
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefeating the trypanosomatid trio: proteomics of the protozoan parasites causing neglected tropical diseases
Anutthaman
Parthasarathy
 a and
Karunakaran
Kalesh
*b
a and
Karunakaran
Kalesh
*b
aRochester Institute of Technology, Thomas H. Gosnell School of Life Sciences, 85 Lomb Memorial Dr, Rochester, NY 14623, USA
bDepartment of Chemistry, Durham University, Lower Mount Joy, South Road, Durham DH1 3LE, UK. E-mail: kalesh.karunakaran@durham.ac.uk
First published on 22nd May 2020
Abstract
Mass spectrometry-based proteomics enables accurate measurement of the modulations of proteins on a large scale upon perturbation and facilitates the understanding of the functional roles of proteins in biological systems. It is a particularly relevant methodology for studying Leishmania spp., Trypanosoma cruzi and Trypanosoma brucei, as the gene expression in these parasites is primarily regulated by posttranscriptional mechanisms. Large-scale proteomics studies have revealed a plethora of information regarding modulated proteins and their molecular interactions during various life processes of the protozoans, including stress adaptation, life cycle changes and interactions with the host. Important molecular processes within the parasite that regulate the activity and subcellular localisation of its proteins, including several co- and post-translational modifications, are also accurately captured by modern proteomics mass spectrometry techniques. Finally, in combination with synthetic chemistry, proteomic techniques facilitate unbiased profiling of targets and off-targets of pharmacologically active compounds in the parasites. This provides important data sets for their mechanism of action studies, thereby aiding drug development programmes.
 Anutthaman Parthasarathy | Anutthaman Parthasarathy studied Chemistry in India (IIT-Madras) and obtained his PhD from the International Max Planck Research School (IMPRS) for Microbiology in Marburg, Germany. He was a postdoctoral fellow at the MPI Marburg and at the Stanford University. After leaving Stanford, he set up and run a non-profit science centre for students in his native India. He is currently a Research Associate at the Rochester Institute of Technology in Rochester, New York, and his current interests include drug discovery, enzymology, microbial metabolism, and the interface of microbiology with chemistry and engineering. |
 Karunakaran Kalesh | Karunakaran Kalesh has an MSc in Chemistry (IIT Madras) and PhD in Chemical Biology (NUS). Following PhD, he was a senior research scientist at AMRI Singapore (2010–2012) and then a Marie Curie Fellow (2012–2014) at Imperial College London. He was an MRC postdoctoral fellow (2014–2015) and a GSK-Imperial College London Engineered Medicines Laboratory joint postdoctoral fellow (2015–2018) before beginning his independent career at Durham University UK in 2019 as an Assistant Professor (Research). His research focusses on developing and applying chemical biology and mass spectrometry-based proteomics methods for identifying and validating new drug targets for leishmaniasis and Chagas disease. |
1. Introduction
The protozoan parasites Leishmania, Trypanosoma cruzi and Trypanosoma brucei, collectively known as the Tritryps, are the etiological agents of the neglected tropical diseases (NTDs) leishmaniasis, Chagas disease and human African trypanosomiasis, respectively.1 Collectively, these three diseases account for the highest rates of mortality amongst all NTDs.2 The World Health Organisation (WHO) lists these NTDs in a group of high priority diseases that require “innovative and intensified disease management (IDM)” on the basis of several factors including poorly understood disease burden, high management cost, lack of appropriate control methods and relatively low investment in research and development.2 Nevertheless, a 90% reduction in the number of African trypanosomiasis cases has been noticed between 2009 and 2018 thanks to the coordinated efforts by control programmes in endemic countries.3 However, a jump of over 26% in leishmaniasis prevalence has been noticed between 2006 and 2016.4 NTDs caused by protozoans continue to be a growing challenge since there is a shortfall in funding and development in this field.5,6 Current treatment options, specifically in the case of leishmaniasis and Chagas disease, are severely limited. For instance, in Chagas disease, the only two available drugs, nifurtimox and benznidazole, suffer from toxicity and limited efficacy.7,8 Similarly, in the case of visceral leishmaniasis, current front-line medications suffer from issues including high cost, teratogenicity, drug resistance, requirement of non-oral administration and prolonged treatment regimens, making treatment in economically poor endemic areas particularly difficult.9–11 There is an urgent need to develop new drugs that can address these shortcomings of the existing ones. An important step towards this goal is the identification and validation of parasite proteins that can act as novel drug targets. This is facilitated by modern proteomics studies that provide a better understanding of the parasite proteome and its functions.Despite the aforementioned difficulties, many recent developments in the “omics” technologies are gradually transforming the research landscape of NTDs caused by protozoan parasites. The Human Genome Project not only created opportunities to treat genetic diseases, but also triggered the development of a number of sequencing technologies such as ChIP-Seq and RNA-Seq, which are able to provide detailed analyses of cell function.12 In general, genomics and transcriptomic techniques allowed the accumulation of large data sets and facilitated increased understanding of the genetic aspects of diseases.13,14 Despite all this promise, they have important shortcomings in the investigation of disease states. Not all genes present in a eukaryotic cell are expressed, since protein synthesis is extensively regulated.15 mRNA levels are only an estimate of the protein abundance and do not necessarily correlate with protein expression.16 This is particularly relevant in the case of Tritryps.17 These kinetoplastids are among the most evolutionarily ancient eukaryotic lineages.18,19 However unlike higher eukaryotes, gene expression in these parasites is almost exclusively controlled by posttranscriptional mechanisms, making DNA or mRNA-based analyses unreliable for protein identification.20–22 An additional handicap is that genomic or transcriptomic profiling cannot identify posttranslational modifications (PTMs), which are often functionally critical.23 Understanding protein–protein interactions and protein localisation is essential for obtaining a more complete picture of cell function, but these issues cannot be addressed satisfactorily by DNA or mRNA-based methods in the Tritryps. Finally, DNA-based approaches cannot effectively answer questions on gene function in systems where biological information is lacking. For example, annotations of up to a half of the open reading frames (ORFs) in Leishmania spp. correspond to “hypothetical proteins”.24 Proteins are the workhorses of the cell, which manifest the end stage of regulation and drive both cellular structure and function, and are thus critical for the understanding of complex processes such as the development of diseases.
Proteomics, which is the systematic analysis of proteins, can provide a more complete picture of global functional protein expression in a cell, tissue or bodily fluid, giving a plethora of information not accessible via other methods.25–27 While early proteomics methods were developed for the characterisation of a small number of proteins, later analytical advances allowed the massive parallel detection and comparative studies of thousands of proteins in a single mass spectrometry (MS) run.28 Unlike earlier times, proteomics MS is now a genuine hypothesis generation tool in conjunction with cellular, molecular and pharmacological methods.29 Today, the investigation of protein complexes and the characterisation of protein pathways via MS methods have become commonplace. More recently, RNA interference (RNAi) has emerged as a common method to disrupt the synthesis of any protein in vivo, offering the means to test the functions of proteins identified by proteomics screens involving hundreds or thousands of candidate proteins.30 However, in the field of trypanosomatid parasitology, RNAi has only been used as a genetic tool in T. brucei. Recent proteomic methods have also enabled pathway analysis and have contributed greatly to closing the gaps in knowledge generated by gene-based methods.31 Consequently, they have facilitated the identification of biomarkers, to support both diagnostics and evidence-based drug design for disease models.32,33 The use of proteomics, however, has its own limitations, especially with respect to the reproducibility of label-free quantifications, necessitating a sufficient number (often more than four) of replications per experimental condition. Additionally, the inherent bias of the MS technique towards abundant proteins sometimes compromises sensitive detection and relative quantification of low-abundant proteins.
This review is not meant to be an exhaustive survey of the proteomics studies of the three protozoan parasites causing NTDs. Instead, this review will highlight the key role that proteomics has played as a particularly relevant technology for shedding light on several aspects of the life process such as life cycle differentiation, stress-adaptation, infectivity and drug resistance of the Tritryps. An overview is provided in Fig. 1. It will also cover important chemical proteomics approaches applied to the three protozoan parasites for the target profiling of pharmacologically active compounds and/or their mechanism of action studies as well as the global profiling and validation of therapeutically relevant PTMs in them.
 | ||
| Fig. 1 Overview of proteomics approaches in Tritryps. The proteome of each life cycle stage of a protozoan parasite of interest (Leishmania spp., Trypanosoma cruzi and Trypanosoma brucei) undergoes characteristic modulations as the parasite responds to a perturbation trigger. The perturbation trigger can either be a natural factor that regulates the development of the parasite (such as changes in pH or temperature, variations in availability of nutrients, and other environmental changes during its transition to a different host species) or a therapeutic intervention such as an inhibitor/growth modulator treatment or a chemical probe treatment. The proteins extracted from a specific life cycle stage of the protozoan parasite of interest following the perturbation window are typically digested to peptides using a protease such as trypsin. The proteomic samples often require additional treatments such as reduction of disulphide bonds, alkylation of free thiols and extensive clean-up or desalting procedures. The relative expression changes in several hundreds to thousands of proteins across different conditions are then measured with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) technique following a suitable quantitative proteomics approach such as label-free quantification or one of the different types of label-based quantification methods. Processing, analyses and visualisation of the large proteomics data sets are carried out using dedicated software programmes. | ||
2. Proteomics approaches in Leishmania
2.1 Leishmaniasis: an introduction
Leishmaniasis is a complex disease caused by about 20 species of protozoan parasites of the genus Leishmania,34 which is transmitted to humans via the bites of blood-sucking sandflies. About 200 million people in over 90 countries in Asia, Africa, Latin America and southern Europe live at the risk of exposure to the parasite,35 with about 2 million new cases of leishmaniasis and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 deaths annually.34 Leishmaniasis affects mainly people experiencing poverty, malnutrition, immune suppression, and pre-existing health issues or people living in deforested areas undergoing rapid urbanisation.34 The disease has three forms: cutaneous, muco-cutaneous and visceral. While the other two forms are persistent and disfiguring, visceral leishmaniasis causes significant mortality and morbidity.35 Current Leishmania therapies are largely inadequate. Only one antileishmanial compound, miltefosine, is available for oral administration. The drug is, however, a known teratogen and therefore unsuitable for use in pregnant women.36 Other frontline treatments such as amphotericin B (and its liposomal formulations) and paromomycin require prolonged treatment regimens and are unsuitable for administration in resource-limited endemic areas.11,37 The old antimonial drugs suffer from widespread resistance and are notorious for their toxicity.38 In general, all currently available antileishmanials suffer from moderate to severe toxicity, resistance and/or clinical relapse.10 Currently there are no new therapeutics for leishmaniasis in clinical development, and only a few in the preclinical stages. Alarmingly, not only is there a scarcity of validated molecular targets for drug development, but also a lack of knowledge on the mechanisms of action of existing molecules approved for clinical use. There is thus a dire need for new treatments, particularly oral drugs, for visceral leishmaniasis.
000–50000 deaths annually.34 Leishmaniasis affects mainly people experiencing poverty, malnutrition, immune suppression, and pre-existing health issues or people living in deforested areas undergoing rapid urbanisation.34 The disease has three forms: cutaneous, muco-cutaneous and visceral. While the other two forms are persistent and disfiguring, visceral leishmaniasis causes significant mortality and morbidity.35 Current Leishmania therapies are largely inadequate. Only one antileishmanial compound, miltefosine, is available for oral administration. The drug is, however, a known teratogen and therefore unsuitable for use in pregnant women.36 Other frontline treatments such as amphotericin B (and its liposomal formulations) and paromomycin require prolonged treatment regimens and are unsuitable for administration in resource-limited endemic areas.11,37 The old antimonial drugs suffer from widespread resistance and are notorious for their toxicity.38 In general, all currently available antileishmanials suffer from moderate to severe toxicity, resistance and/or clinical relapse.10 Currently there are no new therapeutics for leishmaniasis in clinical development, and only a few in the preclinical stages. Alarmingly, not only is there a scarcity of validated molecular targets for drug development, but also a lack of knowledge on the mechanisms of action of existing molecules approved for clinical use. There is thus a dire need for new treatments, particularly oral drugs, for visceral leishmaniasis.
2.2 The Leishmania life cycle
Leishmania spp. have a digenetic life cycle that alternates between intracellular amastigotes in the vertebrate host and multiple extracellular promastigote stages in the blood-sucking sandfly vector. The growth cycle of the parasite in the sandfly begins with weakly motile but highly proliferative procyclic promastigotes in the midgut and culminates in the non-dividing but strongly motile and infectious metacyclic promastigotes. Once inside the vertebrate host, the metacyclic promastigotes transform into non-motile amastigotes, which replicate predominantly inside the immune cells, especially macrophages. Amastigote proliferation eventually leads to host cell rupture, allowing entry into the host's blood stream and infection of other phagocytes. The amastigotes in the host's bloodstream can be taken up by another sandfly during its blood meal. In this case, the amastigotes transform into procyclic promastigotes in the sandfly's midgut, multiply by binary fission and continue the life cycle.2.3 Tracking variations in the parasite proteome for the molecular characterisation of key life processes
In the early days of proteomics, the technique of choice for proteome resolution was two-dimensional gel electrophoresis (2-DE). This method allows separation of proteins in a cell lysate according to their isoelectric points and molecular masses. The resolved proteins are typically visualised by protein staining, for instance by silver staining or by Coomassie blue dye. The first report of Leishmania proteomics employed 2-DE for revealing distinct protein patterns between different L. tropica isolates causing simple cutaneous leishmaniasis and those causing leishmaniasis recidiva, a rare variant of cutaneous leishmaniasis.39 Later, in order to gain insights into the molecular basis of the parasite virulence in cutaneous leishmaniasis, protein patterns of radiolabelled clones of infective and non-infective types generated from a single isolate of L. tropica were compared.40 Although the observed protein patterns of the different clones showed only minor differences, which could partly be attributed to the limited sensitivity of the techniques employed, this study showed the scope of using protein profile differences to classify the infective and non-infective Leishmania isolates. 2-DE proteome patterns have also been demonstrated to be useful for highlighting the differences between two different species of New World Leishmania (L. mexicana and L. braziliensis) and for establishing relatedness between subspecies.41By the 1980s, the combination of 2-DE with MS gained popularity. Several groups employed this combination for gaining insights into the molecular mechanisms of Leishmania differentiation. Nugent et al. used 2-DE to resolve the proteome of axenically differentiated L. mexicana parasites and observed about 2000 protein spots in the different life cycle stages of the parasite.42 Several spots were found to be life cycle-specific with the majority of unique spots showing expression in the infective metacyclic and amastigote stages. MS analyses of selected excised gel spots led to the identification of 47 parasite proteins. Intriguing findings include highly upregulated and preferential expression of proteins involved in protein synthesis such as eukaryotic translation elongation factor-1 alpha (eIF-1α), eukaryotic initiation factor-5 alpha (eIF-5α) and 40S ribosomal protein S2 after differentiation, as well as the exclusive detection of certain heat-shock proteins (HSPs) (e.g. HSP60 and HSP70) in the amastigotes.
