
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of 6-amino-1H-pyrazolo[3,4-d]pyrimidines with in vivo efficacy against visceral leishmaniasis†
Michael G.
Thomas
 a,
Manu
De Rycker
a,
Myriam
Ajakane
b,
Sabrinia D.
Crouch
c,
Lorna
Campbell
a,
Alain
Daugan
b,
Gloria
Fra
d,
César
Guerrero
d,
Claire J.
Mackenzie
a,
Lorna
MacLean
a,
Sujatha
Manthri
a,
Franck
Martin
b,
Suzanne
Norval
a,
Maria
Osuna-Cabello
a,
Jennifer
Riley
a,
Yoko
Shishikura
a,
Juan
Miguel-Siles
c,
Frederick R. C.
Simeons
a,
Laste
Stojanovski
a,
John
Thomas
a,
Stephen
Thompson
a,
Raul F.
Velasco
d,
Jose M.
Fiandor
c,
Paul G.
Wyatt
a,
Kevin D.
Read
a,
Ian H.
Gilbert
*a and
Timothy J.
Miles
*c
a,
Manu
De Rycker
a,
Myriam
Ajakane
b,
Sabrinia D.
Crouch
c,
Lorna
Campbell
a,
Alain
Daugan
b,
Gloria
Fra
d,
César
Guerrero
d,
Claire J.
Mackenzie
a,
Lorna
MacLean
a,
Sujatha
Manthri
a,
Franck
Martin
b,
Suzanne
Norval
a,
Maria
Osuna-Cabello
a,
Jennifer
Riley
a,
Yoko
Shishikura
a,
Juan
Miguel-Siles
c,
Frederick R. C.
Simeons
a,
Laste
Stojanovski
a,
John
Thomas
a,
Stephen
Thompson
a,
Raul F.
Velasco
d,
Jose M.
Fiandor
c,
Paul G.
Wyatt
a,
Kevin D.
Read
a,
Ian H.
Gilbert
*a and
Timothy J.
Miles
*c
aDrug Discovery Unit, Wellcome Centre for Anti-Infectives Research, Division of Biological Chemistry and Drug Discovery, School of Life Sciences, University of Dundee, Dundee DD1 5EH, UK. E-mail: I.H.Gilbert@dundee.ac.uk
bCentre de Recherche, GlaxoSmithKline, Les Ulis, 25,27 Avenue du Quebec, 91140 Villebon sur Yvette, France
cGlobal Health R&D, GlaxoSmithKline, Calle Severo Ochoa, 2, 28760 Tres Cantos, Madrid, Spain. E-mail: tim.j.miles@gsk.com
dGalChimia S.A., Cebreiro s/n, 15823, O Pino, A Coruña, Spain
First published on 6th August 2020
Abstract
Visceral leishmaniasis (VL) affects millions of people across the world, largely in developing nations. It is fatal if left untreated and the current treatments are inadequate. As such, there is an urgent need for new, improved medicines. In this paper, we describe the identification of a 6-amino-N-(piperidin-4-yl)-1H-pyrazolo[3,4-d]pyrimidine scaffold and its optimization to give compounds which showed efficacy when orally dosed in a mouse model of VL.
Introduction
Visceral leishmaniasis (VL) is caused by infection with the protozoan parasites L. donovani and L. infantum and is typically fatal unless treated, infecting around 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–90000 people annually and resulting in a death toll of between 20000–30000.1,2 Current therapies suffer from numerous issues such as high cost, problematic modes of dosing, and toxicity.3 In addition, the global development pipeline for VL is sparse with four additional new chemical entities (NCEs) just entering the early pre-clinical phase.4–7 There is therefore an urgent need for new therapeutic classes to help tackle this neglected disease.
000–90000 people annually and resulting in a death toll of between 20000–30000.1,2 Current therapies suffer from numerous issues such as high cost, problematic modes of dosing, and toxicity.3 In addition, the global development pipeline for VL is sparse with four additional new chemical entities (NCEs) just entering the early pre-clinical phase.4–7 There is therefore an urgent need for new therapeutic classes to help tackle this neglected disease.
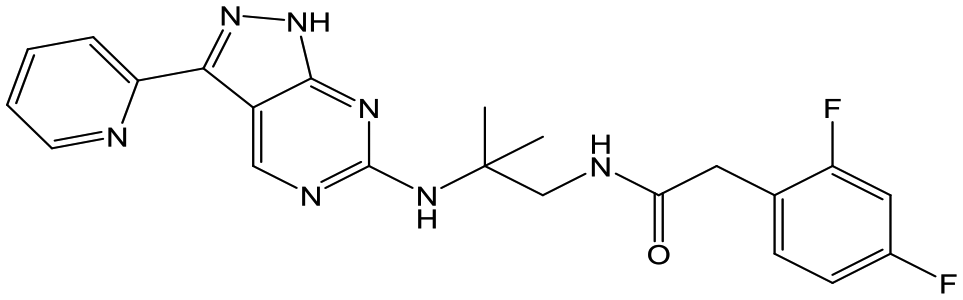
The primary objective of this program was therefore to identify safe, effective, oral, short-course (ideally ≤10 days) drug candidates for VL, in line with the DNDi (Drugs for Neglected Disease initiative) target product profile.8 Due to a lack of validated druggable targets for VL, we focused on chemical series that showed antiparasitic activity in an assay involving parasites growing in mammalian cells, aiming to progress these into in vivo efficacy studies, where they could be bench-marked against miltefosine; this currently being the only available oral therapy for VL. This phenotypic approach was used successfully for the identification of two preclinical candidates.5,7 One of these, DDD853651/GSK3186899 was developed from pyrazolopyrimidine 1, with a key step being elaboration of the cyclohexylamine to a sulfonamide substituted 1,4-trans-cyclohexyldiamine (Fig. 1). In this work, we describe further modifications to the cyclohexylamine, leading to a series of substituted 4-aminopiperidines which displayed efficacy in a mouse model of VL.
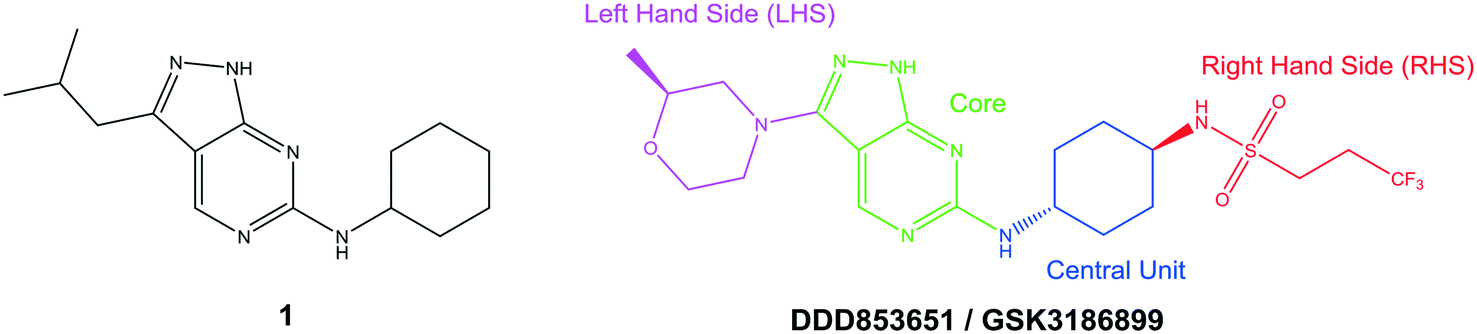 | ||
| Fig. 1 Initial active compound 1 and preclinical candidate DDD853651/GSK3186899. | ||
Results and discussion
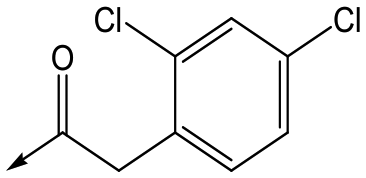
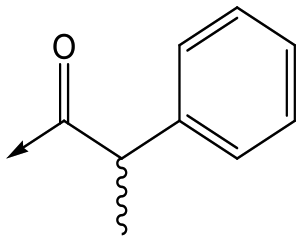
