
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the mechanism of action of pyrrolo[3,2-b]quinoxaline-derivatives as kinase inhibitors†
Andrea
Unzue
a,
Claudia
Jessen-Trefzer
a,
Dimitrios
Spiliotopoulos
b,
Eugenio
Gaudio
c,
Chiara
Tarantelli
c,
Jing
Dong
b,
Hongtao
Zhao
b,
Johanna
Pachmayr
d,
Stefan
Zahler
d,
Elena
Bernasconi
c,
Giulio
Sartori
c,
Luciano
Cascione
 ce,
Francesco
Bertoni
*cf,
Paweł
Śledź
b,
Amedeo
Caflisch
*b and
Cristina
Nevado
*a
ce,
Francesco
Bertoni
*cf,
Paweł
Śledź
b,
Amedeo
Caflisch
*b and
Cristina
Nevado
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zürich, Switzerland. E-mail: cristina.nevado@chem.uzh.ch; Fax: (+41) 446353948; Tel: (+41) 446353945
bDepartment of Biochemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zürich, Switzerland
cInstitute of Oncology Research, Faculty of Biomedical Sciences, USI, Bellinzona, Switzerland
dDepartment of Pharmacy, University of Munich, Butenandstrasse 5-13, 81377 Munich, Germany
eSIB Swiss Institute of Bioinformatics, Lausanne, Switzerland
fOncology Institute of Southern Switzerland (IOSI), Bellinzona, Switzerland
First published on 19th May 2020
Abstract
The X-ray structure of the catalytic domain of the EphA3 tyrosine kinase in complex with a previously reported type II inhibitor was used to design two novel quinoxaline derivatives, inspired by kinase inhibitors that have reached clinical development. These two new compounds were characterized by an array of cell-based assays and gene expression profiling experiments. A global chemical proteomics approach was used to generate the drug-protein interaction profile, which suggested suitable therapeutic indications. Both inhibitors, studied in the context of angiogenesis and in vivo in a relevant lymphoma model, showed high efficacy in the control of tumor size.
Introduction
Drug development is a risky and expensive process1 in which only 8% of the candidates that enter clinical trials are approved by the authorities.2 Late-stage failures of promising drug candidates in clinical development have emphasized the importance of understanding their mode of action.2 As a result, more personalized treatments can be developed, new clinical applications of already approved drugs can be conceived, and a better understanding of off-target effects can be achieved. The application of systems biology approaches, including studies based on gene signatures that inform on pathways and diseases that the drug of interest acts on,3 or chemical proteomics technology, where binding of the molecule of interest is addressed in an unbiased environment to obtain its global target profile, have proven efficient in this regard. In fact, such studies have been applied to several kinase inhibitors including dasatinib, imatinib, nilotinib4 and bosutinib,5 revealing clear differences in the mode of action of these drugs despite being all administered for the treatment of chronic myeloid leukemia (CML).Protein kinases are relevant targets for the treatment of diseases including cancer, inflammation, cardiovascular conditions and immune-related disorders.6 More than 50 small-molecule kinase inhibitors have been approved by the FDA over the last decade.7 In our work, we have focused on the in silico design, synthesis and computational-aided optimization of potent and selective receptor tyrosine kinase inhibitors.8 To date, our approach has yielded nanomolar EphB4 inhibitors whose binding modes, antiproliferative activities and in vivo efficacies have been characterized.9
Here we describe the design, synthesis and in depth biological characterization of the mode of action of two novel type II kinase inhibitors (8a, b) with broader scope of activity. The two compounds showed in vivo anti-tumor activity, especially in models derived from hematological malignancies. The effects of these molecules on gene expression were compared to dasatinib, an FDA approved drug for the treatment of chronic myeloid leukemia.10 Further characterization, including the assessment of their in vitro selectivity, cellular angiogenesis inhibition, as well as a global chemical proteomics approach and in vivo efficacy is reported here.
Design of new type II quinoxaline inhibitors
Within our first generation of quinoxaline EphB4 inhibitors (including type I, I1/2 and II),8d compound 1 proved to be the most potent type I1/2 ligand in vitro inducing a cytostatic effect in an in vivo cancer model. Interestingly, compound 2 represents a type II inhibitor bearing a urea linker that offers an interesting anti-proliferative profile over a cell cancer panel (Fig. 1).8d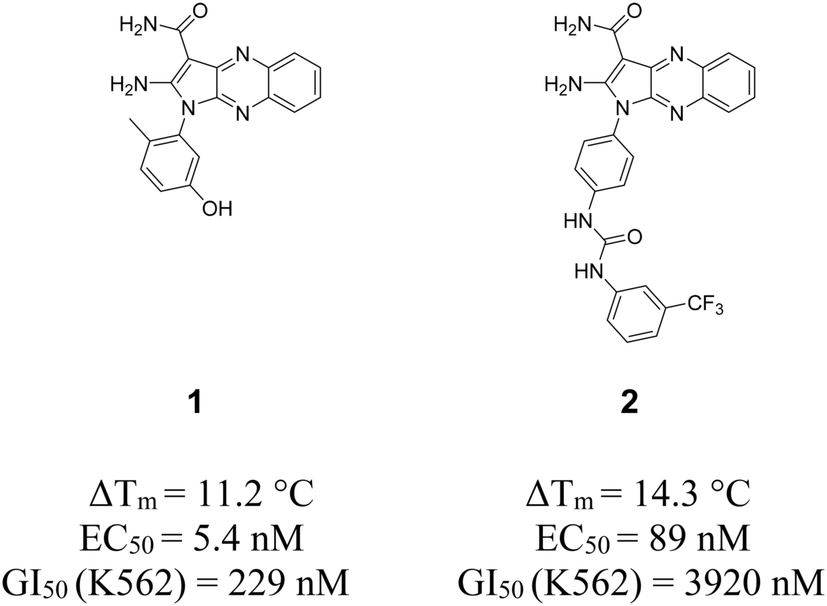 | ||
| Fig. 1 2D structures of previously developed type I1/2 and II quinoxaline inhibitors 1 and 2.8d EC50 values were measured in a cellular phosphorylation assay using mouse embryonic fibroblasts (MEF) that overexpress EphB4. ΔTm values have been determined using differential scanning fluorimetry assay. | ||
The type II binding mode with the Asp-Phe-Gly (DFG)-out conformation of the kinase was confirmed by X-ray diffraction analysis of the catalytic domain of EphA3 in complex with 2 (Fig. 2A, PDB code 4P5Z).8d The pyrrolo[3,2-b]quinoxaline scaffold occupies the ATP binding site with the phenyl substituent located in the hydrophobic pocket that is generated when the kinase adopts an inactive DFG-out conformation. Compound 2 is able to form four hydrogen bonds within the hinge region of the kinase via the amino and amide substituents of the pyrrole ring: the amino substituent at position 2 is involved in a bifurcated hydrogen bond with the side chain hydroxyl of the Thr693 gatekeeper and the backbone carbonyl of Glu694, whereas the amide substituent at position 3 forms two hydrogen bonds with the backbone polar groups of Met696. In addition, the urea linker establishes three extra hydrogen bonds acting as a hydrogen bond acceptor from the Ser757 side chain and the amide backbone of Asp758, and hydrogen bond donor to the side chain of Glu664. The m-CF3-phenyl moiety is located in the hydrophobic pocket originating from the displacement of the Phe765 side chain of the DFG motif in the DFG-out conformation of the kinase.