As only the metacyclic form of the parasite infects vertebrate hosts, some proteomics studies have focussed on identifying the proteins that are differentially expressed in this stage. Mojtahedi et al. employed the 2-DE–MS combination for comparing the proteomes of L. major procyclics and metacyclics.43 Consistent with the high motility and arrested growth of the metacyclic parasites, they observed an increased abundance of several proteins that facilitate parasite motility, whilst the abundance of many proteins involved in protein synthesis decreased. In a more recent study, Moreira et al. applied a liquid-based free-flow electrophoresis method for resolving the proteome of L. infantum wild-type and a pteridine reductase 1 (PTR1) null-mutant in the metacyclic and procyclic stages.44 The PTR1 null-mutant presented reduced levels of intracellular tetrahydrobiopterin, an important growth factor for Leishmania. A reduced level of pterins has been proposed as one of the factors facilitating the metacyclogenesis. MS proteomics revealed significantly altered expression of several structural proteins and metabolic enzymes between the two developmental stages in the contexts of both reduced and normal pterin levels.
Many important molecular processes in the Leishmania life cycle were revealed thanks to the development of axenic promastigote cultures of the parasite. However, long-term maintenance of the parasites in host-free cultures has been shown to affect the infectivity and virulence.45 Nevertheless, Leishmania axenic cultures also provide a convenient means to generate drug resistant strains in the laboratory.46 Intuitively, the comparative proteomic study of a resistant strain against its non-resistant counterpart could reveal proteins associated with drug resistance. Walker et al. applied this concept for deconvoluting the molecular factors associated with resistance to trivalent antimony treatment in L. panamensis strains that were made resistant to the drug in cultures.47 They have identified nine differentially expressed proteins, which include stress-responsive proteins and important metabolic enzymes S-adenosylmethionine synthetase (SAMS) and S-adenosylhomocysteine hydrolase (SAHH). Both SAMS and SAHH play key roles in the pathways upstream of the biosynthesis of trypanothione, a unique thiol metabolite of Kinetoplastida that is implicated in antimony resistance and the parasite's response to oxidative stress.48,49
2.4 Application of quantitative proteomics MS in the study of Leishmania parasites
Conventional 2-DE is now obsolete, with quantitative proteomics MS techniques offering a far better sensitivity of detection, the capability for high-throughput whole proteome analysis in a single MS run and the quantitative comparison of multiple experimental conditions in a single experiment. Quantitative proteomics MS techniques are broadly divided into label-free quantification (LFQ) methods, such as MS1-intensity based methods50 and spectral counting-based methods,51 and several different labelling-based methods. Popular labelling strategies include TMT (tandem mass tag),52 iTRAQ (isobaric tagging for relative and absolute quantitation),53 SILAC (stable isotope labelling by amino acids in cell culture)54 and stable isotope dimethyl labelling.55Proteomics analyses of biological membranes is an underrepresented area even though membrane proteins constitute about 30% of a typical proteome.60 This is because many membrane proteins tend to aggregate and precipitate in the solution, making sample preparation notoriously difficult. However, several membrane proteins have been implicated in crucial roles in Leishmania–host interactions.61 Lynn et al. isolated the membrane proteins of promastigote and amastigote stages of L. mexicana and L. infantum parasites using Percoll density gradients and profiled the differentially expressed proteins using iTRAQ quantitative proteomics MS.62 They identified 107 L. mexicana and 189 L. infantum non-redundant proteins, of which 20–40% showed significantly different abundance levels between the two life cycle stages.
2.4.3.1 Target fishing and mode of action studies. Deconvoluting the mode of action of a pharmacologically active chemical entity is a daunting task in drug discovery programmes. A combination of complementary methods is often required to identify the real target(s) and mode of action. Wyllie et al. recently employed quantitative chemical proteomics MS and in vitro evolution combined with whole-genome analysis67 to identify cyclin dependent kinase 12 (CDK12) (also known as cdc-2-related kinase 12 or CRK12) as the principal molecular target of their preclinical pyrazolopyrimidine compound DDD853651 or GSK3186899 (compound 1 in Fig. 2) for visceral leishmaniasis.68 They first developed a series of analogues (compounds 4–7, Fig. 2) of DDD853651 with a polyethylene glycol (PEG) linker, which after attaching to magnetic beads were used for competitive chemical proteomic pulldown experiments. With compound 4 immobilised on beads, SILAC “light” and “heavy” labelled L. donovani promastigote lysates were used for fishing out the protein binders in the presence and absence respectively of the structurally related competitive inhibitor compound 5. This quantitative proteomics MS experiment identified CDK12 along with several other cyclin-dependent kinases and a few other proteins as selective binders of compound 4. A second round of competitive chemical proteomics experiments using the more potent inhibitors 2 and 3 as competitive binders, and beads derivatised with compounds 4, 6 and 7 also revealed a more or less similar set of selective binders. They subsequently generated drug resistant strains of the parasites and performed whole-genome sequencing, which identified a key mutation in the catalytic site of the CDK12 protein. Additionally, CDK12 overexpressing parasite strains showed increased sensitivity to the compound, indicating that CDK12 is indeed the principal target of the pyrazolopyrimidine compound. In this case, the chemical proteomics alone was not sufficient to identify the major target of the compound. Nevertheless, it helped to narrow down the likely candidates to a small set of proteins to permit subsequent genetic target validation studies.
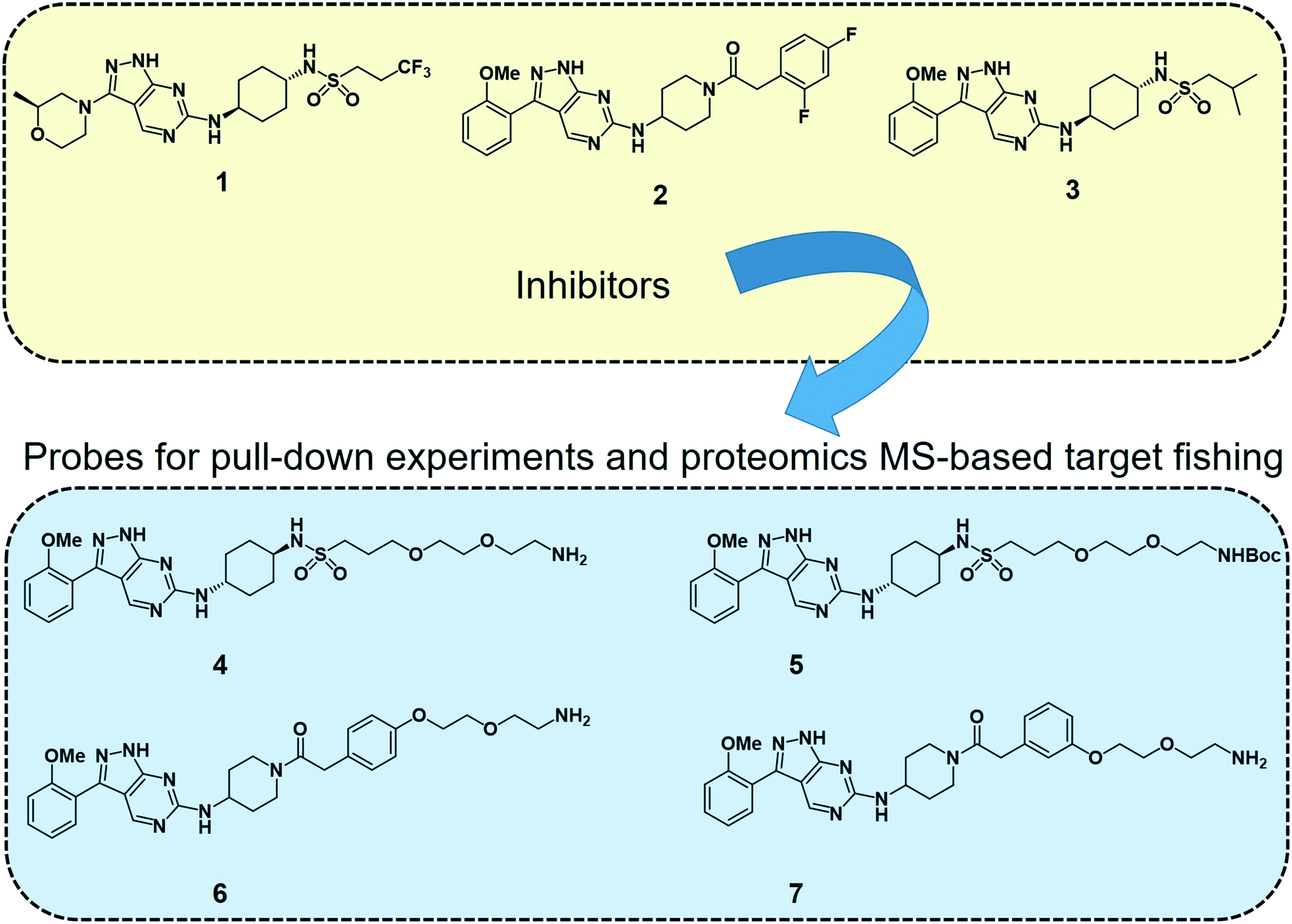 | ||
| Fig. 2 Leishmania CDK12 inhibitors and chemical probes from a pyrazolopyrimidine series used in the proteomics MS-based target profiling and mode of action studies.68 Compound 1 (known as DDD853651 or GSK3186899) is a preclinical developmental compound against visceral leishmaniasis. 2 is another potent compound with an aminopiperidine amide functionality. Compound 3 carries an isobutyl group and 2-methoxyphenyl group in place of the trifluoropropyl and 2-methylmorpholine groups in compound 1. Compounds 4 to 7 are structurally related pyrazolopyrimidines with polyethylene glycol linkers. The primary amino groups in 4, 6 and 7 were used for immobilising the compounds on magnetic beads, facilitating affinity enrichment of the protein binders of the compounds from SILAC-labelled parasite cell lysates, whereas compounds 5, 2 and 3 were used as in-solution competitive binders of the proteins in the affinity-enrichment experiments. | ||
Thermal proteome profiling (TPP) and the related cellular thermal shift assay (CETSA) are complementary to the chemical probe-based target deconvolution methods.69–71 These are based on the principle that binding of a ligand to a protein target often increases the melting temperature (Tm) of that protein. Therefore, when a drug-treated whole-proteome is heated, unbound proteins may denature and precipitate while the ligand-bound protein(s) remain in solution. Typically, samples are treated to different temperatures and the soluble portion of the proteome from each temperature treatment is analysed by LC-MS/MS. It is often used in combination with a quantitative proteomics MS method such as TMT labelling, which permits the defining of the aliquot from each temperature treatment using a specific isobaric label. The target engagement of an N-myristoyltransferase (NMT) inhibitor compound termed DDD1000097 in the L. donovani promastigote proteome was recently unravelled using the TPP method.72 Corpas-Lopez et al. treated L. donovani promastigote lysates with DDD1000097 and the soluble proteome from the aliquots heated to different temperatures was treated with TMT-10plex labels.72 DMSO treatment instead of the inhibitor treatment was used as a control. The data revealed the L. donovani NMT protein as the most significant hit, with a 3.3 °C increase in melting temperature (ΔTm) of the protein following the inhibitor binding. Although currently limited to soluble proteins, TPP and CETSA are useful proteomics methods for the target deconvolution of non-covalent binders.
2.4.3.2 Global profiling of substrates of N-myristoyltransferase in Leishmania. The enzyme NMT that catalyses N-terminal myristoylation of glycine residues in proteins is one of the few solidly validated druggable targets in Leishmania.73 By using a terminal alkyne analogue of myristic acid, Wright et al. metabolically labelled the N-myristoylome of L. donovani promastigotes and axenic amastigotes.74 Affinity enrichment of the alkyne-modified parasite proteins using a multifunctional capture reagent exploiting the copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) click reaction75 enabled LFQ proteomics MS profiling of the parasite N-myristoylome (Fig. 3A). They identified 113 and 49 myristoylated proteins in the promastigotes and amastigotes, respectively. Intriguingly, the majority of the identified proteins did not contain the essential N-terminal glycine residue for N-myristoylation, indicating that some of the observed modifications may have a non-enzymatic origin and (or) that enzymes other than NMT may have contributed to the observed labelling. Therefore, in order to identify the specific NMT substrates in the parasites, the researchers applied the metabolic labelling approach in combination with a chemical knockdown of the NMT protein using inhibitors. Although pre-incubation of the parasites with the NMT inhibitors led to a reduction in the fluorescent labelling, the LFQ proteomics experiments in amastigotes did not reveal a uniform reduction of myristoylation upon inhibitor treatment. However, a subset of the enriched proteome (30 proteins in this study) quantitatively responded to the NMT inhibitors. These proteins all contain the N-terminal glycine residue, indicating that they are substrates of the NMT enzyme in the parasite. This highlights the power of chemical proteomics for characterising the true substrates of an enzyme of potential therapeutic relevance in the parasite.
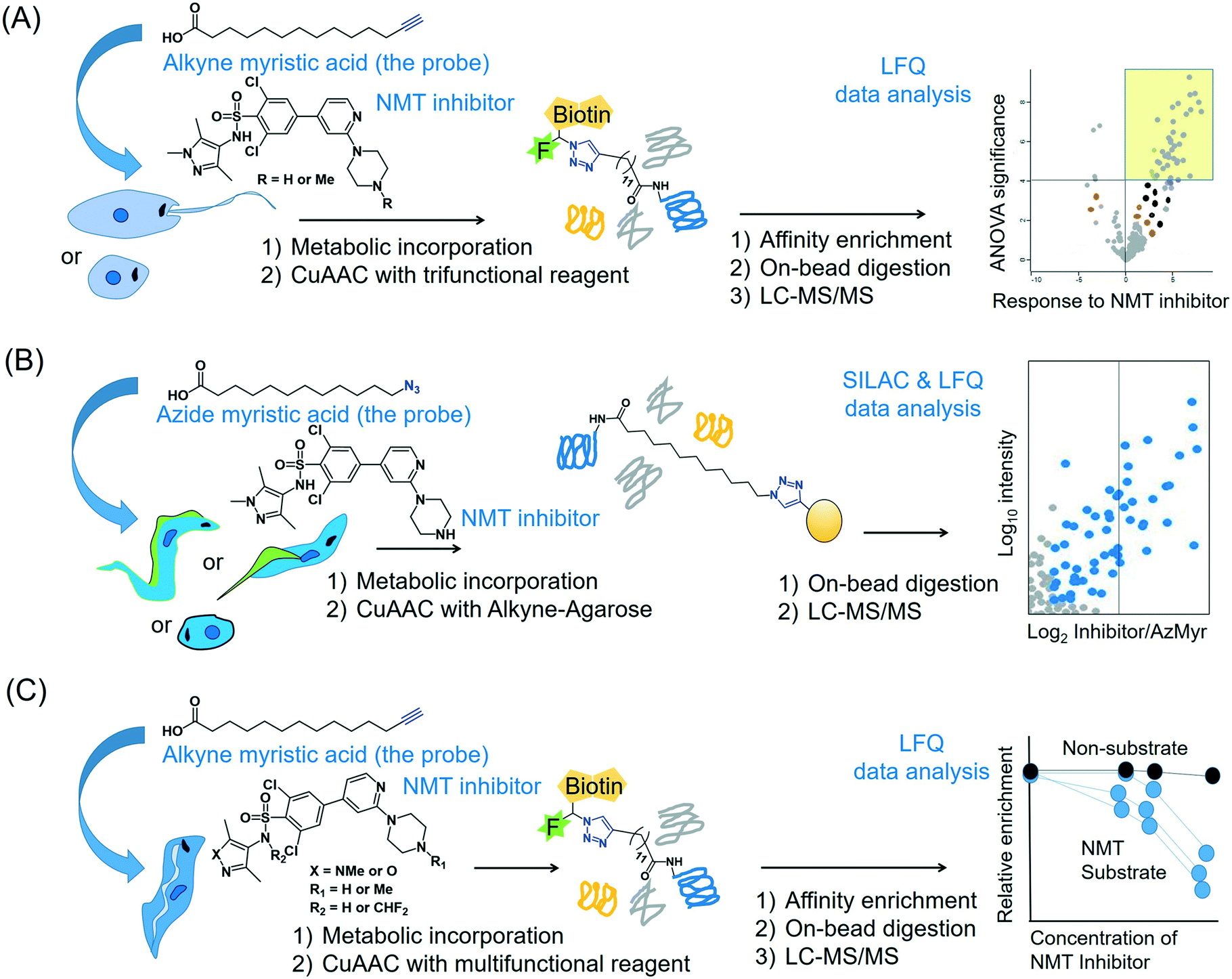 | ||
| Fig. 3 Proteomics MS profiling of substrates of N-myristoyltransferase in (A) Leishmania donovani (promastigotes and amastigotes),74 (B) Trypanosoma cruzi (epimastigotes, trypomastigotes and amastigotes)141 and (C) Trypanosoma brucei (bloodstream form).193 | ||
2.4.3.3 Quantitative profiling of de novo protein synthesis in Leishmania during starvation. Many studies point to the adaptation of Leishmania parasites to nutrient scarcity as playing potentially important roles in the differentiation process.76–78 A method that could accurately and quantitatively profile protein synthesis in the parasite during a period of severe nutrient scarcity holds great potential for elucidating the molecular processes facilitating adaptation to such conditions. Towards this goal, we have recently developed a methodology that combined the BONCAT (bio-orthogonal non-canonical amino acid tagging) technology79 with iTRAQ quantitative proteomics MS in starving L. mexicana promastigotes.80 In this approach, the methionine surrogate, azidohomoalanine (AHA), was metabolically incorporated into newly synthesised proteins (NSPs) of the parasites subjected to nutrient deprivation for different durations. Following cell lysis, the whole cell proteome from each starvation window was separately subjected to click chemistry using a biotin–azide capture reagent, which enabled the selective tagging of the NSPs with biotin. The biotinylated NSPs were affinity-enriched on streptavidin beads, and after stringent washings, the bound proteins were subjected to on-bead tryptic digestion. The tryptic peptides from different starvation windows were labelled with different iTRAQ labels and following the completion of the labelling reactions, the samples were pooled together and analysed by LC-MS/MS (Fig. 4). The multiplexed iTRAQ approach enabled temporally resolved quantitative profiling of over 250 starvation-specific NSPs in the parasite. This study revealed a starvation-specific increase in the abundance of several translation-regulating and stress-responsive proteins, pointing to their important roles in the starvation-adaptation of the parasite. Besides, this study provides a robust methodology for analysing protein synthesis in the parasite in a quantitative and temporally resolved manner.
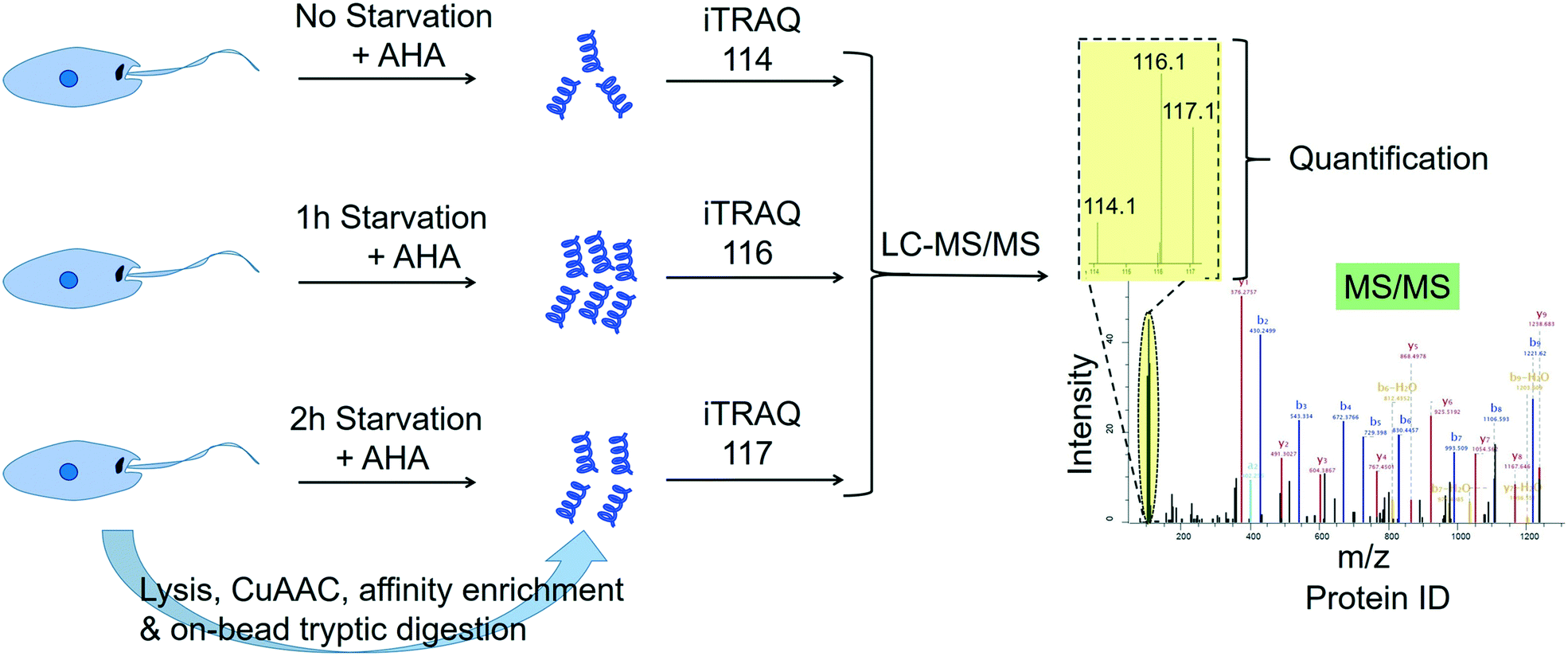 | ||
| Fig. 4 Temporally resolved quantitative proteomics MS profiling of newly synthesised proteins in starving Leishmania mexicana parasites using a BONCAT-click chemistry affinity enrichment-iTRAQ combination.80 | ||
3. Proteomics approaches in Trypanosoma cruzi
3.1 Chagas disease: an introduction
Chagas disease (American trypanosomiasis) is caused by Trypanosoma cruzi (T. cruzi) and is endemic in Latin America, with a wide distribution of cases from the southern US to northern Argentina.81 It is transmitted to humans via blood-sucking triatomine bugs (or “kissing bugs”) and also spreads by migration and blood transfusion.81 Early symptoms may include fever, headaches, swelling at the bite site and lymph node swelling. About one-quarter to a third of patients may develop chronic infections. Some patients develop enlargement of the heart ventricles, oesophagus or colon and digestive or neurological complications up to 30 years after the initial infection.82 Chemotherapy to treat Chagas disease is reliant mainly on benznidazole (Bzn) and nifurtimox (Nx), which were discovered over 40 years ago. They are efficient in treating the acute phase, but of limited use in countering the chronic phase. Both drugs have substantial side effects, need long-term administration and are ineffective against diverse strains, with certain strains being resistant to Bzn.83,84 Thus, improved drugs, especially for the chronic stages, are an urgent need. To make matters worse, only a few compounds are in the clinical assay stage, although diverse natural and synthetic compounds have in vivo and in vitro activity.3.2 The T. cruzi life cycle
In the bloodstream of infected vertebrate hosts, T. cruzi exists in a non-replicative trypomastigote form. When a triatomine vector takes a blood meal from the infected host, the T. cruzi in the blood meal transforms into the epimastigote form inside the bug's midgut. The parasite then travels to the bug's hindgut where it differentiates into infective metacyclic trypomastigotes. When the infected bug takes a blood meal again, it releases these infective trypomastigotes in its faeces near the bite site. Once the trypomastigotes enter the vertebrate host's cell, they differentiate into intracellular amastigotes, which replicate by binary fission. The amastigotes further differentiate into trypomastigotes and enter the bloodstream. T. cruzi is considered a heterogeneous species with a variety of strains in different vectors and mammalian hosts, leading to a high variability in virulence.853.3 Characterising the life cycle stages of T. cruzi
The proteome mapping of T. cruzi has enabled the description of stage-specific protein expression and led to the discovery of some proteins responsible for drug resistance.86–88 In the trypomastigote stage, the parasite relies more on glycolysis than the mitochondrial electron transport chain to meet the energy demand.89 Epimastigotes of the CL Brener reference strain not only had elevated levels of tubulin and heat shock proteins, which were present in multiple isoforms, but also expressed arginine kinase, an enzyme not found in mammals, thus presenting a potentially novel therapeutic target.90 Differential proteomic analysis led to the identification of 22 proteins, mostly metabolic proteins of the glycolytic pathway (enolase, pyruvate kinase and 2,3-bisphosphoglycerate mutase), along with cytoskeletal structural proteins, polypeptide synthesis and chaperones/heat shock proteins.91 Antioxidant defence enzymes were implicated in drug detoxification after prostaglandin F2α synthase was identified as one of the proteins.92 The shotgun proteome of the blood trypomastigote (BT) stage showed that the major groups were trans-sialidase (TS), bioenergetics-related enzymes, chaperones, and cytoskeletal proteins.93 Comparison of the BT proteome with that of the tissue culture-derived trypomastigote (TCDT) or metacyclic trypomastigote (MT) demonstrated that over 2200 proteins were unique to the BT stage and were involved in a variety of cellular processes.93A large-scale comparative proteomic study showed that the metacyclic form had the highest number of proteins expressed, followed by amastigotes, epimastigotes and trypomastigotes.86 The same study also reported that over a third of the total proteome, accounting for over 1000 proteins, consisted of those annotated as “hypothetical”. One notable observation was that the four stages of the parasite might be characterised by distinct energy metabolites, such as histidine during the insect stages and fatty acids during the intracellular amastigotes in the mammal. Another study based on only basic proteins found that epimastigotes may use amino acid metabolism, but TS enzymes and flagellar rod components were specific for trypomastigotes.94 Amastigotes also appeared to be rich in vesicular trafficking proteins, and several important antioxidant enzymes such as ascorbate peroxidase, mitochondrial tryparedoxin peroxidase, trypanothione synthase and iron superoxide dismutase were abundant in the metacyclic form, suggestive of parasite anticipation of an oxidative attack by host phagocytes.86 Proteomic analysis of the trypomastigote stage identified over 1400 proteins, nearly 14% of which were mainly glycophosphatidylinositol (GPI)-anchored surface proteins that may be involved in host cell invasion and immune evasion.95 Epimastigotes and trypomastigotes differed in over 50% of the expressed proteins according to one study, which also identified some candidates with different isoforms involved in metacyclogenesis.96
During metacyclogenesis, the proteome undergoes major changes due to nutritional stress and at different time-points after adhesion. A large-scale proteomic investigation led to the observation of major differences in proteins involved in oxidative stress, protein translation, and metabolic pathways pertaining to proteins, lipids, and carbohydrates.97 Quantitative phosphoproteomic analysis in the same report showed over 7000 phosphorylation sites, of which 260 were regulated differentially, including some potential drug targets such as the enzymes of sterol biosynthesis. A later study reported that during nutritional stress, proteins with various functions, such as fatty acid synthesis and regulation of protein expression, were phosphorylated, which could in turn trigger metacyclogenesis.98 The T. cruzi epimastigote enters the stationary phase from the exponential phase, leading to the early stages of metacyclogenesis. A proteomic comparison of the exponential and stationary phases of epimastigotes was able to quantify over 3000 proteins; ribosomal proteins were upregulated in the exponential phase, whereas proteins involved in autophagy were upregulated in the stationary phase.99 Further, the same report demonstrated the regulation of proteins via N-terminal acetylation during this transition from the exponential to the stationary phase.
The transition from trypomastigotes to amastigotes, known as amastigogenesis, has also been investigated by quantitative proteomics and phosphoproteomics approaches. A number of proteins were regulated in this context, and the majority of regulated proteins were membrane proteins.100 The differentiation of infective trypomastigotes to epimastigotes is termed epimastigogenesis and is among the least studied processes in the trypanosome field. An in vitro method to generate epimastigotes revealed a previously uncharacterised stage called the “recently differentiated epimastigotes”, which resisted complement-mediated lysis in vivo and in vitro.101 Later, proteomic studies identified certain proteins, which were upregulated solely in the recently differentiated epimastigotes, such as ABC transporters and the multidrug resistance protein E.102
3.4 Study of host–T. cruzi interactions
The cell surface of T. cruzi features a dense layer of GPI-anchored molecules, which mediate a variety of interactions of the parasite with its insect vector and mammalian hosts. A comparison of the cell surface proteomes during different stages revealed that the majority of proteins are expressed in more than one stage, but a few are stage specific.100 Further analysis indicated that most of the proteins were membrane-derived and could participate in invasion, adhesion, cell signalling and modifying the host's immune response. The proteomic analysis of membrane-associated fractions from epimastigotes and MT stages showed that over 200 proteins had predicted sites for post-translational lipid modifications, signal sequences or transmembrane domains.103 Interestingly, the MT stage expressed a large set of surface glycoproteins involved in host cell adhesion and invasion, while epimastigotes did not.T. cruzi is an intracellular pathogen and hence, adhesion and invasion of the host cell are crucial processes. Interaction with host cells involves both surface and secreted molecules. Secreted molecules are known to play critical roles in immune evasion, migration inside the host tissues, cell adhesion, communication, differentiation, and parasite proliferation.104 A novel family of surface membrane proteins called TcSMP (T. cruzi surface membrane proteins) conserved in different T. cruzi lineages has been characterised.105 Evidence suggests that the TcSMP are membrane-spanning and might be released into the extracellular environment. They were shown to bind mammalian cells, and trigger Ca2+-dependent signalling and lysosome exocytosis, linking them to host cell invasion. The secretome of T. cruzi consists of extracellular vesicles and soluble proteins not associated with vesicles.106 Metabolic proteins, as well as proteins implicated in cell signalling, parasite survival, nucleic acid packaging and virulence were detected by LFQ proteomics.106 The exoproteome of the TCDT stage was characterised in the Y strain as having 540 proteins; most were suggested to be involved in host cell infections, implying that this parasite has well-developed mechanisms to infect different types of host cells and escape immune attack.107
Proteomics MS holds great potential for the discovery of biomarkers for diagnostic purposes in Chagas disease. The disease has a long incubation time extending to nearly three decades, during which the amastigote forms remain in the host. Therefore, identifying biomarkers derived from secreted parasite molecules in the serum would improve the prospects for a fast and reliable diagnostic assay. A comparative proteomic analysis of the secretomes of two T. cruzi strains, VD and CL Brener, revealed that over 350 of the proteins could be classified into multigene superfamilies, but 94 proteins did not correspond to any known superfamilies.108 Since these proteins were not reported from other T. cruzi strains, they could be potential biomarkers for infection by either the VD or the CL Brener strain. Two other strains, Y and YuYu, also showed distinct proteome profiles of the released vesicles that could be correlated with differences in their virulence and infectivity.109
Proteomics has also been employed for characterising the immune complexome of T. cruzi-infected patients. In a proteomic study on plasma from chronic Chagas disease patients, Ohyama et al. identified 39 antigens derived from T. cruzi and 114 autoantigens from the patients.110 In the same study, more than half of all the patients carried the surface protease GP63 and glucose-6-isomerase antigens derived from the pathogen, as well as six human autoantigens, namely, CD180 antigen, ceruloplasmin, fibrinogen beta chain, fibrinogen beta chain isoform 2 preprotein, isoform gamma-A of the fibrinogen γ-chain and serum paraoxonase. Moreover, two factors, the human complement factor H-related protein 2 and the TS of T. cruzi, occurred most commonly in patients with symptomatic Chagas disease. Another study characterised the absence of peptide fragments of two proteins, apolipoprotein A-1 and fibronectin, in the serum of patients successfully treated with Bzn for congenital Chagas disease.111 Similarly, a proteomic study of two Brazilian strains showed that the expression profiles differed with respect to several proteins, including cruzipain, which is a key virulence factor, and this could be correlated to differences in infectivity determined through in vitro cell-based assays.112 The use of antisense oligonucleotides directed against TS, complement regulatory proteins or E64d in order to inhibit the activity of the cruzipain was found to be effective in reducing the virulence of a highly virulent strain in mouse models.113
The virulence of T. cruzi strains varies widely, and the correct identification of strains is beneficial for diagnostic and treatment purposes. The typing of strains has been accomplished by proteomic methods using tandem MS (MS/MS) libraries.114 This method is independent of the genome data. Moreover, this method reliably identifies the DTU strains and is agnostic towards fragmentation techniques, gradient lengths during chromatographic steps or variations in sample preparation. Immunology techniques have been combined with proteomics in order to search for epitopes, which might propel the development of vaccines. In such an immunoproteome study, the calpain-like protein CAP5.5 was reported to be 23-fold upregulated in the cardiac stage of Chagas disease, compared to the asymptomatic stage.115 In addition, immunoproteomics also led to the identification of the most immunogenic parasite proteins in mouse serum as pyruvate phosphate dikinase, Hsp-85, and β-tubulin. This work also demonstrated that recombinant T. cruzi β-tubulin was sufficient to immunize mice successfully against T. cruzi infection.116
3.5 Posttranslational modifications in T. cruzi
In addition to the protein phosphorylation PTM discussed in section 3.3, several other PTMs have been characterised by proteomics methods in T. cruzi. Selected studies are reviewed herein.Modification of the hydroxyl side chain of serine/threonine residues with a single unit of β-N-acetylglucosamine, termed O-linked β-N-acetylglucosamine (O-GlcNAc) modification, has been implicated in many important cellular processes including the regulation of transcription and translation in higher eukaryotes.125 The O-GlcNAc modification is fundamentally different from other types of glycosylation: whereas the common N- and O-glycosylations are relatively stable PTMs, the O-GlcNAc PTM is dynamic. It is catalysed by the enzyme O-GlcNAc transferase (OGT) and removed by N-acetyl-β-D-glucosamidase (O-GlcNAcase). In a recent study, Torres-Gutiérrez et al. reported the identification of the O-GlcNAc modification in T. cruzi proteins.126 They used the commercial “Click-iT O-GlcNAc enzymatic labeling system”127 for enzymatically labelling and enriching the O-GlcNAc modified proteins from T. cruzi epimastigote lysates. On-bead digestion using a trypsin/Lys-C mixture followed by LC-MS/MS analysis of the eluted peptides identified 1271 putative O-GlcNAclated proteins. As the on-bead digestion protocol led to the retention of O-GlcNAc modified peptides on the resin, on-bead dephosphorylation and β-elimination steps prior to protein digestion were performed on a separate portion of the sample to release the modified peptides. This enabled the identification of six O-GlcNAc modification sites. Many of the putative O-GlcNAclated proteins identified have important biological roles including metabolic functions and stress adaptation.
3.6 Protein–protein interactions in T. cruzi
The combination of proteomics with an assessment of protein–protein interactions, termed “interactomics” forms part of a powerful approach to understanding cellular processes.130 The construction of mutant redox proteins and proteomics of the resulting complexes from pull-down assays with T. cruzi lysates have been used to identify proteins interacting with two major redox enzymes, tryparedoxins I and II (TcTXNI and TcTXNII).131,132 According to these studies, TcTXNI had 15 interacting protein partners, mainly with roles in redox metabolism and protein synthesis/degradation, and TcTXNII had 16 partners involved in a variety of cellular functions. These two enzymes transfer electrons between trypanothione and peroxiredoxins, which is different from the thioredoxin-based electron transfer in mammals and may present novel targets for drug development.133 More recently, two TcI strains of T. cruzi, COL and SYL, were analysed by proteomics, which showed that the antioxidant network of the trypomastigotes was enhanced in comparison to that of a cultured clone.1343.7 Chemical proteomics in T. cruzi
Napthoimidazoles derived from β-lapachone were shown to induce changes in the expression of a number of proteins in bloodstream trypomastigotes, including enzymes such as asparagine synthetase, arginine kinase, enolase, guanine deaminase, succinyl-CoA ligase, ATP synthase subunit B, and methionine sulfoxide reductase, thus paving the way for target-based drug design.138 In epimastigotes, proteomic methods allowed the elucidation of the mechanism of trypanocidal action following napthoimidazole treatment.88 Several pathways impacted were identified in this work, including energy metabolism, protein biosynthesis, chaperone modification, cytoskeletal assembly and ergosterol biosynthesis. Proteomics has also facilitated the study of the possible mechanism of action of trypanocidal natural products. For instance, following piplartine treatment, the expression of two antioxidant enzymes, tryparedoxin peroxidase (TXNPx) and methionine sulfoxide reductase (MSR), was upregulated in T. cruzi, suggesting that these enzymes could be potential therapeutic targets.139
4. Proteomics approaches in T. brucei
4.1 Human African trypanosomiasis: an introduction
Human African trypanosomiasis (HAT), also known as African sleeping sickness, is caused by protozoa of the species Trypanosoma brucei (T. brucei). The disease is endemic in sub-Saharan Africa where it mainly affects poor rural communities; about 55 million people are at risk of contracting it.142T. b. gambiense accounts for the vast majority of cases of HAT in West and Central Africa, while T. b. rhodesiense accounts for the rest, mainly in East and Southern Africa.142 HAT is transmitted to humans via the bite of tsetse flies (Glossina), whereupon the parasites multiply and invade the blood and lymphatic system. The T. b. gambiense variant causes a slow-developing chronic disease in two stages; stage 1 (S1) is the hemolymphatic stage involving fever, headaches, inflammation of the lymph nodes, and simultaneous spleen and liver enlargement.143 Progression to stage 2 (S2), the nervous stage, is characterised by invasion of the central nervous system and neurological symptoms, which can lead to death.144,145 Patients with T. b. gambiense infections in S1 respond to pentamidine, but S2 patients need drugs that can cross the blood–brain barrier. S2 infection can be treated with a nifurtimox–eflornithine combination therapy (NECT) by invasive methods, which are challenging to execute in some areas.146 Both stages can be treated by fexinidazole, which was approved last year.147 However, accurate diagnosis of the stage of HAT will save lives and will need further research. For HAT infection caused by T. b. rhodesiense, only one drug, melarsoprol, is currently available.148 Development of more oral drugs will benefit the efforts to eliminate both forms of HAT.4.2 The T. brucei life cycle
Inside the gut of the tsetse flies, the parasites exist as procyclic trypomastigotes and multiply by binary fission. When leaving the midgut, they differentiate into epimastigotes and migrate to the salivary glands, where a further differentiation to a metacyclic trypomastigote form occurs. When the vector bites the victim, the metacyclic trypomastigotes enter the human bloodstream and transform into bloodstream trypomastigotes, which rapidly multiply by binary fission, and reach various body fluids such as the spinal fluid and the lymph. In the bloodstream, the trypomastigotes proliferate in a long slender (LS) morphological form, which further differentiate, as the parasite-load in the bloodstream increases, into a short stumpy (SS) non-replicative form. The replication arrested SS forms are adapted for transmission to the tsetse flies, where they transform into procyclic promastigotes and continue the life cycle. The LS forms are responsible for the neurological complications arising in S2 of HAT. During this multi-stage life cycle, the parasite adapts by varying its proteome.149–1514.3 Characterising the life cycle stages and host–T. brucei interactions
Global proteome profiling of procyclic and bloodstream stages of T. brucei with SILAC quantitative proteomics MS revealed that the procyclic forms have slower proteome turnover compared to the bloodstream stage; the fastest turnover rates were observed for cell cycle and cytokinesis proteins in both forms.152 Further, lower than average turnover rates were observed for mitochondrial and glycosomal proteins.152 Quantitative SILAC-based proteomics also helped to uncover the stage-specific expression of many proteins, for example, a DEAD box RNA helicase was found to be highly upregulated in the bloodstream form and is thought to be critical for parasite viability and cell cycle progression.153 SILAC-based proteomics MS also showed remodelling of the proteome during three different human-infective forms and the final procyclic stage, with almost 30% of the proteome undergoing changes.154 More specifically, about 40% of the proteins in the plasma membrane, peroxisome, nucleus, mitochondria, microtubule, lysosome and glycosome are upregulated by 2-fold or more while differentiating to the last bloodstream stage (short stumpy or SS). Quantitative proteomics enabled the tracking of over 4000 proteins during this transition and offered clues on how the parasites might evade host defence mechanisms.150Evasion of the mammalian host immune defence is dependent on the antigenic variation of the variant surface glycoprotein (VSG).155 In the bloodstream form, the parasite surface is covered with around ten million copies of a single species of VSG and the sub-telomeric VSG genes are the major virulence determinants. The transcription of VSG at any time occurs from only one of the 20 sub-telomeric expression sites with switching to avoid lysis by the host adaptive immune response. Regulation of virulence gene expression by telomere structures is known in microbial pathogens, wherein those genes are often located next to the telomere.156 Differentiation of the bloodstream form into the procyclic form silences the transcription of the VSG genes. MS of the telomere protein complexes revealed that the complex composition differs between the two forms of the parasite.157 One novel telomere-associated protein, TelAP1, forms a complex with other telomeric proteins and participates in expression site silencing. Metabolomics studies have revealed that the myristate required for incorporation into the VSG can be synthesised by up to four distinct pathways.158 Indeed, multiple redundant biochemical pathways are used by many pathogens, and proteomics may help uncover which pathways and enzymes are involved in each stage.
In combination with subcellular fractionation techniques, proteomics enables biochemical characterisation of subcellular organelles. A nuclear proteome study of T. brucei in the procyclic form has demonstrated that over 750 proteins specifically enriched in the nuclear fraction.159 Motif enrichment analyses detected the presence of the KRxR sequence in many of these identified nuclear proteins, which is proposed to be the nuclear localisation signal for the Kinetoplastida class. Studies have also been carried out for characterising the proteome of the T. brucei lipid rafts.160,161 These are regions of the plasma membrane with high concentrations of cholesterol and glycosphingolipids and are postulated to be hubs that cluster virulence factors.162 The lipid raft proteome of the procyclic form contains proteins, of which functions of around 18% remain unknown.161 In addition, proteins with known or predicted functions also occurred, including Rab-like GTPases (part of the intra-flagellar transport protein or IFT complex), aquaporin, arginine kinase, and calcium dependent cysteine proteases.161 Arginine kinase is a flagellar protein, which plays an important role in infection,163 while aquaporin is a water channel protein which may also transport drugs, localising to the plasma membrane and implicated in the pentamidine resistance of the bloodstream form.164,165 A remarkable 60% of proteins are shared between the procyclic lipid raft proteome and the procyclic flagellar proteome.161 Superoxide dismutase (SOD), a glycosomal protein, was also found in the lipid raft. While its role in T. brucei is not yet elucidated, overexpression of the T. cruzi homologue causes enhanced susceptibility to the drugs Bzn and gentian violet in that organism.166
The potential number of coding sequences in the T. brucei genome is greater than previously thought; a transcriptomic study identified over 1100 sequences under 100 amino acid long, which map onto regions of the genome not containing annotated ORFs.167 Although 993 of these transcripts contain coding sequences corresponding to at least 25 amino acids, their functional expression remained unknown. Nevertheless, a combined application of bioinformatics methods and analysis of previous proteomics data sets successfully identified with high confidence 42 of these small proteins.167 More recently, Crozier et al. employed TMT-10plex quantitative proteomics MS for the analysis of the cell cycle of the procyclic form of T. brucei.168 They quantified the expression levels of over 5000 proteins across the cell cycle in the parasite. Off these, 384 proteins showed cell cycle regulated patterns of expression; 151 of these were hypothetical proteins of unknown function. About 40 of these are considered critical proteins for the parasite survival and therefore could be potential drug targets.
Using proteomics, over 60 human proteins were identified to vary in abundance in the serum, cerebrospinal fluid (CSF), urine and saliva between T. brucei-infected and healthy subjects.169 Two of them, neuroserpin from the CSF and moesin from urine, are considered potential biomarkers for HAT.169 Neuroserpin is predominantly expressed in regions such as the hippocampus and the caudate.170,171 Both those regions are near the choroid plexus, which constitutes a gap in the blood–brain barrier, and is suggested to be the site where the parasite enters the brain.172
Proteomic comparisons of infected and uninfected salivary glands of tsetse flies revealed that highly abundant G. morsitans proteins were downregulated in infected salivary glands.173 If this affects fly performance, it could lead to increased transmission, since it is known that when fly feeding is reduced, biting behaviour increases in order to reach satiety.174 In addition, other proteins advantageous to T. brucei were upregulated, including CamK, Serp-2, SUMO, eight distinct V-type ATPases and amino acid metabolism-related proteins.173 CamK is a key regulator of stage-specific morphological differentiation in all parasitic protozoans,175 while Serp-2 plays a role in Plasmodium parasites to trigger a switch to transmissible sexual stages in mosquitoes.176 SUMO, responsible for SUMOylation discussed earlier in section 3.5.3, provides a quick way for changing the interaction partners and subcellular localisations of proteins, thus contributing to immune evasion.177 Downregulation of regulatory proteins occurred along with upregulation of the ubiquitin proteasome components, suggesting that protein translation/turnover increases leading to metacyclogenesis.173 Among proteins specific to T. brucei, suppression of regulatory proteins and transporters, and the upregulation of a high number of immunity, signal transduction and virulence-related proteins were observed.
4.4 Proteomics of the T. brucei flagellum
T. brucei is an extracellular parasite and all endocytosis and exocytosis are directed through a specific membrane region called the flagellar pocket, in which the majority of invariant surface proteins are considered to be localised.178 Genome mining for proteins with membrane-association is of limited utility since a large number of the predicted proteins are not likely to be surface-localised. A combination of biochemistry, proteomics and bioinformatics was used to characterise the surface proteome, which contained novel proteins, known flagellar pocket proteins and proteins required for parasite survival.179 Transporter-like proteins in the parasite were conserved in well-studied eukaryotes, but receptor-like proteins were mostly specific to trypanosomes. While the said study was able to support the view of a three-compartment cell surface, the mechanisms of protein sorting and targeting are still unclear. No simple localisation signals were found, and the protein architecture type did not correlate with domain localisation.The flagellum plays important roles in T. brucei cell motility, parasite signalling and development, and parasite attachment during invasion of the salivary glands of the tsetse fly, but a sensory role has also been postulated.180 Proteomic analysis of the intact flagellum detected over 200 proteins not previously linked to flagella; out of these, a 14-3-3 protein and eight novel proteins termed FLAM (flagellar member) were experimentally validated.181 Cardiolipin is a mitochondrial and bacterial lipid important for the stability of electron transport complexes.182 Unlike other eukaryotes, the Apicomplexa (which include the malaria parasites) and the Euglenozoa (which include the trypanosomatids) harbour bacterial-type cardiolipin synthases.183 This difference between T. brucei and its hosts, and the fact that it is essential in T. brucei make cardiolipin synthesis an attractive target for therapeutic interventions. Combining the use of knockout mutants for cardiolipin synthase, SILAC and MS, a number of proteins that bind cardiolipin, termed cardiolipin-dependent proteins (CLDP), were discovered.184
4.5 The phosphoproteome of T. brucei
Global phosphoproteomics in T. brucei enabled quantitative comparisons between the procyclic and bloodstream forms and revealed significant differences in phosphorylation patterns between the two forms.185 In the procyclic form, a number of proteins exhibited upregulation of specific phosphorylation sites, including a C14-sterol reductase involved in ergosterol synthesis, a ubiquitin carboxy-terminal hydrolase and PAD2, a carboxylic acid transporter with possible roles in cell differentiation. The bloodstream form upregulates the phosphorylation of other proteins, for example, the RNA recognition motif protein RBP10, which could promote bloodstream mRNA expression. Certain groups of proteins such as ZFPs (zinc finger proteins) and RBPs (RNA binding proteins) which are expected to participate in the posttranscriptional regulation of gene expression were also subject to regulated phosphorylation.Tyrosine-specific phosphoproteomics in T. brucei procyclic forms using a combination of immunoaffinity purification and LC-MS/MS identified over 30 tyrosine-specific phosphorylated proteins involved in protein synthesis, RNA metabolism, energy metabolism, and kinases, including 19 protein kinases and two metabolic kinases.186 In the CMG kinase group, 11 candidates were found to be putative homologues of Leishmania MAPK (mitogen-activated protein kinase). The highly conserved TXY motifs in the activation loops were found fully phosphorylated in eight of the putative TbMAPKs, suggesting that they were activated in the procyclic form of T. brucei.186 The phosphoproteome of the bloodstream form showed that phosphorylated serine/threonine kinases (STE kinases) were present, suggesting signal transduction via MAPK.187 Moreover, the same study indicated that phosphorylation of certain proteins is not stage-specific, since all three CRK isoforms were phosphorylated on the same tyrosine residue. Tyrosine-phosphorylated proteins in both bloodstream and procyclic forms were concentrated in the nucleolus and the cytoskeletal structures (basal body and flagellum), lending support to the notion that the function of signalling molecules is governed by their subcellular localisation. The localisation pattern also underscores the differences between this parasite and higher eukaryotes, such as humans, since kinetoplastids are among the most ancient eukaryotes, probably older than even fungi.19 Further, the localisation of glycogen synthase kinase 3 (GSK3) in the flagellum, along with identified phosphorylation sites in its active loop, indicates that this kinase is active in T. brucei, and suggests that tyrosine phosphorylation may participate in the regulation of flagellum formation and basal body segregation.187 In fact, many organelles such as the basal body, the flagellum, the mitochondrion and the kinetoplast (mitochondrial DNA network) are present in a single copy and must be segregated and duplicated during cell division. Therefore, understanding organelle segregation and duplication could be important for drug development.
Tyrosine-phosphorylated GSK3 has been previously shown to be essential for flagella assembly and maintenance in Chlamydomonas reinhardtii.188 Therefore, GSK3 possibly plays a similar role in T. brucei. The T. brucei genome lacks tyrosine kinase-like kinases, G protein-coupled receptors and proteins with phosphotyrosine-binding domains, but phosphorylation of the members of the STE kinase family and a homologue of the ERK1 from the amoeba Dictyostelium discoideum indicates the presence of a putative ERK1/2 signalling pathway in T. brucei.187 The mechanism or regulation is still unknown and likely novel. Additionally, about a quarter of the manually validated kinase sites were not predicted by bioinformatics, suggesting that T. brucei may contain kinases with novel substrate specificity and previously unrecognised motifs.187
4.6 Chemical proteomics in T. brucei
4.7 Nascent DNA proteomics
The trypanosomatids exhibit unusual DNA replication properties, such as highly divergent origin replication complex (ORC) subunits, an apparent absence of many replication factor homologues and considerably fewer origins of replication than well-studied eukaryotes.194,195 The application of iPOND (isolation of proteins on nascent DNA) technology to T. brucei identified proteins involved in the core replication machinery, transcription, chromatin organisation, and DNA repair that were enriched near an unperturbed active replication fork. About 100 of those proteins were annotated as performing “unknown functions” and potentially specific to trypanosome replication. Follow-up studies on two proteins annotated as Tb927.10.7990 (replication factor C subunit) and Tb927.3.5370 (protein of unknown function) revealed that Tb927.10.7990 is essential since its silencing resulted in a growth defect in procyclic cells. Apart from antigenic variation, unusual replication and posttranscriptional regulation, trypanosomes also display other features such as polycistronic transcription without classical promoters and transcription of certain abundant proteins by RNA polymerase I, underlining the importance of unique nuclear processes in their biology.5. Conclusions
Mass spectrometry-based proteomics has evolved as a powerful technique for studying various aspects of the life cycle of the protozoan parasites causing NTDs. The lack of transcriptional control of gene expression in Tritryps makes protein-based functional studies particularly suitable in these organisms. The differentiation process or life cycle changes is a peculiar aspect of the protozoan biology, the molecular mechanisms of which are largely unknown, despite decades of research using conventional methods of parasitology or biological research methods in general. Unravelling the molecular processes associated with life cycle differentiation holds the keys to understanding the mechanisms of virulence and infectivity. Proteomics MS has so far facilitated the measurement of the differential expression of several hundreds of proteins during the various life cycle stages of the three parasite genera reviewed herein. It is generally assumed that protein interactions in the differentially modulated proteomes detected in the life cycle stages are responsible for the observed phenotypic changes. Deconvoluting the molecular interactions and accurately annotating their functions within the modulated proteome will take significant in silico studies and follow-up biochemical experimental studies. However, a caveat is that the original measurements of the proteome were not made in real-time, as in during the transition of one life cycle stage to another but rather afterwards. Therefore, the currently available proteomics data sets that enabled the differential comparison of life cycle changes, although highly useful, many not have accurately captured all the dynamic perturbations in the proteome occurring at the actual timescale of the differentiation process. Additionally, the protein expression difference is not the only parameter that determines protein function. Rather, a large repertoire of protein modifications including PTMs and changes in the activity of enzymes and dynamic changes in the subcellular localisation of proteins during these processes must be playing important roles. Some progress in this direction has already been accomplished thanks to the recent developments in PTM enrichment strategies and advancements in MS instrumentation. For instance, large-scale profiling of important post- and/or co-translational modifications such as phosphorylation, glycosylation, N-terminal myristoylation and SUMOylation was carried out in the protozoans. Some of the enzymes responsible for these modifications have already been validated as therapeutic targets. Accurate profiling of the target engagement and off-target profiling of pharmacologically active compounds as well as elucidating their mechanisms of action is another stronghold of proteomics MS. A relatively new area termed chemical proteomics that uses a combination of synthetic chemistry tools and quantitative proteomics MS technologies has evolved towards this end. This review has touched upon recent developments in all these areas where proteomics has played important roles.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
KK acknowledges funding from the MRC-Global Challenges Research Fund (Grant number: MR/P027987/1A). AP acknowledges the Thomas H. Gosnell School of Life Sciences at RIT.Notes and references
- M. C. Field, D. Horn, A. H. Fairlamb, M. A. J. Ferguson, D. W. Gray, K. D. Read, M. De Rycker, L. S. Torrie, P. G. Wyatt, S. Wyllie and I. H. Gilbert, Nat. Rev. Microbiol., 2017, 15, 217–231 CrossRef CAS PubMed.
- P. J. Hotez, D. H. Molyneux, A. Fenwick, J. Kumaresan, S. E. Sachs, J. D. Sachs and L. Savioli, N. Engl. J. Med., 2007, 357, 1018–1027 CrossRef CAS PubMed.
- https://www.who.int/neglected_diseases/news/WHO-publishes-guidelines-treatment-sleeping-sickness/en/, (accessed 01 May 2020).
- G. B. D. Disease, I. Injury and C. Prevalence, Lancet, 2017, 390, 1211–1259 CrossRef.
- B. Liese, M. Rosenberg and A. Schratz, Lancet, 2010, 375, 67–76 CrossRef.
- S. L. Reed and J. H. McKerrow, Clin. Infect. Dis., 2018, 67, 323–326 CrossRef PubMed.
- S. Patterson and S. Wyllie, Trends Parasitol., 2014, 30, 289–298 CrossRef CAS PubMed.
- R. Viotti, B. Alarcon de Noya, T. Araujo-Jorge, M. J. Grijalva, F. Guhl, M. C. Lopez, J. M. Ramsey, I. Ribeiro, A. G. Schijman, S. Sosa-Estani, F. Torrico, J. Gascon and N. Latin American Network for Chagas Disease, Antimicrob. Agents Chemother., 2014, 58, 635–639 CrossRef CAS PubMed.
- S. Sundar, A. Singh, M. Rai, V. K. Prajapati, A. K. Singh, B. Ostyn, M. Boelaert, J. C. Dujardin and J. Chakravarty, Clin. Infect. Dis., 2012, 55, 543–550 CrossRef CAS PubMed.
- M. L. den Boer, J. Alvar, R. N. Davidson, K. Ritmeijer and M. Balasegaram, Expert Opin. Emerging Drugs, 2009, 14, 395–410 CrossRef CAS PubMed.
- M. Mueller, K. Ritmeijer, M. Balasegaram, Y. Koummuki, M. R. Santana and R. Davidson, Trans. R. Soc. Trop. Med. Hyg., 2007, 101, 19–24 CrossRef CAS PubMed.
- J. Shendure and E. Lieberman Aiden, Nat. Biotechnol., 2012, 30, 1084–1094 CrossRef CAS PubMed.
- P. D. Wes, I. R. Holtman, E. W. Boddeke, T. Moller and B. J. Eggen, Glia, 2016, 64, 197–213 CrossRef PubMed.
- M. I. McCarthy and D. G. MacArthur, Genome Biol., 2017, 18, 20 CrossRef PubMed.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- S. P. Gygi, Y. Rochon, B. R. Franza and R. Aebersold, Mol. Cell. Biol., 1999, 19, 1720–1730 CrossRef CAS PubMed.
- A. Subramanian and R. R. Sarkar, Genomics, 2015, 106, 232–241 CrossRef CAS PubMed.
- S. M. Beverley, Cell, 1996, 87, 787–789 CrossRef CAS PubMed.
- A. P. Fernandes, K. Nelson and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11608–11612 CrossRef CAS PubMed.
- C. Clayton and M. Shapira, Mol. Biochem. Parasitol., 2007, 156, 93–101 CrossRef CAS PubMed.
- S. Haile and B. Papadopoulou, Curr. Opin. Microbiol., 2007, 10, 569–577 CrossRef CAS PubMed.
- S. Kramer, Mol. Biochem. Parasitol., 2012, 181, 61–72 CrossRef CAS PubMed.
- L. J. Jensen, R. Gupta, N. Blom, D. Devos, J. Tamames, C. Kesmir, H. Nielsen, H. H. Staerfeldt, K. Rapacki, C. Workman, C. A. Andersen, S. Knudsen, A. Krogh, A. Valencia and S. Brunak, J. Mol. Biol., 2002, 319, 1257–1265 CrossRef CAS PubMed.
- P. J. Myler and K. D. Stuart, Curr. Opin. Microbiol., 2000, 3, 412–416 CrossRef CAS PubMed.
- S. Hanash, Nature, 2003, 422, 226–232 CrossRef CAS PubMed.
- G. Marko-Varga and T. E. Fehniger, J. Proteome Res., 2004, 3, 167–178 CrossRef CAS PubMed.
- M. Kavallaris and G. M. Marshall, Med. J. Aust., 2005, 182, 575–579 CrossRef PubMed.
- Z. Zhang, S. Wu, D. L. Stenoien and L. Pasa-Tolic, Annu. Rev. Anal. Chem., 2014, 7, 427–454 CrossRef CAS PubMed.
- B. F. Cravatt, G. M. Simon and J. R. Yates, 3rd, Nature, 2007, 450, 991–1000 CrossRef CAS PubMed.
- R. C. Wilson and J. A. Doudna, Annu. Rev. Biophys., 2013, 42, 217–239 CrossRef CAS PubMed.
- X. Wu, M. A. Hasan and J. Y. Chen, J. Theor. Biol., 2014, 362, 44–52 CrossRef CAS PubMed.
- M. Frantzi, A. Latosinska and H. Mischak, Proteomics: Clin. Appl., 2019, 13, e1800087 Search PubMed.
- P. E. Geyer, L. M. Holdt, D. Teupser and M. Mann, Mol. Syst. Biol., 2017, 13, 942 CrossRef PubMed.
- S. Burza, S. L. Croft and M. Boelaert, Lancet, 2018, 392, 951–970 CrossRef.
- J. Alvar, I. D. Velez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin, M. den Boer and WHO L. C. Team, PLoS One, 2012, 7, e35671 CrossRef CAS PubMed.
- S. Sundar and P. L. Olliaro, Ther. Clin. Risk Manage., 2007, 3, 733–740 CAS.
- D. H. Kim, H. J. Chung, J. Bleys and R. F. Ghohestani, PLoS Neglected Trop. Dis., 2009, 3, e381 CrossRef PubMed.
- S. Sundar and J. Chakravarty, Int. J. Environ. Res. Public Health, 2010, 7, 4267–4277 CrossRef CAS PubMed.
- E. Handman, G. F. Mitchell and J. W. Goding, J. Immunol., 1981, 126, 508–512 CAS.
- E. Handman, R. E. Hocking, G. F. Mitchell and T. W. Spithill, Mol. Biochem. Parasitol., 1983, 7, 111–126 CrossRef CAS PubMed.
- N. G. Saravia, M. A. Gemmell, S. L. Nance and N. L. Anderson, Clin. Chem., 1984, 30, 2048–2052 CrossRef CAS.
- P. G. Nugent, S. A. Karsani, R. Wait, J. Tempero and D. F. Smith, Mol. Biochem. Parasitol., 2004, 136, 51–62 CrossRef CAS PubMed.
- Z. Mojtahedi, J. Clos and E. Kamali-Sarvestani, Exp. Parasitol., 2008, 119, 422–429 CrossRef CAS PubMed.
- W. Moreira, D. Légaré, G. Racine, G. Roy and M. Ouellette, EuPa Open Proteomics, 2014, 4, 171–183 CrossRef CAS.
- P. Pescher, T. Blisnick, P. Bastin and G. F. Spath, Cell. Microbiol., 2011, 13, 978–991 CrossRef CAS PubMed.
- N. Gupta, N. Goyal and A. K. Rastogi, Trends Parasitol., 2001, 17, 150–153 CrossRef CAS PubMed.
- J. Walker, R. Gongora, J. J. Vasquez, J. Drummelsmith, R. Burchmore, G. Roy, M. Ouellette, M. A. Gomez and N. G. Saravia, Mol. Biochem. Parasitol., 2012, 183, 166–176 CrossRef CAS PubMed.
- D. A. Goyeneche-Patino, L. Valderrama, J. Walker and N. G. Saravia, Antimicrob. Agents Chemother., 2008, 52, 4503–4506 CrossRef CAS PubMed.
- R. L. Krauth-Siegel, S. K. Meiering and H. Schmidt, Biol. Chem., 2003, 384, 539–549 CAS.
- X. Wang, S. Shen, S. S. Rasam and J. Qu, Mass Spectrom. Rev., 2019, 38, 461–482 CrossRef CAS PubMed.
- J. Griss, F. Stanek, O. Hudecz, G. Durnberger, Y. Perez-Riverol, J. A. Vizcaino and K. Mechtler, J. Proteome Res., 2019, 18, 1477–1485 CrossRef CAS PubMed.
- A. Thompson, J. Schafer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann, R. Johnstone, A. K. Mohammed and C. Hamon, Anal. Chem., 2003, 75, 1895–1904 CrossRef CAS PubMed.
- S. Wiese, K. A. Reidegeld, H. E. Meyer and B. Warscheid, Proteomics, 2007, 7, 340–350 CrossRef CAS PubMed.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell. Proteomics, 2002, 1, 376–386 CrossRef CAS PubMed.
- J. L. Hsu and S. H. Chen, Philos. Trans. R. Soc., A, 2016, 374, 20150364 CrossRef PubMed.
- E. de Rezende, R. Kawahara, M. S. Pena, G. Palmisano and B. S. Stolf, PLoS Neglected Trop. Dis., 2017, 11, e0006090 CrossRef PubMed.
- J. P. Menezes, T. F. Almeida, A. L. Petersen, C. E. Guedes, M. S. Mota, J. G. Lima, L. C. Palma, G. A. Buck, M. A. Krieger, C. M. Probst and P. S. Veras, Microbes Infect., 2013, 15, 579–591 CrossRef CAS PubMed.
- A. K. Singh, R. K. Pandey, J. L. Siqueira-Neto, Y. J. Kwon, L. H. Freitas-Junior, C. Shaha and R. Madhubala, Infect. Immun., 2015, 83, 1853–1868 CrossRef CAS PubMed.
- A. Isnard, J. G. Christian, M. Kodiha, U. Stochaj, W. R. McMaster and M. Olivier, PLoS Pathog., 2015, 11, e1004776 CrossRef PubMed.
- K. Chandramouli and P. Y. Qian, Hum. Genomics Proteomics, 2009, 2009, 239204 Search PubMed.
- F. Conceicao-Silva and F. N. Morgado, Front. Cell. Infect. Microbiol., 2019, 9, 330 CrossRef CAS PubMed.
- M. A. Lynn, A. K. Marr and W. R. McMaster, J. Proteomics, 2013, 82, 179–192 CrossRef CAS PubMed.
- D. Nandan, S. A. Thomas, A. Nguyen, K. M. Moon, L. J. Foster and N. E. Reiner, PLoS One, 2017, 12, e0170068 CrossRef PubMed.
- L. M. de Pablos, T. R. Ferreira, A. A. Dowle, S. Forrester, E. Parry, K. Newling and P. B. Walrad, Mol. Cell. Proteomics, 2019, 18, 1271–1284 CrossRef CAS PubMed.
- J. Wang, L. Gao, Y. M. Lee, K. A. Kalesh, Y. S. Ong, J. Lim, J. E. Jee, H. Sun, S. S. Lee, Z. C. Hua and Q. Lin, Pharmacol. Ther., 2016, 162, 10–22 CrossRef CAS PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- M. R. Luth, P. Gupta, S. Ottilie and E. A. Winzeler, ACS Infect. Dis., 2018, 4, 301–314 CrossRef CAS PubMed.
- S. Wyllie, M. Thomas, S. Patterson, S. Crouch, M. De Rycker, R. Lowe, S. Gresham, M. D. Urbaniak, T. D. Otto, L. Stojanovski, F. R. C. Simeons, S. Manthri, L. M. MacLean, F. Zuccotto, N. Homeyer, H. Pflaumer, M. Boesche, L. Sastry, P. Connolly, S. Albrecht, M. Berriman, G. Drewes, D. W. Gray, S. Ghidelli-Disse, S. Dixon, J. M. Fiandor, P. G. Wyatt, M. A. J. Ferguson, A. H. Fairlamb, T. J. Miles, K. D. Read and I. H. Gilbert, Nature, 2018, 560, 192–197 CrossRef CAS PubMed.
- H. Franken, T. Mathieson, D. Childs, G. M. Sweetman, T. Werner, I. Togel, C. Doce, S. Gade, M. Bantscheff, G. Drewes, F. B. Reinhard, W. Huber and M. M. Savitski, Nat. Protoc., 2015, 10, 1567–1593 CrossRef CAS PubMed.
- D. Martinez Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef PubMed.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundback, P. Nordlund and D. Martinez Molina, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS PubMed.
- V. Corpas-Lopez, S. Moniz, M. Thomas, R. J. Wall, L. S. Torrie, D. Zander-Dinse, M. Tinti, S. Brand, L. Stojanovski, S. Manthri, I. Hallyburton, F. Zuccotto, P. G. Wyatt, M. De Rycker, D. Horn, M. A. J. Ferguson, J. Clos, K. D. Read, A. H. Fairlamb, I. H. Gilbert and S. Wyllie, ACS Infect. Dis., 2019, 5, 111–122 CrossRef CAS PubMed.
- N. G. Jones, C. M. C. Catta-Preta, A. Lima and J. C. Mottram, ACS Infect. Dis., 2018, 4, 467–477 CrossRef CAS PubMed.
- M. H. Wright, D. Paape, E. M. Storck, R. A. Serwa, D. F. Smith and E. W. Tate, Chem. Biol., 2015, 22, 342–354 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- S. Besteiro, R. A. Williams, L. S. Morrison, G. H. Coombs and J. C. Mottram, J. Biol. Chem., 2006, 281, 11384–11396 CrossRef CAS PubMed.
- N. S. Carter, P. A. Yates, S. K. Gessford, S. R. Galagan, S. M. Landfear and B. Ullman, Mol. Microbiol., 2010, 78, 92–107 CAS.
- J. L. Martin, P. A. Yates, R. Soysa, J. F. Alfaro, F. Yang, K. E. Burnum-Johnson, V. A. Petyuk, K. K. Weitz, D. G. Camp, 2nd, R. D. Smith, P. A. Wilmarth, L. L. David, G. Ramasamy, P. J. Myler and N. S. Carter, PLoS Pathog., 2014, 10, e1003938 CrossRef PubMed.
- D. C. Dieterich, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9482–9487 CrossRef CAS PubMed.
- K. Kalesh and P. W. Denny, PLoS Neglected Trop. Dis., 2019, 13, e0007651 CrossRef PubMed.
- J. A. Perez-Molina and I. Molina, Lancet, 2018, 391, 82–94 CrossRef.
- M. C. P. Nunes, A. Beaton, H. Acquatella, C. Bern, A. F. Bolger, L. E. Echeverria, W. O. Dutra, J. Gascon, C. A. Morillo, J. Oliveira-Filho, A. L. P. Ribeiro, J. A. Marin-Neto, E. American Heart Association Rheumatic Fever, Y. Kawasaki Disease Committee of the Council on Cardiovascular Disease in the, C. Council on, N. Stroke and C. Stroke, Circulation, 2018, 138, e169–e209 CrossRef PubMed.
- B. Zingales, Acta Trop., 2018, 184, 38–52 CrossRef CAS PubMed.
- B. Zingales, R. G. Araujo, M. Moreno, J. Franco, P. H. Aguiar, S. L. Nunes, M. N. Silva, S. Ienne, C. R. Machado and A. Brandao, Mem. Inst. Oswaldo Cruz, 2015, 110, 433–444 CrossRef CAS PubMed.
- A. M. Macedo, C. R. Machado, R. P. Oliveira and S. D. Pena, Mem. Inst. Oswaldo Cruz, 2004, 99, 1–12 CrossRef CAS PubMed.
- J. A. Atwood, 3rd, D. B. Weatherly, T. A. Minning, B. Bundy, C. Cavola, F. R. Opperdoes, R. Orlando and R. L. Tarleton, Science, 2005, 309, 473–476 CrossRef PubMed.
- H. M. Andrade, S. M. Murta, A. Chapeaurouge, J. Perales, P. Nirde and A. J. Romanha, J. Proteome Res., 2008, 7, 2357–2367 CrossRef PubMed.
- R. F. Menna-Barreto, D. G. Beghini, A. T. Ferreira, A. V. Pinto, S. L. De Castro and J. Perales, J. Proteomics, 2010, 73, 2306–2315 CrossRef CAS PubMed.
- R. L. Goncalves, R. F. Barreto, C. R. Polycarpo, F. R. Gadelha, S. L. Castro and M. F. Oliveira, J. Bioenerg. Biomembr., 2011, 43, 651–661 CrossRef CAS PubMed.
- C. A. Pereira, G. D. Alonso, M. C. Paveto, A. Iribarren, M. L. Cabanas, H. N. Torres and M. M. Flawia, J. Biol. Chem., 2000, 275, 1495–1501 CrossRef CAS PubMed.
- A. Parodi-Talice, R. Duran, N. Arrambide, V. Prieto, M. D. Pineyro, O. Pritsch, A. Cayota, C. Cervenansky and C. Robello, Int. J. Parasitol., 2004, 34, 881–886 CrossRef CAS PubMed.
- B. K. Kubata, Z. Kabututu, T. Nozaki, C. J. Munday, S. Fukuzumi, K. Ohkubo, M. Lazarus, T. Maruyama, S. K. Martin, M. Duszenko and Y. Urade, J. Exp. Med., 2002, 196, 1241–1251 CrossRef CAS PubMed.
- G. V. Brunoro, M. A. Caminha, A. T. Ferreira, V. Leprevost Fda, P. C. Carvalho, J. Perales, R. H. Valente and R. F. Menna-Barreto, J. Proteomics, 2015, 115, 58–65 CrossRef CAS PubMed.
- A. D. Magalhaes, S. Charneau, J. Paba, R. A. Guercio, A. R. Teixeira, J. M. Santana, M. V. Sousa and C. A. Ricart, Proteome Sci., 2008, 6, 24 CrossRef PubMed.
- E. S. Nakayasu, T. J. Sobreira, R. Torres, Jr., L. Ganiko, P. S. Oliveira, A. F. Marques and I. C. Almeida, J. Proteome Res., 2012, 11, 237–246 CrossRef CAS PubMed.
- A. Parodi-Talice, V. Monteiro-Goes, N. Arrambide, A. R. Avila, R. Duran, A. Correa, B. Dallagiovanna, A. Cayota, M. Krieger, S. Goldenberg and C. Robello, J. Mass Spectrom., 2007, 42, 1422–1432 CrossRef CAS PubMed.
- J. C. Amorim, M. Batista, E. S. da Cunha, A. C. R. Lucena, C. V. P. Lima, K. Sousa, M. A. Krieger and F. K. Marchini, Sci. Rep., 2017, 7, 9899 CrossRef PubMed.
- A. C. R. Lucena, J. C. Amorim, C. V. de Paula Lima, M. Batista, M. A. Krieger, L. M. F. de Godoy and F. K. Marchini, Cell Stress Chaperones, 2019, 24, 927–936 CrossRef CAS PubMed.
- C. C. Avila, S. N. Mule, L. Rosa-Fernandes, R. Viner, M. J. Barison, A. G. Costa-Martins, G. S. Oliveira, M. M. G. Teixeira, C. R. F. Marinho, A. M. Silber and G. Palmisano, Genes, 2018, 9, 413 CrossRef PubMed.
- R. M. Queiroz, S. Charneau, S. C. Mandacaru, V. Schwammle, B. D. Lima, P. Roepstorff and C. A. Ricart, Mol. Cell. Proteomics, 2014, 13, 3457–3472 CrossRef CAS PubMed.
- E. Rondinelli, R. Silva, J. F. Carvalho, C. M. de Almeida Soares, E. F. de Carvalho and F. T. de Castro, Exp. Parasitol., 1988, 66, 197–204 CrossRef CAS PubMed.
- R. L. Kessler, V. T. Contreras, N. P. Marliere, A. Aparecida Guarneri, L. H. Villamizar Silva, G. Mazzarotto, M. Batista, V. T. Soccol, M. A. Krieger and C. M. Probst, Mol. Microbiol., 2017, 104, 712–736 CrossRef CAS PubMed.
- E. M. Cordero, E. S. Nakayasu, L. G. Gentil, N. Yoshida, I. C. Almeida and J. F. da Silveira, J. Proteome Res., 2009, 8, 3642–3652 CrossRef CAS PubMed.
- H. Soblik, A. E. Younis, M. Mitreva, B. Y. Renard, M. Kirchner, F. Geisinger, H. Steen and N. W. Brattig, Mol. Cell. Proteomics, 2011, 10, M111 010157 CrossRef PubMed.
- N. O. Martins, R. T. Souza, E. M. Cordero, D. C. Maldonado, C. Cortez, M. M. Marini, E. R. Ferreira, E. Bayer-Santos, I. C. Almeida, N. Yoshida and J. F. Silveira, PLoS Neglected Trop. Dis., 2015, 9, e0004216 CrossRef PubMed.
- E. Bayer-Santos, C. Aguilar-Bonavides, S. P. Rodrigues, E. M. Cordero, A. F. Marques, A. Varela-Ramirez, H. Choi, N. Yoshida, J. F. da Silveira and I. C. Almeida, J. Proteome Res., 2013, 12, 883–897 CrossRef CAS PubMed.
- R. M. Queiroz, C. A. Ricart, M. O. Machado, I. M. Bastos, J. M. de Santana, M. V. de Sousa, P. Roepstorff and S. Charneau, Front. Chem., 2016, 4, 42 Search PubMed.
- J. Y. Brossas, J. E. N. Gulin, M. M. C. Bisio, M. Chapelle, C. Marinach-Patrice, M. Bordessoules, G. Palazon Ruiz, J. Vion, L. Paris, J. Altcheh and D. Mazier, PLoS One, 2017, 12, e0185504 CrossRef PubMed.
- K. S. Ribeiro, C. I. Vasconcellos, R. P. Soares, M. T. Mendes, C. C. Ellis, M. Aguilera-Flores, I. C. de Almeida, S. Schenkman, L. K. Iwai and A. C. Torrecilhas, J. Extracell. Vesicles, 2018, 7, 1463779 CrossRef PubMed.
- K. Ohyama, N. T. Huy, H. Yoshimi, N. Kishikawa, J. E. Nishizawa, Y. Roca, R. J. Revollo Guzman, F. U. Velarde, N. Kuroda and K. Hirayama, Parasite Immunol., 2016, 38, 609–617 CrossRef CAS PubMed.
- E. Ruiz-Lancheros, A. Rasoolizadeh, E. Chatelain, F. Garcia-Bournissen, S. Moroni, G. Moscatelli, J. Altcheh and M. Ndao, Open Forum Infect. Dis., 2018, 5, ofy236 Search PubMed.
- S. A. Kikuchi, C. L. Sodre, D. E. Kalume, C. G. Elias, A. L. Santos, M. de Nazare Soeiro, M. Meuser, A. Chapeaurouge, J. Perales and O. Fernandes, Exp. Parasitol., 2010, 126, 540–551 CrossRef CAS PubMed.
- J. San Francisco, I. Barria, B. Gutierrez, I. Neira, C. Munoz, H. Sagua, J. E. Araya, J. C. Andrade, A. Zailberger, A. Catalan, F. Remonsellez, J. L. Vega and J. Gonzalez, Microbes Infect., 2017, 19, 55–61 CrossRef CAS PubMed.
- G. S. de Oliveira, R. Kawahara, L. Rosa-Fernandes, S. N. Mule, C. C. Avila, M. M. G. Teixeira, M. R. Larsen and G. Palmisano, PLoS Neglected Trop. Dis., 2018, 12, e0006351 CrossRef PubMed.
- M. A. Caminha, V. M. B. de Lorena, W. de Oliveira Junior, J. Perales, P. C. Carvalho, D. B. Lima, M. Cavalcanti, S. M. Martins, R. H. Valente and R. F. S. Menna-Barreto, J. Proteomics, 2019, 194, 179–190 CrossRef CAS PubMed.
- F. Montalvao, D. O. Nascimento, M. P. Nunes, C. M. Koeller, A. Morrot, L. M. S. Lery, P. M. Bisch, S. M. R. Teixeira, R. Vasconcellos, L. Freire-de-Lima, M. F. Lopes, N. Heise, G. A. DosReis and C. G. Freire-de-Lima, Front. Immunol., 2018, 9, 671 CrossRef PubMed.
- M. M. Croken, S. C. Nardelli and K. Kim, Trends Parasitol., 2012, 28, 202–213 CrossRef CAS PubMed.
- G. F. Picchi, V. Zulkievicz, M. A. Krieger, N. T. Zanchin, S. Goldenberg and L. M. de Godoy, J. Proteome Res., 2017, 16, 1167–1179 CrossRef CAS PubMed.
- T. C. de Jesus, V. S. Nunes, C. Lopes Mde, D. E. Martil, L. K. Iwai, N. S. Moretti, F. C. Machado, M. L. de Lima-Stein, O. H. Thiemann, M. C. Elias, C. Janzen, S. Schenkman and J. P. da Cunha, J. Proteome Res., 2016, 15, 2039–2051 CrossRef CAS PubMed.
- J. de Oliveira Santos, A. A. Zuma, F. N. de Luna Vitorino, J. P. C. da Cunha, W. de Souza and M. C. M. Motta, Parasitology, 2019, 146, 543–552 CrossRef CAS PubMed.
- C. Ciavaglia Mdo, T. U. de Carvalho and W. de Souza, Biochem. Biophys. Res. Commun., 1993, 193, 718–721 CrossRef PubMed.
- M. E. Giorgi and R. M. de Lederkremer, Carbohydr. Res., 2011, 346, 1389–1393 CrossRef PubMed.
- J. A. Atwood, 3rd, T. Minning, F. Ludolf, A. Nuccio, D. B. Weatherly, G. Alvarez-Manilla, R. Tarleton and R. Orlando, J. Proteome Res., 2006, 5, 3376–3384 CrossRef PubMed.
- M. J. Alves, R. Kawahara, R. Viner, W. Colli, E. C. Mattos, M. Thaysen-Andersen, M. R. Larsen and G. Palmisano, J. Proteomics, 2017, 151, 182–192 CrossRef CAS PubMed.
- X. Yang and K. Qian, Nat. Rev. Mol. Cell Biol., 2017, 18, 452–465 CrossRef CAS PubMed.
- E. Torres-Gutiérrez, Y. Perez-Cervera, L. Camoin, E. Zenteno, M. O. Aquino-Gil, T. Lefebvre, M. Cabrera-Bravo, O. Reynoso-Ducoing, M. I. Bucio-Torres and P. M. Salazar-Schettino, Front. Endocrinol., 2019, 10, 199 CrossRef PubMed.
- H. Hahne, N. Sobotzki, T. Nyberg, D. Helm, V. S. Borodkin, D. M. van Aalten, B. Agnew and B. Kuster, J. Proteome Res., 2013, 12, 927–936 CrossRef CAS PubMed.
- A. Flotho and F. Melchior, Annu. Rev. Biochem., 2013, 82, 357–385 CrossRef CAS PubMed.
- J. C. Bayona, E. S. Nakayasu, M. Laverriere, C. Aguilar, T. J. Sobreira, H. Choi, A. I. Nesvizhskii, I. C. Almeida, J. J. Cazzulo and V. E. Alvarez, Mol. Cell. Proteomics, 2011, 10, M110 007369 CrossRef PubMed.
- I. Bludau and R. Aebersold, Nat. Rev. Mol. Cell Biol., 2020 DOI:10.1038/s41580-020-0231-2.
- D. G. Arias, M. D. Pineyro, A. A. Iglesias, S. A. Guerrero and C. Robello, J. Proteomics, 2015, 120, 95–104 CrossRef CAS PubMed.
- L. Piacenza, M. P. Zago, G. Peluffo, M. N. Alvarez, M. A. Basombrio and R. Radi, Int. J. Parasitol., 2009, 39, 1455–1464 CrossRef CAS PubMed.
- L. Piacenza, G. Peluffo, M. N. Alvarez, J. M. Kelly, S. R. Wilkinson and R. Radi, Biochem. J., 2008, 410, 359–368 CrossRef CAS PubMed.
- M. P. Zago, Y. M. Hosakote, S. J. Koo, M. Dhiman, M. D. Pineyro, A. Parodi-Talice, M. A. Basombrio, C. Robello and N. J. Garg, Infect. Immun., 2016, 84, 1842–1856 CrossRef CAS PubMed.
- A. Trochine, G. Alvarez, S. Corre, P. Faral-Tello, R. Duran, C. I. Batthyany, H. Cerecetto, M. Gonzalez and C. Robello, Exp. Parasitol., 2014, 140, 33–38 CrossRef CAS PubMed.
- B. S. Hall and S. R. Wilkinson, Antimicrob. Agents Chemother., 2012, 56, 115–123 CrossRef CAS PubMed.
- F. Diaz-Viraque, M. L. Chiribao, A. Trochine, F. Gonzalez-Herrera, C. Castillo, A. Liempi, U. Kemmerling, J. D. Maya and C. Robello, Front. Immunol., 2018, 9, 456 CrossRef PubMed.
- G. V. Brunoro, V. M. Faca, M. A. Caminha, A. T. Ferreira, M. Trugilho, K. C. de Moura, J. Perales, R. H. Valente and R. F. Menna-Barreto, PLoS Neglected Trop. Dis., 2016, 10, e0004951 CrossRef PubMed.
- G. A. L. Vieira, M. Silva, L. O. Regasini, F. Cotinguiba, H. J. Laure, J. C. Rosa, M. Furlan and R. M. B. Cicarelli, Braz. J. Infect. Dis., 2018, 22, 208–218 CrossRef PubMed.
- A. J. Roberts, L. S. Torrie, S. Wyllie and A. H. Fairlamb, Biochem. J., 2014, 459, 323–332 CrossRef CAS PubMed.
- A. J. Roberts and A. H. Fairlamb, Sci. Rep., 2016, 6, 31078 CrossRef CAS PubMed.
- P. Buscher, G. Cecchi, V. Jamonneau and G. Priotto, Lancet, 2017, 390, 2397–2409 CrossRef.
- J. R. Franco, P. P. Simarro, A. Diarra and J. G. Jannin, Clin. Epidemiol., 2014, 6, 257–275 Search PubMed.
- S. Mogk, A. Meiwes, S. Shtopel, U. Schraermeyer, M. Lazarus, B. Kubata, H. Wolburg and M. Duszenko, PLoS One, 2014, 9, e91372 CrossRef PubMed.
- S. Mogk, A. Meiwes, C. M. Bosselmann, H. Wolburg and M. Duszenko, Trends Parasitol., 2014, 30, 470–477 CrossRef CAS PubMed.
- A. H. Fairlamb, Trends Parasitol., 2003, 19, 488–494 CrossRef CAS PubMed.
- A. H. Fairlamb, Drugs Today, 2019, 55, 705–712 CrossRef CAS PubMed.
- A. H. Fairlamb and D. Horn, Trends Parasitol., 2018, 34, 481–492 CrossRef CAS PubMed.
- M. D. Urbaniak, M. L. Guther and M. A. Ferguson, PLoS One, 2012, 7, e36619 CrossRef CAS PubMed.
- M. Dejung, I. Subota, F. Bucerius, G. Dindar, A. Freiwald, M. Engstler, M. Boshart, F. Butter and C. J. Janzen, PLoS Pathog., 2016, 12, e1005439 CrossRef PubMed.
- M. M. Shimogawa, E. A. Saada, A. A. Vashisht, W. D. Barshop, J. A. Wohlschlegel and K. L. Hill, Mol. Cell. Proteomics, 2015, 14, 1977–1988 CrossRef CAS PubMed.
- M. Tinti, M. L. S. Guther, T. W. M. Crozier, A. I. Lamond and M. A. J. Ferguson, Wellcome Open Res., 2019, 4, 152 Search PubMed.
- F. Butter, F. Bucerius, M. Michel, Z. Cicova, M. Mann and C. J. Janzen, Mol. Cell. Proteomics, 2013, 12, 172–179 CrossRef PubMed.
- K. Gunasekera, D. Wuthrich, S. Braga-Lagache, M. Heller and T. Ochsenreiter, BMC Genomics, 2012, 13, 556 CrossRef CAS PubMed.
- D. Horn, Mol. Biochem. Parasitol., 2014, 195, 123–129 CrossRef CAS PubMed.
- C. J. Merrick and M. T. Duraisingh, Mol. Microbiol., 2006, 62, 612–620 CrossRef CAS PubMed.
- H. Reis, M. Schwebs, S. Dietz, C. J. Janzen and F. Butter, Nucleic Acids Res., 2018, 46, 2820–2833 CrossRef CAS PubMed.
- T. K. Smith, F. Bringaud, D. P. Nolan and L. M. Figueiredo, F1000Research, 2017, 6, F1000 Faculty Rev-683 Search PubMed.
- C. Goos, M. Dejung, C. J. Janzen, F. Butter and S. Kramer, PLoS One, 2017, 12, e0181884 CrossRef PubMed.
- K. M. Tyler, A. Fridberg, K. M. Toriello, C. L. Olson, J. A. Cieslak, T. L. Hazlett and D. M. Engman, J. Cell Sci., 2009, 122, 859–866 CrossRef CAS PubMed.
- A. I. Sharma, C. L. Olson and D. M. Engman, Pathogens, 2017, 6, E39 CrossRef PubMed.
- A. R. Siafakas, L. C. Wright, T. C. Sorrell and J. T. Djordjevic, Eukaryotic Cell, 2006, 5, 488–498 CrossRef CAS PubMed.
- C. P. Ooi, B. Rotureau, S. Gribaldo, C. Georgikou, D. Julkowska, T. Blisnick, S. Perrot, I. Subota and P. Bastin, PLoS One, 2015, 10, e0133676 CrossRef PubMed.
- J. C. Munday, A. A. Eze, N. Baker, L. Glover, C. Clucas, D. Aguinaga Andres, M. J. Natto, I. A. Teka, J. McDonald, R. S. Lee, F. E. Graf, P. Ludin, R. J. Burchmore, C. M. Turner, A. Tait, A. MacLeod, P. Maser, M. P. Barrett, D. Horn and H. P. De Koning, J. Antimicrob. Chemother., 2014, 69, 651–663 CrossRef CAS PubMed.
- J. D. Unciti-Broceta, J. L. Arias, J. Maceira, M. Soriano, M. Ortiz-Gonzalez, J. Hernandez-Quero, M. Munoz-Torres, H. P. de Koning, S. Magez and J. A. Garcia-Salcedo, PLoS Pathog., 2015, 11, e1004942 CrossRef PubMed.
- N. J. Temperton, S. R. Wilkinson, D. J. Meyer and J. M. Kelly, Mol. Biochem. Parasitol., 1998, 96, 167–176 CrossRef CAS PubMed.
- N. G. Kolev, J. B. Franklin, S. Carmi, H. Shi, S. Michaeli and C. Tschudi, PLoS Pathog., 2010, 6, e1001090 CrossRef PubMed.
- T. W. M. Crozier, M. Tinti, R. J. Wheeler, T. Ly, M. A. J. Ferguson and A. I. Lamond, Mol. Cell. Proteomics, 2018, 17, 1184–1195 CrossRef CAS PubMed.
- J. Bonnet, C. Garcia, T. Leger, M. P. Couquet, P. Vignoles, G. Vatunga, J. Ndung'u, C. Boudot, S. Bisser and B. Courtioux, J. Proteomics, 2019, 196, 150–161 CrossRef CAS PubMed.
- T. Teesalu, A. Kulla, A. Simisker, V. Siren, D. A. Lawrence, T. Asser and A. Vaheri, Thromb. Haemostasis, 2004, 92, 358–368 CrossRef CAS PubMed.
- M. Uhlen, L. Fagerberg, B. M. Hallstrom, C. Lindskog, P. Oksvold, A. Mardinoglu, A. Sivertsson, C. Kampf, E. Sjostedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P. H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Ponten, Science, 2015, 347, 1260419 CrossRef PubMed.
- O. V. Nikolskaia, Y. V. Kim, O. Kovbasnjuk, K. J. Kim and D. J. Grab, Int. J. Parasitol., 2006, 36, 513–519 CrossRef CAS PubMed.
- H. M. Kariithi, S. Boeren, E. K. Murungi, J. M. Vlak and A. M. Abd-Alla, Parasites Vectors, 2016, 9, 424 CrossRef PubMed.
- J. Van Den Abbeele, G. Caljon, K. De Ridder, P. De Baetselier and M. Coosemans, PLoS Pathog., 2010, 6, e1000926 CrossRef PubMed.
- S. B. Ogueta, G. C. Macintosh and M. T. Tellez-Inon, J. Eukaryotic Microbiol., 1998, 45, 392–396 CrossRef CAS PubMed.
- S. Chaubey, M. Grover and U. Tatu, J. Biol. Chem., 2014, 289, 16662–16674 CrossRef CAS PubMed.
- J. S. Seeler and A. Dejean, Nat. Rev. Mol. Cell Biol., 2003, 4, 690–699 CrossRef CAS PubMed.
- M. Engstler, T. Pfohl, S. Herminghaus, M. Boshart, G. Wiegertjes, N. Heddergott and P. Overath, Cell, 2007, 131, 505–515 CrossRef CAS PubMed.
- C. Gadelha, W. Zhang, J. W. Chamberlain, B. T. Chait, B. Wickstead and M. C. Field, Mol. Cell. Proteomics, 2015, 14, 1911–1926 CrossRef CAS PubMed.
- L. Kohl, D. Robinson and P. Bastin, EMBO J., 2003, 22, 5336–5346 CrossRef CAS PubMed.
- I. Subota, D. Julkowska, L. Vincensini, N. Reeg, J. Buisson, T. Blisnick, D. Huet, S. Perrot, J. Santi-Rocca, M. Duchateau, V. Hourdel, J. C. Rousselle, N. Cayet, A. Namane, J. Chamot-Rooke and P. Bastin, Mol. Cell. Proteomics, 2014, 13, 1769–1786 CrossRef CAS PubMed.
- W. Basu Ball, J. K. Neff and V. M. Gohil, FEBS Lett., 2018, 592, 1273–1290 CrossRef CAS PubMed.
- H. F. Tian, J. M. Feng and J. F. Wen, BMC Evol. Biol., 2012, 12, 32 CrossRef CAS PubMed.
- D. Schadeli, M. Serricchio, H. Ben Hamidane, A. Loffreda, A. Hemphill, T. Beneke, E. Gluenz, J. Graumann and P. Butikofer, FASEB J., 2019, 33, 13161–13175 CrossRef CAS PubMed.
- M. D. Urbaniak, D. M. Martin and M. A. Ferguson, J. Proteome Res., 2013, 12, 2233–2244 CrossRef CAS PubMed.
- I. R. Nett, L. Davidson, D. Lamont and M. A. Ferguson, Eukaryotic Cell, 2009, 8, 617–626 CrossRef CAS PubMed.
- I. R. Nett, D. M. Martin, D. Miranda-Saavedra, D. Lamont, J. D. Barber, A. Mehlert and M. A. Ferguson, Mol. Cell. Proteomics, 2009, 8, 1527–1538 CrossRef CAS PubMed.
- N. F. Wilson and P. A. Lefebvre, Eukaryotic Cell, 2004, 3, 1307–1319 CrossRef CAS PubMed.
- R. T. Jacobs, B. Nare, S. A. Wring, M. D. Orr, D. Chen, J. M. Sligar, M. X. Jenks, R. A. Noe, T. S. Bowling, L. T. Mercer, C. Rewerts, E. Gaukel, J. Owens, R. Parham, R. Randolph, B. Beaudet, C. J. Bacchi, N. Yarlett, J. J. Plattner, Y. Freund, C. Ding, T. Akama, Y. K. Zhang, R. Brun, M. Kaiser, I. Scandale and R. Don, PLoS Neglected Trop. Dis., 2011, 5, e1151 CrossRef CAS PubMed.
- D. C. Jones, B. J. Foth, M. D. Urbaniak, S. Patterson, H. B. Ong, M. Berriman and A. H. Fairlamb, PLoS Neglected Trop. Dis., 2015, 9, e0004299 CrossRef PubMed.
- J. A. Frearson, S. Brand, S. P. McElroy, L. A. Cleghorn, O. Smid, L. Stojanovski, H. P. Price, M. L. Guther, L. S. Torrie, D. A. Robinson, I. Hallyburton, C. P. Mpamhanga, J. A. Brannigan, A. J. Wilkinson, M. Hodgkinson, R. Hui, W. Qiu, O. G. Raimi, D. M. van Aalten, R. Brenk, I. H. Gilbert, K. D. Read, A. H. Fairlamb, M. A. Ferguson, D. F. Smith and P. G. Wyatt, Nature, 2010, 464, 728–732 CrossRef CAS PubMed.
- H. P. Price, M. R. Hodgkinson, M. H. Wright, E. W. Tate, B. A. Smith, M. Carrington, M. Stark and D. F. Smith, Biochim. Biophys. Acta, 2012, 1823, 1178–1191 CrossRef CAS PubMed.
- M. H. Wright, D. Paape, H. P. Price, D. F. Smith and E. W. Tate, ACS Infect. Dis., 2016, 2, 427–441 CrossRef CAS PubMed.
- M. Berriman, E. Ghedin, C. Hertz-Fowler, G. Blandin, H. Renauld, D. C. Bartholomeu, N. J. Lennard, E. Caler, N. E. Hamlin, B. Haas, U. Bohme, L. Hannick, M. A. Aslett, J. Shallom, L. Marcello, L. Hou, B. Wickstead, U. C. Alsmark, C. Arrowsmith, R. J. Atkin, A. J. Barron, F. Bringaud, K. Brooks, M. Carrington, I. Cherevach, T. J. Chillingworth, C. Churcher, L. N. Clark, C. H. Corton, A. Cronin, R. M. Davies, J. Doggett, A. Djikeng, T. Feldblyum, M. C. Field, A. Fraser, I. Goodhead, Z. Hance, D. Harper, B. R. Harris, H. Hauser, J. Hostetler, A. Ivens, K. Jagels, D. Johnson, J. Johnson, K. Jones, A. X. Kerhornou, H. Koo, N. Larke, S. Landfear, C. Larkin, V. Leech, A. Line, A. Lord, A. Macleod, P. J. Mooney, S. Moule, D. M. Martin, G. W. Morgan, K. Mungall, H. Norbertczak, D. Ormond, G. Pai, C. S. Peacock, J. Peterson, M. A. Quail, E. Rabbinowitsch, M. A. Rajandream, C. Reitter, S. L. Salzberg, M. Sanders, S. Schobel, S. Sharp, M. Simmonds, A. J. Simpson, L. Tallon, C. M. Turner, A. Tait, A. R. Tivey, S. Van Aken, D. Walker, D. Wanless, S. Wang, B. White, O. White, S. Whitehead, J. Woodward, J. Wortman, M. D. Adams, T. M. Embley, K. Gull, E. Ullu, J. D. Barry, A. H. Fairlamb, F. Opperdoes, B. G. Barrell, J. E. Donelson, N. Hall, C. M. Fraser, S. E. Melville and N. M. El-Sayed, Science, 2005, 309, 416–422 CrossRef CAS PubMed.
- C. Tiengwe, L. Marcello, H. Farr, C. Gadelha, R. Burchmore, J. D. Barry, S. D. Bell and R. McCulloch, PLoS One, 2012, 7, e32674 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |