
Determining order-to-disorder transitions in block copolymer thin films using a self-referencing fluorescent probe†
Zhe
Qiang
a,
Lingqiao
Li
a,
John M.
Torkelson
 ab and
Muzhou
Wang
*a
ab and
Muzhou
Wang
*a
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: mwang@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 13th November 2019
Abstract
The thin film geometry of block copolymers (BCPs) required by many applications often leads to a shift of their order-to-disorder transition temperature (TODT) due to the presence of air–polymer and substrate–polymer interfaces. A challenge associated with measuring BCP thin film TODTs is the lack of a simple, generalizable, non-destructive characterization method, which is of critical importance for exploiting BCP self-assembly in nanotechnology. In this work, we present a non-invasive fluorescence characterization method for determining the TODT of BCP thin films. Fluorescent pyrene molecules were covalently attached at nearly trace levels to one block and used as a probe of the local polymer matrix. The BCP TODT can be determined by a discontinuous change in the intensity ratio I1/I3 of pyrenyl vibronic emission bands as a function of temperature, which is induced by a change of the nanoscopic chemical environment around the pyrene labels at the ODT. When the BCP film thickness is sufficiently small, an increase of TODT compared to bulk is observed. This increase of TODT in thin films is mainly attributed to attractive polymer–substrate interactions, the effects of which can be varied by adjusting the film thickness and substrate surface hydrophobicity. These results not only demonstrate that fluorescence characterization can be used as a generalizable approach for determining BCP thin film ODTs, but also provides additional insights on manipulating functional BCP nanopatterns by controlling the substrate surface functionality.
 Muzhou Wang | Muzhou Wang is an assistant professor in the Department of Chemical and Biological Engineering at Northwestern University, where he leads a group with interests including polymer self-assembly and physics, biomimetic materials, and optics. He was a previously a postdoctoral fellow at the NIST, where he worked with Drs. Jeffrey W. Gilman and J. Alexander Liddle. His Ph.D. is in chemical engineering from MIT, under the supervision of Prof. Bradley D. Olsen. His work has been recognized with several awards including an American Physical Society DPOLY/UKPPG Exchange Lectureship and a National Science Foundation CAREER award. |
Design, System, ApplicationBlock copolymers (BCPs) are utilized in thin film geometries for many applications, including lithography and separations. To design and process these materials, one must know the temperature at which BCPs spontaneously develop nanostructure, i.e., the order-to-disorder transition temperature (TODT), which can differ in thin films relative to bulk. However, ODT characterization is difficult in thin films because bulk measurements are not easily translated to such small sample quantities (μg). This study meets this constraint by using a simple yet highly sensitive fluorescence characterization approach for accurately determining the ODT in BCP thin films. The establishment of this technique enables a quantitative understanding of how to manipulate thin film ODTs by adjusting film thickness and interfacial properties. With the ease in fluorophore labeling, this method can be extended to different polymer chemistries and employed as a facile, universal platform for understanding BCP ODT in thin film systems, which will enable further advancement of BCP nanopatterning technologies. |
Introduction
Block copolymers (BCPs)1–3 have attracted significant attention over recent decades due to their ability to self-assemble into a wide variety of nanostructures useful in a broad array of applications including microelectronics,4–8 templates for porous materials,9,10 drug delivery,11,12 water purification13–15 and optical polarizers.16,17 Key to driving this nanophase segregation process is the immiscibility between different blocks,18,19 which are primarily determined by two major variables: the overall degree of polymerization (N) and the Flory–Huggins interaction parameter (χ) between the two blocks. Application of mean-field theory by Leibler and others predicts that symmetric diblock copolymers can produce ordered nanostructures when the product χN is greater than 10.495.20–24 These theoretical studies hold the critical design rules for generating functional BCP-derived nanopatterns.25–27 As χ is inversely proportional to temperature (T), the transition from the ordered to the disordered state occurs as the temperature increases beyond the order-to-disorder transition temperature TODT,18 where non-favorable enthalpic interactions between opposite blocks is sufficiently weakened and entropy becomes dominant. From a processing perspective, TODT sets an upper limit on the thermal annealing temperature for ordering the BCP nanostructures.28,29While considerable effort has been focused on using BCPs for thin film nanopatterning applications,30–33 their ODT behavior has usually been investigated in the bulk34,35 or solution state.36,37 In thin films, the interactions from the polymer–substrate or air–polymer interfaces often lead to appreciable changes in the TODT and phase behavior compared to the bulk state.38–42 For instance, Green and co-workers reported a significant TODT increase for a lamellae-forming polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) thin film compared to their bulk analogues when the film thickness is less than two domain layers.43 Russell and co-workers observed a thickness-dependent enhancement of TODT of polystyrene-block-polyisoprene (PS-b-PI) thin films when PS domains have a preferential interaction with the substrate.44 Very recently, Ryu and co-workers confirmed that a strong polymer matrix–substrate interfacial interaction increases the TODTs of PS-b-P2VP thin films.45 As the BCP thin film TODT changes as a function of film thickness and substrate surface properties, an accurate determination of TODT is particularly important for the applications that require BCPs with less than a few domain layers thickness.
A challenge associated with measuring thin film TODTs is the lack of a suitable in situ characterization tool. For the bulk BCP systems, researchers have employed many different techniques such as scattering,46–48 rheology,49,50 birefringence51 and differential scanning calorimetry52 for determining their ODTs. However, many of these methods cannot be translated into the thin film geometry mainly due to the instrument limitations in probing very small sample quantities. So far, the most widely used tool for investigating BCP thin film ODTs is grazing incidence small-angle X-ray scattering (GISAXS),53–56 which usually requires access to high-energy synchrotron beamlines. One serious concern associated with using X-ray based techniques is that the ionizing radiation from beam exposure can generate free radicals that initiate crosslinking and/or chain scission. This beam-induced structural damage may impact the measurement accuracy, especially for the time-resolved experiments.57 Thus, a non-destructive characterization method for probing the ODT of BCP thin films is highly desired.
