DOI:
10.1039/C9MD00391F
(Research Article)
RSC Med. Chem., 2020,
11, 102-110
Potent trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio and pentafluorosulfanyl containing (1,3,4-oxadiazol-2-yl)benzamides against drug-resistant Gram-positive bacteria†
Received
7th August 2019
, Accepted 9th November 2019
First published on 16th December 2019
Abstract
According to the Centers for Disease Control and Prevention (CDC), methicillin-resistant Staphylococcus aureus (MRSA) affects about 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients in the US annually and directly causes about 11000 deaths. Therefore, despite the fact that there are several drugs available for the treatment of MRSA, there is a need for new chemical entities. We previously reported that 1,3,4-oxadiazolyl sulfonamide F6 was bacteriostatic and inhibited MRSA strains with a minimum inhibitory concentration (MIC) of 2 μg mL−1. Here, we report the discovery of trifluoromethoxy (OCF3), trifluoromethylsulfonyl (SO2CF3), trifluoromethylthio (SCF3) and pentafluorosulfanyl (SF5) containing (1,3,4-oxadiazol-2-yl)benzamides exhibiting potent antibacterial activities against MRSA [MIC values as low as 0.06 μg mL−1 against linezolid-resistant S. aureus (NRS 119)]. Interestingly, whereas the OCF3 and SO2CF3 containing oxadiazoles were bacteriostatic, the SCF3 and SF5 containing oxadiazoles were bactericidal. They exhibited a wide spectrum of activities against an extensive panel of Gram-positive bacterial strains, including MRSA, vancomycin-resistant Staphylococcus aureus (VRSA), vancomycin-resistant enterococcus (VRE) and methicillin-resistant or cephalosporin-resistant Streptococcus pneumoniae. Furthermore, compounds 6 and 12 outperformed vancomycin in clearing intracellular MRSA in infected macrophages. Moreover, the tested compounds behaved synergistically or additively with antibiotics used for the treatment of MRSA infections.
000 patients in the US annually and directly causes about 11000 deaths. Therefore, despite the fact that there are several drugs available for the treatment of MRSA, there is a need for new chemical entities. We previously reported that 1,3,4-oxadiazolyl sulfonamide F6 was bacteriostatic and inhibited MRSA strains with a minimum inhibitory concentration (MIC) of 2 μg mL−1. Here, we report the discovery of trifluoromethoxy (OCF3), trifluoromethylsulfonyl (SO2CF3), trifluoromethylthio (SCF3) and pentafluorosulfanyl (SF5) containing (1,3,4-oxadiazol-2-yl)benzamides exhibiting potent antibacterial activities against MRSA [MIC values as low as 0.06 μg mL−1 against linezolid-resistant S. aureus (NRS 119)]. Interestingly, whereas the OCF3 and SO2CF3 containing oxadiazoles were bacteriostatic, the SCF3 and SF5 containing oxadiazoles were bactericidal. They exhibited a wide spectrum of activities against an extensive panel of Gram-positive bacterial strains, including MRSA, vancomycin-resistant Staphylococcus aureus (VRSA), vancomycin-resistant enterococcus (VRE) and methicillin-resistant or cephalosporin-resistant Streptococcus pneumoniae. Furthermore, compounds 6 and 12 outperformed vancomycin in clearing intracellular MRSA in infected macrophages. Moreover, the tested compounds behaved synergistically or additively with antibiotics used for the treatment of MRSA infections.
Introduction
On February 27, 2017 the World Health Organization (WHO) published a list of bacteria for which new antibiotics are urgently needed. This first ever list of antibiotic-resistant “priority pathogens”, comprises 12 families of bacteria that pose the greatest threat to human health.1 Methicillin-resistant, vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus were listed as a high priority category that requires development of new antibiotics. S. aureus, a Gram-positive bacterial pathogen, is one of the leading causes of community- and hospital- acquired infection.2 The rise in antimicrobial resistant strains partly contributes to the increasing death rate associated with S. aureus infection.3 For instance, methicillin-resistant S. aureus (MRSA) bacteremia is accompanied by higher mortality rates compared to methicillin-sensitive S. aureus (MSSA) bacteremia.4 Moreover, deadly staphylococcal infections are still a global threat. According to the Centers for Disease Control and Prevention (CDC), more than 119000 people suffered from bloodstream S. aureus infections in the United States in 2017, and nearly 20000 died. Therefore, the need for novel anti-MRSA therapies cannot be overemphasized.
Despite the availability of drugs that are effective against MRSA, such as vancomycin, trimethoprim–sulfamethoxazole, clindamycin, tetracycline, doxycycline, minocycline, daptomycin, rifampin or linezolid,5 about 14% of patients who get serious MRSA infections die.6 The majority of the aforementioned drugs used to treat MRSA have limitations that preclude certain patient class. For example, vancomycin is not appropriate for patients with reduced kidney function (such as elderly or diabetic patients).7 Additionally, S. aureus strains, which are resistant to the most commonly used antibiotics such as vancomycin,7 trimethoprim–sulfamethozole,8 linezolid,9 clindamycin10 and daptomycin11 have also been well documented. Therefore, the increase in resistance to commonly used drugs for MRSA infections, along with growths in both hospital and community acquired MRSA infections demonstrate the vital need for new entities that are active against MRSA.
We previously reported that F6 was active against MRSA with a minimum inhibitory concentration (MIC) of 2 μg mL−12. F6 was found to be bacteriostatic, non-toxic and efficacious in a mouse wound infection model. It was determined in the previous report that the 3,5-dimethylpiperidinyl sulfonamide moiety was essential for antibacterial activity. In this report, we reveal that whereas substitution of the 3,5-dimethylpiperidinyl sulfonamide moiety with most groups abrogated antibacterial activity, substitution with the trifluoromethylsulfonyl, trifluoromethoxy, trifluoromethylthio and pentafluorosulfanyl groups afforded analogs that were more potent than F6.
