
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
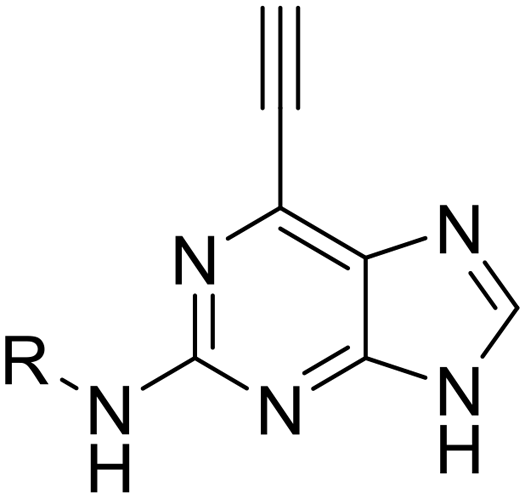
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Arylamino-6-ethynylpurines are cysteine-targeting irreversible inhibitors of Nek2 kinase†
Christopher J.
Matheson‡
a,
Christopher R.
Coxon‡
 a,
Richard
Bayliss
cd,
Kathy
Boxall‡
b,
Benoit
Carbain
a,
Andrew M.
Fry
c,
Ian R.
Hardcastle
a,
Suzannah J.
Harnor
a,
Corine
Mas-Droux
d,
David R.
Newell
e,
Mark W.
Richards
c,
Mangaleswaran
Sivaprakasam
a,
David
Turner
a,
Roger J.
Griffin§
a,
Bernard T.
Golding
a and
Céline
Cano
*a
a,
Richard
Bayliss
cd,
Kathy
Boxall‡
b,
Benoit
Carbain
a,
Andrew M.
Fry
c,
Ian R.
Hardcastle
a,
Suzannah J.
Harnor
a,
Corine
Mas-Droux
d,
David R.
Newell
e,
Mark W.
Richards
c,
Mangaleswaran
Sivaprakasam
a,
David
Turner
a,
Roger J.
Griffin§
a,
Bernard T.
Golding
a and
Céline
Cano
*a
aCancer Research UK Newcastle Drug Discovery Unit, Chemistry, School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, UK. E-mail: celine.cano@ncl.ac.uk; Tel: +44 (0)191 208 7060
bCancer Research UK Cancer Therapeutics Unit, The Institute of Cancer Research, Sutton, UK
cSchool of Molecular and Cellular Biology, The Astbury Centre for Structural Molecular Biology, University of Leeds, UK
dSection of Structural Biology, The Institute of Cancer Research, Sutton, UK
eCancer Research UK Newcastle Drug Discovery Unit, Translational and Clinical Research Institute, Newcastle University Centre for Cancer, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, UK
First published on 22nd May 2020
Abstract
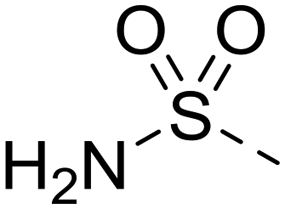
Renewed interest in covalent inhibitors of enzymes implicated in disease states has afforded several agents targeted at protein kinases of relevance to cancers. We now report the design, synthesis and biological evaluation of 6-ethynylpurines that act as covalent inhibitors of Nek2 by capturing a cysteine residue (Cys22) close to the catalytic domain of this protein kinase. Examination of the crystal structure of the non-covalent inhibitor 3-((6-cyclohexylmethoxy-7H-purin-2-yl)amino)benzamide in complex with Nek2 indicated that replacing the alkoxy with an ethynyl group places the terminus of the alkyne close to Cys22 and in a position compatible with the stereoelectronic requirements of a Michael addition. A series of 6-ethynylpurines was prepared and a structure activity relationship (SAR) established for inhibition of Nek2. 6-Ethynyl-N-phenyl-7H-purin-2-amine [IC50 0.15 μM (Nek2)] and 4-((6-ethynyl-7H-purin-2-yl)amino)benzenesulfonamide (IC50 0.14 μM) were selected for determination of the mode of inhibition of Nek2, which was shown to be time-dependent, not reversed by addition of ATP and negated by site directed mutagenesis of Cys22 to alanine. Replacement of the ethynyl group by ethyl or cyano abrogated activity. Variation of substituents on the N-phenyl moiety for 6-ethynylpurines gave further SAR data for Nek2 inhibition. The data showed little correlation of activity with the nature of the substituent, indicating that after sufficient initial competitive binding to Nek2 subsequent covalent modification of Cys22 occurs in all cases. A typical activity profile was that for 2-(3-((6-ethynyl-9H-purin-2-yl)amino)phenyl)acetamide [IC50 0.06 μM (Nek2); GI50 (SKBR3) 2.2 μM] which exhibited >5–10-fold selectivity for Nek2 over other kinases; it also showed > 50% growth inhibition at 10 μM concentration against selected breast and leukaemia cell lines. X-ray crystallographic analysis confirmed that binding of the compound to the Nek2 ATP-binding site resulted in covalent modification of Cys22. Further studies confirmed that 2-(3-((6-ethynyl-9H-purin-2-yl)amino)phenyl)acetamide has the attributes of a drug-like compound with good aqueous solubility, no inhibition of hERG at 25 μM and a good stability profile in human liver microsomes. It is concluded that 6-ethynylpurines are promising agents for cancer treatment by virtue of their selective inhibition of Nek2.
Introduction
The renaissance in the development of inhibitors that bind covalently to a target enzyme either irreversibly or slowly reversibly has yielded several licensed agents, especially in the oncology area. As of 2019, six irreversible protein kinase inhibitors have received FDA approval for use in oncology (afatinib 1, osimertinib, neratinib 2 and ibrutinib, dacomitinib and acalabrutinib, Fig. 1A). Afatanib (1), licensed in 2013 for treatment of metastatic NSCLC through irreversible inhibition of EGFR, and neratinib (2), approved in 2017 for treatment of HER2+ breast cancer, were designed as modifications of gefitinib (3) by installation of electrophilic acrylamide groups that target an active site cysteine. Protein kinases are prominent targets for covalent inhibitors, which owing to the high concentrations of ATP in cells (∼1–10 mM) may be more effective than competitive inhibitors because of irreversible ATP-blockade.1 A range of warhead groups has been employed in irreversible protein kinase inhibitors, for example, acrylamides, alkynes, quinones and epoxides.2 Previously, our group discovered the vinyl sulfone (NU6300) as the first irreversible inhibitor of CDK2 that was shown to react covalently with an active site lysine residue (Lys89) in the selectivity pocket.3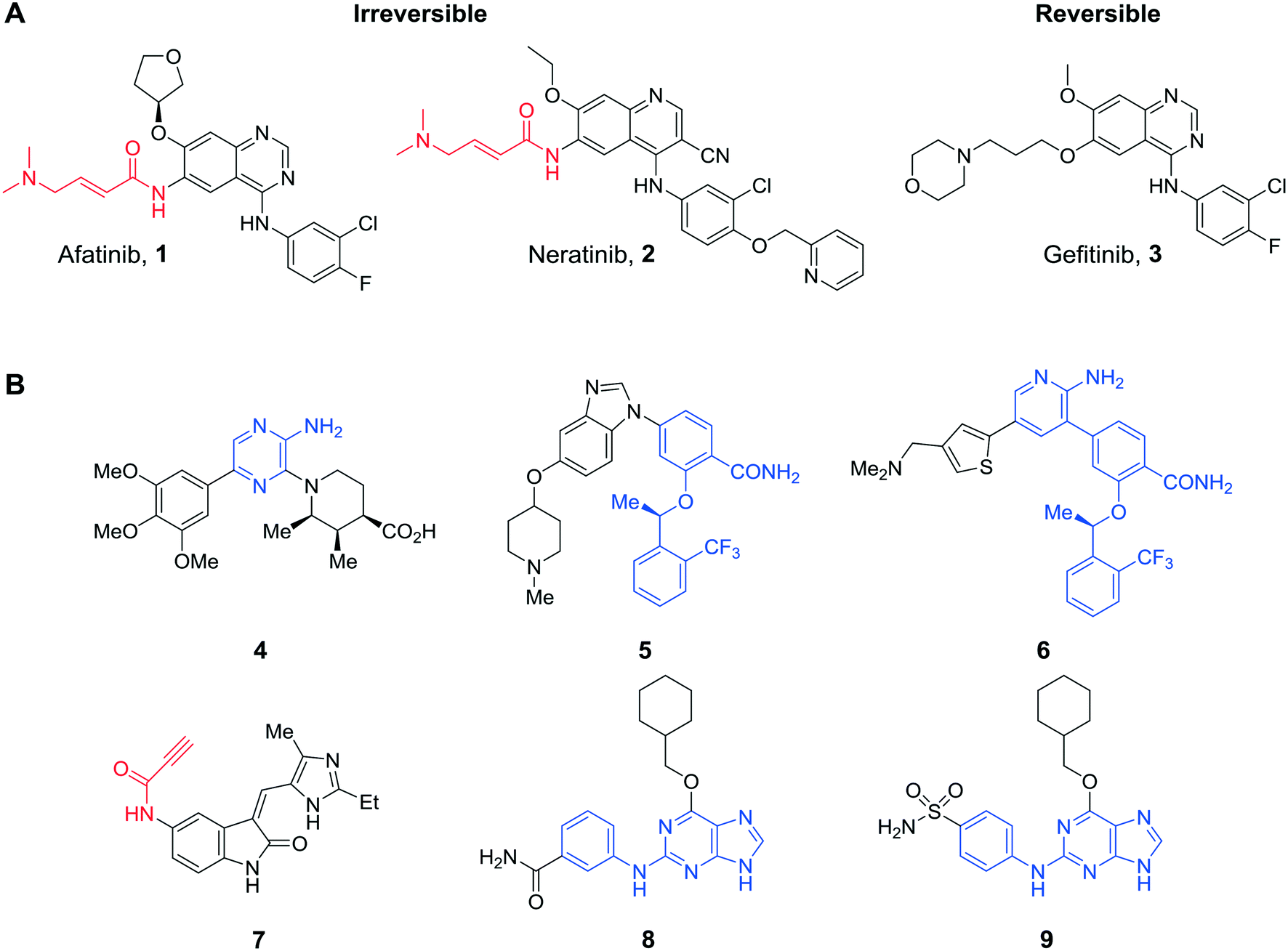 | ||
| Fig. 1 A) Rationalisation of the modification of gefitinib (reversible) to afford afatinib and neratinib (irreversible). B) Reversible and irreversible reported inhibitors of Nek2 kinase. For each case, covalent ‘warhead’ groups are shown in red and core scaffolds in blue. | ||
Nek2 is the closest human homologue of the protein kinase encoded by NIMA (never-in-mitosis gene A) in the fungus Aspergillus nidulans. The human protein is a serine–threonine kinase with multiple functions including a role in the mitotic spindle assembly. Overexpression of Nek2 may result in deregulation of the mitotic machinery leading to chromatid segregation errors, aneuploidy and chromosomal instability, common genetic abnormalities observed in tumour cells. Nek2 has been associated with tumorigenesis through overexpression, which has been linked to Ewing's osteosarcoma, diffuse large B-cell lymphoma, breast, ovarian and colorectal tumours.4–9
Inhibition of Nek2 in a number of tumour cell lines causes growth suppression and apoptosis. Reversible ATP-competitive Nek2 kinase inhibitors have been reported based on different structural classes, including: aminopyrazines (e.g., 4 IC50 = 0.23 μM),10 but with poor Nek2-selectivity over Plk1; benzimidazoles that exhibited >100-fold selectivity for Nek2 over Plk1 (e.g., 5 IC50 = 0.36 μM), yet had poor cellular activity,11 and a hybrid combining a core aminopyrazine moiety with side-chains from the benzimidazole series 6 (IC50 = 0.022 μM)12 (Fig. 1B), which was potent and Nek2-selective. As Nek2 contains a non-catalytic cysteine (Cys22) close to the catalytic domain that is unique to the Nek family, this residue can be targeted for irreversible binding. Only 10 other kinases have a cysteine in a similar position.13 One irreversible Nek2 inhibitor has been published (JH295, 7 IC50 = 0.77 μM),13 which targeted Cys22 via an N-arylpropiolamide Michael acceptor and was reported to be selective against other mitotic kinases, including CDK1, PLK1, Aurora B and Mps1. Herein, we describe the design and synthesis of a new class of selective tool compounds that covalently and irreversibly inhibit the Nek2 protein kinase. Our studies provide an insight into the role of Nek2 in cancer and address the question as to whether Nek2 is truly an attractive target for anti-cancer drug design.
Results and discussion
To improve understanding of the role of Nek2 in cancer, we sought a selective inhibitor of Nek2 by targeting the active site Cys22 residue. We have discovered covalent inhibitors superior to compound 7, which was non-selective within a subset of the kinome investigated here (see ESI†) and was therefore inadequate for further studies. In previous work we developed a series of 6-substituted-2-arylaminopurines that reversibly inhibited Nek2 (IC50 = 0.27–24 μM) and were selective (in some cases >10-fold vs. CDK2).14 This work optimised interactions between the 2-arylamino group and the specificity pocket of the enzyme and revealed that basic or polar substituents e.g. tertiary amines, carboxamides, ureas and sulfonamides conferred good potency (e.g., 8; IC50 = 19 μM (Nek2)). The crystal structure of 8 in complex with Nek2 (Fig. 2A) revealed that the compound binds via a classical hydrogen bonding triplet between the purine N9–H, N3 and C2–NH, and the main chain of kinase hinge region residues Cys89 and Glu87. Furthermore, the 6-alkoxy substituent was adjudged to be non-critical for binding affinity.14 Closer inspection of the structure of the 2-arylamino-6-alkoxypurine 8 bound to Nek2 revealed that the P-loop cysteine (Cys22) is closely positioned to a putative superimposed 6-ethynyl (cyan) substituent (4.4 Å) (Fig. 2A). Moreover, the predicted angle of attack (74°) of cysteine SH at the terminus of the alkyne suggests that only a small realignment of the cysteine is required to achieve maximal orbital overlap with the alkyne π*. Hence, replacement of the 6-position substituent with an ethynyl group was expected to deliver an irreversible inhibitor (Fig. 2B).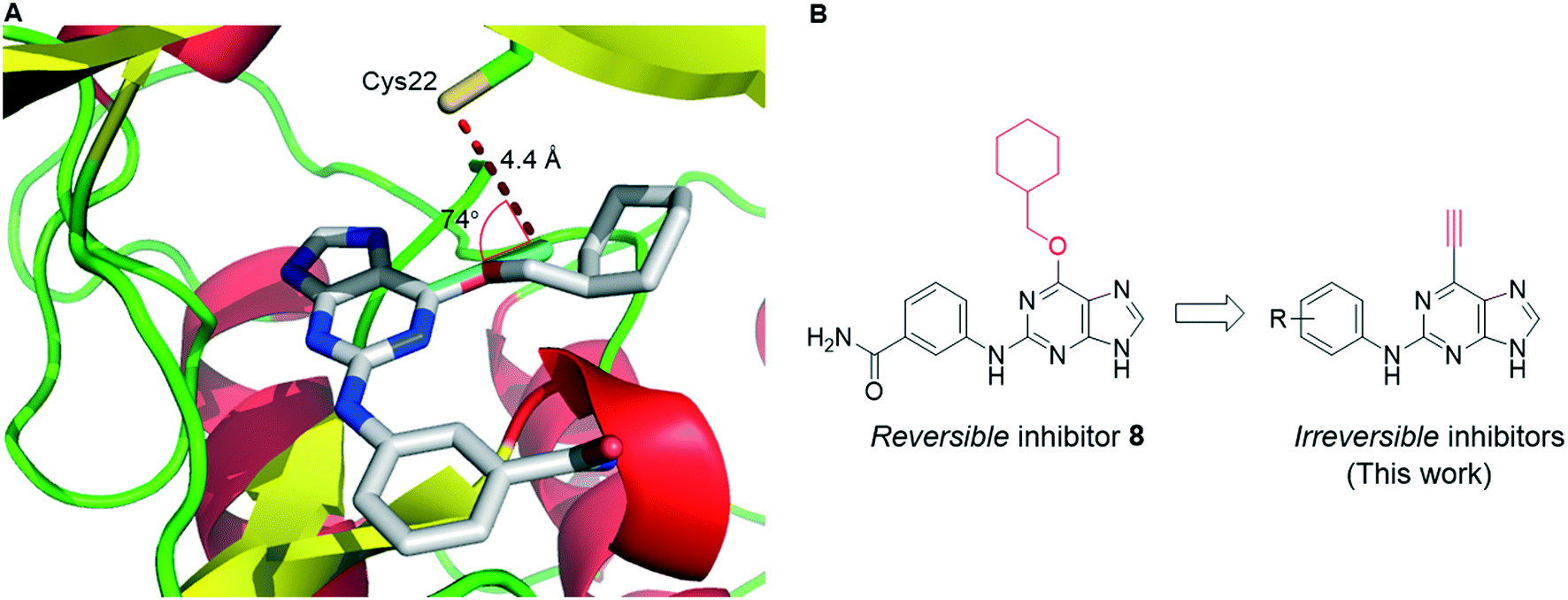 | ||
| Fig. 2 A) X-ray structure (PDB code: 5 M51) of 2-arylamino-6-alkoxypurine 8 (grey) in complex with Nek2, superimposed with a putative 6-ethynylpurine (cyan) reveals that the alkyne is positioned for covalent interaction with cysteine 22. B) The proposed approach to deliver a covalently-reactive Nek2 inhibitor. | ||
Previous reports, including our model study,15 have indicated that 6-ethynyl- and 6-vinyl-purines react via conjugate addition with nitrogen, oxygen and sulfur nucleophiles facilitated in part by the electron deficient purine.16 Such compounds were also found to exhibit profound cytotoxicity in chronic myeloid leukaemia cell lines, which was postulated to arise by attack of nucleophilic amino acid residues on the electrophilic alkyne or alkene.16a
To build upon these foundations, a focussed series of derivatives was prepared that combined side chains known to confer potency in the competitive inhibitor class with a ‘warhead’ capable of covalently reacting with cysteine residues. The synthesis of 2-arylamino-6-ethynyl- and 6-vinyl-purines was therefore undertaken, along with appropriate control compounds, to explore their efficacy as irreversible Nek2 inhibitors and potential applications.
a) Synthesis of 6-ethynylpurine probe molecules
Initially, two 6-ethynylpurines bearing 2-arylamino groups were prepared (Scheme 1) as prototypes for irreversible inhibition of Nek2. Subjecting 2-fluoro-6-chloro-(9-tetrahydropyranyl)-purine (10)17 to Sonogashira cross-coupling conditions afforded near quantitative yields of 11 when 3 mol% of Pd(PPh3)2Cl2 and 2 mol% of CuI were employed with triisopropylsilylethyne (TIPS-ethyne, Scheme 1). The tetrahydropyranyl group was removed by acidic hydrolysis of 11 to give 12 (96% yield) as a central scaffold for further elaboration. Nucleophilic aromatic substitution (SNAr) of the 2-fluoro substituent by aniline nucleophiles under previously established conditions18 gave 2-arylamino-6-triisopropylsilylethynylpurines (13, 14). The silyl group was removed by treatment with fluoride (either TBAF or KF with 18-crown-6) affording 2-arylamino-6-ethynylpurines 23 and 24, albeit in a modest isolated yield. It was noted that if the TIPS moiety was removed from the ethynyl group at an earlier stage, then nucleophilic aromatic substitution at the 2-position of the intermediate purine failed to proceed.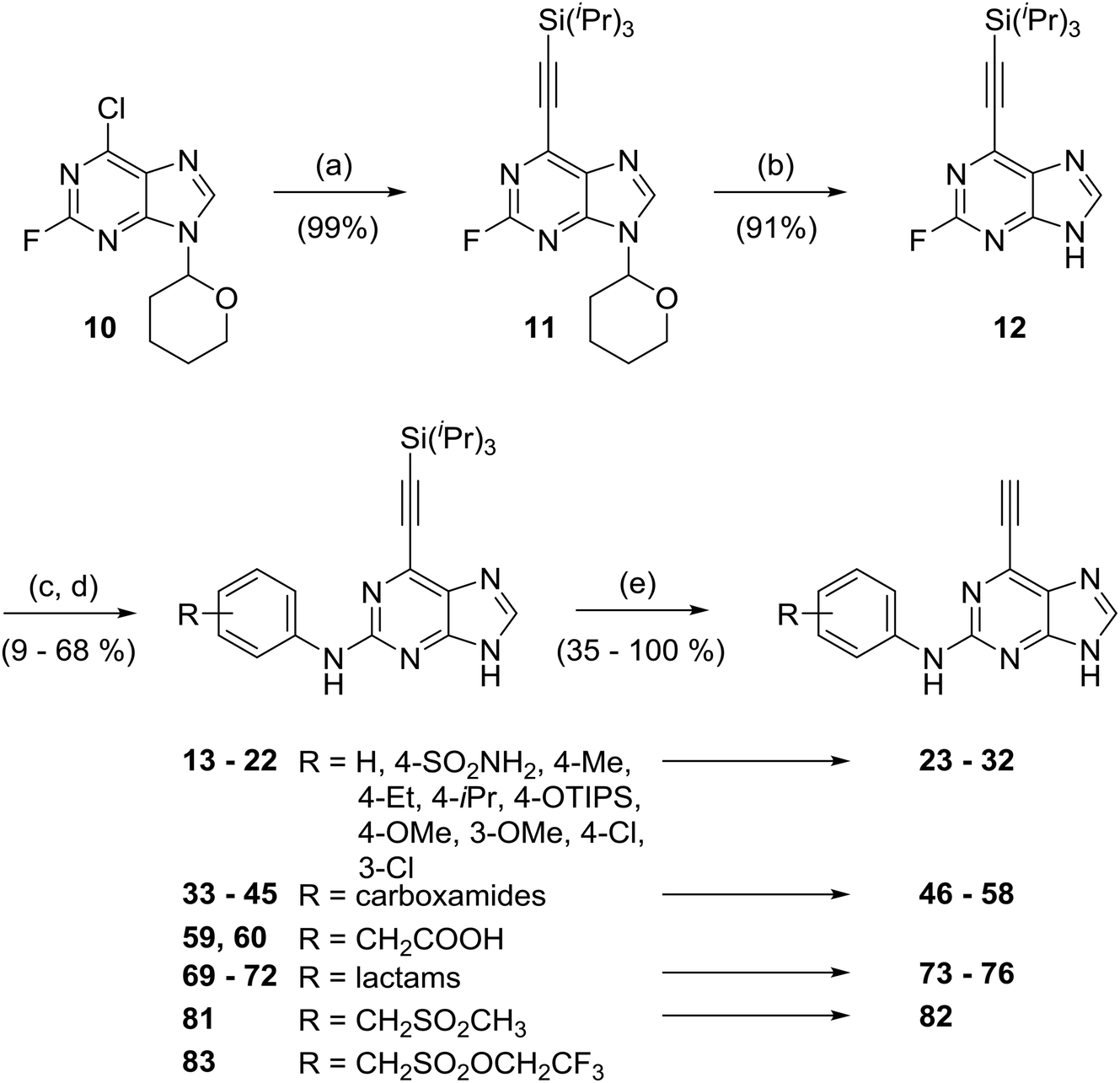 | ||
| Scheme 1 General route to 6-ethynyl-2-arylaminopurines. Reagents and conditions: (a) triisopropylsilylacetylene, Pd(PPh3)Cl2, CuI, Et3N, THF, RT, 18 h; (b) TFA, iPrOH, H2O, 100 °C, 2 h; (c) anilines, TFA, TFE, 80 °C, 18–27 h or MW 140 °C, 2 h; (d) TFA, reflux, 18 h (compounds 33 and 34); (e) TBAF, THF, RT, 5 min or KF, 18-crown-6, THF, RT, 24 h. | ||
To establish preliminary SARs at the 2-position, a series of compounds (25–32) was designed, bearing small alkyl, ether and halide substituents on the 2-arylamino ring, intended to probe the interaction of this structural element with the kinase. To avoid intractable reaction mixtures, potentially reactive heteroatoms (e.g. hydroxyl groups as in compound 18) required protection (e.g. for hydroxyl, TIPS was used and removed concomitant with ethynyl group deprotection) prior to participation in the SNAr step. In previous studies, carboxamides (e.g., 8) and sulfonamides (e.g.9) were generally found to afford the greatest Nek2 inhibitory activity and selectivity.14 The scope of this study was therefore broadened through the synthesis of a number of more complex analogues derived from the corresponding anilines (see ESI† for synthesis of the required anilines). Where necessary, coupling of 4-methoxybenzyl (PMB)-protected aminobenzamides to 2-fluoropurine (12) was followed by removal of the PMB-group using TFA at reflux prior to alkyne-deprotection (Scheme 1). This series of compounds allowed for possible additional interactions between e.g. basic groups of carboxamides (46–58) and the selectivity surface, which was previously found to confer higher potency and selectivity for Nek2 over other related kinases. This series included a carboxamide derivative (58) with a gem-dimethyl group.
Additional carboxamides were generated through coupling of 3- and 4-aminophenylacetic acid to 2-fluoropurine 12 under standard TFA/TFE-mediated conditions. This resulted in the formation of trifluoroethyl esters, which were hydrolysed under basic conditions to the required 3- or 4-substituted acids 59 and 60 without isolation (Scheme 1). The respective carboxylic acids were transformed into carboxamides 66–68 using 1,1′-carbonyldiimidazole (CDI) and the appropriate amine, followed by acidic deprotection of PMB-groups as required, and final removal of the silyl protecting groups (Scheme 2). To explore the consequences of conformational restrictions at the carboxamide side chain, a small series of purines bearing lactam groups (73–76) was prepared by the approach outlined in Scheme 1.
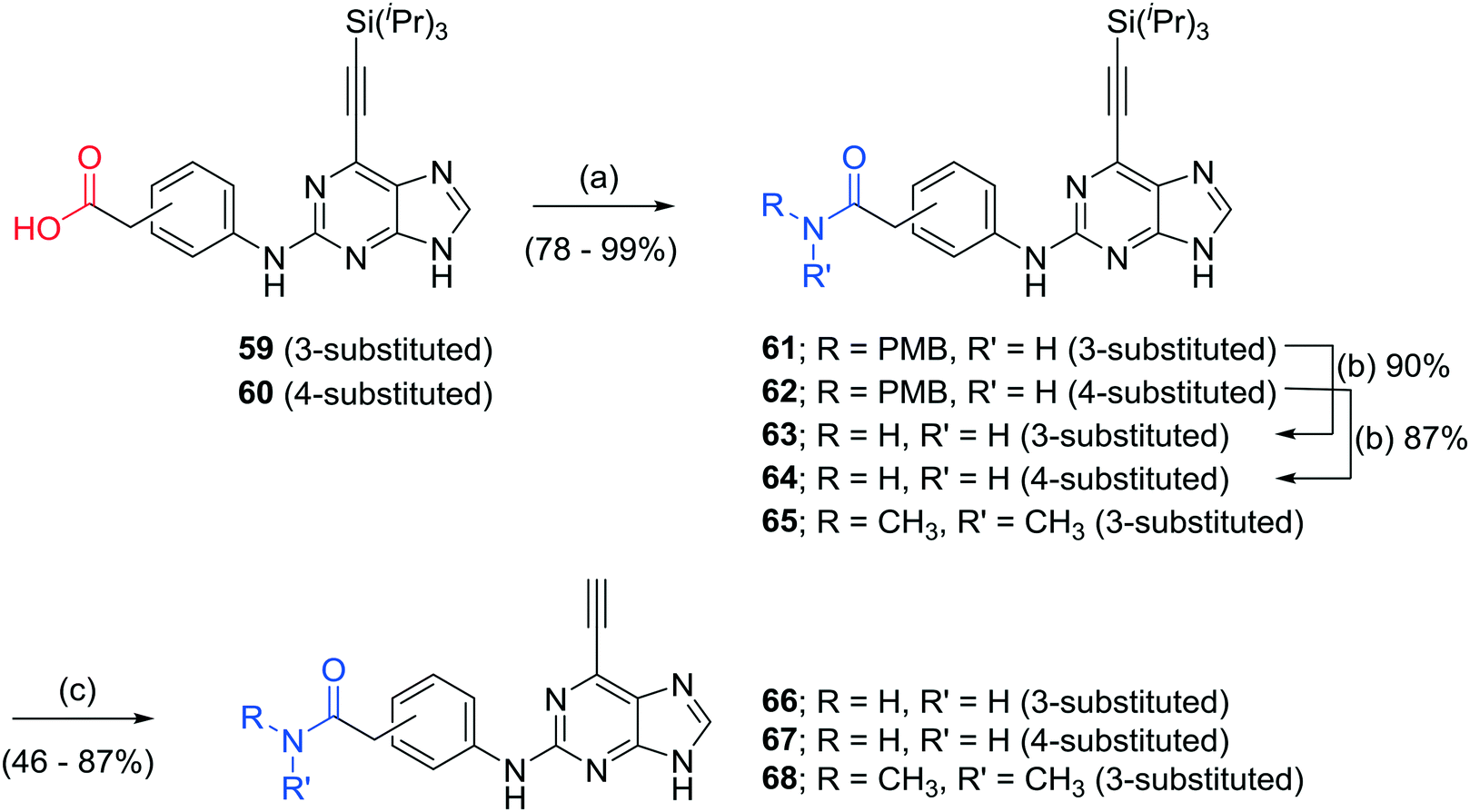 | ||
| Scheme 2 Synthesis of carboxamide derivatives. Reagents and conditions: (a) i) 1 M NaOH, THF, RT, 18 h; ii) RNH2, CDI, DIPEA, DMF, RT, 18 h; (b) TFA, reflux, 72 h; (c) TBAF, THF, RT, 5 min (followed in some cases by Amberlite 15 ion exchange resin, Ca(OH)2, THF, RT, 48 h). | ||
To probe the role of the carboxamide amino group of 66, the ketone analogue 80 was synthesised. A modification of the Dakin–West reaction (conversion of an amino-acid into a ketoamide), can be used to convert a carboxylic acid with an enolisable carbon at the α-position into a methyl ketone. This method was employed to convert 3-nitrophenylacetic acid into 3-nitrophenylacetone by treatment with acetic anhydride in the presence of pyridine (modified Dakin–West reaction19,20). Protection of the carbonyl group of 3-nitrophenylacetone by conversion into a dithiane with 1,3-propanedithiol, was followed by reduction of the nitro group to afford aniline 77. Coupling of 77 to the 2-fluoropurine 12 in the usual manner (Scheme 3) gave 78. Stepwise removal of the dithiane (using [bis(trifluoroacetoxy)iodo]benzene) and silyl (TBAF) protecting groups furnished the target inhibitor (80) via intermediate 79.
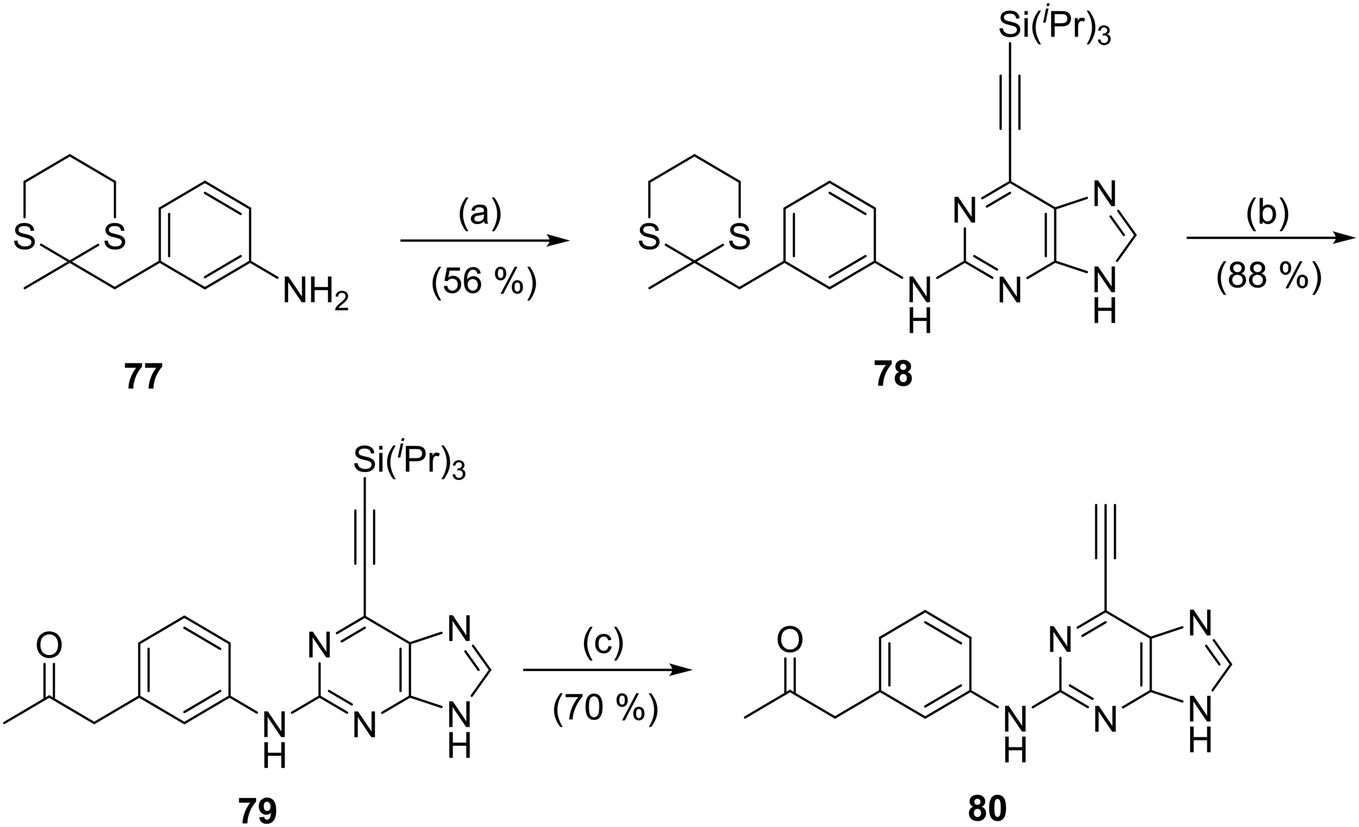 | ||
| Scheme 3 Synthesis of sulfonamides. Reagents and conditions: (a) 12, TFA, TFE, MW 140 °C, 2 h; (b) [bis(trifluoroacetoxy)iodo]benzene, MeOH, H2O, RT, 10 min; (c) i) TBAF, THF, RT 5 min, ii) solid supported TBAF, THF, RT, 48 h. | ||
The homosulfonamide analogues of 24 probing the H-bond donor properties of the sulfonamide were prepared by either i) direct SNAr reaction with the aniline according to Scheme 1, affording the sulfone 82, or ii) from common intermediate, 2,2,2-trifluoroethylsulfonate ester 83 according to Scheme 4.21 For the latter, reaction with amines in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene under microwave heating afforded sulfonamides in good to excellent yield.21 Removal of all protecting groups was completed under the optimised conditions described above to give the target 6-ethynylpurines 87 and 88.
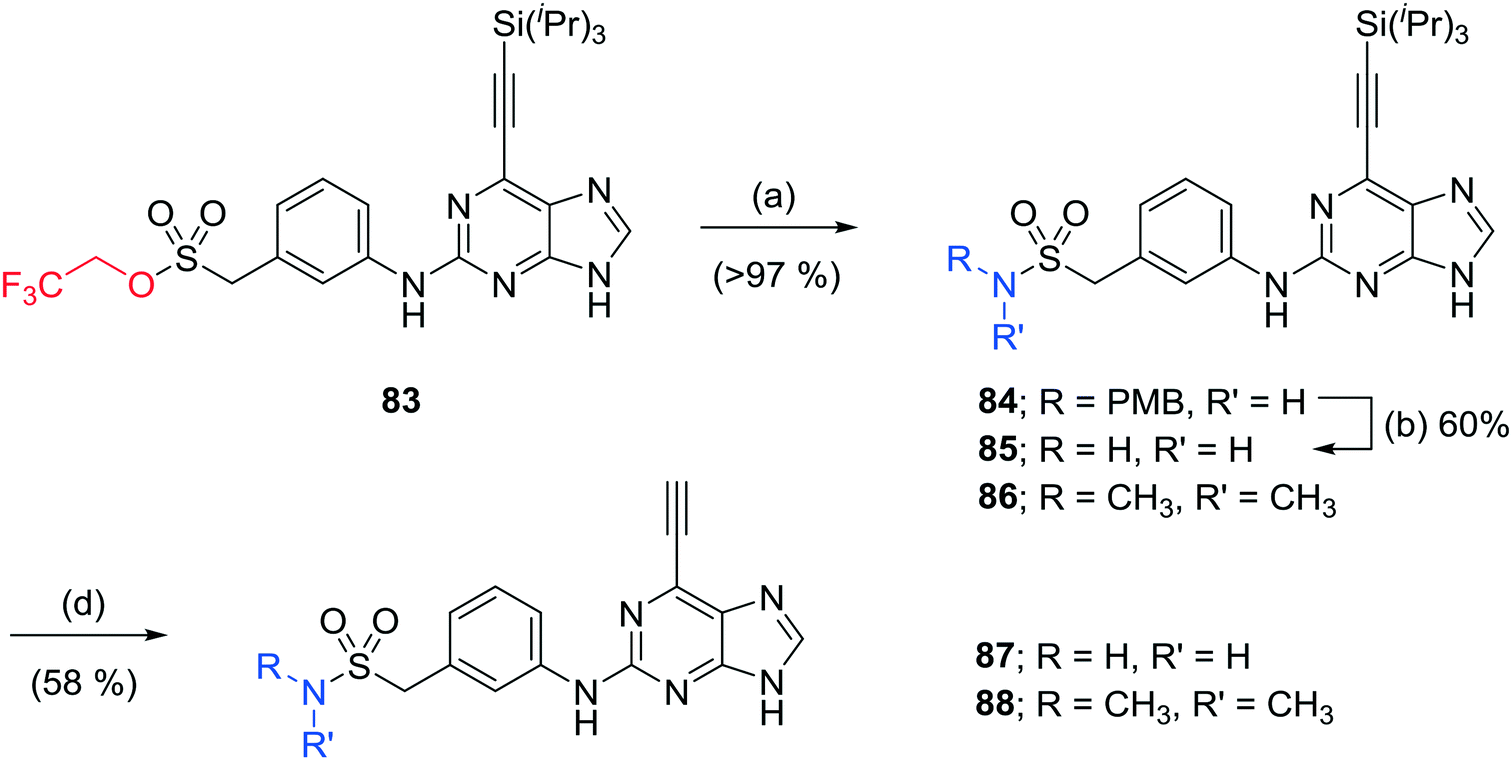 | ||
| Scheme 4 Synthesis of sulfonamides. Reagents and conditions: (a) R2NH, DBU, THF, MW 160 °C, 15 min; (b) TFA, 70 °C, 3 h; (c) TFA, DCM, RT, 18 h; (d) i) TBAF, THF, RT, 5 min, ii) Amberlite 15 ion exchange resin, Ca(OH)2, THF, RT, 48 h. | ||
The replacement of the 6-ethynyl group with a vinyl group was performed to probe the conformation of the conjugate acceptor within the binding pocket of Nek2 and to assess the potential for non-specific reactivity and toxicity. Initial attempts to synthesise the 6-vinylpurine analogues of 23 directly using a vinyltrifluoroborate salt and Suzuki–Miyaura coupling failed. 6-Vinylpurines have been prepared from thioethers by oxidation to the corresponding sulfoxide followed by base-induced elimination.22 In our approach based on an earlier study,23 to 6-vinylpurine (92) conjugate addition of homopiperidine to 6-ethynylpurine (23) furnished the enamine 89 (Scheme 5).14,16d Reduction24 of 89 using sodium cyanoborohydride in the presence of 0.1 M HCl gave tertiary amine 90. N-Oxidation of 90 using m-CPBA gave N-oxide (91), which underwent a Cope β-elimination24 without heating, to give the desired 6-vinylpurine (92) in sufficient yield (30%).
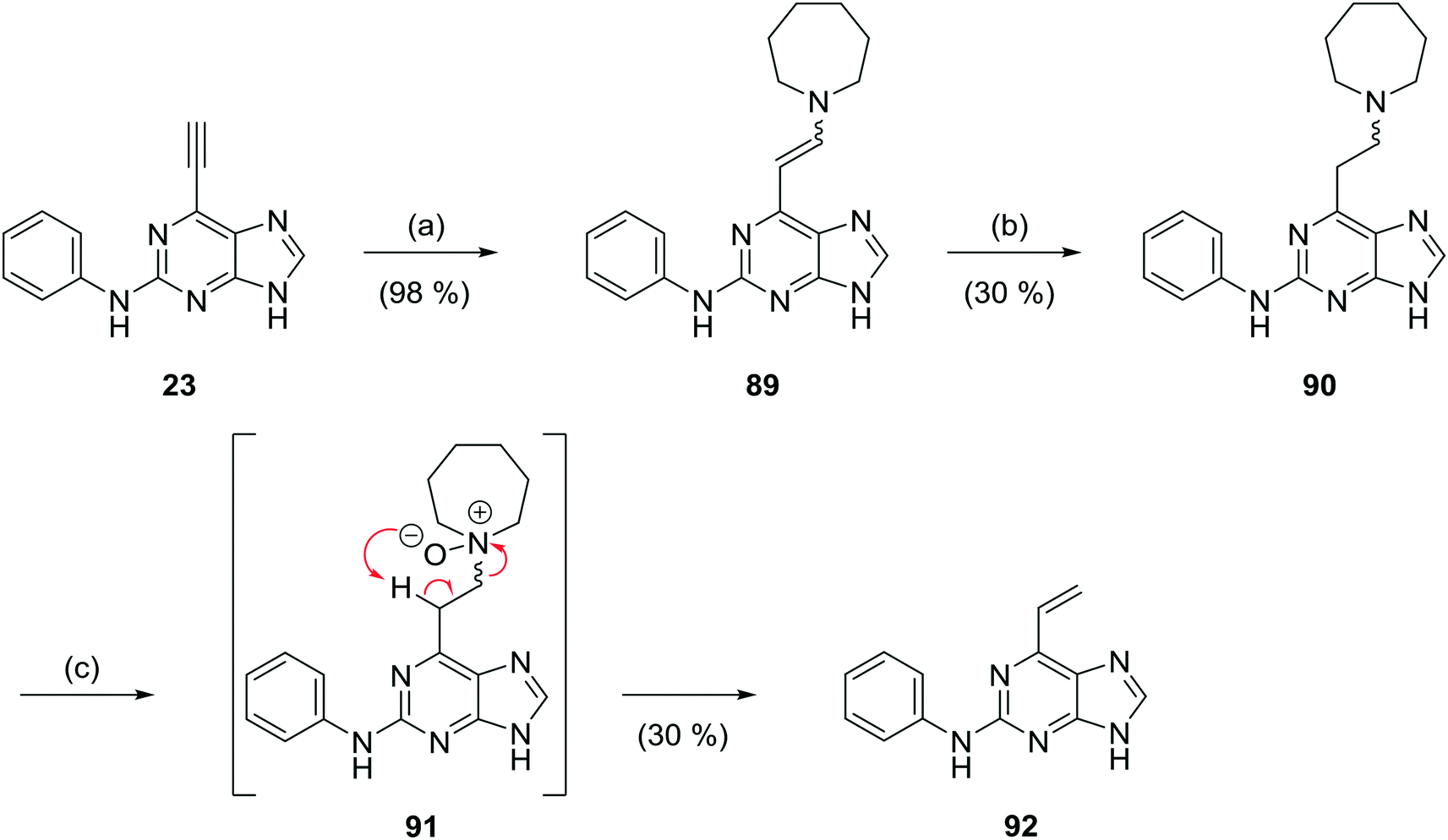 | ||
| Scheme 5 N-Oxidation-β-elimination approach to 6-vinylpurine 92. Reagents and conditions: (a) homopiperidine, THF, MW 100 °C, 10 min; (b) NaBH3CN, TFA, THF, RT, 2 h, or NaBH3CN, 0.1 M HCl, RT, 5 h; (c) m-CPBA, DCM, RT, 2 h. | ||
b) Synthesis of control compounds
The 6-ethylpurine 96 was prepared as a control compound for comparison with the conjugate acceptor 6-ethynylpurine (23). 6-Ethynyl-2-fluoro-9-tetrahydropyranylpurine (93) was directly reduced with hydrogen in the presence of Lindlar's catalyst with quinoline to the fully saturated 6-ethylpurine (94). The THP protecting group was cleaved by acidic hydrolysis followed by treatment with 4-aminobenzenesulfonamide under standard SNAr conditions to produce 96 (Scheme 6).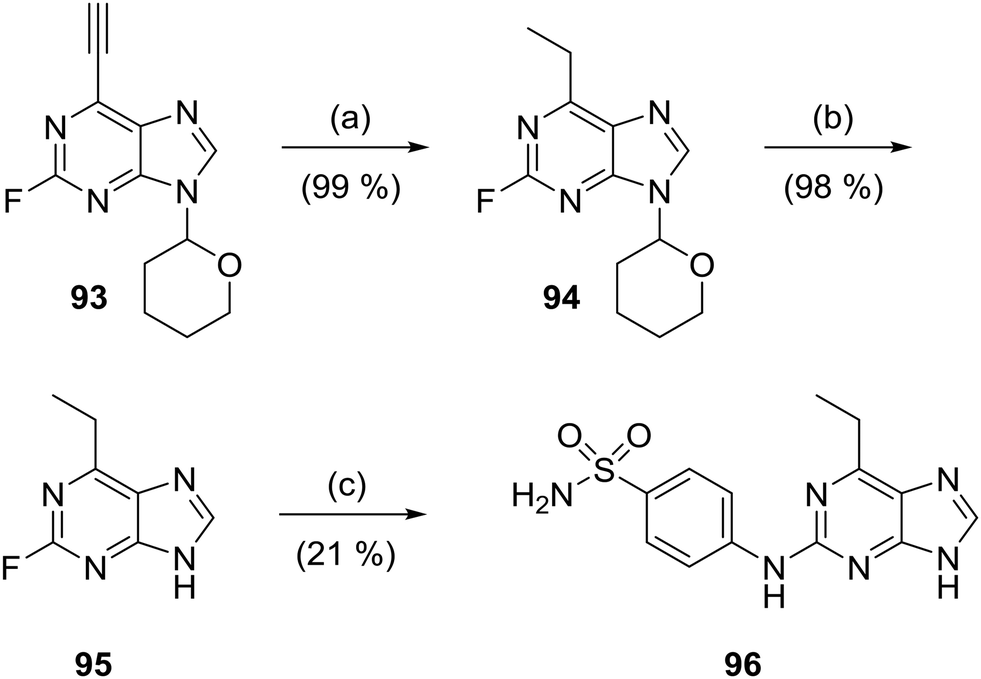 | ||
| Scheme 6 Synthesis of 6-ethylpurine 96. Reagents and conditions: (a) Lindlar's catalyst, quinoline, H2, EtOAc, RT, 2 h; (b) TFA, 2-propanol, water, 100 °C, 2 h; (c) 4-aminobenzenesulfonamide, TFA, TFE, 90 °C, 48 h. | ||
6-Cyanopurine (102) was prepared by heating 2-bromohypoxanthine (97) with aniline to afford N2-phenylguanine (98), which was converted into the 6-chloropurine 99. The purine N-9/N-7 positions were protected as a ∼3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (N-9:N-7) of 4-methoxybenzyl-substituted regioisomers. The major N-9-PMB isomer (100) was isolated in 52% yield and treated with Et4NCN/DABCO to furnish cyanopurine 101, from which the PMB group was removed in hot TFA (Scheme 7).
:1 mixture (N-9:N-7) of 4-methoxybenzyl-substituted regioisomers. The major N-9-PMB isomer (100) was isolated in 52% yield and treated with Et4NCN/DABCO to furnish cyanopurine 101, from which the PMB group was removed in hot TFA (Scheme 7).
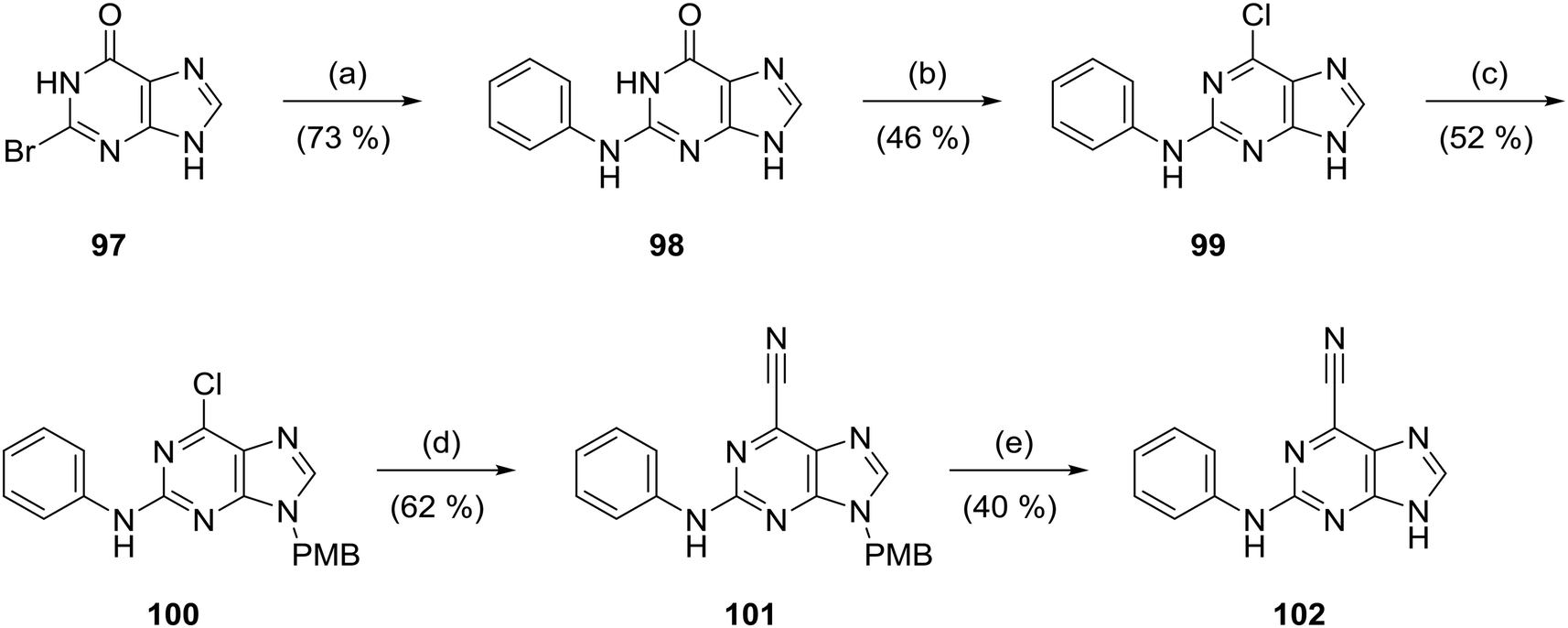 | ||
| Scheme 7 Synthesis of cyanopurine 102. Reagents and conditions: (a) aniline, TFA, TFE, 80 °C, 18 h; (b) PhNMe2, POCl3, 115 °C, 1 h; (c) 4-methoxybenzyl chloride, K2CO3, DMF, 60 °C, 18 h; (d) Et4NCN, DABCO, MeCN, rt, 18 h; (e) TFA, 70 °C, 5 h. | ||
c) Biological evaluation of probes
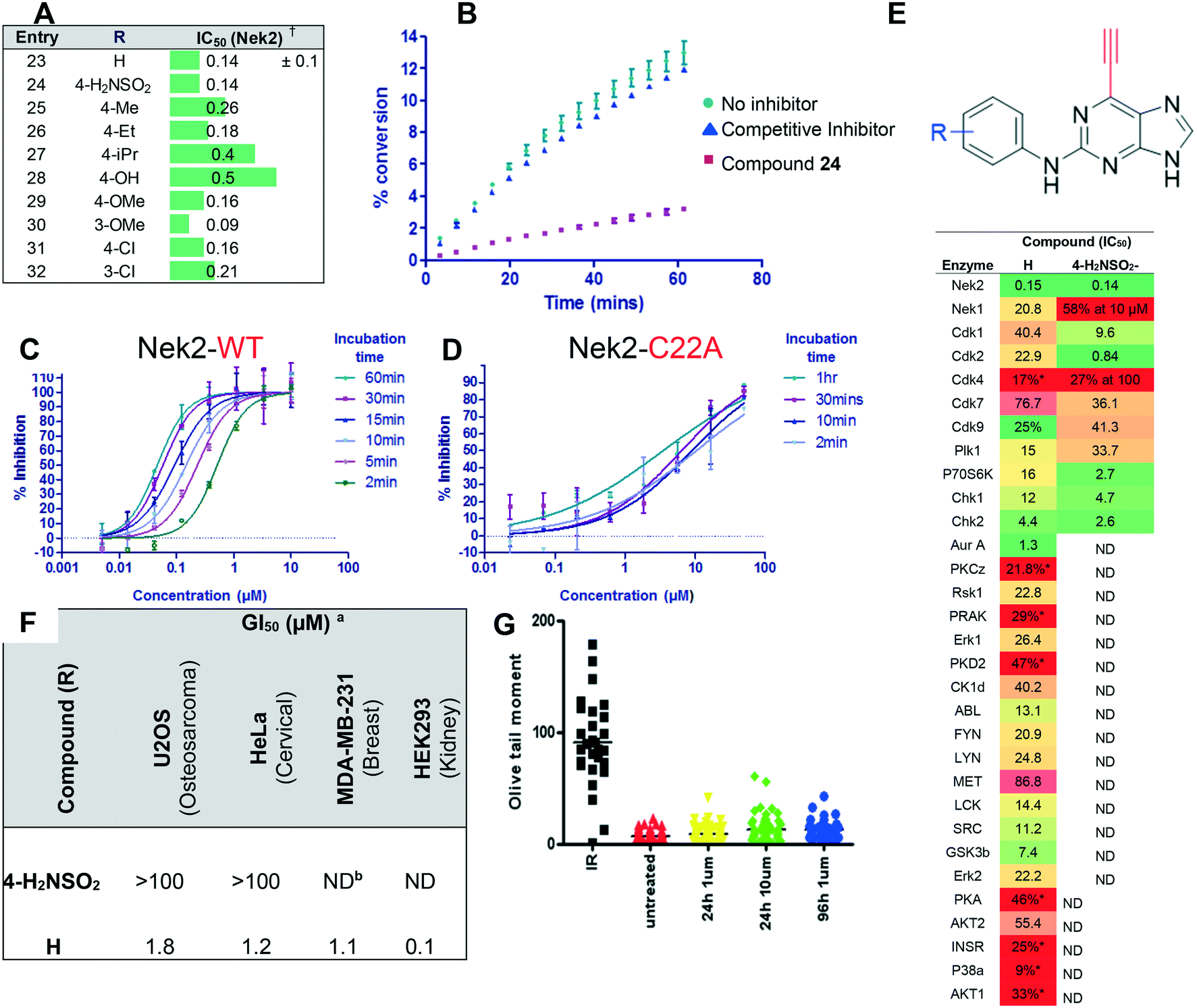 | ||
| Fig. 3 Biochemical and cellular assessment of initial hit compounds 23 and 24 as irreversible inhibitors of Nek2: A) modification of the 2-arylamino motif. †Note: Nek2 IC50 values (uM) determined at 30 μM ATP concentration following a 30 minute incubation. B) Recovery of enzyme activity of Nek2 following dilution of the enzyme–inhibitor complex with ATP for 24 and a known ATP-competitive inhibitor of Nek2. C) and D) dose–response curves as a function of incubation time between 23 and Nek2-WT, and Nek2-C22A. E) Counter-screening of ethylnylpurines 23 and 24 against a panel of protein kinases (note: red = low inhibition, green = high inhibition). F) Cellular growth-inhibition by compounds 23 and 24 (a half maximal growth inhibitory concentration; b not determined). G) Comet assay results for cells treated with 23. | ||
Compounds 23 and 24 were selected as initial probes for determination of the mode of 6-ethynylpurine-mediated inhibition of Nek2. A kinase-inhibition reversibility assay (Fig. 3B) was employed to assess whether Nek2 pre-incubated with an inhibitor, e.g.24 (30 min at 10 × IC50, calculated to afford approximately 91% inhibition) could recover enzymatic activity upon rapid dilution (×100) of the enzyme–inhibitor complex with a solution containing ATP. Following this dilution, a reaction progress curve was generated by monitoring for levels of a phosphorylated substrate (Fig. 3B). Compound 24 afforded only approximately 3% recovery of activity, compared with 11% for a control competitive inhibitor, suggesting that 6-ethynylpurines are indeed covalent binders to Nek2. Further evidence was obtained through the confirmation of time-dependent inhibition of Nek2 kinase activity after treatment with 23 (Fig. 3C). To confirm whether time-dependent inhibition was mediated through Cys22, site-directed mutagenesis of this residue to alanine (Nek2-C22A) was performed, resulting in only weak competitive inhibition by 23 (IC50, Nek2-C22A = 3.5 μM), with no increase in the degree of inhibition being observed with time of incubation (Fig. 3D).
Given the observed cellular activity and apparent selectivity for Nek2 over other kinases, 6-ethynylpurine 23 was considered a suitable starting point from which to conduct modification to further improve inhibitor potency. According to our previously reported findings,14 Nek2 competitive inhibitors of the 6-alkoxypurines class bearing a basic group at the 2-arylamino ring were found to confer good potency and cellular activity. It was considered that this would translate to the irreversible inhibitors class as the competitive binding phase was crucial to achieving targeted and potent inhibition. The parent 6-ethynylpurine irreversible inhibitor (23) was modified at the 2-arylamino to produce a series comprising carboxamides (46–58, 66–68, 73–76) and sulfonamides (87 and 88), which were screened for inhibitory activity against Nek2 (Table 1). In addition, 6-ethynylpurines were evaluated for growth inhibitory activity in vitro against the SKBR3 tumour cell line.
|
|
|||
|---|---|---|---|
| Compound | R | IC50 (μM) Nek2a | GI50 (μM) SKBR3 |
| All IC50 values are results obtained from n = 3 determinations.a Nek2 IC50 values determined at 30 μM ATP concentration following a 30 minute incubation. | |||
| 46 |
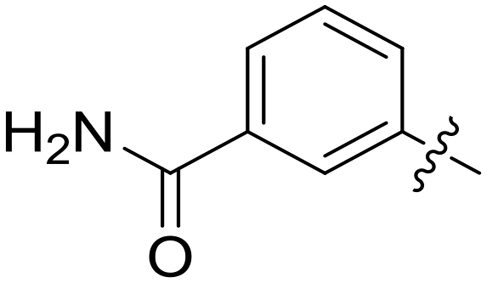
|
0.116 | 0.7 ± 0.1 |
| 47 |
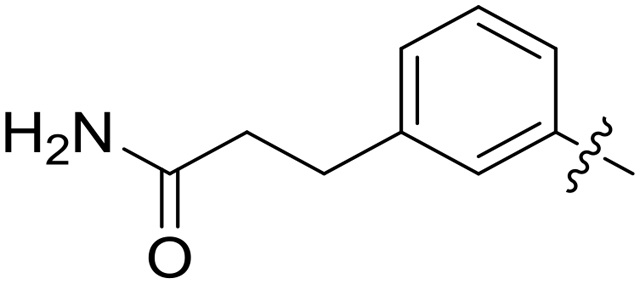
|
0.079 | 3.9 ± 1.3 |
| 48 |
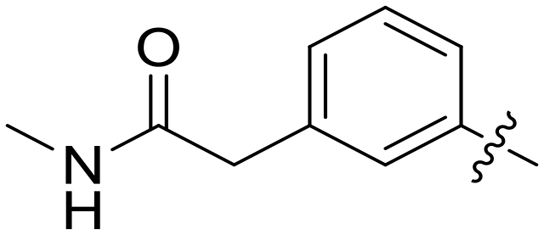
|
0.076 | 2.4 ± 0.1 |
| 49 |

|
0.072 | 4.9 ± 2.4 |
| 50 |
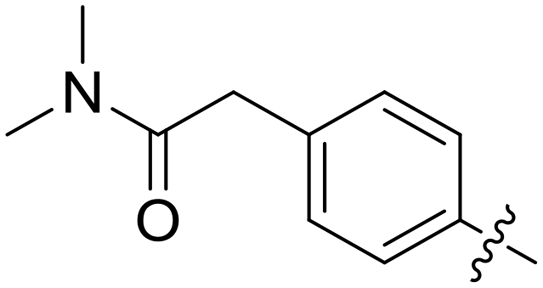
|
0.057 | 1.2 ± 0.6 |
| 51 |
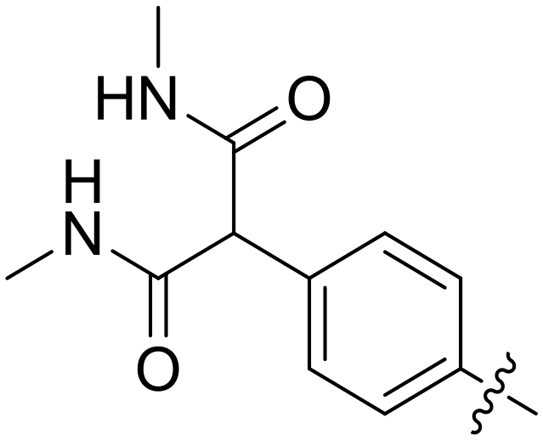
|
0.071 | 87.8 |
| 52 |
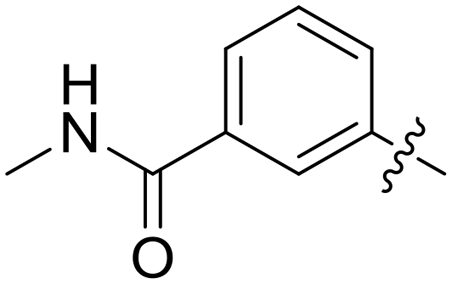
|
0.026 | 0.9 ± 0.1 |
| 53 |
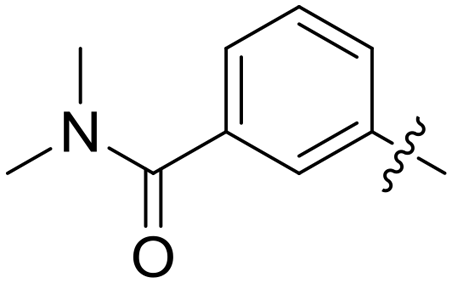
|
0.050 | 0.9 ± 0.1 |
| 54 |
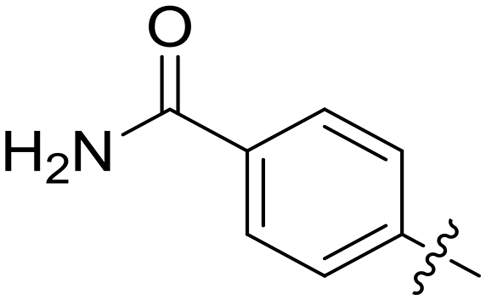
|
0.110 | 7.4 ± 0.1 |
| 55 |
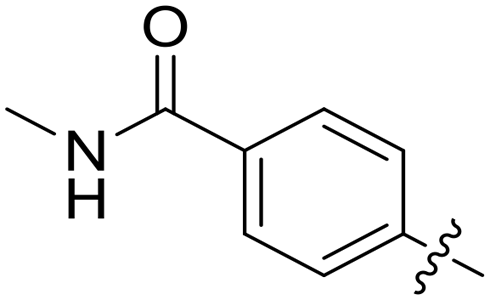
|
0.110 | 4.1 ± 0.5 |
| 56 |
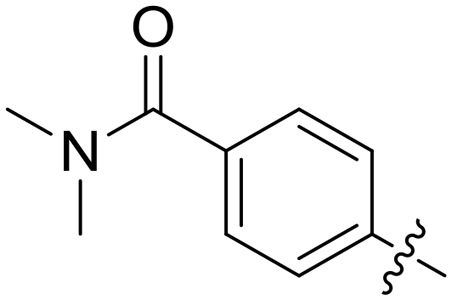
|
0.064 | 1.4 ± 0.4 |
| 57 |
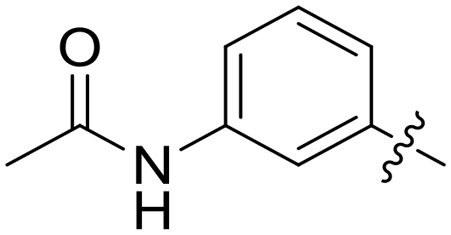
|
0.039 | 0.8 ± 0.5 |
| 58 |
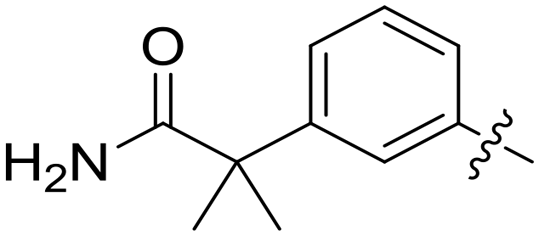
|
0.210 | 1.4 ± 0.6 |
| 66 |
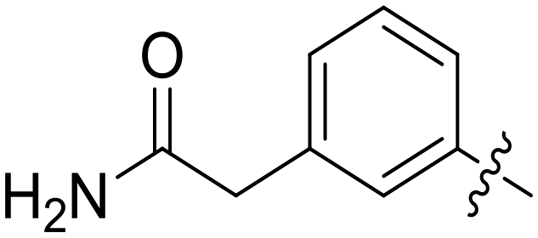
|
0.062 ± 0.01 | 2.2 ± 0.4 |
| 67 |
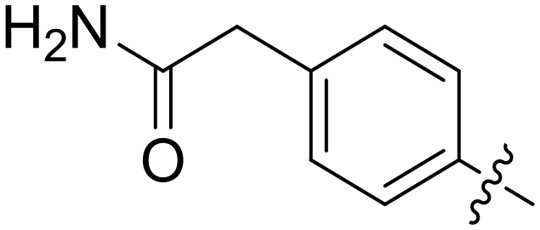
|
0.093 | 8.8 ± 1.8 |
| 68 |
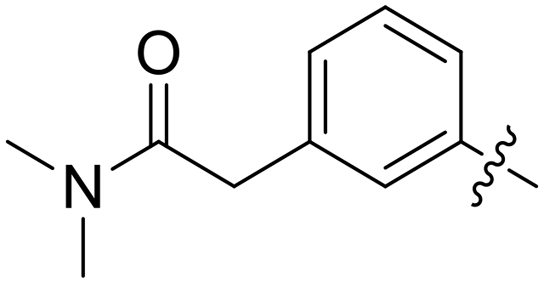
|
0.088 | 0.4 ± 0.2 |
| 73 |
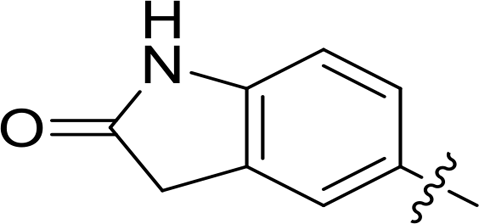
|
0.100 | 10.3 |
| 74 |
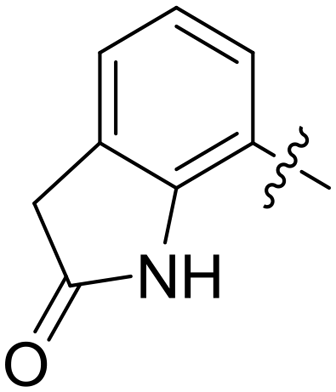
|
0.820 | 1.3 ± 0.2 |
| 75 |
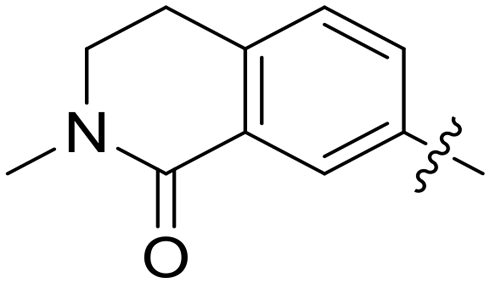
|
0.010 | — |
| 76 |
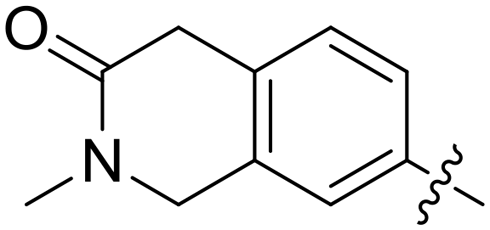
|
0.042 | 0.8 ± 0.1 |
| 80 |
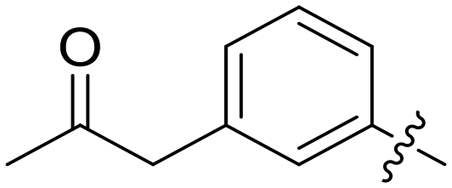
|
0.076 | — |
| 82 |
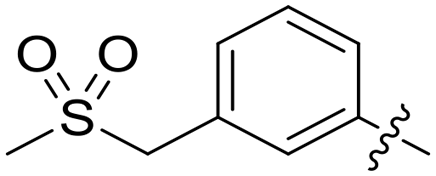
|
0.057 | 1.3 ± 0.0 |
| 87 |
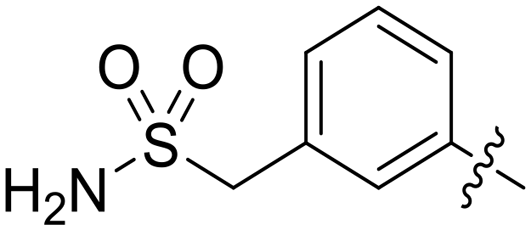
|
0.130 | 4.1 ± 2.2 |
| 88 |
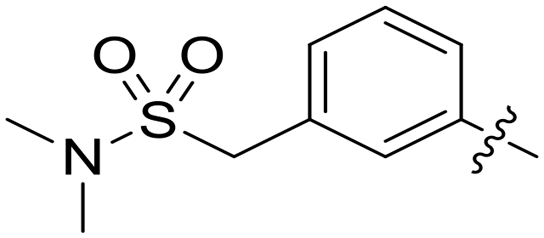
|
0.010 | — |
All purine derivatives tested exhibited inhibitory activity against Nek2 with all but compound 74 possessing an IC50 of less than 0.13 μM. It is evident that the activity of the parent 3-substituted carboxamide 66 (IC50 = 0.062 ± 0.01 μM) is not affected by truncation (46, IC50 = 0.116 μM) or homologation (47, IC50 = 0.079 μM) of the methylene spacer group. Reversing the carboxamide (57, IC50 = 0.039 μM) and complete removal of the heteroatom, through synthesis of ketone 80 (IC50 = 0.076 μM) were inconsequential modifications. Furthermore, moving the 2-arylamino ring substituents from the 3- to the 4-position (e.g., 67; IC50 = 0.093 μM) had little influence on the measured enzyme inhibition. Sequential removal of the carboxamide hydrogen atoms through N-methylation, affording N-methylcarboxamide (48, IC50 = 0.076 μM) and N,N-dimethylcarboxamide (68, IC50 = 0.088 μM) analogues of 66 had little effect on Nek2 inhibitory activity, which was true for both 3- and 4-substituted carboxamides.
Conformational restriction of the carboxamide motif appeared to be somewhat detrimental, with gem-dimethyl carboxamide 58 (IC50 = 0.21 μM) exhibiting a small, approximate 3-fold reduction in inhibitory activity. Moreover, it is likely that the conformational restriction imposed by a bicyclic 2-arylamino system either disrupts favourable or promotes unfavourable interactions within the ATP-binding site of Nek2, as exemplified by compound 74 (IC50 = 0.82 μM). This suggests that the optimal 2-arylamino side-chain characteristics are more linear with respect to the purine scaffold, as indicated by comparison of the activity of 74 with that of the regioisomeric analogue 73 (IC50 = 0.10 μM). In contrast to the carboxamide examples, the N,N-dimethylsulfonamide analogue (88, IC50 = 0.010 μM) lacking an H-bond donor atom conferred a significantly improved potency when compared with the primary sulfonamide (87, IC50 = 0.130 μM). However, this may be simply a function of physicochemical properties e.g., solubility.
Within this compound class, only rather limited SARs are evident, with no discernible trend observed between the activity of compounds against Nek2 and the degree and/or nature of modification of the carboxamide group. Presumably, all compounds excluding 74 exhibit sufficient initial competitive binding affinity for Nek2 to allow the purine to occupy the ATP-binding domain of the kinase and enable subsequent covalent modification of Cys22 to occur. The weaker activity observed for 74 does suggest that it is possible to elucidate SARs for this compound class and that potent activity is not common to all of the 6-ethynylpurines. What these data do reveal, however, is that for irreversible inhibitors in the 6-ethynylpurine class, there is a large degree of tolerance for substitution on the 2-arylamino ring. This indicates that significant structural changes should be tolerated in order to enhance pharmacokinetic/physicochemical properties yet may promote concerns of potential off-target toxicity. Coupled with this, all compounds detailed in Table 1 exhibited growth inhibitory activity in the SKBR3 cell line, yet there appeared to be a poor correlation between the IC50 of compounds in the Nek2 kinase assay and their activity in the SKBR3 cellular GI50 assay. In particular, compound 74 (Nek2; IC50 = 0.82 μM, SKBR3; GI50 = 1.3 ± 0.2 μM) was the least active inhibitor in the Nek2 assay, but proved one of the more potent growth inhibitors in SKBR3 cells. Off-target inhibition or non-specific toxicity are common challenges to overcome within protein kinase inhibitor design, and in particular for irreversible inhibitors. As such, it was considered that these may play a role in the conflicting cellular versus enzyme activity data, despite the promising Comet assay data obtained for 23.
Whilst the carboxamide derivative 66 was identified as amongst the most potent inhibitors in this class and inhibited growth of SKBR3 cell lines (Table 1), the improvement in kinase inhibitory activity did not correlate with the cellular growth inhibitory activity observed in U2OS and HeLa (Table 2) cell lines. It was possible that this was again a consequence of the more polar nature of the 2-arylamino side-chain limiting cellular permeability.
|
|
||||
|---|---|---|---|---|
| Compound | R | IC50 (μM) | GI50 (μM) | |
| Nek2a | U2OS | HeLa | ||
| All IC50 values are results obtained from n = 3 determinations.a Nek2 IC50 values determined at 30 μM ATP concentration following a 30 minute incubation; N.D. not determined. | ||||
| 23 | Ph | 0.14 ± 0.1 | 2.1 ± 1.0 | 1.2 ± 0.5 |
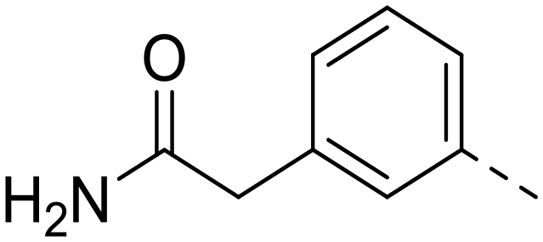
| 66 |
|
0.062 ± 0.01 | 71.0 | 47.1 ± 2.1 |
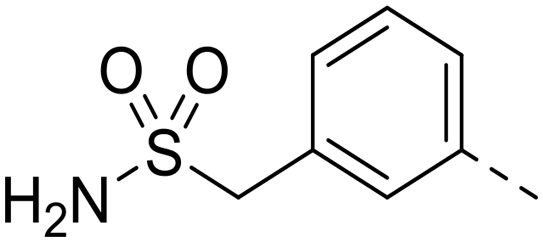
| 87 |
|
0.13 | N.D. | N.D. |
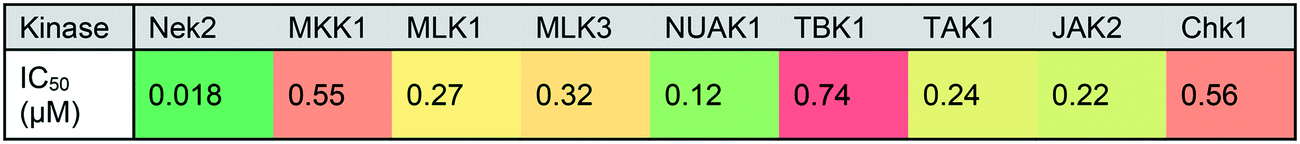
Owing to concerns over lack of kinase selectivity of this compound class, the inhibitory activity of 66 was profiled against a panel of 48 kinases selected from commercially available ProfilerPro® plates at 1 μM inhibitor concentration (see ESI,† Table S1). Only two kinases other than Nek2 were inhibited by greater than 50%, and IC50 values were determined for these: Aurora A (IC50 = 0.08 μM) and BMX (IC50 = 0.83 μM). It is notable that BMX kinase, a member of the EGFR tyrosine kinase family, possesses a cysteine residue within the ATP-binding domain that has been identified as a potential target in the design of irreversible inhibitors.25 In addition, both 23 and 66 were screened against 121 kinases (National Centre for Protein Kinase Profiling, Dundee University) at a single concentration of 1 μM (see ESI,† Table S2). Greater than 50% inhibition of activity was observed for nine of the kinases tested by 66, including Nek2, and IC50 values were determined (Table 3). Encouragingly, the IC50 of 66 against Nek2 was 18 nM in this assay, giving the compound at least 5–10-fold selectivity for Nek2 over other kinases, whilst AurA was only weakly inhibited by 66 in this screen.
|
Interestingly, 23 displayed a better overall selectivity profile for Nek2 compared with 66. In addition to Nek2, only two other kinases (MLK3 and TAK1) were inhibited by greater than 50%. It is possible that the alternative target responsible for the cellular activity of 23 is not included in the screen, which constitutes only a fraction of the human kinome. Conversely, the locus of cellular activity for 23 may not be a kinase.
In keeping with the ultimate objective of developing a targeted therapy, 66 was screened for cell growth inhibition (10 μM, 72 h) against a panel of cell lines to identify specific tumour types where depletion of Nek2 activity is growth inhibitory (Fig. 4A–C). Previous findings in this area indicated that cell lines derived from breast tumours and leukaemias may be particularly sensitive to pharmacological Nek2 inhibition. As was observed previously, the tumour cell lines most sensitive to inhibitor treatment were those derived from breast (MCF7, T47D, ZR75, SKBR3 and MDA-468) and leukaemia (Rec-1 and RIVA), with all of these cell lines exhibiting greater than 50% growth inhibition at a dose of 10 μM 66. GI50 values were determined for these cell lines, along with HeLa and HCT116 (Fig. 4D and E), with the lymphoma Rec-1 (GI50 = 1.6 ± 0.1 μM) and breast cancer SKBR3 (GI50 = 2.2 ± 0.4 μM) cell lines being identified as particularly sensitive to 66 treatment. Furthermore, 66 was also found to be cytotoxic in SKBR3 cells; clonogenic assays with this cell line gave LC50 values in the low micromolar range following pre-incubation for as little as 3 hours prior to subculture (Fig. 4F).
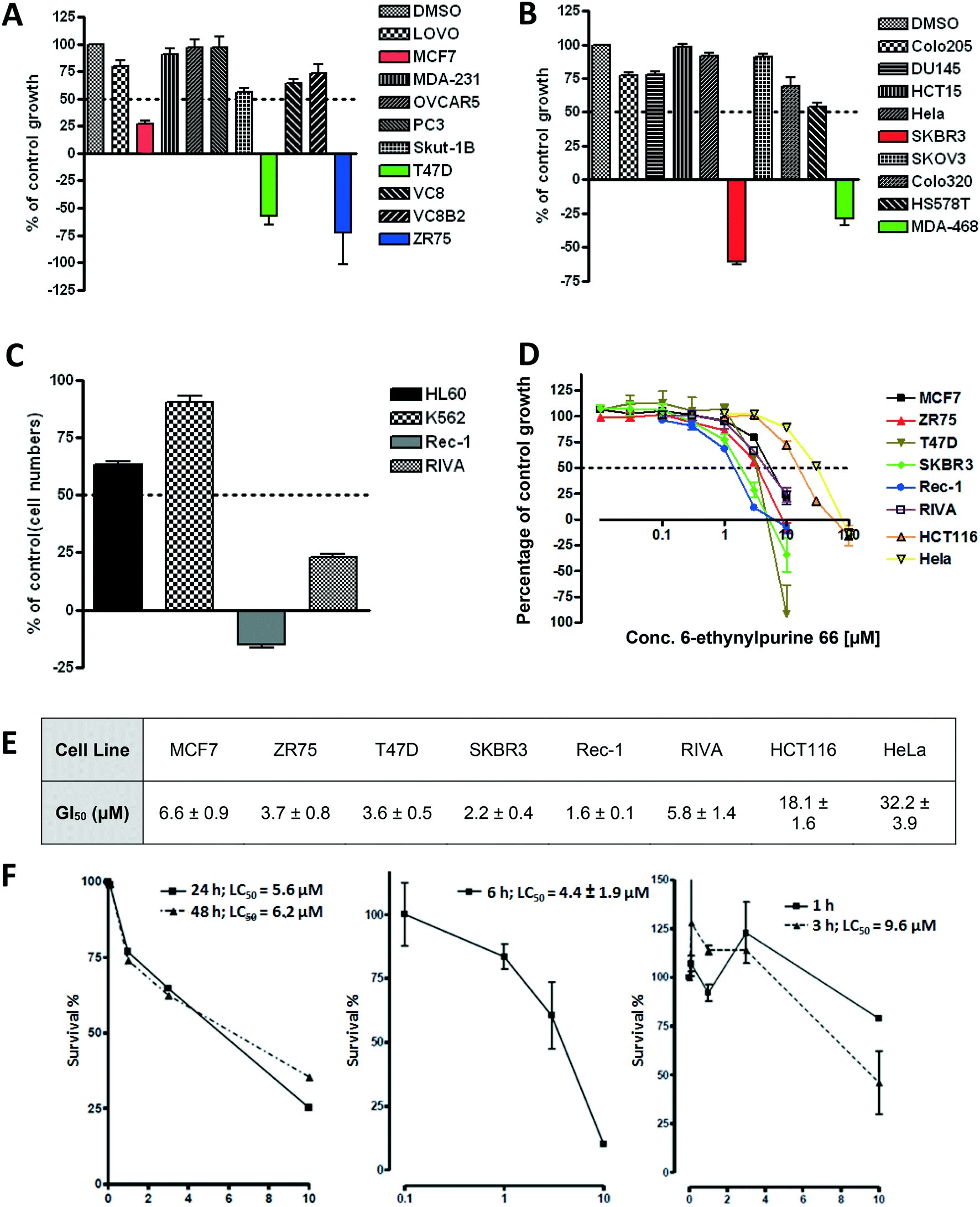 | ||
| Fig. 4 A) Growth inhibition of solid tumour cell lines treated with 10 μM 66 for 72 hours. B) Growth inhibition of solid tumour cell lines treated with 10 μM 66 for 72 hours. C) Growth inhibition of leukaemia cell lines treated with 10 μM 66 for 72 hours. D and E) Growth inhibition of cell lines identified as being sensitive to 66 following 72 h exposure to inhibitor (n = 3). F) Cytotoxicity of 66 in SKBR3 cells following incubation for a range of times. | ||
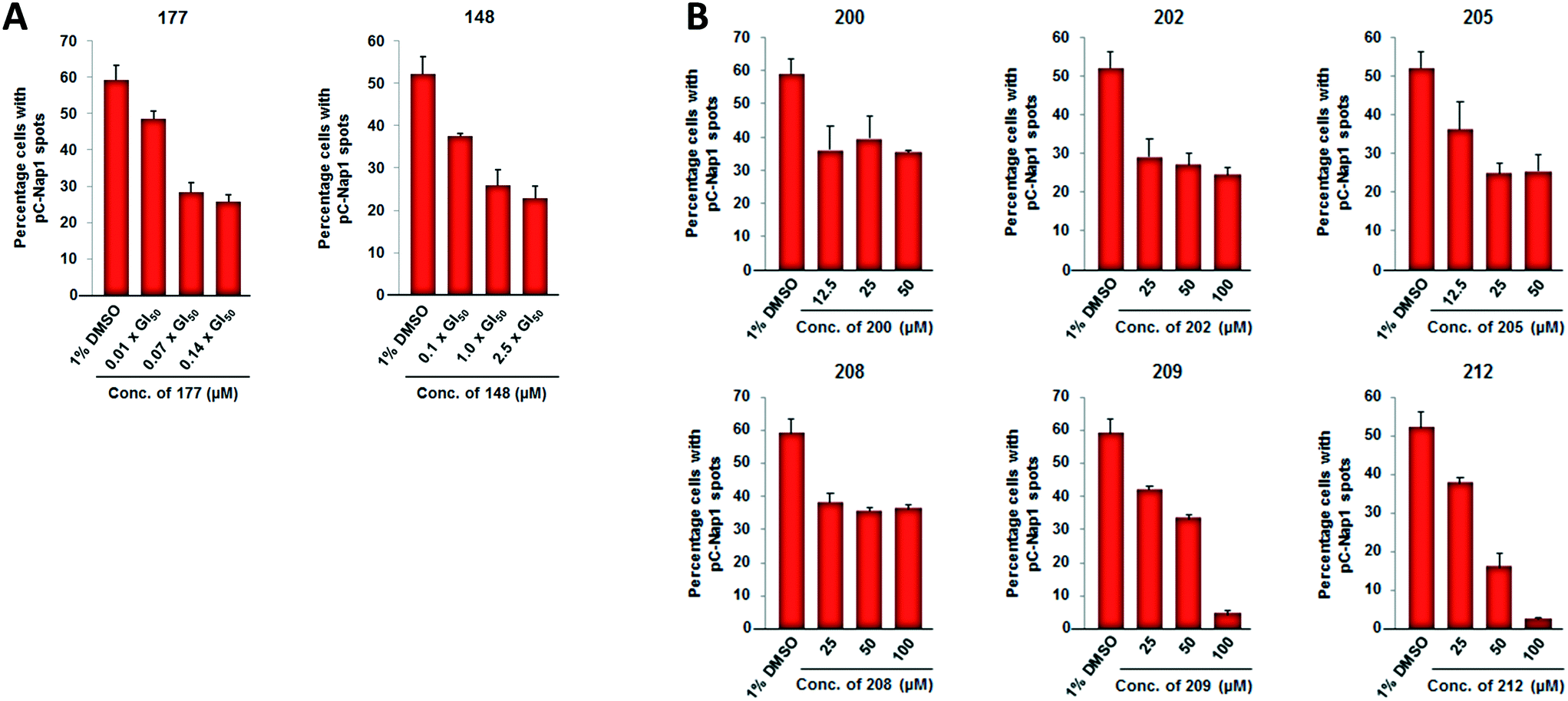 | ||
| Fig. 5 A) Inhibition of C-Nap1 phosphorylation in U2OS cells by 66 compared with 23. B) Dose-dependent reduction of pC-Nap1 levels for selected carboxamides within the 6-ethynylpurine series. | ||
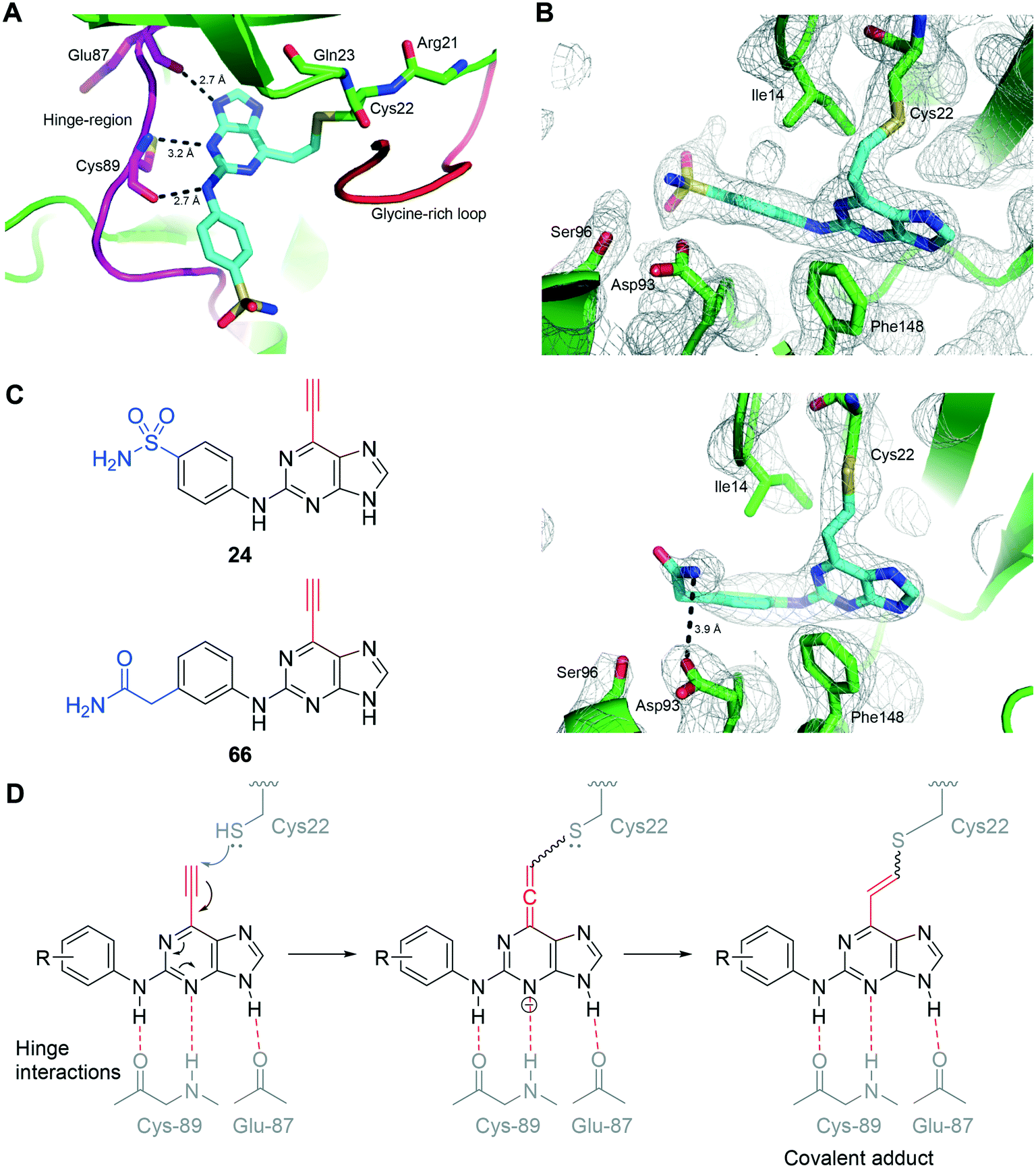 | ||
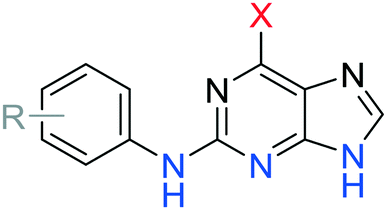
| Fig. 6 Interaction of irreversible inhibitors 24 and 66 with Nek2: A) crystal structure of 24 (cyan) bound to Nek2 indicating interactions with the hinge-region of the kinase (pink) and B) crystal structure of Nek2 (green) in complex with 24 (cyan) including electron density map (2Fo-Fc) contoured at 1.0σ (mesh). C) Crystal structure Nek2 (green) in complex with 66 including electron density map (2Fo-Fc) contoured at 1.0σ (mesh). A potential interaction with the side chain of Asp93 is indicated. D) Proposed mechanism of covalent adduction of cysteine-22 by ethynylpurines. | ||
Perhaps the most intriguing result was related to the 6-vinylpurine 92, which is reported to undergo conjugate addition reactions with thiols, albeit at a slightly slower rate than 6-ethynylpurines.1592 was found to be a very weak Nek2 inhibitor (17% inhibition at 10 μM) compared with ethynyl analogue 23 (IC50 = 0.14 μM). This indicates that the geometry of the conjugate-acceptor substituent at the purine 6-position is critical, underlining the importance of correctly aligning the conjugate acceptor group with Cys22. However, it is possible that due to lower thiol-reactivity, 92 requires a longer incubation time within the Nek2 active site to be equipotent with 23. This outcome is of critical importance and alleviates the fear that unwanted side-reactions would occur indiscriminately.
The saturated 6-ethylpurine (96) is a non-conjugate acceptor, lacking an alkene/alkyne purine 6-substituent required for Michael-type addition reactions. Compound 96 was found to be a weak inhibitor of Nek2 (IC50 = 46.8 μM), but was shown to bind in an identical orientation in the Nek2 active-site as the 6-ethynyl derivative 24 (Fig. 7). The crystal structure of Nek2-96 complex superimposed with Nek2-24 (Fig. 7) revealed that the covalent link between 24 and Cys-22 of Nek2 is accompanied by a relative movement of the N-terminal lobe towards the C-terminal lobe. This movement of the N-terminal lobe is not evident when the competitive inhibitor (96) is in complex with Nek2.
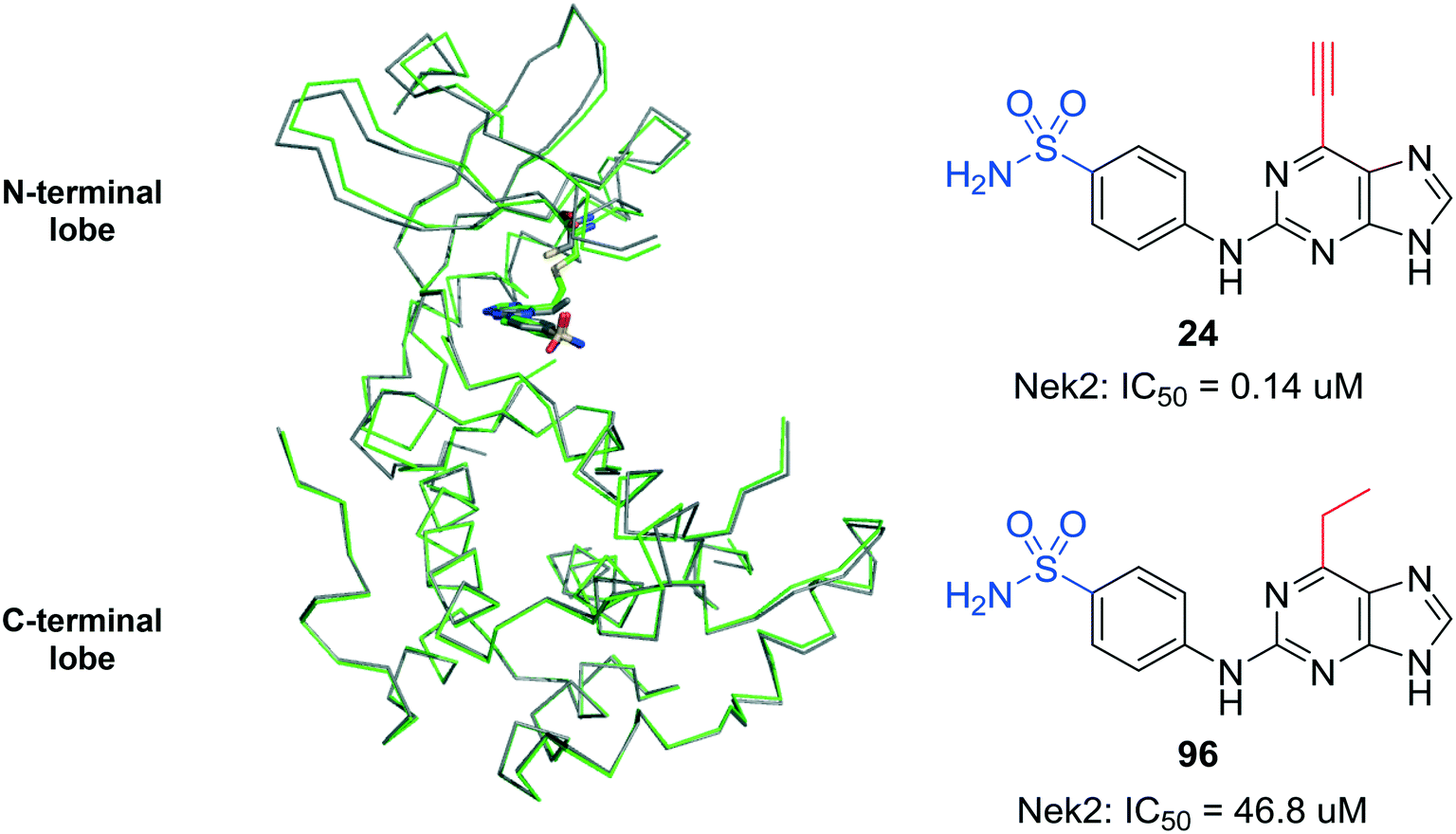 | ||
| Fig. 7 Overlay of the ATP-binding domain of Nek2 in complex with 24 (green) and 96 (grey). | ||
The nitrile group of 102 retains both the linear geometry of the ethynyl group is not expected to undergo conjugate addition by nucleophiles such as thiols in the same manner as 6-ethynylpurines. 102 was measured to be a weak competitive inhibitor of Nek2 (IC50 = 12 μM) and was approximately 200-fold less potent against Nek2 than the equivalent 6-ethynylpurine (23). This result suggested that the competitive binding component only contributes weakly to the activity observed in the 6-ethynylpurine series, assuming that 23, 24 and 102 make similar binding interactions with Nek2. Superposition of the crystal structure of 102 in complex with Nek2 confirmed that a near identical binding-orientation arose compared with the benchmark 6-ethynylpurine 24 (Fig. 8A). The covalent adduction of 24 can clearly be seen by the thioenol ether formed between 24 and Cys22. This is absent in the crystal structure of the cyano compound, indicating that no covalent interaction arose although the same mode of binding of the shared scaffold into the nucleotide-binding pocket of Nek2 was observed and the triplet of hydrogen bonds between the purine and the kinase hinge motif was preserved (Fig. 8B and C). Significantly, in vitro growth inhibition assays showed that 102 (GI50 > 100 μM) has no effect on cell viability in three human tumour cell-lines, WT U2OS, MDA-MB-231 and HeLa cells, as well as in the ‘normal’ human embryonic kidney (Hek293) cell-line.
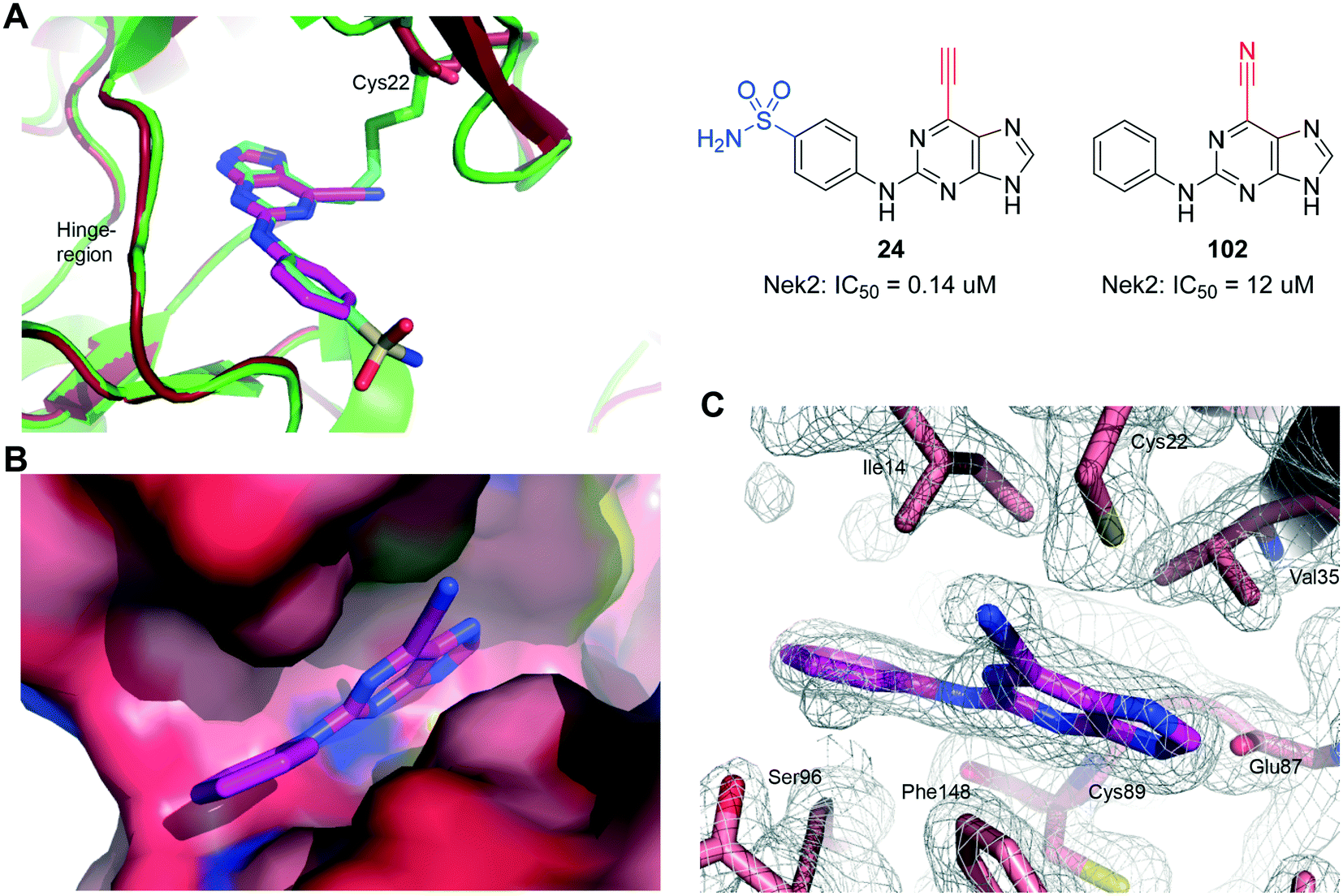 | ||
| Fig. 8 Interaction with competitively binding control compound 102 with Nek2: A) alignment of crystal structures of Nek2 (green) covalently bound to 24 (cyan) and Nek2 (red) non-covalently bound to 102 (magenta). B) Space-filling representation of 102 (magenta) bound into the nucleotide-binding pocket. C) Crystal structure of 102 (magenta) bound to Nek2, including electron density map (2Fo-Fc) contoured at 1.0σ (mesh). Hydrophobic residues lining the pocket, the hinge residues Glu87 and Cys89, and unadducted Cys22 are indicated. | ||
| Microsomal stability | |||||||
|---|---|---|---|---|---|---|---|
| Microsomal stability (human) | Microsomal stability (mouse) | ||||||
| CLint (μL min−1 mg−1) | S.E. CLint | T 1/2 (min) | n | CLint (μL min−1 mg−1) | S.E. CLint | T 1/2 (min) | n |
| 6.21 | 0.681 | 223 | 5 | 10.2 | 1.82 | 136 | 5 |
| Solubility | |||||||
|---|---|---|---|---|---|---|---|
| Estimated precipitation range (μM) | |||||||
| Lower bound | Upper bound | Calculated mid-range | |||||
| 100 | >100 | >100 | |||||
| Caco-2 permeability | |||||||
|---|---|---|---|---|---|---|---|
| Direction | Papp (× 10−6 cm−1) | Mean Papp (× 10−6 cm−1) | SD | n | Mean recovery (%) | ||
| 1 | 2 | ||||||
| A → B | 0.0534 | 0.0421 | 0.0477 | 0.00799 | 2 | 57.6 | |
| B → A | 4.88 | 5.29 | 5.08 | 0.293 | 2 | 64.0 | |
Conclusions
Achieving selectivity between specific members of the same or closely-related protein kinase families is challenging owing to the high degree of structural homology within the catalytic domain. However, targeting specific nucleophilic residues close to the catalytic site provides a means to discriminate. In this context, it is therefore encouraging that over 200 protein kinases contain a targetable cysteine residue close to the catalytic site.2,27–29 Covalent binding of an enzyme inhibitor has the potential advantages of prolonged duration of action and enhanced pharmacological efficacy, which allows lower dosing and reduced off-target effects. However, this advantage may not be relevant for Nek2 protein, which has a short cellular half-life of 45–75 minutes.30 There is concern that covalent binding agents will exhibit non-specific activity and off-target toxicity (e.g. promotion of immune responses31) owing to their perceived ability either to react almost randomly with biological nucleophiles, e.g. proteins and nucleic acids, or to be quenched by glutathione. However, this belief ignores the strict stereoelectronic requirements for efficient reaction of a protein nucleophile with an electrophilic reagent,15 and thus so far adverse properties of covalent inhibitors have been found to be minimal. Nevertheless, the development of covalent inhibitors must recognise the potential for off-target effects and be biomarker-focussed throughout. In this paper we have shown that selective covalent binding to Nek2 can be achieved with ethynylpurines, which exhibit numerous positive features as potential anticancer agents and therefore provide ample justification for the pursuit of this protein target with small molecule inhibitors.Experimental section
Chemicals and solvents
All chemical reagents were purchased from the Aldrich Chemical Company, Apollo Scientific or Alfa Aesar Chemicals and were of the highest available purity. Chemicals were used as supplied with no further treatment. If chemicals used were stated as dry/anhydrous, they were stored in SureSeal™ septum-sealed bottles and removed under an inert nitrogen environment, with the reaction being carried out under the relevant inert atmosphere. Palladium catalysts were stored and measured out under an inert atmosphere.Chromatography
Reaction monitoring and compound identification was aided using thin layer chromatography (TLC) and retardation factor (Rf) values. TLC was conducted with Merck aluminium backed Si F254, NH2 F254s and RP-18 F254s plates. Fluorescent compounds were visualised under short wave (254 nm) UV irradiation. Compound purification was achieved using medium pressure ‘Flash’ column chromatography, with the use of Davisil silica 40–60 μm as the stationary phase, or Biotage automated chromatography using pre-packed silica cartridges. A Biotage SP4 automated flash purification system was used with UV monitoring at 298 nm and compound collection at 254 nm. Biotage KP-NH cartridges were employed for the separation of secondary, tertiary, and heterocyclic amines; using a primary amine (propyl amine) bonded silica. When stated, compounds were purified via semi-preparative HPLC, using an ACE 5 Phenyl 150 × 21.2 mm column using an Agilent 1200 Modular Preparative HPLC system.Analytical techniques
All melting points were determined using a Stuart Scientific SMP3 or a Stuart Scientific SMP40 melting point apparatus and are uncorrected. 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained as solutions in deuterated solvents DMSO-d6, MeOD or CDCl3 using a Bruker Avance III 500 spectrometer recording at 500 MHz. Chemical shifts (δ) are reported in parts per million (ppm) and the spin-multiplicity abbreviated as: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sept (septet), m (multiplet), or br (broad), with coupling constants (J) given in Hertz (Hz). Liquid chromatography – mass spectrometry (LC–MS) was carried out on a Micromass Platform LC running in both positive and negative electrospray mode with a PDA 240–400 nm detector using a Waters Symmetry Shield RP18 3 μm, 4.6 × 20 mm column with a flow rate of 3.0 mL min−1. Alternatively, a Waters Acquity UPLC system was used, with a Waters SQD ESCi source using an Acquity UPLC BEH C18 1.7 μm, 2.1 × 50 mm column with a flow rate of 0.6 mL min−1. The mobile phase used was 0.1 % v/v formic acid (aq.)/MeCN. Fourier transform infrared (FTIR) spectra were obtained using a Bio-Rad FTS 3000MX diamond ATR as a neat sample. Ultraviolet (UV) absorption data were collected using a Hitachi U-2800A spectrophotometer in ethanol. High-resolution mass spectra were performed by the ESPRC UK National Mass Spectrometry Facility, Swansea University, Singleton Park, Swansea, SA2 8PP. The purity of final compounds was assessed by reversed-phase HPLC; all tested compounds were >95% purity. HPLC instrument, Agilent 1200 equipped with a photodiode array detector (190–400 nm). Sample temperature, ambient; injection volume, 5 μL; flow rate, 1 mL min−1. 5% to 100% MeCN gradient over 9 min and an isocratic hold at 100% MeCN for 2.5 min, before returning to initial conditions. Mobile phase A = 0.1% ammonia in water or 0.1% formic acid in water, mobile phase B = MeCN. Column: Waters XSELECT CSH C18, 3.5 μm, 4.6 mm × 150 mm or Waters XTerra RP18, 5 μm, 4.6 mm × 150 mm. Column maintained at ambient temperature.Microwave assisted synthesis
When stated, reactions were carried out under microwave irradiation, in sealed vessels, using a Biotage Initiator Sixty with robotic sample bed. Samples were irradiated at 2.45 GHz, able to reach temperatures of 60–250 °C with a rate of heating at 2–5 °C s−1, and pressures of up to 20 bar.TBAF scavenger resin32
Amberlite 15 Ion exchange resin (SO3H, 100 mL) was loaded into a column and washed with water (400 mL). The column was eluted with sat. calcium hydroxide solution whilst the pH of the eluent was monitored. Once the initially pH neutral eluent became strongly basic, the column was eluted with water until the pH of the eluent returned to neutral. The resin was washed with DCM (300 mL), THF (300 mL) and Et2O (300 mL), before being removed from the column and dried in a vacuum oven at 40 °C.General procedure A: TFA/TFE coupling of anilines with 2-fluoropurines using conventional heating
TFA (2.5–5.0 equiv.) was added to a solution of the purine substrate (1.0 equiv.) and the required aniline (2.0 equiv.) in TFE (10 mL mmol−1). The reaction mixture was heated at reflux for 24 h unless otherwise stated, after which the solution was cooled and evaporated to dryness. The resulting residue was dissolved in EtOAc (10 mL mmol−1) and washed with a saturated aqueous solution of NaHCO3 (5 mL mmol−1) and brine (5 mL mmol−1). The combined aqueous layers were extracted with EtOAc (10 mL mmol−1), and the combined organic extracts were dried (MgSO4) and concentrated in vacuo to give the crude product for chromatographic purification.General procedure B: removal of TIPS-protecting groups using TBAF
TBAF (1.2 equiv.) was added to a solution of the TIPS-protected substrate (1.0 equiv.) in THF (10–20 mL mmol−1). The reaction mixture was stirred at RT for 5 min before being concentrated in vacuo and the crude residue purified by chromatography to afford the target compound.Part 1. To a stirred solution of HBF4 (48% aqueous, 120 mL) at 0 °C, was added 2-amino-6-chloropurine (6.0 g, 35.0 mmol). Over 20 min, a solution of NaNO2 (4.9 g, 70.0 mmol) in water (200 mL) was added dropwise, ensuring the temperature remained close to 0 °C. The pale yellow solution was raised to RT and stirred for 18 h. The resulting solution was neutralised to pH 7 in an ice bath at 0 °C by addition of Na2CO3 (6.00 g) in water (200 mL). The crude material was purified by chromatography on silica (10% MeOH/DCM) to afford 6-chloro-2-fluoropurine as a white crystalline solid (4.52 g, 75%); mp 171–173 °C (lit.,34 mp 174 °C); λmax (EtOH/nm) 393; IR (cm−1) 2964, 2785, 1735, 1581; 1H NMR (500 MHz, DMSO-d6) 8.60 (1H, s, H-8), 13.9 (1H, s, NH-9); LRMS (ES+) m/z 172.6 [M + H]+.
Part 2. 3,4-Dihydropyran (60 μL, 0.58 mmol) was added dropwise over 10 min to a vigorously stirred solution of 6-chloro-2-fluoropurine (100 mg, 0.58 mmol) and (rac)-camphorsulfonic acid (5 mg, 0.02 mmol) in EtOAc (50 mL) at 65 °C. The temperature was maintained at 65 °C for 18 h. The resulting bright yellow solution was neutralised to pH 7 by careful addition of aqueous NH3 solution, until a cloudy suspension persisted. The crude mixture was washed with brine (2 × 30 mL) and the aqueous phase was re-extracted with EtOAc (2 × 30 mL). The combined organic extracts were dried (Na2SO4) and purified by chromatography on silica (30% EtOAc/petrol). The desired compound was isolated as a pale yellow oil which solidified on refrigeration (110 mg, 75%); mp 69–70 °C (lit.33 mp not available); λmax (EtOH/nm) 269; IR (cm−1) 3125, 2954, 2872, 2028, 1577; 1H NMR (300 MHz, DMSO-d6) 1.60 (2H, m, CH2), 1.74 (1H, m, CH), 2.01 (2H, m, CH2), 2.32 (1H, s, CH), 3.71 (1H, t, J = 12.0 Hz, CH), 4.01 (1H, d, J = 12.0 Hz, CH), 5.64 (1H, d, J = 12.0 Hz, CH), 8.25 (1H, s, H-8); LRMS (ES+) m/z 257.7 [M + H]+.
:3 petrol/EtOAc); λmax (EtOH/nm) 302; IR (cm−1) 2945, 2866, 2361, 2000, 1584; 1H NMR (500 MHz, DMSO-d6) 1.12–1.21 (21H, m, Si(CH(CH3)2)3), 8.68 (1H, s, H-8), 13.89 (1H, br, NH-9); HRMS calcd. for C16H24FN4Si (ES+) m/z 319.1749 [M + H]+, found 319.1752.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 6.91–6.96 (1H, m, H-4′), 7.29 (2H, dd, J = 7.6 and 8.0 Hz, H-3′ and H-5′), 7.80 (2H, dd, J = 2.0 and 8.0 Hz, H-2′ and H-6′), 8.30 (1H, s, H-8), 9.70 (1H, s, NH), 13.17 (1H, br, NH-9); HRMS calcd. for C13H10N5 (ES+) m/z 236.0934 [M + H]+, found 236.0931.
C–H), 7.16 (2H, s, SO2NH2), 7.17 (2H, d, J = 9.0 Hz, H-2′ and H-6′), 7.93 (2H, d, J = 9.0 Hz, H-3′ and H-5′), 8.36 (1H, s, H-8); HRMS calcd. for C13H11N6O2S [M + H]+ 315.0664, found 315.0687.
:1 DCM/MeOH) followed by chromatography on reverse phase silica (9:1 MeOH/H2O + 0.1% HCOOH). The desired product was obtained as an orange oil/gum (0.952 g, 64%); Rf 0.32 (9:1 DCM/MeOH); λmax (EtOH/nm) 275; IR (cm−1) 2972, 2360, 2340, 1977, 1702; 1H NMR (500 MHz, DMSO-d6) 1.13–1.22 (21H, m, Si(CH(CH3)2)3), 3.51 (2H, s, CH2CO2H), 6.82–6.85 (1H, m, H-6′), 7.22 (1H, dd, J = 7.7 and 7.8 Hz, H-5′), 7.65–7.68 (1H, m, H-2′), 7.72–7.76 (1H, m, H-4′), 8.25 (1H, s, H-8), 9.71 (1H, s, NH), 12.33 (1H, br, CO2H), 13.10 (1H, br, NH-9); HRMS calcd. for C24H32N5O2Si (ES+) m/z 450.2320 [M + H]+, found 450.2320.
:1 DCM/MeOH) gave the desired compound as a yellow oil/gum (0.238 g, 99%); Rf 0.48 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 277; IR (cm−1) 2360, 2153, 2120, 1980; 1H NMR (500 MHz, DMSO-d6) 1.12–1.25 (21H, m, Si(CH(CH3)2)3), 3.42 (2H, s, COCH2), 3.71 (3H, s, OCH3), 4.20 (2H, d, J = 5.8 Hz, NHCH2), 6.85 (2H, d, J = 8.8 Hz, H-3′′ and H-5′′), 6.86–6.91 (1H, m, H-6′), 7.17 (2H, d, J = 8.8 Hz, H-2′′ and H-6′′), 7.20 (1H, dd, J = 7.9 and 8.0 Hz, H-5′), 7.59–7.62 (1H, m, H-2′), 7.74–7.78 (1H, m, H-4′), 8.24 (1H, s, H-8), 8.44 (1H, t, J = 5.8 Hz, NHCH2), 9.65 (1H, s, NH), 12.06 (1H, br, NH-9); HRMS calcd. for C32H41N6O2Si (ES+) m/z 569.3055 [M + H]+, found 569.3057.
:1 DCM/MeOH) gave the desired compound as a yellow oil/gum (0.170 g, 90%); Rf 0.24 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 276; IR (cm−1) 3282, 2965, 2943, 2362, 1669; 1H NMR (500 MHz, DMSO-d6) 1.13–1.21 (21H, m, Si(CH(CH3)2)3), 3.34 (2H, s, COCH2), 6.83–6.86 (1H, m, H-6′), 6.88 (1H, s, CONHH′), 7.19 (1H, dd, J = 8.5 and 8.7 Hz, H-5′), 7.42 (1H, s, CONHH′), 7.57–7.60 (1H, m, H-2′), 7.72–7.76 (1H, m, H-4′), 8.23 (1H, s, H-8), 9.65 (1H, s, NH), 13.08 (1H, s, NH-9); HRMS calcd. for C24H33N6OSi (ES+) m/z 449.2480 [M + H]+, found 449.2480.
:3 H2O/MeCN), to give the desired compound as a yellow solid (0.084 g, 0.28 mmol, 76%); Rf 0.32 (17:3 DCM/MeOH, KP-NH); mp 250–270 °C (decomposed); λmax (EtOH/nm) 274; IR (cm−1) 3359, 3170, 2921, 2107, 1658; 1H NMR (500 MHz, DMSO-d6) 4.76 (1H, s, CCH), 6.77–6.81 (1H, m, H-6′), 6.83 (1H, s, CONHH′), 7.15 (1H, dd, J = 8.0 and 8.1 Hz, H-5′), 7.38 (1H, s, CONHH′), 7.47–7.50 (1H, m, H-2′), 7.68–7.73 (1H, m, H-4′), 8.19 (1H, s, H-8), 9.52 (1H, s, NH), 13.06 (1H, s, NH-9); HRMS calcd. for C15H13N6O (ES+) m/z 293.1149 [M + H]+, found 293.1145.
:1 MeOH/H2O + 0.1% HCOOH) followed by chromatography on silica (7:3 petrol/EtOAc) to give the desired compound as a yellow/orange oil/gum (0.506 g, 68%); Rf 0.34 (9:1 MeOH/H2O, +0.1% HCOOH, C18); λmax (EtOH/nm) 368, 277; IR (cm−1) 2950, 2365, 2161, 2011, 1967, 1601; 1H NMR (500 MHz, DMSO-d6) 1.13–2.23 (21H, m, Si(CH(CH3)2)3), 4.85 (2H, s, Ar–CH2), 4.95 (2H, q, J = 8.7 Hz, F3CCH2O), 7.00–7.04 (1H, m, H-6′), 7.34 (1H, dd, J = 7.9 and 8.1 Hz, H-5′), 7.74–7.77 (1H, m, H-2′), 7.96–8.00 (1H, m, H-4′), 8.28 (1H, s, H-8), 9.84 (1H, s, NH), 13.12 (1H, br, NH-9); HRMS calcd. for C25H33F3N5O3SSi (ES+) m/z 568.2020 [M + H]+, found 568.2015.
:1 DCM/MeOH) gave the desired compound as a yellow oil (0.224 g, 97%); Rf 0.44 (19:1 DCM/MeOH); λmax (EtOH/nm) 276; IR (cm−1) 2361, 2341, 2162, 1992, 1969, 1609; 1H NMR (500 MHz, DMSO-d6) 1.13–1.22 (21H, m, Si(CH(CH3)2)3), 3.72 (3H, s, OCH3), 4.05 (2H, d, J = 6.0 Hz, NHCH2), 4.23 (2H, s, Ar–CH2), 6.87 (2H, d, J = 8.7 Hz, H-3′′ and H-5′′), 6.91–6.95 (1H, m, H-6′), 7.23 (2H, d, J = 8.7 Hz, H-2′′ and H-6′′), 7.28 (1H, dd, J = 8.0 and 8.2 Hz, H-5′), 7.55 (1H, t, J = 6.0 Hz, NHCH2), 7.72–7.75 (1H, m, H-2′), 7.87–7.91 (1H, m, H-4′), 8.26 (1H, s, H-8), 9.76 (1H, s, NH), 13.06 (1H, s, NH-9); HRMS calcd. for C31H41N6O3SSi (ES+) m/z 605.2725 [M + H]+, found 605.2723.
:1 DCM/MeOH) to give the desired compound as a yellow oil/gum (0.108 g, 60%); Rf 0.21 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 276; IR (cm−1) 3367, 2943, 2865, 2160, 2021, 1606; 1H NMR (500 MHz, DMSO-d6) 1.13–1.23 (21H, m, Si(CH(CH3)2)3), 4.21 (2H, s, Ar–CH2), 6.84 (2H, s, SO2NH2), 6.95–6.98 (1H, br, H-6′), 7.13 (1H, dd, J = 7.9 and 8.0 Hz, H-5′), 7.68–7.71 (1H, m, H-2′), 7.90 (1H, ddd, J = 1.0, 1.9 and 8.0 Hz, H-4′), 8.25 (1H, s, H-8), 9.75 (1H, s, NH), 13.07 (1H, s, NH-9); HRMS calcd. for C23H33N6O2SSi (ES+) m/z 485.2149 [M + H]+, found 485.2147.
:1 DCM/MeOH) to afford the target compound as a pale yellow solid (23 mg, 58%); Rf 0.29 (9:1 DCM/MeOH); mp 190–210 °C (decomposed); λmax (EtOH/nm) 273.5, 359.0; IR (cm−1) 3361, 3248, 2976, 2809, 2707, 2114, 1701, 1610, 1583, 1491; 1H NMR (500 MHz, DMSO-d6) 4.22 (2H, s, SO2CH2), 4.85 (1H, s, CCH), 6.86 (2H, s, SO2NH2), 6.94–6.98 (1H, m, H-6′), 7.30 (1H, dd, J = 7.7 and 8.0 Hz, H-5′), 7.64–7.68 (1H, m, H-2′), 7.88–7.92 (1H, m, H-4′), 8.29 (1H, s, H-8), 9.72 (1H, s, NH), 13.13 (1H, br, NH-9); HRMS calcd. for C14H13N6O2S (ES+) m/z 329.0815 [M + H]+, found 329.0821.
:0.5 DCM/MeOH), yielding the title compound as a yellow solid (69 mg, 98%); mp 135–137 °C (lit.,14 mp 135–137 °C); λmax (EtOH/nm) 360, 238, 254; IR (cm−1) 3030, 2921, 2850, 1629, 1559; 1H NMR (500 MHz, CDCl3) 1.49 (4H, m, homopiperidine CH2), 1.64–1.71 (4H, m, homopiperidine CH2), 3.28–3.38 (4H, m, homopiperidine CH2), 5.49–5.52 (1H, d, J = 15.0 Hz, CH
CH), 6.91–6.96 (1H, m, H-4′), 7.29 (2H, dd, J = 7.6 and 8.0 Hz, H-3′ and H-5′), 7.80 (2H, dd, J = 2.0 and 8.0 Hz, H-2′ and H-6′), 8.30 (1H, s, H-8), 9.70 (1H, s, NH), 13.17 (1H, br, NH-9); HRMS calcd. for C13H10N5 (ES+) m/z 236.0934 [M + H]+, found 236.0931.
C–H), 7.16 (2H, s, SO2NH2), 7.17 (2H, d, J = 9.0 Hz, H-2′ and H-6′), 7.93 (2H, d, J = 9.0 Hz, H-3′ and H-5′), 8.36 (1H, s, H-8); HRMS calcd. for C13H11N6O2S [M + H]+ 315.0664, found 315.0687.
:1 DCM/MeOH) followed by chromatography on reverse phase silica (9:1 MeOH/H2O + 0.1% HCOOH). The desired product was obtained as an orange oil/gum (0.952 g, 64%); Rf 0.32 (9:1 DCM/MeOH); λmax (EtOH/nm) 275; IR (cm−1) 2972, 2360, 2340, 1977, 1702; 1H NMR (500 MHz, DMSO-d6) 1.13–1.22 (21H, m, Si(CH(CH3)2)3), 3.51 (2H, s, CH2CO2H), 6.82–6.85 (1H, m, H-6′), 7.22 (1H, dd, J = 7.7 and 7.8 Hz, H-5′), 7.65–7.68 (1H, m, H-2′), 7.72–7.76 (1H, m, H-4′), 8.25 (1H, s, H-8), 9.71 (1H, s, NH), 12.33 (1H, br, CO2H), 13.10 (1H, br, NH-9); HRMS calcd. for C24H32N5O2Si (ES+) m/z 450.2320 [M + H]+, found 450.2320.
:1 DCM/MeOH) gave the desired compound as a yellow oil/gum (0.238 g, 99%); Rf 0.48 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 277; IR (cm−1) 2360, 2153, 2120, 1980; 1H NMR (500 MHz, DMSO-d6) 1.12–1.25 (21H, m, Si(CH(CH3)2)3), 3.42 (2H, s, COCH2), 3.71 (3H, s, OCH3), 4.20 (2H, d, J = 5.8 Hz, NHCH2), 6.85 (2H, d, J = 8.8 Hz, H-3′′ and H-5′′), 6.86–6.91 (1H, m, H-6′), 7.17 (2H, d, J = 8.8 Hz, H-2′′ and H-6′′), 7.20 (1H, dd, J = 7.9 and 8.0 Hz, H-5′), 7.59–7.62 (1H, m, H-2′), 7.74–7.78 (1H, m, H-4′), 8.24 (1H, s, H-8), 8.44 (1H, t, J = 5.8 Hz, NHCH2), 9.65 (1H, s, NH), 12.06 (1H, br, NH-9); HRMS calcd. for C32H41N6O2Si (ES+) m/z 569.3055 [M + H]+, found 569.3057.
:1 DCM/MeOH) gave the desired compound as a yellow oil/gum (0.170 g, 90%); Rf 0.24 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 276; IR (cm−1) 3282, 2965, 2943, 2362, 1669; 1H NMR (500 MHz, DMSO-d6) 1.13–1.21 (21H, m, Si(CH(CH3)2)3), 3.34 (2H, s, COCH2), 6.83–6.86 (1H, m, H-6′), 6.88 (1H, s, CONHH′), 7.19 (1H, dd, J = 8.5 and 8.7 Hz, H-5′), 7.42 (1H, s, CONHH′), 7.57–7.60 (1H, m, H-2′), 7.72–7.76 (1H, m, H-4′), 8.23 (1H, s, H-8), 9.65 (1H, s, NH), 13.08 (1H, s, NH-9); HRMS calcd. for C24H33N6OSi (ES+) m/z 449.2480 [M + H]+, found 449.2480.
:3 H2O/MeCN), to give the desired compound as a yellow solid (0.084 g, 0.28 mmol, 76%); Rf 0.32 (17:3 DCM/MeOH, KP-NH); mp 250–270 °C (decomposed); λmax (EtOH/nm) 274; IR (cm−1) 3359, 3170, 2921, 2107, 1658; 1H NMR (500 MHz, DMSO-d6) 4.76 (1H, s, CCH), 6.77–6.81 (1H, m, H-6′), 6.83 (1H, s, CONHH′), 7.15 (1H, dd, J = 8.0 and 8.1 Hz, H-5′), 7.38 (1H, s, CONHH′), 7.47–7.50 (1H, m, H-2′), 7.68–7.73 (1H, m, H-4′), 8.19 (1H, s, H-8), 9.52 (1H, s, NH), 13.06 (1H, s, NH-9); HRMS calcd. for C15H13N6O (ES+) m/z 293.1149 [M + H]+, found 293.1145.
:1 MeOH/H2O + 0.1% HCOOH) followed by chromatography on silica (7:3 petrol/EtOAc) to give the desired compound as a yellow/orange oil/gum (0.506 g, 68%); Rf 0.34 (9:1 MeOH/H2O, +0.1% HCOOH, C18); λmax (EtOH/nm) 368, 277; IR (cm−1) 2950, 2365, 2161, 2011, 1967, 1601; 1H NMR (500 MHz, DMSO-d6) 1.13–2.23 (21H, m, Si(CH(CH3)2)3), 4.85 (2H, s, Ar–CH2), 4.95 (2H, q, J = 8.7 Hz, F3CCH2O), 7.00–7.04 (1H, m, H-6′), 7.34 (1H, dd, J = 7.9 and 8.1 Hz, H-5′), 7.74–7.77 (1H, m, H-2′), 7.96–8.00 (1H, m, H-4′), 8.28 (1H, s, H-8), 9.84 (1H, s, NH), 13.12 (1H, br, NH-9); HRMS calcd. for C25H33F3N5O3SSi (ES+) m/z 568.2020 [M + H]+, found 568.2015.
:1 DCM/MeOH) gave the desired compound as a yellow oil (0.224 g, 97%); Rf 0.44 (19:1 DCM/MeOH); λmax (EtOH/nm) 276; IR (cm−1) 2361, 2341, 2162, 1992, 1969, 1609; 1H NMR (500 MHz, DMSO-d6) 1.13–1.22 (21H, m, Si(CH(CH3)2)3), 3.72 (3H, s, OCH3), 4.05 (2H, d, J = 6.0 Hz, NHCH2), 4.23 (2H, s, Ar–CH2), 6.87 (2H, d, J = 8.7 Hz, H-3′′ and H-5′′), 6.91–6.95 (1H, m, H-6′), 7.23 (2H, d, J = 8.7 Hz, H-2′′ and H-6′′), 7.28 (1H, dd, J = 8.0 and 8.2 Hz, H-5′), 7.55 (1H, t, J = 6.0 Hz, NHCH2), 7.72–7.75 (1H, m, H-2′), 7.87–7.91 (1H, m, H-4′), 8.26 (1H, s, H-8), 9.76 (1H, s, NH), 13.06 (1H, s, NH-9); HRMS calcd. for C31H41N6O3SSi (ES+) m/z 605.2725 [M + H]+, found 605.2723.
:1 DCM/MeOH) to give the desired compound as a yellow oil/gum (0.108 g, 60%); Rf 0.21 (19:1 DCM/MeOH, KP-NH); λmax (EtOH/nm) 276; IR (cm−1) 3367, 2943, 2865, 2160, 2021, 1606; 1H NMR (500 MHz, DMSO-d6) 1.13–1.23 (21H, m, Si(CH(CH3)2)3), 4.21 (2H, s, Ar–CH2), 6.84 (2H, s, SO2NH2), 6.95–6.98 (1H, br, H-6′), 7.13 (1H, dd, J = 7.9 and 8.0 Hz, H-5′), 7.68–7.71 (1H, m, H-2′), 7.90 (1H, ddd, J = 1.0, 1.9 and 8.0 Hz, H-4′), 8.25 (1H, s, H-8), 9.75 (1H, s, NH), 13.07 (1H, s, NH-9); HRMS calcd. for C23H33N6O2SSi (ES+) m/z 485.2149 [M + H]+, found 485.2147.
:1 DCM/MeOH) to afford the target compound as a pale yellow solid (23 mg, 58%); Rf 0.29 (9:1 DCM/MeOH); mp 190–210 °C (decomposed); λmax (EtOH/nm) 273.5, 359.0; IR (cm−1) 3361, 3248, 2976, 2809, 2707, 2114, 1701, 1610, 1583, 1491; 1H NMR (500 MHz, DMSO-d6) 4.22 (2H, s, SO2CH2), 4.85 (1H, s, CCH), 6.86 (2H, s, SO2NH2), 6.94–6.98 (1H, m, H-6′), 7.30 (1H, dd, J = 7.7 and 8.0 Hz, H-5′), 7.64–7.68 (1H, m, H-2′), 7.88–7.92 (1H, m, H-4′), 8.29 (1H, s, H-8), 9.72 (1H, s, NH), 13.13 (1H, br, NH-9); HRMS calcd. for C14H13N6O2S (ES+) m/z 329.0815 [M + H]+, found 329.0821.
:0.5 DCM/MeOH), yielding the title compound as a yellow solid (69 mg, 98%); mp 135–137 °C (lit.,14 mp 135–137 °C); λmax (EtOH/nm) 360, 238, 254; IR (cm−1) 3030, 2921, 2850, 1629, 1559; 1H NMR (500 MHz, CDCl3) 1.49 (4H, m, homopiperidine CH2), 1.64–1.71 (4H, m, homopiperidine CH2), 3.28–3.38 (4H, m, homopiperidine CH2), 5.49–5.52 (1H, d, J = 15.0 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.95–6.99 (1H, t, J = 10.5 Hz, H-4′), 7.19 (1H, s, H-8), 7.22–7.26 (2H, dd, J = 9.5, 10.5 Hz, H-3′ and H-5′), 7.32 (1H, br s, NH), 7.44–7.46 (2H, d, J = 9.5 Hz, H-2′ and H-6′), 8.23–8.26 (1H, d, J = 15.0 Hz, CHCH), 12.89 (1H, br s, NH-9); HRMS calcd. for C19H22N6 [M + H]+ 335.1975, found 335.1979.
:0.5 DCM/MeOH) to yield a pale yellow solid (41 mg, 30%); mp 47–49 °C; λmax (EtOH/nm) 326, 277, 238; IR (cm−1) 2928, 2858, 1582; 1H NMR (500 MHz, DMSO-d6) 1.32–1.33 (4H, m, homopiperidine CH2), 1.39–1.40 (4H, m, homopiperidine CH2), 2.53–2.56 (4H, t, J = 5.5 Hz, homopiperidine NCH2), 2.86–2.91 (2H, t, J = 8.0 Hz, CH2), 2.92–2.98 (2H, t, J = 8.0 Hz, CH2), 6.69–6.72 (1H, t, J = 8.5 Hz, H-4′), 7.05–7.08 (2H, dd, J = 7.5 and 8.5 Hz, H-3′ and H-5′), 7.65–7.66 (2H, d, J = 7.5 Hz, H-2′ and H-6′), 7.97 (1H, s, H-8), 9.21 (1H, br s, NH); HRMS calcd. for C19H25N6 [M + H]+ 337.2135, found 337.2138.
:0.5 DCM/MeOH) to yield a yellow solid (6.4 mg, 30%); mp 121–123 °C; UV λmax (EtOH) 270, 207; IR (cm−1) 1578, 1532, 1496, 1439, 1392, 1349; 1H NMR (500 MHz, DMSO-d6) 5.84–5.86 (1H, dd, J = 1.5 and 12.5 Hz, cis CHCHH), 6.86–6.89 (1H, dd, J = 1.5 and 17.5 Hz, trans CHCHH), 7.01–7.04 (1H, t, J = 8.5 Hz, H-4′), 7.08–7.14 (1H, dd, J = 12.5 and 17.5 Hz, CHCH2), 7.24 (1H, s, H-8), 7.27–7.30 (2H, dd, J = 8.5 and 7.5 Hz, H-3′ and H-5′), 7.52–7.54 (2H, d, J = 7.5 Hz, H-2′ and H-6′); HRMS calcd. for C13H12N5 [M + H]+ 238.0509, found 238.0511.
CH), 4.09–4.14 (1H, m, CH), 5.65 (1H, dd, J = 2.6 and 10.8 Hz, NCH), 8.26 (1H, s, H-8); LRMS (ES+) m/z 247.0 [M + H]+.
CH), 6.95–6.99 (1H, t, J = 10.5 Hz, H-4′), 7.19 (1H, s, H-8), 7.22–7.26 (2H, dd, J = 9.5, 10.5 Hz, H-3′ and H-5′), 7.32 (1H, br s, NH), 7.44–7.46 (2H, d, J = 9.5 Hz, H-2′ and H-6′), 8.23–8.26 (1H, d, J = 15.0 Hz, CHCH), 12.89 (1H, br s, NH-9); HRMS calcd. for C19H22N6 [M + H]+ 335.1975, found 335.1979.
:0.5 DCM/MeOH) to yield a pale yellow solid (41 mg, 30%); mp 47–49 °C; λmax (EtOH/nm) 326, 277, 238; IR (cm−1) 2928, 2858, 1582; 1H NMR (500 MHz, DMSO-d6) 1.32–1.33 (4H, m, homopiperidine CH2), 1.39–1.40 (4H, m, homopiperidine CH2), 2.53–2.56 (4H, t, J = 5.5 Hz, homopiperidine NCH2), 2.86–2.91 (2H, t, J = 8.0 Hz, CH2), 2.92–2.98 (2H, t, J = 8.0 Hz, CH2), 6.69–6.72 (1H, t, J = 8.5 Hz, H-4′), 7.05–7.08 (2H, dd, J = 7.5 and 8.5 Hz, H-3′ and H-5′), 7.65–7.66 (2H, d, J = 7.5 Hz, H-2′ and H-6′), 7.97 (1H, s, H-8), 9.21 (1H, br s, NH); HRMS calcd. for C19H25N6 [M + H]+ 337.2135, found 337.2138.
:0.5 DCM/MeOH) to yield a yellow solid (6.4 mg, 30%); mp 121–123 °C; UV λmax (EtOH) 270, 207; IR (cm−1) 1578, 1532, 1496, 1439, 1392, 1349; 1H NMR (500 MHz, DMSO-d6) 5.84–5.86 (1H, dd, J = 1.5 and 12.5 Hz, cis CHCHH), 6.86–6.89 (1H, dd, J = 1.5 and 17.5 Hz, trans CHCHH), 7.01–7.04 (1H, t, J = 8.5 Hz, H-4′), 7.08–7.14 (1H, dd, J = 12.5 and 17.5 Hz, CHCH2), 7.24 (1H, s, H-8), 7.27–7.30 (2H, dd, J = 8.5 and 7.5 Hz, H-3′ and H-5′), 7.52–7.54 (2H, d, J = 7.5 Hz, H-2′ and H-6′); HRMS calcd. for C13H12N5 [M + H]+ 238.0509, found 238.0511.
CH), 4.09–4.14 (1H, m, CH), 5.65 (1H, dd, J = 2.6 and 10.8 Hz, NCH), 8.26 (1H, s, H-8); LRMS (ES+) m/z 247.0 [M + H]+.
Accession codes
X-ray co-crystal structures of Nek2 with compounds 24 (6SGD) and 66 (6SGH), and also with the competitively-binding control compounds 96 (6SGI) and 102 (6SGK) have been deposited with the PDB. Authors will release the atomic coordinates and experimental data upon article publication.Ethical statement
All animal experiments performed were conducted under a UK Government Home Office License in accordance with relevant national laws. All procedures were reviewed and approved by the Newcastle University Animal Welfare Ethical Board and performed according to institutional guidelines.Abbreviations used
| CDI | 1,1′-Carbonyldiimidazole |
| DIPEA | N,N-Diisopropylethylamine |
| HER2 | Human epidermal growth factor receptor 2 |
| MW | Microwave |
| NIMA | Never-in-mitosis gene A |
| PMB | 4-Methoxybenzyl |
| SNAr | Nucleophilic aromatic substitution |
| TFE | 2,2,2-Trifluoroethanol |
Author contributions
C. J. M. and C. R. C. contributed equally to the work described in this paper. C. J. M., synthesis and characterization of compounds; C. R. C., synthesis, characterization of compounds and manuscript preparation; R. B., project conception, experimental design and interpretation, data analysis and manuscript preparation; K. B., design, conduct and interpretation of Nek2 inhibition experiments; B. C., synthesis and characterization of compounds; A. M. F., project conception; I. R. H., project conception; S. J. H., data analysis and manuscript preparation; C. M-D., data collection and analysis of crystal structures; D. R. N., project conception, experimental design and interpretation and data analysis; M. W. R., crystal structure refinement, validation and data deposition; M. S., synthesis and characterization of compounds; D. T., synthesis and characterization of compounds; R. J. G., project conception, experimental design and interpretation and data analysis; B. T. G., experimental design and interpretation, data analysis and manuscript preparation; C. C., experimental design and interpretation, data analysis and manuscript preparation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by a grant from Cancer Research UK (Grant Reference C2115/A21421). RB acknowledges funding from Cancer Research UK (Grant References C24461/A10285, C24461/A23302). The use of the EPSRC Mass Spectrometry Service at the University of Wales (Swansea) is also gratefully acknowledged. Similarly, we thank beamline staff at The Diamond Light Source who provided excellent facilities for data collection.References
- S. R. Klutchko, H. Zhou, R. T. Winters, T. P. Tran, A. J. Bridges, I. W. Althaus, D. M. Amato, W. L. Elliott, P. A. Ellis, M. A. Meade, B. J. Roberts, D. W. Fry, A. J. Gonzales, P. J. Harvey, J. M. Nelson, V. Sherwood, H.-K. Han, G. Pace, J. B. Smaill, W. A. Denny and H. D. H. Showalter, Tyrosine kinase inhibitors. 19. 6-Alkynamides of 4-anilinoquinazolines and 4-anilinopyrido [3, 4-d] pyrimidines as irreversible inhibitors of the erbB family of tyrosine kinase receptors, J. Med. Chem., 2006, 49, 1475 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Developing irreversible inhibitors of the protein kinase cysteinome, Chem. Biol., 2013, 20, 46 CrossRef PubMed.
- E. Anscombe, E. Meschini, R. Mora-Vidal, M. P. Martin, D. Staunton, M. Geitmann, U. H. Danielson, W. A. Stanley, L.-Z. Wang, T. Reuillon, B. T. Golding, C. Cano, D. R. Newell, M. E. M. Noble, S. R. Wedge, J. A. Endicott and R. J. Griffin, Identification and characterization of an irreversible inhibitor of CDK2, Chem. Biol., 2015, 22, 1159 CrossRef CAS PubMed.
- D. H. Wai, K. L. Schaefer, A. Schramm, E. Korsching, F. van Valen, T. Ozaki, W. Boecker, L. Schweigerer, B. Dockhorn-Dworniczak and C. Poremba, Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays, Int. J. Oncol., 2002, 20, 441 CAS.
- S. de Vos, W. K. Hofmann, T. M. Grogan, U. Krug, M. Schrage, T. P. Miller, J. G. Braun, W. Wachsman, H. P. Koeffler and J. W. Said, Gene expression profile of serial samples of transformed B-cell lymphomas, Lab. Invest., 2003, 83, 271 CrossRef CAS PubMed.
- D. G. Hayward, R. B. Clarke, A. J. Faragher, M. R. Pillai, I. M. Hagan and A. M. Fry, The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer, Cancer Res., 2004, 64, 7370 CrossRef CAS PubMed.
- Y. Fang and X. Zhang, Targeting NEK2 as a promising therapeutic approach for cancer treatment, Cell Cycle, 2016, 15, 895 CrossRef CAS PubMed.
- https://depmap.sanger.ac.uk/ .
- M. Marina and H. I. Saavedra, Nek2 and Plk4: prognostic markers, drivers of breast tumorigenesis and drug resistance, Front. Biosci., 2014, 19, 352 CrossRef PubMed.
- D. K. Whelligan, S. Solanki, D. Taylor, D. W. Thomson, K.-M. J. Cheung, K. Boxall, C. Mas-Droux, C. Barillari, S. Burns, C. G. Grummitt, I. Collins, R. L. M. van Montfort, G. W. Aherne, R. Bayliss and S. Hoelder, Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization, J. Med. Chem., 2010, 53, 7682 CrossRef CAS PubMed.
- S. Solanki, P. Innocenti, C. Mas-Droux, K. Boxall, C. Barillari, R. L. M. van Montfort, G. W. Aherne, R. Bayliss and S. Hoelder, Benzimidazole inhibitors induce a DFG-out conformation of never in mitosis gene A-related kinase 2 (Nek2) without binding to the back pocket and reveal a nonlinear structure−activity relationship, J. Med. Chem., 2011, 54, 1626 CrossRef CAS PubMed.
- P. Innocenti, K. M. J. Cheung, S. Solanki, C. Mas-Droux, F. Rowan, S. Yeoh, K. Boxall, M. Westlake, L. Pickard, T. Hardy, J. E. Baxter, G. W. Aherne, R. Bayliss, A. M. Fry and S. Hoelder, Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: Structure–activity relationship, structural biology, and cellular activity, J. Med. Chem., 2012, 55, 3228 CrossRef CAS PubMed.
- M. S. Cohen, C. Zhang, K. M. Shokat and J. Taunton, Structural bioinformatics-based design of selective, irreversible kinase inhibitors, Science, 2005, 308, 1318 CrossRef CAS PubMed.
- C. R. Coxon, C. Wong, R. Bayliss, K. Boxall, K. H. Carr, A. M. Fry, I. R. Hardcastle, C. J. Matheson, D. R. Newell, M. Sivaprakasam, H. Thomas, D. Turner, S. Yeoh, L.-Z. Wang, R. J. Griffin, B. T. Golding and C. Cano, Structure-guided design of purine-based probes for selective Nek2 inhibition, Oncotarget, 2017, 8, 19089 CrossRef PubMed.
- H. Lebraud, C. R. Coxon, V. S. Archard, C. M. Bawn, B. Carbain, C. J. Matheson, D. M. Turner, C. Cano, R. J. Griffin, I. R. Hardcastle, U. Baisch, R. W. Harrington and B. T. Golding, Model system for irreversible inhibition of Nek2: thiol addition to ethynylpurines and related substituted heterocycles, Org. Biomol. Chem., 2014, 12, 141 RSC.
- (a) A. Brathe, L.-L. Gundersen, J. Nissen-Meyer, F. Rise and B. Spilsberg, Cytotoxic activity of 6-alkynyl- and 6-alkenylpurines, Bioorg. Med. Chem. Lett., 2003, 13, 877 CrossRef CAS PubMed; (b) F. Nagatsugi, K. Uemura, S. Nakashima, M. Maeda and S. Sasaki, 6-Vinylated guanosine as a novel cross-linking agent and its versatile synthesis from the 6-O-tosylate by Pd(0)-catalyzed cross-coupling, Tetrahedron Lett., 1995, 36, 421 CrossRef CAS; (c) F. Nagatsugi, K. Uemura, S. Nakashima, M. Maeda and S. Sasaki, 2-Aminopurine derivatives with C6-substituted olefin as novel cross-linking agents and the synthesis of the corresponding β-phosphoramidite precursors, Tetrahedron, 1997, 53, 3035 CrossRef CAS; (d) M. Kuchar, R. Pohl, I. Votruba and M. Hocek, Synthesis of purines bearing functionalized C-substituents by the conjugate addition of nucleophiles to 6-vinylpurines and 6-ethynylpurines, Eur. J. Org. Chem., 2006, 5083 CrossRef CAS.
- C. R. Coxon, E. Anscombe, S. J. Harnor, M. P. Martin, B. Carbain, B. T. Golding, I. R. Hardcastle, L. K. Harlow, S. Korolchuk, C. J. Matheson, D. R. Newell, M. E. M. Noble, M. Sivaprakasam, S. J. Tudhope, D. M. Turner, L.-Z. Wang, S. R. Wedge, C. Wong, R. J. Griffin, J. A. Endicott and C. Cano, Cyclin-dependent kinase (CDK) inhibitors: structure–activity relationships and insights into the CDK-2 selectivity of 6-substituted 2-arylaminopurines, J. Med. Chem., 2017, 60, 1746 CrossRef CAS PubMed.
- B. Carbain, C. R. Coxon, H. Lebraud, K. J. Elliott, C. J. Matheson, E. Meschini, A. R. Roberts, D. M. Turner, C. Wong, C. Cano, R. J. Griffin, I. R. Hardcastle and B. T. Golding, Trifluoroacetic acid in 2, 2, 2-trifluoroethanol facilitates SNAr reactions of heterocycles with arylamines, Chem. – Eur. J., 2014, 20, 2311 CrossRef CAS PubMed.
- H. D. Dakin and R. West, A general reaction of amino acids, J. Biol. Chem., 1928, 78, 91 CAS.
- K.-V. Tran and D. Bickar, Dakin−West synthesis of β-Aryl Ketones, J. Org. Chem., 2006, 71, 6640 CrossRef CAS PubMed.
- C. Wong, R. J. Griffin, I. R. Hardcastle, J. S. Northen, L.-Z. Wang and B. T. Golding, Synthesis of sulfonamide-based kinase inhibitors from sulfonates by exploiting the abrogated SN 2 reactivity of 2, 2, 2-trifluoroethoxysulfonates, Org. Biomol. Chem., 2010, 8, 2457 RSC.
- Y. Taniguchi, Y. Kurose, T. Nishioka, F. Nagatsugi and S. Sasaki, The alkyl-connected 2-amino-6-vinylpurine (AVP) crosslinking agent for improved selectivity to the cytosine base in RNA, Bioorg. Med. Chem., 2010, 18, 2894 CrossRef CAS PubMed.
- R. J. Griffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. Hardcastle, D. R. Newell, M. E. M. Noble, L.-Z. Wang and B. T. Golding, Searching for cyclin-dependent kinase inhibitors using a new variant of the cope elimination, J. Am. Chem. Soc., 2006, 128, 6012 CrossRef CAS PubMed.
- J. Ø. Madsen and P. E. Iversen, The stereochemical course of formic acid reduction of enamines, Tetrahedron, 1974, 30, 3496 CrossRef.
- J. Singh, R. C. Petter and A. F. Kluge, Targeted covalent drugs of the kinase family, Curr. Opin. Chem. Biol., 2010, 14, 475 CrossRef CAS PubMed.
- M. W. Richards, L. O'Regan, C. Mas-Droux, J. M. Y. Blot, J. Cheung, S. Hoelder, A. M. Fry and R. Bayliss, An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9, Mol. Cell, 2009, 36, 560 CrossRef CAS PubMed.
- E. Leproult, S. Barluenga, D. Moras, J. M. Wurtz and N. Winssinger, Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors, J. Med. Chem., 2011, 54, 1347 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Targeting cancer with small molecule kinase inhibitors, Nat. Rev. Cancer, 2009, 9, 28 CrossRef CAS PubMed.
- A. Chaikuad, P. Koch, S. A. Laufer and S. Knapp, The cysteinome of protein kinases as a target in drug development, Angew. Chem., Int. Ed., 2018, 4372 CrossRef CAS PubMed.
- R. S. Hames, S. L. Wattam, H. Yamano, R. Bacchieri and A. M. Fry, APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box, EMBO J., 2001, 20, 7117 CrossRef CAS PubMed.
- J. Uetrecht, Idiosyncratic drug reactions: past, present, and future, Chem. Res. Toxicol., 2007, 21, 84 Search PubMed.
- J. J. Parlow, M. L. Vazque and D. L. Flynn, A mixed resin bed for the quenching and purification of tetrabutylammonium fluoride mediated desilylating reactions, Bioorg. Med. Chem. Lett., 1998, 8, 2391 CrossRef CAS PubMed.
- E. Winzeler, N. S. Gray, D. Han and D. Cheng, Compounds and compositions as kinase inhibitors, WO2008094737A2, 2008 Search PubMed.
- N. S. Gray, S. Kwon and P. G. Schultz, Combinatorial Synthesis of 2,9-Substituted Purines, Tetrahedron Lett., 1997, 38, 1161 CrossRef CAS.
- F. Focher, C. Hildebrand, S. Freese, G. Ciarrocchi, T. Noonan, S. Sangalli, N. Brown, S. Spadari and G. Wright, N2-Phenyldeoxyguanosine: A Novel Selective Inhibitor of Herpes Simplex Thymiding Kinase, J. Med. Chem., 1988, 31, 1496 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinase profiling, assay details, synthetic procedures and characterisation data for all compounds; including aniline precursors. All cell lines were supplied by the American Type Culture Collection, Manassas, Virginia, United States. All animal experiments performed were conducted under a UK Government Home Office License in accordance with relevant national laws. All procedures were reviewed and approved by the Newcastle University Animal Welfare Ethical Board and performed according to institutional guidelines. See DOI: 10.1039/d0md00074d |
| ‡ These authors contributed equally to the work described in this paper. |
| § Deceased 24 September 2014. |
| This journal is © The Royal Society of Chemistry 2020 |