Previous studies have utilized a change in the detailed shape of the emission spectrum from a fluorescent dye or label (e.g., pyrene) to study polymer glass transitions58–62 or to identify the occurrence of phase separation in solution63–65 or bulk.66,67 Here, we extend this fluorescence approach to characterize BCP thin film ODTs by using pyrenyl moieties as labels at trace levels. The temperature dependence of self-referencing peak ratios (I1/I3) from pyrenyl labels is used to report the local polymer hydrophobicity, which can be correlated with the chemistry of their surrounding environment. To examine the applicability of this technique for measuring thin film ODTs, we synthesize cylinder- and lamellae-forming PS-b-PMMA BCPs with accessible TODTs, with the PS blocks containing extremely dilute levels of pyrenyl labels. When the BCP thin film nanostructures are ordered, the pyrenyl labels are surrounded by the PS blocks to which they are chemically attached, and thus the fluorescence spectrum reports this local environment. After increasing temperature beyond the TODT, the shape of the fluorescence spectrum indicates that the pyrenyl labels are locally in the presence of both PS and PMMA units, corresponding to the disordered state of the block copolymer. After establishing this characterization method, we systematically investigate how film thickness and substrate surface chemistry impact BCP thin film TODTs. These results reveal the underlying mechanism for thickness-dependent TODTs of BCP thin films, while further expanding the application of fluorescence characterization techniques to polymers by demonstrating the ability of such techniques to determine the ODT of BCP thin films both simply and accurately.
Experimental section
Materials
All compounds were obtained from Sigma-Aldrich and used as received, unless otherwise noted. Styrene and methyl methacrylate were purified through a basic alumina column to completely remove the inhibitors.General procedure for polymer preparation
All polymers were synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization (Scheme 1). A typical synthetic procedure for preparing PS-b-PMMA BCPs is described as follows. 1.7 mg of 2,2′-azobis(2-methylpropionitrile) (AIBN) (0.01 mmol), 17.3 mg of 2-cyano-2-propyl dodecyl trithiocarbonate (0.05 mmol) and 1.5 g of methyl methacrylate (MMA) (15 mmol) were first dissolved in 5 mL of toluene. The reaction mixture was degassed over three freeze–pump–thaw cycles and then heated to 75 °C for polymerization. After 4 h, the reaction mixture was cooled to room temperature and then precipitated into cold methanol. The solids were collected and dried under vacuum overnight (PMMA: Mn = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1; Đ = 1.11). For the synthesis of PS-b-PMMA, PMMA was used as a macro-RAFT agent for a chain extension reaction. 1.0 g of PMMA, 1.1 mg of AIBN, 2.5 g of styrene and 0.3 mg of 1-pyrenemethyl methacrylate (MaPy) (Toronto Research Chemicals) were dissolved in 5 mL of toluene. The mixture was degassed over three freeze–pump–thaw cycles, and polymerization was performed by heating to 75 °C for 12 h. After polymerization, the mixture was diluted in tetrahydrofuran and then precipitated into cold methanol. The solids were collected and dried under vacuum overnight. The total fluorophore (MaPy) content added during polymerization is less than <0.3 wt%, and UV-vis spectroscopy confirms that the amount incorporated into the polymer did not exceed this concentration (PS-b-PMMA: Mn = 31400 g mol−1; ϕps = 0.30; Đ = 1.23).
800 g mol−1; Đ = 1.11). For the synthesis of PS-b-PMMA, PMMA was used as a macro-RAFT agent for a chain extension reaction. 1.0 g of PMMA, 1.1 mg of AIBN, 2.5 g of styrene and 0.3 mg of 1-pyrenemethyl methacrylate (MaPy) (Toronto Research Chemicals) were dissolved in 5 mL of toluene. The mixture was degassed over three freeze–pump–thaw cycles, and polymerization was performed by heating to 75 °C for 12 h. After polymerization, the mixture was diluted in tetrahydrofuran and then precipitated into cold methanol. The solids were collected and dried under vacuum overnight. The total fluorophore (MaPy) content added during polymerization is less than <0.3 wt%, and UV-vis spectroscopy confirms that the amount incorporated into the polymer did not exceed this concentration (PS-b-PMMA: Mn = 31400 g mol−1; ϕps = 0.30; Đ = 1.23).
 | ||
| Scheme 1 Synthetic routes of RAFT polymerization for synthesizing pyrene-labeled PS-b-PMMA block copolymers. PMMA-CTA was first synthesized in step i and then used as a macro-RAFT agent to extend the PS block (step ii). | ||
For the synthesis of pyrene-labeled homopolymers, a very small amount of MaPy relative to monomers (∼0.3 wt%) was added into the reaction mixture containing monomer, AIBN and RAFT agent. The polymerization and purification conditions (reaction temperature: 75 °C; reaction time: 4 h for PMMA and 12 h for PS) are identical to the above description for block copolymers (PS: Mn = 9500 g mol−1; Đ = 1.18. PMMA: Mn = 23300 g mol−1; Đ = 1.13).
All pyrene-labeled polymers were purified by Soxhlet extraction with methanol for 24 h in order to remove unreacted pyrene molecules (MaPy).
Nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC)
1H NMR of 5 mg mL−1 polymer solutions in CDCl3 (Hg400, 400 MHz Bruker Avance III HD systems) was used to determine the PS-b-PMMA block copolymer compositions. The number-average molecular weight, Mn, and dispersity, Đ, values of the polymers were characterized by GPC with a tetrahydrofuran mobile phase, using an Agilent 1260 Infinity II system with a refractive index detector calibrated with narrow-polydispersity polystyrene standards ranging from 640 g mol−1 to 2300000 g mol−1.
Small angle X-ray scattering (SAXS)
SAXS experiments were performed at the 5-ID-D beamline through the DuPont-Northwestern-Dow Collaborative Access Team at the Advanced Photon Source at Argonne National Laboratory. The measurements were taken upon heating from 145 °C to 181 °C. Bulk BCP samples were sandwiched between 0.03 mm thick Kapton tape and held at each temperature for at least 20 min before a SAXS measurement. The sample-to-detector distance was fixed at 7.5 m, the beam energy was 17.5 keV, and SAXS data was collected by a charged-coupled device (CCD) detector. After background subtraction, the data was reduced by integration over full azimuths to give the intensity as a function of the scattering vector, q.Film preparation and fluorescence characterization
Silicon wafers (University Wafer) were used as the substrate throughout this study. All substrates were cleaned with piranha solution (H2O2:H2SO4 = 3:7, vol:vol) at 95 °C for 12 h, unless otherwise noted. For determining the water contact angle of substrate surfaces, a drop of water (approximately 2 μL) was placed on the substrate and then analyzed using a Kruss DSA100 drop shape analyzer. Thin films were prepared by spin coating 0.5 to 10 wt% solutions of polymer in toluene. Films were annealed at 140 °C for 24 h prior to all characterization. The thickness of BCP films was determined using variable angle spectroscopic ellipsometry (VASE, J.A. Woollam Co., M-2000D) with wavelengths ranging from 420 nm to 1600 nm and incident angles of 55°, 60°, and 65°. The data were fit to a recursive model consisting of silicon substrate and a Cauchy model to determine the film thickness and optical constants.
