
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a novel kinase hinge binder fragment by dynamic undocking
Moira
Rachman
 ab,
Dávid
Bajusz
b,
Anasztázia
Hetényi
c,
Andrea
Scarpino
b,
Balázs
Merő
d,
Attila
Egyed
b,
László
Buday
d,
Xavier
Barril
ae and
György M.
Keserű
*b
ab,
Dávid
Bajusz
b,
Anasztázia
Hetényi
c,
Andrea
Scarpino
b,
Balázs
Merő
d,
Attila
Egyed
b,
László
Buday
d,
Xavier
Barril
ae and
György M.
Keserű
*b
aFacultat de Farmàcia and Institut de Biomedicina, Universitat de Barcelona, Av. Joan XXIII 27-31, 08028 Barcelona, Spain
bMedicinal Chemistry Research Group, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, Budapest 1117, Hungary. E-mail: keseru.gyorgy@ttk.hu
cDepartment of Medical Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
dSignal Transduction and Functional Genomics Research Group, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, Budapest 1117, Hungary
eCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 4th March 2020
Abstract
One of the key motifs of type I kinase inhibitors is their interactions with the hinge region of ATP binding sites. These interactions contribute significantly to the potency of the inhibitors; however, only a tiny fraction of the available chemical space has been explored with kinase inhibitors reported in the last twenty years. This paper describes a workflow utilizing docking with rDock and dynamic undocking (DUck) for the virtual screening of fragment libraries in order to identify fragments that bind to the kinase hinge region. We have identified 8-amino-2H-isoquinolin-1-one (MR1), a novel and potent hinge binding fragment, which was experimentally tested on a diverse set of kinases, and is hereby suggested for future fragment growing or merging efforts against various kinases, particularly MELK. Direct binding of MR1 to MELK was confirmed by STD-NMR, and its binding to the ATP-pocket was confirmed by a new competitive binding assay based on microscale thermophoresis.
Introduction
Protein kinases are a major class of drug targets, with over 30 marketed drugs and 250 drug candidates undergoing clinical studies, mainly but not only in various oncology indications.1 The majority of kinase inhibitors target ATP binding sites (type I), which contain a conserved hinge region. For this reason, one of the main challenges in the development of kinase inhibitors is obtaining selectivity. Another major challenge is avoiding an already congested IP space, as the diversity of hinge binder scaffolds used in type I kinase inhibitors is relatively limited.2 Consequently, these chemotypes have barely been sampled, which prompts the thorough exploitation of their chemical space.3,4 For example, it has been shown recently that only 1% of potential hinge binders are present in known kinase inhibitors.5Fragment-based drug discovery (FBDD) has already been used in many kinase programs4 and has the potential to identify novel fragment-sized hinge binders and specifically “evolve” the fragment for the targeted kinase.6,7 In fact, the first FBDD derived drug on the market, vemurafenib, targets the oncogenic V600E mutant of the B-Raf kinase.8 Structure-based virtual screening approaches have been used effectively for both the identification of novel fragment-sized hinge binders and their optimization. In one case study, Kolb and colleagues screened 730![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds and discovered two ligands with different hinge binder moieties. The initial set was first filtered by kinase hinge binding pharmacophore restraints and the resulting 21418 compounds were docked to the ATP site.9 In another study, Urich and colleagues extracted core fragments from 2.3M commercially available compounds. The resulting unique fragments were filtered for kinase hinge pharmacophores, and were subsequently docked into a panel of protein kinases. This strategy identified a number of hinge binder fragments with no previously reported activity against the investigated kinases.10
000 compounds and discovered two ligands with different hinge binder moieties. The initial set was first filtered by kinase hinge binding pharmacophore restraints and the resulting 21418 compounds were docked to the ATP site.9 In another study, Urich and colleagues extracted core fragments from 2.3M commercially available compounds. The resulting unique fragments were filtered for kinase hinge pharmacophores, and were subsequently docked into a panel of protein kinases. This strategy identified a number of hinge binder fragments with no previously reported activity against the investigated kinases.10
In this work, we used a novel screening strategy to find potent hinge binding fragments. Our dynamic undocking11 based approach identified MR1 (Fig. 1A) that has been experimentally validated against five kinase targets (Table 1). Future fragment growing or merging efforts toward these targets could avoid a congested druggable chemical space (e.g. for JAK2) and could provide a suitable chemical starting point to unmet medical needs (e.g. for MELK). JAK2 belongs to the non-receptor tyrosine kinase family, for which the V617F mutation is known to be implicated in myeloproliferative disorders.12,13 MST3 belongs to the Ste20 serine/threonine protein kinase family, which has been shown to promote proliferation and tumorigenicity.14 Maternal embryonic leucine zipper kinase (MELK) is a serine/threonine kinase belonging to the CAMK family, and was initially found to be expressed in a wide range of early embryonic cellular stages.