For the identification of DDD853651/GSK3186899, which contained a trans-1,4-cyclohexyldiamine, a key strategy was to optimise the balance between potency in the intracellular Leishmania assay (L. donovani in THP-1 cells, referred to as Ld InMac assay9) and solubility.10 In that case, we focused on 2 (Table 1), where the sulfonamide was required for potency but was poorly soluble. We noted that amide 3, whilst inactive, did show improved aqueous solubility (51 vs. 1 μM), so we investigated alternative central units to identify start points which retained activity and showed improved solubility compared to 2. To this end, we examined a 4-aminopiperidine central unit with both sulfonamide and amide substituents (Table 1). In contrast to the trans-1,4-cyclohexyldiamine series, the equivalent sulfonamide was >10-fold less active (5cf.2), whilst amide 4 was >10-fold more active than its comparator (4cf.3) and showed similar aqueous solubility (59 μM cf. 51 μM). We therefore selected 4 as a suitable start-point for further chemistry. As previously discussed,10 in order to progress to in vivo studies we targeted compounds with pEC50 > 5.8, solubility > 100 μM and intrinsic clearance (Cli) < 5.0 ml min−1 g−1 (mouse liver microsomes), so we explored the SAR around 4, initially focusing on the amide substituent. We noted that either lengthening the linker to the aromatic ring, or removing it, led to a loss of activity (6 and 7 respectively). Substitution of the phenylacetamide led to 2,4-difluorinated analogue 8 with an increase in potency compared to 4, whilst other changes, such as the dichloro analogue 9 and α-methyl analogue 10 failed to improve potency, although 10 did show improved aqueous solubility, possibly due to the introduction of a sp3 centre.|
|
|||||
|---|---|---|---|---|---|
| R | Compound | Ld InMaca pEC50 | THP-1a pEC50 | Aqueous solubilityb μM | Cli mousec ml min−1 g−1 |
| a Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes.9 Data are the mean values for n ≥ 3 replicates and standard deviations are ≤0.3. b Aq. solubility is kinetic aqueous solubility (CAD).11 c Cli is mouse liver microsomal intrinsic clearance.10 d ND means not determined. | |||||
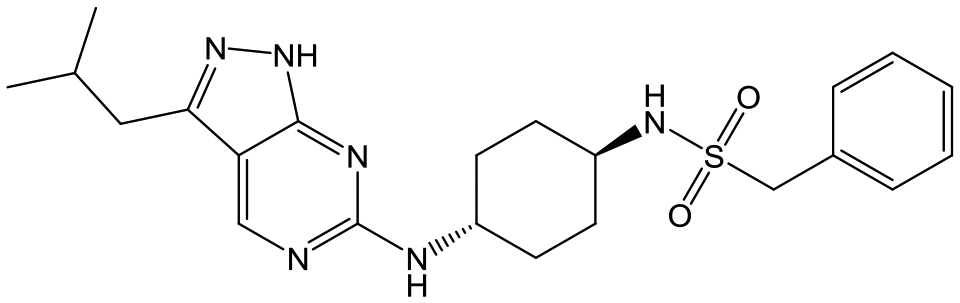
|
2 | 6.2 | <4.3 | <1 | 27 |
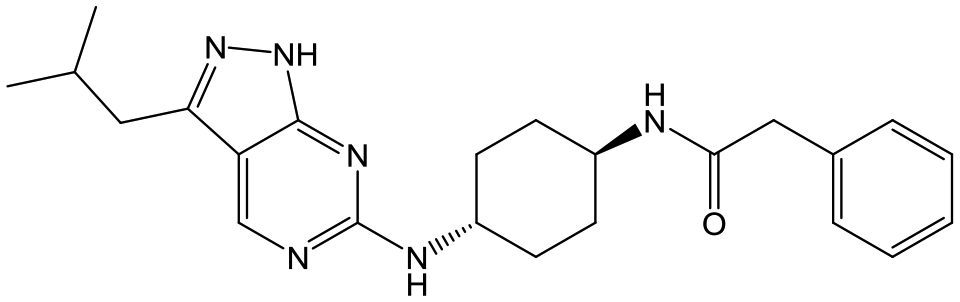
|
3 | <4.3 | <4.3 | 51 | NDd |
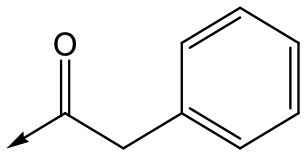
|
4 | 5.1 | <4.3 | 59 | 18 |
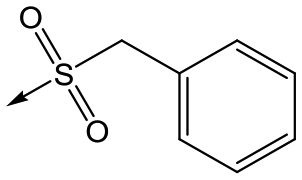
|
5 | 4.9 | <4.3 | 10 | >50 |
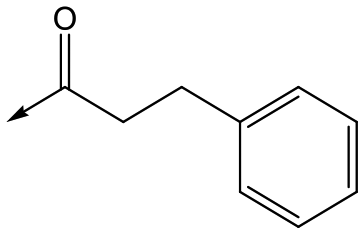
|
6 | 4.7 | <4.3 | NDd | NDd |
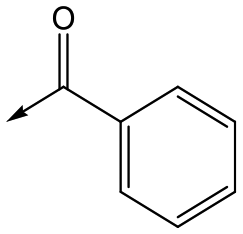
|
7 | <4.3 | <4.3 | 291 | 7 |
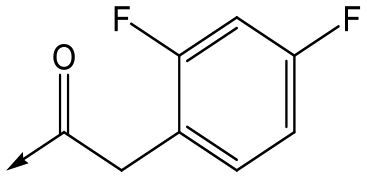
|
8 | 5.8 | <4.3 | 26 | 12 |
|
9 | 5.0 | <4.3 | 26 | 30 |
|
10 (rac) | 5.3 | <4.3 | 140 | NDd |
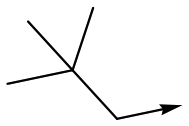
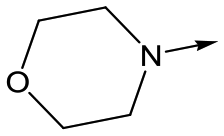
With 2,4-difluorophenylacetamide 8 as the most potent RHS (right hand side, see Fig. 1 in the introduction) identified, we explored the SAR (structure activity relationship) around the LHS (left hand side, see Fig. 1 in the Introduction) to improve solubility and metabolic stability, initially focusing on alkyl substituents (Table 2). Neopentyl 11 was inactive whilst the smaller cyclopropyl group of 12 maintained potency and displayed improved metabolic stability. Changing the cyclopropyl group for tetrahydropyran (THP) led to 13, with a pEC50 value of 6.0, a small improvement in solubility, and excellent metabolic stability. To further improve the physicochemical characteristics of the series, we examined the methylene-linked morpholine 14 which also showed excellent metabolic stability, although it was only weakly active. Removing the methylene linker gave the directly attached morpholine 15, which retained in vitro potency and metabolic stability, whilst switching to O-linked compound 16 gave a drop in activity.
|
|
|||||
|---|---|---|---|---|---|
| R | Compound | Ld InMac pEC50a | THP-1a pEC50 | Aqueous solubilityb μM | Cli mouse,c ml min−1 g−1 |
| a Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes.9 Data are the mean values for n ≥ 3 replicates and standard deviations are ≤0.4. b Aq. solubility is kinetic aqueous solubility (CAD).11 c Cli is mouse liver microsomal intrinsic clearance.10 d ND means not determined. | |||||
|
11 | <4.3 | <4.3 | 27 | 22.0 |
|
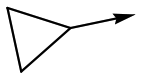
12 | 5.9 | <4.3 | 39 | 5.0 |
|
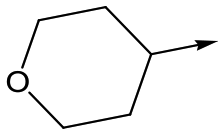
13 | 6.0 | <4.3 | 78 | 0.5 |
|
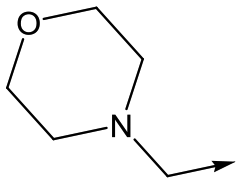
14 | 4.6 | <4.3 | NDd | <0.5 |
|
15 | 6.1 | <4.3 | 14 | 2 |
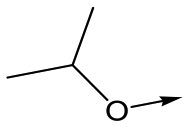
|
16 | 5.2 | <4.3 | 33 | NDd |
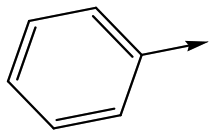
|
17 | 6.5 | <4.3 | <1 | 7.2 |

|
18 | 6.1 | <4.3 | 23 | 6.7 |
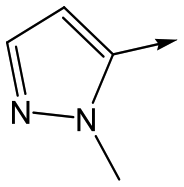
|
19 | 5.5 | 4.7 | 30 | 3.3 |
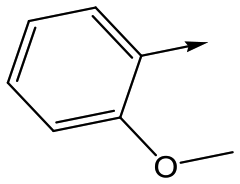
|
20 | 7.1 | 4.4 | <1 | 5.0 |
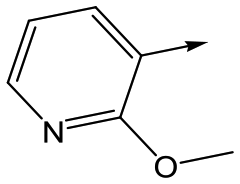
|
21 | 7.1 | <4.3 | 25 | 15 |
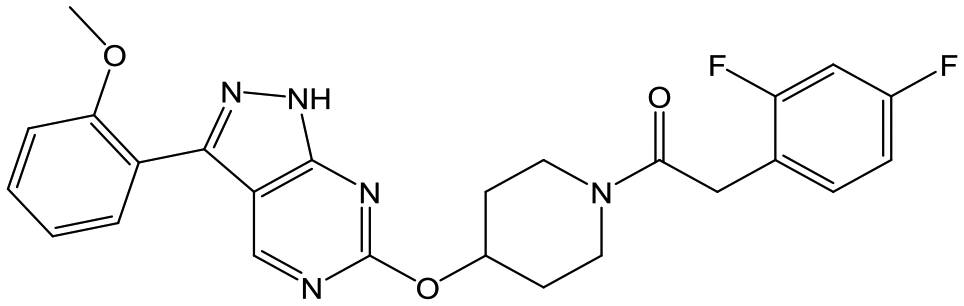
To further explore the SAR around 13, with a cyclic substituent on the LHS, a series of aromatic groups were explored. Phenyl substituted 17 showed an increase in potency compared to 9, although it was hampered by poor solubility; this was improved slightly by moving to the more polar pyridyl analogue 18, and moving to 5-membered heteroaromatics such as 19 also improved metabolic stability, albeit with a 10-fold loss in potency. A variety of other 5 and 6-membered heteroaromatics were explored, but none led to significant improvements (data not shown). Substitution on the aromatic ring of 17 was also investigated, leading to the 2-methoxyphenyl analogue 20, with a pEC50 value of 7.1 in the Ld InMac assay. This was one of the most potent analogues in the series, comparing very favourably with the standard treatments for VL, amphotericin B and miltefosine (pEC50's of 6.7 and 6.1 respectively). In an attempt to further improve solubility, methoxypyridyl analogue 21 was synthesized; whilst potency of 20 was maintained and solubility was improved, poorer microsomal stability was observed.