The fluorescence emission spectra (Photo Technology International fluorimeter in front-face geometry) of polymer thin films were collected from 140 °C to 185 °C at 324 nm excitation, with emission wavelengths from 350 nm to 450 nm (0.5 nm increment with 1 s integration time). The excitation and emission slit widths were 0.5 nm; the bandpass was 1 nm. Films were held at each temperature for 30 min prior to measurement. After subtracting the background noise, the ratio (I1/I3) of the first vibronic band peak intensity (∼378 nm) to the third vibronic band peak intensity (∼388 nm) was used to characterize the order-to-disorder transitions of block copolymer thin films, due to its sensitivity to local molecular hydrophobicity.
Results and discussion
Fig. 1A and B show the fluorescence emission spectra of a 212 nm thick MaPy-labeled polystyrene (PS-py) film and a 208 nm thick MaPy-labeled poly(methyl methacrylate) (PMMA-py) at temperatures from 140 °C to 185 °C. All polymers contain less than <0.3 wt% fluorophore; based on past research,58,61–63,66 their behavior is expected to be identical within error to unlabeled polymers. With increasing temperature, the overall fluorescence intensity decreases due to the enhanced non-radiative decay of the excited-state pyrenyl dye electrons at higher temperature, which competes with their radiative decay to the electronic ground state.57,68 For both PS-py and PMMA-py films, the intensity of the first vibronic peak (I1, associated with higher-energy transitions) of the pyrenyl labels decreases more significantly than the intensity of the third vibronic peak (I3, associated with lower-energy transitions),69,70 which is attributed to the suppression of the molecular caging effect from the reduced local polymer stiffness at higher temperature.71,72 The ratio of peak intensities (I1/I3) of the pyrenyl label fluorescence is also sensitive to the local polarity of the label environment. As shown in Fig. 1C, PMMA-py films exhibit higher I1/I3 values than the PS-py films at all temperatures examined due to the more polar nature of PMMA. In the more polar PMMA environment, the improved dipole–dipole interaction between the polymer matrix and pyrenyl labels enhances the high-energy electron transitions from excited state to ground state, leading to a higher I1/I3 ratio.73,74 | ||
| Fig. 1 Normalized fluorescence emission intensity spectra for (A) a 212 nm-thick MaPy-labeled PS (PS-py) film and (B) a 208 nm-thick MaPy-labeled PMMA (PMMA-py) film. Spectra are shown for 140 °C, 155 °C, 170 °C and 185 °C, from top to bottom. The total fluorescence intensity decreases with increasing temperature. The peak positions of the first and the third vibronic peak (I1 and I3) are indicated in the figure. (C) shows the intensity ratio (I1/I3) as a function of temperature for PMMA-py (black triangles) and PS-py (red circles) films, which can be fit with a linear relationship to describe its temperature dependence. The slope obtained from fitting is −3.8 × 10−3 °C−1 for PS-py (red line) and −1.7 × 10−3 °C−1 for PMMA-py (black line), respectively. | ||
The slope of I1/I3versus temperature also depends on the matrix chemistry: PMMA-py films exhibit a more gradual decrease with increasing temperature compared to PS-py films. For hydrophobic PS films, I1/I3 is dominated by local polymer stiffness.71 However, in a more hydrophilic PMMA matrix, the change of I1/I3 as a function of temperature is attributed to a competition between improved dipole moment (corresponding to an enhanced polarity)75–77 and reduced stiffness at higher temperature. While previous literature suggests that the value of I1/I3 can be influenced by many factors including film thickness, thermal history, and processing conditions (which may vary film by film),57,66,71,72,78 these results indicate that the slope of I1/I3vs. T for certain pyrenyl dyes can be correlated to their local chemical environment. Thus, we hypothesize that the temperature dependence of I1/I3 is suitable to characterize a transition in the matrix surrounding pyrene molecules, which can be triggered by a BCP thin film ODT.
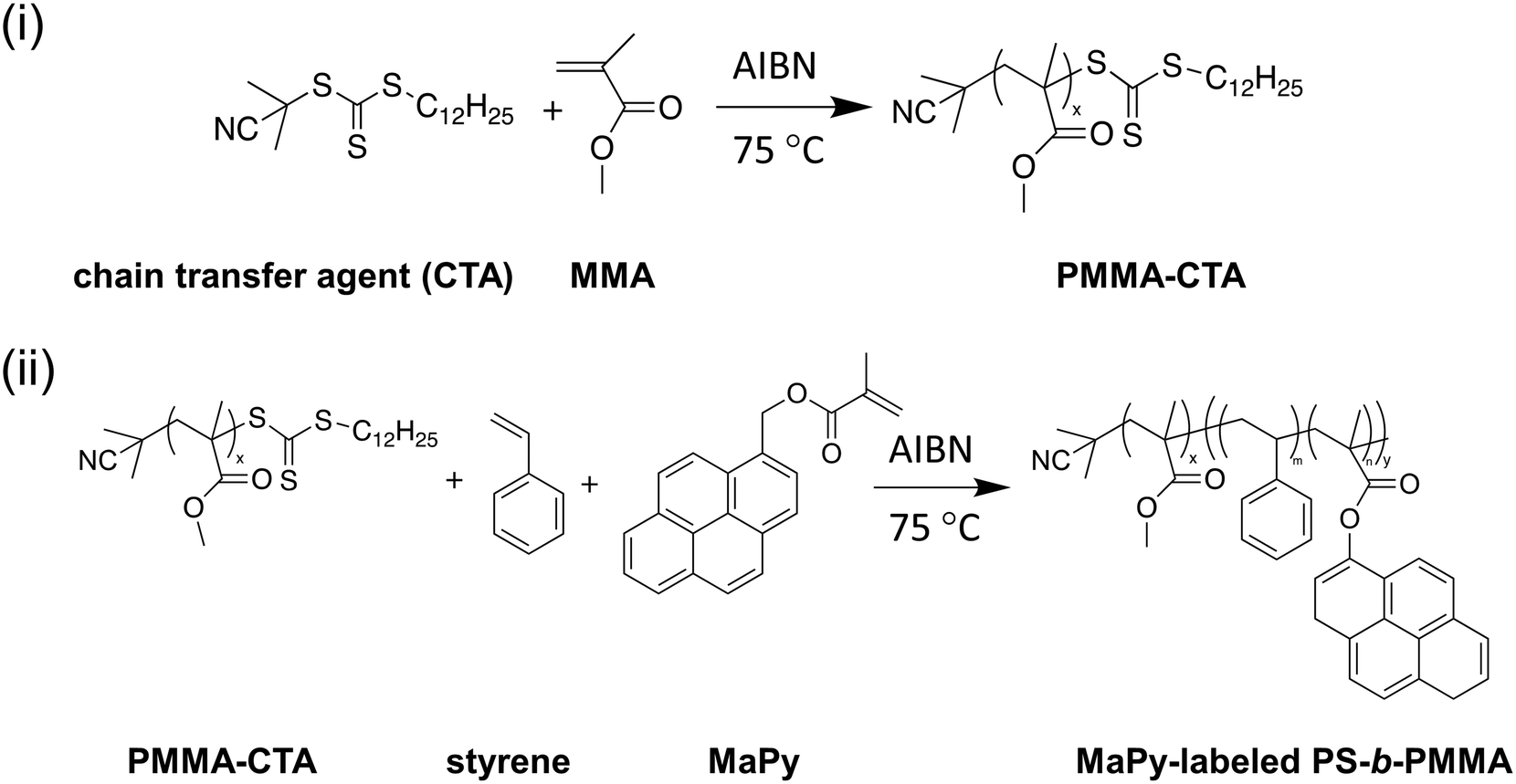
To validate the ability to use pyrene molecules for probing BCP thin film ODTs, two different PS-b-PMMA BCPs were synthesized. In both BCPs, the pyrene labels were covalently attached to the PS block via a random copolymerization strategy at 0.3 wt% relative to styrene, and the molecular weights were chosen to attain accessible TODTs between the glass transition temperature (Tg) and the degradation temperature of each block. Here, C-PSMMA (Mn = 31400 g mol−1; ϕps = 0.30; Đ = 1.23) and L-PSMMA (Mn = 26800 g mol−1; ϕps = 0.54; Đ = 1.24) represent a cylinder-forming PS-b-PMMA and a lamellar-forming PS-b-PMMA, respectively. Both samples were in powder form and annealed at a constant temperature of 140 °C for 24 h prior to measurement. The SAXS profiles for bulk C-PSMMA were measured every 2 °C upon heating. As temperature increases, the primary ordering peak of the bulk C-PSMMA remains discernible until it disappears beyond TODT = 161 °C, indicating a loss of nanostructural order (Fig. 2A). Similarly, the primary ordering peak for bulk L-PSMMA (Fig. 2B) is sharp at low temperatures but disappears above 171 °C. Fig. 2C and D show the inverse of the maximum intensity (1/I(q*)) as a function of inverse temperature (1/T) for both BCPs, and the TODTs can be quantitatively determined from the discontinuities of 1/I(q*),45 which is 161 °C (434 K) for C-PSMMA and 173 °C (446 K) for L-PSMMA, respectively.
 | ||
| Fig. 2 SAXS intensity profiles for (A) C-PSMMA and (B) L-PSMMA, which are cylinder- and lamellae-forming PS-b-PMMA block copolymers. Both samples were in powder form for SAXS measurements. The SAXS profiles were measured at different temperatures as indicated in the figure, and were vertically shifted in order to avoid overlapping. The domain spacing was determined by d = 2π/q*, which was 20.8 nm for C-PSMMA and 19.6 nm for L-PSMMA. Inverse of the maximum intensity at the first ordering peak as a function of the inverse temperature for two films are shown in (C) for C-PSMMA and (D) for L-PSMMA, with discontinuities indicating TODT of 161 °C and 173 °C. | ||
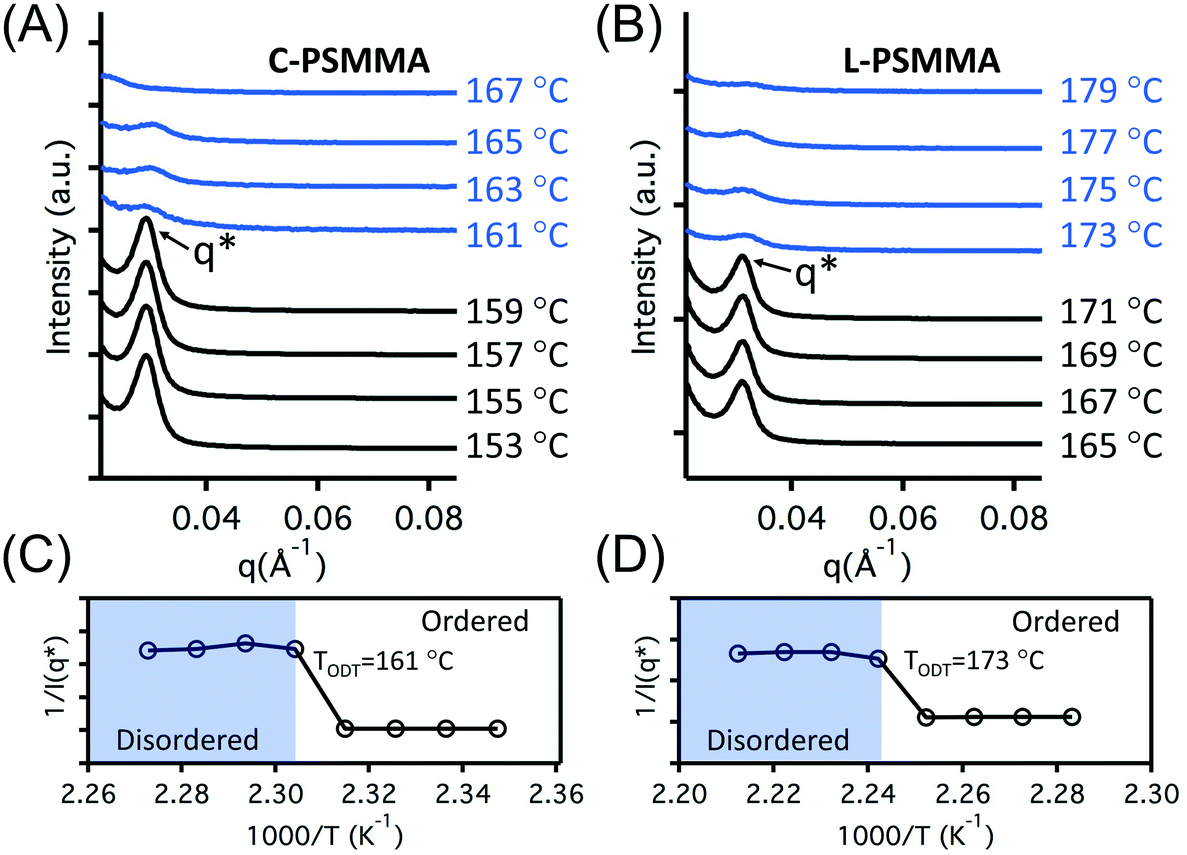
The TODTs of bulk PS-b-PMMA samples determined by SAXS provide an important reference for the remainder of our study. For investigation of the TODT in thin films, a 398 nm-thick C-PSMMA film and a 403 nm-thick L-PSMMA film were prepared, both corresponding to approximately 20 domain layers (20L0). A series of emission spectra of these two films were recorded as a function of temperature (Fig. 3). The changes in the pyrenyl label emission spectra with increasing temperature are a response to changes in local environment as previously discussed. For C-PSMMA and L-PSMMA, the intensities of the first vibronic band peak (I1) and the third vibronic band peak (I3) are both easily visible at 150 °C and gradually decrease with increasing annealing temperature. Upon surpassing a threshold temperature, an obvious change in the emission spectral shape of PS-b-PMMA thin films is observed, leading to a nearly indiscernible I3 peak for both BCPs. This change is attributed to a shift in the local environment around the pyrenyl labels, which are locally surrounded by PS blocks at temperatures below the ODT but enter a matrix containing both PS and PMMA blocks as the BCPs become disordered.
 | ||
| Fig. 3 Normalized pyrene fluorescence emission intensity at 324 nm excitation for (A) a 398 nm-thick C-PSMMA film and (B) a 403 nm-thick L-PSMMA film. Spectra are shown from 150 °C to 185 °C at every 5 °C, from top to bottom. Pyrene was covalently attached to the PS block. The total fluorescence intensity decreases with increasing temperature. The peak positions of the first and the third vibronic peak (I1 and I3) are indicated in the figure. | ||
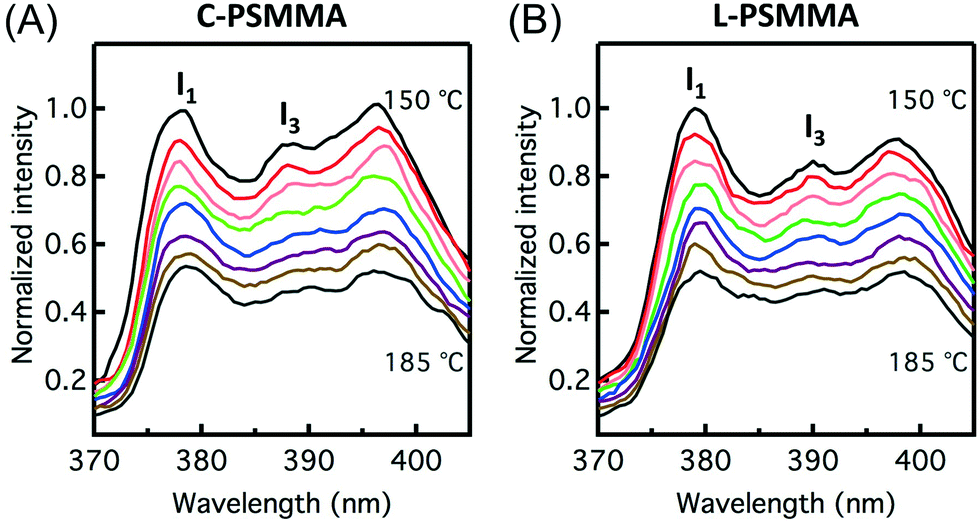
In order to precisely determine the TODTs for the PS-b-PMMA thin films shown in Fig. 3, values of I1/I3 are plotted against temperature (Fig. 4). At lower temperatures in the ordered state, the negative slopes (−4.2 × 10−3 °C−1 for C-PSMMA and −5.1 × 10−3 °C−1 for L-PSMMA) are similar to the previous result for homopolymer PS-py films (−3.8 × 10−3 °C−1, Fig. 1A), suggesting that the pyrenyl labels are positioned within the ordered PS nanodomains of the BCPs. As temperature is increased, discontinuities in both the values and slope of I1/I3 were observed; these discontinuities are expected based on our hypothesized response of pyrenyl dyes to an order-to-disorder transition of BCP thin films. Once the nanostructure becomes disordered, the PS and PMMA blocks become miscible and mixed. Thus, even though the pyrenyl dyes remain covalently bound to the PS block, they now experience a local environment that contains contributions from both the PS and PMMA blocks. For both BCPs, the TODT values can be determined directly from this fluorescence discontinuity at 161 °C (C-PSMMA) and 172 °C (L-PSMMA), respectively. Both TODTs from fluorescence characterization are in excellent agreement with the TODTs of bulk samples determined from SAXS (Fig. 2), indicating that the temperature dependence of I1/I3 from the pyrenyl label fluorescence provides a robust characterization approach for determining the ODTs of block copolymer thin films.
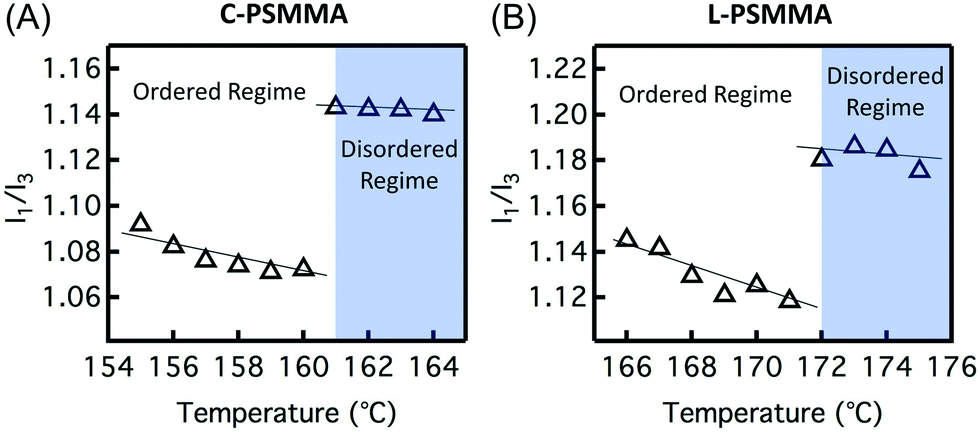 | ||
| Fig. 4 Vibronic band intensity ratio (I1/I3) as a function of temperature for (A) a 398 nm-thick C-PSMMA film and (B) a 403 nm-thick L-PSMMA film. The disordered regime is identified in blue color. A linear regression was used to fit the data in both the ordered and disordered regimes for polymer films. | ||
This simple fluorescence technique can be used to investigate the effect of film thickness on the BCP TODT by examining films with varying thickness from 30L0 down to 1L0. For film thicknesses above 10L0, the TODTs of C-PSMMA thin films are very close to their bulk TODT value (Fig. 5A). When the C-PSMMA film thickness is below 10L0 (∼210 nm), the TODT increases with decreasing thickness, indicating that nanoconfinement effects become significant. The thinnest C-PSMMA film (∼20 nm), corresponding to only one BCP domain layer, exhibits the highest TODT of 185 °C (∼24 °C higher than its bulk analogue). This enhanced TODT effect of BCP thin films is caused by the strong attractive interfacial interaction between the PMMA matrix and the substrate surface. The hydroxyl groups on the silicon substrate surface after piranha treatment lead to hydrogen bonding with the PMMA matrix,79 which can increase the effective χ of BCP thin films and enhance their nanophase separation. Hydrogen bonding interactions have been used to improve the ordering in BCPs in many examples of BCP/nanoparticles and polymer blends.80–85 For L-PSMMA films shown in Fig. 5B, the TODT begins to increase with decreasing thickness when the film thickness is 15L0. This slight increase (2 °C higher than bulk value) suggests that the critical thickness (n0) for a thickness-dependent TODT might be higher for lamellar than cylindrical structures due to the attractive polymer–substrate interactions associated with adjacent layers. A similar and more obvious morphology-dependent TODT enhancement for PS-b-P2VP thin films was recently observed by Ryu and co-workers, indicating that the planar interfacial interactions from the substrate are able to propagate deeper into the multilayer structures of lamellar domains through the BCP film thickness than cylindrical structures.45 For the monolayer L-PSMMA films, the TODT difference compared to the bulk sample (25 °C) is close to the C-PSMMA.
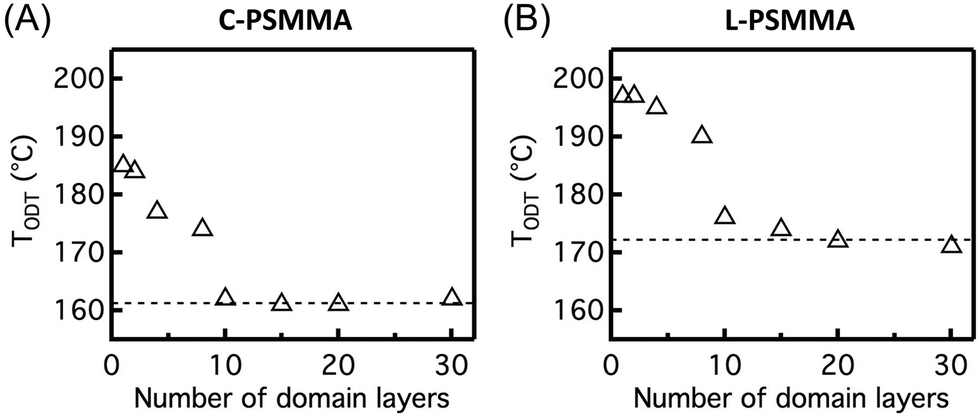 | ||
| Fig. 5 BCP thin film TODT as a function of the number of domain layers for (A) C-PSMMA and (B) L-PSMMA. Bulk TODTs (161 °C for C-PSMMA and 173 °C for L-PSMMA) determined by SAXS are indicated using the dashed line in the plots. | ||
We further quantified the effect of hydrogen bonding interactions on BCP thin film TODTs by preparing C-PSMMA films (∼20 nm thickness) on Si substrates with different hydrophobicities. By varying the duration of piranha treatment or attaching perfluorinated groups to the Si surface (see ESI†), we obtained different hydrophobicities of the bare substrate surface, as indicated by water contact angles ranging from ∼0 ° to ∼90 °. When the monolayer C-PSMMA BCP film is deposited onto the most hydrophilic substrate, the TODT exhibits a maximum increase of ∼24 °C compared to the bulk sample (Fig. 6). By decreasing the hydrophilicity of the substrate surface, as shown by a water contact angle of ∼90 ° prior to BCP deposition, the TODT difference between monolayer film and bulk sample decreases to only 4 °C. This relationship confirms that the polymer–substrate interaction is an important mechanism responsible for the TODT enhancement in BCP thin film, which becomes more influential as the film thickness decreases. These results also provide additional insight for generating nanostructured thin films from BCPs that cannot nanophase separate in bulk by manipulating the substrate surface chemistry.
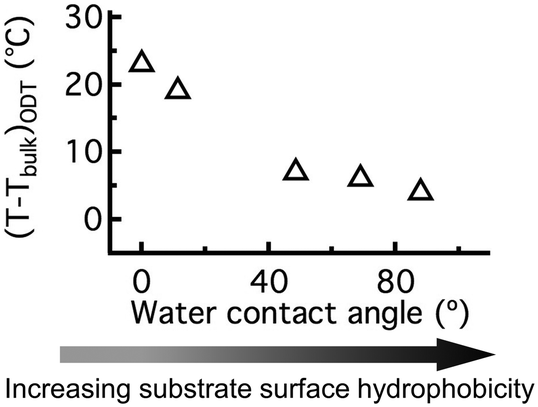 | ||
| Fig. 6 The temperature difference between the bulk TODT and the monolayer thin film TODT of C-PSMMA as a function of substrate-surface water contact angle. | ||
Conclusions
We have demonstrated the application of a simple fluorescence characterization approach for determining the order-to-disorder transition temperature of block copolymer thin films. The temperature dependence of the pyrenyl dye vibronic band fluorescence intensity ratio I1/I3 is sensitive to the local polymer matrix hydrophobicity, which can be used as a self-referencing method to correlate with the chemistry of its nanoscopic environment. Pyrenyl dyes were chemically attached at nearly trace levels onto one block of the copolymer and used as probes of the ODT. A discontinuous change of I1/I3 with increasing temperature is used to identify the ODT of BCP thin films, because the pyrenyl labels are locally surrounded by one block in an ordered nanostructure but are mixed with both blocks above the ODT. Two PS-b-PMMA copolymers were used as model systems, and fluorescence measurements show excellent agreement with SAXS data when the film thickness approaches bulk conditions. When thickness is sufficiently small, an increase of TODT is observed due to the preferential hydrogen bonding interactions between the substrate surface and the PMMA blocks. This polymer–substrate interfacial effect becomes more pronounced as film thickness decreases, which leads to a maximum ∼25 °C higher TODT for a monolayer of lamellae-forming BCP films compared to bulk. The effect of nanoconfinement on thin film TODT was also investigated as a function of substrate surface hydrophobicity, and we found that the TODT enhancement becomes less significant when BCP is deposited on more hydrophobic substrate surfaces. This fluorescence characterization technique is expected to be generalizable to many other BCP systems for accurately determining their ODTs as long as the chemically distinct blocks exhibit discernable differences in repeat unit polarity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. Q. and M. W. acknowledge the Donors of the American Chemical Society Petroleum Research Foundation and L. L. acknowledges an ISEN Fellowship and a Northwestern Terminal-Year Fellowship for partial support of this research. The authors thank Profs. Julia Kalow and Kenneth Shull of Northwestern University for access to equipment and useful discussions. SAXS data was collected with the help of Tyler Heyl and Steven Weigand. Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I, DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. SAXS data was collected using an instrument funded by the National Science Foundation under Award Number 0960140.References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- Y. Tseng and S. B. Darling, Polymer, 2010, 2, 470 CAS.
- J. H. Kim, H. M. Jin, G. G. Yang, K. H. Han, T. Yun, J. Y. Shin, S.-J. Jeong and S. O. Kim, Adv. Funct. Mater., 2019, 1902049 CrossRef.
- Y. S. Jung, J. H. Lee, J. Y. Lee and C. A. Ross, Nano Lett., 2010, 10, 3722 CrossRef CAS PubMed.
- C. A. Ross, Y. S. Jung, V. P. Chuang, J. G. Son, K. W. Gotrik, R. A. Mickiewicz, J. K. W. Yang, J. B. Chang, K. K. Berggren, J. Gwyther and I. Manners, Proc. SPIE, 2010, 7637, 76370H CrossRef.
- J. Y. Cheng, C. A. Ross, V. Z.-H. Chan, E. L. Thomas, R. G. H. Lammertink and G. J. Vancso, Adv. Mater., 2001, 13, 1174 CrossRef CAS.
- S. O. Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411 CrossRef CAS.
- J. Y. Kim, H. M. Jin, S.-J. J. T. Chang, B. H. Kim, S. K. Cha, J. S. Kim, D. O. Shin, J. Y. Choi, J. H. Kim, G. G. Yang, S. Jeon, Y.-G. Lee, K. M. Kim, J. Shin and S. O. Kim, Nanoscale, 2018, 10, 100–108 RSC.
- S. Wang, P. Tangvijitsakul, Z. Qiang, S. M. Bhaway, K. Lin, K. A. Cavicchi, M. D. Soucek and B. D. Vogt, Langmuir, 2016, 32, 4077 CrossRef CAS.
- H. Ahn, S. Park, S. Kim, P. J. Yoo, D. Y. Ryu and T. P. Russell, ACS Nano, 2014, 8, 11745 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844 CrossRef CAS PubMed.
- S. H. Chen, C. Willis and K. R. Shull, J. Membr. Sci., 2017, 544, 388 CrossRef CAS.
- S. P. Nunes, Macromolecules, 2016, 49, 2905 CrossRef CAS.
- W. A. Philip, B. O'neill, M. Rodwogin, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2010, 2, 847 CrossRef.
- S. Y. Kim, J. Gwyther, I. Manner, P. M. Chaikin and R. A. Register, Adv. Mater., 2014, 26, 791 CrossRef CAS.
- B. H. Kim, S. J. Park, H. M. Jin, J. Y. Kim, S.-W. Son, M.-H. Kim, C. M. Koo, J. Shin, J. U. Kim and S. O. Kim, Nano Lett., 2015, 15, 1190 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602 CrossRef CAS.
- J. Qin, P. Grzywacz and D. C. Morse, J. Chem. Phys., 2011, 135, 89402 CrossRef PubMed.
- T. M. Beardsley and M. W. Matsen, Phys. Rev. Lett., 2016, 217801 CrossRef PubMed.
- J. Glaser, P. Medapuram, T. M. Beardsley, M. W. Matsen and D. C. Morse, Phys. Rev. Lett., 2014, 63802, 1 Search PubMed.
- G. H. Fredrickson and E. Helfand, J. Chem. Phys., 1987, 87, 697 CrossRef CAS.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Wilson, Macromolecules, 2014, 47, 2 CrossRef CAS.
- L. Wan, R. Ruiz, H. Gao, K. C. Patel, T. R. Albrecht, J. Yin, J. Kim, Y. Cao and G. Lin, ACS Nano, 2015, 9, 7506 CrossRef CAS.
- C. Sinturel, F. S. Bates and M. A. Hillmyer, ACS Macro Lett., 2015, 4, 1044 CrossRef CAS.
- H. M. Jin, D. Y. Park, S.-J. Jeong, G. Y. Lee, J. Y. Kim, J. H. Mun, S. K. Cha, J. Lim, J. S. Kim, K. H. Kim, K. J. Lee and S. O. Kim, Adv. Mater., 2017, 29, 1700595 CrossRef.
- H. M. Jin, S. H. Lee, J. Y. Kim, S.-W. Son, B. H. Kim, H. K. Lee, J. H. Mun, S. K. Cha, J. S. Kim, P. F. Nealey, K. J. Lee and S. O. Kim, ACS Nano, 2016, 10, 3435 CrossRef CAS.
- J. N. L. Albert and T. H. Epps, Mater. Today, 2010, 13, 24 CrossRef CAS.
- R. A. Segalman, Mater. Sci. Eng., R, 2005, 48, 191 CrossRef.
- C. Jin, B. C. Olsen, E. J. Luber and J. M. Buriak, Chem. Mater., 2017, 29, 176 CrossRef CAS.
- Z. Qiang, S. A. Akolawala and M. Wang, ACS Macro Lett., 2018, 7, 566 CrossRef CAS.
- T. P. Russell, R. P. Hjelm and P. A. Seeger, Macromolecules, 1990, 23, 890 CrossRef CAS.
- D. P. Sweat, M. Kim, S. R. Larson, J. W. Choi, Y. Choo, C. O. Osuji and P. Gopalan, Macromolecules, 2014, 47, 6687 CrossRef CAS.
- L. Leibler, H. Orland and J. C. Wheeler, J. Chem. Phys., 1983, 79, 3550 CrossRef CAS.
- P. Linse, J. Chem. Phys., 1993, 97, 13896 CrossRef CAS.
- E. Kim, H. Ahn, D. Y. Ryu, J. Kim and J. Cho, Macromolecules, 2009, 42, 8385 CrossRef CAS.
- C. Shin, H. Ahn, E. Kim, D. Y. Ryu, J. Huh, K. Kim and T. P. Russell, Macromolecules, 2008, 41, 9140 CrossRef CAS.
- I. Park, S. Park, H. Park, T. Chang, H. Yang and C. Y. Ryu, Macromolecules, 2006, 39, 315 CrossRef CAS.
- S. Park, K. Koo, K. Kim, H. Ahn, B. Lee, C. Park and D. Y. Ryu, Polymer, 2015, 60, 32 CrossRef CAS.
- P. Mansky, O. K. C. Tsui, T. P. Russell and Y. Gallot, Macromolecules, 1992, 32, 4832 CrossRef.
- A. Arceo and P. F. Green, J. Phys. Chem. B, 2005, 109, 6958 CrossRef CAS.
- A. Menelle, T. P. Russell, S. H. Anastasiadis, S. K. Satija and C. F. Majkrzak, Phys. Rev. Lett., 1992, 68, 67 CrossRef CAS.
- Y. Kim, D. Yong, W. Lee, S. Jo, H. Ahn, J. Kim and D. Y. Ryu, Macromolecules, 2018, 51, 8550 CrossRef CAS.
- Y. Zhao, E. Sivaniah and T. Hashimoto, Macromolecules, 2008, 41, 9948 CrossRef CAS.
- N. Sakamoto and T. Hashimoto, Macromolecules, 1995, 28, 6825 CrossRef CAS.
- M. Gopinadhan, P. W. Majewski, Y. Choo and C. O. Osuji, Phys. Rev. Lett., 2013, 110, 1 CrossRef PubMed.
- C. D. Han and J. Kim, J. Polym. Sci., Part B: Polym. Phys., 1987, 25, 1741 CrossRef CAS.
- C. D. Han and J. Kim, Macromolecules, 1989, 22, 283 Search PubMed.
- B. A. Garetz, M. C. Newstein, H. J. Dai, S. V. Jonnalagadda and N. P. Balsara, Macromolecules, 1993, 26, 3151 CrossRef CAS.
- V. P. Voronov, V. M. Buleiko, V. E. Podneks, I. W. Hamley, J. P. A. Fairclough, A. J. Ryan, S.-M. Mai, B.-X. Liao and C. Booth, Macromolecules, 1997, 30, 6674 CrossRef CAS.
- P. Muller-Buschbaum, Eur. Polym. J., 2016, 81, 470 CrossRef.
- A. Hexemer and P. Mu, IUCrJ, 2015, 106 CrossRef CAS PubMed.
- D. Posselt, J. Zhang, D. Smilgies, A. V. Berezkin, I. I. Potemkin and C. M. Papadakis, Prog. Polym. Sci., 2017, 66, 80 CrossRef CAS.
- B. Lee, I. Park, J. Yoon, S. Park, J. Kim, K. Kim, T. Chang and M. Ree, Macromolecules, 2005, 38, 4311 CrossRef CAS.
- S. A. Vaselabadi, D. Shakarisaz, P. Ruchhoeft, J. Strzalka and G. E. Stein, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1074 CrossRef CAS.
- C. J. Ellison and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2745 CrossRef CAS.
- C. J. Ellison and J. M. Torkelson, Nat. Mater., 2003, 2, 695 CrossRef CAS PubMed.
- D. Christine, R. A. Register and R. D. Priestley, Phys. Rev. Lett., 2018, 121, 247801 CrossRef PubMed.
- M. K. Mundra, C. J. Ellison, P. Rittigstein and J. M. Torkelson, Eur. Phys. J.: Spec. Top., 2007, 141, 143 Search PubMed.
- S. Kim, S. A. Hewlett, C. B. Roth and J. M. Torkelson, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 30, 83 CrossRef CAS PubMed.
- C. M. Evans, K. J. Henderson, J. D. Saathoff, K. R. Shull and J. M. Torkelson, Macromolecules, 2013, 46, 4131 CrossRef CAS.
- J. Aguiar, P. Carpena, J. A. Monlina-Bolivar and C. C. Ruiz, J. Colloid Interface Sci., 2003, 258, 116 CrossRef CAS.
- E. D. Goddard, N. J. Turro, P. L. Kuo and K. P. Anathapadmanabhan, Langmuir, 1985, 1, 352 CrossRef CAS.
- C. B. Roth and J. M. Torkelson, Macromolecules, 2007, 40, 3328 CrossRef CAS.
- A. Kriisa, S. S. Park and C. B. Roth, J. Polym. Sci., Part B: Polym. Phys., 2011, 50, 250 CrossRef.
- R. D. Priestley, L. J. Broadbelt and J. M. Torkelson, Macromolecules, 2005, 38, 654 CrossRef CAS.
- D. S. Karpovich and G. J. Blanchard, J. Phys. Chem., 1995, 99, 3951 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039 CrossRef CAS.
- S. Askar and J. M. Torkelson, Polymer, 2016, 99, 417 CrossRef CAS.
- S. Askar, C. M. Evans and J. M. Torkelson, Polymer, 2015, 76, 113 CrossRef CAS.
- C. Tedeschi, H. Mohwald and S. Kristein, J. Am. Chem. Soc., 2001, 67, 954 CrossRef PubMed.
- E. A. Prado, S. B. Yamaki, T. D. Z. Atvars, O. E. Zimerman and R. G. Weiss, J. Phys. Chem. B, 2000, 104, 5905 CrossRef CAS.
- R. Salovey, J. Polym. Sci., 1961, 153, S7 CrossRef.
- T. M. Birshtein, A. A. Merku'yeva and A. N. Goryunov, Polym. Sci. U.S.S.R., 1983, 25, 143 CrossRef.
- M. Shima, M. Sato, M. Atsumi and K. Hatada, Polym. J., 1994, 26, 579 CrossRef CAS.
- S. Kim and J. M. Torkelson, Macromolecules, 2011, 44, 4546 CrossRef CAS.
- P. Dixit, X. Chen, J. Miao, S. Divakaran and R. Preisser, Appl. Surf. Sci., 2007, 253, 8639 CrossRef.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. J. Frechet, A. P. Alivisatos and T. Xu, Nat. Mater., 2009, 8, 975 Search PubMed.
- D. Song, L. Yin, Y. Gai, N. S. Colella, C. Li, X. Liu, S. Gido and J. J. Watkins, J. Am. Chem. Soc., 2015, 137, 3771 CrossRef CAS PubMed.
- Y. Lin, V. K. Daga, E. R. Anderson, S. P. Gido and J. J. Watkins, J. Am. Chem. Soc., 2011, 133, 6513 CrossRef CAS PubMed.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868 RSC.
- A. J. Wilson, Soft Matter, 2007, 3, 409 RSC.
- J. Ruokolainen, M. Torkkeli, R. Serimaa, E. Komanschek, G. Brinke and O. Ikkala, Macromolecules, 1997, 30, 2002 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00091g |
| This journal is © The Royal Society of Chemistry 2020 |