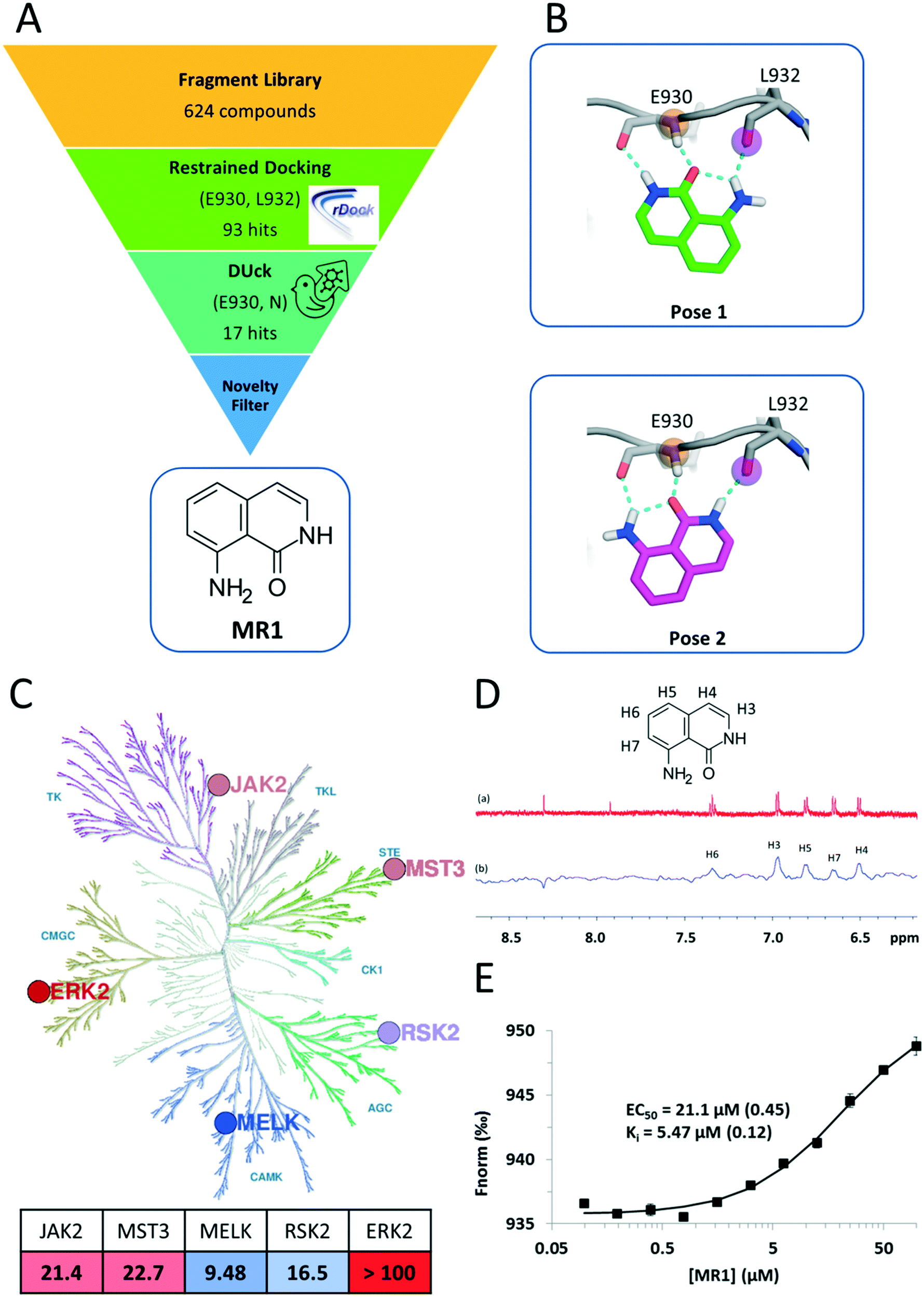 | ||
| Fig. 1 A) Summary of the virtual screening workflow. B) Predicted binding modes of MR1 in the JAK2 ATP pocket. Both binding modes utilize tridentate H-bond interactions with the backbone carbonyl and amide groups of the hinge residues E930 and L932. C) Kinase inhibitory profile of MR1 overlaid on the kinase phylogenetic tree28 (coloured blue to red, from most active to least active). The table summarizes the respective IC50 values in μM units, with the same colouring scheme (illustration reproduced courtesy of Cell Signaling Technology, Inc., http://www.cellsignal.com). D) 1H NMR spectrum of MR1 (a) and saturation transfer difference NMR spectrum of MR1 in the presence of MELK (b). E) In the competitive, MST-based binding assay, titration of MELK with MR1 reveals concentration-dependent binding with a Ki value of 5.47 μM and confirms the ATP-pocket as the location of binding. | ||
More recently, MELK has been identified to be present in several human cancers and stem cell populations with a unique spatial and temporal pattern, which suggests a prominent role in cell cycle control, cell proliferation, apoptosis, cell migration, cell renewal, embryogenesis, oncogenesis, and cancer treatment resistance and recurrence.16 ERK2 is part of the Ras/Raf/MEK/ERK pathway, which is often overactivated in a very wide range of cancers, while RSK2 is a downstream effector of this pathway and phosphorylates substrates involved in transcription, translation, cell cycle regulation, and cell survival.15
Results and discussion
Here, we present a novel and potent hinge binding fragment, MR1 (Fig. 1A), that was discovered through virtual screening (VS) of our in-house fragment library19 using kinase hinge pharmacophore information in a hierarchical VS strategy (Fig. 1A). Our initial screening has been performed on JAK2 as a prototypic protein kinase with a large number of type I inhibitors. It is well known that the kinase hinge region exhibits a pronounced H-bond pattern with ATP or ATP-site competitors and that these interactions are imperative for type I kinase inhibitors to bind. As such, a typical VS strategy would incorporate such information by ensuring that the retained ligands maintain these features through the use of e.g. pharmacophoric restraints. Here, we employ an additional filter that emphasizes the importance of the defined H-bonds through dynamic undocking (DUck11), which estimates the robustness of H-bonds. DUck is an orthogonal method to approaches meant to predict binding free energies. Instead, it assesses structural stability through evaluation of the robustness of H-bonds. The VS strategy consisted of docking with rDock20 using pharmacophoric restraints to ensure that the features necessary for H-bonding with the hinge were present, then using DUck to estimate the resistance to rupture of these H-bonds. This strategy led to the discovery of MR1, which was tested on five kinases from the major branches of the kinase phylogenetic tree (Fig. 1C), each with several relevant indications (Table 1). MST3 and MELK, in particular, have only few known potent inhibitors.MR1 was found to inhibit four of the target kinases with IC50 values in the low to mid-micromolar range (corresponding to ligand efficiencies between 0.54–0.59),21 which is in line with its small size and other hinge binders of this complexity.7 ERK2 was the only kinase that was not inhibited; one possible reason behind this is the higher ATP concentration (100 μM) in the ERK2 inhibition assay, as compared to the rest of the kinases (10–50 μM), resulting in a stronger competition of ATP toward the binding site. The assessment of the binding mode of MR1 was based on the docking and DUck scores. Two plausible solutions were generated (Fig. 1B), both of which adopt a tridentate interaction with the hinge. This was verified also for MELK: here, pose 2 is clearly preferred (WQB value of 7.7 kcal mol−1, vs. 1.6 kcal mol−1 for pose 1). The ability of fragments to bind simultaneously in multiple orientations has been reported in numerous occasions, and can even be favourable from an entropic perspective.22 Depending on how the fragment is then elaborated, it freezes into one binding mode or the other,23,24 which will likely stay conserved during the elaboration process25 (this also means that the mentioned entropic gain will be lost, but ideally, this is compensated by a larger favourable enthalpy decrease due to additional secondary interactions between the protein and the elaborated ligand). The tridentate interaction is not often observed within kinases (a bidentate interaction being the most common), but – as supported by the inhibition data – a third interaction allows for high versatility.26 This can be constrained at the optimization phase for introducing family-specific substituents. In fact, it is not the hinge binder but rather its decoration that determines kinase selectivity. Consequently, selective kinase inhibitors can stem from an unselective fragment.7,27
In addition to identifying MR1, we have observed a stepwise enrichment of known hinge binders along the workflow: while the whole fragment library contained known hinge binders in 14.4% of the compounds (90 out of 624), this ratio was 64.7% for the virtual hits after the DUck calculations (11 out of 17). This can be considered as a retrospective validation of the presented virtual screening workflow.
Next, we aimed to confirm the direct and ATP-competitive binding of MR1 experimentally. For this purpose, we have selected MELK, as MR1 displayed the strongest inhibitory activity against this kinase. Direct binding to MELK was confirmed by an STD-NMR measurement (Fig. 1D), while the binding site was validated with a competitive MST-based (microscale thermophoresis) assay developed in our lab (Fig. 1E).
The assay is based on the displacement of a fluorescently labelled type I reference ligand (see the Experimental section) and it has additionally enabled us to quantify the dissociation constant of MR1 from MELK as 5.47 μM. Its high ligand efficiency (LE = 0.67) nominates MR1 as a viable starting point for optimizing ATP-site kinase inhibitors.
Experimental
Virtual screening
The structures were prepared using MOE 2016,29 by removing water and cofactors, capping the termini and gaps, and for protonation with default settings. For the virtual screening, the JAK2 PDB structure, 3E64 (chain A) was used.30 For docking, the cavity was defined in the prepared structure by the reference ligand method, using the crystallized ligand as reference. The fragment library consisted of 624 compounds and was prepared with LigPrep,31 so that ligands above 300 Da would be ignored, at most eight stereoisomers, six tautomers and eight ring conformers would be generated and lastly, probable ionization states within the pH range of six to eight would be generated.The prepared library was docked with pharmacophoric restraints at the hinge region, namely, Glu930, O and Leu932, N. The pharmacophore was defined as a 2 Ångström radius around the mentioned receptor atoms. If the feature did not adhere to the positional constraints, rDock would assign a positive (unfavourable) pharmacophore restraint score, for which the cutoff was set to 0.5. Furthermore, a high-throughput VS (HTVS) protocol was implemented, which consisted of three stages, for which at every stage the number of docking runs increases, and the rDock “SCORE.INTER” filter becomes stricter. The filter was adapted to scores expected for fragment-sized molecules which resulted in 93 compounds in total. DUck was performed on this set of compounds, pulling from Leu932, N (pulling from Glu930, O did not result in any additional filtering). The first step for a DUck simulation is the definition of the chunk (a part of the protein structure) that represents the local environment surrounding the residue interacting with the ligand. When selecting residues for the chunk, the following guidelines were considered: I) selecting as few residues as possible to reduce computational time, II) residues were not selected if they would block the ligand from exiting the pocket during the simulations based on the directionality of the H-bond, III) residues were not removed if this would lead to the possibility of solvent entering the pocket from areas other than where the ligand is exiting, and lastly IV) preserving the local environment of the interacting atoms in the already prepared structures. The sequence gaps created during the process of selecting the chunk residues were capped. For this, each section of residues was split into separate chains, and the termini of each chain were acetylated or methylated. Lastly, the chunk was checked for clashes possibly created during the capping of the chains. The chunk included the following residues: 853–859, 861–865, 879–882, 898, 902, 911, 912, 927, 929–941, 976, 978, 980–984 and 993–996. After production of the chunk, DUck performs I) automatic ligand parameterization in MOE, II) minimization, III) equilibration, and IV) two SMD simulations (at two different temperatures, 300 K and 325 K), in which the distance between the interacting atoms in the ligand and protein is increased from 2.5 to 5.0 Å, and V) if the WQB value (work necessary to break the H-bond) in the previous step reaches a pre-defined threshold, then the system is sampled by a short unbiased MD simulation, after which the resulting new structures are fed into steps IV) and V); the last two steps being repeated in a finite number of cycles (replicas).11 Steps II) to V) were performed with GPU-based pmemd.cuda in AMBER. Here, up to five replicas of steps IV) and V) were performed, during which a WQB threshold of 6 kcal mol−1 (work necessary to break the H-bond) was used, so that the simulations were discontinued if the measured WQB in any replica was below the threshold. If five runs were completed, the lowest obtained WQB value was used, which resulted in 17 compounds that surpassed the threshold. Finally, we checked the novelty of these fragments against known kinase inhibitors by substructure searches, which ultimately led to the selection of compound MR1 for experimental validation.
To suggest a binding pose for MELK, MR1 was docked in all liganded MELK structures available. Both poses in Fig. 1B were identified and submitted to a DUck simulation, pulling from the NH atom in the hinge (Cys 89 N). Chunks were derived by selecting residues within 10 Å from Cys89 N.
To check for known hinge binders in the fragment library, all kinase inhibitors found in the ATP binding site were downloaded from the KLIFS database.32 The cores (5 Angstrom from the hinge NH group) were extracted from known binders from KLIFS and from the compounds in the fragment library. A substructure search was performed to find which of the fragment cores in the library were present in the known hinge binder cores.
Kinase inhibition assay
MR1 (purity 97% by LC/MS) was tested against the target kinases with the Z′-LYTE kinase inhibition assay (Life Technologies). The assay employs a fluorescence-based format and is based on the different sensitivities of phosphorylated and nonphosphorylated peptides to proteolytic cleavage. A suitable peptide substrate is labelled with two fluorophores, forming a FRET pair. After incubating the kinase + peptide + test compound mixture for an hour, a development reaction is carried out. Any peptide that was not phosphorylated by the kinase is cleaved, disrupting the resonance energy transfer between the FRET pair. The reaction progress is quantified based on the ratio of the detected emission at 445 nm (coumarin) and 520 nm (fluorescein), i.e. the ratio of cleaved vs. intact peptides. A more detailed description of the assay is available on the website of Life Technologies.33 IC50 values were determined from 10-pt titration measurements (with duplicate datapoints) using the SelectScreen™ Biochemical Kinase Profiling Service available at Life Technologies.NMR measurements
The human MELK protein (Q14680) DNA was obtained from OriGene and the kinase–uba domain (1–337) sequence was cloned into a modified pET vector encoding an N-terminal His-tag. The recombinant protein was expressed in E. coli BL21 pLysS cells, harvested by centrifugation and purified by immobilized metal affinity chromatography and anion exchange chromatography. 1H and STD-NMR measurements were performed using a 600 MHz Bruker Avance III spectrometer equipped with a 5 mm cryo-TXI (1H, 13C, 15N) probe with z-gradient at 298 K. The MELK protein and MR1 were dissolved in a 10% (v/v) D2O and H2O mixture containing 20 mM Tris buffer (pH 8.0), 260 mM NaCl, 1 mM TCEP and 5% glycerol. Spectra were acquired with water suppression using excitation sculpting with the pulsed gradient scheme. For the 1H and STD measurements, the MELK and MR1 concentrations were 2.14 and 50 μM, respectively. As a reference, STD experiments were also performed without the target, containing the ligand species alone.STD-NMR spectra were acquired using a series of 40 equally spaced 50 ms Gaussian-shaped pulses for selective saturation of the protein, with a total saturation time of 2 s and a 50 ms spinlock to suppress protein signals. The frequency of the on-resonance saturation was set at 1.0 ppm and the off-resonance saturation frequency was set at 40.0 ppm. A total of 2000 scans were collected for each pseudo-2D experiment.
Microscale thermophoresis binding assay
Microscale thermophoresis measurements were conducted on a NanoTemper Monolith NT.115 device, using the red fluorescence channel. MELK was stored and applied in a buffer consisting of 20 mM Tris, 260 mM NaCl, 1 mM TCEP and 5% glycerol, at a pH of 8.0. The MR1 stock was prepared in DMSO and diluted into the protein buffer, with the final DMSO concentrations not exceeding 1%. We have applied the ligand displacement assay principle to detect the thermophoresis of the fluorescently labeled MELK reference ligand Kinase Tracer 236 (ThermoFisher, cat. no. PV5592) at increasing concentrations of MR1. A titration curve was acquired with serial 1:1 dilutions starting from 100 μM ligand concentration (with 0.56 μM protein, 50 nM Kinase Tracer 236, 60% LED power, 20% MST power), with 11 datapoints. Each datapoint was acquired in triplicate. The primary result was a relative EC50 value of MR1 against MELK (21.1 μM), which was converted to a Ki value of 5.47 μM, using the following formula: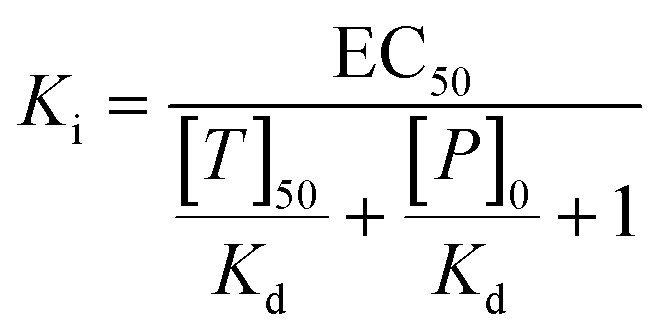 | (1) |
Conclusions
Here, we report the discovery of a novel hinge binder using a new computational screening protocol based on dynamic undocking. 8-Amino-2H-isoquinolin-1-one (MR1) was found to inhibit kinases from four major branches of the kinase phylogenetic tree, with low to mid-micromolar potencies (translating to ligand efficiencies between 0.54–0.59). All of these kinases are relevant clinical targets, mostly for oncological indications. In particular, MST3 and MELK have few known potent inhibitors, presenting the opportunity to create novel chemical matter for these kinases with a less congested IP space. In addition to its versatile inhibitory profile, direct binding of MR1 to MELK was confirmed by STD-NMR, and its binding to the ATP site was validated by an MST-based (microscale thermophoresis) ligand displacement assay developed as part of this work. Together, these findings nominate MR1 as a viable fragment starting point for a range of type I kinase inhibitor programs.Abbreviations
| AGC | PKA, PKG and PKC containing families |
| ATP | Adenosine triphosphate |
| CAMK | Ca2+/calmodulin-dependent protein kinases |
| CMGC | CDK, MAPK, GSK3 and CLK containing families |
| DMSO | Dimethyl sulfoxide |
| DUck | Dynamic undocking |
| ERK2 | Common alternative name for mitogen-activated protein kinase 1 (MAPK1) |
| JAK2 | Janus kinase 2 |
| LC/MS | Liquid chromatography/mass spectrometry |
| MD | Molecular dynamics |
| MELK | Maternal embryonic leucine zipper kinase |
| MST3 | Common alternative name for serine/threonine-protein kinase 24 (STK24) |
| NMR | Nuclear magnetic resonance |
| RSK2 | Ribosomal protein S6 kinase alpha-3 |
| SMD | Steered molecular dynamics |
| STD | Saturation transfer difference |
| STE | Yeast sterile 7-, 11- and 20-homologue kinases |
| TK | Tyrosine kinases |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Kitti Koprivanacz and Tamás Szimler for their help in protein expression and purification. This study was supported by the MSCA ITN FRAGNET (project 6758993) grant to M. R., A. S., X. B. and G. M. K., by the National Research, Development and Innovation Office of Hungary (OTKA K116904), the MCIU/AEI/FEDER, UE (SAF2015-68749-R and RTI2018-096429-B-I00), the Catalan government (2014 SGR 1189), and the National Research, Development and Innovation Fund of Hungary (HunProEx 2018-1.2.1-NKP-2018-00005).Notes and references
- D. Fabbro, S. W. Cowan-Jacob and H. Moebitz, Br. J. Pharmacol., 2015, 172, 2675–2700 CrossRef CAS PubMed.
- D. Bajusz, G. G. Ferenczy and G. M. Keserű, Curr. Top. Med. Chem., 2017, 17, 2235–2259 CrossRef CAS PubMed.
- S. Klaeger, S. Heinzlmeir, M. Wilhelm, H. Polzer, B. Vick, P. A. Koenig, M. Reinecke, B. Ruprecht, S. Petzoldt, C. Meng, J. Zecha, K. Reiter, H. Qiao, D. Helm, H. Koch, M. Schoof, G. Canevari, E. Casale, S. Re Depaolini, A. Feuchtinger, Z. Wu, T. Schmidt, L. Rueckert, W. Becker, J. Huenges, A. K. Garz, B. O. Gohlke, D. P. Zolg, G. Kayser, T. Vooder, R. Preissner, H. Hahne, N. Tõnisson, K. Kramer, K. Götze, F. Bassermann, J. Schlegl, H. C. Ehrlich, S. Aiche, A. Walch, P. A. Greif, S. Schneider, E. R. Felder, J. Ruland, G. Médard, I. Jeremias, K. Spiekermann and B. Kuster, Science, 2017, 358, eaan4368 CrossRef PubMed.
- P. N. Mortenson, V. Berdini and M. O'Reilly, Methods Enzymol., 2014, 548, 69–92 CAS.
- H. Zhao and A. Caflisch, Bioorg. Med. Chem. Lett., 2015, 25, 2372–2376 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- P. Bamborough, M. J. Brown, J. A. Christopher, C. Chung and G. W. Mellor, J. Med. Chem., 2011, 54, 5131–5143 CrossRef CAS PubMed.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. Dandrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, Nature, 2010, 467, 596–599 CrossRef CAS PubMed.
- P. Kolb, C. B. Kipouros, D. Huang and A. Caflisch, Proteins: Struct., Funct., Bioinf., 2008, 73, 11–18 CrossRef CAS PubMed.
- R. Urich, G. Wishart, M. Kiczun, A. Richters, N. Tidten-luksch, D. Rauh, B. Sherborne, P. G. Wyatt and R. Brenk, Chem. Biol., 2013, 8, 1044–1052 CAS.
- S. Ruiz-Carmona, P. Schmidtke, F. J. Luque, L. Baker, N. Matassova, B. Davis, S. Roughley, J. Murray, R. Hubbard and X. Barril, Nat. Chem., 2017, 9, 201–206 CrossRef CAS PubMed.
- E. J. Baxter, L. M. Scott, P. J. Campbell, C. East, N. Fourouclas, S. Swanton, G. S. Vassiliou, A. J. Bench, E. M. Boyd, N. Curtin, M. A. Scott, W. N. Erber, T. Avis, A. Barthorpe, G. Bignell, M. Blow, L. Brackenbury, G. Buck, S. Clegg, J. Clements, J. Cole, H. Davies, S. Edkins, K. Gray, M. Gorton, S. O'Meara, K. Halliday, R. Harrison, W. Haynes, K. Hills, C. Hunter, D. Jones, V. Kosmidou, R. Laman, R. Lugg, A. Parker, J. Perry, R. Petty, A. Small, H. Solomon, P. Stephens, Y. Stephens, C. Stevens, R. Smith, P. Tarpey, C. Tofts, J. Varian, S. West, S. Widaa, S. Bamford, A. Butler, E. Dawson, E. Dicks, K. Edwards, S. Forbes, C. Greenman, J. Hinton, A. Menzies, K. Raine, R. Shepherd, J. Teague, A. Yates, R. Wooster, A. Futreal, M. Stratton and A. R. Green, Lancet, 2005, 365, 1054–1061 CrossRef CAS.
- R. L. Levine, M. Wadleigh, J. Cools, B. L. Ebert, G. Wernig, B. J. P. Huntly, T. J. Boggon, I. Wlodarska, J. J. Clark, S. Moore, J. Adelsperger, S. Koo, J. C. Lee, S. Gabriel, T. Mercher, A. D'Andrea, S. Fröhling, K. Döhner, P. Marynen, P. Vandenberghe, R. A. Mesa, A. Tefferi, J. D. Griffin, M. J. Eck, W. R. Sellers, M. Meyerson, T. R. Golub, S. J. Lee and D. G. Gilliland, Cancer Cell, 2005, 7, 387–397 CrossRef CAS PubMed.
- C. Cho, K. Lee, W. Chen and C. Wang, Oncotarget, 2016, 7, 14586–14604 Search PubMed.
- K. A. Casalvieri, C. J. Matheson, D. S. Backos and P. Reigan, Trends Cancer, 2017, 3, 302–312 CrossRef CAS PubMed.
- R. Ganguly, A. Mohyeldin, J. Thiel, H. I. Kornblum, M. Beullens and I. Nakano, Clin. Transl. Med., 2015, 4, 11 CrossRef PubMed.
- A. I. Ojesina, L. Lichtenstein, S. S. Freeman, C. S. Pedamallu, I. Imaz-Rosshandler, T. J. Pugh, A. D. Cherniack, L. Ambrogio, K. Cibulskis, B. Bertelsen, S. Romero-Cordoba, V. Treviño, K. Vazquez-Santillan, A. S. Guadarrama, A. A. Wright, M. W. Rosenberg, F. Duke, B. Kaplan, R. Wang, E. Nickerson, H. M. Walline, M. S. Lawrence, C. Stewart, S. L. Carter, A. McKenna, I. P. Rodriguez-Sanchez, M. Espinosa-Castilla, K. Woie, L. Bjorge, E. Wik, M. K. Halle, E. A. Hoivik, C. Krakstad, N. B. Gabiño, G. S. Gómez-Macías, L. D. Valdez-Chapa, M. L. Garza-Rodríguez, G. Maytorena, J. Vazquez, C. Rodea, A. Cravioto, M. L. Cortes, H. Greulich, C. P. Crum, D. S. Neuberg, A. Hidalgo-Miranda, C. R. Escareno, L. A. Akslen, T. E. Carey, O. K. Vintermyr, S. B. Gabriel, H. A. Barrera-Saldaña, J. Melendez-Zajgla, G. Getz, H. B. Salvesen and M. Meyerson, Nature, 2014, 506, 371–375 CrossRef CAS PubMed.
- L. Fang, W. Lu, H. H. Choi, S.-C. J. Yeung, J.-Y. Tung, C.-D. Hsiao, E. Fuentes-Mattei, D. Menter, C. Chen, L. Wang, J. Wang and M.-H. Lee, Cancer Cell, 2015, 28, 183–197 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- S. Ruiz-Carmona, D. Alvarez-Garcia, N. Foloppe, A. B. Garmendia-Doval, S. Juhos, P. Schmidtke, X. Barril, R. E. Hubbard and S. D. Morley, PLoS Comput. Biol., 2014, 10, e1003571 CrossRef PubMed.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- G. C. P. van Zundert, B. M. Hudson, S. H. P. de Oliveira, D. A. Keedy, R. Fonseca, A. Heliou, P. Suresh, K. Borrelli, T. Day, J. S. Fraser and H. van den Bedem, J. Med. Chem., 2018, 61, 11183–11198 CrossRef CAS PubMed.
- S. Malhotra and J. Karanicolas, J. Med. Chem., 2017, 60, 128–145 CrossRef CAS PubMed.
- D. Erlanson, Pract. Fragm. blog, 2017 Search PubMed.
- M. N. Drwal, G. Bret, C. Perez, C. Jacquemard, J. Desaphy and E. Kellenberger, J. Med. Chem., 2018, 61, 5963–5973 CrossRef CAS PubMed.
- L. Xing, J. Klug-Mcleod, B. Rai and E. A. Lunney, Bioorg. Med. Chem., 2015, 23, 6520–6527 CrossRef CAS PubMed.
- S. L. Posy, M. A. Hermsmeier, W. Vaccaro, K. H. Ott, G. Todderud, J. S. Lippy, G. L. Trainor, D. A. Loughney and S. R. Johnson, J. Med. Chem., 2011, 54, 54–66 CrossRef CAS PubMed.
- M. Chartier, T. Chénard, J. Barker and R. Najmanovich, PeerJ, 2013, 1, e126 CrossRef PubMed.
- Mol. Oper. Environ. CCG ULC 2016.08, 1010 Sherbooke St. West, Suite #910, Montr. QC, Canada, H3A 2R7, 2018.
- S. Antonysamy, G. Hirst, F. Park, P. Sprengeler, F. Stappenbeck, R. Steensma, M. Wilson and M. Wong, Bioorg. Med. Chem. Lett., 2009, 19, 279–282 CrossRef CAS PubMed.
- Schrödinger Release 2016-4, LigPrep, Schrödinger, LLC, New York, NY, 2016.
- A. J. Kooistra, G. K. Kanev, O. P. J. van Linden, R. Leurs, I. J. P. de Esch and C. de Graaf, Nucleic Acids Res., 2016, 44, D365–D371 CrossRef CAS PubMed.
- Z'-LYTE Kinase Assay Kits. http//www.lifetechnologies.com/hu/en/home/life-science/drug-discovery/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/z-lyte.html (Accessed 14 Oct 2019).
| This journal is © The Royal Society of Chemistry 2020 |