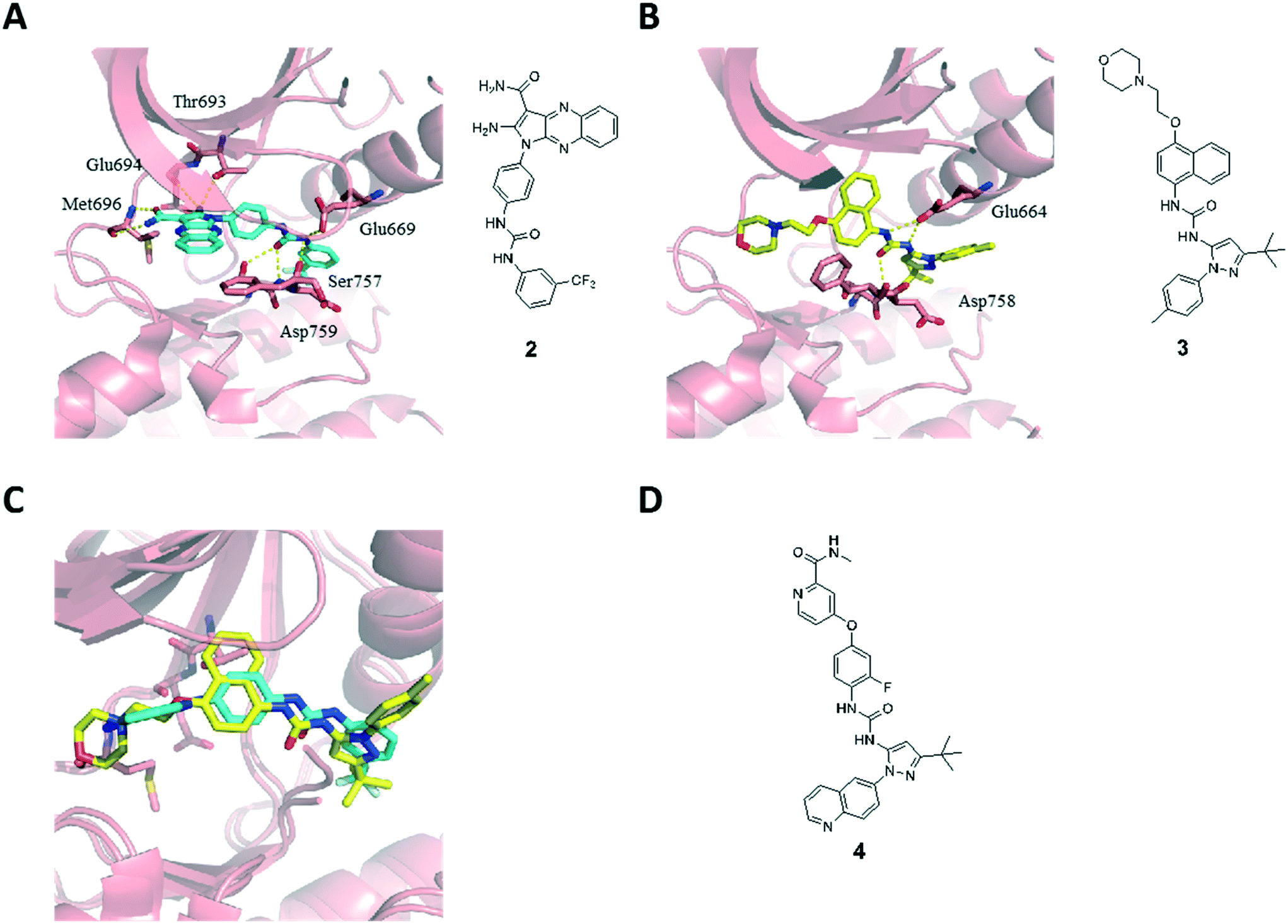 | ||
| Fig. 2 Crystal structures of the catalytic domain of the tyrosine kinase EphA3 in complex with the low-nanomolar inhibitors 2 (A) and 3 (B) and superposition of the two inhibitors 2 and 3 (C). The ATP binding site of the EphA3 kinase is shown in pink ribbons while the side chains mentioned in the text and the inhibitors are shown by sticks in blue and yellow for compounds 2 and 3, respectively. (D) 2D structure of the BCR-ABL1 inhibitor DCC-2036 (4). | ||
We have also solved the X-ray structure of the clinical candidate BIRB796 (3) in complex with the catalytic domain of EphA3 (Fig. 2B, PDB code 4TWN).11 Compound 3 is a selective p38 MAP kinase inhibitor developed by Boehringer Ingelheim for the treatment of rheumatoid arthritis and other inflammatory conditions including Crohn's disease and psoriasis that has undergone several clinical trials.3a,12 Compound 3 is a type II kinase inhibitor that is characterized by a pyrazole ring bearing a tert-butyl substituent nested within the hydrophobic pocket. Similar to our type II inhibitor 2, the urea linker in inhibitor 3 establishes a hydrogen bond network with Asp758 and Glu664. This structure inspired further modifications of compound 1. In particular, the superposition of compounds 2 and 3 reveals an overall similar binding mode and a clear overlap of the urea linker and its substituents (Fig. 2C), which prompted us to introduce a tert-butyl substituted pyrazole scaffold within our quinoxaline inhibitors series. We decided to synthesize not only the 4-methyl phenyl substituted pyrazole ring present in compound 3, but also a quinoline substituted one mimicking compound DCC-2036 (4),13 a BCR-ABL1 inhibitor that underwent clinical trials for the treatment of chronic myeloid leukemia (Fig. 2D).14
Synthesis of new type II quinoxaline inhibitors
The synthesis of the novel type II quinoxaline derivatives bearing the tert-butyl pyrazole moiety is shown in Scheme 1. Compound 5 was prepared according to previously reported procedures by condensation of commercially available 2,3-dichloroquinoxaline with malononitrile in the presence of sodium hydride.15 The substitution of the chlorine at position 3 with synthetically prepared anilines 6a, b (ref. 16) followed by cyclization afforded intermediates 7a, b (ref. 17) (Scheme 1), which were then hydrolyzed under strong acidic conditions affording the final compounds 8a, b.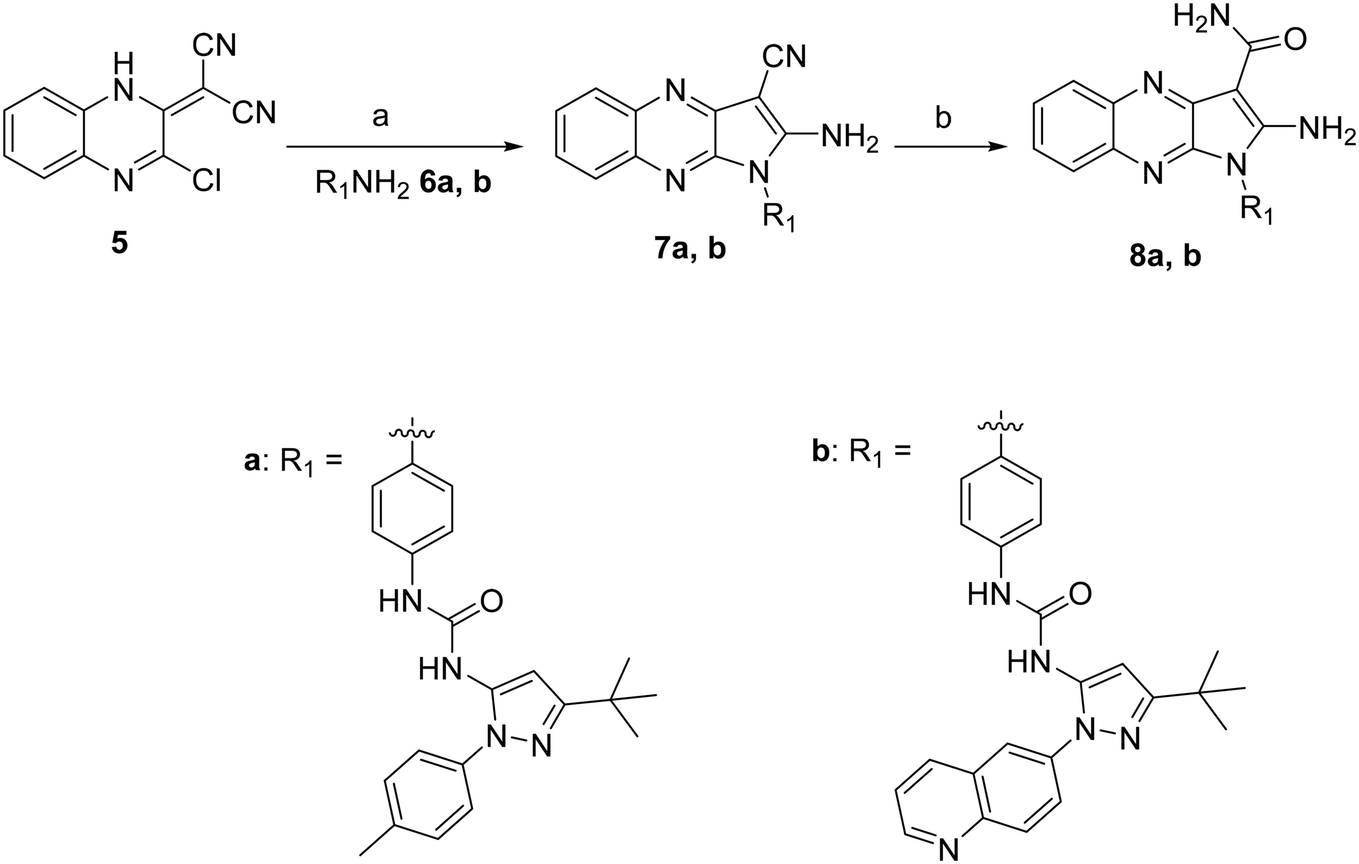 | ||
| Scheme 1 Reagents and reaction conditions: a) 6a, b (1.2 equiv.), DMF, 80 °C, 12 h, 55–62%. b) H2SO4, 25 °C, 30 min, 89–90%. | ||
Potency assessment: differential scanning fluorimetry and EC50 determination
The binding of the two novel type II quinoxaline inhibitors 8a, b towards the tyrosine kinases EphA3 and EphB4 was first studied in vitro and compared to our first generation quinoxaline inhibitors 1 and 2.8d Differential scanning fluorimetry (DSF) was used to measure the increase in thermal stability of EphA3 upon ligand binding.18 Compounds 1 and 2 exhibited pronounced thermal shifts of 11.2 and 14.3 °C respectively, while the new inhibitors 8a, b showed even higher values (17.3 and 19.3 °C respectively, Table 1). However, when compounds 8a, b were then tested in cellular phosphorylation assays on MEF cells transfected with Myc-tagged human EphB4, their potency dropped compared to initial compound series to EC50 values of 360 and 1100 nM, respectively (Table 1).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Type of binding | R1 | R2 | R3 | R4 | Thermal shift (°C)a | Cellular EC50b (nM) |
| a Average values of triplicate measurements. The standard deviation is smaller than 0.5 °C. b Cellular EC50 values were measured in a cellular phosphorylation assay using MEF cells overexpressing EphB4 at Proqinase. | |||||||
| 1 | I1/2 | Me | H | OH | H | 11.2 | 5.4 |
| 2 | II | H |

|
H | H | 14.3 | 89 |
| 8a | II | H |
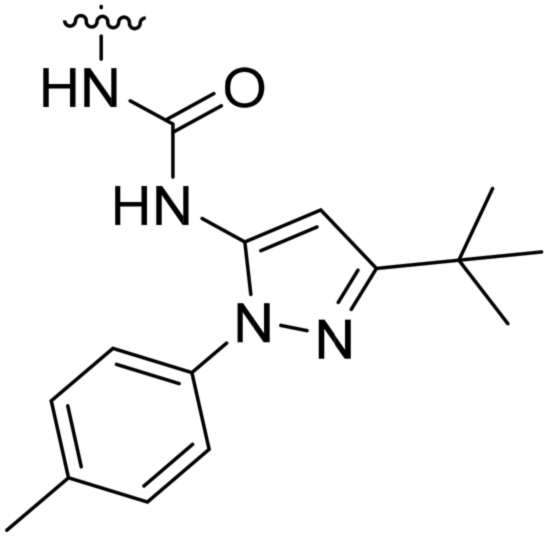
|
H | H | 17.3 | 360 |
| 8b | II | H |
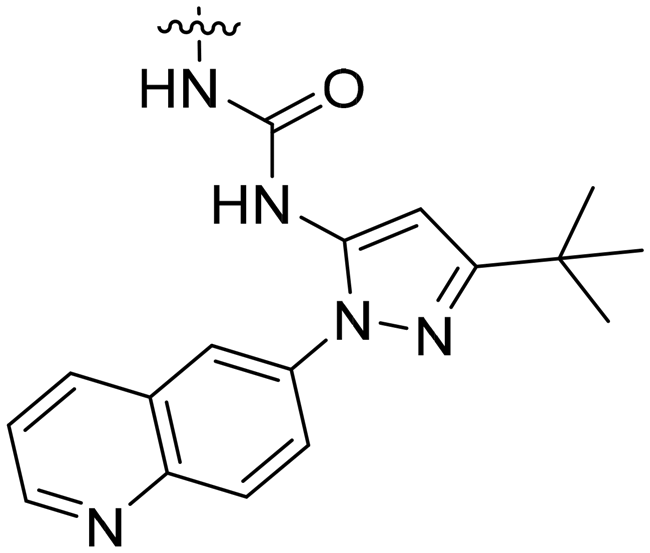
|
H | H | 19.3 | 1100 |
Selectivity profiles from biochemical assays
Given the potency loss observed towards EphB4 in the cell based assays for compounds 8a, b (Table 1), we decided to assess the influence of the new urea substituents on the selectivity over a broad panel of kinases. The selectivity profile was obtained by an in vitro competition binding assay that reports on binding affinity with no ATP required, using recombinant kinases (KINOMEscan at DiscoveRx).19 The selectivity panel consisted of 453 human kinases, 58 of which were disease related mutant kinases (mainly of ABL1, EGFR, and PIK3CA). Single dose measurements were carried out at 1 μM concentration of the ligand.Compounds 8a and 8b present a very similar selectivity profile that differs from that obtained for compound 1 (Fig. 3). Quantitatively, 60 kinases (14 of which are disease-related mutants) are more strongly inhibited by compounds 8a, b, where the difference between the affinity of 8a or 8b and compound 1 is at least 60% (expressed as% with respect to DMSO control). A total of 31 out of the 46 non-mutant kinases are serine/threonine-specific protein kinases, more than half of them belonging to the CMGC kinase family, and several are CDK kinases, which are involved in critical cellular processes.20 This data clearly suggests a broader selectivity profile for compounds 8a, b, which are also able to inhibit clinically relevant mutant kinases including ABL1, EGFR, FLT3, KIT, and MET. Interestingly, the activity towards EphB2, 3 and 4 kinases drops significantly in compounds 8a, b, in line with the EC50 values obtained in cells overexpressing EphB4 (Table 1).
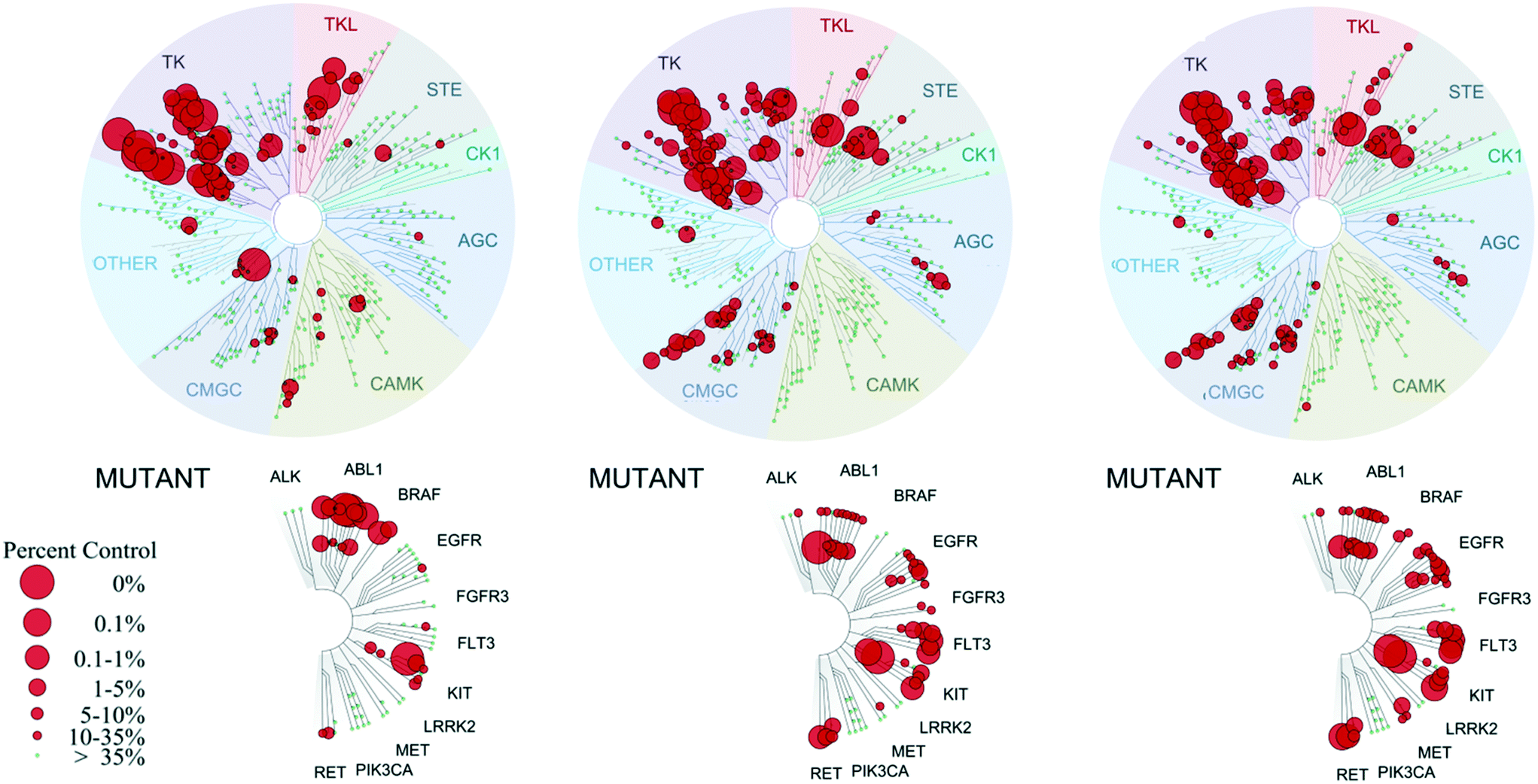 | ||
| Fig. 3 Selectivity profiles of compound 1 (left), 8a (center) and 8b (right) tested on a panel of 453 protein kinases at DiscoveRx. Measurements were performed at a concentration of 1 μM of the inhibitor. The affinity is defined with respect to a DMSO control. The dendrogram was obtained from KinomeScan using the KinomeTree software.19 | ||
Anti-proliferative activity and gene expression profiling
Compounds 8a and 8b were screened against the NCI-60 cancer cell line panel and compared to our previously developed quinoxalines (1, 2) and FDA-approved kinase inhibitors dasatinib and imatinib (Fig. 4). The two compounds presented a broader anti-proliferative activity than compounds 1 and 2 (which is in line with their broader selectivity profiles) and a significant correlation in terms of GI50 values across the different cell lines. The leukemia K-562 cell line was particularly sensitive towards the two optimized type II quinoxaline inhibitors, showing remarkably good GI50 values (31 and <10 nM, respectively).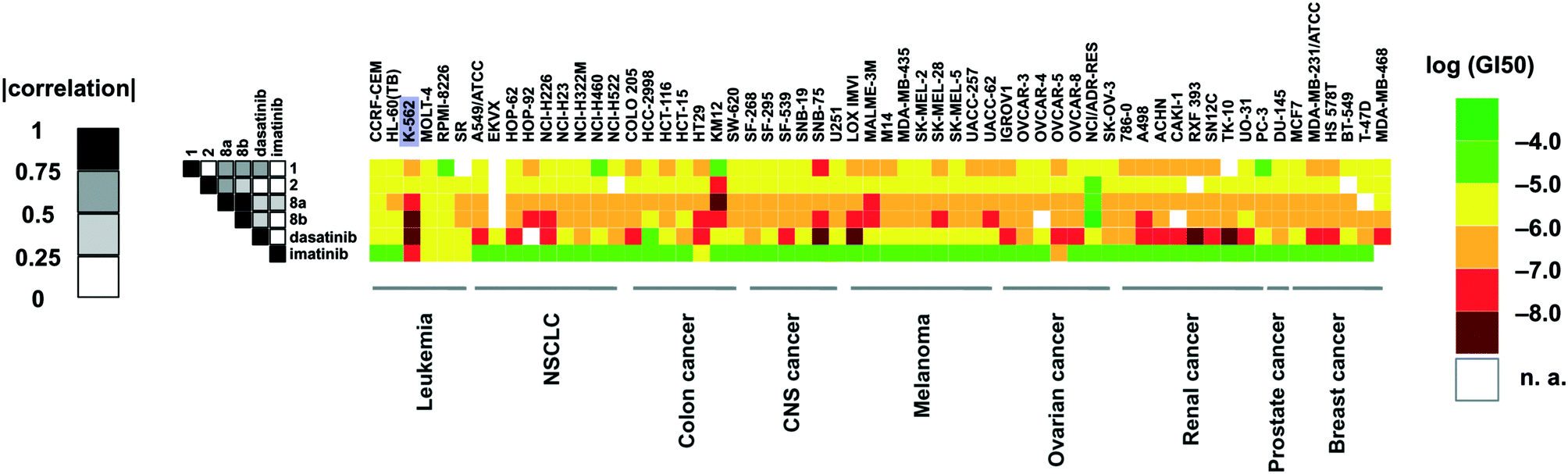 | ||
| Fig. 4 The antiproliferative activity of quinoxalines 1, 2, 8a and 8b is shown together with the growth inhibition of two FDA approved anticancer drugs (dasatinib and imatinib) evaluated on the NCI-60 cancer cell line panel. The growth inhibition is shown as a matrix with cell lines and compounds arranged horizontally and vertically, respectively. The legend bar shows the color coding, which reflects the log(GI50) value, with red and green indicating high and low toxicity, respectively. The grey scale on the left hand side of the figure shows the absolute value of the correlation of the compounds' GI50 values over for the different cell lines. | ||
In an effort to understand the mode of action of our quinoxaline inhibitors, we compared the transcriptome of K562 cells (one of the most sensitive cell lines towards our compounds, Fig. 4) after 6 hours of exposure to DMSO, as vehicle, to 1 μM of compound 8b or 1 μM of dasatinib, for reference (ESI† – RNA-Sequencing Table). Compound 8b and dasatinib caused very similar changes in the transcriptome of the cells (Fig. 5A). Compound 8b downregulated MYC and E2F targets, genes known to be involved in oxidative phosphorylation, unfolded protein response, proteasome pathway, PI3K/AKT/mTOR signaling, spliceasome and DNA repair (ESI† – RNA-Seq Table compound 8b). In contrast, genes negatively regulated by RAS were enriched among the transcripts upregulated by compound 8b (Fig. 5B). The gene expression signature of compound 8b overlapped with that of dasatinib on tumor infiltrating macrophages, and with PI3K/mTOR inhibitors in different tumor models (Fig. 5B) (ESI† – RNA-Seq Table compound 8b). Same signatures were observed in dasatinib treated cells (ESI† – RNA-Seq Table dasatinib).
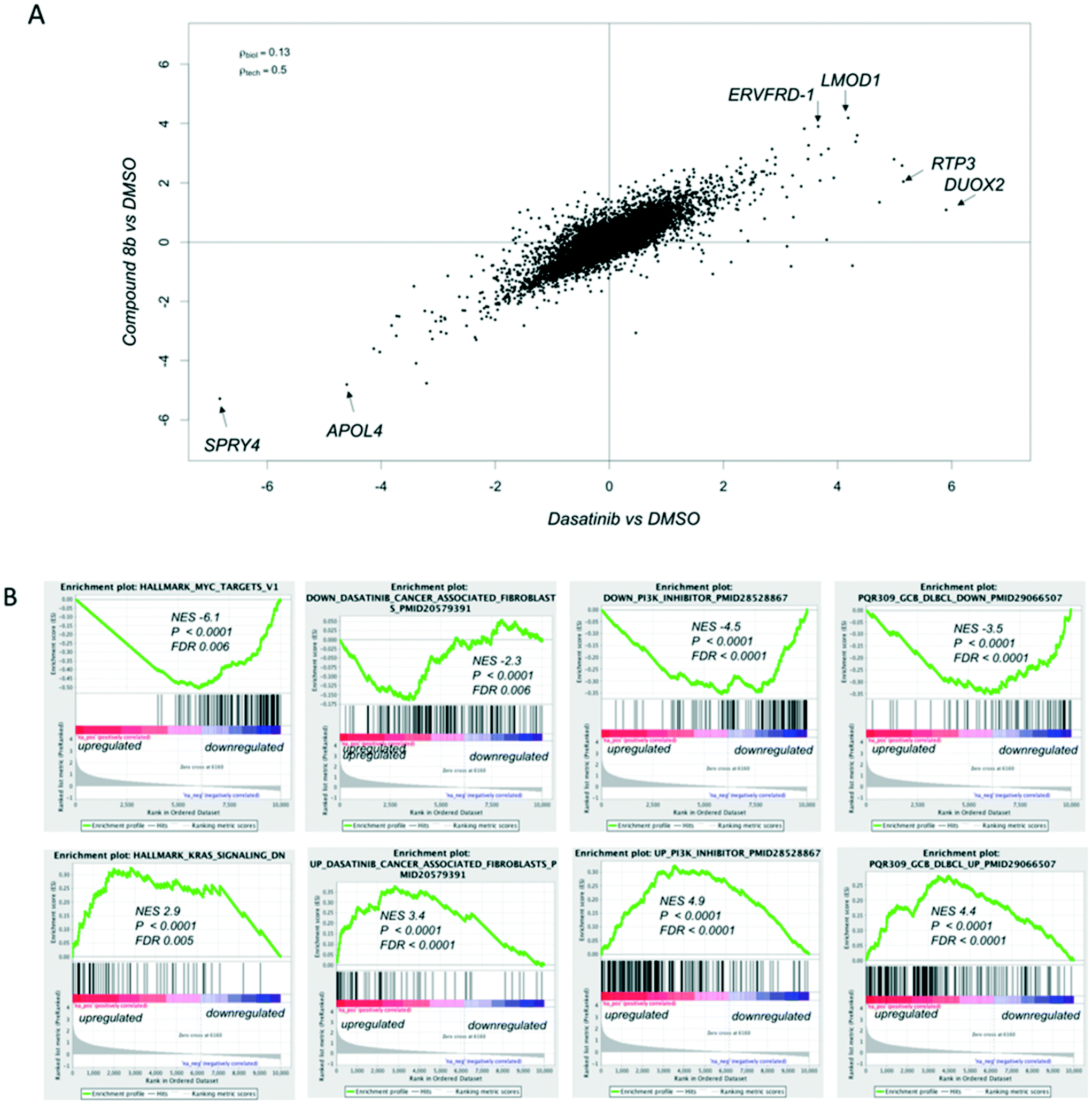 | ||
Fig. 5 Transcriptome analysis of CML K562 cells exposed to compounds 8b or to dasatinib. (A) Scatter plots by log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 fold gene expression changes in compound 8b/DMSO-treated K652 vs. dasatinib/DMSO-treated K562 cells. (B) Representative GSEA plots illustrating the transcriptional expression signature enrichment in genes downregulated (upper panel) and upregulated (lower panel) after exposure to DMSO or compound 8b in K562 cells. Green line represents the enrichment score; bars in the middle portion of the plots show where the members of the gene set appear in the ranked list of genes; positive or negative ranking metric indicate respectively correlation or inverse correlation with the profile; FDR, false discovery rate; P, probability value; NES, normalized enrichment score. 2 fold gene expression changes in compound 8b/DMSO-treated K652 vs. dasatinib/DMSO-treated K562 cells. (B) Representative GSEA plots illustrating the transcriptional expression signature enrichment in genes downregulated (upper panel) and upregulated (lower panel) after exposure to DMSO or compound 8b in K562 cells. Green line represents the enrichment score; bars in the middle portion of the plots show where the members of the gene set appear in the ranked list of genes; positive or negative ranking metric indicate respectively correlation or inverse correlation with the profile; FDR, false discovery rate; P, probability value; NES, normalized enrichment score. | ||
Chemical proteomics
We decided to perform a chemical proteomics experiment on our best quinoxaline inhibitor 8a aiming to obtain the global proteomic profiles of this molecule in an unbiased cellular setting. This approach required the synthesis of a tethered inhibitor containing a suitable functional group that would allow the attachment to a resin in a covalent fashion. Based on the X-ray structure of compound 2 (Fig. 2A) we envisioned that the introduction of a linker at the 7 position of the quinoxaline moiety would be tolerated due to its solvent accessibility. Thus, the analogue of compound 8a, bearing a primary amino group suitable for immobilization, was synthesized, affording compound 13 (Scheme 2).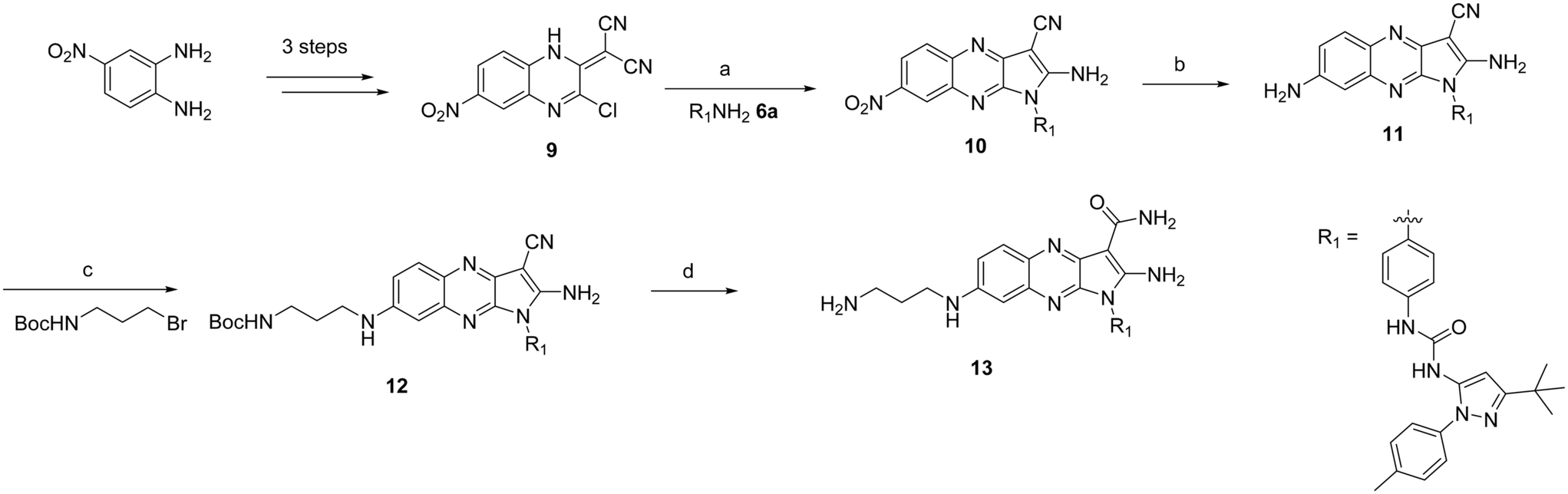 | ||
| Scheme 2 Reagents and reaction conditions: a) 6a, DMF, 80 °C, 4.5 h, 71%. b) H2, Pd/C, MeOH, rt, 4 h, 70%. c) tert-Butyl (3-bromopropyl)carbamate, KI, DIPEA, DMF, 80 °C, 6 h, 29%. d) H2SO4, 25 °C, 30 min, quantitative yield. | ||
Intermediate 9 was prepared in three steps as previously reported starting from the commercially available 4-nitrobenzene-1,2-diamine.15b,21 The substitution of the chlorine at position 3 with synthetically prepared aniline 6a (ref. 16) followed by cyclization in DMF at 80 °C afforded intermediate 10.17 The nitro group was reduced in the presence of Pd on activated charcoal and a hydrogen atmosphere yielding the desired aniline 11 in good yield, which was further reacted with tert-butyl (3-bromopropyl)carbamate to yield intermediate 12. In the final step, strong acidic conditions were employed resulting in the hydrolysis of the nitrile group and removal of the Boc protecting group in quantitative yield, affording the intermediate 13.
The inhibitory activity of 13 was then compared to its parent compound 8a by measuring the ABL2 kinase activity in a competition binding assay (Fig. 6).19 Crucially, no activity loss was observed for compound 13, making it a suitable probe for our chemical proteomics study. More importantly, the selectivity profile of compound 13 linearly correlates to its parent compound 8a as determined at 1 μM concentration in a panel of 453 kinases with an R2 value of 0.8 (Fig. 6C) by a competition assay.19
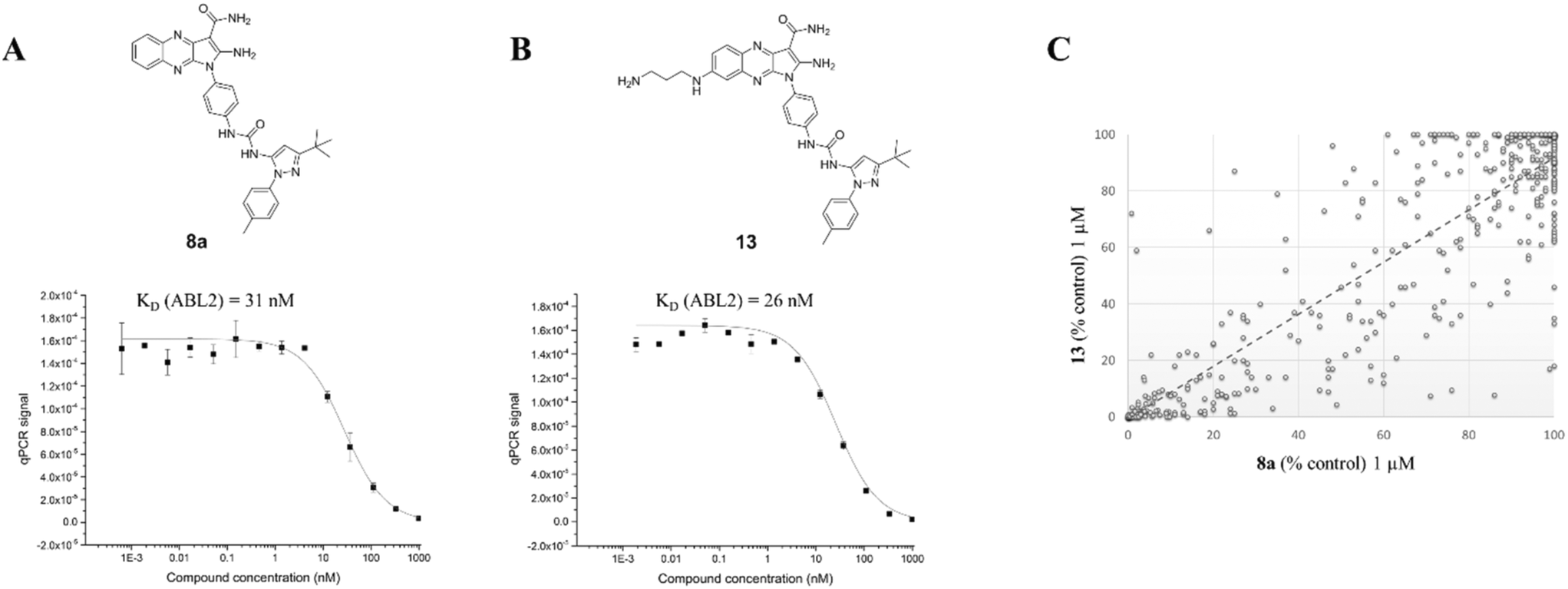 | ||
| Fig. 6 (A and B) In vitro inhibitory activity towards ABL2 kinase of compounds 8a and 13. (C) Probe compound 13 and its parent compound 8a show a strong correlation across a 453 kinase panel at 1 μM concentration, demonstrating the validity of compound 13 as a probe for chemical proteomic studies. | ||
The tethered analogue 13 was covalently bound to agarose polymer beads and incubated with the cell lysate of K562 leukemia cells (beads treated with DMSO were used as control). The beads were subsequently loaded onto spin columns, washed, and the protein binders were eluted with formic acid as previously reported.5b Among the 250 proteins identified by MS analysis as pulled down with immobilized compound 13 (Table S1†), there were 16 kinases (Table 2), five of which turned out also as targets in the selectivity profile analysis (LYN, BTK, YES, CSK and ABL2), plus PRKDC, MTOR, AGK, PI4KA (Table 2).
| Gene name | Description | Fold changea |
|---|---|---|
| a The peptide count average of the two duplicate measurements is given. | ||
| PRKDC | DNA-dependent protein kinase catalytic subunit | 6172 |
| LYN | Tyrosine-protein kinase Lyn | 1721 |
| ERLIN2 | Erlin-2 | 1620 |
| YES1 | Tyrosine-protein kinase yes | 1020 |
| BTK | Tyrosine-protein kinase BTK | 1216 |
| MTOR | Serine/threonine-protein kinase mTOR | 1116 |
| CSK | Tyrosine-protein kinase CSK | 1115 |
| AGK | Acylglycerol kinase, mitochondrial | 912 |
| CDK1 | Cyclin-dependent kinase 1 | 6.6 |
| NDKA | Nucleoside diphosphate kinase A | 6 |
| GLPK | Glycerol kinase | 5.5 |
| ABL2 | Abelson tyrosine-protein kinase 2 | 5 |
| PDXK | Pyridoxal kinase | 5 |
| NDKB | Nucleoside diphosphate kinase B | 5 |
| PI4KA | Phosphatidylinositol 4-kinase alpha | 5 |
| SQSTM1 | Sequestosome-1 | 5 |
Four of the major interactors of quinoxaline 13, LYN, BTK, CSK and YES1, are nanomolar binders already identified in the in vitro selectivity profile (Fig. 3). LYN, YES1, BTK and CSK are inhibited to 0.5, 1.9, 3.3 and 0.65% control respectively by our small molecule 13 (0.85, 9.6, 0.95 and 0.9% control, by the parent compound 8a respectively) at 1 μM concentration. The 250 human proteins belonged to pathways related to protein secretion, oxidative phosphorylation, DNA repair, MYC and E2F targets, IL6/JAK/STAT3 signaling, glycolysis, mitotic spindle, and mTORC1 signaling.
Inhibitors 8a and 8b in preclinical models of lymphoma
The engagement of target kinases LYN, BTK and mTOR by quinoxaline inhibitors demonstrated by the proteomics data prompted the investigation of their activity against lymphomas (not included in the NCI-60 panel), for which these three kinases are highly relevant. Anti-proliferative activity of 8a and 8b was assessed, in parallel to dasatinib, in eight B-cell lymphoma cell lines derived from mantle cell lymphoma (MCL) (Jeko1, MAVER1), chronic lymphocytic leukemia (MEC-1, PCL-12) and diffuse large B cell lymphoma (DLBCL) of the activated B cell like (ABC) (RI-1, SU-DHL-2) or of the germinal center B cell (GCB) (VAL, SU-DHL-4). Compounds were active with median GI50 of 600 nM (95% C.I. 300–1065 nM) and 250 nM (95% C.I. 100–1049 nM), respectively and their anti-tumor activity appeared more potent than dasatinib (median IC50 = 1325 nM, 95% C.I. 100–16625 nM), in particular in four of the cell lines (MAVER1, SU-DHL-2, VAL and MEC1) (Fig. 7A).
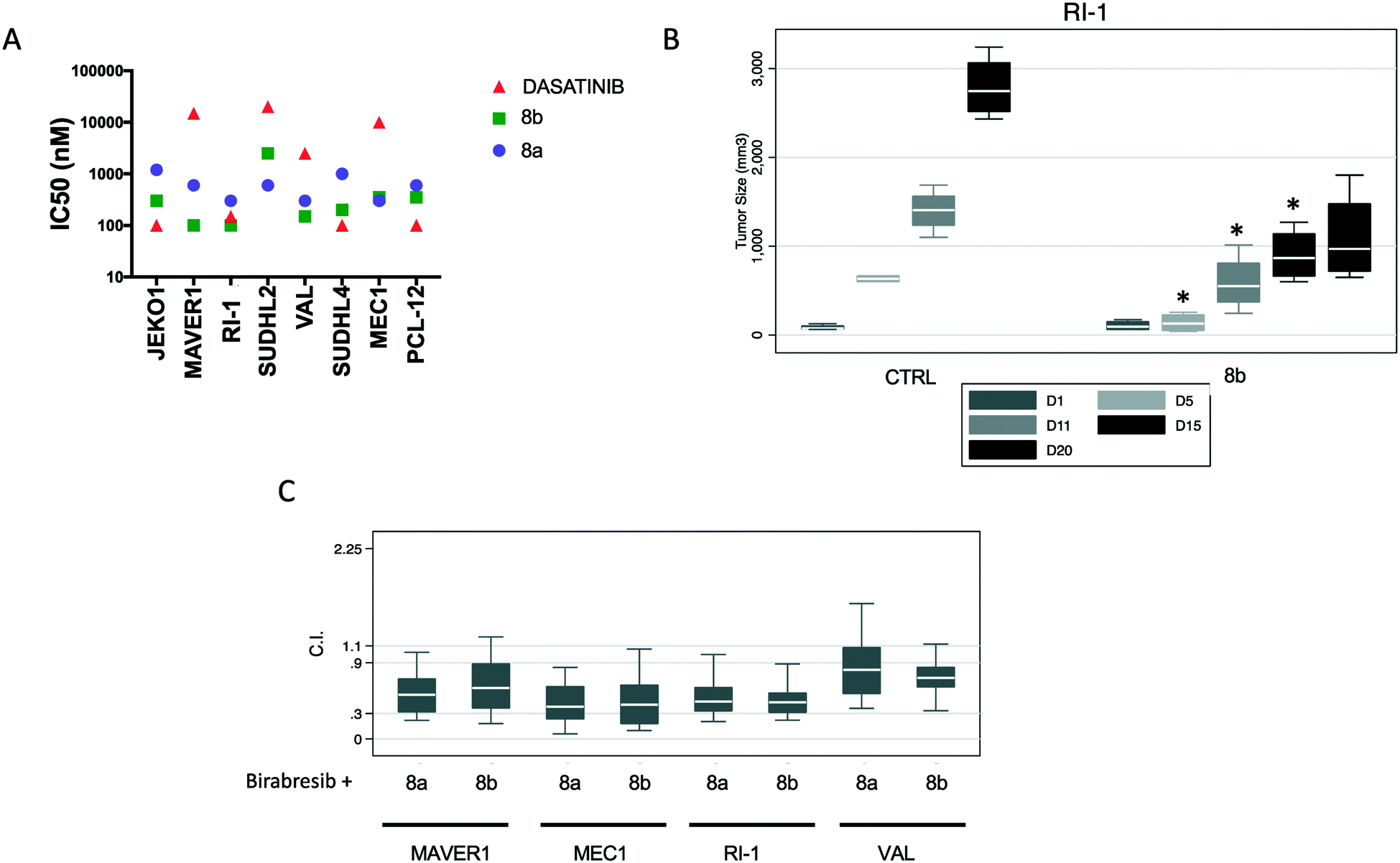 | ||
| Fig. 7 Anti-tumor activity of compounds 8a and 8b in lymphoma models. (A) Eight B-cell lymphoma cell lines derived from mantle cell lymphoma (MCL) (Jeko1, MAVER1), chronic lymphocytic leukemia (MEC-1, PCL-12) and diffuse large B cell lymphoma (DLBCL) of the activated B cell like (ABC) (RI-1, SU-DHL-2) or of the germinal center B cell (GCB) (VAL, SU-DHL-4) were exposed to increased concentration of 8b or 8a for 72 h. (B) Effects of 8b in a xenograft model of ABC-DLBCL. NOD-Scid mice subcutaneously inoculated with RI-1 cells (15 × 10 (ref. 6)) were split in three groups respectively treated with 8a (100 mg kg−1, IP, n = 4, data not shown), 8b (100 mg kg−1, IP, n = 4) and control vehicle (n = 4). Y-Axis, tumor volume in mm3. X-Axis, days (D) of treatment. For compound 8bvs. CTRL, D5-D15, p < 0.05. In each box-plot, the line in the middle of the box represents the median and the box extends from the 25th to the 75th percentile (interquartile range, IQ); the whiskers extend to the upper and lower adjacent values (i.e., ±1.5 IQ); outside values have been omitted from the figure. (C) Values of the combination index (C.I.) of 8a or 8b with birabresib in ABC-DLBCL and MCL cell lines. | ||
The anti-lymphoma activity of compounds 8a and 8b was then assessed using a mouse xenograft model with the ABC DLBCL RI-1 cell line. Compound 8a turned out to be very toxic already after the first dose, due to swelling of the area surrounding the injection and body-weight loss, and had to be stopped although the body weight started to recover after two weeks without treatment. Inhibitor 8b was administered once every 5 days and was well tolerated with no general toxicity or body-weight loss. After two weeks of treatment, xenografts treated with 8b were three times smaller than the control group or than the group treated with the initial single dose of 8a at the end of the experiment (day 15) (Fig. 7B).
Dasatinib as well as inhibitors of BTK or mTOR are synergistic when combined with BET bromodomain inhibitors.22 Thus, we combined compounds 8a and 8b with birabresib (MK-8628/OTX015), a BET bromodomain inhibitor with preclinical and early clinical anti-lymphoma activity.22b,23 Synergism, i.e., combination index (CI) values smaller than 1.0, was observed with the combination of compound 8a or 8b with birabresib in all the four cell lines that were studied (Fig. 7C).
Cellular angiogenesis assay
The implication of numerous kinases in angiogenesis24 led us to examine the efficacy of our inhibitors on tube formation via an in vitro matrigel angiogenesis assay. Angiogenesis, the process of vascular growth by sprouting from pre-existing vessels, is a key player during tumor growth and metastasis in the context of several diseases including cancer, macular degeneration, diabetic retinopathy and arthritis.3a A remarkable effect was observed for the novel quinoxaline 8a (65, 90, and 100% inhibition of tube length, total branching points and total loops respectively, Fig. 8) in comparison to our previously developed inhibitors 1 (9, 23 and 21% inhibition of tube length, total branching points and total loops respectively) and 2 (5, 9 and 13% respectively).9 We hypothesize that the promising angiogenesis inhibition results obtained for compound 8a could be due to its ability to inhibit several CDK kinases (in contrast to compound 1, Fig. 3), as CDK inhibitors have been reported to block angiogenesis in vitro and in vivo.25 Further, these results are in line with those previously reported for dasatinib and related TKI inhibitors.26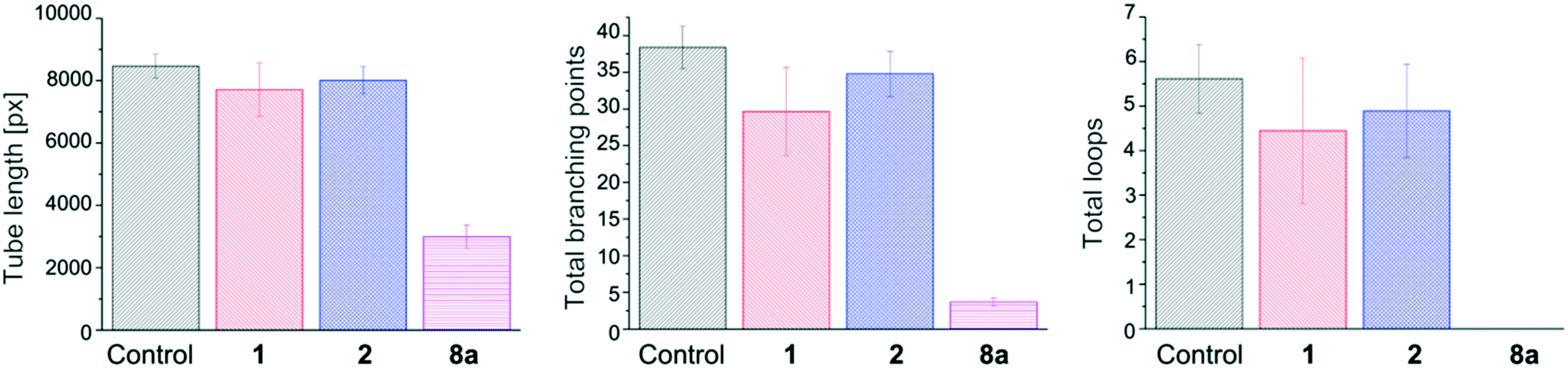 | ||
| Fig. 8 In vitro angiogenesis assay on tube formation. Compounds were incubated with human umbilical vein endothelial cells (HUVEC) cells seeded on matrigel for 15 h. Tube length (left), total branching points (middle) and total loops (right) were quantified. All measurements were done at least in triplicate, and their SEM values are indicated as error bars. | ||
Conclusions
Two quinoxaline-containing chimeric kinase inhibitors 8a and 8b were designed based on the available X-ray structures of EphA3-inhibitor complexes through compound merging. They inhibit mainly tyrosine kinases and show broader spectrum of activity than the parent compound 1, as well as high anti-proliferative activity against several cancer cell lines, as demonstrated in the NCI-60 cell line panel screening. Immortalised myelogenous leukemia cell line K562 was shown to be particularly sensitive and was selected for further investigations to elucidate the mode of action of compounds 8a–b. Transcriptomic profiling revealed orthogonal effects of compounds 1 and 8b on gene expression, with the latter closely resembling the effect profile of dasatinib, the SRC-family protein tyrosine kinase inhibitor. By conducting a chemical proteomics study we identified 17 kinases as potential binders of compounds 8a, including the known dasatinib targets LYN, BTK, CSK and YES1. Together, these studies have inspired two possible applications of the developed chimeric compounds. Demonstrated ability of our inhibitors to inhibit the CDKs prompted us to investigate effect of compound 8a on inhibiting angiogenesis in vitro, showing remarkable improvement compared to the parent compound 1. In turn, their engagement with the kinases LYN, BTK and mTOR, which are relevant in lymphomas, triggered in vitro and in vivo studies on compound 8a–b in various lymphoma models. Both compounds have shown submicromolar IC50 values in five lymphoma cell lines. A significant decrease in tumor size is observed in an RI-1 lymphoma xenograft mouse model at a dose of 100 mg kg−1 of compound 8b, without any associated mean body weight loss. These results indicate promise for therapeutic use of developed inhibitors in vivo. Further evaluation of compound 8b in a xenograft mice model established with a more responsive cell line than the RI-1, such as the K562 human chronic myeloid leukemia cell line, might demonstrate even greater extent of tumor size control.Accession codes
PDB codes for EphA3 in complex with the inhibitors 2 and 3 are 4P5Z and 4TWN, respectively.Author contributions
CN, AC, FB and SZ conceived and designed the experiments. Experimental work was conducted by AU, CJ-T, DS, EG, CT, JD, JL, EB, GS, LC and PS. Crystallographic and computational work was conducted by PS, HZ and JD. The manuscript was written and edited by CN, AC, FB, PS, AU. CJ-T and EG.Ethical statement
Mice maintenance and animal experiments were performed under institutional guidelines established for the Animal Facility at The Institute of Research in Biomedicine (IRB, Bellinzona, CH). and with study protocols approved by the local Cantonal Veterinary Authority (No. TI-20-2015).Abbreviations
| Eph | Erythropoietin-producing human hepatocellular carcinoma receptor |
| DFG | Aspartate-phenylalanine-glycine |
| ATP | Adenosine triphosphate |
| SPR | Surface plasmon resonance |
| DMF | Dimethylformamide |
| TBTU | N,N,N′,N′-Tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate |
| DIPEA | Diisoproylethylamine |
| DCM | Dichloromethane |
| THF | Tetrahydrofuran |
| HOBt | Hydroxybenzotriazole |
| EDC | N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide |
| FRET | Fluorescence-resonance energy transfer |
| Abl | Abelson murine leukemia viral oncogene homologue |
| EGFR | Epidermal growth factor receptor |
| DDR | Discoidin domain receptor |
| Lck | Lymphocyte-specific kinase |
| CDK | Cyclin dependent kinase |
| DMSO | Dimethyl sulfoxide |
| MEF | Mouse embryonic fibroblasts |
| VEGF | Vascular endothelial growth factor |
| HUVEC | Human umbilical vein endothelial cells |
| NCI | National Cancer Institute |
| DTP | Developmental Therapeutics Program |
| IP | Intraperitoneal |
Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank Dr. Erich Brunner for interesting discussions regarding the chemical proteomic experiments. We also acknowledge and thank the NIH/National Cancer Institute for in vitro and in vivo evaluations of compounds under the Developmental Therapeutics Program (DTP), and in particular Dr. Stephen L. White of the NIH/NCI Drug Synthesis & Chemistry Branch for information and discussions relating to the in vivo studies. The authors would like to thank the Swiss National Science Foundation and the Swiss Cancer League (Krebsliga) for financial support (to AC and CN) and the Gelu Foundation (to FB). PŚ is a recipient of UZH Entrepreneur Fellowship in Biotechnology.References
- R. E. Hubbard, Mol. BioSyst., 2005, 1, 391 Search PubMed.
- Editorial article: Mechanism matters, Nat. Med., 2010, 16, 347, DOI:10.1038/nm0410-347.
- (a) J. Lamb, E. D. Crawford, D. Peck, J. W. Modell, I. C. Blat, M. J. Wrobel, J. Lerner, J. P. Brunet, A. Subramanian, K. N. Ross, M. Reich, H. Hieronymus, G. Wei, S. A. Armstrong, S. J. Haggarty, P. A. Clemons, R. Wei, S. A. Carr, E. S. Lander and T. R. Golub, Science, 2006, 313, 1929 CrossRef CAS; (b) J. Lamb, Nat. Rev. Cancer, 2007, 7, 54 CrossRef CAS.
- U. Rix, O. Hantschel, G. Duernberger, L. L. R. Rix, M. Planyavsky, N. V. Fernbach, I. Kaupe, K. L. Bennett, P. Valent, J. Colinge, T. Kocher and G. Superti-Furga, Blood, 2007, 110, 4055 CrossRef CAS.
- (a) L. L. R. Rix, U. Rix, J. Colinge, O. Hantschel, K. L. Bennett, T. Stranzl, A. Muller, C. Baumgartner, P. Valent, M. Augustin, J. H. Till and G. Superti-Furga, Leukemia, 2009, 23, 477 CrossRef PubMed; (b) N. V. Fernbach, M. Planyavsky, A. Muller, F. P. Breitwieser, J. Colinge, U. Rix and K. L. Bennett, J. Proteome Res., 2009, 8, 4753 CrossRef CAS PubMed.
- (a) G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912 CrossRef CAS; (b) P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309 CrossRef CAS PubMed.
- (a) R. Li and J. A. Stafford, Kinase Inhibitor Drugs, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009 CrossRef; (b) J. Blanc, R. Geney and C. Menet, Anti-Cancer Agents Med. Chem., 2013, 13, 731 CrossRef CAS PubMed; (c) A. Kontzias, A. Kotlyar, A. Laurence, P. Changelian and J. J. O'Shea, Curr. Opin. Pharmacol., 2012, 12, 464 CrossRef CAS PubMed; (d) Z. O'Brien and M. F. Moghaddam, Expert Opin. Drug Metab. Toxicol., 2013, 9, 1597 CrossRef; (e) F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353 CrossRef CAS.
- (a) K. Lafleur, D. Huang, T. Zhou, A. Caflisch and C. Nevado, J. Med. Chem., 2009, 52, 6433 CrossRef CAS PubMed; (b) H. T. Zhao, J. Dong, K. Lafleur, C. Nevado and A. Caflisch, ACS Med. Chem. Lett., 2012, 3, 834 CrossRef CAS PubMed; (c) K. Lafleur, J. Dong, D. Huang, A. Caflisch and C. Nevado, J. Med. Chem., 2013, 56, 84 CrossRef CAS PubMed; (d) A. Unzue, J. Dong, K. Lafleur, H. T. Zhao, E. Frugier, A. Caflisch and C. Nevado, J. Med. Chem., 2014, 57, 6834 CrossRef CAS PubMed; (e) M. Xu, A. Unzue, J. Dong, D. Spiliotopoulos, C. Nevado and A. Caflisch, J. Med. Chem., 2016, 59, 1340 CrossRef CAS PubMed.
- A. Unzue, K. Lafleur, H. Zhao, T. Zhou, J. Dong, P. Kolb, J. Liebl, S. Zahler, A. Caflisch and C. Nevado, Eur. J. Med. Chem., 2016, 112, 347 CrossRef CAS PubMed.
- M. Brave, V. Goodman, E. Kaminskas, A. Farrell, W. Timmer, S. Pope, R. Harapanhalli, H. Saber, D. Morse, J. Bullock, A. Men, C. Noory, R. Ramchandani, L. Kenna, B. Booth, J. Gobburu, X. Jiang, R. Sridhara, R. Justice and R. Pazdur, Clin. Cancer Res., 2008, 14, 352 CrossRef CAS.
- J. Dong, H. T. Zhao, T. Zhou, D. Spiliotopoulos, C. Rajendran, X. D. Li, D. Z. Huang and A. Caflisch, ACS Med. Chem. Lett., 2015, 6, 79 CrossRef CAS PubMed.
- (a) C. Pargellis, L. Tong, L. Churchill, P. F. Cirillo, T. Gilmore, A. G. Graham, P. M. Grob, E. R. Hickey, N. Moss, S. Pav and J. Regan, Nat. Struct. Biol., 2002, 9, 268 CrossRef CAS PubMed; (b) J. Regan, S. Breitfelder, P. Cirillo, T. Gilmore, A. G. Graham, E. Hickey, B. Klaus, J. Madwed, M. Moriak, N. Moss, C. Pargellis, S. Pav, A. Proto, A. Swinamer, L. Tong and C. Torcellini, J. Med. Chem., 2002, 45, 2994 CrossRef CAS.
- W. W. Chan, S. C. Wise, M. D. Kaufman, Y. M. Ahn, C. L. Ensinger, T. Haack, M. M. Hood, J. Jones, J. W. Lord, W. P. Lu, D. Miller, W. C. Patt, B. D. Smith, P. A. Petillo, T. J. Rutkoski, H. Telikepalli, L. Vogeti, T. Yao, L. Chun, R. Clark, P. Evangelista, L. C. Gavrilescu, K. Lazarides, V. M. Zaleskas, L. J. Stewart, R. A. Van Etten and D. L. Flynn, Cancer Cell, 2011, 19, 556 CrossRef CAS PubMed.
- https://clinicaltrials.gov/ was accessed on the 21st of March 2016.
- (a) E. F. Pratt and J. Kereszte, J. Org. Chem., 1967, 32, 49 CrossRef CAS; (b) C. A. Obafemi and W. Pfleiderer, Molecules, 2004, 9, 223 CrossRef CAS.
- For further details see the ESI†.
- H. Otomasu, S. Ohmiya, T. Sekuguch and H. Takahash, Chem. Pharm. Bull., 1970, 18, 2065 CrossRef CAS.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212 CrossRef CAS PubMed.
- M. A. Fabian, W. H. Biggs, 3rd, D. K. Treiber, C. E. Atteridge, M. D. Azimioara, M. G. Benedetti, T. A. Carter, P. Ciceri, P. T. Edeen, M. Floyd, J. M. Ford, M. Galvin, J. L. Gerlach, R. M. Grotzfeld, S. Herrgard, D. E. Insko, M. A. Insko, A. G. Lai, J. M. Lelias, S. A. Mehta, Z. V. Milanov, A. M. Velasco, L. M. Wodicka, H. K. Patel, P. P. Zarrinkar and D. J. Lockhart, Nat. Biotechnol., 2005, 23, 329 CrossRef CAS PubMed.
- M. Malumbres, E. Harlow, T. Hunt, T. Hunter, J. M. Lahti, G. Manning, D. O. Morgan, L. H. Tsai and D. J. Wolgemuth, Nat. Cell Biol., 2009, 11, 1275 CrossRef CAS PubMed.
- J. Deng, E. Feng, S. Ma, Y. Zhang, X. Liu, H. Li, H. Huang, J. Zhu, W. Zhu, X. Shen, L. Miao, H. Liu, H. Jiang and J. Li, J. Med. Chem., 2011, 54, 4508 CrossRef CAS PubMed.
- (a) C. Xu, K. A. Buczkowski, Y. Zhang, H. Asahina, E. M. Beauchamp, H. Terai, Y. Y. Li, M. Meyerson, K. K. Wong and P. S. Hammerman, Mol. Cancer Ther., 2015, 14, 2382 CrossRef CAS PubMed; (b) M. Boi, E. Gaudio, P. Bonetti, I. Kwee, E. Bernasconi, C. Tarantelli, A. Rinaldi, M. Testoni, L. Cascione, M. Ponzoni, A. A. Mensah, A. Stathis, G. Stussi, M. E. Riveiro, P. Herait, G. Inghirami, E. Cvitkovic, E. Zucca and F. Bertoni, Clin. Cancer Res., 2015, 21, 1628 CrossRef CAS PubMed; (c) A. Stathis and F. Bertoni, Cancer Discovery, 2018, 8, 24 CrossRef CAS PubMed.
- S. Amorim, A. Stathis, M. Gleeson, S. Iyengar, V. Magarotto, X. Leleu, F. Morschhauser, L. Karlin, F. Broussais, K. Rezai, P. Herait, C. Kahatt, F. Lokiec, G. Salles, T. Facon, A. Palumbo, D. Cunningham, E. Zucca and C. Thieblemont, Lancet Haematol., 2016, 3, e196 CrossRef.
- (a) K. J. Gotink and H. M. W. Verheul, Angiogenesis, 2010, 13, 1 CrossRef CAS PubMed; (b) R. H. Adams, Semin. Cell Dev. Biol., 2002, 13, 55 CrossRef CAS PubMed; (c) M. Jeltsch, V. M. Leppanen, P. Saharinen and K. Alitalo, Cold Spring Harbor Perspect. Biol., 2013, 5 Search PubMed.
- (a) J. Liebl, V. Krystof, G. Vereb, L. Takacs, M. Strnad, P. Pechan, L. Havlicek, M. Zatloukal, R. Furst, A. M. Vollmar and S. Zahler, Angiogenesis, 2011, 14, 281 CrossRef CAS PubMed; (b) J. Liebl, S. B. Weitensteiner, G. Vereb, L. Takacs, R. Furst, A. M. Vollmar and S. Zahler, J. Biol. Chem., 2010, 285, 35932 CrossRef CAS PubMed.
- A. Gover-Proaktora, G. Granota, M. Pasmanik-Chor, O. Pasvolsky, S. Shapira, O. Raza, P. Raananic and A. Leader, Leuk. Lymphoma, 2018, 60(1), 189 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures for the synthesis, characterization and biological evaluation of all reported compounds can be found in the ESI. See DOI: 10.1039/d0md00049c |
| This journal is © The Royal Society of Chemistry 2020 |