
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence4-Nitrophenyl activated esters are superior synthons for indirect radiofluorination of biomolecules†
Mohammad B.
Haskali
 *ab,
Ashleigh L.
Farnsworth
cd,
Peter D.
Roselt
a and
Craig A.
Hutton
cd
*ab,
Ashleigh L.
Farnsworth
cd,
Peter D.
Roselt
a and
Craig A.
Hutton
cd
aThe Centre for Molecular Imaging and Translational Research Laboratory, The Peter MacCallum Cancer Centre, Melbourne, Australia. E-mail: mo.haskali@petermac.org
bSir Peter MacCallum Department of Oncology, The University of Melbourne, Victoria 3010, Australia
cSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia
dBio21 Molecular Science and Biotechnology Institute, The University of Melbourne, VIC 3010, Australia
First published on 7th July 2020
Abstract
Indirect radiolabelling has for a long time been the mainstay strategy for radiofluorination of biomolecules. Acylation of biomolecules through the use of an 18F-labelled activated ester is a standard method for indirect radiolabelling. However, the preparation of 18F-labelled activated esters is typically a complex and multistep procedure. Herein, we describe the use of 4-nitrophenyl (PNP) activated esters to rapidly prepare 18F-labelled acylation synthons in one step. Furthermore, we present a comparative study of PNP activated esters and the commonly utilised 2,3,5,6-tetrafluorphenyl (TFP) activated esters under direct radiofluorination conditions and demonstrate their relative acylation behaviour. We demonstrate the superiority of PNP esters under direct radiofluorination conditions with favourable acylation kinetics.
Molecular imaging is transforming modern medicine through the use of radiopharmaceuticals that allow specific physiological processes occurring within the body to be detected at the cellular level.1 Positron emission tomography (PET) is a common molecular imaging technique that has had a pronounced influence on personalized healthcare.2 Radiopharmaceuticals utilised for PET imaging are typically labelled with short lived radionuclides including fluorine-18 (18F), carbon-11 (11C) and gallium-68 (68Ga).3,4 In particular, the use of fluorine-18 to radiolabel peptides combines favourable pharmacology with the ideal physicochemical and imaging characteristics of fluorine-18.5–8 However, the preparation of 18F-labelled peptides remains challenging and time consuming with multistep and laborious processes often required. Typically, sensitive biomolecules are radiolabelled with fluorine-18 indirectly, necessitating the use of a pre-labelled synthon (prosthetic groups) as they cannot tolerate the harsh radiofluorination conditions and are often thought to be inert to direct radiofluorination.9 [18F]Fluoroacylation is a common method used for indirect radiolabelling of biomolecules.10 [18F]Fluoroacylation synthons are typically prepared in multiple steps through the radiofluorination of an appropriate stable