Having demonstrated that variations to the RHS (Table 1) and LHS (Table 2) could deliver compounds with a good balance of potency, solubility and metabolic stability, we switched our attention to the core pyrazolopyrimidine and the central unit (Table 3). Although we later elucidated the target of the series as being Leishmania-CRK12 (cyclin related kinase 12),5 at this point we were reliant on phenotypic drug discovery strategies to drive compound design. We were aware that intermolecular hydrogen bonding between the donor–acceptor pairs within the core was likely to be detrimental to solubility (Fig. 2), as was the planarity of the compounds.10 We therefore synthesized a set of analogues to probe the SAR around the various hydrogen bond donor–acceptor pairs in the core, and also examined a further series of central units to identify any that could maintain potency whilst adding more flexibility and three-dimensional character.
| Compound | Comparator | Ld InMac pEC50a | THP-1 pEC50a | |
|---|---|---|---|---|
| a Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes.9 Data are the mean values for n ≥ 3 replicates and standard deviations are ≤0.3. | ||||
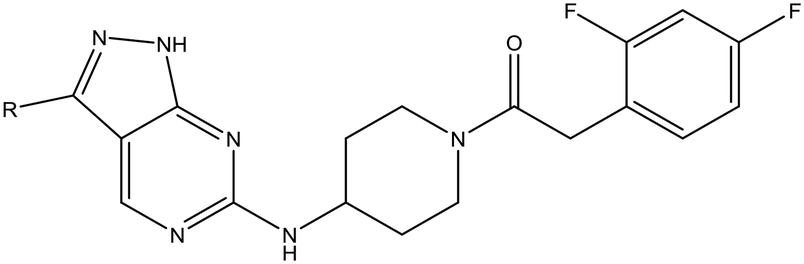
|
22 | 9 | <4.3 | <4.3 |
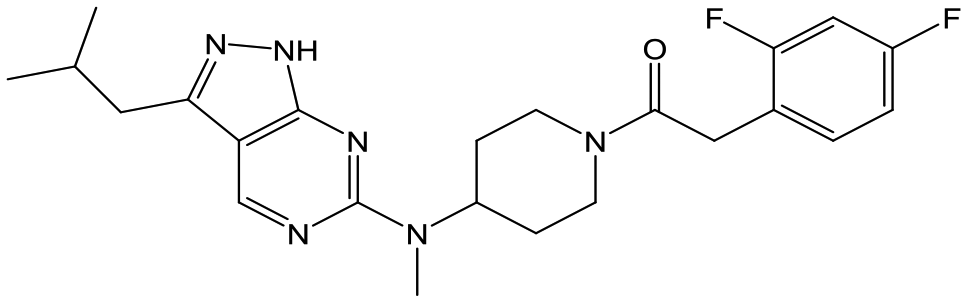
|
23 | 20 | 4.7 | <4.3 |
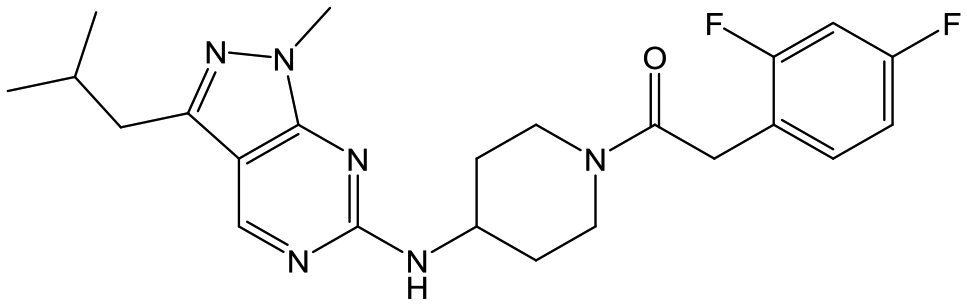
|
24 | 9 | <4.3 | <4.3 |
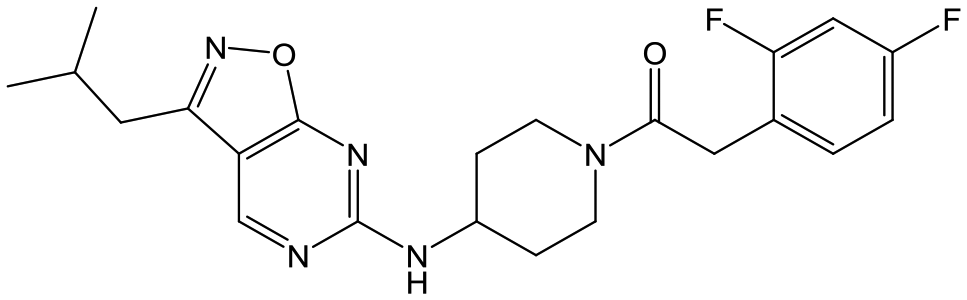
|
25 | 9 | <4.3 | <4.3 |
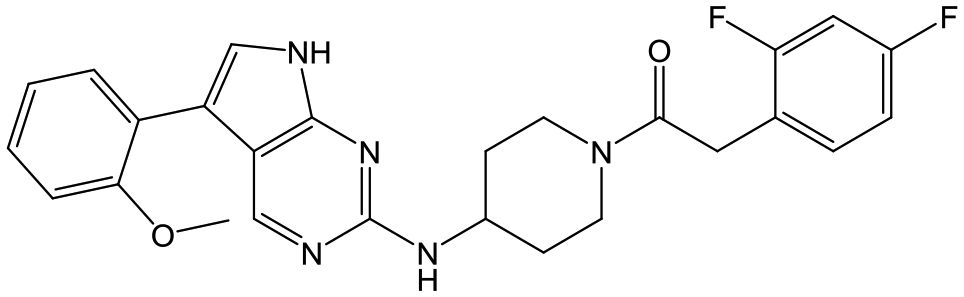
|
26 | 20 | <4.3 | <4.3 |
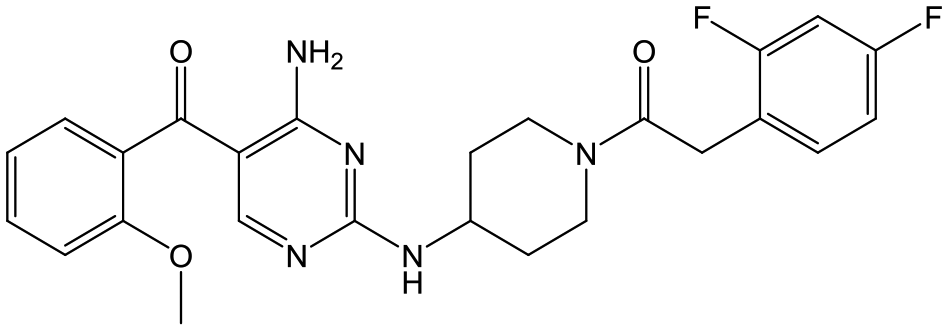
|
27 | 20 | 5.2 | 5.0 |
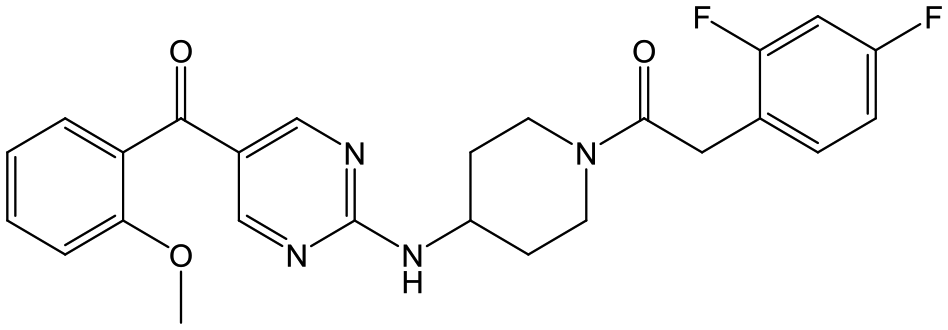
|
28 | 20 | 5.2 | <4.3 |
|
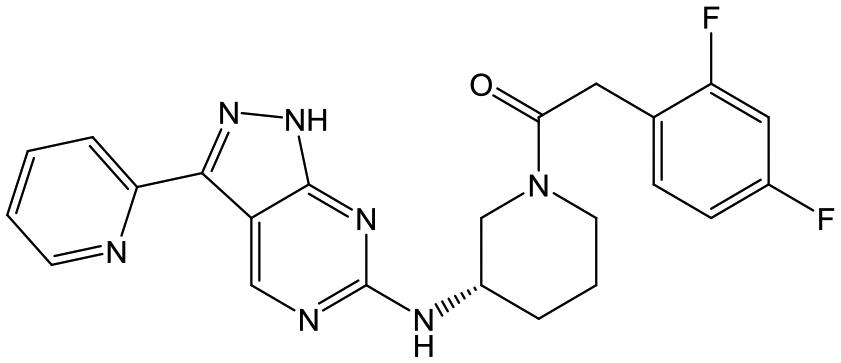
29 (S) | 18 | 4.9 | <4.3 |
|
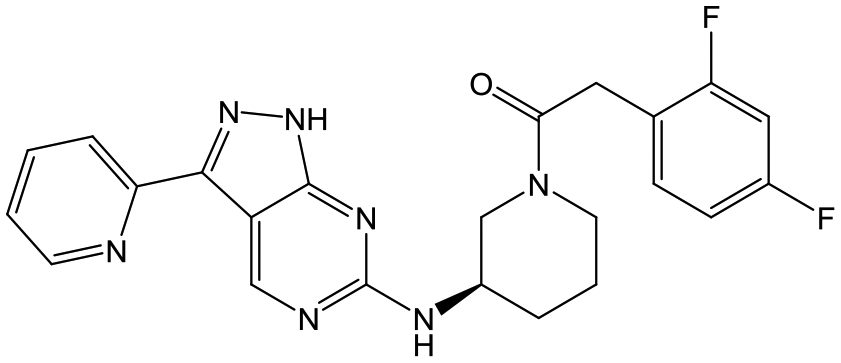
30 (R) | 18 | <4.3 | <4.3 |
|
31 (cis) | 20 | 4.5 | 4.6 |
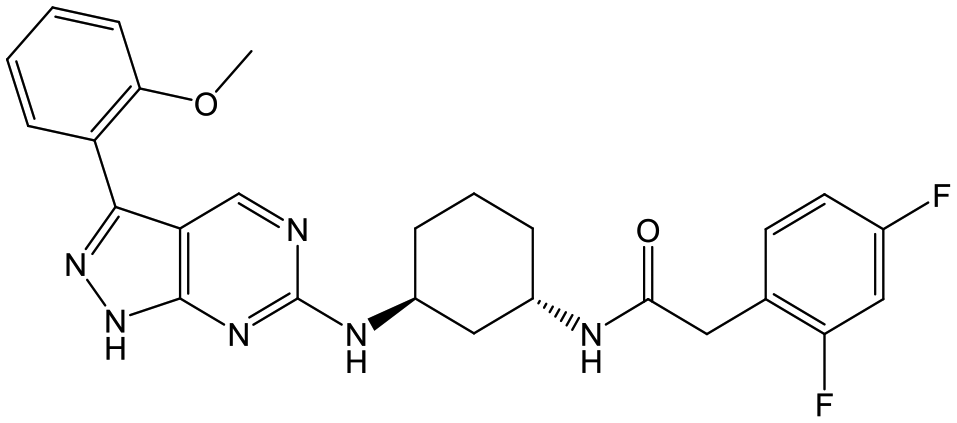
|
32 (trans) | 20 | 4.8 | 4.5 |
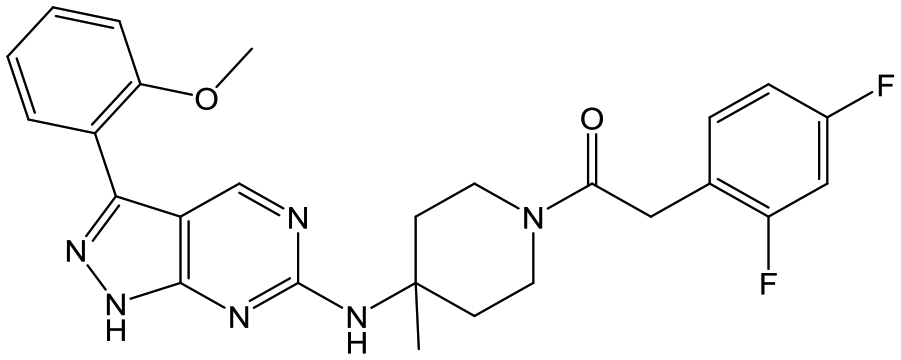
|
33 | 20 | 5.4 | 4.4 |
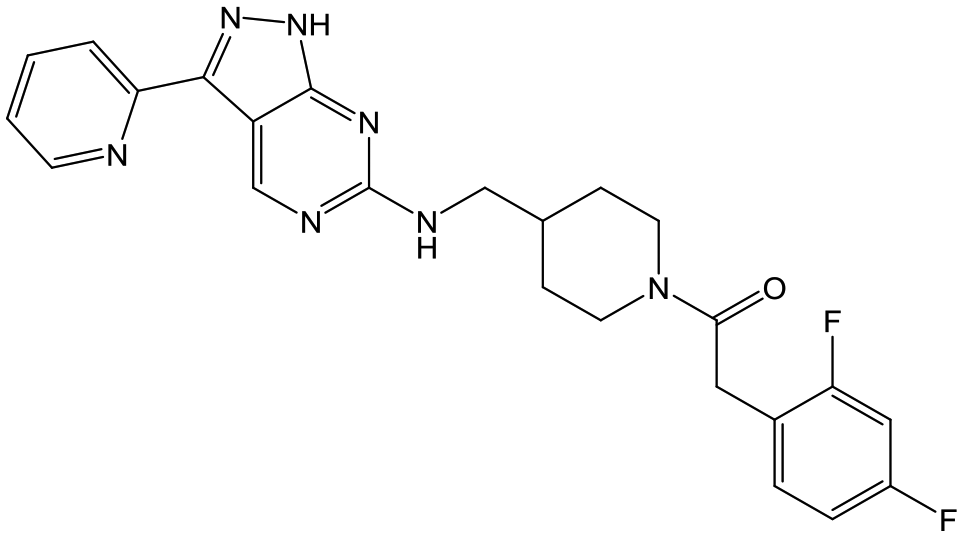
|
34 | 18 | <4.3 | <4.3 |
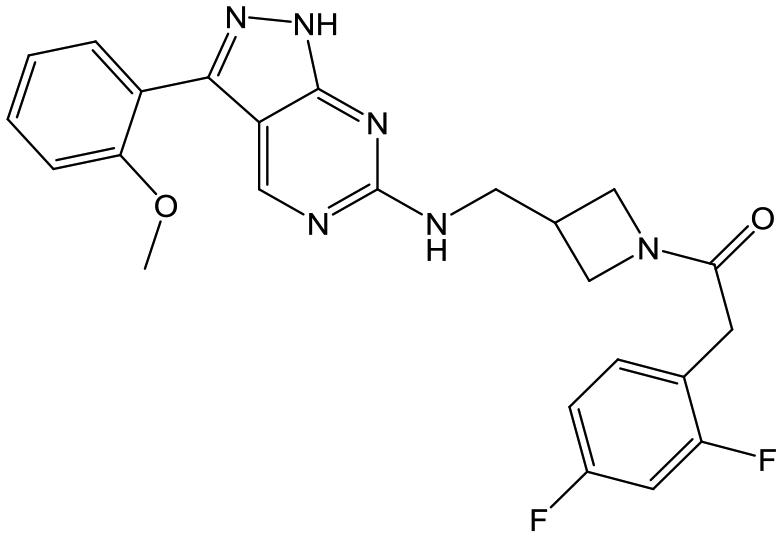
|
35 | 20 | <4.3 | <4.3 |
|
36 | 18 | <4.3 | <4.3 |
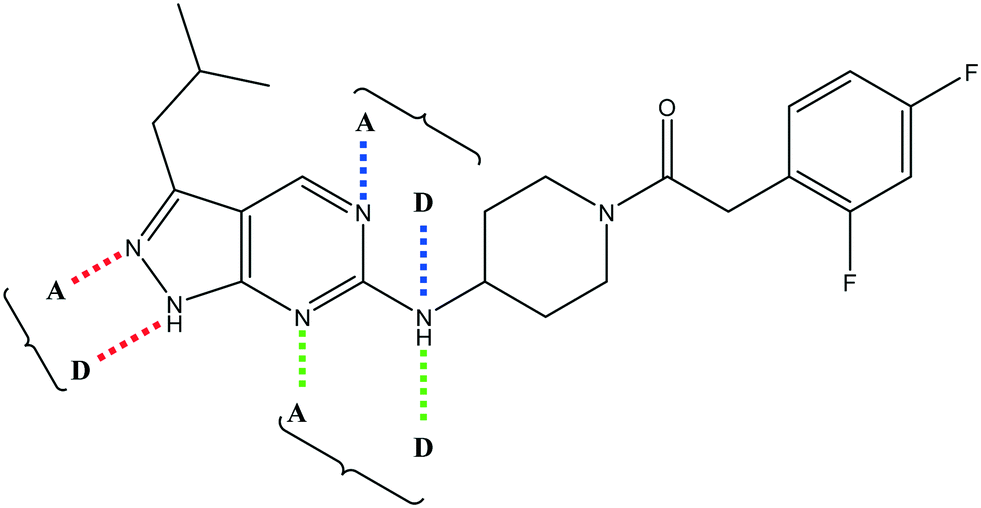 | ||
| Fig. 2 Hydrogen bond donor–acceptor pairs in 8. | ||
Alkylation of the aminopyrimidine N–H or replacement by oxygen to give 22 and 23 respectively, led to a >10-fold reduction in potency compared to its' matched molecular pair (compounds 9, 18 and 20 were all used as comparators for this exercise). Additionally, alkylation of the pyrazole N–H (24) as well as replacement of the pyrazole with isoxazole (25) or pyrrole (26) all led to a >10-fold loss of potency. The pyrazole ring was opened to give pyridimines 27 and 28, leading to decreased potency and also toxicity against the host THP-1 cells in the case of 27.
Switching attention to the central unit, 3-aminopiperidine enantiomers 29 and 30 and the 1,3-diaminocyclohexyl analogues 31 and 32 were either weakly active or inactive, as was methylated aminopiperidine 33. More flexible linkers such as aminomethylpiperidine 34, aminomethylcyclobutyl 35 and ethylenediamine 36 were also inactive. Further diverse linkers, with numerous substituents (amides and sulfonamides) were tested, but all proved to be inactive beyond the already identified piperidines and 1,4-trans-cyclohexylamines (data not shown).
Refocusing on 20, where the introduction of the 2-methoxyphenyl LHS gave a significant improvement in potency, further RHS piperidine substituents were explored (Table 4). Introduction of the trifluoropropylsulfonamide from the preclinical candidate (DDD853651/GSK3186899) did give a potent compound (37) although with very poor solubility and intrinsic clearance; this was similar for carbamate linked compounds 38 and 39. Conversely, urea linked compounds such as 40 maintained reasonable solubility and metabolic stability but reduced potency. Also, directly attaching a heterocycle led to 41 with very good potency but low solubility. Finally we examined a further set of amides, identifying cyclopropylmethyl analogue 42 with good potency and metabolic stability, and 3,4-difluorophenyl analogue 43 with improved potency and solubility compared to 20.
|
|
|||||
|---|---|---|---|---|---|
| R | Compound | Ld InMac pEC50a | THP-1 pEC50a | Aqueous solubilityb μM | Cli mouse,c ml min−1 g−1 |
| a Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes.9 Data are the mean values for n ≥ 3 replicates and standard deviations are ≤0.4. b Aq. solubility is kinetic aqueous solubility (CAD).11 c Cli is mouse liver microsomal intrinsic clearance.10 d ND means not determined. | |||||
|
20 | 7.1 | 4.4 | <1 | 5.0 |
|
37 | 6.7 | <4.3 | <1 | 14 |
|
38 | 6.3 | 4.4 | 24 | 14 |
|
39 | 6.4 | <4.3 | NDd | 9 |
|
40 | 5.7 | <4.3 | 79 | 2 |
|
41 | 6.8 | <4.3 | 412 | 3.4 |
|
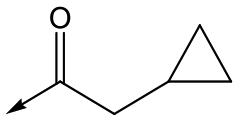
42 | 6.5 | 4.4 | 22 | 1.6 |
|
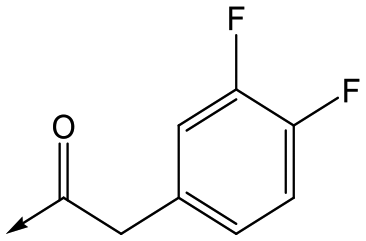
43 | 7.6 | 4.8 | 53 | 6.3 |
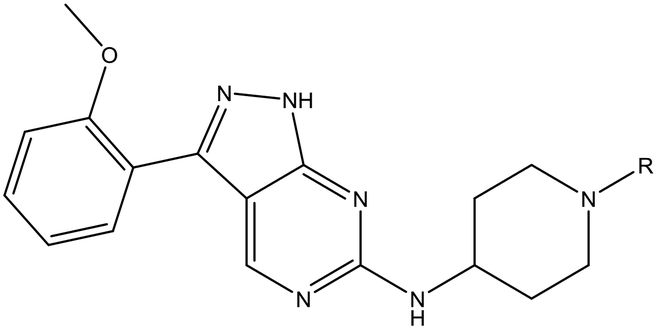
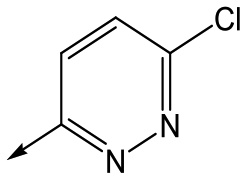
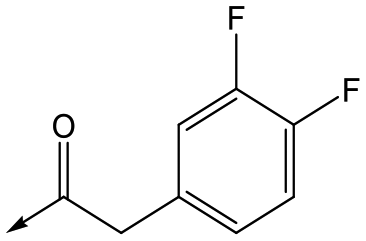
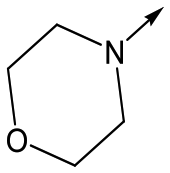
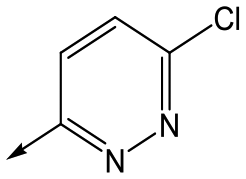
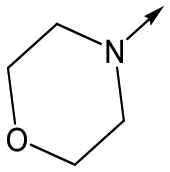
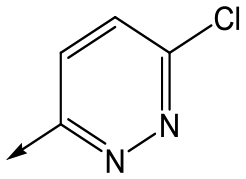
Utilising the SAR understanding we had developed, a set of analogues were synthesized that combined the most interesting LHS and RHS (Table 5). Maintaining the cyclopropylmethyl amide of 42 led to the THP (44), 2-pyridyl (45) and morpholine (46) analogues that were only moderately potent but were much more soluble, with 44 also having very good metabolic stability. Maintaining the 3,4-difluorophenyl RHS led to THP 47 and morpholine 48, both of which had reasonable potency, high solubility and good metabolic stability. Finally, the chloropyridazine RHS led to THP analogue 49 which was reasonably potent and morpholine 50 which again showed good potency, solubility and metabolic stability.
|
|
||||||
|---|---|---|---|---|---|---|
| R1 | R2 | Compound | Ld InMac pEC50a | THP-1 pEC50a | Aqueous solubilityb μM | Cli mousec ml min−1 g−1 |
| a Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes.9 Data are the mean values for n ≥ 3 replicates and standard deviations are ≤0.4. b Aq. solubility is kinetic aqueous solubility (CAD).11 c Cli is mouse liver microsomal intrinsic clearance.10 d ND means not determined. | ||||||
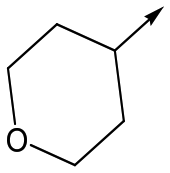
|
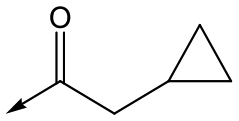
|
44 | 5.1 | <4.3 | ≥496 | 0.8 |
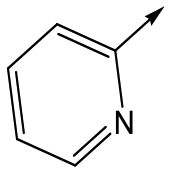
|
|
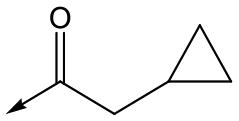
45 | 5.3 | <4.3 | ≥392 | NDd |
|
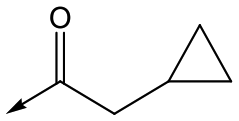
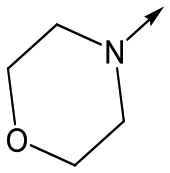
|
46 | 4.8 | <4.3 | NDd | NDd |
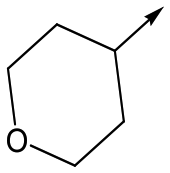
|
|
47 | 6.0 | <4.3 | ≥429 | 3.1 |
|
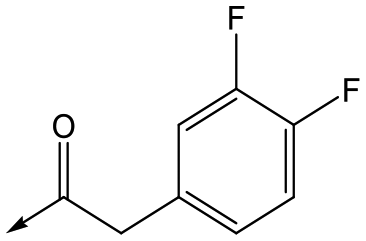
|
48 | 6.1 | <4.3 | ≥465 | 1.6 |
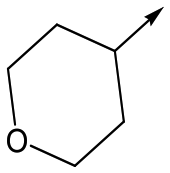
|
|
49 | 5.8 | <4.3 | 6312 | NDd |
|
|
50 | 6.2 | <4.3 | 12112 | 0.6 |
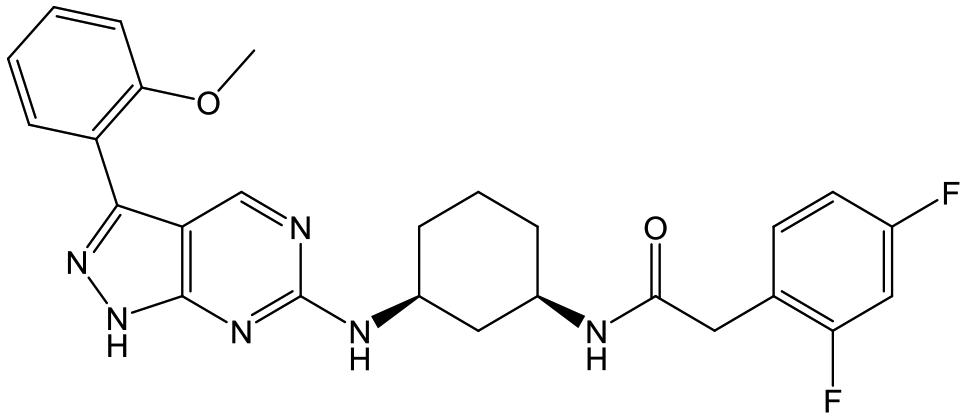
To select the most suitable compounds to progress to in vivo studies, the fasted state simulated intestinal fluid (FaSSIF) solubility of 20, 42, 48 and 50 was measured,13 as this had proved to be successful for triaging compounds in the 1,4-trans-cyclohexyldiamine series.1020, the most potent compound with Cli ≤ 5.0 ml min−1 g−1, and 42, which had moderate aqueous solubility, both had FaSSIF solubility of <1 μg ml−1, whereas 48 and 50 had FaSSIF solubilities of 97 and 88 μg ml−1 respectively. On this basis, 48 and 50 were progressed into orally dosed pharmacokinetic studies in female Balb-c mice (Table 6), with both having sufficient Cmax (the highest concentration of a drug in the blood) and AUC (area under the curve) to progress into the previously reported mouse efficacy model of VL.10
| Compound | Dose mg kg−1 | AUC ng min ml−1 | C max ng ml−1 | T max h |
|---|---|---|---|---|
| Vehicle was 0.5% weight/volume (w/v) hydroxypropylmethylcellulose with 0.4% volume/volume (v/v) Tween 80 and 0.5% v/v benzyl alcohol. Tmax is the time where the highest concentration of drug in the blood is achieved. | ||||
| 48 | 100 | 1253767 |
5765 | 1 |
| 50 | 100 | 2388964 |
5229 | 4 |
Both compounds were dosed orally for 5 days (Table 7), with 48 giving 60% reduction of parasite liver load at the lower dose, and 98% reduction at the higher dose, and 50 giving 17% reduction at the lower dose and 57% at the higher dose. For 48, this met our criteria for progression towards candidate selection and it was therefore profiled more fully.
| Compound | Dosing regimen | % Suppression of parasite load |
|---|---|---|
| Vehicle was 0.5% w/v hydroxypropylmethylcellulose with 0.4% v/v Tween 80 and 0.5% v/v benzyl alcohol. b.i.d. means twice (bis in die) a day. PO (Per Os) means oral administration. | ||
| 48 | 30 mg kg−1b.i.d. for 5 days PO | 60 |
| 48 | 100 mg kg−1b.i.d. for 5 days PO | 98 |
| 50 | 25 mg kg−1b.i.d. for 5 days PO | 17 |
| 50 | 50 mg kg−1b.i.d. for 5 days PO | 57 |
At this point, the target of the series was demonstrated to be the parasitic kinase Leishmania-CRK12, so to fully understand kinase selectivity 48 was further profiled against a panel of 140 human kinase receptors at 10 μM concentration.14 From this, only 4 kinases were inhibited at >40% (ERK8 [extracellular signal-regulated kinases], p38α and p38β MAPK [mitogen-activated protein kinases] and CDK2 [cyclin dependant kinase]). Full IC50 curves were generated for these, showing that the kinases most affected were ERK8 (IC50 = 0.11 μM) and p38β MAPK (IC50 = 0.55 μM). We didn't consider these results to preclude further development of 48.
