
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress toward the asymmetric synthesis of carbon-substituted piperazine pharmacophores and oxidative related heterocycles
Plato A.
Magriotis†

Department of Pharmacy, Laboratory of Medicinal Chemistry, University of Patras, Rio26504, Greece. E-mail: pmagriotis@upatras.gr
First published on 22nd May 2020
Abstract
The important requirement for approval of a new drug, in case it happens to be chiral, is that both enantiomers of the drug should be studied in detail, which has led synthetic organic and medicinal chemists to focus their attention on the development of new methods for asymmetric synthesis especially of relevant saturated N-heterocycles. On the other hand, the piperazine ring, besides defining a major class of saturated N-heterocycles, has been classified as a privileged structure in medicinal chemistry, since it is more than frequently found in biologically active compounds including several marketed blockbuster drugs such as Glivec (imatinib) and Viagra (sildenafil). Indeed, 13 of the 200 best-selling small molecule drugs in 2012 contained a piperazine ring. Nevertheless, analysis of the piperazine substitution pattern reveals a lack of structural diversity, with almost every single drug in this category (83%) containing a substituent at both the N1- and N4-positions compared to a few drugs having a substituent at any other position (C2, C3, C5, and C6). Significant chemical space that is closely related to that known to be biologically relevant, therefore, remains unexplored. In order to explore this chemical space, efficient and asymmetric syntheses of carbon-substituted piperazines and related heterocycles must be designed and developed. Initial, recent efforts toward the implementation of this particular target are in fact the subject of this review.
 Plato A. Magriotis | Since 2006, Plato A. Magriotis, Ph.D., has been an Associate Professor of Medicinal Chemistry in the Department of Pharmacy at the University of Patras in Greece and a Research Affiliate with the Department of Chemistry at New York University. Magriotis received his Ph.D. in Chemical Biology with Professor Francis Johnson at Stony Brook University in 1983 and did postdoctoral work at Harvard University with Nobel Laureate Professor E. J. Corey. His career started at West Virginia University and continued at Merck & Co. as well as New York University in the U.S., prior to his return to Greece. Magriotis' research program focuses on the development of new methodology for the synthesis of relevant pharmacophores applied in drug discovery. |
Introduction
Heterocyclic small molecules are of exceedingly high importance in medicinal chemistry and drug discovery. Recently, the focus has shifted from purely aromatic N-heterocycles to substituted saturated N-heterocycles that have been indicated as the most valuable scaffolds for the development of new pharmaceuticals.1 Naturally, the latter entail a higher number of stereogenic centers. Consequently, new methods that allow practical access to these scaffolds with both absolute and relative stereocontrol are of very high and continuous interest. Furthermore, the important requirement for approval of a new drug, in case it happens to be chiral, is that both enantiomers of the drug should be studied in detail,2 which has led synthetic organic and medicinal chemists to focus their attention on the development of new methods for asymmetric syntheses especially of relevant saturated N-heterocycles.On the other hand, the piperazine ring, besides defining a major class of saturated N-heterocycles, has been classified as a privileged structure in medicinal chemistry,3 since it is more than frequently found in biologically active compounds including several marketed blockbuster drugs such as Glivec (imatinib) and Viagra (sildenafil).4 Actually, an analysis of all U.S. FDA approved small molecule drugs found that 21% contained saturated 6-membered N-heterocycles with an additional heteroatom (N, piperazines; O, morpholines; S, thiomorpholines).5 Indeed, 13 of the 200 best-selling small molecule drugs in 2012 contain a piperazine ring.6 In the vast majority of these molecules, however, the piperazine ring is not substituted on any of its carbon atoms. Specifically, analysis of the piperazine substitution pattern reveals a lack of structural diversity, with almost every single drug in this category (83%) containing a substituent at both the N1- and N4-positions compared to a few drugs having a substituent at any other position (C2, C3, C5, and C6).5
Significant chemical space that is closely related to that known to be biologically relevant, therefore, remains unexplored. In order to explore this chemical space, efficient and asymmetric syntheses of carbon-substituted piperazines and related heterocycles must be designed and developed.4,5 Initial, recent efforts toward the implementation of this particular target are in fact the subject of this review since recent, elegant racemic approaches to piperazine synthesis have been properly reviewed.7
Since piperazine derivatives have been reported to elicit a broad spectrum of pharmacological activities including antidepressant, anticancer, anthelmintic, antibacterial, antifungal, antimycobacterial, antimalarial, antituberculant, anticonvulsant,4a and anti-AIDS,8 one can easily comprehend that the sky will be the limit, as far as novel drug development is concerned, once asymmetric syntheses of this substituted heterocycle will be fully developed.4,9 Importantly, marine bis-tetrahydroisoquinoline alkaloids, including ecteinascidins (1 and 2), renieramycins (3 and 4), and saframycins (5 and 6, Fig. 1), besides sharing a densely functionalized common pentacyclic scaffold (A–E rings), they also feature a central C-polysubstituted piperazine as in the ecteinascidins (1 and 2, Fig. 1, ring C).
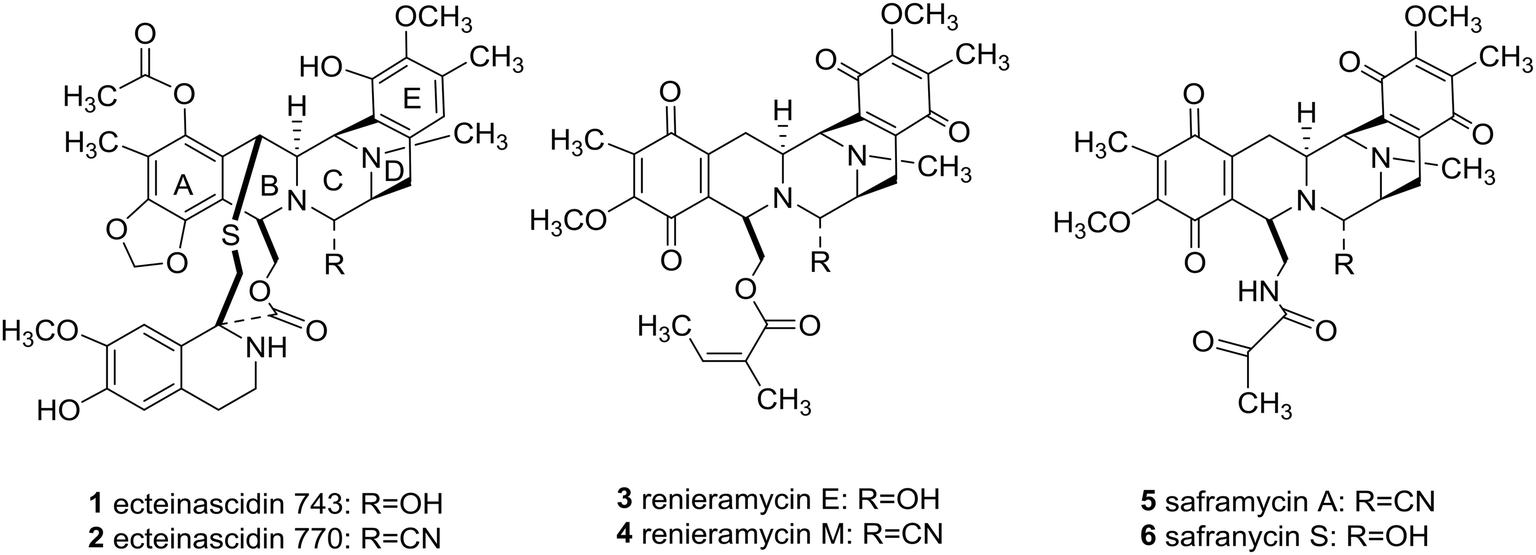 | ||
| Fig. 1 Marine bistetrahydroisoquinoline alkaloids. | ||
Significantly, the unique marine natural product ecteinascidin-743 (1, Et-743 also known as trabectedin or Yondelis™), isolated from the Caribbean tunicate Ecteinascidia turbinate,10 is a commercially available drug against soft-tissue sarcoma approved by the European Commission (EMEA) in 200711 and the U.S. Food and Drug Administration (FDA) in 2015,12 and is also undergoing clinical trials for breast, prostate, ovarian, and pediatric sarcomas in other countries, due to its extraordinarily potent cytotoxicity activities against several rodent tumors and human tumor xenografts with in vitro IC50 values in the 0.1–1 ng mL−1 range.13 The complex molecular architecture, the remarkable biological activity, and the restricted natural availability (1.0 g from about 1.0 ton of tunicate) made Et-743 an exceedingly attractive target for total synthesis, and eight synthetic approaches to the stereochemically complex antitumor drug Et-743 completed between 1996 and 2015 have been reviewed.13b,14 Additionally, a convergent formal synthesis and a scalable total synthesis of Et-743 appeared in the literature in 2019.15 2-Oxopiperazines (piperazinones) and 2,5-diketopiperazines are heterocycles related to piperazine by oxidation and they themselves represent key structural units of a great number of natural products such as agelastatins (7–10), pseudotheonamide A1 (11), and epicoccin G (12), as well as haematocin (13), respectively (Fig. 2).16
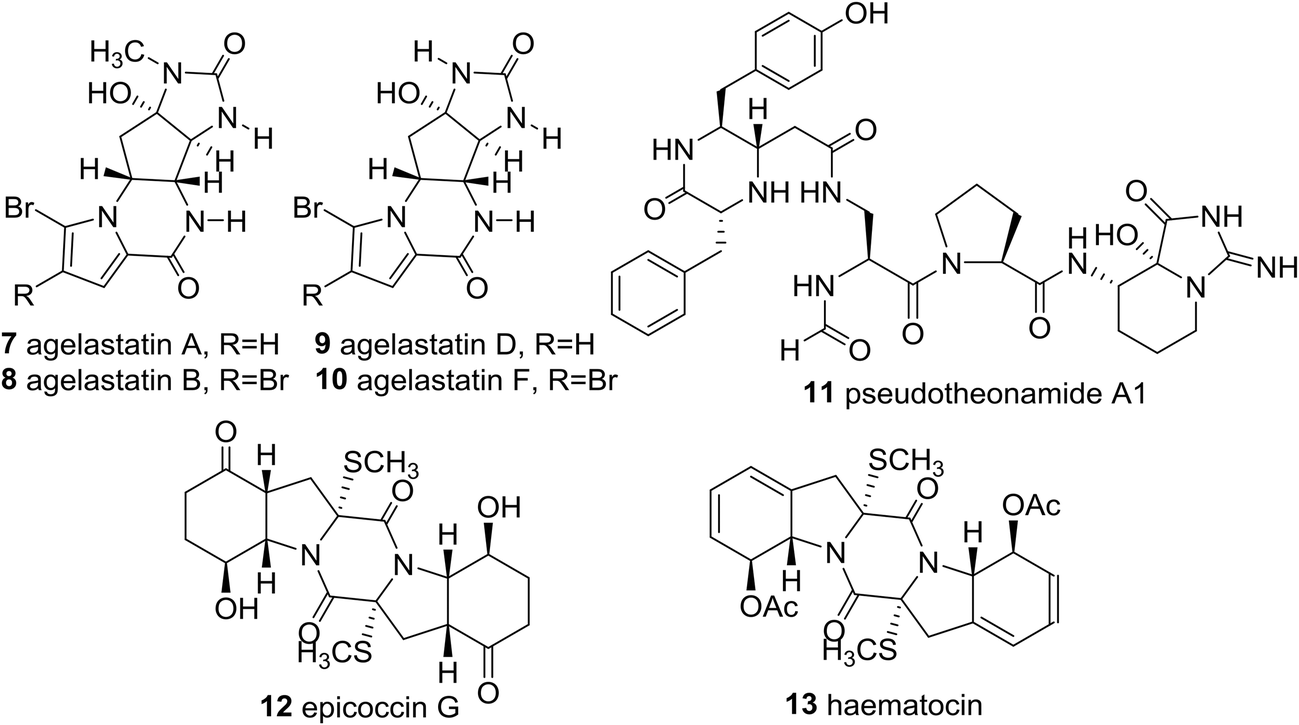 | ||
| Fig. 2 2-Oxopiperazine and 2,5-diketopiperazine containing natural products. | ||
Catalytic asymmetric syntheses of carbon-substituted piperazines and related heterocycles
A. Catalytic enantioselective approaches
Interestingly, SAR studies on piperazinone derivatives as a novel class of potent HCV NS4B inhibitors have been reported,17 and a catalytic enantioselective synthesis of piperazinones from aldehydes was recently reported, entailing a four-step reaction sequence, not requiring any intermediate purification, and employing MacMillan's third-generation catalyst 14 and chloroquinone as the chlorinating reagent.18 Accordingly, mild catalytic asymmetric α-chlorination of heptanal was coupled with Pinnick oxidation leading to homochiral α-chloroheptanoic acid 15. Nucleophilic substitution of the latter by N,N′-dibenzylethylenediamine (16) furnished the desired protected 2-oxopiperazine 17 in good overall yield under specific conditions (Scheme 1).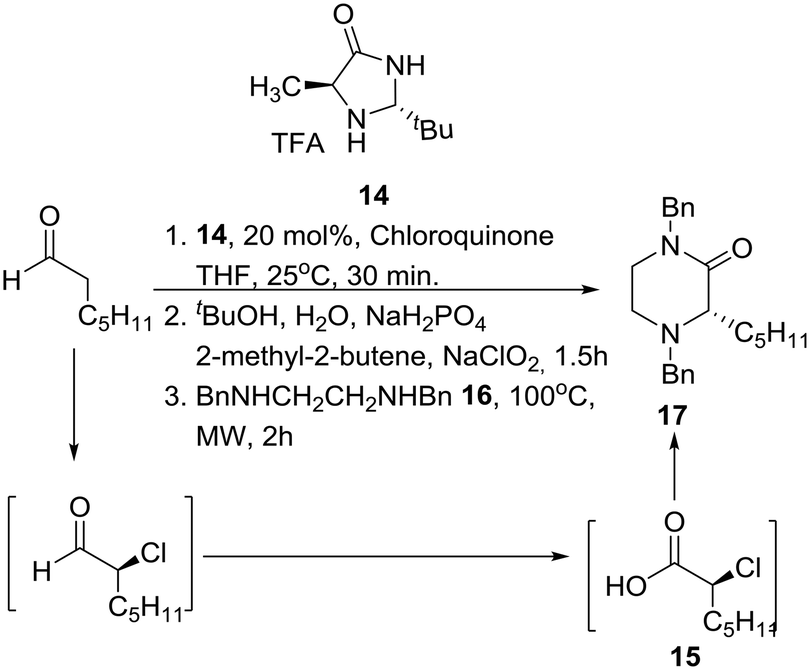 | ||
| Scheme 1 Kokotos' catalytic asymmetric synthesis of 2-oxopiperazines. | ||
A few years prior to this advance, a general, enantioselective synthesis of protected morpholines and piperazines was disclosed by Lindsley and O'Reilly based on Jørgensen's catalytic enantioselective α-chlorination of aldehydes followed by reduction to their corresponding 2-chloro alcohols (Scheme 2).19 Thus, homochiral-pyrrolidine catalyzed α-chlorination of aliphatic aldehydes followed by reduction to their corresponding 2-chloro alcohols, conversion to their respective triflates, chemoselective displacement with ambident nucleophiles 18 or 19, and finally internal SN2 reaction of the chloride with inversion of configuration provided the desired morpholines 20 and piperazines 21, respectively.
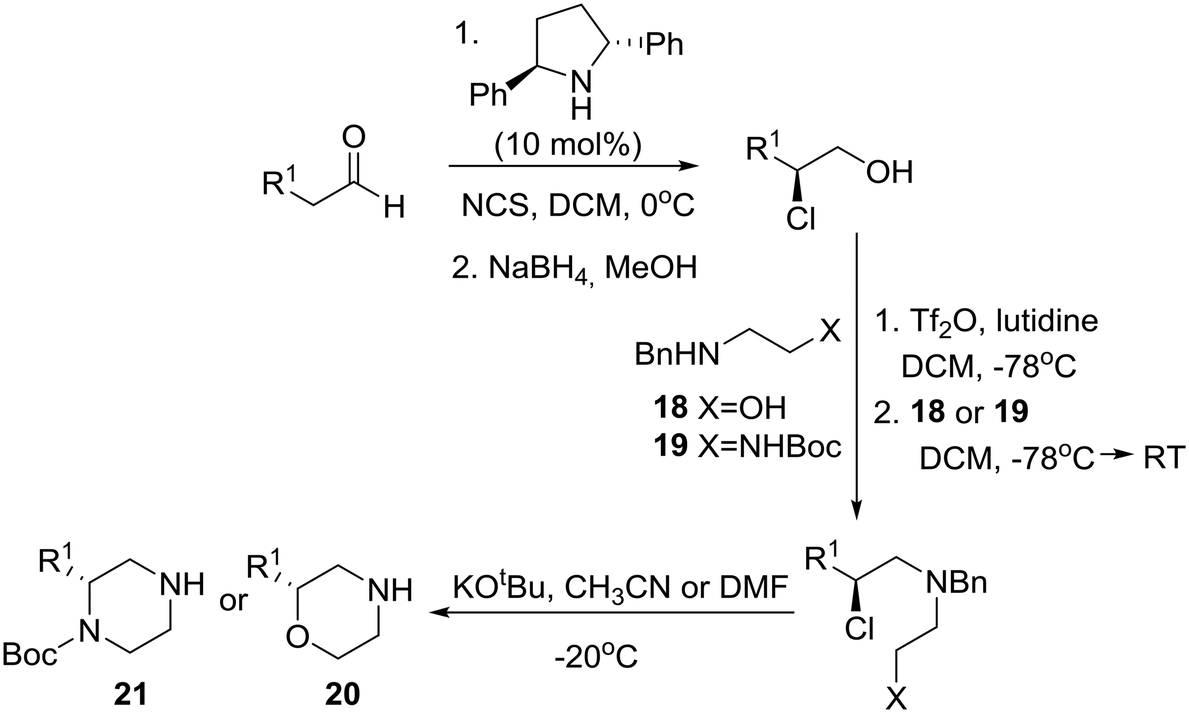 | ||
| Scheme 2 Lindsley's catalytic asymmetric syntheses of morpholines and piperazines. | ||
A little later, the group of Laurel Schafer and coworkers in the U.S., after having developed a catalytic asymmetric synthesis of morpholines, used mechanistic insights from this process to realize a catalytic enantioselective synthesis of piperazines based on tandem hydroamination and asymmetric transfer hydrogenation reactions employing catalysts 22 and 23, respectively, as described in Scheme 3.20
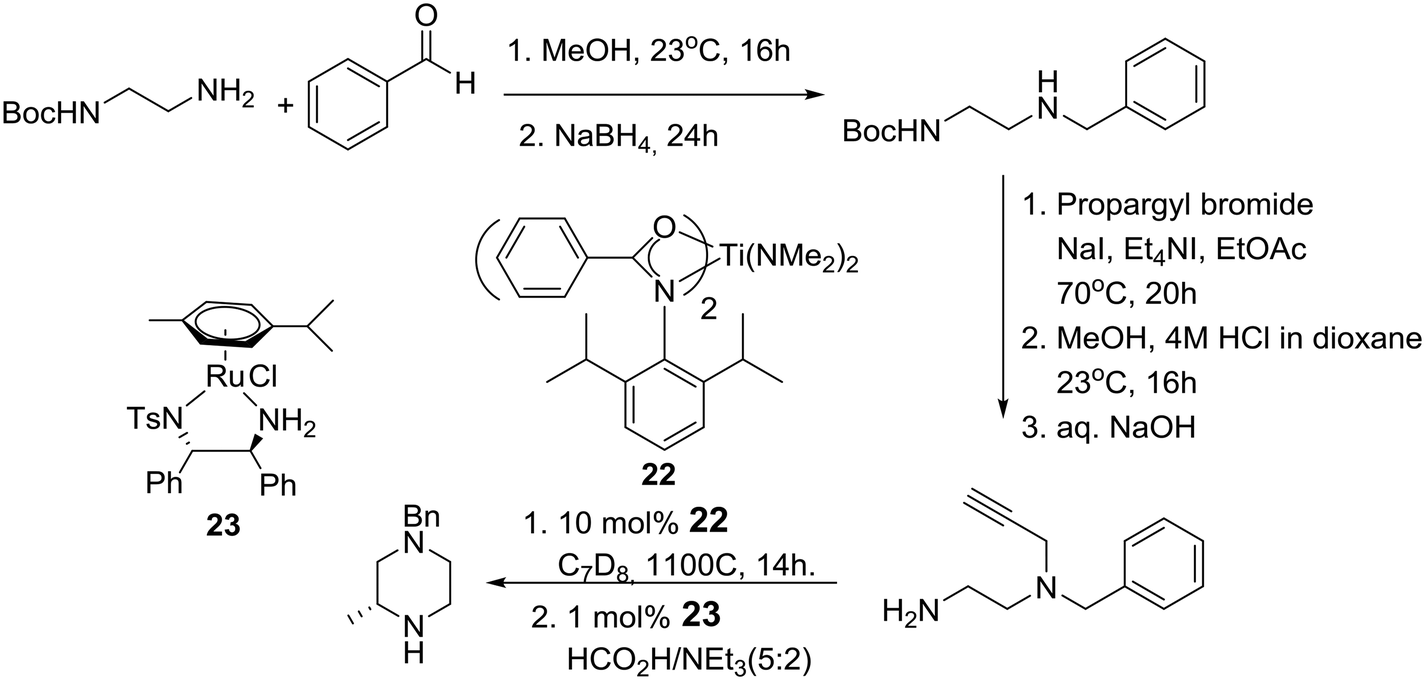 | ||
| Scheme 3 Schafer's catalytic asymmetric synthesis of piperazines. | ||
On the other hand, palladium-catalyzed asymmetric tandem allylic substitution of (Z)-1,4-bis(isopropoxycarbonyloxy)-2-butene (24) with the nucleophile 1,2-bis-(benzylamino)ethane (16), employing 2-(phosphinophenyl)pyridine (25) as a chiral ligand, furnished optically active 1,4-dibenzyl-2-vinylpiperazine (26) in 88% yield and 86% ee as described in eqn (1).21
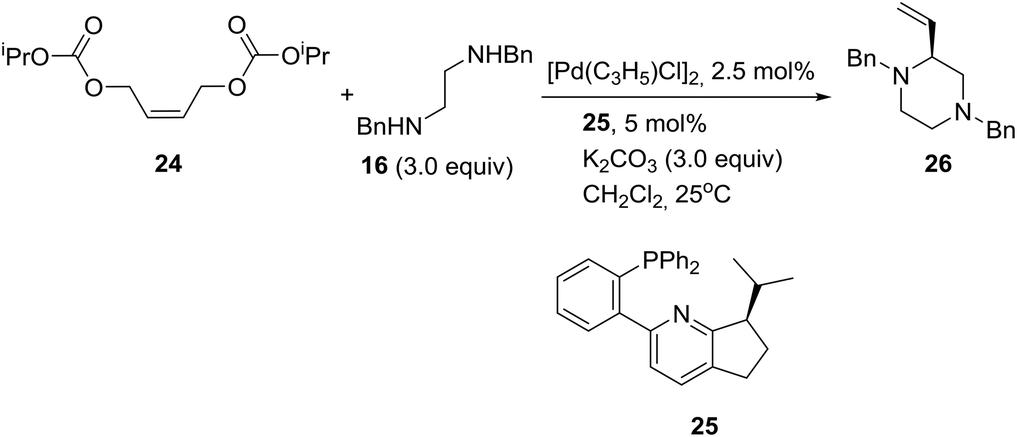 | (1) |
Reducing the unprecedented chemical space regarding carbon-substitution on piperazines, elucidated in the Introduction section, the group of Professor Stoltz in the U.S. reported the catalytic enantioselective synthesis of α-tertiary piperazin-2-ones and thereby piperazines by employing catalytic asymmetric allylation. Thus, the asymmetric palladium-catalyzed decarboxylative allylic alkylation of differentially N-protected piperazin-2-ones allowed the synthesis of a variety of highly enantioenriched tertiary piperazin-2-ones (eqn (2)). Deprotection and reduction afforded the corresponding tertiary piperazines, which were employed for the synthesis of medicinally important analogues such as imatinib analogues.22 Thus, treatment of α-tertiary piperazinone ester 27 with tris(4,4′-methoxydibenzylideneacetone)-dipalladium(0) ([pd2(pmdba)3]) at 5 mol% loading and (S)-(CF3)3-t-BuPHOX ligand 28 at 12.5 mol% loading in a 0.014 M solution of warm toluene produced the desired α-tertiary allylated product 29 in 77% yield and 96% ee (eqn (2)).
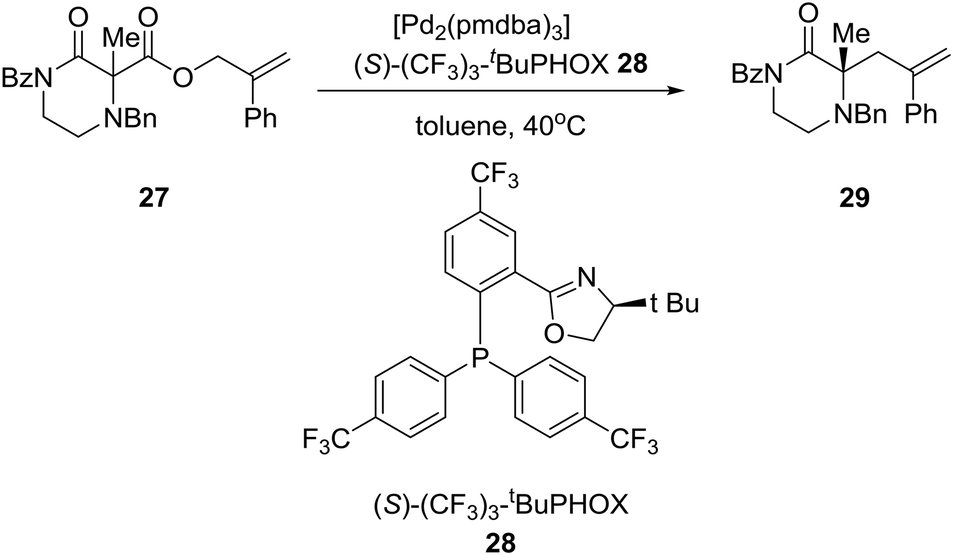 | (2) |
A few years later, the group of Ruijter in Holland described a related process involving an intramolecular Tsuji–Trost reaction of Ugi adducts 3023 for the synthesis of spiro-diketopiperazines 31 [eqn (3), in >90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 enantiomer ratios (er)], which display diverse biological activities, including neuroprotective properties, antiinflammatory activity, and antiproliferative effects against drug-resistant human cancer cell lines.24 Obviously, the high er values were achieved by employing the optimized homochiral ligand 32.
:10 enantiomer ratios (er)], which display diverse biological activities, including neuroprotective properties, antiinflammatory activity, and antiproliferative effects against drug-resistant human cancer cell lines.24 Obviously, the high er values were achieved by employing the optimized homochiral ligand 32.
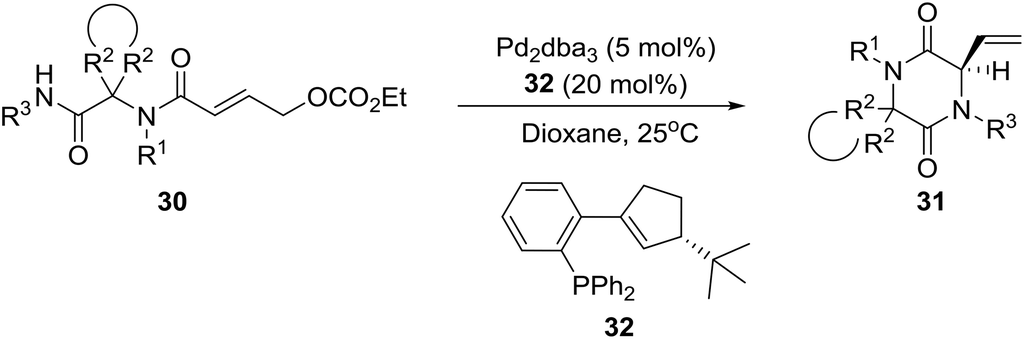 | (3) |
B. Catalytic diastereoselective approaches
A highly diastereoselective intramolecular palladium-catalyzed hydroamination reaction as a key step in a modular synthesis of 2,6-disubstituted piperazine 3325 has been reported employing an aminoalkene 34 synthesized from a homochiral cyclic sulfamidate 35 (eqn (4)).26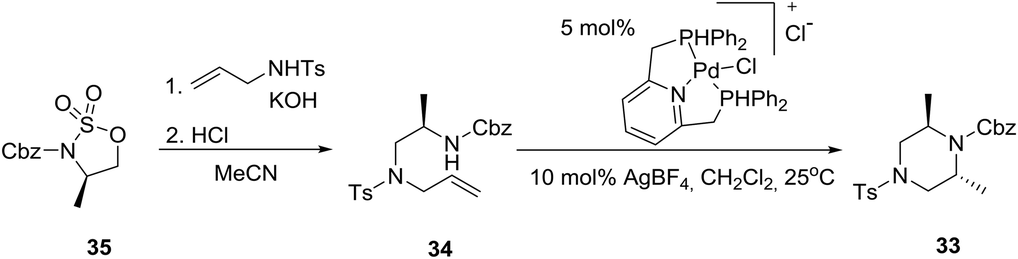 | (4) |
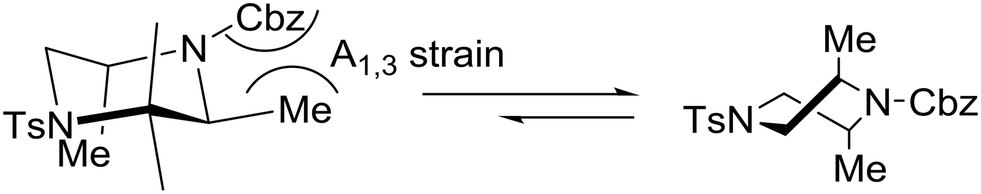
The relative stereochemistry of the piperazine product was determined to be trans by single-crystal X-ray diffraction of 33. Remarkably, the preferred conformation of the piperazine ring was found to be a twist-boat conformation rather than the typical chair conformation. Obviously, this preference is a result of the allylic strain between the methyl substituents and the carbamate protecting group. In the chair conformation, one of the two methyl substituents is forced to adopt an equatorial position adjacent to the carbamate group, which would result in a strong A1,3-interaction shown below.27 Given the borderline of piperazine derivatives between small molecules and peptides,23 the latter concept may find important implications in drug design.
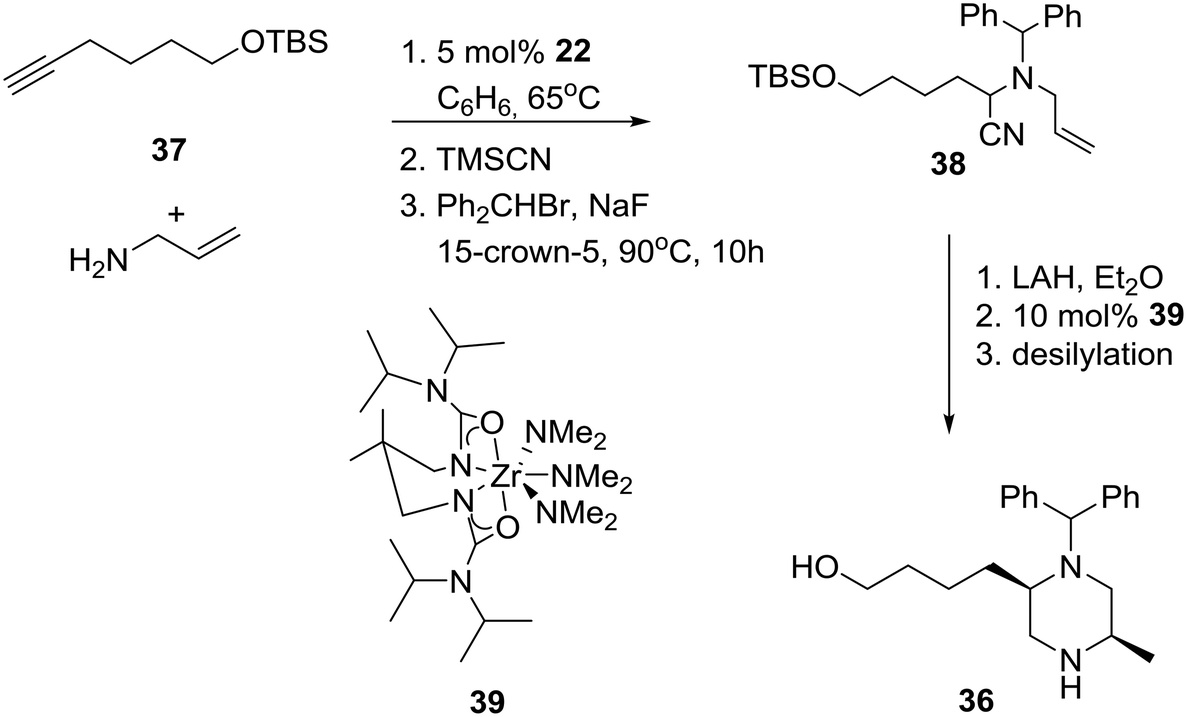
Cognizant of their previous results, outlined in the Catalytic enantioselective approaches section, the group of Professor Schafer proceeded to apply their process to the diastereoselective synthesis of 2.5-disubstituted-N-benzhydryl-piperazine 36 (Scheme 4) as a selective N-type Ca2+ channel blocker aimed at the inhibition of spinal neurotransmitter release and the attenuation of afferent pain signals.28
 | ||
| Scheme 4 Schafer's catalytic diastereoselective synthesis of piperazines. | ||
Specifically, anti-Markovnikov regioselective intermolecular hydroamination of terminal alkyne 37 with allylamine, followed by a modified Strecker reaction with trimethylsilyl cyanide and alkylation with benzhydryl bromide, produced α-aminonitrile 38.29 Nitrile reduction of the latter to the corresponding substituted diaminoalkene provided the requisite piperazine 36 in about 15% overall yield after ring-closure using Zr-ureate intramolecular aminoalkene hydroamination catalyst 39 and deprotection of the TBS group. Presumably, the reduced yield of this N-benzhydryl substituted piperazine 36 is due to the increased steric bulk of the benzhydryl substituent versus the benzyl-substituted amines implicated in the more efficient original synthesis of related cis-2,5-disubstituted piperazines.30
Chiral-pool assisted asymmetric syntheses of carbon-substituted piperazines and related heterocycles
A. Chiral auxiliary based
The asymmetric synthesis of (R)-(+)-2-methypiperazine (40) has been reported from R-(−)-phenylglycinol, as the chiral auxiliary, and N-Boc glycine via the protected 2-oxopiperazine 41 as shown in Scheme 5.31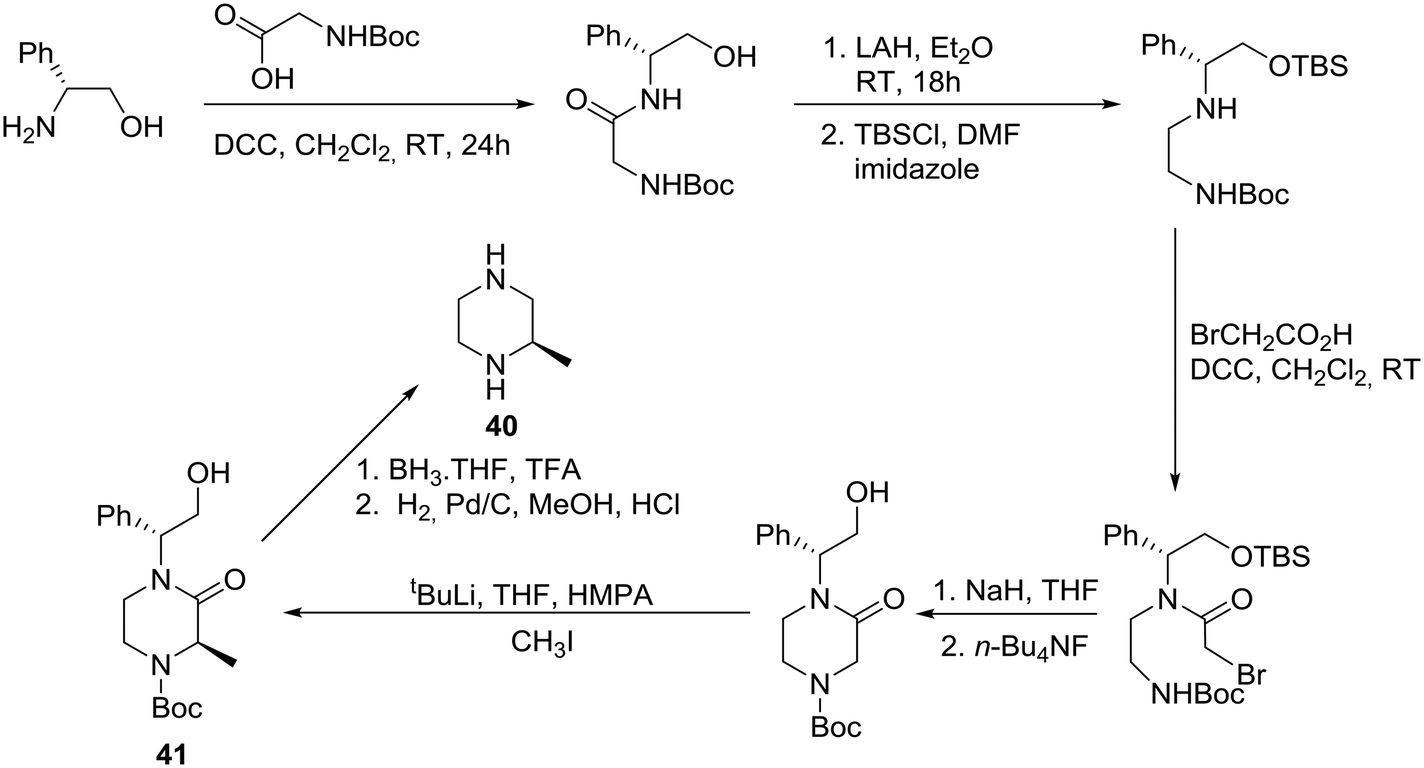 | ||
| Scheme 5 Asymmetric synthesis of 2-methyl piperazine 40. | ||
Accordingly, (R)-(−)-phenylglycinol was condensed with N-Boc glycine in the presence of DCC to give the expected amide, reduction of which followed by protection of the hydroxyl group provided the corresponding silyl ether in 66% overall yield. Selective condensation with bromoacetic acid was achieved in the presence of DCC, followed by cyclization to deliver the requisite 2-oxopiperazine after desilylation. Diastereoselective methylation of the latter proceeded smoothly and furnished 2-oxopiperazine 41 in 80% yield and >90% de. Finally, decarbonylation and debenzylation resulted in the desired (R)-(+)-2-methylpiperazine (40) in 63% overall yield.
The synthesis of homochiral cis- and trans-2-phenyl-3-(trifluoromethyl)piperazines involving diastereoselective nucleophilic addition of the Ruppert–Prakash reagent (TMSCF3) to homochiral α-amino sulfinylimines, derived from (R)-phenylglycinol and bearing Ellman's auxiliary, was reported by a group in Spain.32 This methodology allowed an entry to the unknown and stereochemically defined trifluoromethylated piperazines as building blocks in drug discovery due to the noticeable effect of the CF3 group on the metabolic stability, basicity, and lipophilicity of the piperazine core.
As shown in Scheme 6, this synthesis began with protection of (R)-phenylglycinol as the corresponding bis-p-methoxybenzyl (PMB) derivative R-42. Swern oxidation, followed by condensation of the resulting α-amino aldehyde with (S)-(−)-2-methyl-2-propanesulfinamide in the presence of titanium(IV) ethoxide as a dehydrating agent, yielded enantiomerically pure tert-butylsulfinimine (R,Ss)-43 ([α]25D +121.8). After careful optimization of the key diastereoselective trifluoromethylation, it was found that dropwise addition of TMSCF3 to a solution of Ellman's imine 43 and finely ground tetramethylammonium fluoride (TMAF) as an activator of the Ruppert–Prakash reagent in THF at −35 °C produced the protected diamine (R,R,Ss)-44 as a single diastereomer. Next, the tert-butanesulfinyl group was removed upon exposure to HCl in refluxing methanol in a sealed tube, giving rise to the monoprotected diamine (R,R)-45. Employment of the closed vessel proved essential for the partial unmasking of the benzylic amine, a prerequisite for the subsequent enclosure of both nitrogens into the piperazine ring (Scheme 6). Thus, the reaction of primary amine 45 with 2-chloroacetyl chloride followed by 6-exo-tet cyclization of the resulting chloroderivative 46 provided an (R,R)-oxopiperazine, consecutive reductions of which furnished the desired cis-disubstituted (R,R)-piperazine 47.32 Interestingly, employment of commercially available (R)-(+)-2-methyl-2-propanesulfinamide and a slightly different final deprotection sequence yielded the corresponding trans-disubstituted (R,S)-piperazine.32
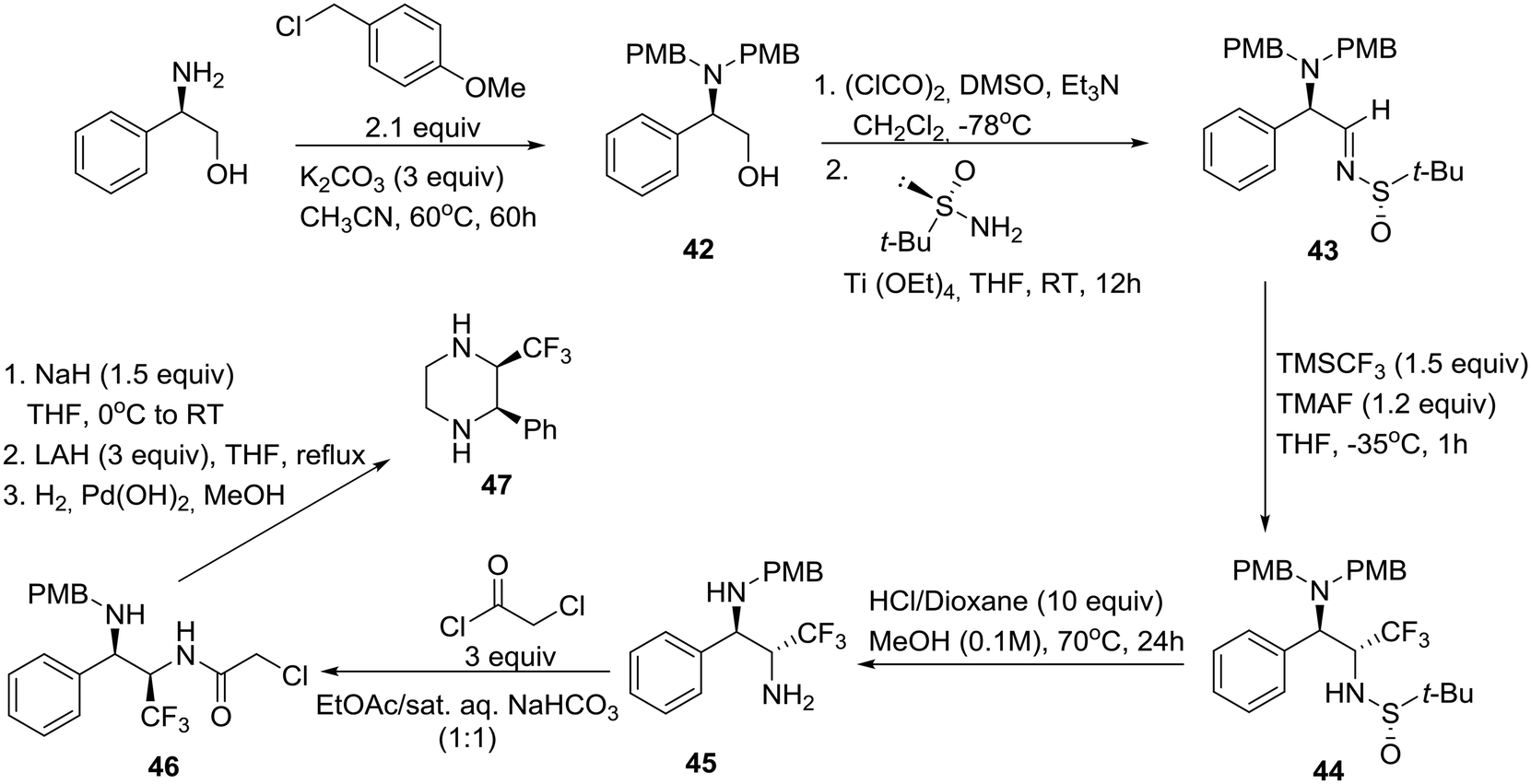 | ||
| Scheme 6 Asymmetric synthesis of cis-2-phenyl-3-(trifluoro-methyl)piperazine 47 as a building block for drug discovery. | ||
By exploring previously reported highly diastereoselective [3 + 2] cycloaddition of nonracemic p-tolylsulfinimines 48 and iminoesters 49 providing enantiopure imidazolidines 50 and thereby vicinal diaminoalcohols 51 by reduction (Scheme 7),33 the group of Viso and coworkers in Spain reported the efficient conversion of these homochiral products into both chiral piperazinones and piperazines as exemplified in Scheme 8.34
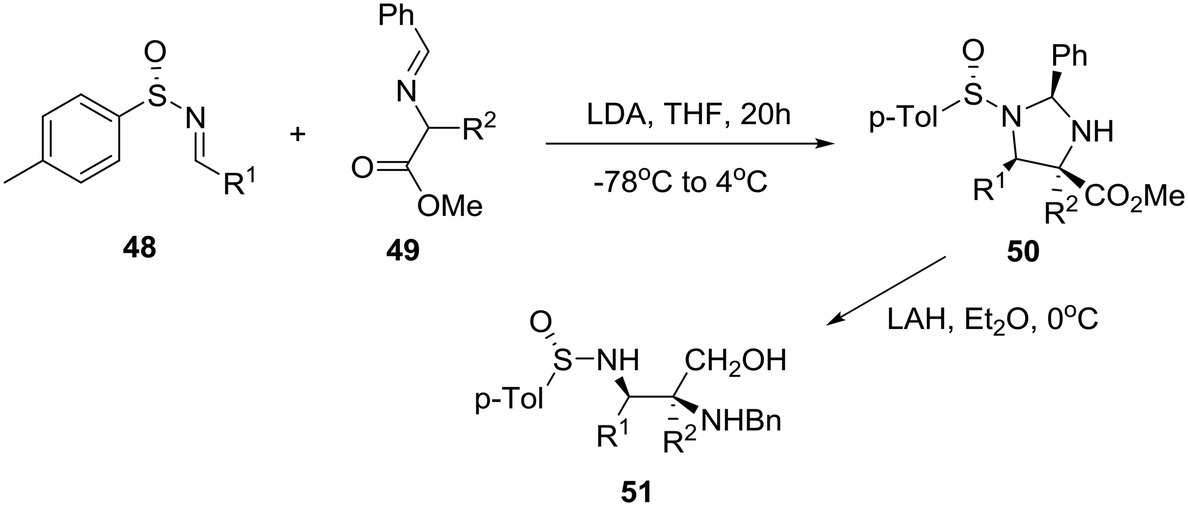 | ||
| Scheme 7 Asymmetric synthesis of enantiopure vicinal diaminoalcohols 51. | ||
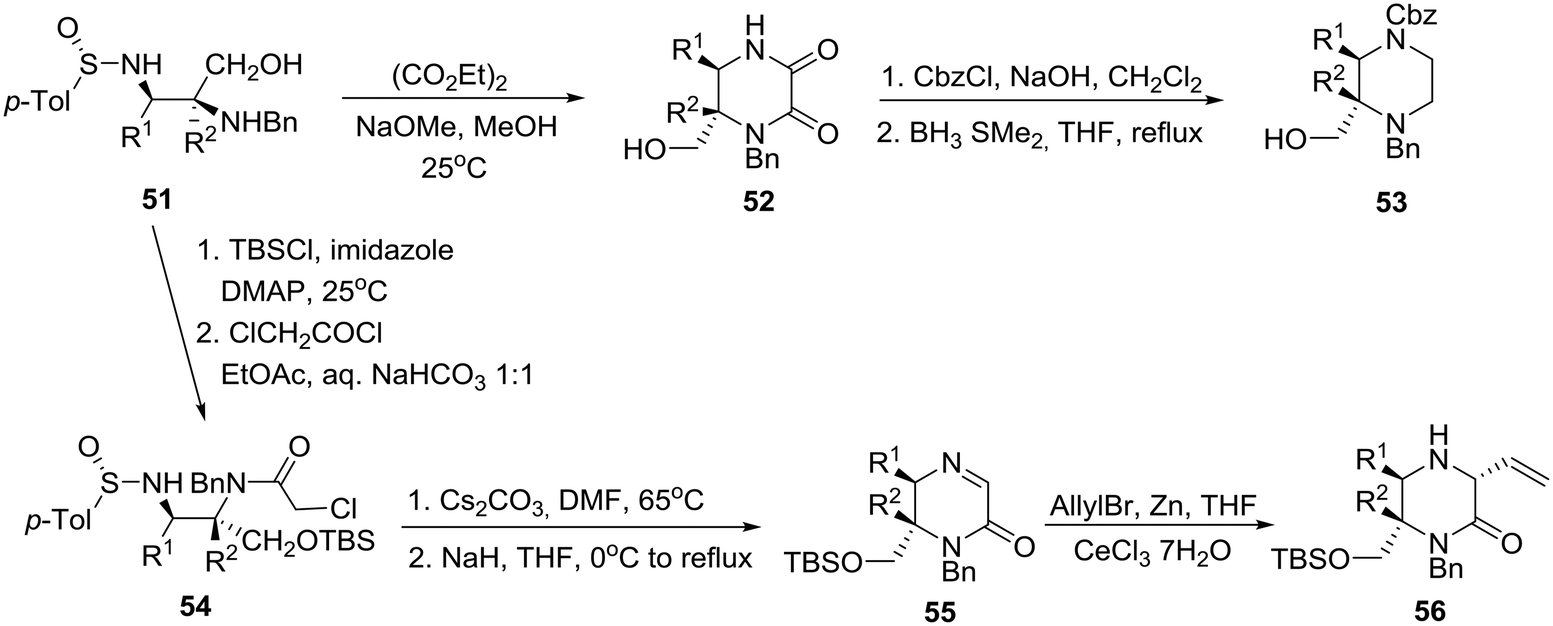 | ||
| Scheme 8 Asymmetric synthesis of disubstituted piperazine 53 and trisubstituted piperazinone 56. | ||
Specifically, treatment of diaminoalcohol 51 with diethyl oxalate and sodium methoxide in methanol gave rise to 2,3-diketopiperazinone 52, borane reduction of which furnished enantiopure, orthogonally protected 2,3-disubstituted piperazine 53. On the other hand, exposure of 51 to TBSCl and chloroacetyl chloride followed by cesium carbonate in DMF at 65 °C and sodium hydride at 0 °C to reflux produced imino ketopiperazine 55 through the intermediacy of 54. Finally, addition of an allyl organocerium reagent to 5535 gave rise to homochiral trisubstituted piperazinone 56 in 87% yield (Scheme 8).
B. Chiral amino acid or carbohydrate pool based
Borrowing from the chirality of 4-O-trifloxy-2,3-anhydro-ribopypanosides and employing N,N′-disubstituted ethylenediamines such as 16 (Scheme 1), the group of Voelter in Germany reported the synthesis of chiral tetrasubstituted piperazines of potential pharmacological interest as shown in eqn (5).36 Thus, simultaneous nucleophilic attack of the cis-oriented epoxy triflate 57 by the two nitrogen atoms of N,N′-dibenzyl ethylenediamine 16 (Scheme 1) afforded the pyrano-fused tetrasubstituted piperazine 58 in about 80% yield (eqn (5)).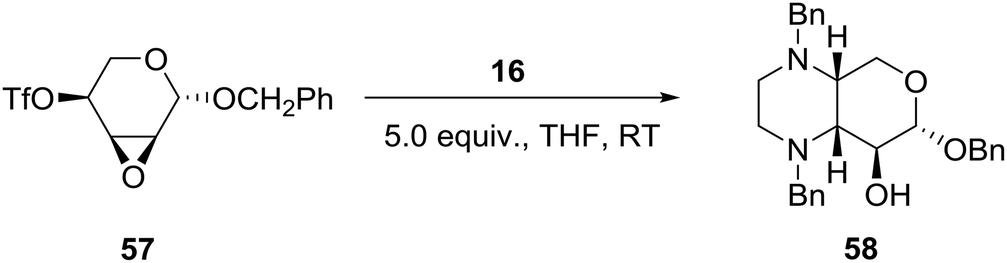 | (5) |
Exploiting the chiral pool, the group of Panda, at the Central Drug Research Institute, Lucknow in India, reported the synthesis of cis-2,5-disubstituted homochiral piperazines by the regioselective ring-opening of natural amino acid-derived chiral aziridines 59 by natural amino acid methyl ester hydrochloride salts 60 (Scheme 9).37 In practice, treatment of (S)-aziridine 59 with (S)-amino acid methyl ester hydrochloride salt 60 in the presence of an equivalent amount of triethylamine and boron trifluoride etherate complex in tetrahydrofuran at 60 °C for four days produced the desired N-tosyl diamine 61 in good yields (25–66%, seven examples, Scheme 9). Protection of the secondary amine in 61, with mesyl chloride followed by reduction of the methyl ester, produced protected diamino-alcohol 62, intramolecular Mitsunobu cyclization of which furnished the desired cis-2,5-disubstituted enantiopure piperazine 63. A related solid-phase synthesis of selectively N-protected homochiral 2,5-disubstituted piperazines has been reported by Franzyk and coworkers in Denmark, featuring a regioselective microwave-assisted aminolysis of resin-bound aziridines with ethanolamine followed by intramolecular Fukuyama–Mitsunobu cyclization.38
 | ||
| Scheme 9 Asymmetric synthesis of cis-2,5-disubstituted piperazine 63. | ||
Earlier, the group of Wolfe had reported a Pd-catalyzed carboamination approach to the construction of cis-2,6-disubstituted N-aryl piperazines based on the use of unprotected homochiral amino acids as starting materials.39 Specifically, phenylation of the amino group followed by amidation of the carboxyl group with N-(benzyl)allylamine employing a 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT) reagent40 and reduction furnished the desired carboamination substrates 64 that readily underwent Pd-catalyzed transformation with an aryl bromide to furnish cis-2,6-disubstituted N-aryl piperazines 65 in 50–63% yields and 98–99% ee (Scheme 10). Notably, the use of other reagents such as DCC/HOBT or CDI instead of DEPBT resulted in partial racemization to afford products with 85–90% ee. The working hypothesis for cis-piperazine formation involves cyclization via a transition state (shown as an insert in Scheme 10) in which the N–Ph group is rotated such as N is pyramidalized. This eliminates the A1,3-interaction and allows the pseudoequatorial orientation of R1 leading to the cis-2,6-disubstituted stereoisomers.27
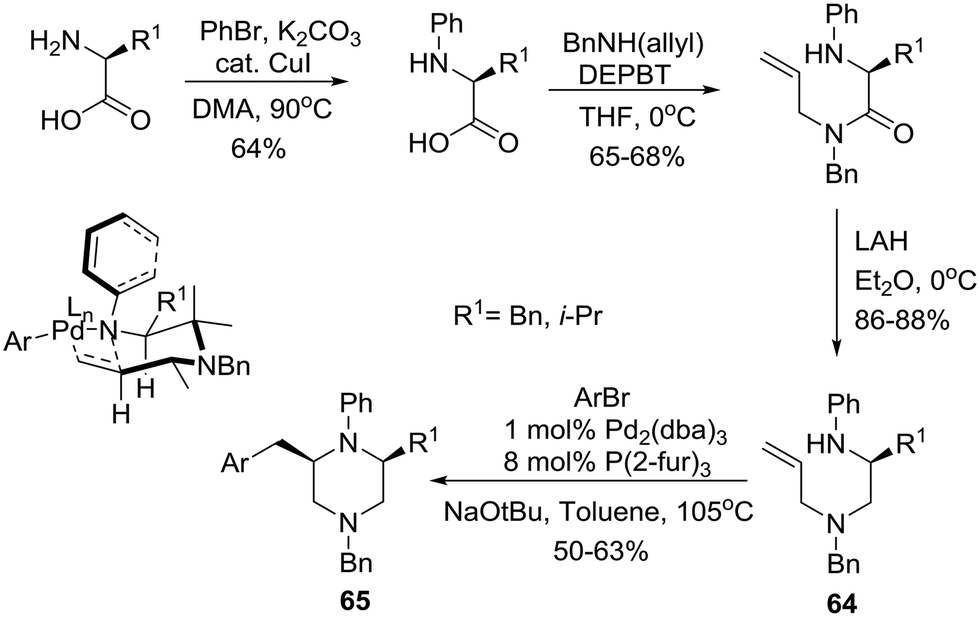 | ||
| Scheme 10 Asymmetric synthesis of cis-2,6-disubstituted N-aryl piperazines 65. | ||
As a part of research in a structure–activity relationship (SAR) study involving the discovery of novel GABAA-based anxiolytic agents, a group at the Upjohn Company became interested in preparing analogues incorporating homochiral 2,6-polymethylated piperazines such as 66. The syntheses involved the employment of an assortment of starting materials readily available from the chiral pool as exemplified in Scheme 11.41 Specifically, the conversion of N-t-Boc-L-alanine to the corresponding dibenzylamide followed by partial deprotection and reduction gave diamine (+)-67 in >98% ee. Notably, complete decomplexation of the amine from borane required heating at reflux in aqueous KOH. Next alkylation of 67 with methyl (R)-2-[(trifluoromethanesulfonyl)oxy]propionate, generated by the sequential treatment of methyl (R)-lactate with trifluoromethanesulfonic anhydride and 2,6-lutidine, proceeded smoothly to furnish ester (−)-68, monodebenzylation and cyclization of which provided interesting piperazin-2-one (+)-69. Lithium aluminum hydride (LAH) reduction of 69 followed by hydrogenolysis of the remaining benzyl group with Perlman's catalyst gave rise to the desired (2S, 6S)-2,6-dimethylpiperazine (66) in an overall yield of 44% and >98% ee.
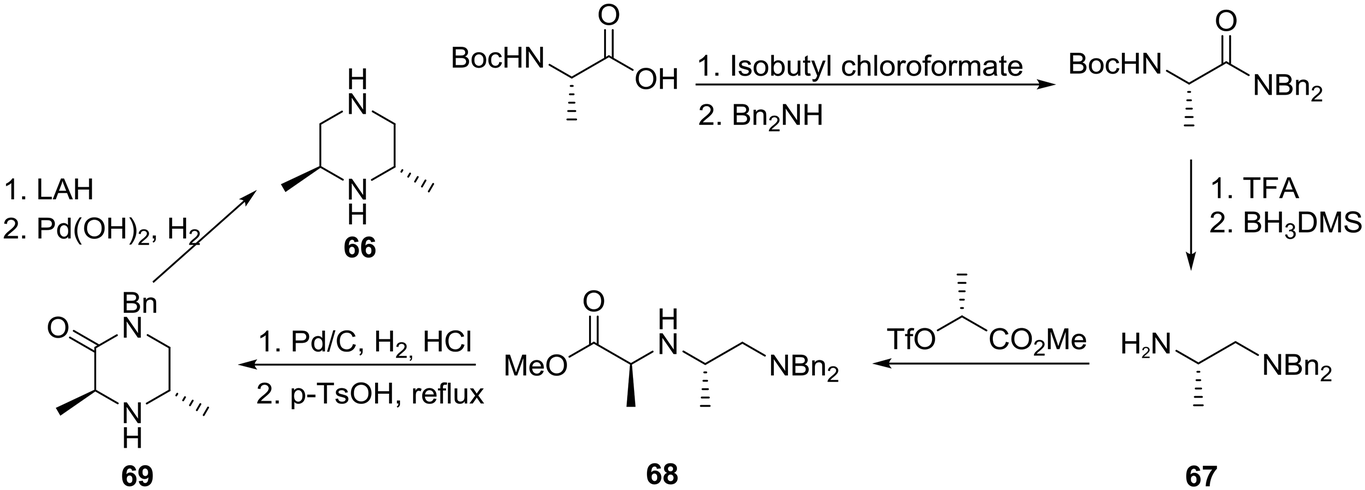 | ||
| Scheme 11 Asymmetric synthesis of 2,6-dimethyl piperazine 66. | ||
Research efforts at Merck around the beginning of this century led to the identification of MB243, a potent and selective small-molecule agonist of the MC4R representing one of the five cloned melanocortin receptor subtypes (MC1R–MC5R) which are part of a family of seven-transmembrane G-protein-coupled receptors.42
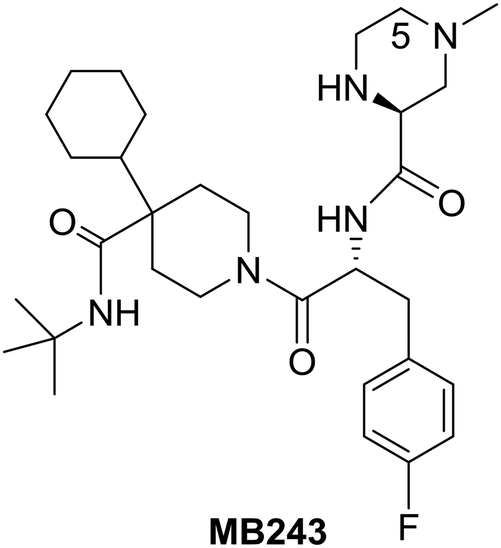
Although MB243 was shown to stimulate erectile activity and reduce food intake in rats, further development of this compound was discontinued due to its potential for bioactivation as seen in vitro through extensive covalent binding in rat and human liver microsomes. NMR analysis of the isolated adducts suggested that bioactivation occurred via initial oxidation at C-5 on the piperazine ring. Consequently, it was speculated that blocking the site of activation via alkylation on the piperazine ring should reduce bioactivation. Accordingly, the 5-alkylated piperazine compounds were prepared in a linear fashion as shown in Scheme 12.42
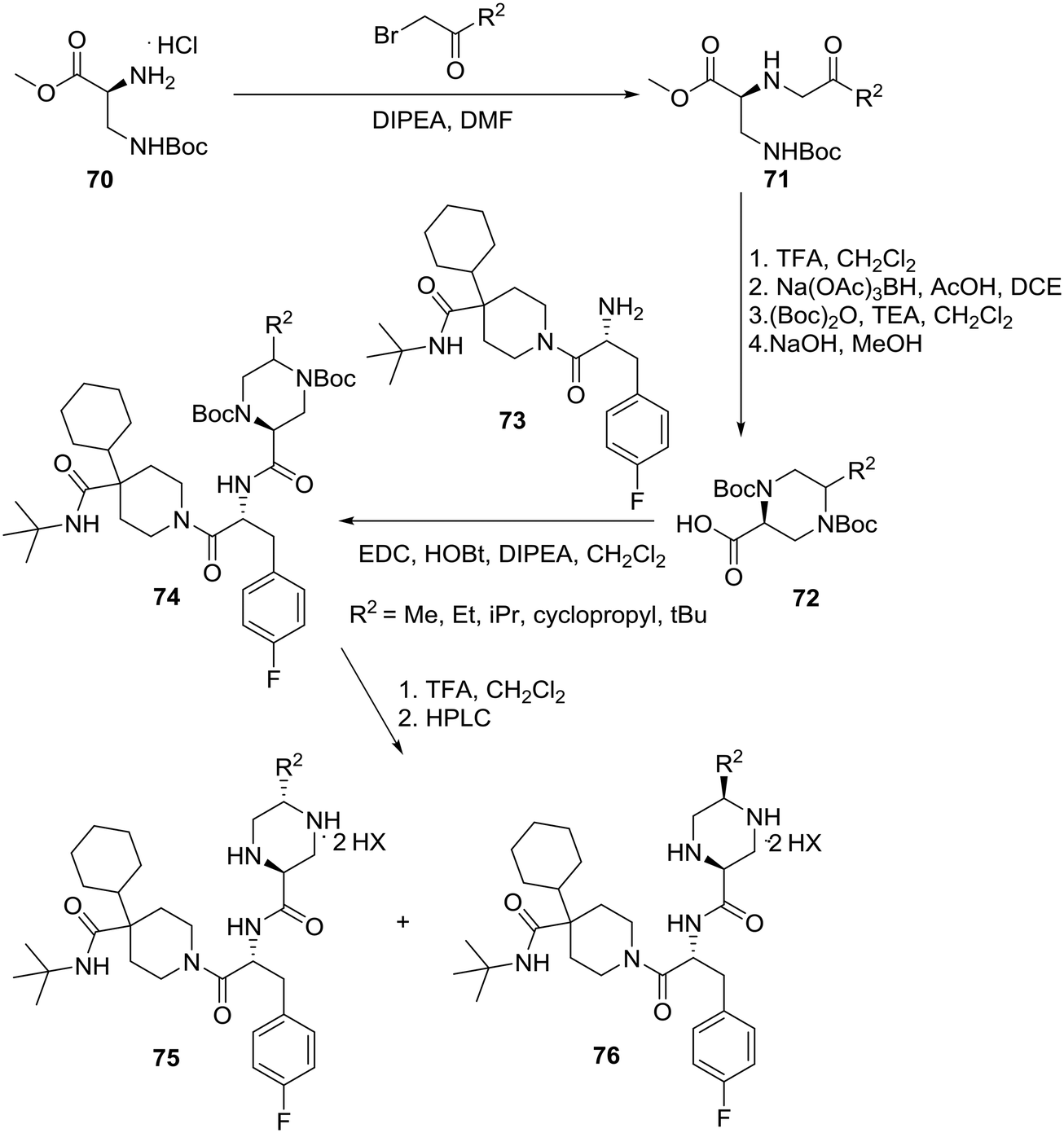 | ||
| Scheme 12 Merck asymmetric synthesis of 2-piperazinecarboxamides as potent and selective MC4R agonists. | ||
Starting with commercially available methyl N-β-Boc-L-α,β-diaminopropionate hydrochloride (70), conversion to diamine 71 was achieved via alkylation with various substituted α-bromoketones. Removal of the Boc group followed by intramolecular reductive amination provided piperazine of which protection of both secondary amines followed by hydrolysis of the ester yielded the 5-alkylated-2-piperazinecarboxylic acids 72. Coupling of piperazines 72 with 1-[(2R)-2-amino-3-(4-fluoro-phenyl)-1-oxopropyl]-4-cyclohexyl-N-(1,1-dimethylethyl)-4-piperidine carboxamide (73) and deprotection furnished dipeptides 74 as 2:1–10:1 mixtures of trans:cis diastereomers that were separated using preparative reverse-phase HPLC to give trans-piperazines 75 and cis piperazines 76 as ditrifluoroacetic acid salts (Scheme 12).
An interesting intramolecular [3 + 2]-cycloaddition of an azide to a C–C double bond as a novel approach to piperazines was reported by a group in Russia.43 The readily available amino alcohols 77 were converted to their corresponding N-o-nitrobenzenesulfonyl derivatives 78, allylation of which yielded the unstable to polymerization N-allyl compounds 79 which were kept in dilute solutions (MeCN) to promote the intramolecular cyclization to the relatively stable triazolines 80. The target 2-(chloromethyl)piperazines 81 were prepared from 80 by reactions with selected acyl, carbamoyl, and alkyl halides. Notably, the best yields (76–89%) were obtained with CbzCl as shown in eqn (6).
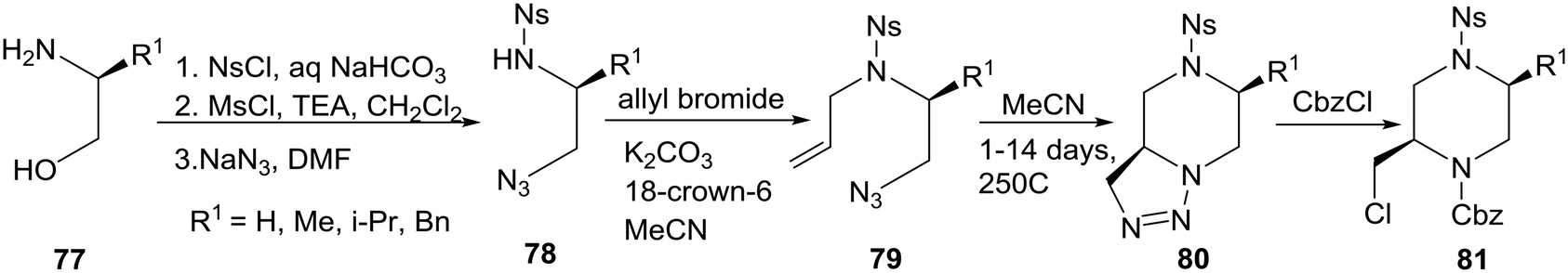 | (6) |
The asymmetric synthesis of 4-formyl-1-(ω-haloalkyl)-β-lactams 84 (Scheme 13) and their transformation into functionalized homochiral piperazines were reported by the group of De Kimpe in Belgium.44 The synthesis of chiral 4-formyl-1-(ω-haloalkyl)-β-lactams was performed by means of a slightly modified four-step literature procedure as shown in Scheme 13. (R)-Glyceraldehyde acetonide 82 was condensed with 2-chloroethylamine (in situ prepared from the corresponding hydrohalide salt using 3 equiv. of Et3N) in dichloromethane in the presence of MgSO4, and the resulting imine was used as a substrate for the Staudinger reaction using benzyloxy- or methoxyacetyl chloride to construct β-lactam 83 in a highly diastereoselective fashion. The latter azetidin-2-one 83 could be easily converted to the requisite (3R, 4R)-4-formyl-β-lactam 84 by consecutive hydrolysis and oxidation (Scheme 13).44
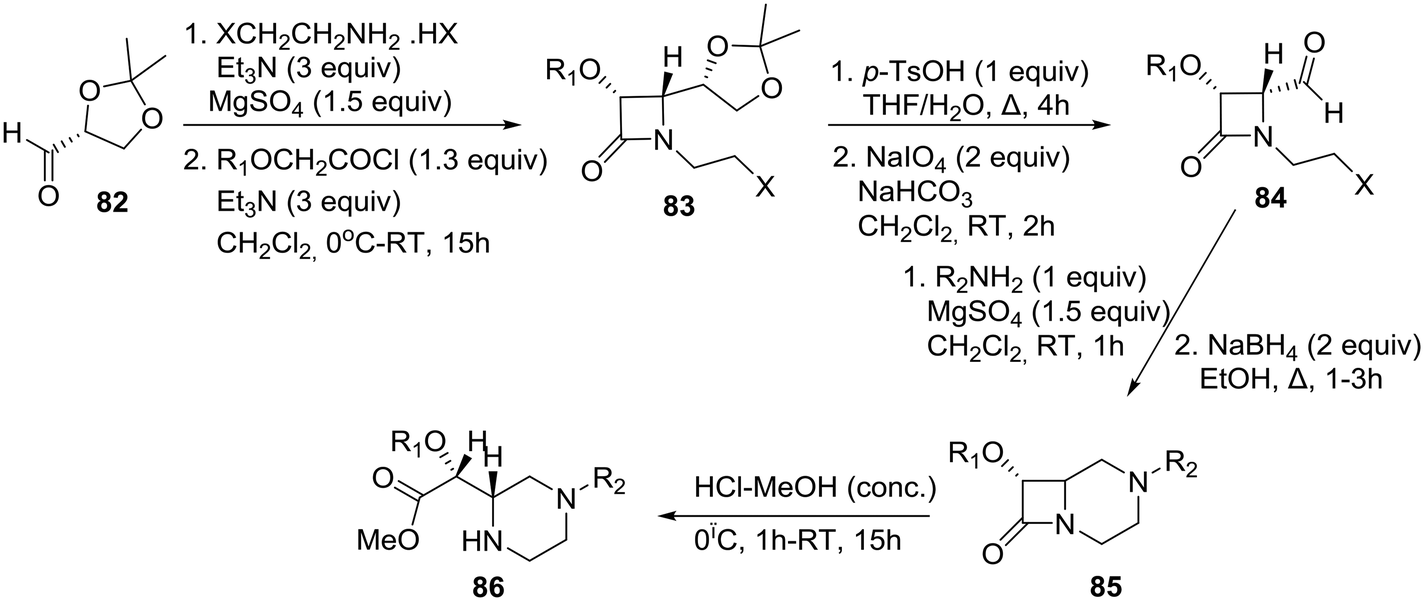 | ||
| Scheme 13 De Kimpe demonstration of the β-lactam synthon methodology toward the asymmetric synthesis of piperazines. | ||
Intramolecular imination of 4-formyl-1-(2- and 3-haloalkyl)azetidin-2-one 84 upon treatment with 1 equiv. of a primary amine in CH2Cl2 in the presence of MgSO4 and subsequent reduction afforded bicyclic β-lactam 85, which was then treated with HCl in methanol to provide novel methyl (R)-[(S)-piperazin-2-yl]acetate 86 in good yield through acid-catalyzed methanolysis of the β-lactam ring. Therefore, the β-lactam synthon methodology,45 according to which β-lactams can be employed as useful intermediates for organic synthesis and as synthons for various biologically active compounds, has been amply demonstrated in its homochiral version by this particular example.46
An interesting synthetic strategy was described recently, by the group of Panda at the Central Drug Research Institute in India, for the construction of (S)-amino acid-derived homochiral cis-2,5-disubstituted piperazines. Cu-Catalyzed spontaneous regioselective ring-opening and ring-closing of non-activated N-tosyl aziridines as well as Pd-mediated N–C bond formation from N-tosyl brominated amino-acid derivatives are the key steps for accessing the disubstituted piperazines.47 Although both routes are effective, the Pd-catalyzed process is more favorable than the Cu-catalyzed process, furnishing both symmetrical and unsymmetrical piperazines as illustrated in eqn (7). Mechanistic pathways for the synthesis of symmetrical and unsymmetrical piperazines have been proposed.47
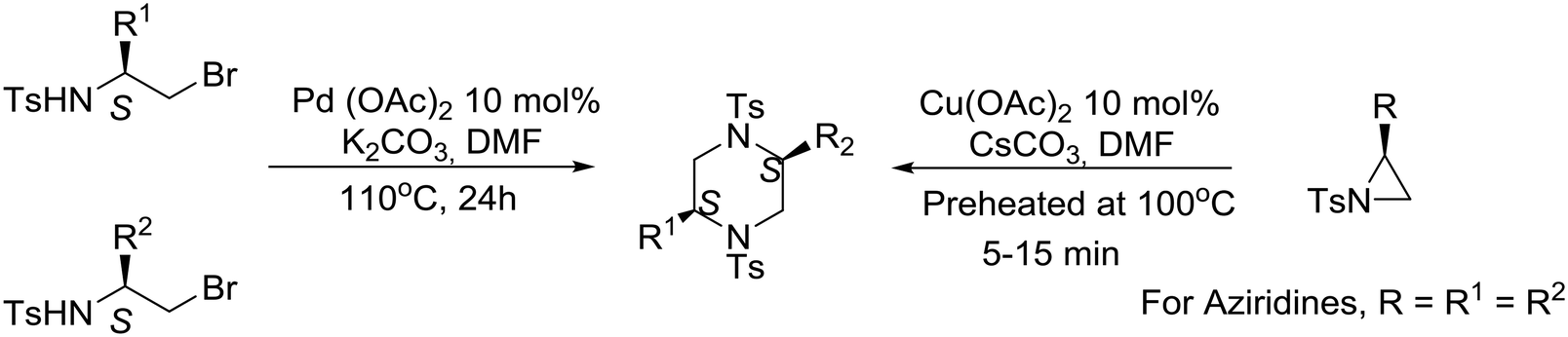 | (7) |
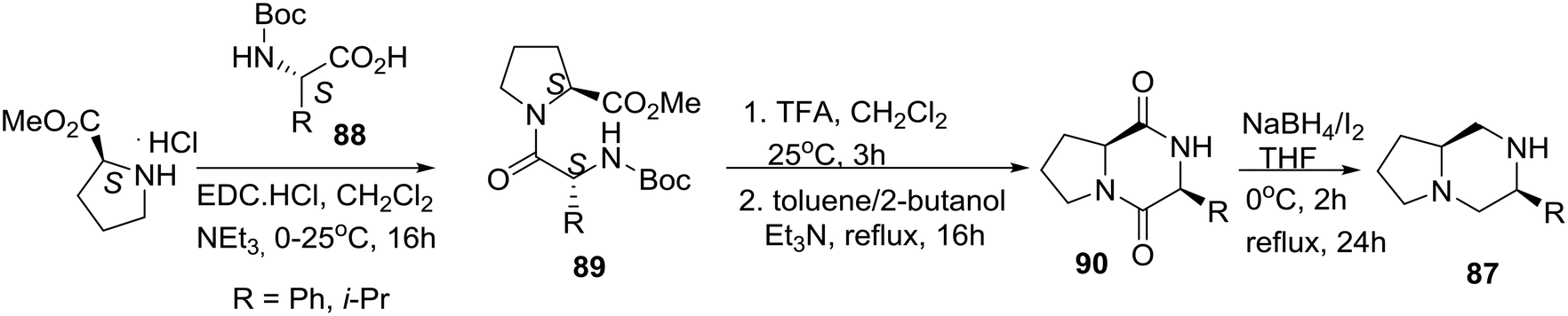
As noted above, a classic method for accessing carbon-substituted piperazines is the reduction of 2,5-diketopiperazines generated via peptide cyclodimerization or by the multicomponent Ugi reaction.23,48 Highlighting their reduction reagent (NaBH4/I2), the group of Periasamy in India reported the synthesis of the homochiral bicyclic piperazine 87 derived from L-proline (eqn (8)).49
 | (8) |
L-Proline methyl ester hydrochloride salt was coupled with N-Boc protected amino acid derivatives 88 to prepare dipeptides 89, which after TFA treatment and cyclization afforded diketopiperazine derivatives 90. Efficient (68–74% yields) reduction of the latter with the NaBH4/I2 reagent gave the desired enantiomerically pure bicyclic piperazines 87.49
Finally, capitalizing on prior knowledge obtained through studies on the intramolecular aza-Michael reaction,50 the group of Santini and Young at Baylor College of Medicine in the U.S. reported the synthesis of homochiral 5- and 6-substituted-piperazine-2-acetic acid esters as intermediates for library production.51 In the case of 2,5-disubstituted piperazines, a divergent six-step synthesis was developed in which homochiral amino alcohols 77 (eqn (6)) were transformed, with high diastereoselectivity, into either cis or trans 5-substituted piperazine-2-acetic acid esters that could be chromatographically rendered diastereomerically homogeneous.51a The synthesis proceeded via a three step construction of non-isolable N-nosyl aziridines 91 which entered two pathways: the cis selective Boc pathway and the trans selective TFA pathway (Scheme 14).51a
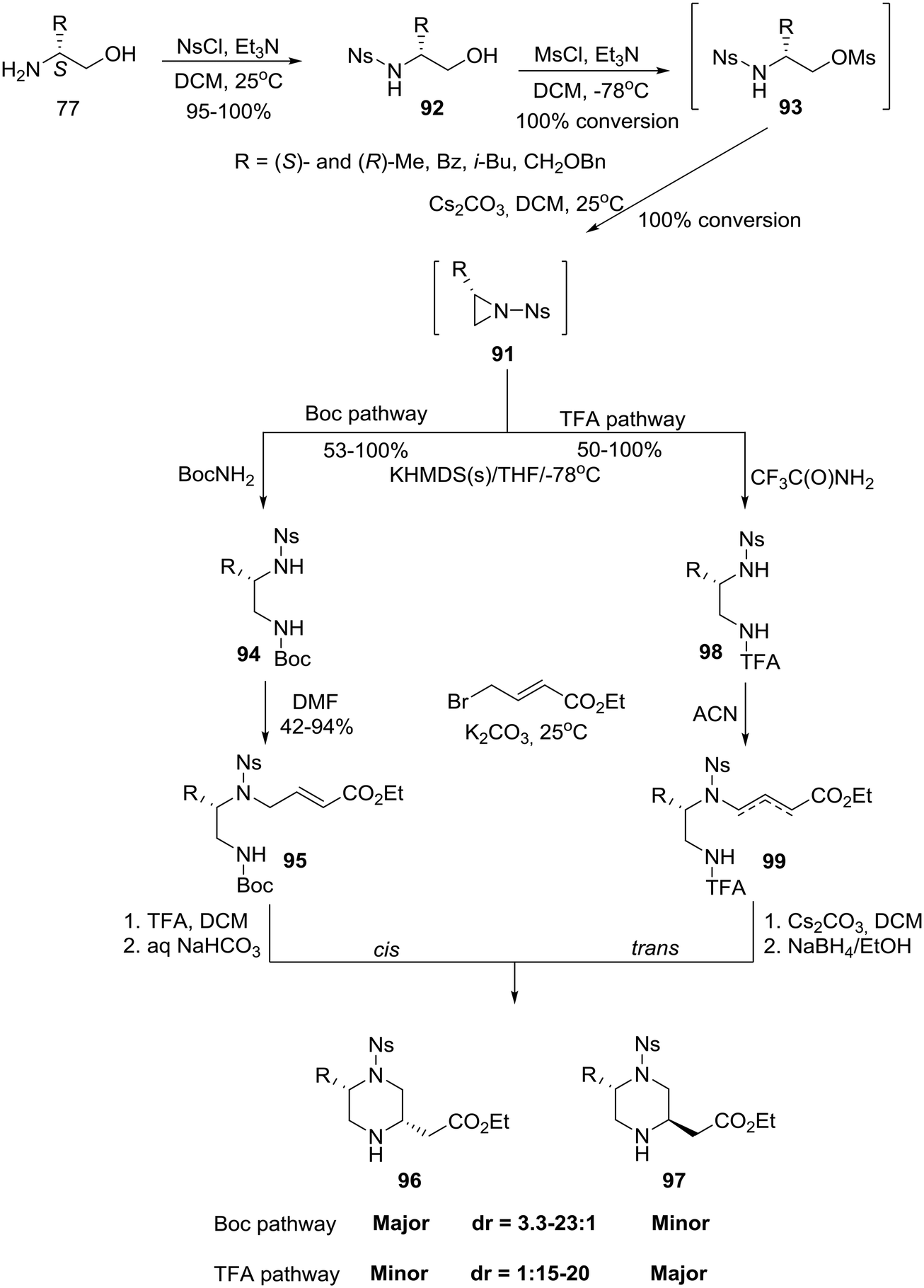 | ||
| Scheme 14 General synthetic scheme for the asymmetric construction of cis and trans 5-alkyl-substituted piperazine-2-acetic acid esters. | ||
Specifically, amino alcohols 77 (eqn (6)) were converted to their N-nosylated derivatives 92, respectively, by exposure to NsCl/TEA in DCM at ambient temperature. The crude products were treated with MsCl/TEA in DCM to give the mesylated intermediates 93 as determined by LC-MS. Mesylates 93 were isolated in crude form by extractive workup and immediately advanced without further characterization. Accordingly, treatment with Cs2CO3 in DCM effectively generated the N-nosyl aziridines 91, as determined by LC-MS, which were isolated as crude solutions given the instability of the N-nosyl aziridine in neat form.51a Treatment of aziridines 91 with the preformed potassium salt of BocNH2 in THF at −78 °C directly afforded the regiospecifically ring-opened, orthogonally protected diamines 94 that were in turn regioselectively alkylated with ethyl 4-bromocrotonate to obtain the cyclization precursors 95. Deprotection with TFA/DCM followed by bicarbonate neutralization furnished a mixture of piperazines 96-cis and 97-trans (dr cis:trans 3.3–23:1). Using this route, however, with a dr of >5:1, would not enable the authors to obtain enough of the minor trans diastereomer to meet their goals. They, therefore, surmised that having a substituted nucleophilic nitrogen would increase the steric demand of the aza-Michael donor, a consequence that might alter the diastereoselectivity of the cyclization. Thus, they pursued a base-promoted anionic cyclization of a carbamate, namely the TFA pathway: treatment of aziridines 91 with the preformed potassium salt of CF3C(O)NH2 afforded the differentially substituted diamines 98. Alkylation of diamines 98 with ethyl 4-bromocrotonate proceeded more cleanly in ACN (acetonitrile) than in DMF (dimethylformamide) and produced a 1.5:1 [{2,3}:{3,4}] mixture of the olefin isomers 99 which were individually advanced through the base-promoted cyclization. Gratifyingly, piperazines 97-trans were obtained after removal of the TFA group (Scheme 14).
On the other hand, an efficient four-step synthesis allowed transformation of six homochiral amino acids into 6-substituted piperazine-2-acetic acid esters as diastereomeric mixtures of which cis and trans products could be chromatographically separated (Scheme 15).51b
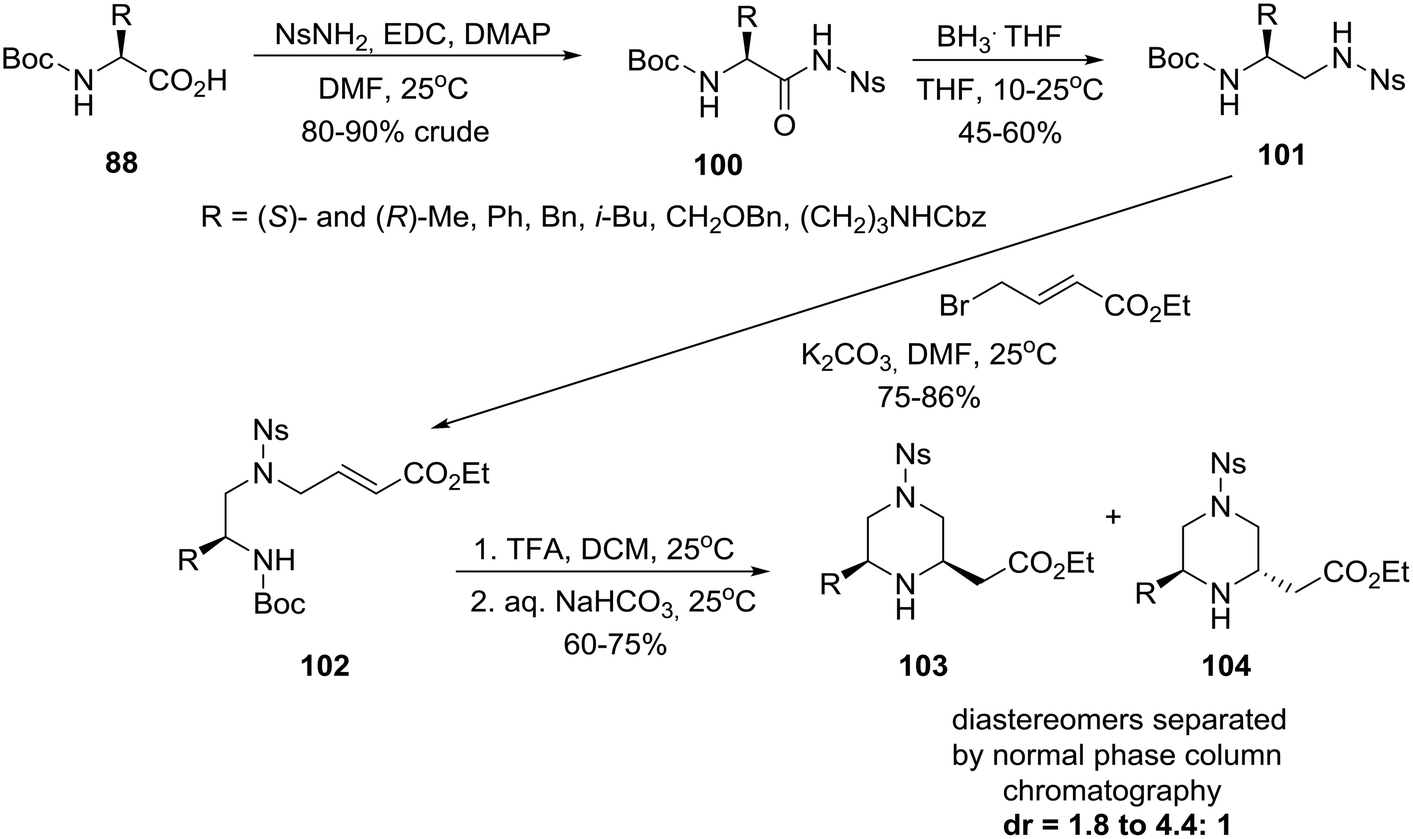 | ||
| Scheme 15 Asymmetric construction of cis and trans 6-alkyl-substituted piperazine-2-acetic acid esters. | ||
Thus, homochiral amino acids 88 (eqn (8)) were converted to nosylamides 100 by the action of EDC/DMAP in the presence of NsNH2. Pleasingly, the nosyl amides 100, recovered in crude form by extractive workup, could be directly reduced by a BH3THF complex to the differentially protected diamines 101, alkylation of which with ethyl 4-bromo-E-crotonate, as described in Scheme 15, gave acrylates 102. Removal of the N-Boc protecting group with TFA/DCM followed by neutralization cleanly afforded a mixture of piperazines from which diastereomers 103cis and 104trans were isolated by chromatography.51b
To complete the series, for the synthesis of homochiral 3-substituted-piperazine-2-acetic acid esters as intermediates for library production, the group of Santini and Young at Baylor College of Medicine adopted an “inter” retrosynthetic strategy entailing the formation of a second stereocenter by an intermolecular aza-Michael reaction of ethanolamine to an acrylate also derived from a homochiral amino acid 88 (Scheme 16).52
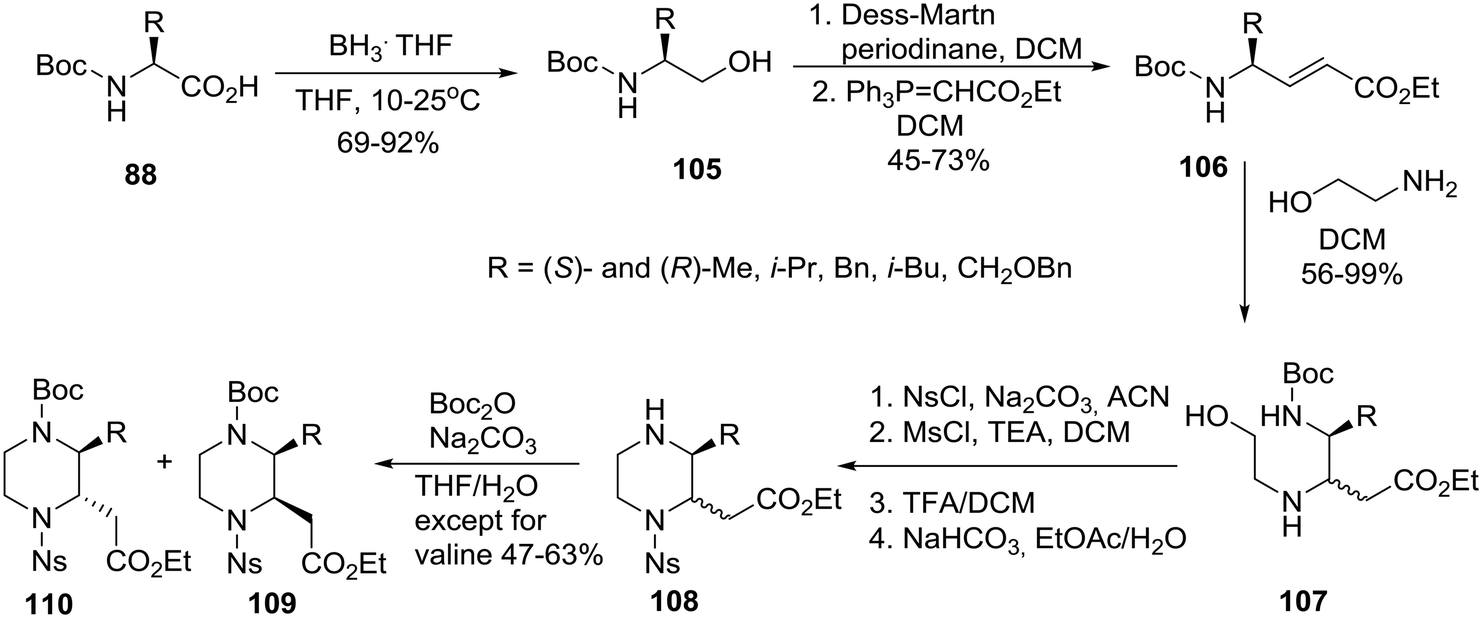 | ||
| Scheme 16 Asymmetric synthesis of cis and trans 3-alkyl-substituted piperazine-2-acetic acid esters. | ||
The homochiral N-Boc amino acids 88 were reduced to their corresponding alcohols 105 by exposure to the BH3THF complex. Subsequent oxidation using the Dess–Martin conditions generated the respective aldehydes (not shown, no loss of enantiomeric purity) that were immediately treated with (carbethoxymethylene)triphenylphosphorane to afford the acrylates 106 all having a trans geometry. The latter were treated with ethanolamine (4 equiv.) to bring about the aza-Michael reaction which occurred at ambient temperature with modest diastereoselectivity affording mixtures of diastereomers 107, protection of which with NsCl and reaction with MsCl/TEA gave the O-mesylated intermediates (not shown) that were deprotected with TFA to furnish the piperazines 108 after neutralization. The separation of some piperazines proved to be difficult at this point, but Boc reprotection of the nitrogen at the 4-position rendered the remaining diastereomers 109cis and 110trans separable by normal-phase column chromatography.52 In the trans products, substituents at C2 and C3 are diaxial to minimize the allylic 1,3-strain with the nosyl amide as well as to avoid steric clashes that would occur between adjacent diequatorial groups.27 The strategic preparation and separation of diastereomeric mixtures proved to be key to the efficient implementation of this systematic chemical diversity (SCD) strategy.51,52
Conclusion and outlook
As mentioned in the Introduction section, despite the ubiquity of piperazine-containing therapeutics, the structural, regiochemical, and stereochemical diversity of these compounds along the piperazine's carbon framework is remarkably limited. Notwithstanding the recent progress on asymmetric synthesis of piperazines described in this review, the development of a general method for the enantioselective construction of carbon-substituted piperazine cores has yet to be achieved. This observation provokes the query of whether stereochemically diverse piperazines represent a class of inconsequential compounds or whether current synthetic limitations are hindering access to these stereochemically more elaborate targets.53 Nevertheless, efforts toward the development of a general, catalytic enantioselective synthetic approach for carbon-substituted piperazines are underway in this laboratory.Conflicts of interest
There is no conflict of interest to declare.References
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) C.-V. T. Vo, M. U. Luescher and J. W. Bode, Nat. Chem., 2014, 6, 310 CrossRef CAS PubMed; (c) C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075 CrossRef CAS; (d) S.-Y. Hsieh and J. W. Bode, Org. Lett., 2016, 18, 2098 CrossRef CAS PubMed.
- S. C. Stinson, Chem. Eng. News, 2001, 79(20), 45 CrossRef.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- (a) M. Shaquiquzzaman, G. Verma, A. Marella, M. Akhter, W. Akhtar, M. F. Khan, S. Tasneem and M. M. Alam, Eur. J. Med. Chem., 2015, 102, 487 CrossRef CAS PubMed; (b) Z. Ye, K. E. Gettys and M. Dai, Beilstein J. Org. Chem., 2016, 12, 702 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- T. James, P. MacLellan, G. M. Burlsem, I. Simpson, J. A. Grant, S. Warriner, V. Sridharan and A. Nelson, Org. Biomol. Chem., 2014, 12, 2584 RSC.
- K. E. Gettys, Z. Ye and M. Dai, Synthesis, 2017, 49, 2589 CrossRef CAS.
- R. Nathans, H. Cao, N. Sharova, A. Ali, M. Sharkey, R. Stranska, M. Stevenson and T. M. Rana, Nat. Biotechnol., 2008, 26, 1187 CrossRef CAS PubMed.
- A. K. Rathi, R. Syed, H.-S. Shin and R. V. Patel, Expert Opin. Ther. Pat., 2016, 26, 777 CrossRef CAS PubMed.
- (a) K. L. Rinehart, T. G. Holt, N. L. Fregeau, J. G. Stroh, P. A. Keifer, F. Sun, L. H. Li and D. G. Martin, J. Org. Chem., 1990, 55, 4512 CrossRef CAS; (b) K. L. Rinehart, Med. Res. Rev., 2000, 20, 1 CrossRef CAS PubMed.
- (a) C. Cuevas and A. Francesch, Nat. Prod. Rep., 2009, 26, 322 RSC; (b) T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69 CrossRef CAS PubMed.
- A. Barone, D.-C. Chi, M. R. Theoret, H. Chen, K. J. He, D. Kufrin, W. S. Helms, S. Subramaniam, H. Zhao, A. Patel, K. B. Goldberg, P. Keegan and R. Pazdur, Clin. Cancer Res., 2017, 23, 7448 CrossRef CAS PubMed.
- (a) S. Hegde and M. Schmidt, Annu. Rep. Med. Chem., 2008, 43, 455 CAS; (b) V. H. Le, M. Inai, R. M. Williams and T. Kan, Nat. Prod. Rep., 2015, 32, 328 RSC.
- “Synthetic Approaches to the Stereochemically Complex Antitumor Drug Ecteinascidin-743: A Marine Natural Product by the Name Yondelis® or Trabectidin” P. A. Magriotis, in Stereochemistry and Global Connectivity: The Legacy of Ernest L. Eliel, ed. H. N. Chenget al., ACS Symposium Series, American Chemical Society, Washington, DC, 2017, ch. 5, vol. 2, pp. 61–78 Search PubMed.
- (a) J. Jia, R. Chen, Y. Jia, H. Gu, Q. Zhou and X. Chen, J. Org. Chem., 2019, 84, 13696 CrossRef CAS PubMed; (b) W. He, Z. Zhang and D. Ma, Angew. Chem., Int. Ed., 2019, 58, 3972 CrossRef CAS PubMed.
- (a) M. Movassaghi, D. S. Siegel and S. Ham, Chem. Sci., 2010, 1, 561 RSC; (b) Y. Nakao, A. Masuda, S. Matsunaga and N. Fusetani, J. Am. Chem. Soc., 1999, 121, 2425 CrossRef CAS; (c) T. Shioiri and N. Irako, Chem. Lett., 2002, 130 CrossRef CAS; (d) A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS PubMed; (e) P. Thesmar and O. Baudoin, J. Am. Chem. Soc., 2019, 141, 15779 CrossRef CAS PubMed.
- R. Kakarla, J. Liu, D. Naduthambi, W. Chang, R. T. Mosley, D. Bao, H. M. Micolochick Steuer, M. Keilman, S. Bansal, A. M. Lam, W. Seibel, S. Neilson, P. A. Furman and M. J. Sofia, J. Med. Chem., 2014, 57, 2136 CrossRef CAS PubMed.
- (a) N. Kaplaneris, C. Spyropoulos, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2016, 18, 5800 CrossRef CAS PubMed; (b) M. P. Brochu, S. P. Brown and D. W. C. MacMillan, J. Am. Chem. Soc., 2004, 126, 4108 CrossRef CAS PubMed.
- (a) M. C. O'Reilly and C. W. Lindsley, Org. Lett., 2012, 14, 2910 CrossRef PubMed; (b) M. Marigo, S. Bachmann, N. Halland, A. Braunton and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 5507 CrossRef CAS PubMed.
- Y. Y. Lau, H. Zhai and L. L. Schafer, J. Org. Chem., 2016, 81, 8696 CrossRef CAS PubMed.
- K. Ito, Y. Imahayashi, T. Kuroda, S. Eno, B. Saito and T. Katsuki, Tetrahedron Lett., 2004, 45, 7277 CrossRef CAS.
- K. M. Korch, C. Eidamshaus, D. C. Bohenna, S. Nam, D. Horne and B. M. Stoltz, Angew. Chem., Int. Ed., 2015, 54, 179 CrossRef CAS PubMed.
- A. Dömling and Y. Huang, Synthesis, 2010, 2859 CrossRef.
- N. Faltracco, S. Cotogno, C. M. L. Vande Velde and E. Ruijter, J. Org. Chem., 2019, 84, 12058 CrossRef PubMed.
- B. M. Cochran and F. E. Michael, Org. Lett., 2008, 10, 329 CrossRef CAS PubMed.
- J. J. Posakony, J. R. Grierson and T. J. Tewson, J. Org. Chem., 2002, 67, 5164 CrossRef CAS PubMed.
- F. Johnson, Chem. Rev., 1968, 68, 375 CrossRef CAS.
- A. Borzenko, H. Pajouhesh, J.-L. Morrison, E. Tringham, T. P. Snutch and L. L. Schafer, Bioorg. Med. Chem. Lett., 2013, 23, 3257 CrossRef CAS PubMed.
- (a) A. V. Lee and L. L. Schafer, Synlett, 2006, 2973 CAS; (b) A. V. Lee, M. Sajitz and L. L. Schafer, Synthesis, 2009, 97 CAS.
- H. Zhai, A. Borzenko, Y. Y. Lau, S. H. Ahn and L. L. Schafer, Angew. Chem., Int. Ed., 2012, 51, 12219 CrossRef CAS PubMed.
- V. Schanen, C. Riche, A. Chiaroni, J.-C. Quirion and H.-P. Husson, Tetrahedron Lett., 1994, 35, 2533 CrossRef CAS.
- M. Sánchez-Roselló, O. Delgado, N. Mateu, A. A. Trabanco, M. Van Gool and S. Fustero, J. Org. Chem., 2014, 79, 5887 CrossRef PubMed.
- A. Viso, R. F. De la Pradilla, A. Garcia, C. Guerrero-Strachan, M. Alonso, M. Tortosa, A. Flores, M. Martinez-Ripoli, I. Fonseca, I. André and A. Rodríguez, Chem. – Eur. J., 2003, 9, 2867 CrossRef CAS PubMed.
- A. Viso, R. F. De la Pradilla, A. Flores, A. Garcia, M. Tortosa and M. L. Lopez-Rodriguez, J. Org. Chem., 2006, 71, 1442 CrossRef CAS PubMed.
- A. Music and D. Didier, Synlett, 2019, 30, 1843 CrossRef CAS.
- R. J. Abdel-Jalil, R. A. Al-Qawasmeh, Y. Al-Abed and W. Voelter, Tetrahedron Lett., 1998, 39, 7703 CrossRef CAS.
- K. Samanta and G. Panda, Chem. – Asian J., 2011, 6, 189 CrossRef CAS PubMed.
- L. K. Ottesen, C. A. Olsen, M. Witt, J. W. Jaroszewski and H. Franzyk, Chem. – Eur. J., 2009, 15, 2966 CrossRef CAS PubMed.
- J. S. Nakhla and J. P. Wolfe, Org. Lett., 2007, 9, 3279 CrossRef CAS PubMed.
- H. Li, X. Jiang, Y.-h. Ye, C. Fan, T. Romoff and M. Goodman, Org. Lett., 1999, 1, 91 CrossRef CAS PubMed.
- J. W. Mickelson, K. L. Belonga and E. J. Jacobsen, J. Org. Chem., 1995, 60, 4177 CrossRef CAS.
- B. L. Palucki, M. K. Park, R. P. Nargund, R. Tang, T. MacNeil, D. H. Weinberg, A. Vongs, C. I. Rosenblum, G. A. Doss, R. R. Miller, R. A. Stearns, Q. Peng, C. Tamvakopoulos, L. H. T. Van der Ploeg and A. A. Patchett, Bioorg. Med. Chem. Lett., 2005, 15, 1993 CrossRef CAS PubMed.
- T. V. Lukina, S. I. Sviridov, S. V. Shorshnev, G. G. Alexandrov and A. E. Stepanov, Tetrahedron Lett., 2005, 46, 1205 CrossRef CAS.
- S. Dekeukeleire, M. D'hooghe, M. Vanwalleghem, W. Van Brabandt and N. De Kimpe, Tetrahedron, 2012, 68, 10827 CrossRef CAS.
- (a) I. Ojima and F. Delaloge, Chem. Soc. Rev., 1997, 26, 377 RSC; (b) C. Palomo, J. M. Aizpurua, I. Ganboa and M. Oiarbide, Pure Appl. Chem., 2000, 72, 1763 CAS; (c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437 CrossRef CAS PubMed.
- (a) P. A. Magriotis, Angew. Chem., Int. Ed., 2001, 40, 4377 CrossRef CAS; (b) P. A. Magriotis, Eur. J. Org. Chem., 2014, 2647 CrossRef CAS.
- S. K. Manna and G. Panda, RSC Adv., 2013, 3, 18332 RSC.
- M. Stucchi, S. Cairati, R. Cetin-Atalay, M. S. Christodoulou, G. Grazioso, G. Pescitelli, A. Silvani, D. C. Yildirim and G. Lesma, Org. Biomol. Chem., 2015, 13, 4993 RSC.
- V. Harish and M. Periasamy, Tetrahedron: Asymmetry, 2017, 28, 175 CrossRef CAS.
- C. Zhong, Y. Wang, A. W. Hung, S. L. Schreiber and D. W. Young, Org. Lett., 2011, 13, 5556 CrossRef CAS PubMed.
- (a) P. Jain, I. O. Raji, S. Chamakouri, K. R. MacKenzie, B. T. Ebright, C. Santini and D. W. Young, J. Org. Chem., 2019, 84, 6040 CrossRef CAS PubMed; (b) S. Chamakouri, P. Jain, S. K. R. Guduru, J. W. Arney, K. R. MacKenzie, C. Santini and D. W. Young, J. Org. Chem., 2018, 83, 6541 CrossRef PubMed.
- S. K. R. Guduru, S. Chamakuri, I. O. Raji, K. R. MacKenzie, C. Santini and D. W. Young, J. Org. Chem., 2018, 83, 11777 CrossRef PubMed.
- J. B. MacManus, N. P. R. Onuska, M. S. Jeffreys, N. C. Goodwin and D. A. Nicewicz, Org. Lett., 2020, 22, 679 CrossRef PubMed.
Footnote |
| † This review is dedicated to Professor Francis Johnson (Stony Brook University) on the occasion of his ninetieth birthday and in acknowledgement of his immense contributions to Medicinal Chemistry. |
| This journal is © The Royal Society of Chemistry 2020 |