ester, hydrolysis of the ester, activation of the resulting acid to form an acylating agent and final HPLC purification of the synthon.8,10–12 This multi-step process is then followed by the reaction of the isolated [18F]fluoroacylation synthon with a biomolecule followed by a second HPLC purification to prepare the corresponding radiopharmaceutical. The use of TFP pre-activated diaryliodoniumbenzoate (4) was recently employed as a direct precursor to the TFP activated 4-[18F]fluorobenzoate synthon [18F]6 (Fig. 2), though the radiosynthesis proceeded in less than 1% yield when standard radiofluorination conditions (containing KHCO3/Kryptofix mixture) were employed.13 The yield of ester [18F]6 was enhanced to 24% under less basic conditions (no addition of KHCO3/Kryptofix).13 Recently, we demonstrated the direct, one-step radiofluorination of peptides using 18F-labelled pre-activated acylation agent 4-nitrophenyl 2-[18F]fluoropropionate ([18F]1).14–16 A similar approach has been reported for the preparation of 2,3,5,6-tetrafluorophenyl 6-[18F]fluoronicotinate ([18F]2) (Fig. 1).17,18
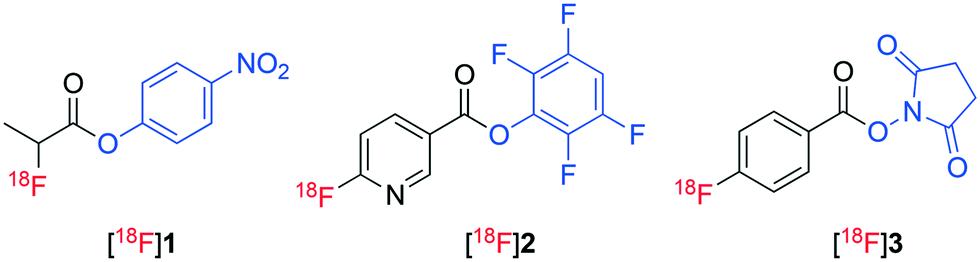 | ||
| Fig. 1 [18F]-labelled acylating agents [18F]1, [18F]2 and [18F]3. | ||
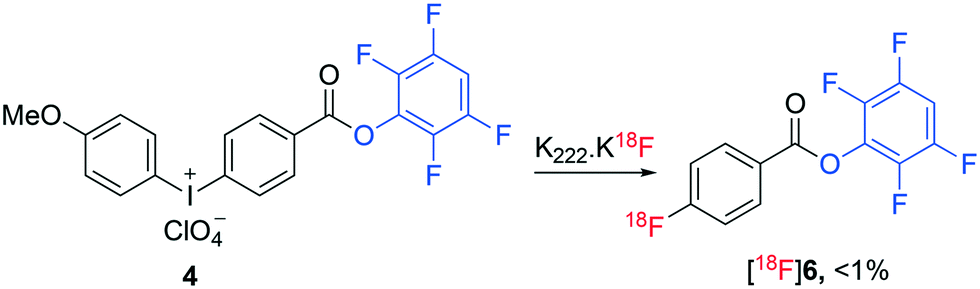 | ||
| Fig. 2 Reported radiosynthesis of ester [18F]6 by direct radiofluorination using K222·K[18F]F complex.13 | ||
Herein, we evaluate the stability of PNP and TFP esters to direct radiofluorination conditions for the preparation of 4-[18F]fluorobenzoate and 6-[18F]fluoronicotinate synthons. Furthermore, we report the relative stability and acylation activities of PNP and TFP pre-activated synthons. This study demonstrates the application of PNP active esters of 18F-labelled synthons for subsequent radiolabelling of biomolecules.
We first attempted the synthesis of PNP and TFP esters of both 4-methoxyphenyliodonium benzoate and 6-trimethylammonium nicotinate, 16–19, as precursors for direct radiofluorination. Esterification of 4-iodobenzoic acid 8 and 6-bromonicotinic acid 9 with 4-nitrophenol 10 and 2,3,5,6-tetrafluorophenol 11 afforded the corresponding PNP and TFP esters 12–15, respectively (Fig. 3). Notably, the PNP esters 12 and 13 were formed in very high yield and were readily recrystallized immediately after work up to afford pure products. In comparison, the TFP esters 14 and 15 lacked the crystallinity of the PNP analogues making them less convenient to handle. Iodo-benzoates 12 and 14 were oxidized by mCPBA and converted to the corresponding (4-methoxyphenyl)-(phenyl)iodonium tosylates 16 and 17. Bromo-nicotinates 13 and 15 were treated with 1 M trimethylamine in THF to form the corresponding ammonium species 18 and 19. The formation of esters 16–19 proceeded readily in high yields and on gram scale, making them highly accessible precursors. However, TFP ester 19 required careful handling and storage as it was found to be particularly sensitive to hydrolysis.
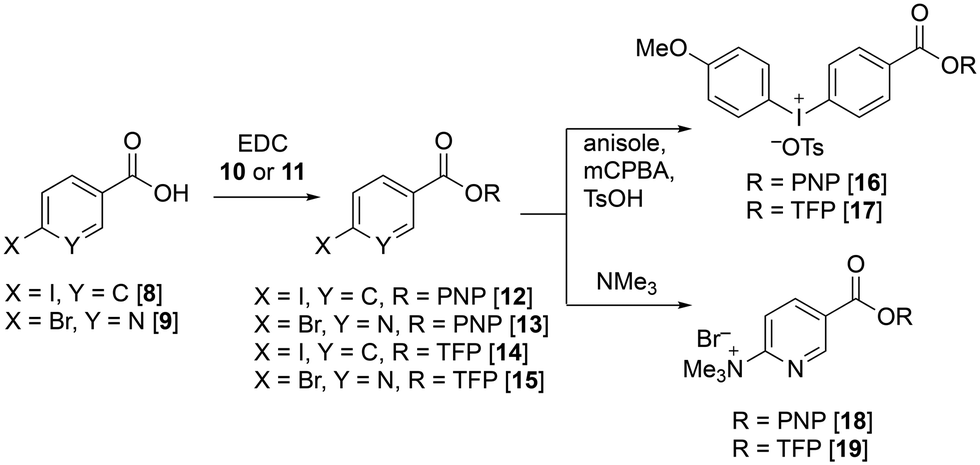 | ||
| Fig. 3 The synthesis of precursors 16–19 from benzoic acid 8 and nicotinic acid 9. | ||
Esters 16–19 were treated with [18F]fluoride in the form of K222·K[18F]F complex at 100 °C in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amyl alcohol (4:6) using potassium bicarbonate (for 18 and 19) or potassium oxalate (for 16 and 17) as accompanying bases. PNP esters 16 and 18 were readily soluble in the DMSO:t-amyl alcohol (TAA) mixture and were stable to the radiofluorination conditions leading to 94 ± 2% (n = 3) (58% isolated yield, decay corrected to SOS) and 85 ± 3% (n = 3) (42% isolated yield, decay corrected to SOS) of the corresponding fluoronicotinate and fluorobenzoate PNP esters, [18F]20 and [18F]21, respectively. The yields of esters [18F]20 and [18F]21 (94 ± 2% and 85 ± 3%) were determined based on HPLC analysis of the crude product. The recovery of radioactivity from the analytical column was always above 85%. The formation of [18F]21 demonstrates the first one-step synthesis of an activated ester of [18F]fluorobenzoate in high and reproducible yield. The yields of fluorobenzoate PNP ester [18F]21 were strongly dependent on the mass of precursor 16 used; furnishing 0%, 68% and 88% yields when employing 5 mg, 10 mg and 20 mg of diaryliodonium precursor 16, respectively. No such correlation was observed for the nicotinate PNP ester [18F]20, with the greater reactivity of the nicotinate 18 towards nucleophilic aromatic substitution resulting in high yields of 20 on all scales. With excellent yields of esters [18F]20 and [18F]21 obtained, no further optimisation was undertaken.19
:amyl alcohol (4:6) using potassium bicarbonate (for 18 and 19) or potassium oxalate (for 16 and 17) as accompanying bases. PNP esters 16 and 18 were readily soluble in the DMSO:t-amyl alcohol (TAA) mixture and were stable to the radiofluorination conditions leading to 94 ± 2% (n = 3) (58% isolated yield, decay corrected to SOS) and 85 ± 3% (n = 3) (42% isolated yield, decay corrected to SOS) of the corresponding fluoronicotinate and fluorobenzoate PNP esters, [18F]20 and [18F]21, respectively. The yields of esters [18F]20 and [18F]21 (94 ± 2% and 85 ± 3%) were determined based on HPLC analysis of the crude product. The recovery of radioactivity from the analytical column was always above 85%. The formation of [18F]21 demonstrates the first one-step synthesis of an activated ester of [18F]fluorobenzoate in high and reproducible yield. The yields of fluorobenzoate PNP ester [18F]21 were strongly dependent on the mass of precursor 16 used; furnishing 0%, 68% and 88% yields when employing 5 mg, 10 mg and 20 mg of diaryliodonium precursor 16, respectively. No such correlation was observed for the nicotinate PNP ester [18F]20, with the greater reactivity of the nicotinate 18 towards nucleophilic aromatic substitution resulting in high yields of 20 on all scales. With excellent yields of esters [18F]20 and [18F]21 obtained, no further optimisation was undertaken.19
The performance of the analogous TFP esters 17 and 19 was then investigated under similar conditions. Nicotinate TFP ester 19 afforded [18F]2 in variable yields (33–75%), with significant amounts of decomposition observed. This variability suggested the possible instability of the TFP activated ester [18F]2. Benzoate TFP ester 17 was insoluble in the DMSO:amyl alcohol mixture, and was sparingly soluble in neat DMSO. Subjecting benzoate TFP ester 17 to direct radiofluorination in neat DMSO resulted in <1% yield of [18F]6, a similar result to that reported by Richarz et al.13 However, when the insoluble mixture of precursor 17 in DMSO:amyl alcohol was heated with K222·K[18F]F complex, ester [18F]6 was formed in 31% after 10 minutes. No further studies were conducted with ester 17 or product [18F]6 due to their relative intolerance to direct radiofluorination conditions and low solubility (Fig. 4).
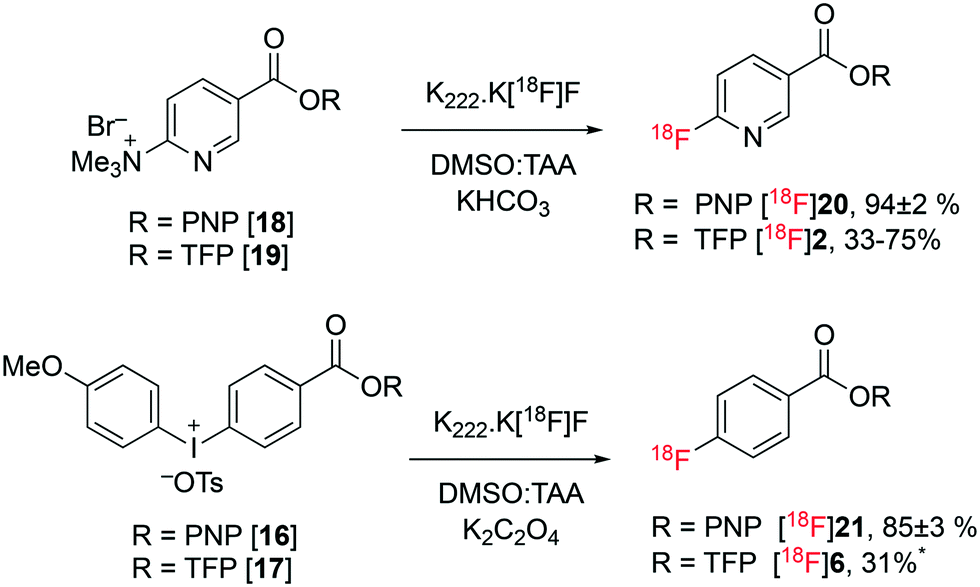 | ||
| Fig. 4 Radiofluorination of esters 16–19 to form the corresponding synthons [18F]2, [18F]6, [18F]20 and [18F]21. Yields of [18F]20 and [18F]21 presented as average of n = 3 ± standard deviation. *Labelling attempted only once. | ||
Next, the stability of [18F]20, [18F]21 and [18F]2 in DMSO at room temperature was assessed. TFP ester [18F]2 was found to undergo complete hydrolysis in less than one hour. In contrast, both PNP esters [18F]20 and [18F]21 esters were found to be remarkably stable, with no hydrolysis detected over nine hours. Indeed, the susceptibility of [18F]2 to hydrolysis and subsequent formation of by-products is well established.17,18
The relative acylation propensities of esters [18F]20, [18F]21 and [18F]2 were then examined with benzylamine 22 as a substrate. Varying concentrations of benzylamine 22 in DMSO (0.14–9.2 mM) containing 0.36 mM triethylamine were treated with [18F]20, [18F]21 and [18F]2 for 10 min at room temperature (Fig. 5). In order to understand the relative acylation potential of these esters, it was essential to correct for their decomposition in neat DMSO (control reaction). Since only TFP-ester [18F]2 underwent decomposition in the control reaction, only the resulting amidated product [18F]23 was influenced by this correction affording slightly higher acylation yields. PNP esters [18F]20 and [18F]21 underwent rapid acylation of benzylamine affording 93–100% yields of amidated products [18F]23 and [18F]24, respectively, in the presence of 9.2 mM benzylamine 22. The yields gradually decreased to 5–13% of amidated products [18F]23 and [18F]24 at 0.14 mM benzylamine 22. The TFP ester [18F]2 afforded the amidated product [18F]23 in 73% yield when treated with 9.2 mM benzylamine 22, which decreased to 10% with 0.14 mM benzylamine. Moreover, the yield of amides generated from TFP-benzoate [18F]2 were less consistent than the yields generated from the PNP esters [18F]20 and [18F]21. The higher and more reproducible yields of amide products generated from the PNP esters [18F]20 and [18F]21 are presumed to be a direct consequence of their higher stability in solution.
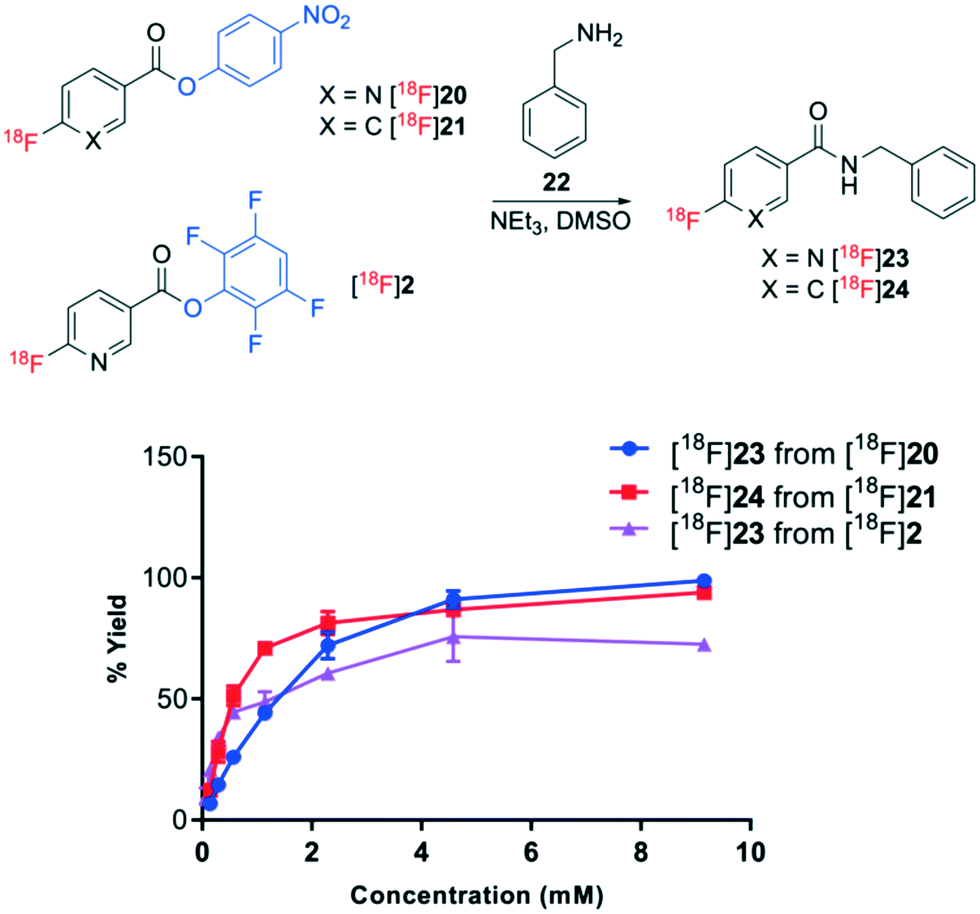 | ||
| Fig. 5 Acylation of esters [18F]20, [18F]21 and [18F]2 with benzylamine 22 to afford amidated products [18F]23 and [18F]24. % yield is the average of n = 3 with standard deviation bars shown. | ||
These results highlight the superiority of PNP esters as activated intermediates for the direct [18F]fluoroacylation of biomolecules. This study reflects the importance of the relative stability of activated esters as synthons to facilitate efficient [18F]fluoroacylation.
We next applied the PNP ester synthons [18F]20 and [18F]21 to the direct radiolabelling of RGD-peptide c(RGDyK) 25. Both esters [18F]20 and [18F]21 were quantitatively consumed at room temperature over 10 min to form the corresponding radiolabelled peptides [18F]26 and [18F]27, respectively (Fig. 6). Radiolabelled peptides [18F]26 and [18F]27 were obtained in >98% radiochemical purity and with high molar activity (170 and 211 GBq μmol−1, respectively) at end of synthesis as determined by analytical HPLC (using UV absorption at 220 nm).
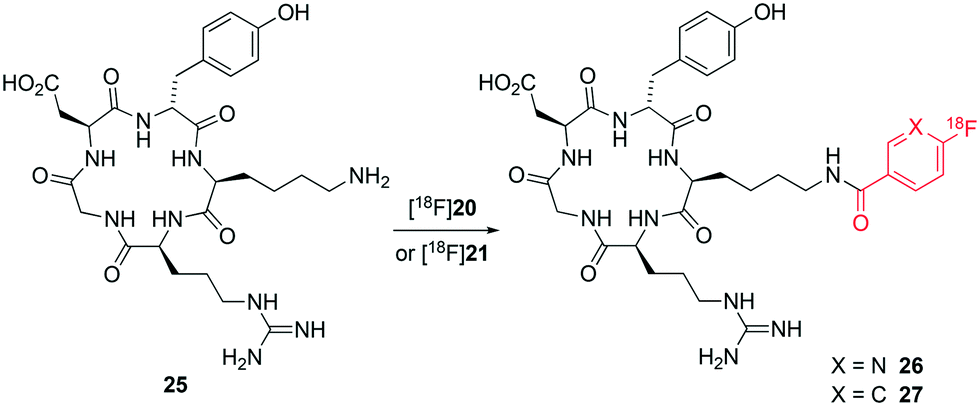 | ||
| Fig. 6 Radiolabelling of c(RGDyK) peptide 25 with PNP esters [18F]20 and [18F]21. | ||
Peptides [18F]26 and [18F]27 were prepared in ≥98% conversion from the corresponding synthons [18F]20 and [18F]21. The overall reaction time, purification and formulation of peptides [18F]26 and [18F]27 was achieved in less than 90 minutes on an iPHASE Flexlab automated radiochemistry module. This is in stark contrast with previously reported conditions that employ multistep procedures (>3 radiochemical steps) for the preparation of synthons, leading to overall synthesis time of >130 minutes and ultimately affording fluorine-18 labelled c(RGDyK) in low overall yields and sub-optimal automation procedures.20,21
Conclusions
In conclusion, PNP esters of [18F]fluoronicotinate [18F]21 and [18F]fluorobenzoate [18F]20 were formed under direct radiofluorination conditions at elevated temperatures. TFP-nicotinate [18F]2 and TFP-benzoate [18F]6 were found to be less tolerable of direct radiofluorination conditions employed. Moreover, the stability of the PNP esters renders them more effective at acylation in comparison to their TFP-counterparts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MH acknowledges funding from the Peter MacCallum Cancer Centre through Peter MacCallum Cancer Foundation Grant #1728. The authors acknowledge the support of the Bio21 Molecular Science and Biotechnology Institute MMSP facility.Notes and references
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- R. Pither, Expert Rev. Mol. Diagn., 2003, 3, 703–713 CrossRef CAS PubMed.
- B. Gulyás and C. Halldin, Q. J. Nucl. Med. Mol. Imaging, 2012, 56, 173–190 Search PubMed.
- M. Fani, J. P. André and H. R. Maecke, Contrast Media Mol. Imaging, 2008, 3, 53–63 CrossRef CAS PubMed.
- S. Okarvi, Eur. J. Nucl. Med., 2001, 28, 929–938 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- M. Fani, H. R. Maecke and S. M. Okarvi, Theranostics, 2012, 2, 481–501 CrossRef CAS PubMed.
- S. Richter and F. Wuest, Molecules, 2014, 19, 20536–20556 CrossRef PubMed.
- O. Morris, M. Fairclough, J. Grigg, C. Prenant and A. McMahon, J. Labelled Compd. Radiopharm., 2019, 62, 4–23 CrossRef CAS PubMed.
- H. J. Wester and M. Schottelius, Fluorine-18 Labeling of Peptides and Proteins, in PET Chemistry, ed. Schubiger P. A., Lehmann L. and Friebe M., Springer, Berlin, Heidelberg, 2007, vol. 64, pp. 79–111 Search PubMed.
- M. Kuchar, M. Pretze, T. Kniess, J. Steinbach, J. Pietzsch and R. Löser, Amino Acids, 2012, 43, 1431–1443 CrossRef CAS PubMed.
- G. Vaidyanathan and M. R. Zalutsky, Nat. Protoc., 2006, 1, 1655–1661 CrossRef CAS PubMed.
- R. Richarz, P. Krapf, F. Zarrad, E. A. Urusova, B. Neumaier and B. D. Zlatopolskiy, Org. Biomol. Chem., 2014, 12, 8094–8099 RSC.
- M. B. Haskali, P. D. Roselt, J. A. Karas, W. Noonan, C. W. Wichmann, A. Katsifis, R. J. Hicks and C. A. Hutton, J. Labelled Compd. Radiopharm., 2013, 56, 726–730 CrossRef CAS PubMed.
- M. B. Haskali, D. Denoyer, W. Noonan, C. Culinane, C. Rangger, N. Pouliot, R. Haubner, P. D. Roselt, R. J. Hicks and C. A. Hutton, Mol. Pharmaceutics, 2017, 14, 1169–1180 CrossRef CAS PubMed.
- M. B. Haskali, P. D. Roselt, R. J. Hicks and C. A. Hutton, J. Labelled Compd. Radiopharm., 2018, 61, 61–67 CrossRef CAS PubMed.
- D. E. Olberg, J. M. Arukwe, D. Grace, O. K. Hjelstuen, M. Solbakken, G. M. Kindberg and A. Cuthbertson, J. Med. Chem., 2010, 53, 1732–1740 CrossRef CAS PubMed.
- R. A. Davis, C. Drake, R. C. Ippisch, M. Moore and J. L. Sutcliffe, RSC Adv., 2019, 9, 8638–8649 RSC.
- R. P. Baum and H. R. Kulkarni, Theranostics, 2012, 2, 437–447 CrossRef CAS PubMed.
- R. Haubner, B. Kuhnast, C. Mang, W. A. Weber, H. Kessler, H.-J. Wester and M. Schwaiger, Bioconjugate Chem., 2004, 15, 61–69 CrossRef CAS PubMed.
- D. Thonon, D. Goblet, E. Goukens, G. Kaisin, J. Paris, J. Aerts, S. Lignon, X. Franci, R. Hustinx and A. Luxen, Mol. Imaging Biol., 2011, 13, 1088–1095 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00140f |
| This journal is © The Royal Society of Chemistry 2020 |