A further in vivo study demonstrated that 48 had a mean liver:blood ratio of 4.8:1 which could prove beneficial, as the liver is one of the major sites of parasitic burden in VL infections.248 was also progressed to a rat dose-escalation study in order to determine whether exposures higher than the efficacious exposure could be achieved (Table 8); this would be critical in order to progress the compound to rat toxicology studies. In this case, increasing the dose from 100 to 300 mg kg−1 led to only a 1.2-fold increase in Cmax and a 1.7-fold increase in AUC(0-Tlast) suggesting that it would be challenging to determine a therapeutic index in future toxicological studies.
| Dose (mg kg−1) | C max (ng ml−1) | T max (h) | AUC (0-Tlast) (ng h ml−1) |
|---|---|---|---|
| Vehicle was 0.5% w/v hydroxypropylmethylcellulose. | |||
| 10 | 229 | 0.67 | 488 |
| 100 | 5520 | 6.00 | 50621 |
| 300 | 6760 | 6.67 | 84647 |
In order to synthesise the compounds described, a number of different synthetic routes were utilized, as outlined in Schemes 1–4. Scheme 1 shows the synthetic route that was used to access compounds 4–10, where the readily accessible 2-chloro-4-methoxy pyrimidine 5110 was treated with the Boc (tert-butyloxycarbonyl protecting group) protected aminopyrimidine 52 to give 53, which was deprotected under acidic conditions to give 54. This was subsequently coupled with a relevant acid then cyclized with hydrazine to yield analogues 4–7 and 9–10. Alternately, an elaborated aminopiperidine could be coupled with 51 to give 55a (R = benzylsulfonyl) or 55b (R = 2,4-difluorobenzoyl) which were cyclized to give compounds 5 and 8 respectively.
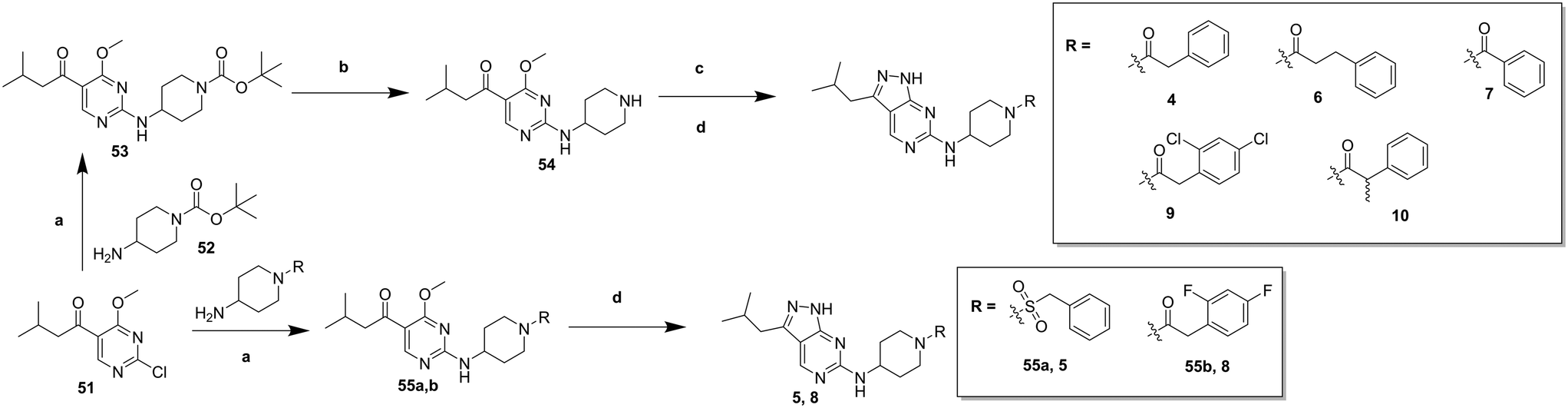 | ||
| Scheme 1 Synthesis of compounds 4–10. (a) N,N-Diisopropylethylamine (DIPEA), butan-1-ol; (b) trifluoroacetic acid (TFA), dichloromethane (DCM), 97% (over 2 steps); (c) R–COOH, N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate (TBTU), DIPEA, DCM; (d) hydrazine hydrate, butan-1-ol, 30–56% (over 2 steps). | ||
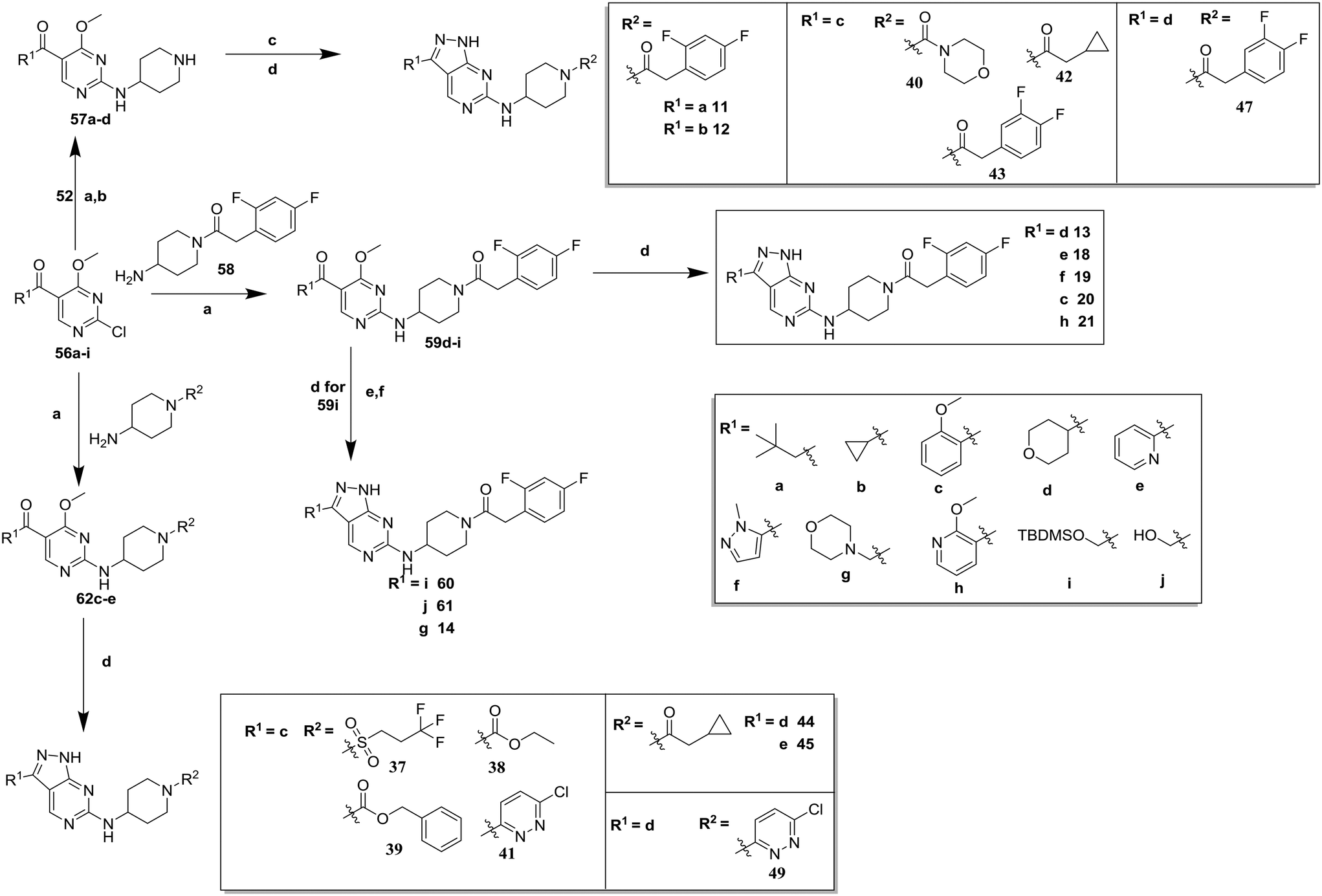 | ||
| Scheme 2 Synthesis of compounds 11–21, 37–45, 47 and 49. (a) DIPEA, butan-1-ol; (b) TFA, DCM, 47–83% (over 2 steps); (c) R2–COOH, TBTU, DIPEA, dimethylformamide (DMF); (d) hydrazine hydrate, butan-1-ol, 22–48% (over 2 steps); (e) tetra-n-butylammonium fluoride (TBAF), THF, 86%; (f) methanesulfonyl chloride (MsCl), DIPEA, acetonitrile (MeCN) then morpholine, 18%. | ||
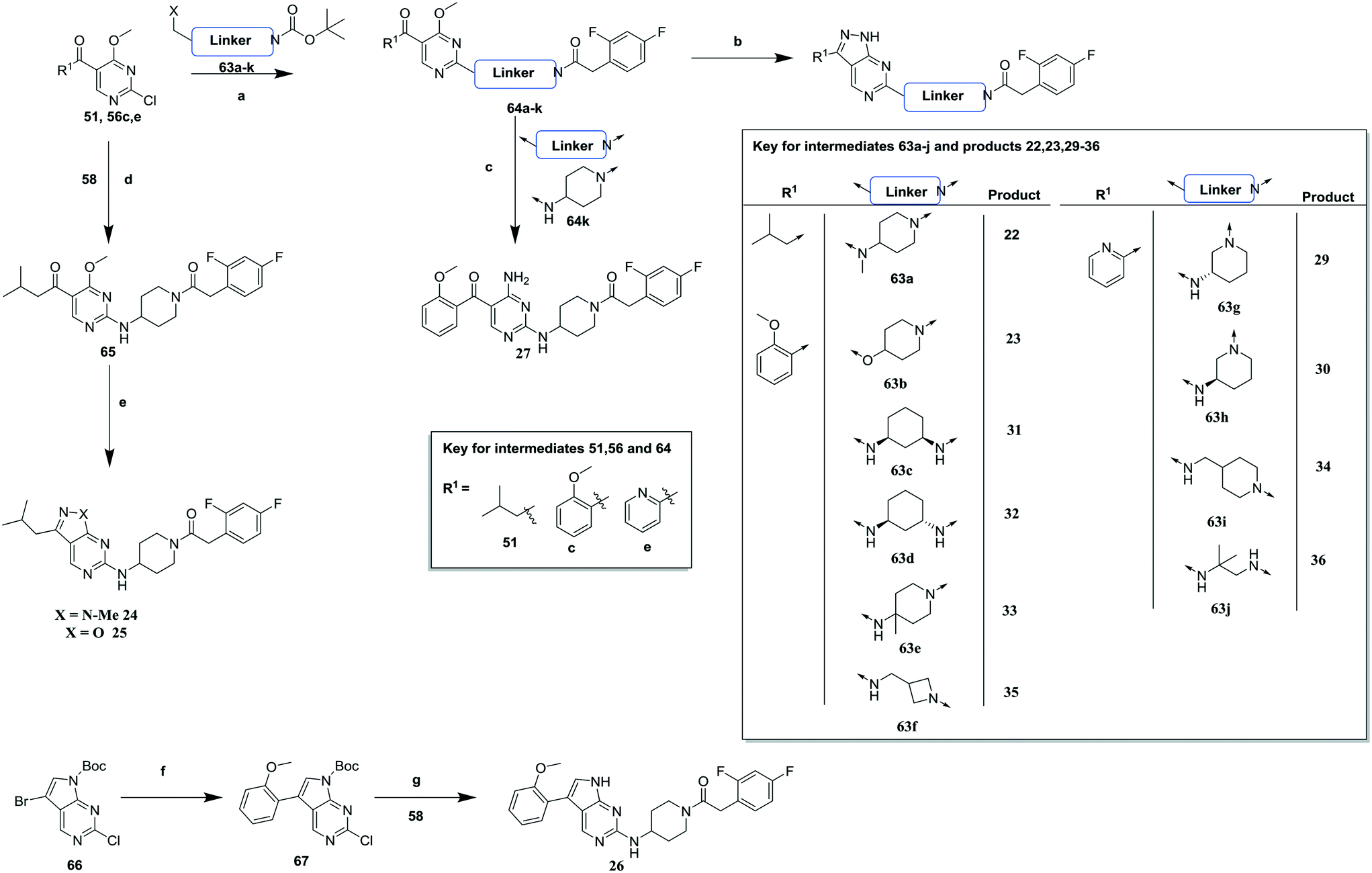 | ||
| Scheme 3 Synthesis of compounds 22, 23, 26, 29–36. (a) (i) DIPEA, butan-1-ol (ii) TFA, DCM (iii) TBTU, DIPEA, DCM; (b) hydrazine hydrate, butan-1-ol, 14–43% (over steps a and b); (c) ammonium hydroxide, ethanol (EtOH), 83%; (d) DIPEA, butan-1-ol; (e) (i) methylhydrazine, DIPEA, butan-1-ol (for 24), 18% (over 2 steps) (ii) hydroxylamine. Hydrogen chloride (HCl), DIPEA, butan-1-ol (for 25), 10% (over 2 steps); (f) (i) (2-methoxyphenyl)boronic acid, potassium phosphate (K3PO4), bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II), MeCN (ii) Boc2O, sodium hydride (NaH), THF, 92%; (g) DIPEA, N-methyl-2-pyrrolidone (NMP), 17%. | ||
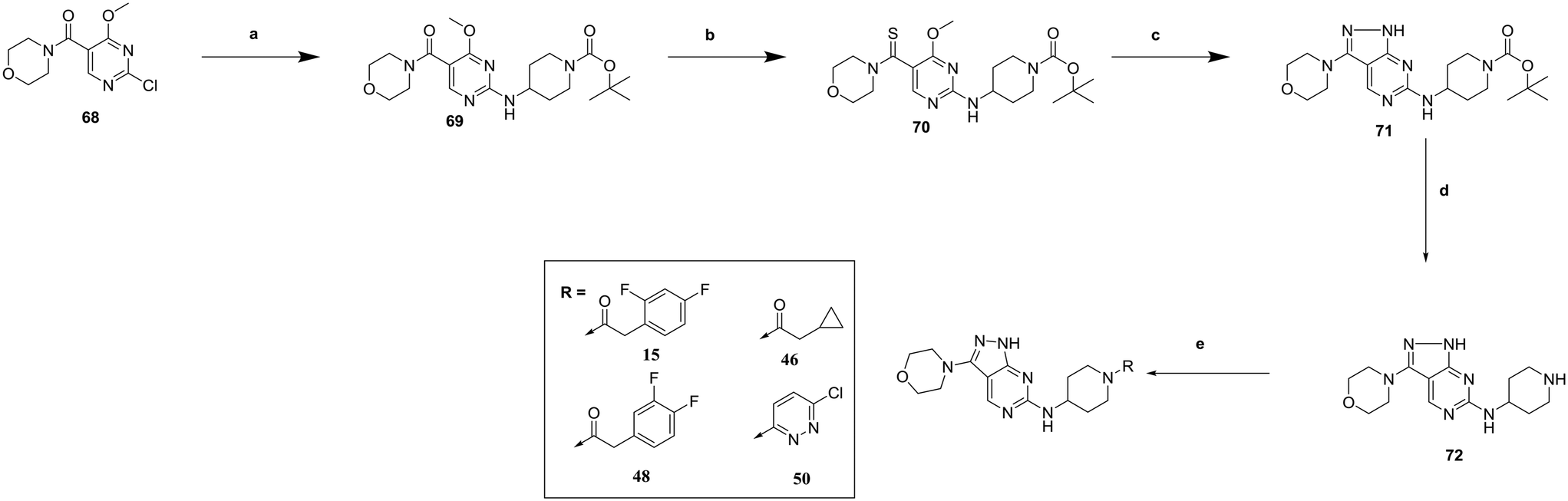 | ||
| Scheme 4 Synthesis of 15, 46, 48 and 50. (a) 1-Boc-4-aminopiperidine, DIPEA, butan-1-ol, 67%; (b) Lawessons reagent, THF, 86%; (c) hydrazine hydrate, 1,4-dioxane, 94% (d) TFA/DCM, 100% (e) (i) R–COOH, TBTU, DIPEA, DMF (for 15, 46 and 48) (ii) 3,6-dichloropyridazine, DIPEA, EtOH (for 50), 36–43%. | ||
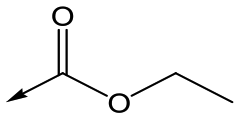
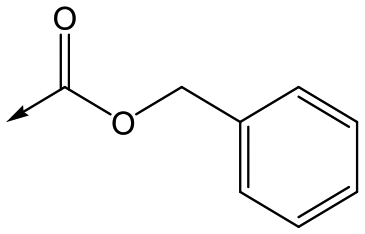
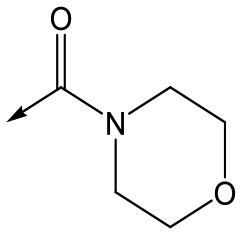
Compounds 11–21, with the 2,4-difluorophenacetamide RHS and variations to the LHS were synthesized according to Scheme 2, such that 56, with an appropriate R-group (R1 = a–d in table), was coupled to 52 and the resulting intermediate Boc-deprotected by treatment with TFA to give 57a–d. 57a and 57b were coupled to 2,4-difluorophenylacetic acid and subsequently cyclized with hydrazine to give 11 and 12. Compounds 40, 42, 43 and 47 were synthesized by a similar procedure using 57c or d and a suitable acid. Alternately, 56d–g were coupled directly with the elaborated aminopiperidine 58 to give 59d–g and cyclized with hydrazine to give compounds 13 and 18–21. 14 was synthesized by a similar route, but after 59i was cyclised to give 60, deprotection of the intermediate TBDMS alcohol gave 61 which was mesylated and displaced with morpholine. Finally, by choosing suitably substituted aminopiperidines alongside the relevant 56, intermediates 62c–e were generated and cyclized to give compounds 37–39, 41, 44, 45 and 49.
Scheme 3 highlights the synthetic routes to compounds with variations to the linking group between the pyrimidine core and the RHS amide (22–36). For compounds 22, 23 and 29–36, a suitably substituted 2-chloro-4-methoxy pyrimidine (51 or 56c, e) was treated with the relevant mono-Boc protected diamine 63a–j. These were subsequently deprotected and coupled with 2,4-difluorophenylacetic acid to give 64a–j which were cyclized with hydrazine to give the relevant compound. Alternatively, treatment of 64k with ammonia gave the monocyclic analogue 27. For compounds 24 and 25, 65 was generated from 56a and 58 then cyclized with either methylhydrazine or hydroxylamine to give 24 and 25 respectively. Replacement of the pyrazolopyrimidine scaffold with a 7H-pyrrolo[2,3-d]pyrimidine required a separate synthesis, whereby Boc protected 66 was cross-coupled with 2-methoxyphenylboronic acid and subsequently Boc-reprotected to give 67 which was coupled with 58 and then Boc deprotected to give 26.
Finally, the 3-morpholinopyrazole analogues 15, 46, 48 and 50 were synthesized via cyclisation of relevant thioamides as previously described,10 and shown in Scheme 4. 68 was therefore treated with 52 to give 69. Treatment with Lawessons reagent led to thioamide 70 which was cyclized with hydrazine to give 71, with subsequent Boc deprotection yielding 72. This was coupled with the relevant acid to give 15, 46 and 48, or treated with 3,6-dichloropyridazine to give 50.
In summary, an aminopiperidine subseries of the previously disclosed pyrazolopyrimidines was identified as having potent in vitro antileishmanial activity. SAR exploration demonstrated that the core and central unit were optimal for activity, whilst optimization of the RHS and LHS led to 48, with in vivo efficacy suitable for progression towards preclinical candidate selection. Whilst further profiling of 48 showed that it had a good safety profile, with only relatively weak inhibition of four human kinases highlighted, a dose escalation study in rats showed that 48 had infra-linear increase in exposure; this, alongside a minimum efficacious dose of 100 mg kg−1 meant that the compound was not progressed further. Despite this, the aminopiperidine subseries demonstrated the potential to deliver an alternative Leishmania CRK12 inhibitor for the treatment of VL.
Ethical statements
Mouse and rat pharmacokinetics
All animal studies were ethically reviewed and carried out in accordance with Animals (Scientific Procedures) Act 1986 and the GlaxoSmithKline (GSK)/Dundee University policies on the care, welfare, and treatment of animals.In vivo efficacy
All regulated procedures, at the University of Dundee, on living animals were carried out under the authority of a project license issued by the Home Office under the Animals (Scientific Procedures) Act 1986, as amended in 2012 (and in compliance with EU Directive EU/2010/63). License applications will have been approved by the University's Ethical Review Committee (ERC) before submission to the Home Office. The ERC has a general remit to develop and oversee policy on all aspects of the use of animals on University premises and is a subcommittee of the University Court, its highest governing body.Conflicts of interest
The following authors have shares in GlaxoSmithKline: M. A., S. D. C., A. D., F. M., J. M.-S., J. M. F., P. G. W., K. D. R., and T. J. M. The other authors declare no competing interests.Acknowledgements
Funding for this work was provided by Wellcome (no. 092340 and 100476). We thank Gina MacKay, Darren Edwards and Dan Fletcher for assistance with performing nuclear magnetic resonance (NMR) and mass spectrometry (MS) analyses, Raul Fernandez Velasco and the Galchimia chemistry team for synthesizing key compounds, and Alastair Pate, Francesco Gastaldello and James Burkinshaw for data management.Notes and references
- World Health Organization (WHO) Leishmaniasis Fact Sheet, World Health Organization: Geneva, 2018, (accessed April 2020), http://www.who.int/mediacentre/factsheets/fs375/en/.
- Drugs for Neglected Diseases Leishmaniasis factsheet, Drugs for Neglected Diseases initative (DNDi), June 2018, (accessed April 2020), https://www.dndi.org/diseases-projects/leishmaniasis/.
- S. Burza, S. L. Croft and M. Boelaert, Lancet, 2018, 392, 951–970 CrossRef.
- Drugs for Neglected Diseases initative Portfolio, Drugs for Neglected Diseases initative (DNDi), December 2019, (accessed April 2020), https://www.dndi.org/diseases-projects/portfolio/.
- S. Wyllie, M. G. Thomas, S. Patterson, S. Crouch, M. De Rycker, R. Lowe, S. Gresham, M. D. Urbaniak, T. D. Otto, L. Stojanovski, F. R. C. Simeons, S. Manthri, L. M. MacLean, F. Zuccotto, N. Homeyer, H. Pflaumer, M. Boesche, L. Sastry, P. Connolly, S. Albrecht, M. Berriman, G. Drewes, D. W. Gray, S. Ghidelli-Disse, S. Dixon, J. M. Fiandor, P. G. Wyatt, M. A. J. Ferguson, A. H. Fairlamb, T. J. Miles, K. D. Read and I. H. Gilbert, Nature, 2018, 560, 192–197 CrossRef CAS.
- S. Khare, A. S. Nagle, A. Biggart, Y. H. Lai, F. Liang, L. C. Davis, S. W. Barnes, C. J. N. Mathison, E. Myburgh, M.-Y. Gao, J. R. Gillespie, X. Liu, J. L. Tan, M. Stinson, I. C. Rivera, J. Ballard, V. Yeh, T. Groessl, G. Federe, H. X. Y. Koh, J. D. Venable, B. Bursulaya, M. Shapiro, P. K. Mishra, G. Spraggon, A. Brock, J. C. Mottram, F. S. Buckner, S. P. S. Rao, B. G. Wen, J. R. Walker, T. Tuntland, V. Molteni, R. J. Glynne and F. Supek, Nature, 2016, 537, 229–233 CrossRef CAS.
- S. Wyllie, S. Brand, M. G. Thomas, M. De Rycker, C. Chung, I. Pena, R. P. Bingham, J. A. Bueren-Calabuig, J. Cantizani, D. Cebrian, P. D. Craggs, L. Ferguson, P. Goswami, J. Hobrath, J. Howe, L. Jeacock, E. Ko, J. Korczynska, L. MacLean, S. Manthri, M. S. Martinez, L. Mata-Cantero, S. Moniz, A. Nühs, M. Osuna-Cabello, E. Pinto, J. Riley, S. Robinson, P. Rowland, F. R. C. Simeons, Y. Shishikura, D. Spinks, L. Stojanovski, J. Thomas, S. Thompson, E. Viayna-Gaza, R. J. Wall, F. Zuccotto, D. Horn, M. A. J. Ferguson, A. H. Fairlamb, J. M. Fiandor, J. Martin, D. W. Gray, T. J. Miles, I. H. Gilbert, K. D. Read, M. Marco and P. G. Wyatt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(19), 9318–9323 CrossRef CAS.
- Drugs for Neglected Diseases initative Target Product Profile, Drugs for Neglected Diseases initative (DNDi), 2019, (accessed April 2020), https://www.dndi.org/diseases-projects/leishmaniasis/.
- M. De Rycker, I. Hallyburton, J. Thomas, L. Campbell, S. Wyllie, D. Joshi, S. Cameron, I. H. Gilbert, P. G. Wyatt, J. A. Frearson, A. H. Fairlamb and D. W. Gray, Antimicrob. Agents Chemother., 2013, 57(7), 2913–2922 CrossRef CAS.
- M. G. Thomas, M. De Rycker, M. Ajakane, S. Albrecht, A. I. Álvarez-Pedraglio, M. Boesche, S. Brand, L. Campbell, J. Cantizani-Perez, L. A. T. Cleghorn, R. C. B. Copley, S. D. Crouch, A. Daugan, G. Drewes, S. Ferrer, S. Ghidelli-Disse, S. Gonzalez, S. L. Gresham, A. P. Hill, S. J. Hindley, R. M. Lowe, C. J. MacKenzie, L. MacLean, S. Manthri, F. Martin, J. Miguel-Siles, V. L. Nguyen, S. Norval, M. Osuna-Cabello, A. Woodland, S. Patterson, I. Pena, M. T. Quesada-Campos, I. H. Reid, C. Revill, J. Riley, J. R. Ruiz-Gomez, Y. Shishikura, F. R. C. Simeons, A. Smith, V. C. Smith, D. Spinks, L. Stojanovski, J. Thomas, S. Thompson, T. Underwood, D. W. Gray, J. M. Fiandor, I. H. Gilbert, P. G. Wyatt, K. D. Read and T. J. Miles, J. Med. Chem., 2019, 62(3), 1180–1202 CrossRef CAS.
- M. W. Robinson, A. P. Hill, S. A. Readshaw, J. C. Hollerton, R. J. Upton, S. M. Lynn, S. C. Besley and B. J. Boughtflower, Anal. Chem., 2017, 89(3), 1772–1777 CrossRef CAS.
- A. P. Hill and R. J. Young, Aq. Solubility for 41 and 50 was measured in a different assay (CLND) which gives broadly similar results, Drug Discovery Today, 2010, 15(15–16), 648–655 CrossRef CAS.
- S. Klein, AAPS J., 2010, 12(3), 397–406 CrossRef CAS.
- Premier screen kinase panel, International Center for kinase Profiling, Protein Phosphorylation and Ubiquitination Unit, University of Dundee.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00203h |
| This journal is © The Royal Society of Chemistry 2020 |