
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn investigation of the antileishmanial properties of semi-synthetic saponins†
Orlagh
Anderson‡
 a,
Joseph
Beckett‡
a,
Carla C.
Briggs‡
a,
Liam A.
Natrass
ab,
Charles F.
Cranston
a,
Elizabeth J.
Wilkinson
c,
Jack H.
Owen
c,
Rhodri
Mir Williams
c,
Angelos
Loukaidis
c,
Marc E.
Bouillon
c,
Deiniol
Pritchard
d,
Martina
Lahmann
c,
Mark S.
Baird
d and
Paul W.
Denny
*a
a,
Joseph
Beckett‡
a,
Carla C.
Briggs‡
a,
Liam A.
Natrass
ab,
Charles F.
Cranston
a,
Elizabeth J.
Wilkinson
c,
Jack H.
Owen
c,
Rhodri
Mir Williams
c,
Angelos
Loukaidis
c,
Marc E.
Bouillon
c,
Deiniol
Pritchard
d,
Martina
Lahmann
c,
Mark S.
Baird
d and
Paul W.
Denny
*a
aDepartment of Biosciences and Centre for Global Infectious Diseases, Durham University, Stockton Road, Durham, DH1 3LE, UK. E-mail: p.w.denny@durham.ac.uk; Tel: +44 (0)191 3343983
bDepartment of Chemistry and Centre for Global Infectious Diseases, Durham University, Stockton Road, Durham, DH1 3LE, UK
cDepartment of Chemistry, School of Natural Science, Bangor University, Gwynedd LL57 2UW, UK
dNaturiol Bangor Ltd, Alun Roberts Building, Bangor University, Gwynedd LL57 2UW, UK
First published on 9th June 2020
Abstract
Leishmaniasis is a neglected tropical disease caused by insect-vector borne protozoan parasites of the, Leishmania species. Whilst infection threatens and affects millions of the global poor, vaccines are absent and drug therapy limited. Extensive efforts have recently been made to discover new leads from small molecule synthetic compound libraries held by industry; however, the number of new chemical entities identified and entering development as anti-leishmanials has been very low. This has led to increased interest in the possibility of discovering naturally derived compounds with potent antileishmanial activity which may be developed towards clinical applications. Plant-derived triterpenoid and steroidal saponins have long been considered as anti-microbials and here we describe an investigation of a library of 137 natural (9) and semi-synthetic saponins (128) for activity against Leishmania mexicana, a causative agent of cutaneous leishmaniasis. The triterpenoid sapogenin, hederagenin, readily obtained in large quantities from Hedera helix (common ivy), was converted into a range of 128 derivatives. These semi-synthetic compounds, as well as saponins isolated from ivy, were examined with a phenotypic screening approach to identify potent and selective anti-leishmanial hits. This led to the identification of 12 compounds, including the natural saponin gypsogenin, demonstrating high potency (ED50 < 10.5 μM) against axenic L. mexicana amastigotes, the mammalian pathogenic form. One of these, hederagenin disuccinate, was sufficiently non-toxic to the macrophage host cell to facilitate further analyses, selectivity index (SI) > 10. Whilst this was not active in an infected cell model, the anti-leishmanial properties of hederagenin-derivatives have been demonstrated, and the possibility of improving the selectivity of natural hederagenin through chemical modification has been established.
Introduction
Leishmaniasis is one of the 20 neglected tropical diseases (NTD), and is endemic in over 90 countries, affecting approximately 12 million people per year, with over one billion people living at risk of disease.1 Causative Leishmania species are sand fly borne kinetoplastid protozoan parasites2 with infection leading to a wide spectrum of disease, from self-healing but scarring cutaneous leishmaniasis (CL) to fatal visceral disease (VL). The diversity of disease is dependent on the parasite species, genetic background and host immunity.3 Driven by elimination efforts in south Asia, the global burden of VL has decreased substantially in the past decade. However, in the same time period, CL cases have increased dramatically (0.7–1.0 million per year) largely due to forced migration in conflict zones.4 Against this backdrop, no vaccine is available and treatment relies entirely on a limited number of less than ideal drugs. Recently, public–private partnerships have seen industrial scale (>1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000) compound libraries screened for anti-leishmanials either phenotypically5,6 or target-based.7 All these initiatives focused on VL, leaving the most common form of leishmaniasis, CL, as a neglected NTD. Treatment of CL largely relies on the pentavalent antimonials – sodium stibogluconate (Pentostam) and meglumine antimoniate (Glucantime).8,9 Both drugs have been in clinical use for over 70 years despite their severe side-effects,10 parenteral administration,11 and the emergence of drug resistance.12 Diamidine pentamidine13 and amphotericin B (Fungizone)14 are second-line CL drugs, but are similarly associated with severe side-effects and Leishmania resistance has been observed under laboratory conditions.15
000000) compound libraries screened for anti-leishmanials either phenotypically5,6 or target-based.7 All these initiatives focused on VL, leaving the most common form of leishmaniasis, CL, as a neglected NTD. Treatment of CL largely relies on the pentavalent antimonials – sodium stibogluconate (Pentostam) and meglumine antimoniate (Glucantime).8,9 Both drugs have been in clinical use for over 70 years despite their severe side-effects,10 parenteral administration,11 and the emergence of drug resistance.12 Diamidine pentamidine13 and amphotericin B (Fungizone)14 are second-line CL drugs, but are similarly associated with severe side-effects and Leishmania resistance has been observed under laboratory conditions.15
The severe issues with the drugs used to treat CL demand the discovery and development of new effective therapies. Natural products are the basis of traditional ‘folk’ therapies, including for leishmaniasis.16,17 The active compounds from these include chalcones,18–21 flavonoids,22,23 alkaloids24 and terpenoids.25,26 Further, the anti-leishmanial amphotericin B is a natural product polyene, which was isolated from Streptomyces nodosus as an antifungal in 1955.27 With reference to this history, it is logical to revisit the natural world in the search for much needed anti-leishmanials.28
Saponins are a diverse class of compounds produced by many plants, and some marine organisms, via the mevalonic acid pathway. They consist of an aglycone unit (sapogenin) linked to between one and three carbohydrate moieties.29,30 Depending on the nature of the sapogenin, saponins are divided into two main classes – triterpenoid and steroidal. Triterpenoid saponins mainly occur in dicotyledonous angiosperms and comprise sapogenin moieties of six isoprene units, usually arranged as five rings, and bear structurally diverse carbohydrate groups.31 The less varied and widespread steroidal saponins consist primarily of a five-ring furostane or a six-ring spirostane skeleton and are almost exclusively produced by monocotyledonous angiosperms.32 The structural diversity of saponins is associated with a wide range of bioactivities. As secondary metabolites, their role in plants lies primarily in protection from herbivory and pathogen attack.29 However, saponins also possess a variety of pharmaceutical properties, from anti-inflammatory and anti-cancer to vaccine adjuvant activity.33,34 Several previous studies have identified saponins with anti-leishmanial activity.35–51
The beneficial effects of extracts from Hedera helix (common ivy), and in some cases of the discrete saponins extracted, have been widely reported. These include anti-inflammatory properties,52–54 anti-asthma activity55,56 and applications in cough medicines.57 More recently, anti-viral58 and anti-cancer properties20,59 have been reported. Extracts also show potent anti-fungal properties,60,61 but of particular interest were the reports of anti-leishmanial saponins extracted from H. helix.36,43,50,53,62 Extracted and purified monodesmoside triterpenoid saponins (α-, β- and δ-hederin) demonstrated activity against both promastigote (insect stage) and intracellular amastigote (mammalian stage) forms of both L. tropica and L. infantum.43
The saponins of common ivy are primarily based on a triterpene skeleton, hederagenin (Fig. 1), in which the alcohols or the acid are linked to sugars. Base hydrolysis on isolation of the saponins leads to the removal of the acid-bound sugars, and acid hydrolysis to the removal of all sugars. It is possible to isolate large quantities of saponins from both ivy leaf and fruit,63 based primarily on a single sapogenin, hederagenin. It is therefore of interest to determine whether the biological activity of these saponins, and of hederagenin, can be optimised by simple chemical modification to provide novel, plant based, solutions for application in medicine or agriculture. As part of a wider study of chemically modified saponins, we now describe the evaluation of the anti-leishmanial effects of 128 semi-synthetic molecules obtained by modification of the hederagenin core, together with nine natural product controls isolated from ivy extracts.
