Swaidan, E. Litwiller and I. Pinnau, J. Membr. Sci., 2015, 490, 321–327 CrossRef CAS.
- J. R. Wiegand, Z. P. Smith, Q. Liu, C. T. Patterson, B. D. Freeman and R. Guo, J. Mater. Chem. A, 2014, 2, 13309–13320 RSC.
- S. Luo, Q. Liu, B. Zhang, J. R. Wiegand, B. D. Freeman and R. Guo, J. Membr. Sci., 2015, 480, 20–30 CrossRef CAS.
- S. Luo, J. R. Wiegand, P. Gao, C. M. Doherty, A. J. Hill and R. Guo, J. Membr. Sci., 2016, 518, 100–109 CrossRef CAS.
- J. R. Weidman, S. Luo, J. M. Breier, P. Buckley, P. Gao and R. Guo, Polymer, 2017, 126, 314–323 CrossRef CAS.
- J. R. Weidman, S. Luo, C. M. Doherty, A. J. Hill, P. Gao and R. Guo, J. Membr. Sci., 2017, 522, 12–22 CrossRef CAS.
- T. Li, J. Liu, S. Zhao, Z. Chen, H. Huang, R. Guo and Y. Chen, J. Membr. Sci., 2019, 585, 282–288 CrossRef CAS.
- T. Li, H. Huang, L. Wang and Y. Chen, RSC Adv., 2017, 7, 40996–41003 RSC.
- J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1991, 32, 840–850 CrossRef CAS.
- J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1991, 32, 2618–2625 CrossRef CAS.
- J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1992, 33, 1701–1711 CrossRef CAS.
- C. L. Aitken, W. J. Koros and D. R. Paul, Macromolecules, 1992, 25, 3651–3658 CrossRef CAS.
- Y. Dai, M. D. Guiver, G. P. Robertson and Y. S. Kang, Macromolecules, 2005, 38, 9670–9678 CrossRef CAS.
- C. L. Aitken, W. J. Koros and D. R. Paul, Macromolecules, 1992, 25, 3424–3434 CrossRef CAS.
- Y. Dai, M. D. Guiver, G. P. Robertson, Y. S. Kang, K. J. Lee and J. Y. Jho, Macromolecules, 2004, 37, 1403–1410 CrossRef CAS.
- N. T. Tsui, A. J. Paraskos, L. Torun, T. M. Swager and E. L. Thomas, Macromolecules, 2006, 39, 3350–3358 CrossRef CAS.
- Y. He, F. M. Benedetti, S. Lin, C. Liu, Y. Zhao, H. Ye, T. Van Voorhis, M. G. De Angelis, T. M. Swager and Z. P. Smith, Adv. Mater., 2019, 31, 1807871 CrossRef PubMed.
- Y. Zhao, Y. He and T. M. Swager, ACS Macro Lett., 2018, 7, 300–304 CrossRef CAS.
- C. G. Bezzu, M. Carta, A. Tonkins, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Adv. Mater., 2012, 24, 5930–5933 CrossRef CAS PubMed.
- C. G. Bezzu, M. Carta, M.-C. Ferrari, J. C. Jansen, M. Monteleone, E. Esposito, A. Fuoco, K. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, J. Mater. Chem. A, 2018, 6, 10507–10514 RSC.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesaña-Gándara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 CrossRef CAS PubMed.
- R. Swaidan, M. Al-Saeedi, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2014, 47, 5104–5114 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230 RSC.
- P. Budd, K. Msayib, C. Tattershall, B. Ghanem, K. Reynolds, N. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- M. D. Guiver and Y. M. Lee, Science, 2013, 339, 284–285 CrossRef CAS PubMed.
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed.
- F. Alghunaimi, B. Ghanem, N. Alaslai, R. Swaidan, E. Litwiller and I. Pinnau, J. Membr. Sci., 2015, 490, 321–327 CrossRef CAS.
- I. Rose, M. Carta, R. Malpass-Evans, M.-C. Ferrari, P. Bernardo, G. Clarizia, J. C. Jansen and N. B. McKeown, ACS Macro Lett., 2015, 4, 912–915 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, J. D. Selbie and D. Fritsch, Adv. Mater., 2008, 20, 2766–2771 CrossRef CAS PubMed.
- Y. Rogan, L. Starannikova, V. Ryzhikh, Y. Yampolskii, P. Bernardo, F. Bazzarelli, J. C. Jansen and N. B. McKeown, Polym. Chem., 2013, 4, 3813 RSC.
- Y. Rogan, R. Malpass-Evans, M. Carta, M. Lee, J. C. Jansen, P. Bernardo, G. Clarizia, E. Tocci, K. Friess, M. Lanč and N. B. McKeown, J. Mater. Chem. A, 2014, 2, 4874–4877 RSC.
- M. Lee, C. G. Bezzu, M. Carta, P. Bernardo, G. Clarizia, J. C. Jansen and N. B. McKeown, Macromolecules, 2016, 49, 4147–4154 CrossRef CAS.
- B. S. Ghanem, R. Swaidan, E. Litwiller and I. Pinnau, Adv. Mater., 2014, 26, 3688–3692 CrossRef CAS PubMed.
- M. A. Abdulhamid, H. W. H. Lai, Y. Wang, Z. Jin, Y. C. Teo, X. Ma, I. Pinnau and Y. Xia, Chem. Mater., 2019, 31, 1767–1774 CrossRef CAS.
- H. W. H. Lai, S. Liu and Y. Xia, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3075–3081 CrossRef CAS.
- H. W. H. Lai, Y. C. Teo and Y. Xia, ACS Macro Lett., 2017, 6, 1357–1361 CrossRef CAS.
- S. Liu, Z. Jin, Y. C. Teo and Y. Xia, J. Am. Chem. Soc., 2014, 136, 17434–17437 CrossRef CAS PubMed.
- B. S. Ghanem, R. Swaidan, X. Ma, E. Litwiller and I. Pinnau, Adv. Mater., 2014, 26, 6696–6700 CrossRef CAS PubMed.
- J. R. Weidman, S. Luo, Q. Zhang and R. Guo, Ind. Eng. Chem. Res., 2017, 56, 1868–1879 CrossRef CAS.
- Z. P. Madzarevic, S. Shahid, K. Nijmeijer and T. J. Dingemans, Sep. Purif. Technol., 2019, 210, 242–250 CrossRef CAS.
- M. R. Coleman and W. J. Koros, J. Membr. Sci., 1990, 50, 285–297 CrossRef CAS.
- H. Borjigin, Q. Liu, W. Zhang, K. Gaines, J. S. Riffle, D. R. Paul, B. D. Freeman and J. E. McGrath, Polymer, 2015, 75, 199–210 CrossRef CAS.
- Y. Zhuang, J. G. Seong, Y. S. Do, H. J. Jo, M. J. Lee, G. Wang, M. D. Guiver and Y. M. Lee, Macromolecules, 2014, 47, 3254–3262 CrossRef CAS.
- S. Kim and Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32 CrossRef CAS.
- S. Luo, Q. Zhang, L. Zhu, H. Lin, B. A. Kazanowska, C. M. Doherty, A. J. Hill, P. Gao and R. Guo, Chem. Mater., 2018, 30, 5322–5332 CrossRef CAS.
- S. Luo, Q. Zhang, T. K. Bear, T. E. Curtis, R. K. Roeder, C. M. Doherty, A. J. Hill and R. Guo, J. Membr. Sci., 2018, 551, 305–314 CrossRef CAS.
- S. H. Han, N. Misdan, S. Kim, C. M. Doherty, A. J. Hill and Y. M. Lee, Macromolecules, 2010, 43, 7657–7667 CrossRef CAS.
- I. M. Hodge, Science, 1995, 267, 1945–1947 CrossRef CAS PubMed.
- L. C. E. Strum, Polym. Eng. Sci., 1977, 17, 165–173 CrossRef.
- J. M. Hutchinson, Prog. Polym. Sci., 1995, 20, 703–760 CrossRef CAS.
- Y. Huang and D. R. Paul, Polymer, 2004, 45, 8377–8393 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 5565–5575 CrossRef CAS.
- Y. Huang and D. R. Paul, Macromolecules, 2006, 39, 1554–1559 CrossRef CAS.
- Y. Huang, X. Wang and D. R. Paul, J. Membr. Sci., 2006, 277, 219–229 CrossRef CAS.
- D. Cangialosi, V. M. Boucher, A. Alegría and J. Colmenero, Soft Matter, 2013, 9, 8619 RSC.
- L. M. Robeson, W. F. Burgoyne, M. Langsam, A. C. Savoca and C. F. Tien, Polymer, 1994, 35, 4970–4978 CrossRef CAS.
- K. Nagai, B. D. Freeman and A. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 1222–1239 CrossRef CAS.
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS.
- R. Swaidan, M. Al-Saeedi, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2014, 47, 5104–5114 CrossRef CAS.
- P. Budd, N. McKeown, B. Ghanem, K. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS.
- V. Loianno, Q. Zhang, S. Luo, R. Guo and M. Galizia, Macromolecules, 2019, 52, 4385–4395 CrossRef CAS.
- V. Loianno, S. Luo, Q. Zhang, R. Guo and M. Galizia, J. Membr. Sci., 2019, 574, 100–111 CrossRef CAS.
- R. D. Crist, Z. Huang, R. Guo and M. Galizia, 2019, In preparation.
- National Academies of Sciences, Engineering, and Medicine, A Research Agenda for Transforming Separation Science, The National Academies Press, Washington, DC, 2019, DOI:10.17226/25421.
| This journal is © The Royal Society of Chemistry 2020 |