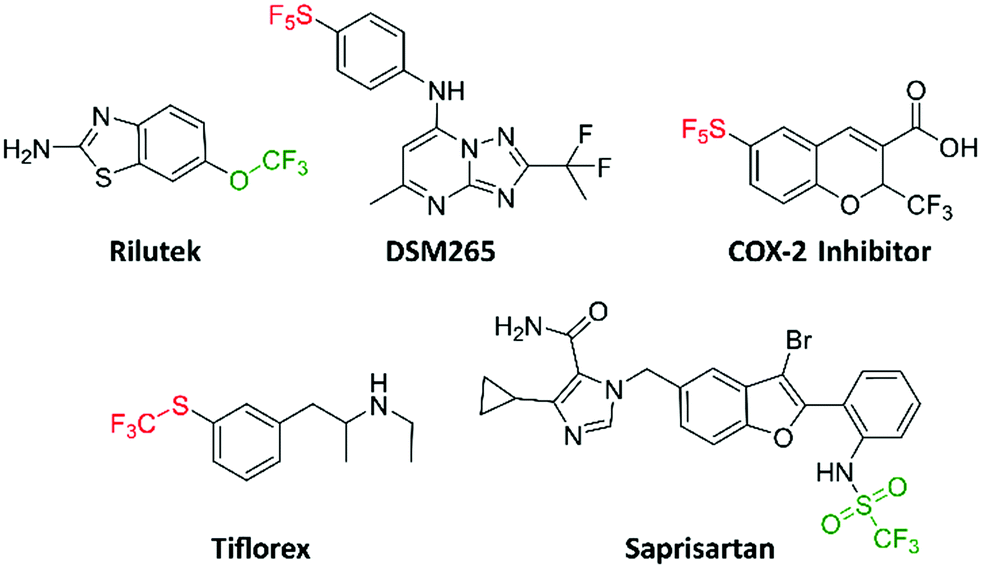
A few bioactive compounds, which also bear the trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio and pentafluorosulfanyl groups have been reported (Fig. 1). DSM265 (a new antimalarial drug) completed phase IIa clinical trials (NCT02123290).13 DSM265 is orally bioavailable with favorable pharmacokinetics, revealing that the pentafluorosulfanyl group is appropriate to append to potential drugs. Likewise, the pentafluorosulfanyl containing COX-2 inhibitor has been shown to reduce inflammation and pain in vivo.14 As for trifluoromethylthio-containing compounds, triflorex is an FDA approved drug for the treatment of anorexia.15 Triflorex is given orally, demonstrating that the trifluoromethylthio group can be adequately substituted on potential orally bioavailable compounds. The trifluoromethylsulfonyl and trifluoromethoxy groups are increasingly being introduced into bioactive compounds. For example, Rilutek (FDA approved for the treatment of amyotrophic lateral sclerosis (ALS)) and saprisartan (approved for hypertension) contain the trifluoromethoxy and trifluoromethylsulfonyl groups respectively (Fig. 1).16,17
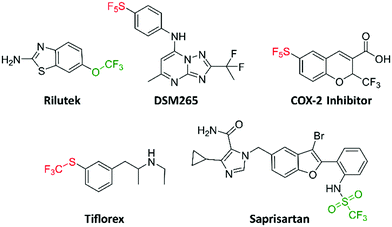 |
| | Fig. 1 Trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio, and pentafluorosulfanyl containing bioactive molecules. | |
Results and discussion
Synthesis of (1,3,4-oxadiazol-2-yl)benzamides and evaluation of their antibacterial activity
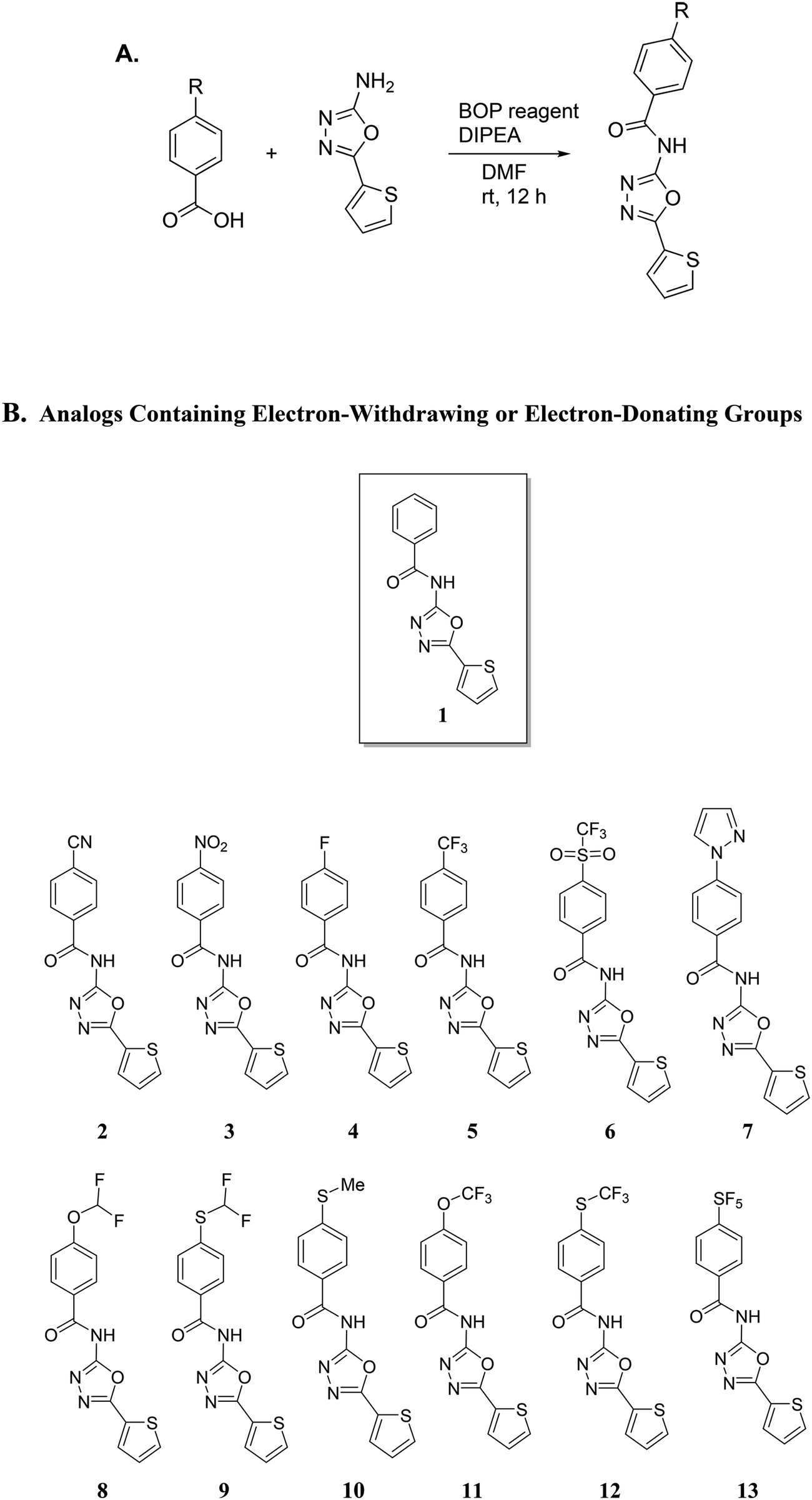
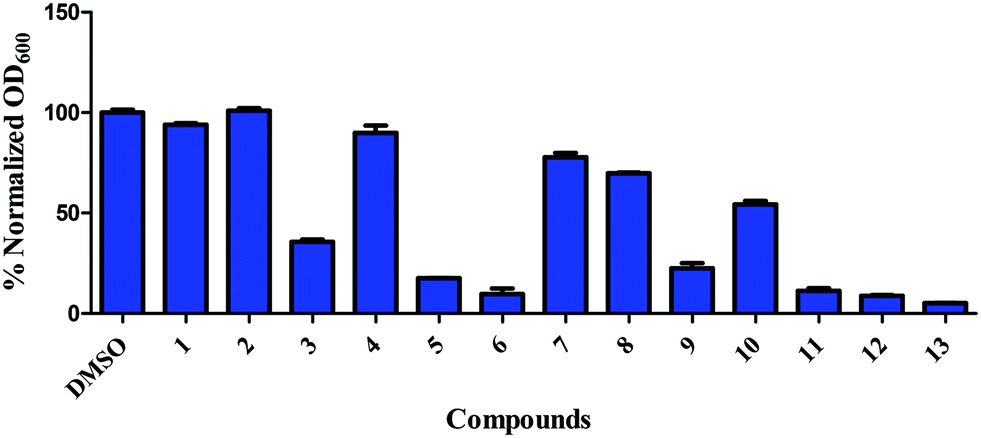
Our group has been interested in discovering novel chemotypes that are active against drug-resistant bacteria. F6 and its analogs were readily made in one step via amide coupling. Coupling reagents such as 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), hydroxybenzotriazole (HOBt), or (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were either unsuccessful or low-yielding whereas using benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) reagent resulted in product yield up to 43%. Using methylithium to deprotonate the amine, followed by coupling with the acid chloride also afforded products in up to 25% yield. Having developed the methodology to make F6, we applied the BOP reagent to make a series of analogs bearing electron-withdrawing and donating groups (Fig. 2).18 These compounds were initially screened for their ability to inhibit the growth of S. aureus at 16 μg mL−1 (Fig. 3). For compounds that showed inhibitory activity, we determined the MIC (Table 1). From these results, compounds bearing, trifluoromethyl (5), trifluoromethylsulfonyl (6), trifluoromethoxy (11), trifluoromethylthio (12), and pentafluorosulfanyl (13) groups were active but not others (Fig. 3 and Table 1). Interestingly, it appears that electron-withdrawing ability alone did not dictate antibacterial activity. For example, compounds bearing 4-cyano (2) -nitro (3), and -fluoro (4) groups had low to moderate activity. Furthermore, compounds containing hydrophilic electron donating groups (7) were not active (Fig. 3). The degree of fluorination also had an effect on antibacterial activity for sulfur and oxygen containing compounds. For instance, MICs for methylthio (10), difluoromethylthio (9), trifluoromethylthio (12), and pentafluorosulfanyl (13) were 8 μg mL−1, 4 μg mL−1, 0.5 μg mL−1, and 0.5 μg mL−1 respectively. MICs for compounds 8 and 11 were 32 μg mL−1 and 1 μg mL−1 respectively.
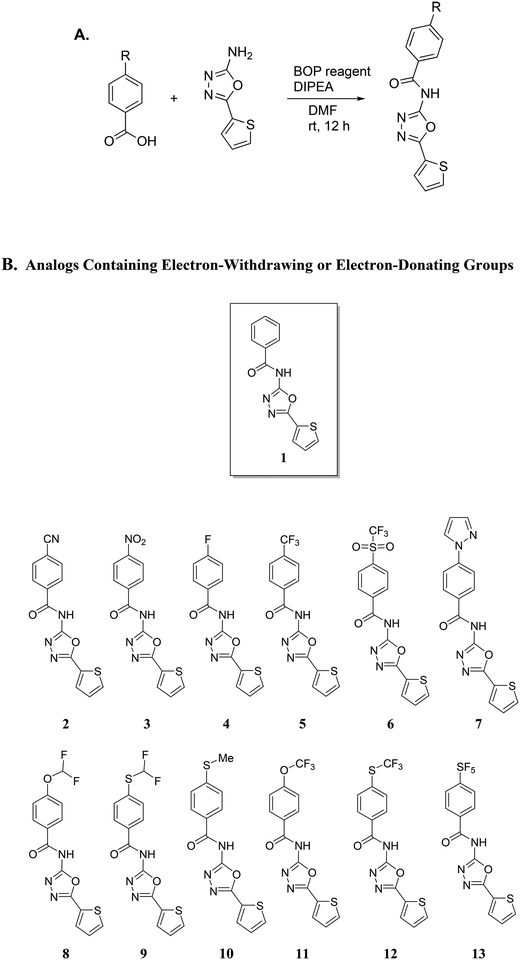 |
| | Fig. 2 A. Synthetic scheme for the synthesis of analogs using BOP reagent. BOP = Benzotriazol-1-yloxy-tris (dimethylamino)phosphonium hexafluorophosphate; DIPEA = N,N-diisopropylethylamine; DMF = dimethylformamide. B. Analogs containing either electron-withdrawing or electron-donating groups. | |
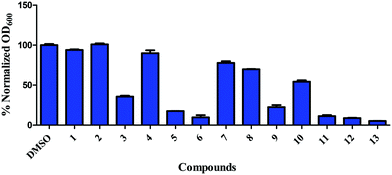 |
| | Fig. 3 Inhibition of growth of S. aureus ATCC 25923 by 1,3,4-(oxadiazol-2-yl)benzamides. S. aureus, at early exponential growth, was treated with either DMSO or 16 μg mL−1 of compounds and OD600 was measured after 24 h. Error bars represent standard error of the mean of duplicates. | |
Table 1 Initial screening (MIC, in μg mL−1) of (1,3,4-oxadiazol-2-yl)benzamides against Staphylococcus aureus ATCC 25923 and methicillin-resistant Staphylococcus aureus (MRSA) ATCC 33592
| Compound/control antibiotic |
S. aureus ATCC 25923 |
MRSA ATCC 33592 |
|
3
|
4 |
8 |
|
4
|
32 |
32 |
|
5
|
2 |
2 |
|
6
|
0.5 |
0.5 |
|
8
|
32 |
32 |
|
9
|
4 |
4 |
|
10
|
4 |
4 |
|
11
|
1 |
1 |
|
12
|
0.5 |
0.5 |
|
13
|
0.25 |
0.5 |
| Vancomycin |
1 |
1 |
| Linezolid |
2 |
2 |
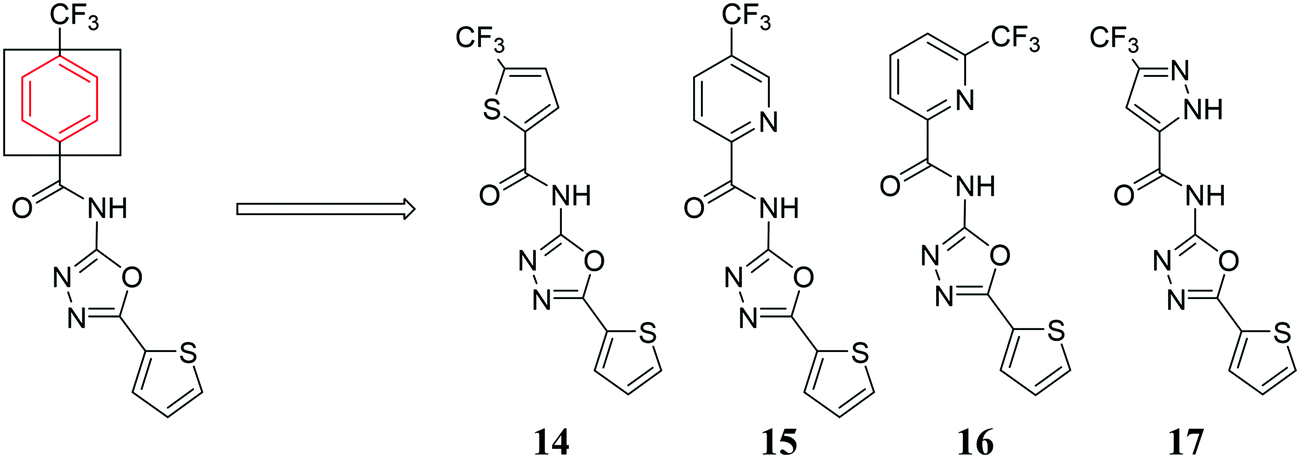
After identifying compounds 5, 6, 11, 12, and 13 as being potent against MRSA, we next investigated other trifluoromethyl-substituted heteroaromatic compounds (Fig. 4). Unfortunately, this series was not as active as our original hit molecules (compare MICs for 5, 6, 11, 12, and 13, Table 1, with compounds 14–17, Table 2), demonstrating that the phenyl group is needed for optimal antibacterial activity.
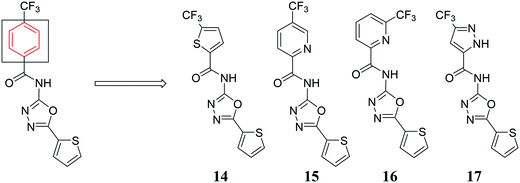 |
| | Fig. 4 Trifluoromethyl-substituted heteroaromatic compounds synthesized. | |
Table 2 Initial screening (MIC, in μg mL−1) of other trifluoromethyl-substituted heteroaromatic compounds against Staphylococcus aureus ATCC 25923 and MRSA ATCC 33592
| Compound |
S. aureus ATCC 25923 |
MRSA ATCC 33592 |
|
14
|
4 |
2 |
|
15
|
64 |
32 |
|
16
|
16 |
16 |
|
17
|
>64 |
>64 |
Compounds 6, 11, 12, & 13 are not active against Gram-negative bacteria
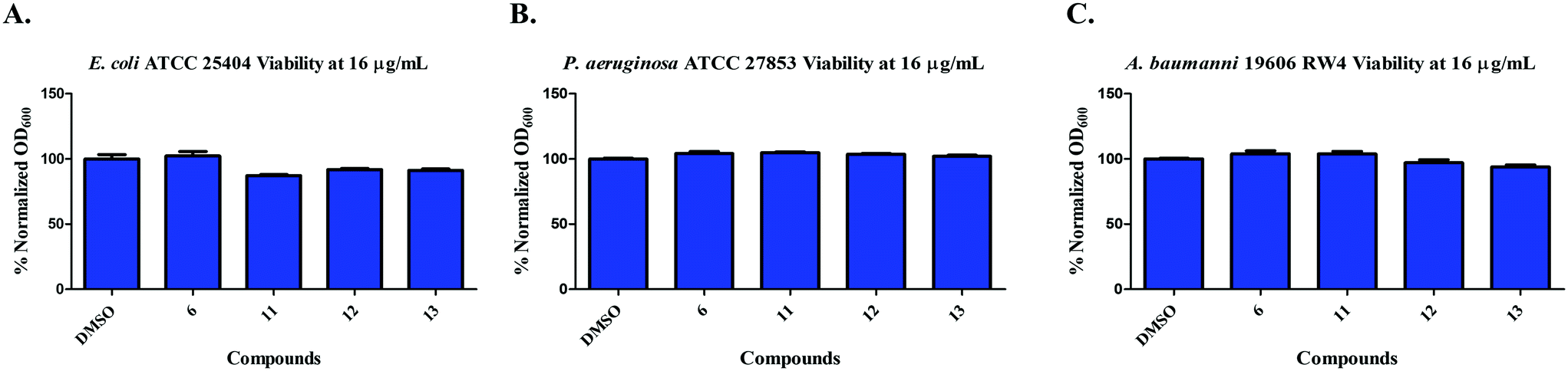
We next moved to test if compounds 6, 11, 12, and 13 were effective against Gram-negative bacteria. Thus, we tested the compounds viability against clinically relevant Gram-negative bacterial strains. Compounds 6, 11, 12, and 13 did not inhibit the growth of Escherichia coli ATCC 25404, Pseudomonas aeruginosa ATCC 27853, and Acinetobacter baumannii 19606 RW4 at concentrations of 16 μg mL−1 (Fig. 5).
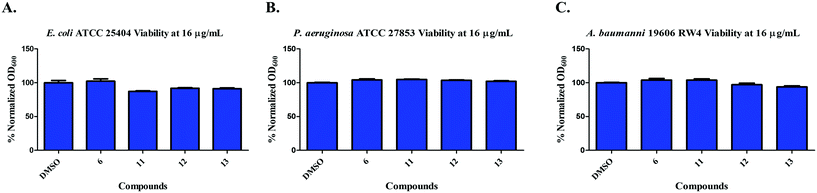 |
| | Fig. 5 Inhibition of growth of A) E. coli ATCC 25404; B) P. aeruginosa ATCC 27853; C) A. baumannii ATCC 19606 by compounds 6, 11, 12, & 13 at 16 μg mL−1. | |
Comprehensive antibacterial profile of compounds 6, 11, 12 and 13 against multidrug-resistant Gram-positive clinical strains
After the initial screening of compounds 6, 11, 12 and 13, we assessed their antibacterial profile against a panel of multidrug-resistant staphylococcal isolates. Compound 6 inhibited growth of the tested strains at concentrations ranging from 4 to 8 μg mL−1. Compounds 11, 12 and 13 exhibited potent activities, inhibiting the tested multidrug-resistant staphylococcal strains at concentrations ranging from 0.06 to 2 μg mL−1 (Table 3). Furthermore, compounds 6, 11, 12 and 13 maintained the same potent activities against other clinically relevant Gram-positive bacterial species including vancomycin-resistant enterococci (VRE), multidrug-resistant Streptococcus pneumoniae and Listeria monocytogenes (Table 4). Compounds 11, 12 and 13 exhibited potent activities against S. epidermidis, which represents the most common source of infections on implanted medical prosthetic devices due to its ability to form strongly adherent biofilms that have intrinsic resistance to antibiotics and the host defense systems.20,21 Moreover, the compounds kept their superiority over vancomycin against vancomycin-resistant enterococci (VRE). VRE is indicated as the leading cause of nosocomial infections in the USA, causing about 20–30% of hospital-acquired infections and the second major cause of such infections across the world.22 Moreover, according to the World of Health Organization (WHO), vancomycin-resistant E. faecium is categorized as one of twelve bacterial pathogens that urgently need the development of new therapeutics and alternative strategies to combat their infections.23 Additionally, compounds 6, 11, 12 and 13 exhibited a potent activities against S. pneumoniae, inhibiting the tested strains at concentrations ranging from 0.125 to 2 μg mL−1. S. pneumoniae is categorized by CDC as a serious threat bacterium that requires prompt and sustained action to overcome its problems. It is associated with 1200000 infections yearly with about 7000 deaths in the USA alone. S. pneumoniae is responsible for an estimated $96 million in medical costs per year and is the leading cause of bacterial pneumonia and meningitis in the US. It is also a major cause of bloodstream, ear and sinus infections.24
Table 3 The minimum inhibitory concentrations (MICs in μg mL−1) of compounds 6, 11, 12 and 13 against methicillin-sensitive Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Staphylococcus aureus (VRSA) strains
| Bacterial strainsa |
Compounds/control antibiotics |
|
6
|
11
|
12
|
13
|
Vancomycin |
Linezolid |
|
See ESI for sources of bacterial strains.
|
| Methicillin-sensitive Staphylococcus aureus ATCC 6538 |
8 |
1 |
0.5 |
0.5 |
1 |
0.5 |
| Methicillin-sensitive Staphylococcus aureus NRS 107 |
4 |
1 |
0.25 |
0.125 |
1 |
0.5 |
| MRSA NRS 119 |
8 |
1 |
0.5 |
0.06 |
1 |
>64 |
| MRSA NRS382 (USA100) |
8 |
1 |
0.5 |
0.5 |
2 |
1 |
| MRSA NRS383 (USA200) |
8 |
2 |
0.25 |
1 |
1 |
1 |
| MRSA NRS384 (USA300) |
4 |
0.5 |
0.5 |
0.5 |
1 |
1 |
| MRSA NRS123 (USA400) |
4 |
1 |
0.5 |
0.25 |
1 |
1 |
| MRSA NRS 385 (USA500) |
8 |
2 |
2 |
0.5 |
2 |
1 |
| MRSA NRS 386 (USA700) |
4 |
1 |
0.5 |
0.125 |
1 |
1 |
| MRSA NRS 387 (USA800) |
4 |
2 |
0.125 |
0.25 |
2 |
1 |
| MRSA NRS 483 (USA1000) |
4 |
1 |
0.5 |
0.25 |
2 |
1 |
| MRSA NRS 484 (USA1100) |
4 |
0.5 |
0.5 |
0.5 |
2 |
2 |
| VRSA 10 |
4 |
1 |
0.5 |
0.25 |
>64 |
1 |
| VRSA 12 |
8 |
1 |
0.5 |
0.125 |
>64 |
1 |
Table 4 The minimum inhibitory concentrations (MICs in μg mL−1) of compounds 6, 11, 12 and 13 against a panel of clinically important Gram-positive bacterial pathogens including Staphylococcus epidermidis, Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus faecium and Listeria monocytogenes
| Bacterial strainsa |
Compounds/control antibiotics |
|
6
|
11
|
12
|
13
|
Vancomycin |
Linezolid |
|
See ESI for sources of bacterial strains.
|
|
Staphylococcus epidermidis NRS101 |
4 |
0.5 |
0.25 |
0.06 |
2 |
1 |
| Cephalosporin- resistant Streptococcus pneumoniae ATCC 51916 |
2 |
2 |
0.25 |
0.25 |
0.5 |
1 |
| Methicillin- resistant Streptococcus pneumoniae ATCC 700677 |
1 |
1 |
0.25 |
0.125 |
1 |
1 |
|
Enterococcus faecalis ATCC 51299 (VRE) |
4 |
8 |
4 |
2 |
32 |
1 |
|
Enterococcus faecium ATCC 700221 (VRE) |
4 |
4 |
4 |
2 |
>64 |
1 |
|
Listeria monocytogenes ATCC 19111 |
2 |
2 |
0.5 |
0.125 |
0.5 |
0.5 |
Compounds 6, 11, 12 and 13 are highly tolerable to human cell lines
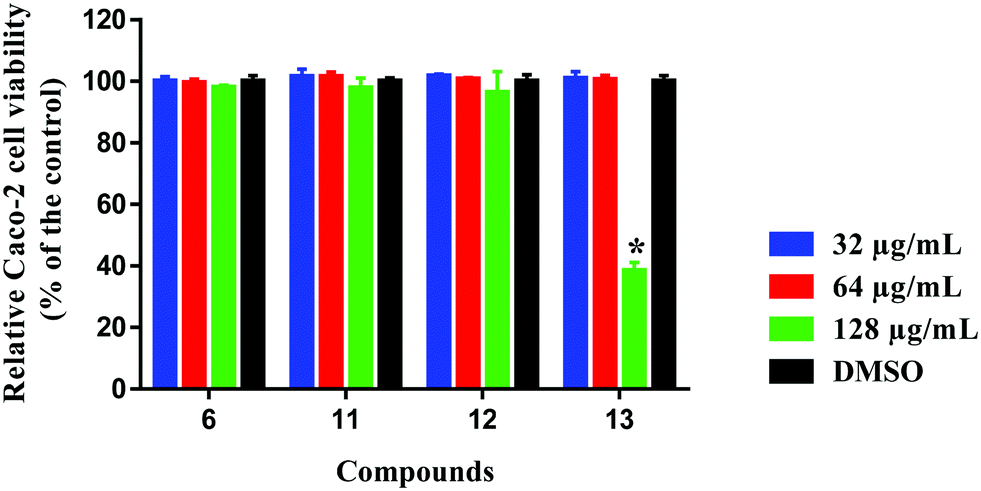
Selectivity towards prokaryotic cells is an essential attribute for any antibiotic candidate. In this regard, compound 6, 11, 12 and 13 were assessed for toxicity to mammalian cells, and they exhibited excellent safety profiles against human colorectal cells (Caco-2) (Fig. 6). Compounds 6, 11 and 12 were highly tolerable to Caco-2 cells at concentrations higher than 128 μg mL−1. This concentration is 256-times higher than the compounds' corresponding MIC values against MRSA ATCC 33592 used in the initial screening. Compound 13 was non-toxic to Caco-2 cells up to 64 μg mL−1, which is 128-times higher than the compound's MIC against MRSA ATCC 33592.
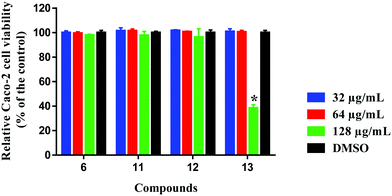 |
| | Fig. 6 Toxicity analysis of compounds 6, 11, 12 and 13 (tested in quadruplicates at 32, 64 and 128 μg mL−1) against human colorectal cells (Caco-2) using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. Results are presented as percent viable cells relative to DMSO (negative control to determine a baseline measure for the cytotoxic impact of each compound). The absorbance values represent an average of four samples analyzed for each compound. Error bars represent sample standard deviation values. The data were analyzed via two-way ANOVA with post hoc Dunnett's test for multiple comparisons (P < 0.05). Caco-2 (ATCC HTB-37) was obtained from the American type culture collection (ATCC) (Manassas, VA, USA). | |
Compounds 6, 11, 12, & 13 do not lyse red blood cells
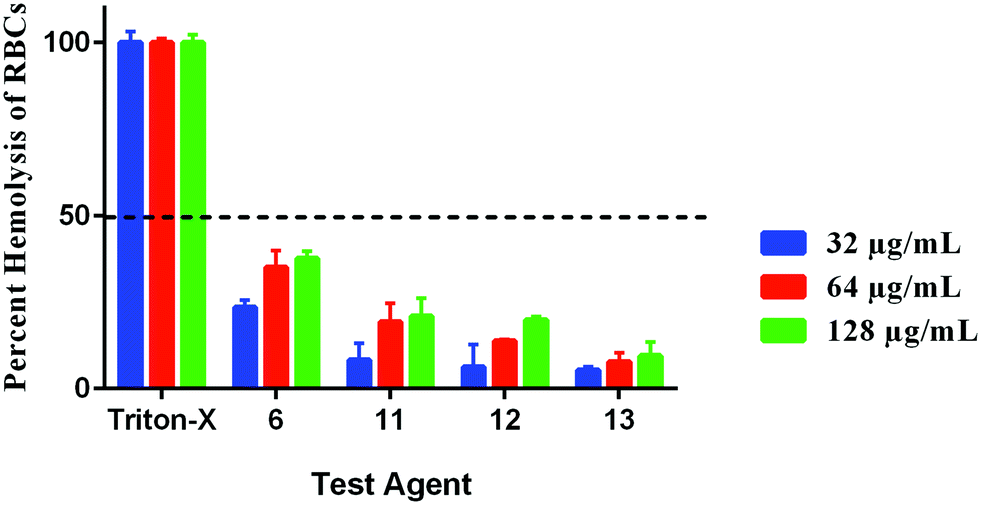
Hemolysis can have serious implications on organ function. For instance, hemolysis has been shown to cause reduced nitrous oxide signaling, resulting in higher systolic, diastolic and mean arterial blood pressure, as well as cardiovascular and renal dysfunction, inflammation, thrombosis, and enhanced susceptibility to infections.25 Thus, we sought to determine if our compounds would lyse RBCs. All compounds were safe to RBCs at concentrations higher than 128 μg mL−1 as presented by their HC50 values; the concentration of each compound causing 50% hemolysis to RBCs. Compound 6 caused around 37% hemolysis to RBCs at the concentration of 128 μg mL−1 (256 × MIC). Its hemolytic activity dropped to 35% and 23% at the concentrations of 64 and 32 μg mL−1, respectively. Compounds 11, 12, and 13 caused little hemolysis (20%, 19% and 9%, respectively at the concentration of 128 μg mL−1) with minimal hemolytic effect at the lower tested concentrations (Fig. 7). Therefore, these results demonstrate that compounds 6, 11, 12, and 13 will have a minor hemolytic activity to human RBCs when used clinically to treat bacterial infections.
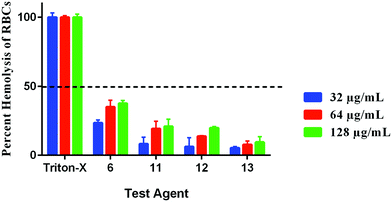 |
| | Fig. 7 Hemolytic activity of compounds 6, 11, 12, and 13 (in triplicate) against human RBCs. The results are presented as percent RBCs hemolysis for each compound relative to triton-X (positive control showing complete hemolysis of RBCs). The absorbance values represent an average of three samples analyzed for each compound. Error bars represent sample standard deviation values. Single donor human red blood cells (RBCs) were purchased from Innovative Research (MI, USA). | |
Compounds 6 & 11 are bacteriostatic, while compounds 12 & 13 are bactericidal against MRSA
With the growing interest in drugs containing trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio, and pentafluorosulfanyl groups,26,27 we proceeded to analyze other aspects of antibacterial activity for compounds 6, 11, 12, and 13. We previously revealed that the 3,5-dimethylpiperidinyl sulfonamide, F6 is bacteriostatic.12 Thus, we measured the minimum bactericidal concentration (MBC) for 6, 11, 12, and 13 against MRSA ATCC 33592. Curiously, compounds 12 and 13 were bactericidal, whereas compounds 6 and 11 were bacteriostatic. Compounds' 12 and 13 MBC values were one-fold higher than their corresponding MICs. On the other hand, the MBC values of compounds 6 and 11 were more than 3-folds higher than their corresponding MICs (Table 3).
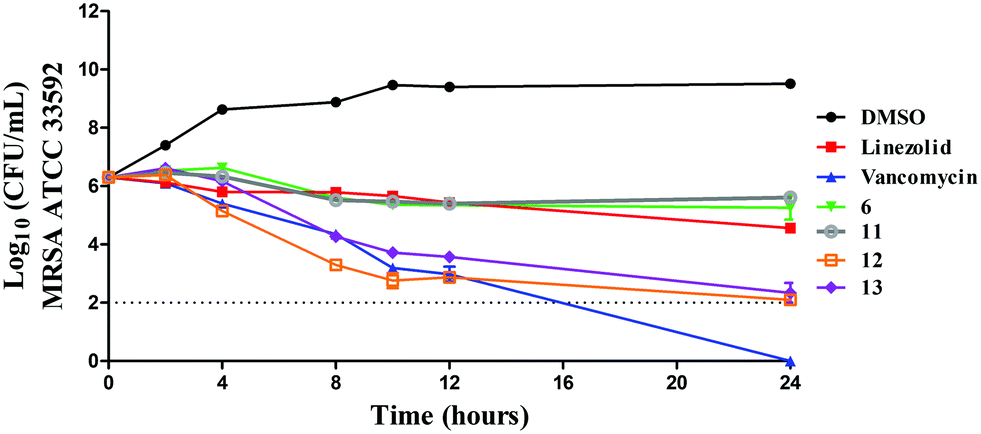
In order to confirm this mode of killing against MRSA, a time-kill kinetics assay was performed against MRSA ATCC 33592 (Fig. 8). In concurrence with the MBC results (Table 5), compounds 6 and 11 exhibited a bacteriostatic activity while compounds 11 and 12 exhibited a bactericidal activity. After 24 hours, compounds 6 and 11 caused a 1.04- and 0.69-log10-reduction in the bacterial count, which was similar to linezolid that caused about 1.7-log10-reduction. In contrast, compounds 12 and 13 caused a 4.2- and 3.96-log10-reduction in CFU mL−1 respectively, which was similar to vancomycin. There are a few examples in the literature whereby researchers have replaced the CF3, NO2 or t-Butyl moieties in drugs or lead compounds with the SF5 unit.19 Here, we provide a cautionary tale that such replacements are not always conservative and could indeed change the mode of action of or response to a drug or lead compound. Future work, beyond the scope of this manuscript, will investigate the specific modes of actions of these compounds.
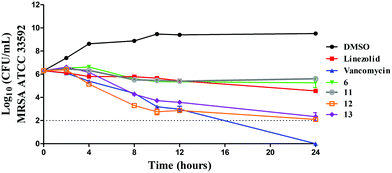 |
| | Fig. 8 Time-kill kinetics analysis of compounds against MRSA ATCC 33592 using vancomycin and linezolid as control antibiotics. | |
Table 5 The minimum bactericidal concentration (MBC, in μg mL−1) of compounds 6, 11, 12, & 13 and control antibiotics against MRSA ATCC 33592
| Compounds/control antibiotics |
MBC (μg mL−1) |
|
6
|
>128 |
|
11
|
>128 |
|
12
|
1 |
|
13
|
1 |
| Vancomycin |
1 |
| Linezolid |
>128 |
Compounds 6 and 12 significantly kill intracellular MRSA
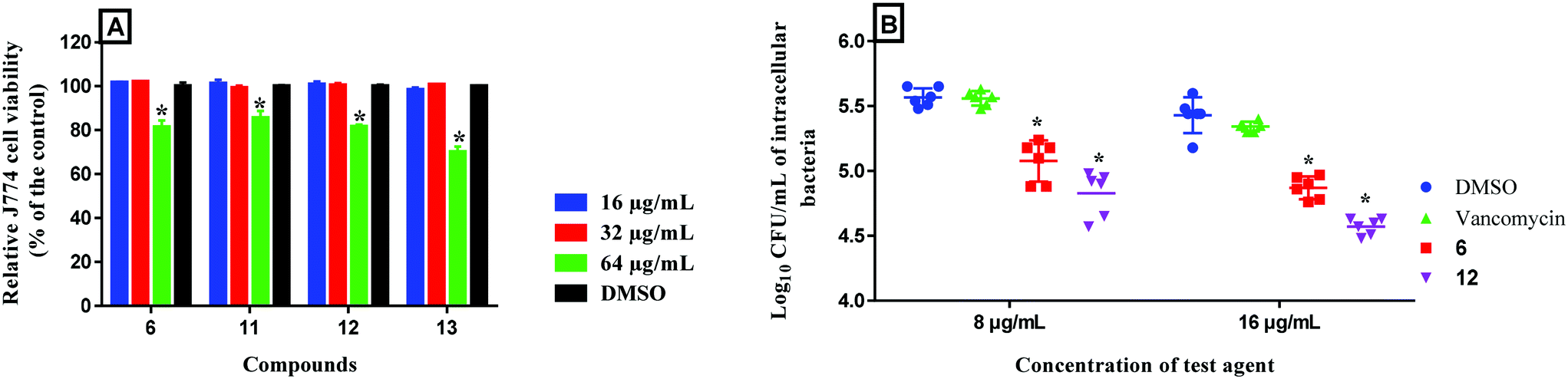
The rapid and potent antibacterial activity of the (1,3,4-oxadiazol-2-yl)benzamide compounds propelled us to investigate whether these compounds could gain entry inside macrophages infected with MRSA to reduce the burden of intracellular bacteria. S. aureus is a highly successful pathogenic bacteria due to extensive release of different virulence factors and its ability to evade host innate immune responses. S. aureus produces toxins, like leukocidin A/B, that are able to specifically target and kill phagocytes.28 Additionally, it has been reported that intracellular MRSA can replicate within the phagolysosome after phagocytosis by macrophages thus permitting the organism to survive, escape from phagocytes, disseminate and cause chronic and persistent infections.29 Most antibiotics, such as linezolid, β-lactams, vancomycin, aminoglycosides and oritavancin, are unable to target intracellular bacteria.30 Consequently, antibacterial compounds capable of gaining entry inside infected macrophages are needed. In this regard, we tested the activity of compounds 6 (bacteriostatic compound) and 12 (bactericidal compound) in clearing intracellular MRSA. First, we tested the toxicity profile of compounds 6, 11, 12 and 13 against murine macrophages (J774), used for the intracellular infection experiment, and they showed excellent safety profile (Fig. 9A) where they were tolerable at a concentration as high as 32 μg mL−1. Therefore, we tested two of the compounds at 8 and 16 μg mL−1. As depicted in Fig. 9B, after 24 hours incubation, compound 6 generated a 0.5- and 0.6-log10 reduction of intracellular MRSA at 8 and 16 μg mL−1 respectively. On the other hand, compound 12 effectively reduced the intracellular MRSA count where it generated 0.7- and 0.9-log10 reduction of intracellular MRSA at 8 and 16 μg mL−1 respectively. This activity was superior to vancomycin which was unable to reduce the burden of intracellular MRSA. This result indicates that (1,3,4-oxadiazol-2-yl)benzamides are advantageous over vancomycin in the ability to reduce the burden of intracellular MRSA within infected macrophages.
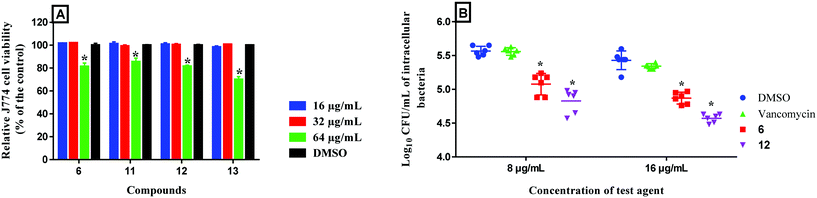 |
| | Fig. 9 A) Toxicity analysis of compounds 6, 11, 12 and 13 (tested in triplicate at 16, 32 and 64 μg mL−1) against murine macrophage (J774) cells using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. The results are presented as percent viable cells relative to DMSO (negative control to determine a baseline measure for the cytotoxic impact of each compound). The absorbance values represent an average of three samples analyzed for each compound. Error bars represent sample standard deviation values. The data were analyzed via two-way ANOVA with post-hoc Dunnett's test for multiple comparisons to determine statistical difference between the values obtained for each compound and DMSO (*, P < 0.05). B) Effect of compounds 6 and 12 and vancomycin to reduce intracellular MRSA present inside murine macrophages (J774). Data are presented as log10 colony forming units of MRSA USA400 per mL inside infected murine macrophages after treatment with 8 and 16 μg mL−1 of either compounds 6, 12 or vancomycin (tested in triplicates) for 24-hours. Data were analyzed via two-way ANOVA, with post hoc Dunnet's test for multiple comparisons (P < 0.05), utilizing GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). The asterisk (*) represents a significant difference between the treatment of J774 cells with compounds 6 or 12 in comparison to vancomycin. J774 cell line (ATCC TIB-67) was obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA). | |
Synergistic interactions of compounds 6, 11, 12, & 13 with standard antibiotics
Combination therapy is commonly used in the clinic to treat bacterial infections. For instance, beta-lactams like piperacillin, ceftaroline, and oxacillin are consistently combined with either vancomycin or daptomycin for the treatment of MRSA infections.31 Therefore, we investigated the synergistic interactions of our compounds with 4 antibiotics. Interestingly, we discovered that compounds 12 and 13 synergized with methicillin, reducing methicillin's MIC from 128 μg mL−1 to 32 μg mL−1 against MRSA ATCC33592 (Table 6) with a fractional inhibitory concentration index (FICI) of 0.5. Compounds 6 and 11 showed additivity when combined with methicillin, with compound 6 reducing methicillin's MIC to 4 μg mL−1. Furthermore, compound 11 demonstrated synergy with daptomycin, reducing its MIC from 0.5 μg mL−1 to 0.125 μg mL−1. Daptomycin can cause nephrotoxicity and hepatotoxicity32 so the potential to reduce the dosage needed for daptomycin could be clinically important. Compounds 6, 12, and 13 demonstrated additivity with daptomycin but these same compounds did not synergize with either linezolid or vancomycin (Table 6).
Table 6 The cumulative fractional inhibitory concentration index (∑FICI) range of compounds 6, 11, 12, and 13 in combination with antibiotics against MRSA ATCC 33592. ∑FICI was interpreted as follows: ∑FICI of ≤0.5 is considered to demonstrate synergy (SYN). An ∑FICI of >0.5–1.25 was categorized as additive (ADD). ∑FICI of >1.25–4 was considered as indifference (IND), while ∑FICI values of >4 were categorized as antagonistic
| Entry |
Antibiotic |
MIC alone |
Combination MIC |
∑FICI |
SYN/ADD/IND |
Combination MIC |
∑FICI |
SYN/ADD/IND |
Combination MIC |
∑FICI |
SYN/ADD/IND |
Combination MIC |
∑FICI |
SYN/ADD/IND |
| Antibiotic |
Antibiotic |
6
|
Antibiotic |
11
|
Antibiotic |
12
|
Antibiotic |
13
|
| 1 |
Linezolid |
2 |
2 |
0.0625 |
1.03 |
ADD |
2 |
0.25 |
1.25 |
ADD |
2 |
0.0625 |
1.06 |
ADD |
2 |
0.5 |
1.50 |
IND |
| 2 |
Methicillin |
128 |
4 |
1 |
0.53 |
SYN |
16 |
0.5 |
0.63 |
ADD |
32 |
0.25 |
0.50 |
SYN |
32 |
0.25 |
0.50 |
SYN |
| 3 |
Daptomycin |
0.50 |
0.25 |
0.25 |
0.63 |
ADD |
0.125 |
0.25 |
0.50 |
SYN |
0.25 |
0.125 |
0.63 |
ADD |
0.25 |
0.25 |
0.75 |
ADD |
| 4 |
Vancomycin |
1 |
1 |
0.0625 |
1.03 |
ADD |
1 |
0.0625 |
1.06 |
ADD |
0.5 |
0.5 |
1.00 |
ADD |
0.5 |
0.5 |
1.00 |
ADD |
Conclusion
We have identified promising trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio, and pentafluorosulfanyl (1,3,4-oxadiazol-2-yl)benzamides with potent antibacterial activity against MRSA, VRSA and S. pneumoniae. Additionally, they showed acceptable tolerability to human cell lines and did not lyse RBCs. Furthermore, compounds 6 and 12 outperformed vancomycin in clearing intracellular MRSA inside infected macrophages. Moreover, compounds 12 and 13 behaved synergistically when combined with methicillin exhibiting ∑FICI of 0.50 against MRSA ATCC 33592 while compound 11 behaved synergistically with daptomycin exhibiting ∑FICI of 0.50 against the same strain. Consequently, these compounds could be potentially used alone or in combination with antibiotics against multidrug-resistant staphylococci. Overall, this work adds to the increasing number of reports that have attempted to address the anti-bacterial resistance issue with novel small molecules.33–45
Funding sources
NMR and MS data were acquired by the NMR and MS facilities supported by NIH P30 CA023168. We thank Purdue University for funding.
Conflicts of interest
There are no conflicts to declare.
References
- World Health Organization, WHO publishes list of bacteria for which new antibiotics are urgently needed, 2017.
- S. Y. C. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603 CrossRef CAS PubMed.
- H. F. Chambers and F. R. Deleo, Nat. Rev. Microbiol., 2009, 7, 629–641 CrossRef CAS PubMed.
- S. E. Cosgrove, G. Sakoulas, E. N. Perencevich, M. J. Schwaber, A. W. Karchmer and Y. Carmeli, Clin. Infect. Dis., 2003, 36, 53–59 CrossRef PubMed.
- R. C. Moellering, Jr., Clin. Infect. Dis., 2008, 46, 1032–1037 CrossRef PubMed.
-
T. Frieden, 2013, https://www.cdc.gov/drugresistance/threat-report-2013/.
- R. V. Rasmussen, V. G. Fowler, Jr., R. Skov and N. E. Bruun, Future Microbiol., 2011, 6, 43–56 CrossRef CAS PubMed.
- P. Huovinen, Clin. Infect. Dis., 2001, 32, 1608–1614 CrossRef CAS PubMed.
- A. Azhar, S. Rasool, A. Haque, S. Shan, M. Saeed, B. Ehsan and A. Haque, J. Med. Microbiol., 2017, 66, 1328–1331 CrossRef CAS PubMed.
- G. K. Mangala, P. N. S. Rao, G. Vishwanath, K. Suresh and V. Vijayanath, J. Pure Appl. Microbiol., 2012, 6, 407–410 Search PubMed.
- M. Roch, P. Gagetti, J. Davis, P. Ceriana, L. Errecalde, A. Corso and A. E. Rosato, Front Microbiol., 2017, 8, 2303 CrossRef PubMed.
- C. Opoku-Temeng, G. A. Naclerio, H. Mohammad, N. Dayal, N. S. Abutaleb, M. N. Seleem and H. O. Sintim, Eur. J. Med. Chem., 2018, 155, 797–805 CrossRef CAS PubMed.
- T. Fan, W. Meng, Y. Xiao and X. Zhang, Tetrahedron Lett., 2017, 58, 4473–4475 CrossRef CAS.
- Y. M. Zhang, Y. C. Wang, C. He, X. R. Liu, Y. Z. Lu, T. T. Chen, Q. Pan, J. F. Xiong, M. Q. She, Z. C. Tu, X. C. Qin, M. K. Li, M. D. Tortorella and J. J. Talley, J. Med. Chem., 2017, 60, 4135–4146 CrossRef CAS PubMed.
- P. B. Curtis-Prior and M. Prouteau, Int. J. Obes., 1983, 7, 575–581 CAS.
- G. Bensimon, L. Lacomblez and V. Meininger, N. Engl. J. Med., 1994, 330, 585–591 CrossRef CAS PubMed.
- A. J. Forhead, J. K. Jellyman, K. Gillham, J. W. Ward, D. Blache and A. L. Fowden, J. Endocrinol., 2011, 208, 137–145 CrossRef CAS PubMed.
-
M. W. Harrold and R. M. Zavod, Basic Concepts in Medicinal Chemistry, American Society of Health-System Pharmacists, Inc., Bethesda, MD, 1st edn, 2013 Search PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- M. Otto, Nat. Rev. Microbiol., 2009, 7, 555 CrossRef CAS PubMed.
- H. A. Khan, A. Ahmad and R. Mehboob, Asian Pac. J. Trop. Biomed., 2015, 5, 509–514 CrossRef.
- R. Rajamuthiah, B. B. Fuchs, A. L. Conery, W. Kim, E. Jayamani, B. Kwon, F. M. Ausubel and E. Mylonakis, PLoS One, 2015, 10, e0124595 CrossRef PubMed.
-
CDC, Control and Prevention, Antibiotic resistance threats in the United States, 2013, Centres for Disease Control and Prevention, US Department of Health and Human Services, 2013 Search PubMed.
- F. Rapido, J. Geophys. Res. Space Physics, 2017, 15, 218–221 Search PubMed.
- M. F. Sowaileh, R. A. Hazlitt and D. A. Colby, ChemMedChem, 2017, 12, 1481–1490 CrossRef CAS PubMed.
- G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941–951 CrossRef CAS PubMed.
- J. H. Melehani, D. B. James, A. L. DuMont, V. J. Torres and J. A. Duncan, PLoS Pathog., 2015, 11, e1004970 CrossRef PubMed.
- R. S. Flannagan, B. Heit and D. E. Heinrichs, Cell. Microbiol., 2016, 18, 514–535 CrossRef CAS PubMed.
- P. Vanderauwera, G. Prinz and G. Petrikkos, Infection, 1991, 19, S216–S223 CrossRef CAS PubMed.
- R. Bartash and P. Nori, Int. J. Infect. Dis., 2017, 63, 7–12 CrossRef CAS PubMed.
- G. Abraham, D. Finkelberg and L. M. Spooner, Ann. Pharmacother., 2008, 42, 719–721 CrossRef CAS PubMed.
- A. Antonoplis, X. Zang, M. A. Huttner, K. K. L. Chong, Y. B. Lee, J. Y. Co, M. R. Amieva, K. A. Kline, P. A. Wender and L. Cegelski, J. Am. Chem. Soc., 2018, 140, 16140–16151 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, H. Tan, Y. S. Kim, S. Jin, H. Luesch and R. W. Huigens, 3rd, J. Med. Chem., 2018, 61, 3962–3983 CrossRef CAS PubMed.
- W. M. Huggins, W. T. Barker, J. T. Baker, N. A. Hahn, R. J. Melander and C. Melander, ACS Med. Chem. Lett., 2018, 9, 702–707 CrossRef CAS PubMed.
- W. Kim, A. D. Steele, W. Zhu, E. E. Csatary, N. Fricke, M. M. Dekarske, E. Jayamani, W. Pan, B. Kwon, I. F. Sinitsa, J. L. Rosen, A. L. Conery, B. B. Fuchs, P. M. Vlahovska, F. M. Ausubel, H. Gao, W. M. Wuest and E. Mylonakis, ACS Infect. Dis., 2018, 4, 1540–1545 CrossRef CAS PubMed.
- B. Li, S. Ni, F. Mao, F. Chen, Y. Liu, H. Wei, W. Chen, J. Zhu, L. Lan and J. Li, J. Med. Chem., 2018, 61, 224–250 CrossRef CAS PubMed.
- H. K. Lui, W. Gao, K. C. Cheung, W. B. Jin, N. Sun, J. W. Y. Kan, I. L. K. Wong, J. Chiou, D. Lin, E. W. C. Chan, Y. C. Leung, T. H. Chan, S. Chen, K. F. Chan and K. Y. Wong, Eur. J. Med. Chem., 2019, 163, 95–115 CrossRef CAS PubMed.
- C. Opoku-Temeng, N. Dayal, J. Miller and H. O. Sintim, RSC Adv., 2017, 7, 8288–8294 RSC.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- I. Rowe, M. Guo, A. Yasmann, A. Cember, H. O. Sintim and S. Sukharev, Int. J. Mol. Sci., 2015, 16, 17909–17932 CrossRef CAS PubMed.
- J. R. Smith, J. Yim, S. Rice, K. Stamper, R. Kebriaei and M. J. Rybak, Antimicrob. Agents Chemother., 2018, 62, e00101-18 CrossRef PubMed.
- J. Wang and H. O. Sintim, Chem. – Eur. J., 2011, 17, 3352–3357 CrossRef CAS PubMed.
- K. R. Robinson, J. J. Mills and J. G. Pierce, ACS Med. Chem. Lett., 2019, 10, 374–377 CrossRef CAS PubMed.
- A. Q. Cusumano and J. G. Pierce, Bioorg. Med. Chem. Lett., 2018, 28, 2732–2735 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of compounds (1H, 13C NMR and HRMS). Bacterial strains, media, cell lines and reagents used. Protocols for the determination of the MICs and MBCs against bacteria, synergy of compounds with standard antibiotics, in vitro cytotoxicity analysis of compounds 6, 11, 12 and 13 against human colorectal and murine macrophage cells, hemolysis assay, time-kill kinetics analysis, intracellular infection of J774 cells with MRSA and treatment with compounds 6 and 12. Table of bacterial isolates used in this study. See DOI: 10.1039/c9md00391f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 bc and
Herman O.
Sintim
bc and
Herman O.
Sintim