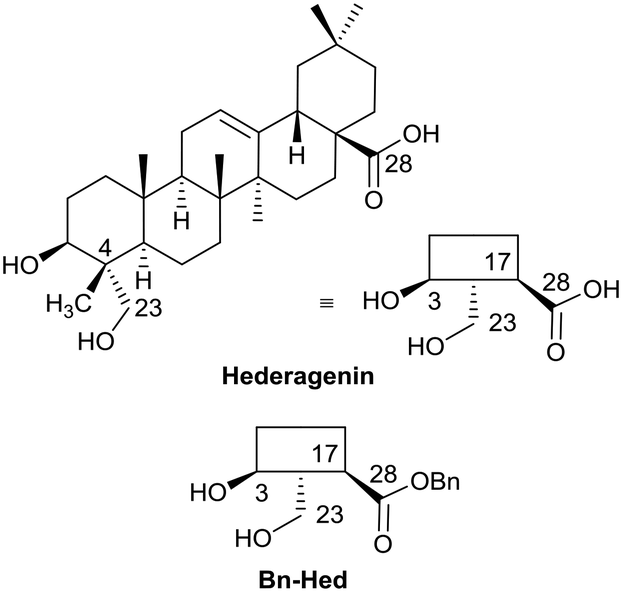 | ||
| Fig. 1 Short-hand structures for hederagenin and its benzyl ester (Bn-Hed) with relevant numbering. | ||
Results and discussion
Compound screening scheme
The library of hederagenin based compounds (137), either isolated from Hedera helix or synthesised from hederagenin obtained by extraction and hydrolysis of ivy saponins, were screened as illustrated in Fig. 2.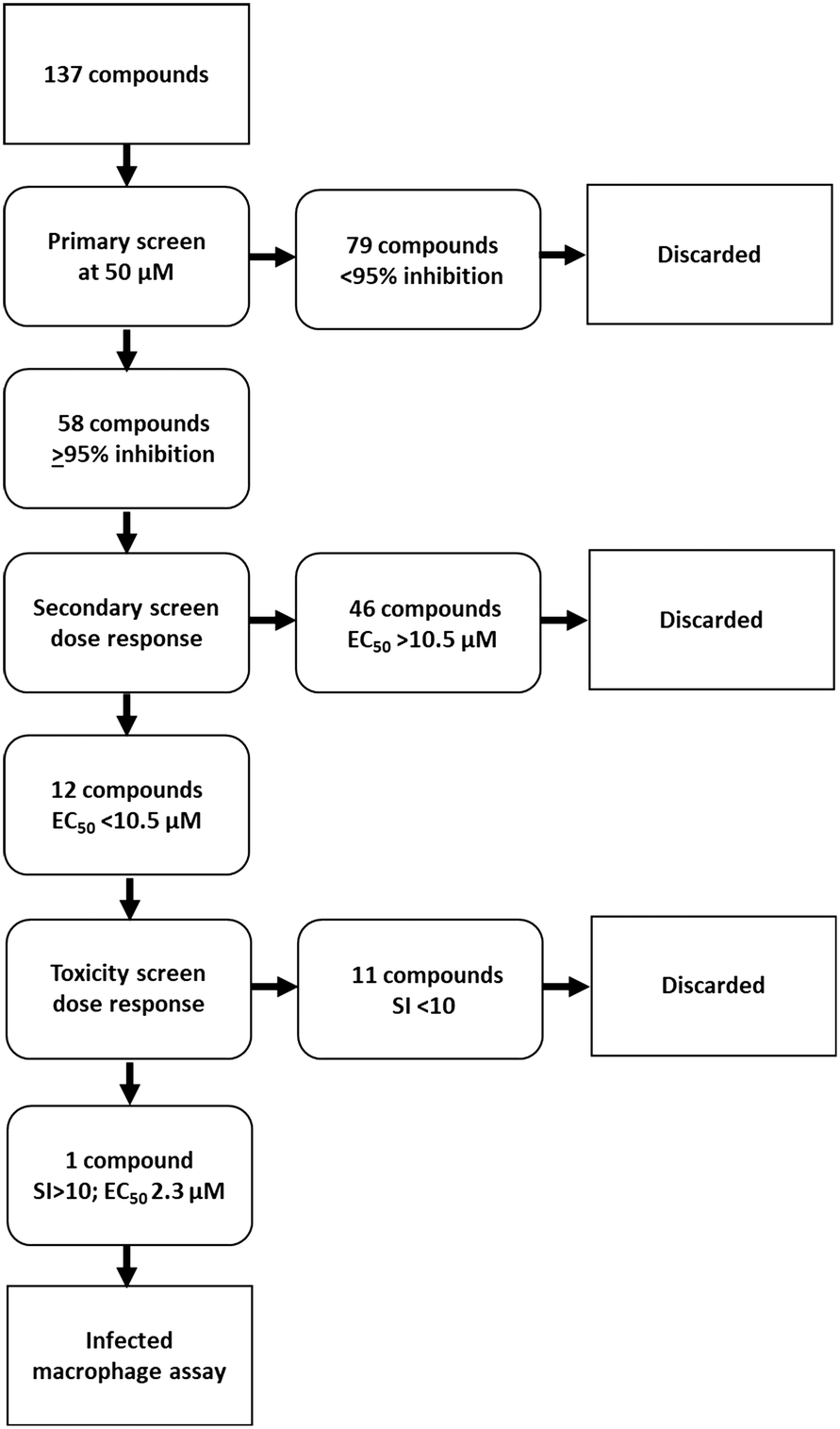 | ||
| Fig. 2 Screening scheme applied to compound library. | ||
All available compounds were subjected to a primary screen against mammalian stage L. mexicana axenic amastigotes as described below. These data and the structures of all compounds tested are provided in the ESI† (Table S1). Compounds that achieved ≥95% inhibition were taken forward to a secondary dose response screen which identified 12 highly active compounds for further examination.
Syntheses of compounds taken forward to the toxicity screen
The syntheses of key compounds, those 12 taken through to toxicity screening (Fig. 2) and not described elsewhere, are reported here (NMR data in ESI†). The 3,23-O-isopropylidene protected hederagenin MC-014 (ref. 64) was converted into the corresponding isocyanate (MC0-15) by a standard method. This intermediate was converted into a large number of derivatives (Table S1†). In particular, MC-033 and MC0-57 were prepared respectively by reaction with L-proline, or with pyrrolidine followed by hydrolysis (Scheme 1).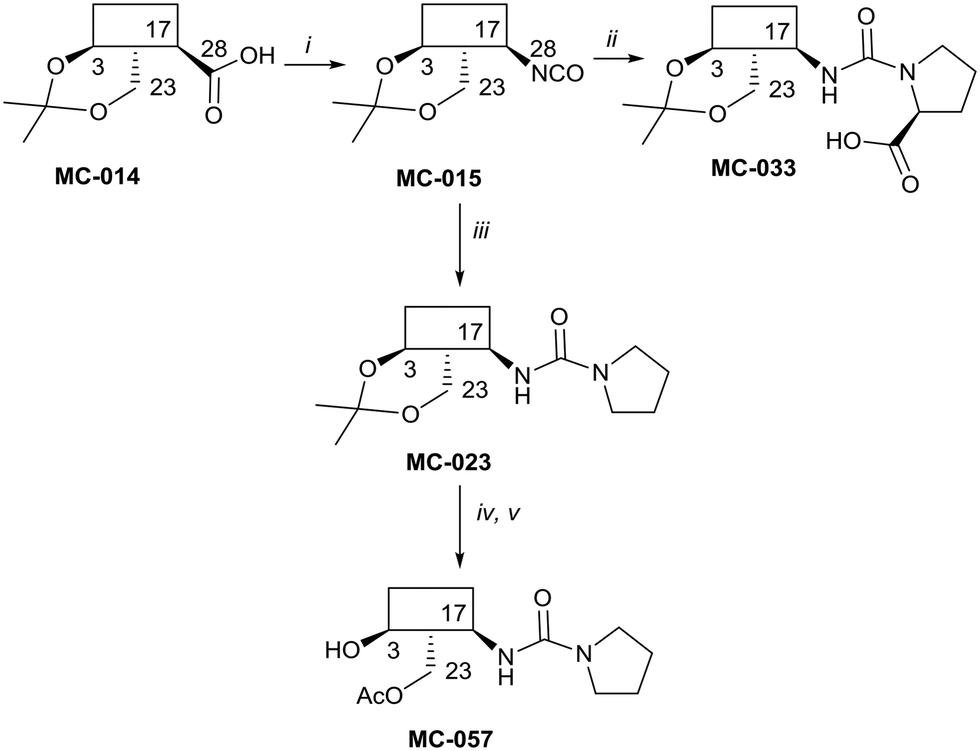 | ||
| Scheme 1 i. DPPA, Et3N, tol, 90 °C, 80%; ii. L-proline, EtOH, reflux to rt, quant.; iii. pyrrolidine, tol, 88%; iv. HCl(aq), DCM/H2O, quant; v. AcCl, DMAP, Et3N, tol, 5 °C, 56%. | ||
Hederagenin methyl ester (1)65 was converted into a series of 2,23-O-acetal derivatives, including, after ester hydrolysis, MC-071 (Scheme 2).
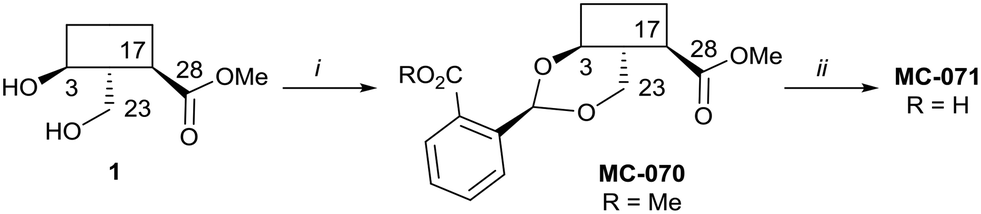 | ||
| Scheme 2 i. Methyl 2-formylbenzoate, PTSA (cat), DCM, quant.; ii. LiOH; THF/MeOH/H2O (4:1:1), 65 °C, 86%. | ||
Anemoclemosides66 are natural products featuring a unique open chain sugar moiety, linked to the sapogenin through an 3,23-O-acetal. However, they share the same common hederagenin aglycone. In this work a series of anemoclemosides (Table S1†), including the natural compound anemoclemoside A,64 were prepared from hederagenin. The total synthesis of this series of compounds will be described elsewhere (Lahmann and Bouillon, in preparation), but many of them have been screened in this work. The key compound IVL-81 was prepared by peracetylation of anemoclemoside A (Scheme 3).
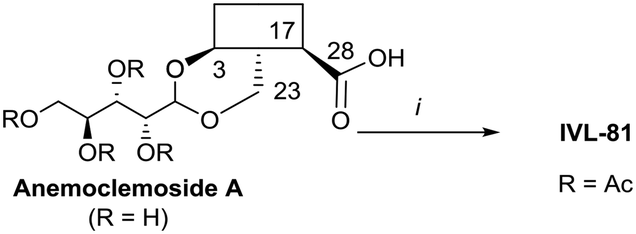 | ||
| Scheme 3 i. Ac2O, pyridine, 90 °C, 95%. | ||
With the intention of removing the enzymatically cleavable glycosidic linkage and thus increase in vivo stability, a series of C-glycosides was also prepared (Table S1†). As an alternative, acetal formation via the oxidised 6-position of monosaccharides was also performed. The key compound IVL-106 was obtained after oxidation67 of the tri-O-benzyl protected methyl glucoside 2 to the corresponding aldehyde 3, followed by acetal formation with Bn-Hed (Fig. 1) to give 4, and subsequent global deprotection (Scheme 4).
 | ||
| Scheme 4 i. (COCl)2, Et3N, DMSO/DCM, −78 °C, 87%; ii. Bn-Hed, PTSA (cat), DCM, 78%; iii. Pd(OH)2/C, H2, MeOH/MTBE, 92%. | ||
While the sugars in most natural saponins occur in their pyranose form, it was of interest to evaluate a derivative with a furanosyl residue. Thus, IVL-104 was prepared from an L-fucose derived propenyl derivative 5 by oxidation,68 prior to acetal formation with Bn-Hed and global deprotection (Scheme 5). Since it was not possible to separate the pseudo anomers at any step of the synthesis, the diastereomeric mixture was used for biological evaluation.
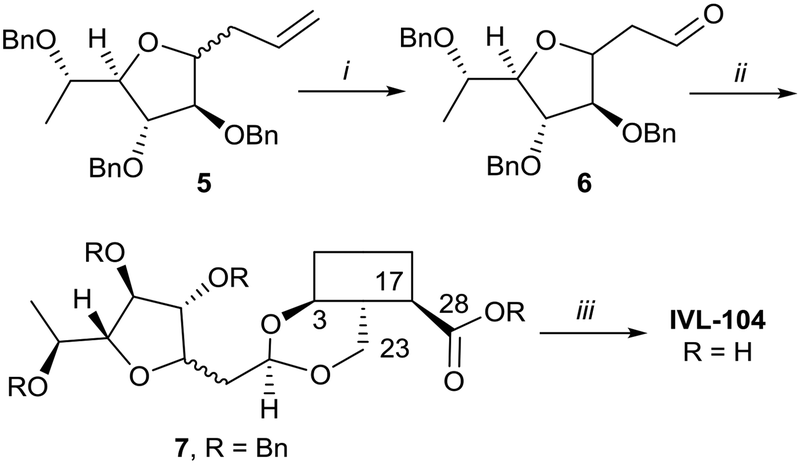 | ||
| Scheme 5 i. 1. O3, DCM −75 °C, 2. PPh3, 75%; ii. Bn-Hed, PTSA (cat), DCM, 95%; iii. Pd(OH)2/C, H2, MeOH/MTBE, 80%. | ||
Gypsogenin (IVL-9) is a saponin closely related to hederagenin,69 and for this work, it was synthesised from hederagenin by a protection, oxidation deprotection sequence (Scheme 6).
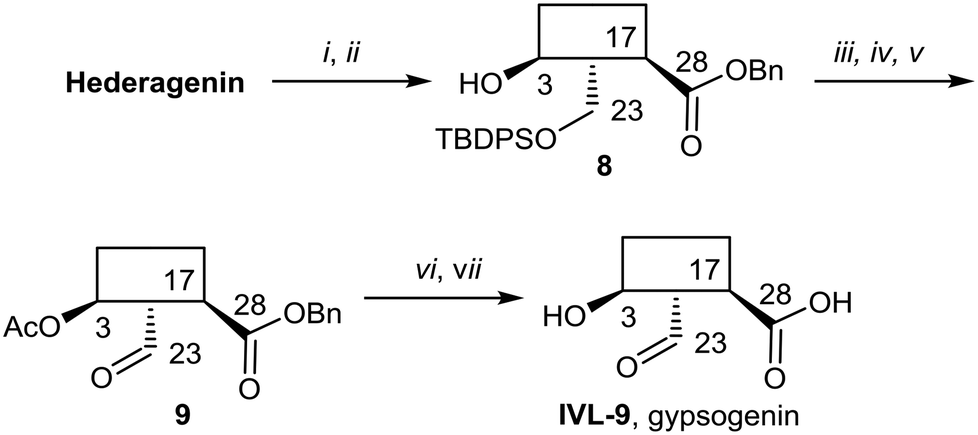 | ||
| Scheme 6 i. BnBr, K2CO3, DMF, 25 °C, 75% (S1); ii. TBDPSCl, imidazole, DCM, 0 °C to rt, quant. (S1); iii. Ac2O, pyridine, DCM, rt to 90 °C, 95%; iv. TBAF/AcOH, THF, 50 °C to 100 °C, 27%; v. TEMPO, BAIB, DCM, 67%; vi. NaOH, DME/THF/H2O, 25 °C, 83%; vii. Pd(OH)2/C, H2, MeOH/EtOAc, 80%. | ||
Screening of modified and natural saponins
|
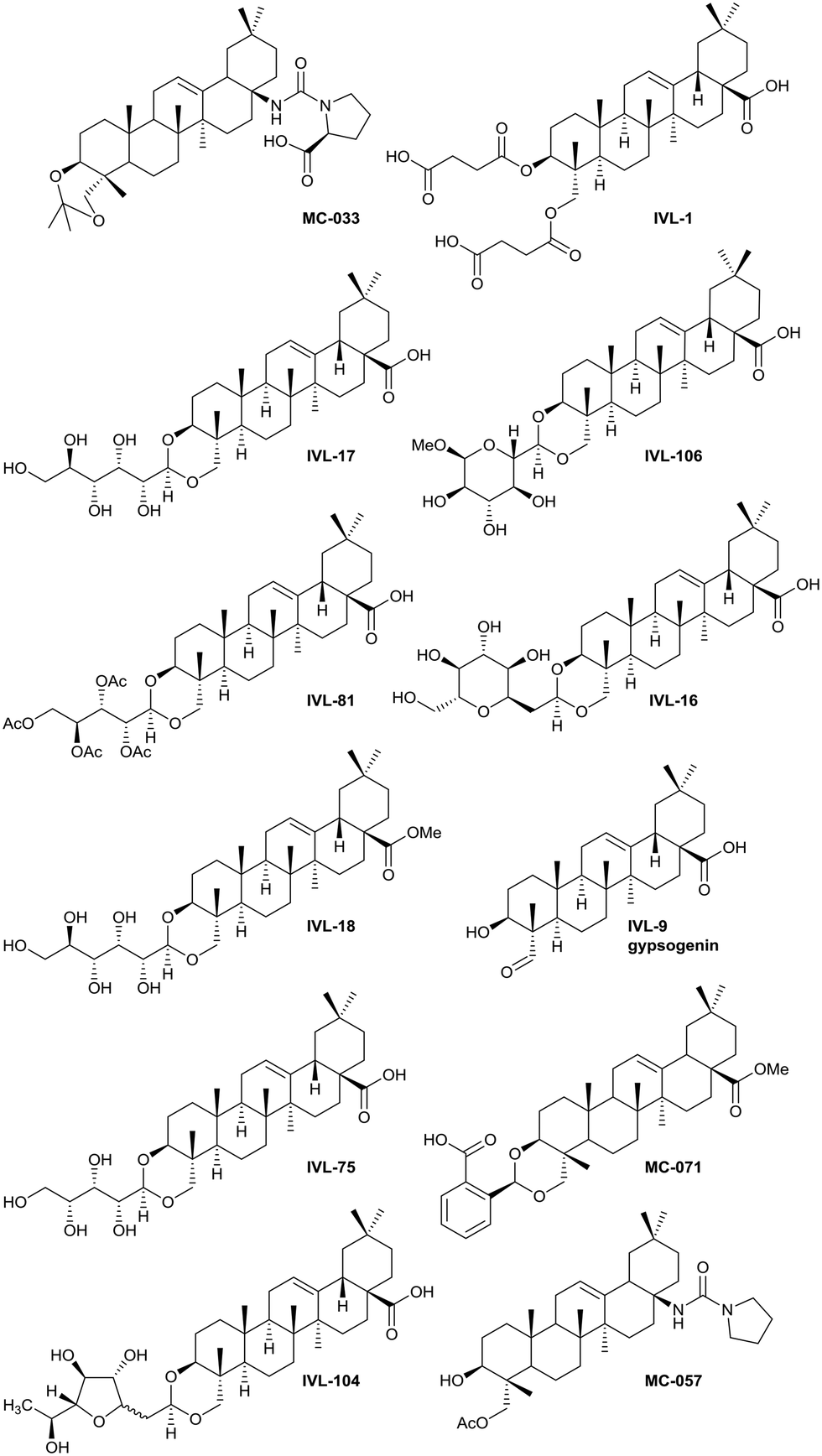 | ||
| Fig. 3 Hit compound structures selected from dose response study. | ||
Previous analyses of the anti-leishmanial activity of isolated and purified hederagenins (including α-hederin and related hederin and hederacoside molecules) demonstrated that they are membrane disruptors of all life cycle stages of the parasite, but also exert similar activity against mammalian cells leading to toxicity.36,50 Similar toxicity issues have been noted with respect to anti-leishmanial peptides and peptoids which are both classes of membrane disrupter.70,72–75 Therefore, the ability to modify hederagenin to form non-toxic, yet anti-leishmanial, hederagenin disuccinate (IVL-1; Fig. 3) is an important finding. However, although the axenic amastigote screen was performed with the clinically relevant parasite stage, this assay has limitations. Drug penetration of the host cell is not evaluated, neither is activity in the phagolysosomal environment in which the parasite resides.76 Previous studies have reported a lack of correlation between compounds selected in axenic form screening and intracellular amastigote assays.77 Therefore, IVL-1 (hederagenin disuccinate) was taken forward to an infected cell assay (Fig. 2 and Table 1, columns 8 and 9). Unfortunately, while passing all initial screens, IVL-1 was inactive in the infected cell assay (ED50 > 50 μM), that is against Leishmania amastigotes within the macrophage host. Nonetheless, the ability to develop a non-toxic anti-leishmanial hederagenin-derivative is an important step forward for further development; medicinal chemistry and/or pharmaceutics may improve the profile of this compound to facilitate activity against the most clinically relevant, intra-cellular form of Leishmania.
Structure activity relationships
To further consider the hederagenin derivatives as possible anti-leishmanial therapeutics, it is important to understand the relationship between structure and activity.In total 137 compounds have been screened for this work. Although sharing the common hederagenin core, the library is diverse and can be broadly divided into 8 subgroups according to functionalities. These the groups are: the natural products, including the hederosides (group 1), the anemoclemosides (2), other 3,23-hederagenin acetals (3), the hederagenin esters (4), compounds in which both the acid and the diol are protected (as esters and acetals respectively) – protected hederagenins (5), the ureas (6), the triacids (7), and a small number non-classified derivatives, principally intermediates to the synthesis of other compounds (8). Representative results for the various groups are summarised as follows:
Most of the natural saponins evaluated in this study reached the cut-off point in the primary screen, however only gypsogenin (IVL-9) was taken forward following secondary screening. It is the only aldehyde in the compound library, which may explain its efficacy, and its toxicity (Table 1). As observed for other saponins, its bioactivity as an anti-inflammatory and cytotoxic compound has been associated with the presence of the carbohydrate component.78 However, gypsogenin has also been demonstrated to trigger the apoptotic pathway in cancer cells.79 Of the synthetically modified hederagenins, three types were most active anti-leishmanial: group 6 – a urethane modification in position C-28; group 2 – an acid at C-28 and a sugar moiety at C-3 and/or C-23, to resemble natural monodesmotic saponins; and group 7, IVL-1, with a terminal acid substituent at each of these positions.
Saponins are well known to disrupt membranes through the interaction with sterols.80 As such, the amphiphilicity and the stereochemistry introduced by saccharide moieties may be of importance. An overall trend was that removal of polarity from both sides of the aglycone (the C-3, C-26 and C-28 positions) reduced the anti-leishmanial efficacy. Interestingly, the bidesmosidic natural compound hederacoside D (IVL-15) was inactive, suggesting that a balanced distribution of polarity over the saponin is important. While some C-28 esters passed the primary screen cut-off, only the saponin-like methyl ester (e.g.IVL-18) was selected after the secondary assay. The C-28 methyl ester incorporating a carboxylic acid group on the C-3,23 acetal (MC-071), was also selected. Conversely, reducing position C-28 to the corresponding alcohol (MC-029) resulted in low activity (below primary screen threshold). An interesting group of compounds in this context are the xylose based anemoclemosides analogues. Whilst the C-28 acids IVL-17 and IVL-75 were carried through to the final screes, then excluded by virtue of low selectivity (SI 6.7 and 3.0 respectively), the corresponding C-28 alcohol IVL-90 failed at the secondary assay stage. The C-28 methyl ester IVL-89 was excluded after primary screening. This demonstrated that the loss of the acid group in C-28 could not be compensated by keeping an unprotected sugar unit on the C3, C23 position, indicating that subtle variations in the structure could be relevant. There was at least some link between activity and stereochemistry in that only a minority of the cyclised compounds derived from xylose (e.g.IVL-106) reached the primary screen threshold, compared to the glucosyl, arabinosyl and rhamnosyl derivatives. In general, the anemoclemosides resemble the natural monodesmosides such as the α- and δ-hederins, with some added flexibility due to the open chain presentation of the sugar moiety, and anti-leishmanial activity is at least partly dependent on amphiphilicity and the stereochemistry introduced by saccharide moieties. Against this background, the activity of IVL-81, in which the sugar was protected as a tetra-acetate, is surprising and indicated that there could be subtle reasons for anemoclemoside anti-leishmanial activity.
A large number of the library members (46) contained the urea unit, a well-established ‘drug-like’ pharmacore, and most of these were active in the primary screen (Table S1†). It is interesting to note that the one with the lowest ED50 (MC-033) was the only example to have a free acid substituent on the urea (Fig. 3).
Conclusions
Given the paucity of safe and effective therapies for CL, and a rising number of cases in an increasing geographic area, the search for novel anti-leishmanials is as urgent as ever. Reflecting the absence of fully validated and assay formulated targets in these parasites, the focus has been on phenotypic screening for new lead compounds.81 Natural product discovery forms part of this global effort and novel anti-leishmanials from these sources are discovered almost always as a result of target-blind screening.16,17,28Novel triterpenoid saponins isolated from Maesa argentea leaves whilst active against infective stage L. infantum were also cytotoxic at a similar range.39 Whilst consideration of delivery mechanisms may mitigate against toxicity,51 other triterpenoid saponins have more promising profiles. For example, maesabalide II, again from isolated Maesa argentea leaves, demonstrated potent and specific anti-leishmanial (L. infantum) activity and promising in vivo activity.41,42 Furthermore, triterpenoid saponins isolated from Careya arborea leaves44 and the lower stem parts of Astragalus oleifolius48 showed specific activity against infective stage L. donovani. Together these data indicated that triterpenoid saponins (including hederagenins) may be a source of drug leads from leishmaniasis. However, in many cases these natural products are isolated in small quantities and only after a significant effort separating them from other components. In contrast, hederagenin can be readily isolated from Hedera helix (common ivy) in large quantities and in this study we utilized a phenotypic screening approach to identify anti-leishmanial semi-synthetic triterpenoid saponins based on hederagenin.
The most selective (non-toxic to the mammalian macrophage host cell) anti-leishmanial compound, IVL-1, structurally differs substantially from all other 136 compounds in the library screened. This tri-acid, the disuccinate of hederagenin, is known to be active in improving the growth of ruminants,82 and it and its salts have been reported to have value as anti-depressant drugs.83 It will be of interest therefore to further evaluate the efficacy of this molecule and examine other di- or tri-acidic species. It is also of interest to determine whether the presence of three acid groups have a specific effect on its bioavailability. However, to facilitate further development IVL-1 must be developed to show activity against the parasites when they are residing with the host cell (intra-macrophage). The use of both liposomal and nano-particle formulations have been shown to improve efficacy of natural product anti-leishmanials and it is feasible that either or both of these approaches may facilitate the uptake of IVL-1 into the macrophage as well as improving bioavailability.16,84–86 Subsequently, to aid rational development towards the clinic, an understanding of the mode of action is vital.87–90 Mass spectrometry-based metabolomics has been applied to investigate this for both clinical drugs and experimental natural compounds, although the results of these studies can be difficult to interpret.28,91
Notably all but one, the natural cyclic peptide amphotericin B, of the current clinical anti-leishmanials lacks a well-defined mechanism of action.16,91 Mammalian cells harbour cholesterol as the primary sterol, in contrast fungi and Leishmania spp possess ergosterol.91 This difference is exploited for both anti-fungal and anti-leishmanial therapy by amphotericin B which selectively sequesters ergosterol.91 Following this precedent, saponins are known to disrupt membranes via interaction with sterols80 and, like amphotericin B, the selective nature of IVL-1 may be due to the presence of ergosterol rather than cholesterol in the plasma membrane of Leishmania. This requires further investigation and, notably, amphotericin B resistant Leishmania have been selected and demonstrated by whole genome sequencing to harbour mutations in the ergosterol biosynthetic pathway.92,93 Similar analyses could, in future, reveal the mode of action of the highly selective disuccinate of hederagenin, IVL-1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has, in part, received funding from UK Research and Innovation via the Global Challenges Research Fund under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1. The compounds studied in this work were prepared as part of an IUK grant (102101). EJW, JHO, RMW and AL carried out chemical synthetic work as undergraduates at Bangor University. All the biological experimentation was carried out by OA, JB, CCB, LAN and CFC when they were undergraduate students at Durham University.References
- WHO, https://www.who.int/leishmaniasis/en/, 2019.
- K. Stuart, R. Brun, S. Croft, A. Fairlamb, R. E. Gurtler, J. McKerrow, S. Reed and R. Tarleton, J. Clin. Invest., 2008, 118, 1301–1310 CrossRef CAS PubMed.
- R. Reithinger, J. C. Dujardin, H. Louzir, C. Pirmez, B. Alexander and S. Brooker, Lancet Infect. Dis., 2007, 7, 581–596 CrossRef PubMed.
- S. Burza, S. L. Croft and M. Boelaert, Lancet, 2018, 392, 951–970 CrossRef.
- S. Khare, A. S. Nagle, A. Biggart, Y. H. Lai, F. Liang, L. C. Davis, S. W. Barnes, C. J. Mathison, E. Myburgh, M. Y. Gao, J. R. Gillespie, X. Liu, J. L. Tan, M. Stinson, I. C. Rivera, J. Ballard, V. Yeh, T. Groessl, G. Federe, H. X. Koh, J. D. Venable, B. Bursulaya, M. Shapiro, P. K. Mishra, G. Spraggon, A. Brock, J. C. Mottram, F. S. Buckner, S. P. Rao, B. G. Wen, J. R. Walker, T. Tuntland, V. Molteni, R. J. Glynne and F. Supek, Nature, 2016, 537, 229–233 CrossRef CAS PubMed.
- I. Pena, M. Pilar Manzano, J. Cantizani, A. Kessler, J. Alonso-Padilla, A. I. Bardera, E. Alvarez, G. Colmenarejo, I. Cotillo, I. Roquero, F. de Dios-Anton, V. Barroso, A. Rodriguez, D. W. Gray, M. Navarro, V. Kumar, A. Sherstnev, D. H. Drewry, J. R. Brown, J. M. Fiandor and J. Julio Martin, Sci. Rep., 2015, 5, 8771 CrossRef CAS PubMed.
- J. L. Norcliffe, J. G. Mina, E. Alvarez, J. Cantizani, F. de Dios-Anton, G. Colmenarejo, S. G. Valle, M. Marco, J. M. Fiandor, J. J. Martin, P. G. Steel and P. W. Denny, Sci. Rep., 2018, 8, 3938 CrossRef PubMed.
- S. L. Croft and G. H. Coombs, Trends Parasitol., 2003, 19, 502–508 CrossRef CAS PubMed.
- L. Kedzierski, A. Sakthianandeswaren, J. M. Curtis, P. C. Andrews, P. C. Junk and K. Kedzierska, Curr. Med. Chem., 2009, 16, 599–614 CrossRef CAS PubMed.
- F. Chappuis, S. Sundar, A. Hailu, H. Ghalib, S. Rijal, R. W. Peeling, J. Alvar and M. Boelaert, Nat. Rev. Microbiol., 2007, 5, 873–882 CrossRef CAS PubMed.
- C. Demicheli, R. Ochoa, J. B. da Silva, C. A. Falcao, B. Rossi-Bergmann, A. L. de Melo, R. D. Sinisterra and F. Frezard, Antimicrob. Agents Chemother., 2004, 48, 100–103 CrossRef CAS PubMed.
- S. L. Croft, S. Sundar and A. H. Fairlamb, Clin. Microbiol. Rev., 2006, 19, 111–126 CrossRef CAS PubMed.
- P. G. Bray, M. P. Barrett, S. A. Ward and H. P. de Koning, Trends Parasitol., 2003, 19, 232–239 CrossRef CAS PubMed.
- C. P. Thakur, R. K. Singh, S. M. Hassan, R. Kumar, S. Narain and A. Kumar, Trans. R. Soc. Trop. Med. Hyg., 1999, 93, 319–323 CrossRef CAS PubMed.
- C. Di Giorgio, F. Faraut-Gambarelli, A. Imbert, P. Minodier, M. Gasquet and H. Dumon, J. Antimicrob. Chemother., 1999, 44, 71–76 CrossRef CAS PubMed.
- R. L. Charlton, B. Rossi-Bergmann, P. W. Denny and P. G. Steel, Parasitology, 2018, 145, 219–236 CrossRef PubMed.
- P. E. Cockram and T. K. Smith, J. Nat. Prod., 2018, 81, 2138–2154 CrossRef CAS PubMed.
- J. C. Aponte, D. Castillo, Y. Estevez, G. Gonzalez, J. Arevalo, G. B. Hammond and M. Sauvain, Bioorg. Med. Chem. Lett., 2010, 20, 100–103 CrossRef CAS PubMed.
- P. Boeck, C. A. Bandeira Falcao, P. C. Leal, R. A. Yunes, V. C. Filho, E. C. Torres-Santos and B. Rossi-Bergmann, Bioorg. Med. Chem., 2006, 14, 1538–1545 CrossRef CAS PubMed.
- M. Chen, S. B. Christensen, T. G. Theander and A. Kharazmi, Antimicrob. Agents Chemother., 1994, 38, 1339–1344 CrossRef CAS PubMed.
- T. F. de Mello, H. R. Bitencourt, R. B. Pedroso, S. M. Aristides, M. V. Lonardoni and T. G. Silveira, Exp. Parasitol., 2014, 136, 27–34 CrossRef CAS PubMed.
- E. R. da Silva, C. Maquiaveli Cdo and P. P. Magalhaes, Exp. Parasitol., 2012, 130, 183–188 CrossRef CAS PubMed.
- B. Mittra, A. Saha, A. R. Chowdhury, C. Pal, S. Mandal, S. Mukhopadhyay, S. Bandyopadhyay and H. K. Majumder, Mol. Med., 2000, 6, 527–541 CAS.
- B. B. Mishra, R. K. Singh, A. Srivastava, V. J. Tripathi and V. K. Tiwari, Mini-Rev. Med. Chem., 2009, 9, 107–123 CrossRef CAS PubMed.
- D. C. Arruda, F. L. D'Alexandri, A. M. Katzin and S. R. Uliana, Antimicrob. Agents Chemother., 2005, 49, 1679–1687 CrossRef CAS PubMed.
- S. R. M. S. do Socorro, R. R. Mendonca-Filho, H. R. Bizzo, I. de Almeida Rodrigues, R. M. Soares, T. Souto-Padron, C. S. Alviano and A. H. Lopes, Antimicrob. Agents Chemother., 2003, 47, 1895–1901 CrossRef PubMed.
- J. D. Dutcher, Dis. Chest, 1968, 54(Suppl 1), 296–298 CrossRef PubMed.
- A. J. Mbekeani, R. S. Jones, M. Bassas Llorens, J. Elliot, C. Regnault, M. P. Barrett, J. Steele, B. Kebede, S. K. Wrigley, L. Evans and P. W. Denny, Int. J. Parasitol.: Drugs Drug Resist., 2019, 11, 118–128 CAS.
- J. M. Augustin, V. Kuzina, S. B. Andersen and S. Bak, Phytochemistry, 2011, 72, 435–457 CrossRef CAS PubMed.
- S. T. Mugford, T. Louveau, R. Melton, X. Qi, S. Bakht, L. Hill, T. Tsurushima, S. Honkanen, S. J. Rosser, G. P. Lomonossoff and A. Osbourn, Plant Cell, 2013, 25, 1078–1092 CrossRef CAS PubMed.
- T. Moses, K. K. Papadopoulou and A. Osbourn, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 439–462 CrossRef CAS PubMed.
- S. Ur Rahman, M. Ismail, M. Khurram, I. Ullah, F. Rabbi and M. Iriti, Molecules, 2017, 22, 2156 CrossRef PubMed.
- B. W. Greatrex, A. M. Daines, S. Hook, D. H. Lenz, W. McBurney, T. Rades and P. M. Rendle, ChemistryOpen, 2015, 4, 740–755 CrossRef CAS PubMed.
- S. G. Sparg, M. E. Light and J. van Staden, J. Ethnopharmacol., 2004, 94, 219–243 CrossRef CAS PubMed.
- R. C. de Paula, S. M. da Silva, K. F. Faria, F. Frezard, C. P. S. Moreira, K. Foubert, J. C. D. Lopes, P. R. V. Campana, M. P. Rocha, A. F. Silva, C. G. Silva, L. Pieters and V. L. Almeida, J. Ethnopharmacol., 2019, 232, 155–164 CrossRef PubMed.
- F. Delmas, C. Di Giorgio, R. Elias, M. Gasquet, N. Azas, V. Mshvildadze, G. Dekanosidze, E. Kemertelidze and P. Timon-David, Planta Med., 2000, 66, 343–347 CrossRef CAS PubMed.
- M. C. Duarte, G. S. Tavares, D. G. Valadares, D. P. Lage, T. G. Ribeiro, L. M. Lage, M. R. Rodrigues, A. A. Faraco, M. Soto, E. S. da Silva, M. A. Chavez Fumagalli, C. A. Tavares, J. P. Leite, J. S. Oliveira, R. O. Castilho and E. A. Coelho, Exp. Parasitol., 2016, 166, 21–28 CrossRef CAS PubMed.
- A. Dutta, A. Ghoshal, D. Mandal, N. B. Mondal, S. Banerjee, N. P. Sahu and C. Mandal, J. Med. Microbiol., 2007, 56, 1196–1204 CrossRef CAS PubMed.
- K. Foubert, T. Gorella, A. Faizal, P. Cos, L. Maes, S. Apers, D. Geelen and L. Pieters, Planta Med., 2016, 82, 1568–1575 CrossRef CAS PubMed.
- N. Germonprez, L. Maes, L. Van Puyvelde, M. Van Tri, D. A. Tuan and N. De Kimpe, J. Med. Chem., 2005, 48, 32–37 CrossRef CAS PubMed.
- L. Maes, N. Germonprez, L. Quirijnen, L. Van Puyvelde, P. Cos and D. Vanden Berghe, Antimicrob. Agents Chemother., 2004, 48, 2056–2060 CrossRef CAS PubMed.
- L. Maes, D. Vanden Berghe, N. Germonprez, L. Quirijnen, P. Cos, N. De Kimpe and L. Van Puyvelde, Antimicrob. Agents Chemother., 2004, 48, 130–136 CrossRef CAS PubMed.
- B. Majester-Savornin, R. Elias, A. M. Diaz-Lanza, G. Balansard, M. Gasquet and F. Delmas, Planta Med., 1991, 57, 260–262 CrossRef CAS PubMed.
- D. Mandal, N. Panda, S. Kumar, S. Banerjee, N. B. Mandal and N. P. Sahu, Phytochemistry, 2006, 67, 183–190 CrossRef CAS PubMed.
- S. M. Mohamed, E. Y. Bachkeet, S. A. Bayoumi, S. Jain, S. J. Cutler, B. L. Tekwani and S. A. Ross, Fitoterapia, 2015, 107, 114–121 CrossRef CAS PubMed.
- A. L. Moreira, D. B. Scariot, B. L. Pelegrini, G. L. Pessini, T. Ueda-Nakamura, C. V. Nakamura and I. C. P. Ferreira, Evid. Based Complement. Alternat. Med., 2017, 2017, 5620693 Search PubMed.
- H. A. Oketch-Rabah, S. F. Dossaji, S. B. Christensen, K. Frydenvang, E. Lemmich, C. Cornett, C. E. Olsen, M. Chen, A. Kharazmi and T. Theander, J. Nat. Prod., 1997, 60, 1017–1022 CrossRef CAS PubMed.
- M. Ozipek, A. A. Donmez, I. Calis, R. Brun, P. Ruedi and D. Tasdemir, Phytochemistry, 2005, 66, 1168–1173 CrossRef PubMed.
- F. Rezaee, B. Zolfaghari and M. S. Dinani, Res. Pharm. Sci., 2018, 13, 469–475 CrossRef PubMed.
- O. Ridoux, C. Di Giorgio, F. Delmas, R. Elias, V. Mshvildadze, G. Dekanosidze, E. Kemertelidze, G. Balansard and P. Timon-David, Phytother. Res., 2001, 15, 298–301 CrossRef CAS PubMed.
- H. Van de Ven, M. Vermeersch, A. Matheeussen, J. Vandervoort, W. Weyenberg, S. Apers, P. Cos, L. Maes and A. Ludwig, Int. J. Pharm., 2011, 420, 122–132 CrossRef CAS PubMed.
- A. Gepdiremen, V. Mshvildadze, H. Suleyman and R. Elias, Phytomedicine, 2005, 12, 440–444 CrossRef CAS PubMed.
- Y. Lutsenko, W. Bylka, I. Matlawska, O. Gavrylyuk, M. Dworacka, A. Krawczyk and V. Brytska, Acta Pol. Pharm., 2017, 74, 1159–1166 CAS.
- A. Rai, Indian J. Pharm. Sci., 2013, 75, 99–102 CrossRef PubMed.
- A. B. Hocaoglu, O. Karaman, D. O. Erge, G. Erbil, O. Yilmaz, B. Kivcak, H. A. Bagriyanik and N. Uzuner, Iran. J. Allergy, Asthma Immunol., 2012, 11, 316–323 Search PubMed.
- S. Zeil, U. Schwanebeck and C. Vogelberg, Phytomedicine, 2014, 21, 1216–1220 CrossRef CAS PubMed.
- M. Stauss-Grabo, S. Atiye, A. Warnke, R. S. Wedemeyer, F. Donath and H. H. Blume, Phytomedicine, 2011, 18, 433–436 CrossRef CAS PubMed.
- J. Song, S. G. Yeo, E. H. Hong, B. R. Lee, J. W. Kim, J. Kim, H. Jeong, Y. Kwon, H. Kim, S. Lee, J. H. Park and H. J. Ko, Biomol. Ther., 2014, 22, 41–46 CrossRef CAS PubMed.
- J. Wang, H. Deng, J. Zhang, D. Wu, J. Li, J. Ma and W. Dong, Phytother. Res., 2020, 34, 601–611 CrossRef CAS PubMed.
- T. A. Prescott, L. P. Rigby, N. C. Veitch and M. S. Simmonds, Phytochemistry, 2014, 101, 116–120 CrossRef CAS PubMed.
- O. Rosca-Casian, C. Mircea, L. Vlase, A. M. Gheldiu, D. T. Teuca and M. Parvu, Acta Biol. Hung., 2017, 68, 196–207 CrossRef CAS PubMed.
- H. Hooshyar, S. Talari and F. Feyzi, Jundishapur J. Microbiol., 2014, 7, e9432 Search PubMed.
- M. S. Baird and D. Preskett, European Pat., EP2234479, 2007 Search PubMed.
- J. Sun, X. Han and B. Yu, Org. Lett., 2005, 7, 1935–1938 CrossRef CAS PubMed.
- G. S. R. Subba Rao, P. Kondaiah, S. K. Singh, P. Ravanan and M. B. Sporn, Tetrahedron, 2008, 64, 11541–11548 CrossRef CAS PubMed.
- X.-C. Li, C.-R. Yang, Y.-Q. Liu, R. Kasai, K. Ohtani, K. Yamasaki, K. Miyahara and K. Shingu, Phytochemistry, 1995, 39, 1175–1179 CrossRef CAS.
- D. Rouzaud and P. Sinaÿ, J. Chem. Soc., Chem. Commun., 1983, 1353–1354, 10.1039/C39830001353.
- O. Cortezano-Arellano, C. A. Meléndez-Becerra, F. Cortés, F. Sartillo-Piscil and A. Cordero-Vargas, Carbohydr. Res., 2014, 393, 51–59 CrossRef CAS PubMed.
- H. I. Ciftci, S. E. Ozturk, T. F. S. Ali, M. O. Radwan, H. Tateishi, R. Koga, Z. Ocak, M. Can, M. Otsuka and M. Fujita, Biol. Pharm. Bull., 2018, 41, 570–574 CrossRef CAS PubMed.
- F. L. Chadbourne, C. Raleigh, H. Z. Ali, P. W. Denny and S. L. Cobb, J. Pept. Sci., 2011, 17, 751–755 CrossRef CAS PubMed.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- H. L. Bolt, G. A. Eggimann, P. W. Denny and S. L. Cobb, MedChemComm, 2016, 7, 799–805 RSC.
- S. L. Cobb and P. W. Denny, Curr. Opin. Invest. Drugs, 2010, 11, 868–875 CAS.
- G. A. Eggimann, H. L. Bolt, P. W. Denny and S. L. Cobb, ChemMedChem, 2015, 10, 233–237 CrossRef CAS PubMed.
- G. A. Eggimann, K. Sweeney, H. L. Bolt, N. Rozatian, S. L. Cobb and P. W. Denny, Molecules, 2015, 20, 2775–2785 CrossRef PubMed.
- L. M. Alcantara, T. C. S. Ferreira, F. R. Gadelha and D. C. Miguel, Int. J. Parasitol.: Drugs Drug Resist., 2018, 8, 430–439 Search PubMed.
- M. De Rycker, I. Hallyburton, J. Thomas, L. Campbell, S. Wyllie, D. Joshi, S. Cameron, I. H. Gilbert, P. G. Wyatt, J. A. Frearson, A. H. Fairlamb and D. W. Gray, Antimicrob. Agents Chemother., 2013, 57, 2913–2922 CrossRef CAS PubMed.
- M. Wang, Z. Zhan, Y. Xiong, Y. Zhang and X. Li, Fitoterapia, 2019, 139, 104360 CrossRef CAS PubMed.
- S. Emirdağ-Öztürk, T. Karayıldırım, A. Çapcı-Karagöz, Ö. Alankuş-Çalışkan, A. Özmen and E. Poyrazoğlu-Çoban, Eur. J. Med. Chem., 2014, 82, 565–573 CrossRef PubMed.
- M. C. Jamur and C. Oliver, Methods Mol. Biol., 2010, 588, 63–66 CrossRef PubMed.
- I. Pena, M. P. Manzano, J. Cantizani, A. Kessler, J. Alonso-Padilla, A. I. Bardera, E. Alvarez, A. Rodriquez, D. W. Gray, M. Navarro, V. Kumar, A. Shaerstnev, D. H. Drewry, J. R. Brown, J. M. Fiandor and J. J. Martin, Sci. Rep., 2015, 5, 8771 CrossRef CAS PubMed.
- B. Radek, C. Wehrli, D. Preskett, M. E. Bouillon, S. Duval, M. E. Ramos, J. R. Al-dulayymi, C. J. Newbold, S. W. Strawson, M. Baird and M. Lahmann, Patent, WO2016193309A1, 2016 Search PubMed.
- R. Ma, Q. Shi, R. Zhou, Q. Zhou and J. Huang, Patent, WO2013174245A1, 2012 Search PubMed.
- C. B. de Mattos, D. F. Argenta, L. Melchiades Gde, M. N. Cordeiro, M. L. Tonini, M. H. Moraes, T. B. Weber, S. S. Roman, R. J. Nunes, H. F. Teixeira, M. Steindel and L. S. Koester, Int. J. Nanomed., 2015, 10, 5529–5542 Search PubMed.
- E. C. Torres-Santos, J. M. Rodrigues, Jr., D. L. Moreira, M. A. Kaplan and B. Rossi-Bergmann, Antimicrob. Agents Chemother., 1999, 43, 1776–1778 CrossRef CAS PubMed.
- D. O. Escrivani, M. V. Lopes, F. Poletto, S. R. Ferrarini, A. J. Sousa-Batista, P. G. Steel, S. S. Guterres, A. R. Pohlmann and B. Rossi-Bergmann, Nanomedicine, 2020, 24, 102121 CrossRef CAS PubMed.
- P. W. Denny, Expert Opin. Drug Discovery, 2018, 13, 1153–1160 CrossRef CAS PubMed.
- P. W. Denny, Parasitology, 2018, 145, 111–115 CrossRef PubMed.
- P. W. Denny and P. G. Steel, J. Biomol. Screening, 2015, 20, 56–63 CrossRef CAS PubMed , [pii].
- J. L. Norcliffe, E. Alvarez-Ruiz, J. J. Martin-Plaza, P. G. Steel and P. W. Denny, Parasitology, 2014, 141, 8–16 CrossRef CAS PubMed.
- E. G. Armitage, A. Q. I. Alqaisi, J. Godzien, I. Pena, A. J. Mbekeani, V. Alonso-Herranz, A. Lopez-Gonzalvez, J. Martin, R. Gabarro, P. W. Denny, M. P. Barrett and C. Barbas, Antimicrob. Agents Chemother., 2018, 62, e02095–e02117 CrossRef CAS PubMed.
- R. Mwenechanya, J. Kovarova, N. J. Dickens, M. Mudaliar, P. Herzyk, I. M. Vincent, S. K. Weidt, K. E. Burgess, R. J. S. Burchmore, A. W. Pountain, T. K. Smith, D. J. Creek, D. H. Kim, G. I. Lepesheva and M. P. Barrett, PLoS Neglected Trop. Dis., 2017, 11, e0005649 CrossRef PubMed.
- A. W. Pountain, S. K. Weidt, C. Regnault, P. A. Bates, A. M. Donachie, N. J. Dickens and M. P. Barrett, PLoS Neglected Trop. Dis., 2019, 13, e0007052 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00123f |
| ‡ Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |