
Structure-based design and analysis of SuFEx chemical probes
Lyn H.
Jones
 *a and
Jeffery W.
Kelly
b
*a and
Jeffery W.
Kelly
b
aCenter for Protein Degradation, Dana-Farber Cancer Institute, 360 Longwood Avenue, Boston, MA 02215, USA. E-mail: lyn_jones@dfci.harvard.edu
bDepartments of Chemistry and Molecular Medicine, The Scripps Research Institute, La Jolla, CA, USA
First published on 6th January 2020
Abstract
The discerning reactivity of sulfur(VI)-fluoride exchange (SuFEx) chemistry has enabled the context-specific labeling of protein binding sites by chemical probes that incorporate these versatile warheads. Emerging information from protein-probe structures and proteomic mapping experiments is helping advance our understanding of the protein microenvironment that dictates the reactivity of targetable amino acid residues. This review explores these new findings that should influence the future rational design of SuFEx probes for a multitude of applications in chemical biology and drug discovery.
General introduction
Covalent engagement of protein binding sites by small molecules armed with electrophilic warheads has led to the development of irreversible inhibitor drugs and chemical probes with improved pharmacological duration and selectivity. Most covalent inhibitors incorporate cysteine-reactive electrophiles, such as acrylamides, because cysteine is the most nucleophilic residue and its reactivity is often enhanced in binding sites through neighboring amino acids that lower the thiol pKa (stabilization of the negatively charged thiolate).1 Unfortunately, cysteine is of low frequency in small molecule binding sites, and thus often is not available for targeting.2 Novel chemistries are required to enable the development of drugs and probes that engage residues beyond cysteine.3In the 1960s Fahrney and Gold discovered that sulfonyl fluoride containing probes react with the catalytic serine in protease enzymes.4,5 Subsequently, Baker leveraged the reactivity of sulfonyl fluorides by converting reversible ligands for several enzymes into irreversible covalent inhibitors through sulfonyl fluoride incorporation.6–11 Colman similarly used the warhead to develop covalent adenosine probes that mapped reactive nucleophilic amino acids in ATP binding sites in proteins.12,13 In fact, research using sulfonyl fluorides fortuitously discovered that the electrophile could target several nucleophilic residues in protein binding sites, including tyrosine, lysine, serine and threonine.14,15 Aryl sulfonyl fluorides in particular possess sufficient aqueous stability and binding site dependent amino acid reactivity that makes the weak electrophile ideal for applications in chemical biology and covalent chemical probe development.14 One reason for the site-specific labeling could be that hydrogen-bonding to the fluorine leaving group is required for enhancing the electrophilicity of the sulfur atom.15 Additionally, a recent crystal structure of a sulfonyl fluoride fragment inhibitor with human neutrophil elastase revealed hydrogen-bonding interactions between a proximal histidine residue and the sulfonyl oxygen atom that presumably further enhances sulfur electrophilicity and anchors the ligand to facilitate targeted serine adduction.16 Aryl fluorosulfates possess attenuated reactivity compared to their corresponding sulfonyl fluoride congeners and have also started to receive attention in chemical biology and medicinal chemistry covalent probe design.17
This review explores SuFEx chemical probes from the perspective of the microenvironment of the binding site they engage and how this influences amino acid reactivity. Rationally designed SuFEx covalent inhibitors have started to appear in recent years and an improved understanding of the protein structural features that increase the targetable nature of amino acids beyond cysteine will likely have considerable impact in future chemical probe design.
Rational design of sulfonyl fluoride probes
a) Lysine targeting
An early report of rational development of SuFEx chemical probes described covalent stabilizers of transthyretin (TTR), that have therapeutic utility by preventing aggregation known to cause polyneuropathy and cardiomyopathy. Structure-based design of sulfonyl fluoride-containing ligands recognizing the thyroxin binding site were developed (e.g.1, BAOD-mSF, Fig. 1a) to engage lysine at position 15 in the protein, which is pKa-perturbed due to the presence of a neighbouring Lys-15′ residue.18 The importance of appropriately positioning the electrophile to target the nucleophilic lysine was highlighted by the lack of TTR conjugation with the para-SF isomer. This result demonstrates the significance of the templated reactivity of the sulfonyl fluoride warhead, that differs from non-specific reactivity observed for more reactive and less discerning electrophiles.3 The structural information gleaned in this study (PDB 4FI6, Fig. 1) suggested that the proton of the Lys15-Glu54 salt bridge may activate the SF electrophile by hydrogen-bonding to the leaving fluoride ion.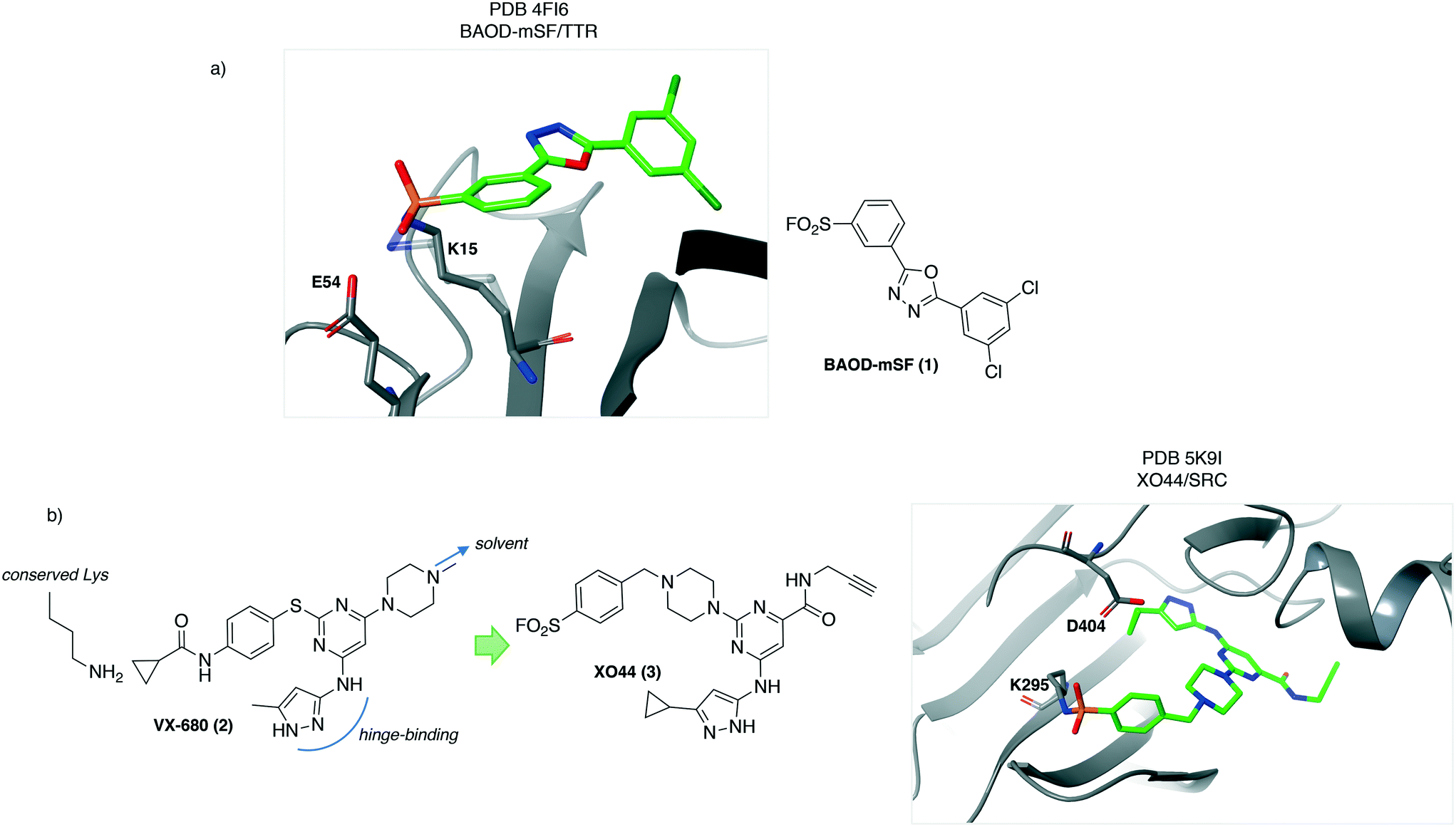 | ||
| Fig. 1 a) Crystal structure of the sulfonyl fluoride probe BAOD-mSF (1) with transthyretin. b) Design of the alkyne-containing, covalent kinase probe XO44 (3) based on the promiscuous inhibitor VX-680 (2) and a crystal structure of XO44 with SRC kinase. | ||
Lysine targeting has also enabled the development of activity-based probes. Kinase ATP binding sites are very similar across the family, making the design of selective inhibitors challenging. Chemoproteomic technologies have emerged to measure kinase inhibitor selectivity in lysate.19,20 Inspired by the discovery that the sulfonyl fluoride ATP-probe FSBA12,21 is a covalent inhibitor of kinases through labeling of the conserved lysine residue,22 a cell-permeable kinase probe was developed to measure ATP-site occupancy in live cells for the first time.23 The crystal structure of the promiscuous kinase inhibitor VX-680 (2) with AurA24 was chosen as the starting point for the design of a covalent probe bearing the SF warhead. The tri-substituted pyrimidine core allowed for correct positioning of the hinge-binding motif, the SF electrophile to target the conserved lysine and an alkyne reporter functionality (Fig. 1b). The latter was installed in a region directed towards solvent so as not to clash with kinase binding. This work yielded XO44 (3, Fig. 1b) which was used to measure the selectivity of kinase inhibitors such as dasatinib in cells using competition-based chemoproteomics. The salt bridge formed between the conserved lysine (K295) and the aspartate residue D404 could facilitate lysine reactivity in SRC (PDB 5K9I, Fig. 1b).25 The crystal structure also reveals hydrogen-bonding from the backbone N–H of Phe278 to the sulfonyl oxygen atom that may further enhance the sulfonylation reaction.
In a related study, a sulfonyl fluoride probe based on the EGFR inhibitor erlotinib was designed to target the conserved lysine in the kinase in a site-specific manner showing that the approach is a general strategy to develop functionalized covalent chemical probes.26
b) Tyrosine targeting
The mRNA decapping scavenger enzyme DcpS was identified as the putative target of diaminoquinazoline (DAQ) compounds that were shown to be active in phenotypic models of spinal muscular atrophy. To validate DcpS as the relevant target of DAQs in human primary cells, a covalent occupancy biomarker probe containing a click handle was developed.27 A cocrystal structure with a reversible DAQ inhibitor showed an absence of cysteines in the binding pocket.28 However, proximal tyrosine residues appeared to be targetable using the sulfonyl fluoride warhead. The electrophile was rationally installed into the DAQ template to engage Tyr113 (4, SF-o1 and 5, SF-m1) and Tyr143 (6, SF-p1), and specific protein conjugation was confirmed by peptide mapping and X-ray co-crystallization studies (PDB codes 4QDE, 4QEB and 4QDV respectively, Fig. 2). These were the first examples of rationally targeting tyrosine residues in a protein binding site. Tyrosine reactivity appeared to be enhanced by neighbouring basic residues that facilitate phenol-OH deprotonation (His139 and Lys142 are proximal to Tyr113 and Tyr143 respectively, Fig. 2). Additionally, hydrogen bonding from sulfonyl oxygen atoms to neighboring tyrosine phenols may enhance SuFEx reactivity. The SF-p1-DcpS structure revealed a near-by small pocket that could accommodate a terminal alkyne motif suitable for copper(I)-mediated azide alkyne cycloaddition (or ‘click’) chemistry.29,30 The resulting probe SF-p1-yne (7, Fig. 2) was used to label DcpS in live cells, and subsequent lysis enabled bioorthogonal click chemistry with biotin-azide, allowing for target enrichment and analysis by Western blot. Pre-incubation of cells with the parent inhibitor competed labeling and isolation of DcpS by SF-p1-yne in a dose-responsive manner, permitting a quantitative assessment of target occupancy. The technology enabled further pharmacokinetic–pharmacodynamic studies that led to the development of inhibitors with improved drug-like properties.31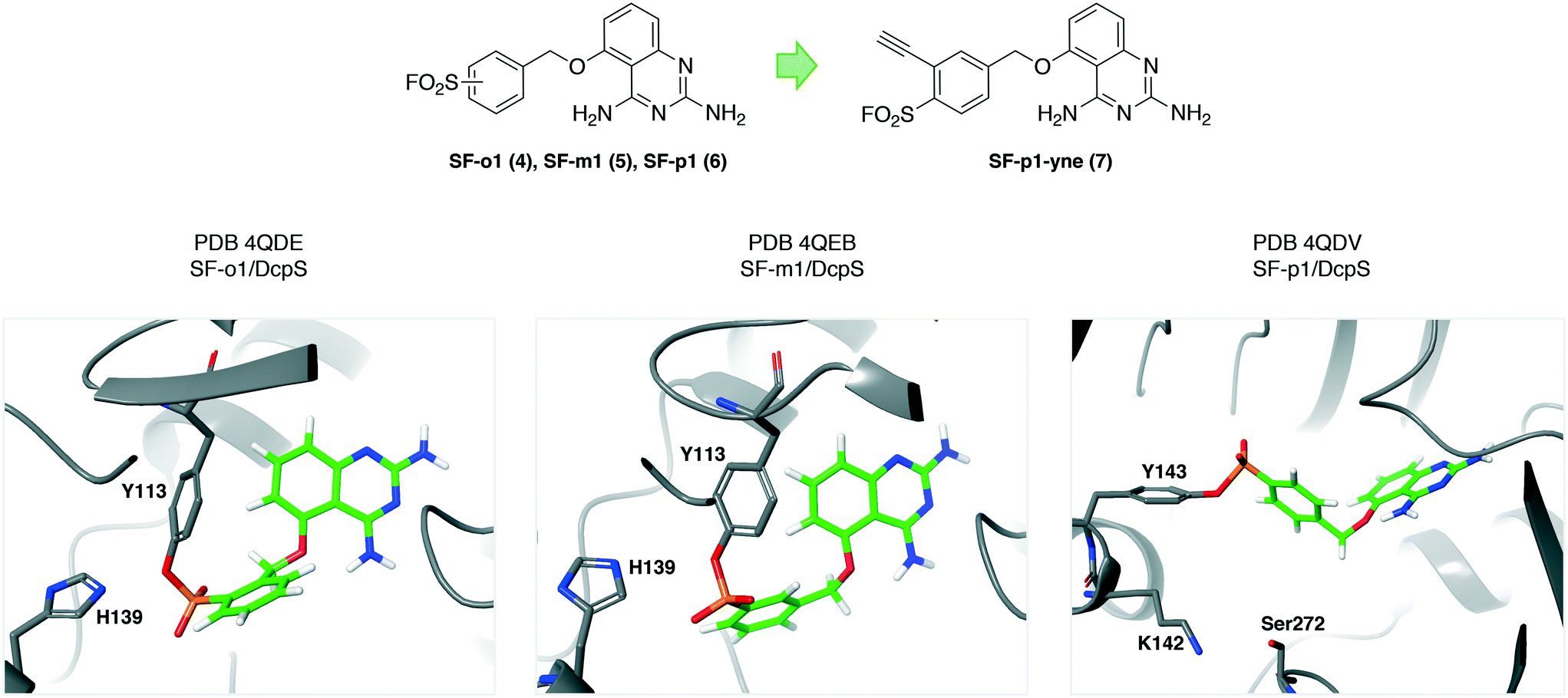 | ||
| Fig. 2 Sulfonyl fluoride covalent inhibitors of the mRNA decapping scavenger enzyme DcpS. These were the first compounds rationally designed to target specific tyrosine residues in proteins.27,32 Proximal basic amino acid residues (histidine and lysine) likely enhance tyrosine reactivity. This work led to the development of an occupancy biomarker probe SF-p1-yne (7) that was used in key preclinical pharmacokinetic–pharmacodynamic studies.31,33 | ||
Similarly, a sulfonyl fluoride covalent inhibitor of the splicing kinase SRPK1 was developed through rational targeting of an ATP-site tyrosine.34 The ALK inhibitor drug alectinib was shown to also target SRPK1 and a cocrystal structure highlighted the proximity of Tyr227 to the morpholine group of the ligand (PDB 5XV7, Fig. 3). Although not reported, the neighboring hydrophobic leucine residues (Fig. 3) may create a microenvironment that enhances the reactivity of the tyrosine hydroxy functionality.35 Peptide mapping mass spectrometry confirmed adduct formation of the probe SRPKIN-1 (8, Fig. 3) with Tyr227 of SRPK1. SRPKIN-1 possessed adequate properties to be able to potently block angiogenesis in a mouse model of age-related macular degeneration (dose-dependent effects observed at 50 and 300 nM).
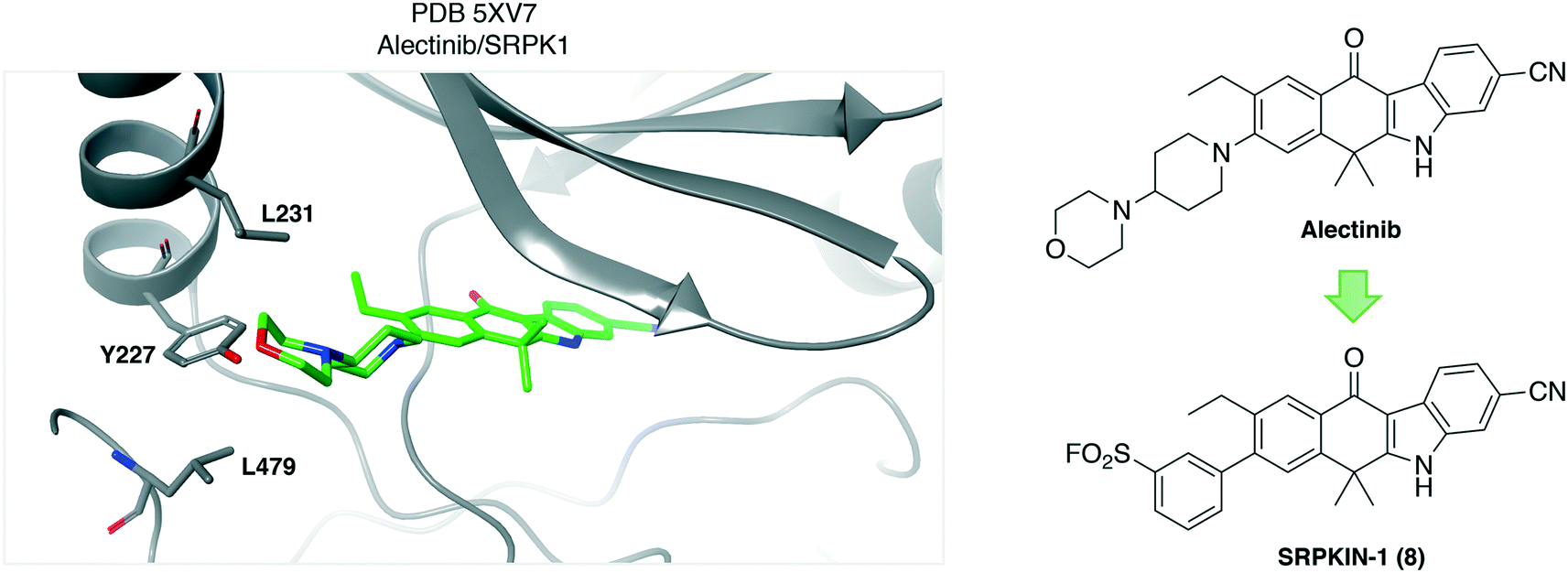 | ||
| Fig. 3 Crystal structure of alectinib with the kinase SRPK1 showing the proximity of Tyr227 to the ligand. Neighboring leucine residues may enhance the reactivity of the tyrosine OH functionality. | ||
Protein–protein interactions
Protein–protein interactions (PPIs) are challenging targets for drug discovery due to the large and flat interfacial surface. Often, inhibitors of PPIs are peptidic in nature as they mimic the natural interaction.36 A linear peptidic inhibitor (9) of the interaction between the proinflammatory interleukin-17A (IL-17A) and its receptor (IL-17RA) was identified previously and converted into a sulfonyl fluoride probe (10) in order to identify the binding site (Fig. 4).37 Peptide 10 was shown to covalently inhibit the protein–protein interaction by FRET, and peptide mapping experiments confirmed engagement of Tyr85 in IL-17A. Docking of the probe into the binding site, and crystallography studies of 9 with IL-17A showed that the linear peptide adopted a U shape in the binding site. These studies enabled structure-based design of macrocycle 11 with significantly enhanced potency.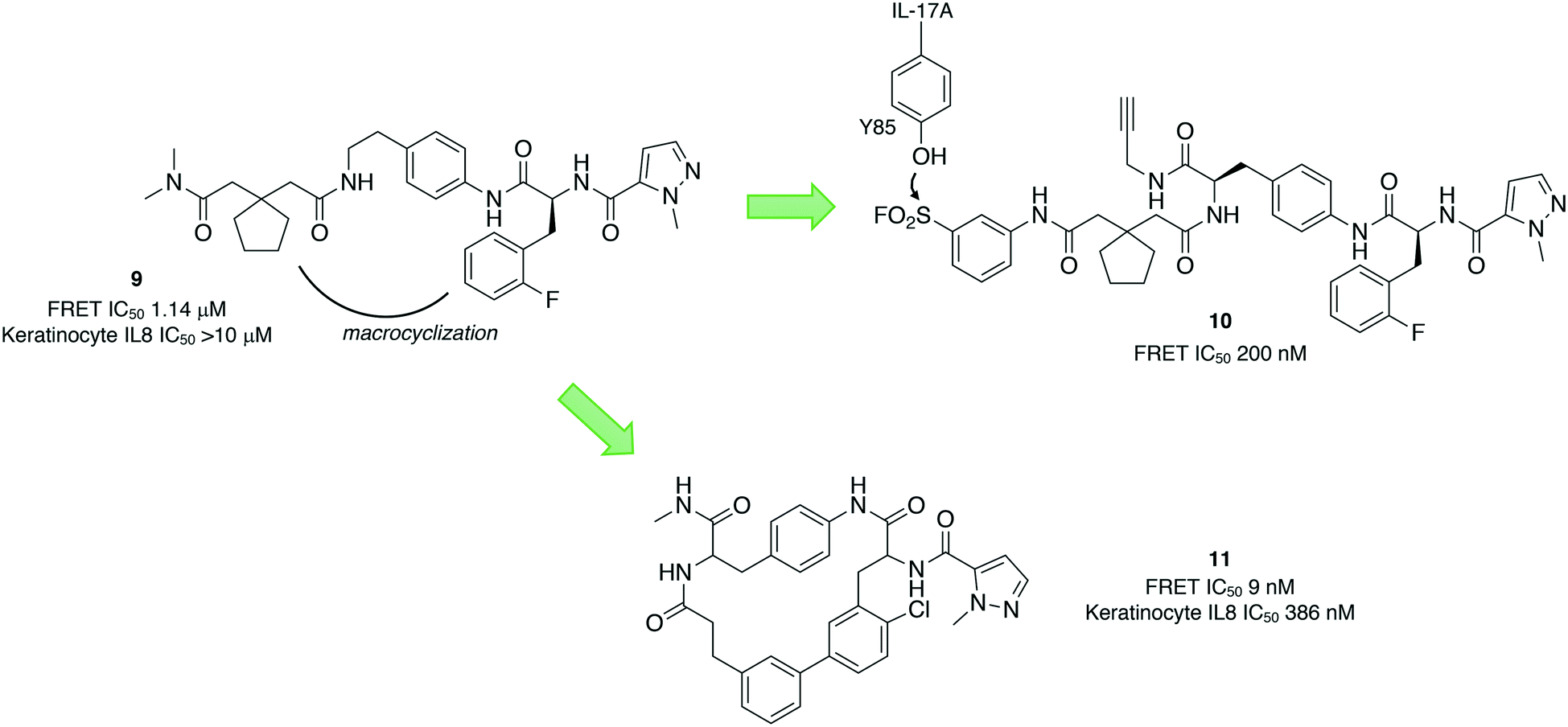 | ||
| Fig. 4 Structure-based design of the macrocyclic IL-17A inhibitor 11. The sulfonyl fluoride probe 10 was developed to identify the binding site of 9 using peptide mapping MS that facilitated medicinal chemistry optimization. | ||
Because tyrosine residues are enriched at protein interfaces, sulfonyl fluoride warheads are well-suited to the development of inhibitors of PPIs.38 Another recent example illustrated the potential of this approach by incorporating the sulfonyl fluoride electrophile into a previously identified biaryl fragment ligand BA-1 (12) of the antiapoptotic protein B-cell lymphoma extra-large (Bcl-xL), that yielded the first covalent inhibitors of the protein. Sulfonyl fluoride BA-SF1 (13) was designed to target tyrosine 101 of the BH3 domain-binding groove, a residue that is not present in the related protein Bcl-2. The crystal structure (PDB 6RNU) shows there is a proximal arginine residue to the tyrosine that presumably increases the phenol OH reactivity (Fig. 5). Subsequent elaboration of the scaffold provided BA-SF2 (14) that was found to covalently inhibit Bcl-xL, with selectivity over Bcl-2 as expected. The alkyne-containing probe BA-SF2-yne (15) was also developed that may have utility in further target occupancy experiments.
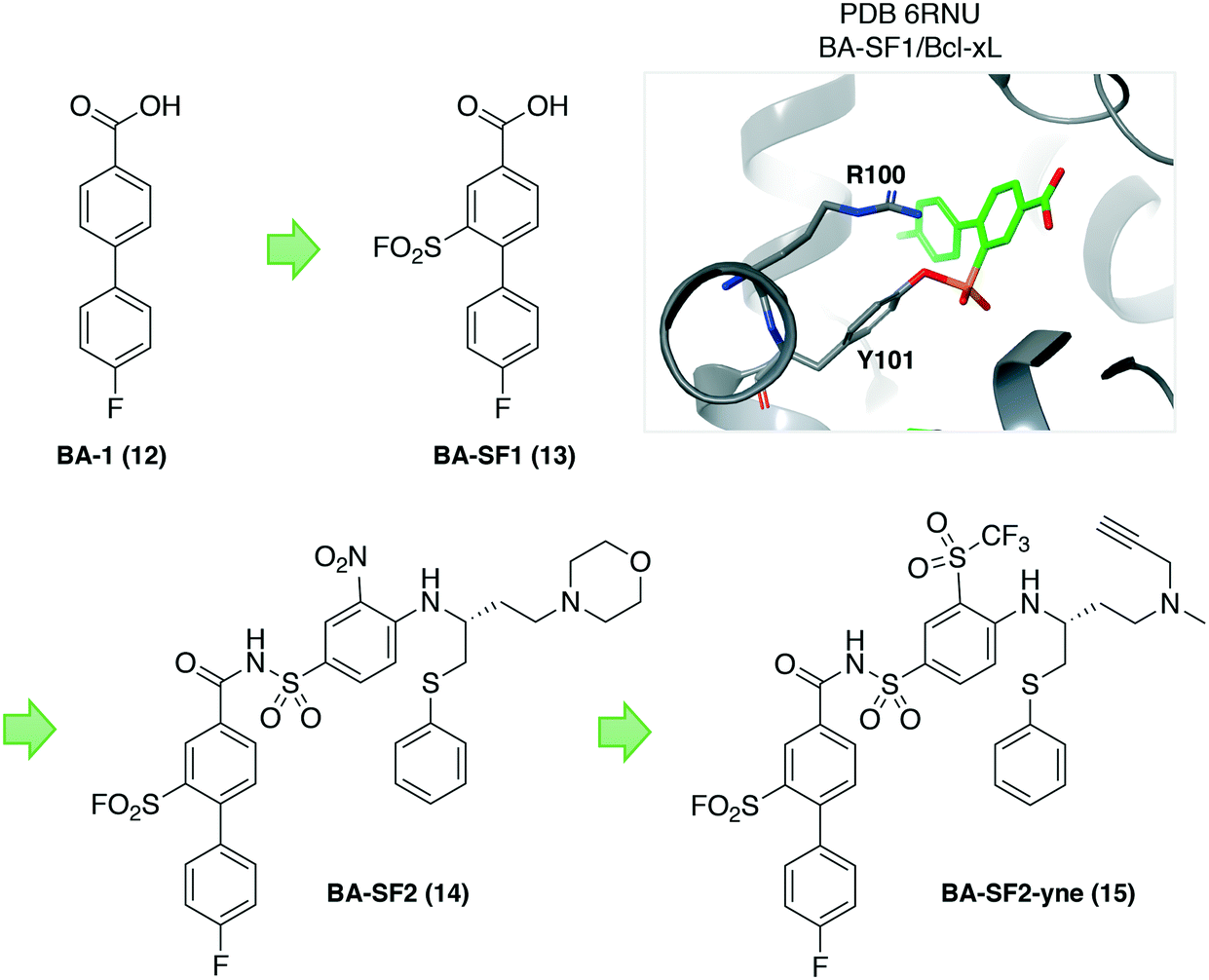 | ||
| Fig. 5 Design of covalent sulfonyl fluoride probes targeting Tyr101 in Bcl-xL. A proximal arginine residue appears to facilitate tyrosine sulfonylation. | ||
Membrane proteins
Structural biology of integral membrane proteins is challenging due to their low expression, conformational flexibility and inherent instability. Small molecule ligands are often used to stabilize the protein under investigation, to facilitate crystallization and structural biology studies. Covalent ligands are particularly useful in this regard due to their ability to substantially increase complex stability.Using the sulfonyl fluoride integration strategy, DU172 (16, Fig. 6) was developed some time ago as an irreversible inhibitor of the adenosine A1 receptor (A1-AR).39 More recently, DU172 was used to strongly stabilize A1-AR by 16 °C, enabling subsequent crystallization of the complex to 3.2 Å resolution (PDB 5UEN, Fig. 6).40 Tyr271 was identified unambiguously as the site of covalent attachment to the protein. There appears to be hydrogen bonding from Asn70 to the sulfonyl oxygen atom that could facilitate complex formation. Although many of the key interacting residues between DU172 and A1-AR are conserved in the related receptor A2A-AR, the structure revealed a considerably different binding pocket (A1-AR is a wide-open cavity while that of A2A-AR is narrower) that helps explain the selectivity of the probe for A1-AR. The crystal structure was subsequently used in combination with Gaussian accelerated molecular dynamics simulations to understand the binding mode of positive allosteric modulators (PAMs) of the receptor.41 The structure would also appear to be a very useful aid in the design of selective A2A-AR antagonists, currently a focus in cancer immunotherapy, with A1-AR being the anti-target in this case.
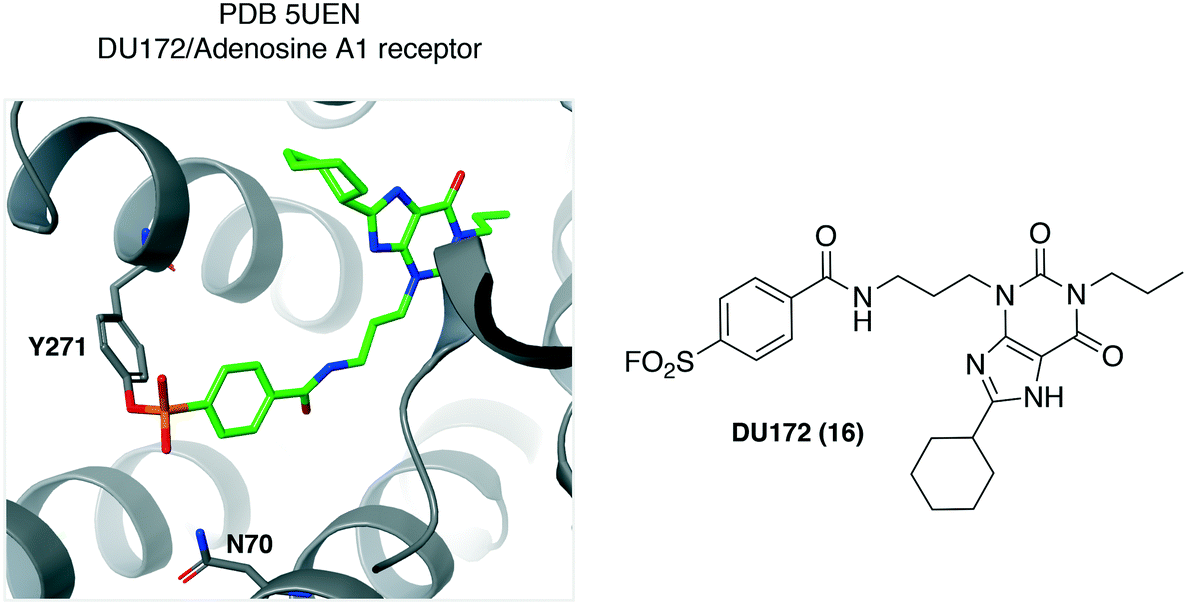 | ||
| Fig. 6 The adenosine A1 receptor irreversible antagonist DU172 (16) was found to engage the Tyr271 residue. | ||
Recently, a similar strategy was used to convert an A3-AR reversible antagonist into a covalent inhibitor through the installation of a sulfonyl fluoride warhead.42 Although a cocrystal structure was not reported, site-directed mutagenesis was used to show that the Tyr265 residue (analogous to the A1-AR Tyr271) was engaged by the probe (shown to be a significantly less potent inhibitor of the Tyr265Phe mutant). This work may lead to the crystallization of A3-AR which has yet to be reported. Likewise, a covalent sulfonyl fluoride-containing PAM of the metabotropic glutamate receptor 2 (mGlu2) was developed, but this time the probe was found to engage a threonine residue (Thr791) using SDM, setting the stage for future structural biology studies.43
Fluorosulfate probes
Fluorosulfates have emerged as useful SuFEx motifs, but with improved stability over their sulfonyl fluoride congeners.44 As a result, the fluorosulfate electrophile may find utility in drug design. An interesting application of fluorosulfate chemistry was reported recently in a strategy termed ‘inverse drug discovery’.45 Briefly, drug-like (Lipinski's rule of 5 compliant)46 probes incorporating a fluorosulfate warhead and terminal alkyne reporter were exposed to cells and their interacting proteins enriched and identified using affinity chromatography MS-based proteomics. AQ-FS-yne (17) was found to engage the enzyme glutathione S-transferase omega (GSTO1) and a crystal structure with the probe that lacks the click handle (AQ-FS, 18) revealed conjugation to the Tyr229 residue (reactivity potentially enhanced by the proximal Lys57 residue, PDB 5UEH, Fig. 7). Another example from this study showed that the probe BAI-FS (19) engaged a lysine in nucleoside diphosphate kinase A (NME1), which may be a catalytic residue involved in DNA cleavage and thus intrinsically reactive (PDB 5UI4). In a related experiment, an aryl fluorosulfate probe 20 bearing a click handle was found to label analogous tyrosine residues in a series of intracellular lipid binding proteins (CRABP2, FABP4 and FABP5).47 The reactive tyrosine in these proteins is flanked by arginine residues that reduced the pKa of the phenol-OH. These results demonstrate how a chemistry-driven strategy can enable the identification of tractable biological targets of therapeutic interest.45,48 | ||
| Fig. 7 Fluorosulfate chemical probes engage nucleophilic amino acid residues in protein binding sites. | ||
Sometimes the adduct formed between the protein and a fluorosulfate probe may be unstable and react further. Interestingly, the fluorosulfate analogue of the sulfonyl fluoride TTR ligands, BAOD-FS (21) appears to engage the reactive K15, and the resulting conjugate undergoes hydrolysis to form the sulfated lysine product (PDB 4YDM, Fig. 7).49
As discussed, the fluorosulfate warhead is less reactive than the sulfonyl fluoride electrophile. For this reason the fluorosulfate congener of the DcpS inhibitor FS-p1 (22, Fig. 7) was prepared and shown to have improved drug-like properties over the sulfonyl fluoride (higher metabolic and chemical stability) as expected.44 However, the fluorosulfate FS-p1 did not react with neighboring tyrosine, lysine or even histidine residues, but had instead engaged a noncatalytic serine residue elsewhere in the binding site (Ser272, Fig. 2), as shown by peptide mapping MS. The serine residue is surrounded by several basic amino acid residues that enhance reactivity of the hydroxyl group (cf. the hydrolase catalytic serine). These results illustrate the importance of conformation in determining amino acid selectivity because the addition of a single oxygen atom to the sulfonyl fluoride SF-p1 probe hindered accommodation of the fluorosulfate motif resulting in a considerably different binding pose.
Conclusions
SuFEx chemistry has been harnessed in medicinal chemical biology to develop probes for a diverse range of proteins. The microenvironment of the protein binding site undoubtedly influences the reactivity of the amino acid side chains that engage the sulfonyl fluoride or fluorosulfate electrophiles. We hope that the structural analyses described here provide some insights into the structural requirements for targeting specific residues en route to developing useful chemical probes and, potentially, covalent drugs. A recent assessment of chemoproteomics experiments using sulfonyl fluoride probes identified many adducted tyrosines and lysines in proteins for which crystal structures are available.27 These data show that reactive tyrosine residues are usually proximal to basic amino acid residues (lysine, arginine and histidine) while most lysines are proximal to acidic residues (glutamate and aspartate) in-line with the analysis here.50 Other SuFEx-reactive amino acid residues include serine, threonine and histidine. Broadly, tyrosine appears to be more readily adducted than lysine (noticeable not only in the proteomics experiments, but also in co-crystal structures when tyrosine and lysine residues are both present in binding sites and proximal to the warhead). However, a suitable trajectory between the amino acid residue and the incoming SuFEx warhead appears to be the dominating factor that determines amino acid preference. There also appear to be instances of hydrogen bonding to the sulfonyl oxygen atom or the F leaving group that may enhance the electrophilicity of the sulfur atom and anchor the motif to accelerate templated conjugation.The utility of sulfur(VI) fluoride exchange chemical biology continues to grow. SuFEx probes are used to map hyper-reactive amino acid residues in proteins, facilitate crystallization of membrane proteins, and enable target validation and occupancy biomarker technologies. Recent studies have leveraged SuFEx chemistry to crosslink proteins (via tyrosine, lysine and histidine) as a way to map protein–protein interactions in cells.51,52 Future applications will include the identification of allosteric binding sites and target identification from phenotypic screens by incorporating these electrophiles into hit compounds. The exciting potential for the incorporation of fluorosulfates into drugs merits exploration.44,45,53 The structural insights provided here may facilitate rational prospective targeting of amino acid residues in proteins beyond cysteine using sulfur(VI) fluoride exchange chemistry.
Conflicts of interest
LHJ receives research funding from Deerfield.References
- L. B. Poole, Free Radical Biol. Med., 2015, 80, 148–157 CrossRef CAS PubMed.
- N. A. Khazanov and H. A. Carlson, PLoS Comput. Biol., 2013, 9, e1003321 CrossRef PubMed.
- L. H. Jones, Angew. Chem., Int. Ed., 2018, 57, 9220–9223 CrossRef CAS PubMed.
- A. M. Gold and D. Fahrney, Biochemistry, 1964, 3, 783–791 CrossRef CAS.
- A. M. Gold, Biochemistry, 1965, 4, 897–901 CrossRef CAS PubMed.
- B. R. Baker and G. J. Lourens, J. Med. Chem., 1967, 10, 1113–1122 CrossRef CAS PubMed.
- B. R. Baker and G. J. Lourens, J. Med. Chem., 1968, 11, 38–41 CrossRef CAS PubMed.
- B. R. Baker and J. A. Kozma, J. Med. Chem., 1968, 11, 656–661 CrossRef CAS PubMed.
- B. R. Baker and H. U. Siebenneick, J. Med. Chem., 1971, 14, 802–805 CrossRef CAS PubMed.
- B. R. Baker and M. Cory, J. Med. Chem., 1971, 14, 805–808 CrossRef CAS PubMed.
- B. R. Baker, Annu. Rev. Pharmacol., 1970, 10, 35–50 CrossRef CAS PubMed.
- P. K. Pal, W. J. Wechter and R. F. Colman, Biochemistry, 1975, 14, 707–715 CrossRef CAS PubMed.
- R. F. Colman, Annu. Rev. Biochem., 1983, 52, 67–91 CrossRef CAS PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18808–18814 CrossRef CAS PubMed.
- L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584–586 CrossRef CAS PubMed.
- N. P. Grimster, S. Connelly, A. Baranczak, J. Dong, L. B. Krasnova, K. B. Sharpless, E. T. Powers, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 5656–5668 CrossRef CAS.
- M. P. Patricelli, A. K. Szardenings, M. Liyanage, T. K. Nomanbhoy, M. Wu, H. Weissig, A. Aban, D. Chun, S. Tanner and J. W. Kozarich, Biochemistry, 2007, 46, 350–358 CrossRef CAS.
- M. Bantscheff, D. Eberhard, Y. Abraham, S. Bastuck, M. Boesche, S. Hobson, T. Mathieson, J. Perrin, M. Raida, C. Rau, V. Reader, G. Sweetman, A. Bauer, T. Bouwmeester, C. Hopf, U. Kruse, G. Neubauer, N. Ramsden, J. Rick, B. Kuster and G. Drewes, Nat. Biotechnol., 2007, 25, 1035–1044 CrossRef CAS.
- M. P. Kamps, S. S. Taylor and B. M. Sefton, Nature, 1984, 310, 589–592 CrossRef CAS PubMed.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- B. Zhao, A. Smallwood, J. Yang, K. Koretke, K. Nurse, A. Calamari, R. B. Kirkpatrick and Z. Lai, Protein Sci., 2008, 17, 1791–1797 CrossRef CAS.
- N. Jura, X. Zhang, N. F. Endres, M. A. Seeliger, T. Schindler and J. Kuriyan, Mol. Cell, 2011, 42, 9–22 CrossRef CAS PubMed.
- O. Fadeyi, M. D. Parikh, M. Z. Chen, R. E. Kyne, A. P. Taylor, I. O'Doherty, S. E. Kaiser, S. Barbas, S. Niessen, M. Shi, S. L. Weinrich, J. C. Kath, L. H. Jones and R. P. Robinson, ChemBioChem, 2016, 17, 1925–1930 CrossRef CAS PubMed.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- J. Singh, M. Salcius, S. W. Liu, B. L. Staker, R. Mishra, J. Thurmond, G. Michaud, D. R. Mattoon, J. Printen, J. Christensen, J. M. Bjornsson, B. A. Pollok, M. Kiledjian, L. Stewart, J. Jarecki and M. E. Gurney, ACS Chem. Biol., 2008, 3, 711–722 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- A. Gopalsamy, A. Narayanan, S. Liu, M. D. Parikh, R. E. Kyne, O. Fadeyi, M. A. Tones, J. J. Cherry, J. F. Nabhan, G. LaRosa, D. N. Petersen, C. Menard, T. L. Foley, S. Noell, Y. Ren, P. M. Loria, J. Maglich-Goodwin, H. Rong and L. H. Jones, J. Med. Chem., 2017, 60, 3094–3108 CrossRef CAS PubMed.
- L. H. Jones, H. Xu and O. O. Fadeyi, Methods Enzymol., 2019, 622, 201–220 Search PubMed.
- J. J. Cherry, C. J. DiDonato, E. J. Androphy, A. Calo, K. Potter, S. K. Custer, S. Du, T. L. Foley, A. Gopalsamy, E. J. Reedich, S. M. Gordo, W. Gordon, N. Hosea, L. H. Jones, D. K. Krizay, G. LaRosa, H. Li, S. Mathur, C. A. Menard, P. Patel, R. Ramos-Zayas, A. Rietz, H. Rong, B. Zhang and M. A. Tones, PLoS One, 2017, 12, e0185079 CrossRef PubMed.
- J. M. Hatcher, G. Wu, C. Zeng, J. Zhu, F. Meng, S. Patel, W. Wang, S. B. Ficarro, A. L. Leggett, C. E. Powell, J. A. Marto, K. Zhang, J. C. Ki Ngo, X. D. Fu, T. Zhang and N. S. Gray, Cell Chem. Biol., 2018, 25, 460–470.e466 CrossRef CAS.
- E. L. Mehler, M. Fuxreiter, I. Simon and E. B. Garcia-Moreno, Proteins, 2002, 48, 283–292 CrossRef CAS.
- X. Ran and J. E. Gestwicki, Curr. Opin. Chem. Biol., 2018, 44, 75–86 CrossRef CAS.
- S. Liu, L. A. Dakin, L. Xing, J. M. Withka, P. V. Sahasrabudhe, W. Li, M. E. Banker, P. Balbo, S. Shanker, B. A. Chrunyk, Z. Guo, J. M. Chen, J. A. Young, G. Bai, J. T. Starr, S. W. Wright, J. Bussenius, S. Tan, A. Gopalsamy, B. A. Lefker, F. Vincent, L. H. Jones, H. Xu, L. R. Hoth, K. F. Geoghegan, X. Qiu, M. E. Bunnage and A. Thorarensen, Sci. Rep., 2016, 6, 30859 CrossRef CAS PubMed.
- L. H. Jones, A. Narayanan and E. C. Hett, Mol. BioSyst., 2014, 10, 952–969 RSC.
- A. R. Beauglehole, S. P. Baker and P. J. Scammells, J. Med. Chem., 2000, 43, 4973–4980 CrossRef CAS PubMed.
- A. Glukhova, D. M. Thal, A. T. Nguyen, E. A. Vecchio, M. Jörg, P. J. Scammells, L. T. May, P. M. Sexton and A. Christopoulos, Cell, 2017, 168, 867–877.e813 CrossRef CAS PubMed.
- Y. Miao, A. Bhattarai, A. T. N. Nguyen, A. Christopoulos and L. T. May, Sci. Rep., 2018, 8, 16836 CrossRef PubMed.
- X. Yang, J. P. D. van Veldhoven, J. Offringa, B. J. Kuiper, E. B. Lenselink, L. H. Heitman, D. van der Es and A. P. IJzerman, J. Med. Chem., 2019, 62, 3539–3552 CrossRef CAS PubMed.
- M. L. J. Doornbos, X. Wang, S. C. Vermond, L. Peeters, L. Pérez-Benito, A. A. Trabanco, H. Lavreysen, J. M. Cid, L. H. Heitman, G. Tresadern and A. P. IJzerman, J. Med. Chem., 2019, 62, 223–233 CrossRef CAS PubMed.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 CrossRef CAS PubMed.
- L. H. Jones, Bioorg. Med. Chem., 2019, 27, 3451–3453 CrossRef CAS PubMed.
- A. Baranczak, Y. Liu, S. Connelly, W. G. Du, E. R. Greiner, J. C. Genereux, R. L. Wiseman, Y. S. Eisele, N. C. Bradbury, J. Dong, L. Noodleman, K. B. Sharpless, I. A. Wilson, S. E. Encalada and J. W. Kelly, J. Am. Chem. Soc., 2015, 137, 7404–7414 CrossRef CAS PubMed.
- L. H. Jones, Mol. BioSyst., 2016, 12, 1728–1730 RSC.
- N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 4995–4999 CrossRef CAS PubMed.
- B. Yang, H. Wu, P. D. Schnier, Y. Liu, J. Liu, N. Wang, W. F. DeGrado and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11162–11167 CrossRef CAS PubMed.
- C. Baggio, P. Udompholkul, L. Gambini, A. F. Salem, J. Jossart, J. J. P. Perry and M. Pellecchia, J. Med. Chem., 2019, 62, 9188–9200 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |