
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallophthalocyanines in a ternary photoactive layer (P3HT:MPc:PC70BM) for bulk heterojunction solar cells†
Denisha
Gounden
 a,
Michael N.
Pillay
a,
Timo
Raab
b,
Nolwazi
Nombona
c,
Lukas
Schmidt-Mende
b and
Werner E.
van Zyl
*a
a,
Michael N.
Pillay
a,
Timo
Raab
b,
Nolwazi
Nombona
c,
Lukas
Schmidt-Mende
b and
Werner E.
van Zyl
*a
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa. E-mail: vanzylw@ukzn.ac.za
bFaculty of Physics, University of Konstanz, Konstanz, Germany
cDepartment of Chemistry, University of Pretoria, Pretoria, 0028, South Africa
First published on 16th October 2020
Abstract
Novel bulk heterojunction (BHJ) organic photovoltaic (OPV) solar cells have been fabricated by introducing a series of metallophthalocyanines (MPcs) into a blend of a poly(3-hexylthiophene-2,5-diyl) (P3HT) and [6,6]-phenyl C70 butyric acid methyl ester (PC70BM) photoactive layer. Sixteen MPcs have been synthesised with due consideration of the metal type and valency, functionalised ligand moieties and their respective molecular energy levels. The MPcs were expected to act as an electron cascade material that improves the photovoltaic performance in a two-fold manner: first, to provide an efficient energy level offset between the polymeric donor (P3HT) and the fullerene-based derivative acceptor (PC70BM) to enhance charge transfer and reduce recombination within the OPV solar cells, and second, to harvest a greater portion of the solar spectrum as MPcs are associated with absorption near the near-infrared region. BHJ OSCs were fabricated in the following inverted configuration: ITO/TiO2/P3HT:MPc:PC70BM/WO3/Ag and were investigated for their optimum photovoltaic performance. The BHJ OSCs comprising P3HT, PC70BM and the manganese MPc (MnPc) series photoactive layer displayed the best PCEs in the range of 2.36–2.62% which were approximately 47.5–63.7% higher than those of the binary reference BHJ OSCs.
Introduction
Organic solar cells (OSCs) have been promulgated as promising candidates for 3rd-generation PV devices due to their abundant, lightweight, and flexible materials and low-cost solution processing techniques for manufacturing.1,2 Importantly, the energy invested in manufacturing OSCs can be recovered in a shorter time period than it took to produce it while causing minimal damage to the environment.3,4 Improvements towards the power conversion efficiencies (PCEs) of OSCs, in particular, BHJ OSCs, have been exponential, with Zhu et al. (2020) recently reporting a PCE close to 17%.5 The BHJ structure is considered an effective strategy for obtaining high-performance solar cells due to the bi-continuous interpenetrating network between donor and acceptor materials that provides superior exciton dissociation and charge carrier mobilities in the active layer.6 The PV device performance is generally characterised by its PCE, which depends on several vital processes. These processes include (i) harvesting a greater portion of the solar spectrum by the active layer to generate more excitons,7 (ii) effective exciton dissociation and charge transport at the donor/acceptor interface to prevent recombination,8 and (iii) interfacial modification to provide efficient charge carrier collection by the electrodes.9 Therefore, chemical and physical engineering are necessary for PCE improvement. Using a ternary structure approach is one method to improve the PV performance in comparison to conventional polymer–fullerene binary systems. Various materials, including dyes,10 small organic molecules,11 polymers12 and graphene-based materials13 can be used as the additional third component to offer synergistic effects towards PV performance. In particular, the selection of an additive component can provide optical properties that can extend the absorption range of the active layer, improve the morphology of the active layer at the donor/acceptor interface for superior exciton dissociation and assist with efficient charge transport.14,15Metallophthalocyanines are suitable ternary components due to their most basic photoinduced sensitizer features as well as valuable hole transport properties that originate from their tuneable conjugated framework.16,17 As a ternary component, MPcs display inherently strong absorption within the red region of the solar spectrum and may contribute to higher exciton generation in conjunction with P3HT.18 They spontaneously self-assemble into thin films that favour molecular π–π stacking that can be advantageous towards the active layer.19 In this study, traditional and novel tetra-substituted, solution-processable MPcs have been synthesised as potential ternary additives to a pre-existing binary system comprising PC70BM and P3HT (Fig. 1) to form an overall “ternary system” to improve the performance of the BHJ cells. The MPcs synthesised in this work can be classified according to the valency of the central metals within the Pc framework and the various moieties placed at their peripheral (β) positions. Four different transition metals were examined, cobalt (Co), nickel (Ni), titanium (Ti) and manganese (Mn). The planar coordinating Co and Ni metal centres and the axially coordinating Ti and Mn metal centres were synthesised in conjunction with four different bulky substitutes placed at the β positions.
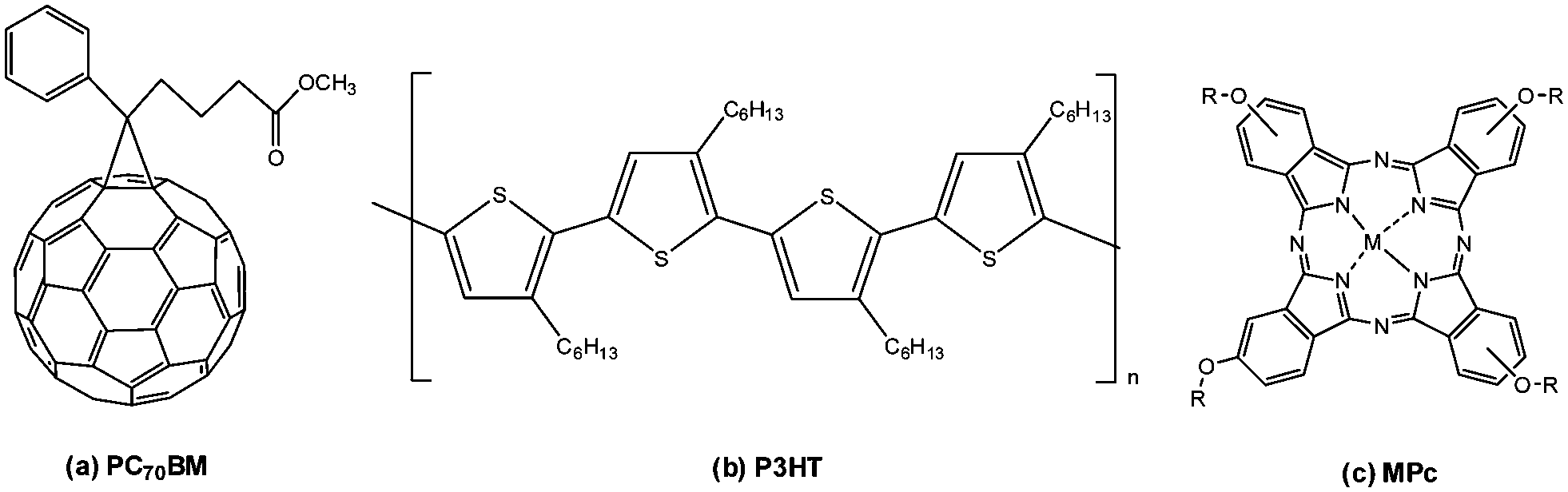 | ||
| Fig. 1 Molecular structure of the organic materials used in the photoactive layer: (a) PC70BM, (b) P3HT and (c) MPcs. | ||
The most important criterion for the integration of MPcs as the additive layer between P3HT and PC70BM is the alignment of their respective energy levels for the effective transfer of charge carriers. The HOMO and the LUMO of the MPcs need to be located between the corresponding energy levels of the electron-accepting (PC70BM) and electron-donating (P3HT) materials to form a cascading energy level alignment to improve charge transfer. The experimental energy levels for the MPcs were determined using UV-vis spectroscopy and photoelectron spectroscopy in air (PESA) and compared well to the theoretical energy levels as derived from density functional theory (DFT) studies.
Results and discussion
Synthetic procedures
Scheme 1 provides the general synthetic pathway followed to obtain the four phthalonitrile derivative precursors (2a–2d) and the series of MPcs (3a–3d, 4a–4d, 5a–5d, and 6a–6d). Briefly, the phthalonitrile derivatives were synthesised in dry DMF, to which anhydrous K2CO3 acted as a base-catalyst towards the nucleophilic aromatic nitro-displacement of 4-nitrophthalonitrile with either R = (a) 4-tert-butylphenol; (b) 2-naphthol; (c) 3-hydroxyacetaphenone or (d) 3-hydroxybenzoic acid.20,21 The cyclotetramerisation of the phthalonitrile derivatives into the corresponding MPcs was carried out under reflux in a high boiling point solvent (i.e. 1-pentanol) in the presence of the respective metal salt and cyclizing catalyst, DBU. The resulting MPcs showed excellent solubility in organic solvents CH3Cl, DCM and THF presumably as a result of functionalization at peripheral (β) positions (2 or 3), (9 or 10), (16 or 17) and (23 or 24). Naturally, the resultant metal phthalocyanines are a mixture of positional isomers.22 Purification of the MPcs was achieved using a variety of methods including TLC, column chromatography and Soxhlet extraction. The characterisation of previously reported (3a, 3b, 3d, 4a, 4b, 4d, 5a, 6b) and, to our knowledge, novel (6a, 5b, 3c, 4c, 5c, 6c, 5d, 6d) complexes involved a combination of techniques such as elemental analysis, FT-IR, UV-vis and MALDI-TOF. All the results agree with the proposed structures outlined in Scheme 1.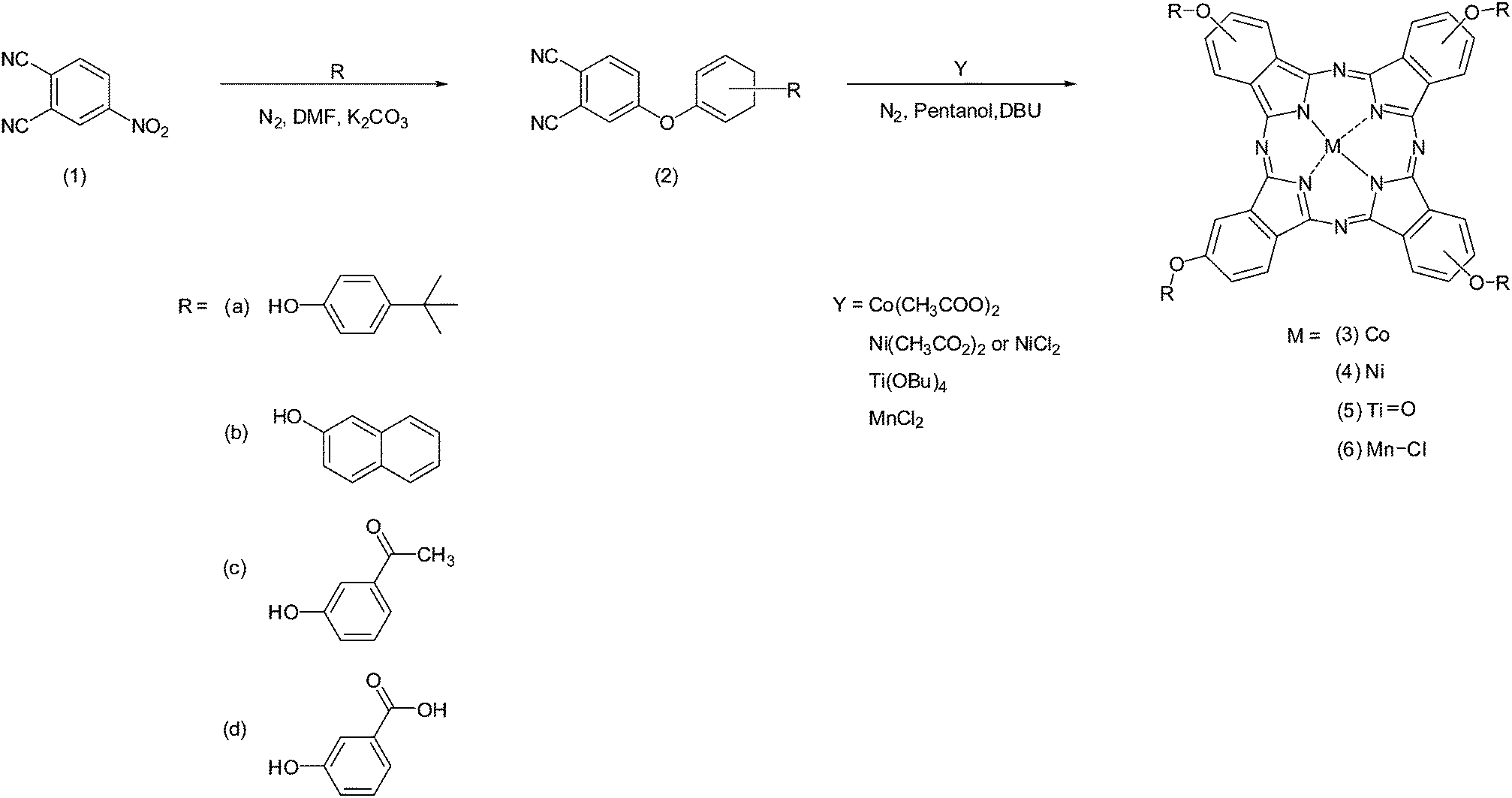 | ||
| Scheme 1 General synthetic pathway for the synthesis of MPcs: 3a–d, 4a–d, 5a–d and 6a–d. | ||
Characterisation of MPcs
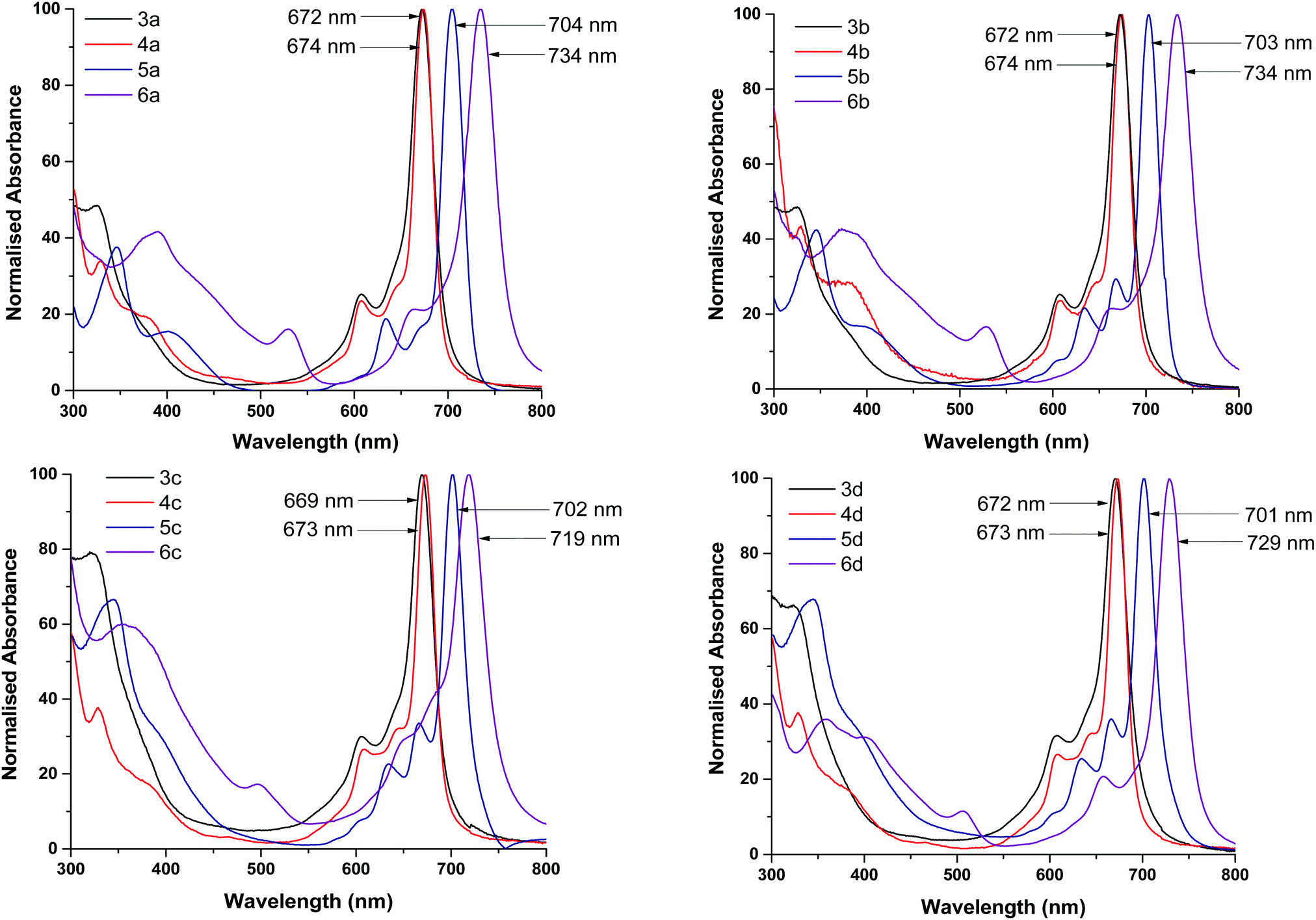 | ||
| Fig. 2 UV-vis absorption spectra of MPc complexes (3a–3d, 4a–4d, 5a–5d and 6a–6d) in solution (CH3Cl or THF). | ||
The absorption spectra of Pcs have been studied extensively by Edward and Gouterman who have reported identifiable Q and B bands.23 These electronic absorption responses originate from the extended aromatic system of the Pc skeleton and are responsible for the chromophore behaviour of the Pcs.24 Typical UV-vis spectra were observed for all Pc complexes. The intense single Q bands seen in the red region were obtained as a result of excitation from the highest occupied molecular orbital (HOMO, a1u) to the lowest unoccupied molecular orbital (LUMO, eg) while B bands appearing at lower energies in the blue region were as a result of transitions from a2u and b2u to the eg orbitals.25 The Q and B bands are characteristic to Gouterman's 4-orbital model corresponding to the D4h symmetry of a metal porphyrin and responsible for the intense blue-green colours of the MPcs.26 The Q bands of the axially coordinated TiPc and MnPc complexes lie between 704 nm and 734 nm and have a bathochromic shift in comparison to the planar Co and Ni complexes whose Q bands lie between 669 and 674 nm. These shifts are due to changes in the electronic distribution of the Pc framework resulting in the increase or decrease of the HOMO–LUMO energy gap. The electronic distribution in the macrocyclic structure is influenced by both the central metal and electron-donating or withdrawing nature of the surrounding substituents. The bathochromic shift of the Q bands for the TiPc and MnPc complexes correspond to the lowering of the HOMO–LUMO energy gap whilst the hypsochromic shift of the Q bands in the Co and Ni complexes corresponds to the increase of the HOMO–LUMO energy gap.
This result is reaffirmed by the calculated energy gap, see Table 1. There is no noticeable difference in Q band shifts, except for the MnPc complexes when drawing comparisons between different substituents containing the same metal centre. Halogenation often results in dramatic changes in the spectroscopic properties of Pcs.27–30 The Q and B bands of chloro-substituted Pcs undergo a redshift in comparison to non-halogenated Pcs, which indicates that the central metal together with the axially coordinated Cl halogen atom has a greater influence over the Eg than the substituents. Absorption bands appearing in the 360–400 nm region for the Ni and TiPc complexes are due to charge transfers between the central metal and substituted ligands.31 The MnPc complexes display broad bands in the 480–550 nm range that are most likely due to charge transfer from the electron-rich ring to the electron-poor metal centre and have been reported in numerous other studies.32–34 No absorption bands are observed in these regions of the Co complexes. Using spin coating, thin films of the MPcs were cast onto glass substrates to obtain the solid-state UV-vis absorption spectra (ESI†). In the solid-state, all of the maximum absorption peaks of the MPcs undergo varying degrees of blue shifting and to a larger extent peak broadening due to intermolecular π–π stacking within the film.35,36 The number of double bonds within the MPc crystal structure of the films results in an overlap of π-orbitals between the molecules allowing for energy transfer throughout the entire structure.37 Peak broadening in thin films is considered advantageous for OSC applications as it facilitates charge carrier mobility and conductivity.38–40
| Metal | Substituent | No. of electrons in the outer d-orbital | Experimental | Theoretical | ||||
|---|---|---|---|---|---|---|---|---|
| E g (eV) | LUMO (eV) | HOMO (eV) | E g (eV) | LUMO (eV) | HOMO (eV) | |||
| Co | a | 3d7 | 1.77 | −4.63 | −2.86 | 2.15 | −4.75 | −2.61 |
| b | 1.77 | −5.02 | −3.25 | 2.07 | −4.79 | −2.72 | ||
| c | 1.78 | −5.05 | −3.27 | 1.69 | −4.30 | −2.61 | ||
| d | 1.77 | −4.90 | −3.13 | 2.06 | −5.15 | −3.09 | ||
| Ni | a | 3d8 | 1.78 | −4.64 | −2.86 | 2.19 | −4.75 | −2.56 |
| b | 1.78 | −4.97 | −3.19 | 2.04 | −4.79 | −2.74 | ||
| c | 1.79 | −5.00 | −3.22 | 2.06 | −5.09 | −3.03 | ||
| d | 1.79 | −4.85 | −3.06 | 2.20 | −5.22 | −3.03 | ||
| Ti | a | 3d2 | 1.70 | −4.86 | −3.16 | 2.08 | −4.90 | −2.82 |
| b | 1.70 | −4.95 | −3.25 | 1.64 | −4.85 | −3.21 | ||
| c | 1.69 | −5.22 | −3.53 | 1.41 | −5.00 | −3.59 | ||
| d | 1.70 | −4.90 | −3.20 | 2.09 | −5.27 | −3.18 | ||
| Mn | a | 3d5 | 1.61 | −5.06 | −3.45 | 1.49 | −4.85 | −3.35 |
| b | 1.61 | −5.20 | −3.59 | 1.49 | −4.90 | −3.41 | ||
| c | 1.64 | −5.41 | −3.77 | 1.51 | −5.23 | −3.71 | ||
| d | 1.63 | −5.33 | −3.70 | 1.47 | −5.26 | −3.78 | ||
Experimental energy levels (bandgap and HOMO energy levels)
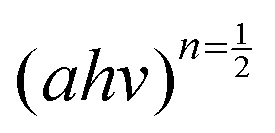 against energy (eV) and can be viewed in the ESI.† The Eg values were obtained by extrapolation of
against energy (eV) and can be viewed in the ESI.† The Eg values were obtained by extrapolation of 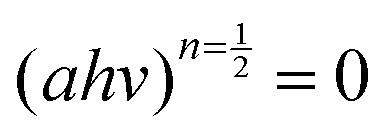 . Table 1 indicates that the optical Eg assumes the following decreasing trend for all substituents: NiPc > CoPc > TiPc > MnPc.
. Table 1 indicates that the optical Eg assumes the following decreasing trend for all substituents: NiPc > CoPc > TiPc > MnPc.
The Eg values for the planar MPcs are slightly larger than the axially coordinated MPcs, with the MnPcs displaying the lowest Eg values. The slight structural modifications brought about by the different moieties on the MPcs do not significantly influence the bandgap. Hence the differences observed in the Eg values appear to originate from the central metal. The CoPcs and NiPcs have similar Eg, which is possibly due to the outermost orbital of the metals being almost filled, reflecting in similar electrostatic behaviour of these complexes.35
Theoretical energy levels
The theoretical HOMO, LUMO and Eg values are recorded in Table 1 and the optimised Cartesian coordinates of the MPcs structures can be viewed in the ESI.† The experimental energy level values compare well to the theoretical energy level values with slight deviations. On comparison with the experimental Eg, the theoretical Eg is found to be higher with deviations of <0.3 eV.Thermogravimetric analysis (TGA)
The thermal stability of the 16 MPcs was investigated by TGA. The planar Co and Ni Pcs show thermal degradation over two or three relatively stable stages while the axially coordinated complexes, TiPcs and MnPcs, show thermal degradation between three and five stages (ESI†). For comparative purposes, the thermal degradation of these metal complexes will be discussed in three broad temperature profiles as specified in Table S8 in the ESI.† In stage I, a minimal weight loss (between 5–10%) was observed as a result of evaporation of residual moisture and possibly the loss of groups attached to peripheral ligands.43 Stage II is identified with a major weight loss and the “major decomposition temperatures” are listed in Table S8 (ESI†). During stage II, the CoPcs and NiPcs lost between 60 and 75% of their weight, whereas the TiPcs and MnPcs showed greater weight loss between 80 and 98%. Kalvelage et al. (2000) carried out investigations on the pyrolysis of CoPcs and FePcs at 600 °C. They reported that the pyrolysis products contained ∼90% phthalimides, ∼8% phthalonitriles and the remaining 2% was recognized as non-cyclic nitrogen-containing and nitrogen-free compounds.44 Using various methods such as mass spectrometry (MS) and gas chromatography (GC-MS), Achar et al. (2007) also described the thermal decomposition of CoPc between 500 °C and 1000 °C and found that the main pyrolysis products reported were benzonitriles, cyanogens, hydrogen cyanide and phthalonitriles.30These results suggest that the weight loss taking place in stage II results from the partial or full decomposition of the macrocyclic structure, where mostly lower weight atoms containing nitrogen are lost from the metal Pc structure.45 The planar coordinated MPcs appear to have better thermal stability as they have higher decomposition temperatures and show lower weight loss during stage II in comparison to the axially coordinated MPcs. This may be due to higher bonding energies of the Co and Ni metal centres with the phthalocyanine skeleton as these metals have almost complete 3d outer orbitals.46 In Stage III, little to no weight loss was observed and this indicates the destruction of the macrocyclic skeleton, leaving behind only the metallic centres. The TGA study provides evidence that the MPcs display sufficient thermal stability to act as additives in a BHJ solar cell device.
Bulk heterojunction solar cell fabrication and testing
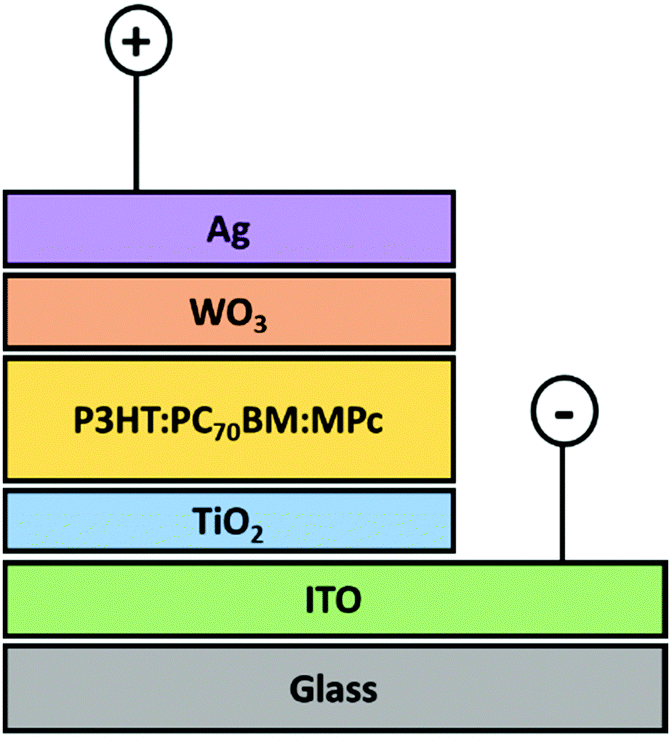 | ||
| Fig. 3 Schematic representation of the triple blended bulk heterojunction device planar configuration. | ||
The synthesised MPcs were incorporated into a blend of PC70BM and P3HT as a modifier to improve the PCEs of the BHJ cells. The addition of an additive will be beneficial only if the LUMO of the MPcs is located below the LUMO of the P3HT donor and above the LUMO of the PC70BM acceptor.47 The energy levels of the BHJ OSC components are shown in Table 2.
The estimated HOMO and LUMO of the P3HT are −5.0 eV and −2.75 eV, respectively. When incident photons excite an electron–hole pair within P3HT, it is expected that the electron will flow towards the cathode (i.e. ITO) via the structured energy cascade made up of the subsequent lower-lying LUMOs of MPc, PC70BM and TiO2 layers. The photogenerated holes can flow to the anode (i.e. Ag) through the WO3 HTM as the HOMO of the WO3 will ensure efficient extraction of the generated holes. Based on these estimated energy levels, the LUMOs of the MPcs match that of the PC70BM (−3.8 eV) and will allow for the effective transfer of electrons.
The energy alignment also ensures that any photogenerated charge carriers emanating from the MPc complexes can flow through the system towards the respective electrodes. Fig. 4a–d show the concise energy level diagrams for the ternary BHJ system used in this study and demonstrate that all sixteen MPcs show the appropriate HOMO–LUMO energy level cascading arrangement to ensure charge transfer and to specifically promote better exciton dissociation at the P3HT:PC70BM interface throughout the blended active layer. Despite the similar band gaps between the sixteen MPcs, the TiPcs (5a–5d) and MnPcs (6a–6d) display lower-lying HOMO–LUMO levels than the CoPcs and NiPcs. Further assessment of the MPc energy levels indicates that the (4a–4d) MPcs (i.e. NiPcs) are similar to that of the (3a–3d) MPcs (i.e. CoPcs) and MPcs substituted with the 2-naphthol group (i.e.3b, 4b and 5b) are similar to the MPcs substituted with the 4-tert-butylphenol group (i.e.3a, 4a and 5a). To eliminate redundancy and potential overlap in results, all the NiPcs (4a–4d) as well as the 3b, 4b, 5b and 6b MPcs were not incorporated in the ternary blended BHJ cells and thus only nine MPcs of the sixteen were used as additives and evaluated under solar simulations.
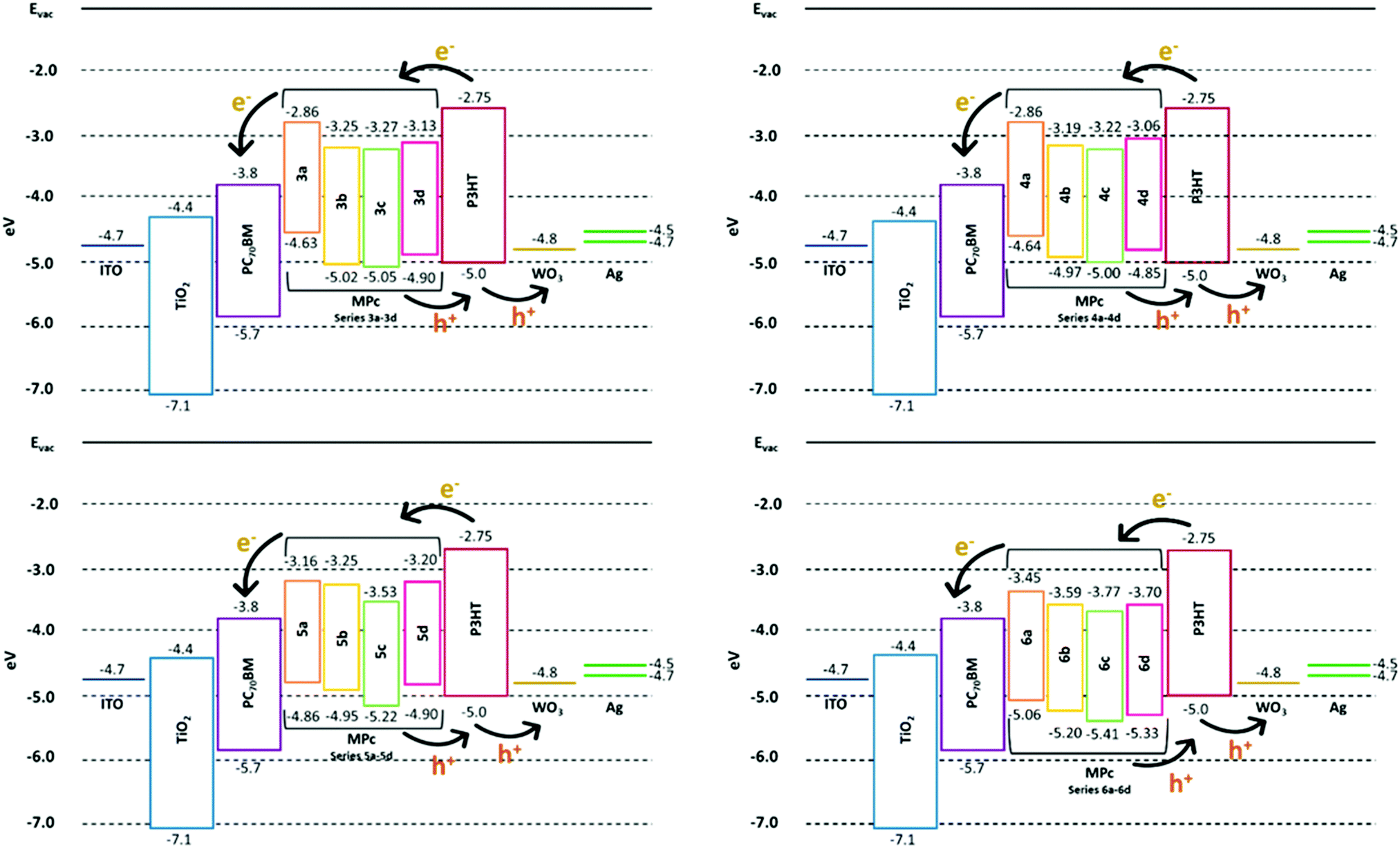 | ||
| Fig. 4 Energy level diagrams of the ternary BHJ system with the CoPc (3a–3d), NiPc (4a–4d), TiPc (5a–5d) and MnPc (6a–6d) complexes as additives. | ||
Optimisation studies: UV-vis spectroscopy
Solid-state UV-vis absorption spectra of the respective ternary blends at varying MPc wt% for 3d, 5d and 6d with a comparative over-lay of the reference P3HT:PC70BM binary blend are shown in Fig. 5, see ESI† for other ternary systems. As expected from the spin-coated films, peaks observed from the solid-state absorption analysis are much broader than the well-defined peaks seen from solution analysis due to intermolecular stacking and as stated previously, advantageous in OPV applications.54 The binary blend shows maximum absorbance at λmax = 465 nm, which is a typical λmax for P3HT films, and there is little to no absorbance in the 650–800 nm region as expected.55 The absorption spectra of the ternary blend are dominated by the P3HT features except for the 650–800 nm region for the TiPcs and MnPcs. The addition of the TiPcs (5a, 5b and 5d) and MnPcs (6a, 6b and 6d) introduced absorption within the 650–800 nm region as a consequence of their Q-bands. These observations correlate with the UV-vis spectra in Fig. 2 and Fig. S10 (ESI†), where the Q bands of the individual TiPcs and MnPcs are observed in the 700 nm region. In the presence of P3HT and PC70BM, a noticeable redshift of the Q bands was observed. One of the advantages of combining MPcs into the photoactive layer is the collective light absorption of the P3HT and MPcs. The increased light-harvesting is expected to enhance photogenerated charge carriers and improve charge transport due to their compatible energy level alignment and π–π interactions between the materials.55 In contrast, the CoPcs exhibited Q bands in the lower 600 nm region and this absorption overlapped with the absorption spectrum of the binary blend and therefore cannot be seen in the spectra. Therefore, only the axially coordinated MPcs were able to expand the absorption window of the blend significantly. Also, all ternary blends displayed improved absorbance within the 300–450 nm region in comparison to the binary blend. Well-defined Q bands of the TiPcs and MnPcs appear to increase with increasing wt%, and as such, ternary blends consisting of the 15 wt%, MPc provided the highest absorbance within the 650–800 nm region. This could indicate that even at the highest wt%, the effects of aggregation did not hinder the absorbance by the MPcs in the 650–800 nm region thereby establishing that the optimum ratio of the ternary P3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC70BM:MPc blend is 1:1.2:0.15 in this study.
:PC70BM:MPc blend is 1:1.2:0.15 in this study.
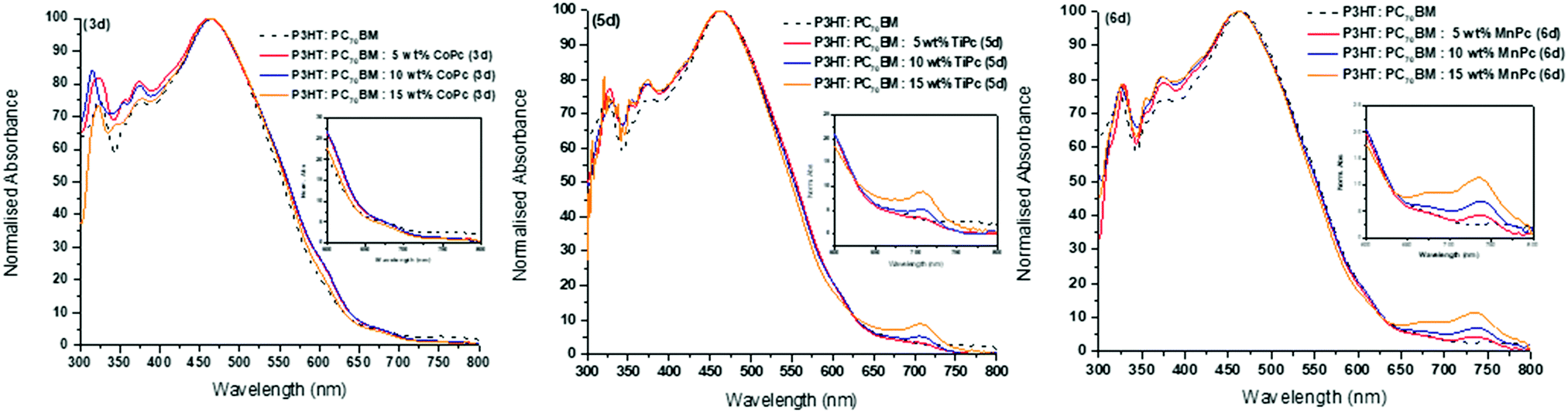 | ||
| Fig. 5 UV-vis spectra of CoPc (3d), TiPc (5d) and MnPc (6d) complexes for optimisation studies. | ||
Photovoltaic performance of ternary BHJ cells
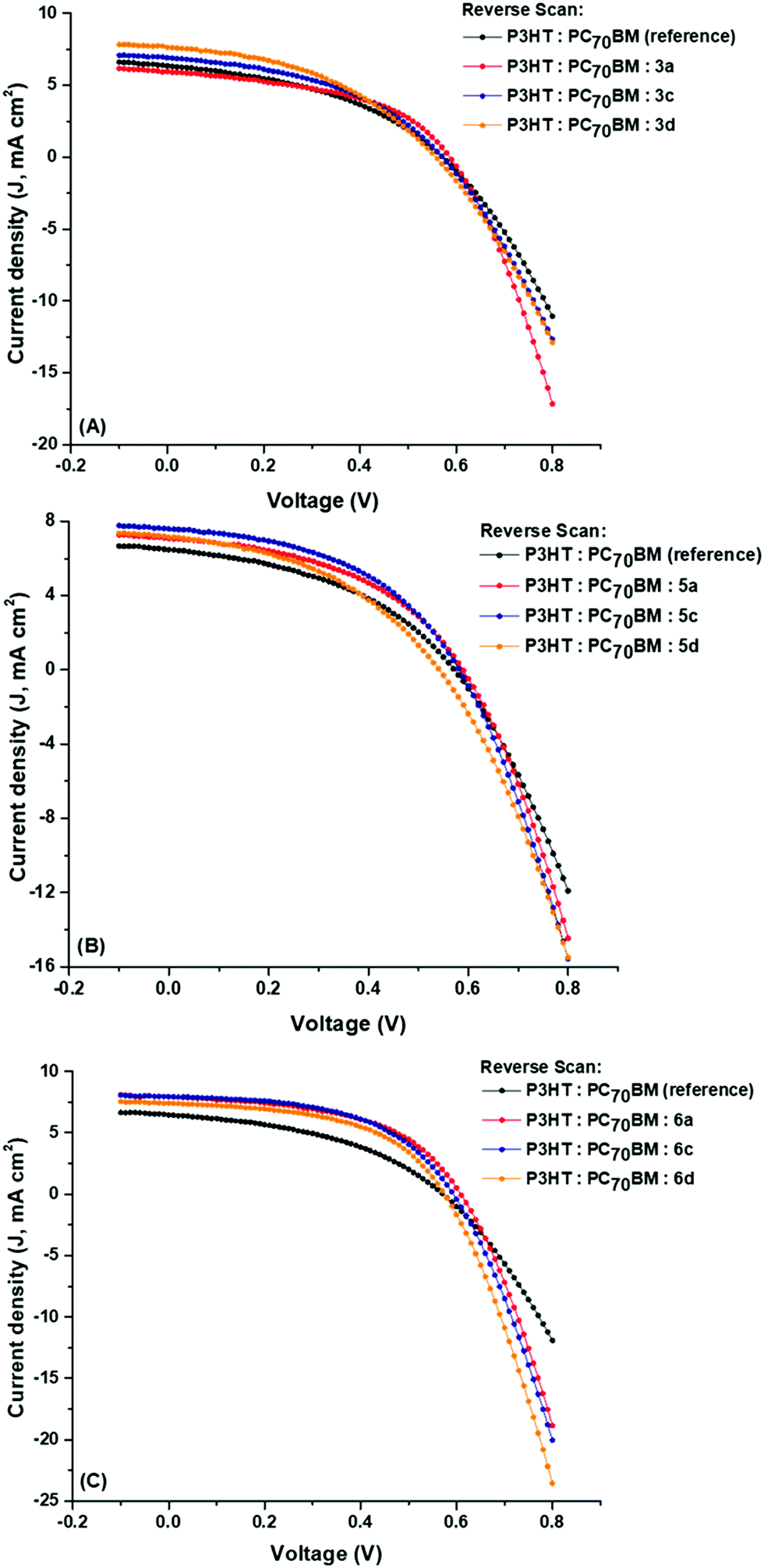
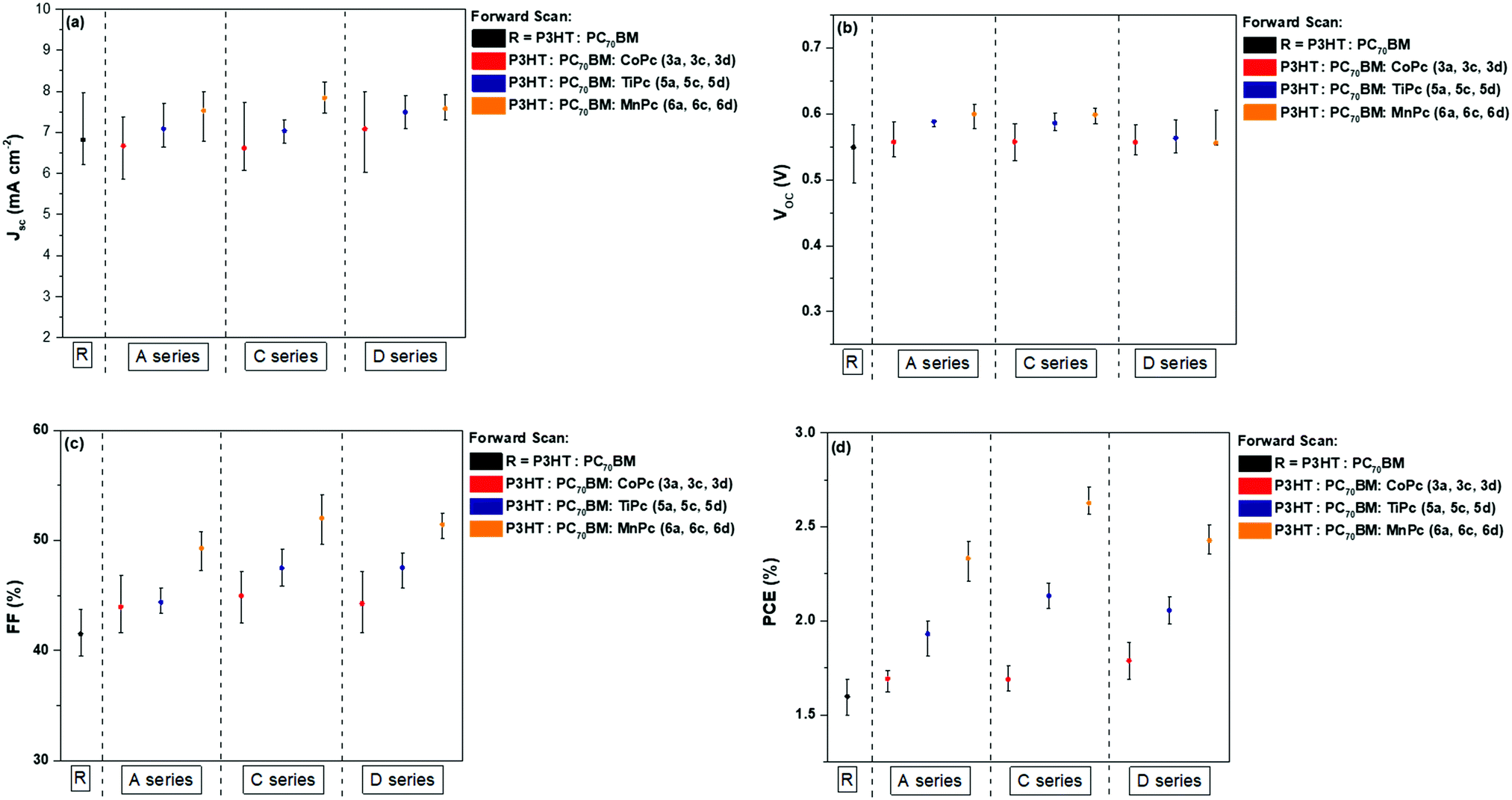
Table 3 summarises the parameters of the reference and the nine ternary BHJ cells where the values are averaged from 10 devices. The J–V curves are shown in Fig. 6, while comparative visual representations of each parameter were constructed and are shown in Fig. 7. Parameters that are used to characterise the performance of the BHJ cells such as the current-density (JSC), the open-circuit voltage (VOC), the fill factor (FF) and the power conversion efficiency (PCE) are all determined from the current density–voltage (J–V) curves as seen below. The J–V curves allow us to study the current generated in the BHJ cells with reference to the area of the active layer. The reference binary BHJ cells displayed the following results: VOC = 0.54 V, JSC = 6.81 mA cm−2, FF = 41.1% and PCE = 1.60%. After introducing the axially coordinated TiPcs and MnPcs, a noticeable increase in the JSC and FF values was recorded. The percent increase in the JSC values for the TiPcs and the MnPcs ranged between 2.71–9.44% and 9.56–13.14% in comparison to the reference device. The higher JSC and FFs produced improved PCEs ranging from 1.93–2.09% and 2.36–2.62% for the TiPcs and MnPcs, respectively.| Ternary blends | Photovoltaic parameters | ||||
|---|---|---|---|---|---|
| P3HT:PC70BM: MPc | Series | J SC (mA cm−2) | V OC (V) | FF (%) | PCE (%) |
| a Data averaged from 10 cells each. | |||||
| P3HT:PC70BM | Ref | 6.81 ± 0.63 | 0.54 ± 0.02 | 41.1 ± 2.36 | 1.60 ± 0.05 |
| P3HT:PC70BM:CoPc | 3a | 6.66 ± 0.50 | 0.56 ± 0.02 | 43.9 ± 1.96 | 1.69 ± 0.03 |
| 3c | 6.62 ± 0.57 | 0.55 ± 0.02 | 44.9 ± 1.76 | 1.69 ± 0.04 | |
| 3d | 7.08 ± 0.54 | 0.56 ± 0.01 | 44.2 ± 2.41 | 1.79 ± 0.07 | |
| P3HT:PC70BM:TiPc | 5a | 7.08 ± 0.36 | 0.56 ± 0.02 | 44.3 ± 0.88 | 1.93 ± 0.06 |
| 5c | 7.36 ± 0.33 | 0.59 ± 0.01 | 47.5 ± 1.28 | 2.15 ± 0.05 | |
| 5d | 7.52 ± 0.32 | 0.57 ± 0.01 | 46.2 ± 2.07 | 2.00 ± 0.04 | |
| P3HT:PC70BM:MnPc | 6a | 7.53 ± 0.41 | 0.60 ± 0.01 | 48.7 ± 2.12 | 2.36 ± 0.07 |
| 6c | 7.84 ± 0.27 | 0.60 ± 0.01 | 52.0 ± 1.57 | 2.62 ± 0.07 | |
| 6d | 7.58 ± 0.23 | 0.55 ± 0.08 | 51.4 ± 0.85 | 2.42 ± 0.05 | |
 | ||
| Fig. 6 J–V curves (forward scans) of BHJ OSCs with ternary components (A) 3a, 3c, 3d, (B) 5a, 5c, 5d and (C) 6a, 6c, 6d. | ||
 | ||
| Fig. 7 Comparative graphs (forward scans) for performance parameters (a) JSC, (b) VOC (c) FF and (d) PCE of BHJ OSCs with MPcs (3a, 3c, 3d, 5a, 5c, 5d and 6a, 6c, 6d) as ternary components. | ||
When comparing between ternary BHJ cells, devices consisting of MnPcs provided the best results, displaying the most improved PCEs between ternary devices. The higher PCE may be explained as follows:
(a) Evaluation of the MnPc molecular orbitals: The comparatively lower-lying LUMOs of the MnPcs could have provided a more efficient route for electron transfer from the P3HT donor. The difference between the LUMO of the acceptor and donor materials is often considered the driving force for charge separation.56,57 Also, the energy cascade provided by the MnPc additives could act as a suppressant towards charge recombination of the P3HT.
(b) Increased JSC values: Higher JSC values may have stemmed from increased absorption at longer wavelengths in comparison to the binary blend. The photogenerated charge carriers will subsequently dissociate between the donor and acceptor interface created by the MPcs and increased the resultant JSC values.
(c) Increased JSC values: The absorption co-efficient (ε) of the nine MPcs was also evaluated to determine the possible effects on the PV parameters. As seen in Table S3 (ESI†), the ε values for CoPcs 3a, 3c and 3d are reported to be 3.54 M−1 cm−1, 3.47 M−1 cm−1 and 3.46 M−1 cm−1 respectively. The ε values are comparatively lower than the ε values obtained for the TiPcs and MnPcs. The MnPcs, 6a, 6c and 6d produced the highest ε values of 3.69 M−1 cm−1, 3.75 M−1 cm−1 and 3.75 M−1 cm−1. The higher ε values indicate that the TiPcs and to a greater extent the MnPcs absorb more light than the CoPcs at the maximum wavelength, where the electronic transitions are between the a1u → eg orbitals. The ability to absorb more light may have contributed to the higher JSC values obtained for the MnPcs.
The VOC values of the reference cells showed minimal to no change when compared to the VOC values of the CoPc and TiPcs but did show a noticeable difference when compared to the MnPcs. One of the factors that have reportedly influenced the VOC parameter is the offset between the HOMO of the donor material and the LUMO of the acceptor material.58 Acting as an additive between the donor and acceptor materials, the significantly lower LUMO of the MnPcs may have contributed to the slightly higher VOC values. When comparing the three different MnPcs (6a, 6c and 6d), the 6c MnPc with the lowest-lying LUMO displayed the highest averaged PCE of 2.62% ± 0.07. This is approximately 63.7% higher than the PCE attained for the reference cell. However, the PCE obtained by the 6a and 6c complexes was not significantly lower, as they produced PCEs of 2.36 ± 0.07 and 2.42 ± 0.05, respectively. Although the substituents shifted the HOMO–LUMO energy levels to varying degrees, they did not have a major impact on the overall cell performance. The slight structural differences between the three moieties functionalised onto the MPcs did not outweigh the influence of the central metal on the cell performance as cell performance varied substantially between metals rather than between the moieties. This was also observed for the CoPcs and TiPcs.
Photocurrent hysteresis is a well-reported phenomenon in thin-film organic films and molecular devices; the forward and reverse scans for the reference and ternary BHJ cells display minor hysteresis (ESI†). The BHJ parameters, JSC, VOC, FF and PCE, for the reverse scans compare superbly with those of the forward scans.
External quantum efficiency (EQE)
To gain further insight into the origin of the increased JSC values upon the addition of the axially coordinated MPcs, the incident photon-to-conversion efficient (IPCE) or the EQE spectra were recorded for the reference and ternary devices, Fig. 8. The EQE measurements reflect the absorption that occurs throughout the solar cell device, and is represented as the ratio between the number of charge carriers collected to the number of incident photons and can be represented by the following equation: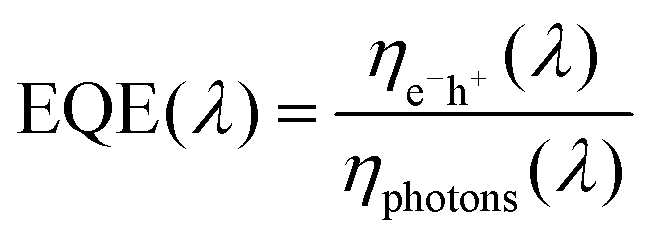
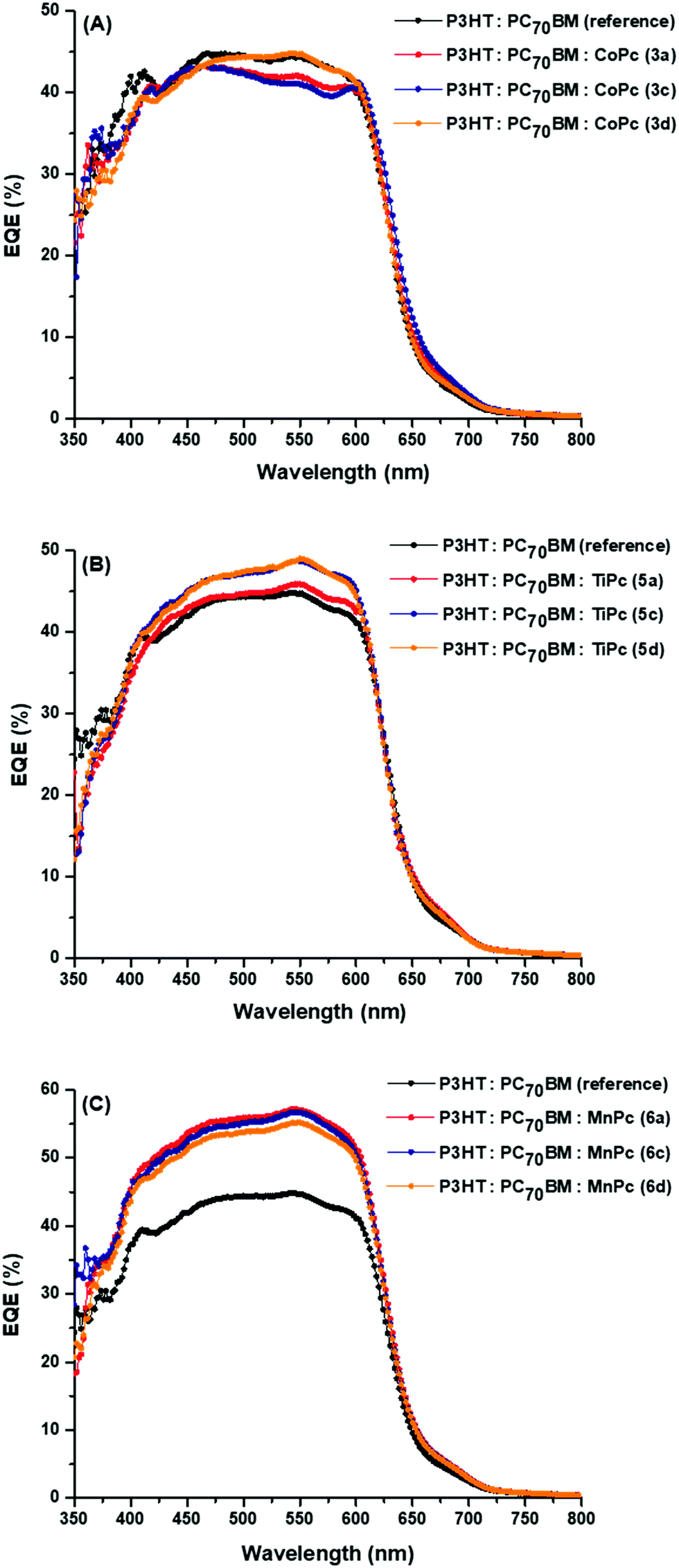 | ||
| Fig. 8 EQE spectra of the BHJ OSCs with ternary components (A) 3a, 5a, 6a (B) 3c, 5c, 6c and (C) 3d, 5d, 6d. | ||
The P3HT:PC70BM reference cell shows peaks at ∼360 nm, ∼410 nm, ∼468 nm and ∼540 nm. The peaks at 360 nm and 410 nm are associated with PC70BM absorption while the latter two peaks are associated with P3HT absorption. As expected, the EQE spectra of the CoPc ternary devices are similar to that of the reference cells but have less pronounced peaks in the 400 nm region. These spectra also indicated that at 540 nm, maximum EQE values between 41% and 45% were recorded for the CoPc ternary BHJ cells, which was close to the 45% reported for the reference cell.
There appears to be no absorption in the 750–800 nm region. This is in contrast with the UV-vis absorption data, as the axially coordinated MPcs displayed noticeable absorption in the IR-region. This suggests that despite absorption in the IR-region, the axially coordinated MPcs in the donor–acceptor blend do not efficiently contribute to photocurrent generation because the separated charges originating from the axial MPcs seem to recombine. This voids the abovementioned explanation in points (b) and (c) put forward to explain the observed increase in PCEs. In addition, certain MPcs (3a, 3d, 5a and 5d) have higher HOMO levels in comparison to P3HT (−5.0 eV) and possibly hinder hole extraction by the P3HT donor. The absence of efficient hole extraction can contribute to charge recombination within the MPc molecules and furthermore lends itself towards the argument that the MPcs were unable to contribute to photocurrent due to recombination of its charge carriers. This is not the case for the LUMO levels of the MPcs, which has the ability to extract and transfer photogenerated electrons to the PC70BM acceptor.
However, the EQE value at 540 nm corresponding to the P3HT absorption significantly increased up to 8% and 20% for the TiPc and MnPc ternary BHJ cells, respectively, suggesting that the TiPcs and more so the MnPcs either improve the charge transfer between the P3HT and PC70BM or hinder the charge recombination from the excitons emanating from the P3HT donor.59,60 This indicates that the MnPcs have the ability to improve the charge extraction and transport at the interface of the donor–acceptor materials.61 Lee et al. (2011) carried out a comparative study between (i) MPc complexes that were synthetically attached to the tail of the P3HT polymer and (ii) MPcs complexes that were mixed into the P3HT:PC70BM as done in this study. The attached MPcs were able to absorb incident light and add to the overall photocurrent generation.
Their corresponding IPCE spectrum also showed the contribution of excitons in the 650–800 nm region as a result of the MPc. Similar to this study, the IPCE spectra of the MPc complexes that were simply mixed into the photoactive blend did not contribute towards photocurrent generation but did assist in improving the charge transfer, and it was suggested that the MPc may have aggregated in the simple blend of P3HT:PC70BM and therefore did not perform as well.62
Stability testing
Although high efficiencies are desirable, the reported values usually represent the initial performance of the OPV cells, therefore monitoring PCE degradation over time is also of importance. The energy output of any solar cell device is the product of its efficiency and lifetime stability. Consequently, the device stability of OPV cells is a vital parameter that is often inferior to that of inorganic silicon-based solar cells that are currently in the market.Though stability testing can last as long as 1000 hours and more, in this study, the PCEs of the nine ternary devices were recorded over a seven-day testing period, see Fig. 9.
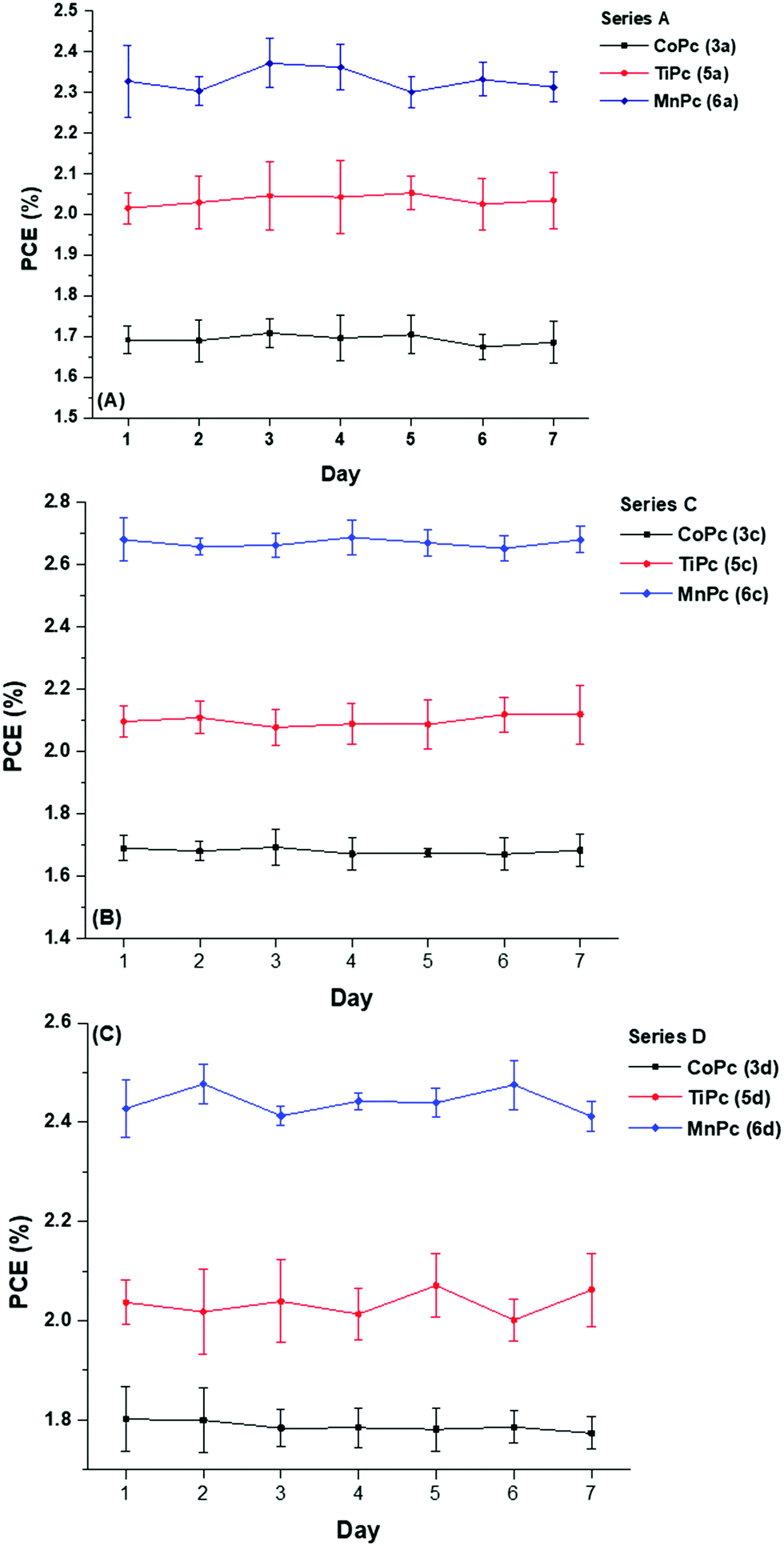 | ||
| Fig. 9 Stability testing of the of BHJ OSCs with ternary components (A) 3a, 5a, 6a (B) 3c, 5c, 6c and (C) 3d, 5d, 6d. | ||
The stability tests were carried out under identical conditions to the initial PV measurements (Table 3), i.e. under standard test conditions (100 mW cm2 under air mass, AM 1.5 global at 289 K, without encapsulation). When not in use, the cells were housed in ordinary plastic dividers and stored at 25 °C. During the seven-day testing period, the recorded results showed little to no deviation when compared to the initially reported values. Device instability can occur due to an array of complex phenomena that take place at any of the layers and/or interfacial layers such as the diffusion of oxygen and water,63 oxidation and eventual rusting of metal electrodes64 and thermal and photoinduced degradation of the active layer.65
Despite the relatively consistent PCE output from the nine BHJ devices over the seven days, longer testing times should be considered to gain a holistic outlook of any potential threats to device stability. In the wake of extensive perovskite solar cell (PSC) research directed towards tackling stability issues, even though the PCEs obtained from PSCs are often comparatively higher than OPV cells, the stability of these BHJ cells under ambient conditions is favourable when compared to the stability of PSC devices developed in recent studies.66,67
Experimental
Materials
Acetic anhydride (Ac2O), acetone (C3H6O), 25% ammonia solution (NH4OH), chloroform (CHCl3), chlorobenzene (C6H5Cl), ethyl acetate (C4H8O2), formamide (HCONH2), 32% hydrochloric acid (HCl), methanol (MeOH, CH3OH), propan-2-ol (IPA, C3H8O) and tetrahydrofuran (THF, C4H8O) were obtained from Merck. Dimethylformamide (DMF, C3H7NO) and 1-pentanol (C5H12O) were obtained from Sigma-Aldrich and dried using molecular sieves before use. Manganese(II) chloride dihydrate (MnCl2·H2O), nickel(II) acetate (Ni(CH3CO2)2·4H2O) and nickel chloride hexahydrate (NiCl2·6H2O) were oven-dried at 100 °C for 24 hours and were stored in a desiccator before use. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), 2-naphthol (C10H8O), 3-hydroxyacetaphenone (C8H8O2), 3-hydroxybenzoic acid (C7H6O3), 4-tert-butylphenol (C10H14O), cobalt acetate tetrahydrate (Co(CH3CO2)2·4H2O), high purity silica 60 gel (63–200 μm), potassium carbonate (K2CO3), silica gel TLC 60 F254 sheets, thionyl chloride (SOCl2), titanium butoxide (Ti(OBu)4), urea (CH4N2O) and zinc powder were purchased from Sigma-Aldrich. Indium tin oxide (ITO) glass substrates (56 × 56 × 1.1 mm; thickness ∼1200–1600 Å; transparency >84%; resistance 15 Ω sq−1) were purchased from Luminescence Tech. Corp, titanium oxide (TiO2) sol–gel was purchased from Solaronix and silver metal pellets (Ag, 99.999% purity) and tungsten oxide (WO3, 99.99% purity) was purchased from Testbourne Ltd.Equipment
Thin-layer chromatography was performed on silica gel 60 F254 sheets. Column chromatography was performed on silica gel 60 (63–200 μm) with various solvent systems. Elemental analysis of carbon, hydrogen, nitrogen and oxygen for the precursor materials was carried out on an Elementar Vario EL cube (Germany). Solution processable UV-vis absorption spectra were recorded on a Shimadzu UV-2250 spectrophotometer (Japan) while an Agilent Technologies Cary 5000 UV-vis-NIR spectrophotometer (USA) was used to record the absorption spectra of solid-state samples. Fourier transform infrared (FT-IR) spectra were obtained using a PerkinElmer Spectrum 100 FT-IR Spectrometer (USA), equipped with a diamond crystal ATR accessory. Mass spectroscopy data were obtained using a Bruker AutoFLEX III smart-beam MALDI-TOF mass spectrometer (Germany), with an α-cyano-4-hydroxycinnamic acid matrix in the positive ion mode. The thermal characteristics of the MPcs were measured using a PerkinElmer Simultaneous Thermal Analyzer (STA 6000) (USA) under N2 conditions, heating to a maximum of 850 °C at a rate of 10 °C min−1. The work function and ionization potentials of the materials were estimated using photoelectron spectroscopy in air (PESA) on an AC-2 from Riken Keiki, (Japan). The electron transport material and ternary blended photoactive layer were deposited onto the ITO substrates using a LabSpin spin coater from SUSS Microtec (Germany) in a GS Glovebox (Systemtechnik, Germany) under N2 filled glove box conditions (moisture content < 10 ppm). The hole transport material and anode layers were thermally evaporated with a TVA-01 thermal evaporator from THEVA (Germany). The evaporation for the HTM and anode material was carried out at a rate of ∼0.1 Å s−1 whilst maintaining a pressure above 4.0 × 10−6 mbar. J–V and EQE measurements were performed using a Keithley 2400 Source Meter (USA) using a self-designed Matlab program68 under AM 1.5 G (100 mA cm−2) and ambient conditions, without encapsulation. The BHJ cells were illuminated by using a LOT Quantum Design 300 W Xenon solar simulator (Germany). The active area was determined at 0.133 cm2 by attaching a shadow mask onto the solar cell device. A series of neutral density filters from Thorlabs (USA) were fixed onto a motorised wheel and mounted in the beam path to adjust the intensity of the simulated incident light. Light intensities were calibrated using a certified silicon reference solar cell (Frauenhofer Institute, Germany) equipped with a KG-5 filter. A motorised stage allows for one cell (with three pixels) to be measured at a time. The EQE measurements were performed under short circuit conditions. See ESI† for detailed fabrication and characterisation parameters.Computational details
The geometry optimisations of the MPcs were carried out by implementing the B3LYP density functional with the 6-31G(d) basis set as applied within the Gaussian 09 software.69 The B3LYP exchange–correlation density functional uses Becke's method via his B88 exchange functional and the Lee–Yang Parr correlation functional, which incorporates a combination of semi-empirical Hartree–Fock and DFT exchange.70–73 The molecular modelling of the MPcs complexes and post modelling analyses of the molecular orbitals were carried out using the GaussView 6.0.16 package.74Synthetic procedures
(1) 4-Nitrophthalonitrile. Yield: 15.0 g (60%). Colour: Cream. IR (νmax cm−1): 2239 (–CN); 1535 (NO2 asym.); 1345 (NO2 sym.). Elemental analysis (%) – calculated for C8H3N3O2: C 55.50, H 1.75, N 24.27; found: C 54.78, H 1.95, N 23.41.
(2a) 4-tert-Butylphenoxy phthalonitrile. Compounds (1) 4-nitrophthalonitrile (1.00 g, 5.78 mmol) and (a) 4-tert-butylphenol (1.02 g, 6.76 mmol) were dissolved in dry DMF (11 mL) with finely ground, anhydrous K2CO3 (1.28 g, 9.26 mmol). After 4, 12 and 24 hours, the reaction was supplemented with K2CO3 (0.06 g, 0.43 mmol). The reaction was left to stir for a total of 72 hours, after which an excess of distilled H2O was added and stirred for an additional 30 minutes. The precipitated compound (2a) was vacuum filtered and recrystallised from MeOH. Yield: 0.58 g (36%). Colour: Cream. IR (νmax cm−1): 2232 (–CN); 1241 (C–O–C). Elemental analysis (%) – calculated for C18H16N2O: C 78.24, H 5.84, N 10.14, O 5.79; found: C 77.89, H 6.048, N 10.09, O 6.67.
(2b) 4-(2-Naphthoxy) phthalonitrile. Compound (2b) was prepared according to the same procedure described for compound (2a) by using compound (b) 2-naphthol (1.09 g, 6.93 mmol) in place of compound (a). Yield: 0.51 g (33%). Colour: Brown. IR (νmax cm−1): 2232 (–CN); 1244 (C–O–C). Elemental analysis (%) – calculated for C18H10N2O: C 79.99, H 3.73, N 10.36, O 5.92; found: C 79.31, H 3.80, N 10.31, O 7.04.
(2c) 4-(3-Acetylphenoxy) phthalonitrile. Compound (2c) was prepared according to the same procedure described for compound (2a) by using compound (c) 3-hydroxyacetaphenone (0.92 g, 6.76 mmol) in place of compound (a). Yield: 0.50 g (33%). Colour: Light Green. IR (νmax cm−1): 1248 (C–O–C); 2234 (–CN). Elemental analysis (%) – calculated for C16H10N2O2: C 73.27, H 3.84, N 10.68, O 12.20; found: C 72.65, H 3.83, N 10.74 O 12.88.
(2d) 3-(3,4-Dicyanophenoxy) benzoic acid. Compound (2d) was prepared according to a similar procedure described for compound (2a) with modifications to the reaction conditions. Compound (d) 3-hydroxybenzoic acid (0.93 g, 6.76 mmol) was used in place of compound (a). The reaction was stirred for a total of 120 hours (5 days), after which 600 mL of distilled H2O was added and left to stir for 30 minutes. After that, HCl (32%) was added dropwise until a pH of 1 was obtained, causing the precipitation of the product. The acidified solution was allowed to stir for a further 15 minutes and was subsequently vacuum filtered. Yield: 0.45 g (0.30%). Colour: yellow-cream. IR (νmax cm−1): 1247 (C–O–C); 2236 (–CN). Elemental analysis (%): calculated for C15H8N2O3: C 66.18, H 3.05, N 10.60 O 20.16. Found: C 64.58, H 2.95, N 11.27 O 21.15.
(3a) 4β-(4-tert-Butylphenoxy) cobalt(II) phthalocyanine. To a 50 mL round bottom flask, compound 2a (0.5 g, 1.81 mmol), cobalt(II) acetate (0.10 g, 0.401 mmol) and DBU (5 drops) were refluxed with dry pentanol (10 mL) at 160 °C for 8 hours. After cooling to room temperature, MeOH (30 mL) was added to the intense blue slurry and was left to stir for 30 minutes to maximise product precipitation. The blue product was vacuum filtered and left to air dry overnight. The crude product was purified by column chromatography with silica gel and a C6H14/CH3Cl (1
:5) mixture as the eluent. Yield: 400 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1166. Found: 1167 [M − H]+.
(3b) 4β-(4-(2-Naphthoxy)) cobalt(II) phthalocyanine. Complex (3b) was prepared according to the same procedure described for compound (3a) by using compound (2b) 4-(2-naphthoxy) phthalonitrile (0.40 g, 1.48 mmol) in place of compound (2a). The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 225 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1142. Found: 1143 [M − H]+.
(3c) 4β-(4-(3-Acetylphenoxy)) cobalt(II) phthalocyanine. Complex (3c) was prepared according to the same procedure described for compound (3a) by using compound (2c) 4-(3-acetylphenoxy) phthalonitrile (0.40 g, 1.52 mmol) in place of compound (2a). The crude product was purified by column chromatography with silica gel and a THF/C4H8O2 (1
:2) mixture as the eluent. Yield: 157 mg. Colour: Green. MALDI-TOF-MS (m/z): calculated: 1109. Found: 1110 [M − H]+.
(3d) 4β-(3-(3,4-Dicyanophenoxy) benzoic acid) cobalt(II) phthalocyanine. To a 50 mL round bottom flask, compound (2d) (0.4 g, 1.51 mmol), cobalt(II) acetate (0.10 g, 0.401 mmol) and DBU (5 drops) were refluxed with dry pentanol (10 mL) at 160 °C for 9 hours. After cooling to room temperature, MeOH (30 mL) was added to the reaction mixture and was left to stir for 30 minutes to maximise product precipitation. The product was vacuum filtered and left to air dry overnight. The crude product showed solubility in CH3Cl and THF only in the presence of HCl (1 drop, 32%). Column chromatography with silica gel was performed using a gradient solvent system of C6H14 and THF from 0% to100% of THF. Yield: 164 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1117. Found: 1118 [M − H]+.
(4a) 4β-(4-tert-Butylphenoxy) nickel(II) phthalocyanine. Complex (4a) was prepared according to the same procedure described for compound (3a) by using nickel(II) acetate (0.10 g, 0.402 mmol) in place of cobalt(II) acetate. The solid product was subjected to Soxhlet extraction with C4H8O2 for 24 hours to remove unreacted starting materials. Yield: 402 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1166. Found: 1167 [M − H]+.
(4b) 4β-(4-(2-Naphthoxy)) nickel(II) phthalocyanine. Complex (4b) was prepared according to the same procedure described for compound (3a) by using nickel(II) acetate (0.10 g, 0.402 mmol) in place of cobalt(II) acetate and compound (2b) 4-(2-naphthoxy) phthalonitrile (0.40 g, 1.48 mmol) in place of compound (2a). The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with C4H8O2 as the eluting solvent. Yield: 232 mg. Colour: Green. MALDI-TOF-MS (m/z): calculated: 1142. Found: 1142 [M]+.
(4c) 4β-(4-(3-Acetylphenoxy)) nickel(II) phthalocyanine. Complex (4c) was prepared according to the same procedure described for compound (3a) by using nickel(II) acetate (0.10 g, 0.402 mmol) in place of cobalt(II) acetate and compound (2c) 4-(3-acetylphenoxy) phthalonitrile (0.40 g, 1.52 mmol) in place of compound (2a). The crude product was purified by column chromatography with silica gel and a C4H8O2/MeOH (1
:1) mixture as the eluent. The crude product was purified by column chromatography with silica gel and a C6H14/DCM (1:2) mixture as the eluent. Yield: 150 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1109. Found: 1110 [M − H]+.
(4d) 4β-(3-(3,4-Dicyanophenoxy) benzoic acid) nickel(II) phthalocyanine. Complex (4d) was prepared according to the same procedure described for compound (3d) by using nickel(II) chloride (0.10 g, 0.772 mmol) in place of cobalt(II) acetate. After 4 hours of refluxing, HCl (5 drops, 32%) was added to the reaction mixture, changing the colour of the mixture to intense blue. The reaction was left to reflux for complete 9 hours. After cooling to room temperature, MeOH (30 mL) was added to the reaction mixture and was left to stir for 30 minutes to maximise product precipitation. The product was repeatedly washed using MeOH and vacuum filtered and left to air dry overnight. Yield: 155 mg. Colour: Blue. MALDI-TOF-MS (m/z): calculated: 1117. Found: 1118 [M − H]+.
(5a) 4β-(4-tert-Butylphenoxy) titanium(IV) oxide phthalocyanine. Complex (5a) was prepared according to the same procedure described for compound (3a) by using titanium butoxide (0.2 mL, 0.586 mmol) in place of cobalt(II) acetate and adding urea (70 mg, 1.165 mmol) to the reaction. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. Yield: 406 mg (52%). Colour: Green. MALDI-TOF-MS (m/z): calculated: 1171. Found: 1172 [M − H]+.
(5b) 4β-(4-(2-Naphthoxy)) titanium(IV) oxide phthalocyanine. Complex (5b) was prepared according to the same procedure described for compound (3a) by using titanium butoxide (0.2 mL, 0.586 mmol) in place of cobalt(II) acetate, compound (2b) 4-(2-naphthoxy) phthalonitrile (0.40 g, 1.48 mmol) in place of compound (2a) and adding urea (70 mg, 1.165 mmol) to the reaction. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. Yield: 345 mg. Colour: Green. MALDI-TOF-MS (m/z): calculated: 1147. Found: 1147 [M]+.
(5c) 4β-(4-(3-Acetylphenoxy)) titanium(IV) oxide phthalocyanine. Complex (4c) was prepared according to the same procedure described for compound (3a) by using titanium butoxide (0.2 mL, 0.586 mmol) in place of cobalt(II) acetate and compound (2c) 4-(3-acetylphenoxy) phthalonitrile (0.40 g, 1.52 mmol) in place of compound (2a) and adding urea (70 mg, 1.165 mmol) to the reaction. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 233 mg. Colour: Green. MALDI-TOF-MS (m/z): calculated: 1114. Found: 1115 [M − H]+.
(5d) 4β-(3-(3,4-Dicyanophenoxy) benzoic acid) titanium(IV) oxide phthalocyanine. Complex (5d) was prepared according to the same procedure described for compound (3d) by using titanium butoxide (0.2 mL, 0.586 mmol) in place of cobalt(II) acetate and adding urea (70 mg, 1.165 mmol) to the reaction. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. Yield: 167 mg. Colour: Green. MALDI-TOF-MS (m/z): calculated: 1122. Found: 1123 [M − H]+.
(6a) 4β-(4-tert-Butylphenoxy) manganese(III) chloride phthalocyanine. Complex (6a) was prepared according to the same procedure described for compound (3a) by using manganese(II) chloride (0.10 g, 0.618 mmol) in place of cobalt(II) acetate. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 390 mg. Colour: Dull Green. MALDI-TOF-MS (m/z): calculated: 1197. Found: 1162 [M − Cl]+.
(6b) 4β-(4-(2-Naphthoxy)) manganese(III) chloride phthalocyanine. Complex (6b) was prepared according to the same procedure described for compound (3a) by using manganese(II) chloride (0.10 g, 0.618 mmol) in place of cobalt(II) acetate and compound (2b) 4-(2-naphthoxy) phthalonitrile (0.40 g, 1.48 mmol) in place of compound (2a). The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 362 mg (52%). Colour: Brown. MALDI-TOF-MS (m/z): calculated: 1173. Found: 1138 [M − Cl]+.
(6c) 4β-(4-(3-Acetylphenoxy)) manganese(III) chloride phthalocyanine. Complex (6c) was prepared according to the same procedure described for compound (3a) by using manganese(II) chloride (0.10 g, 0.618 mmol) in place of cobalt(II) acetate and compound (2c) 4-(3-acetylphenoxy) phthalonitrile (0.40 g, 1.52 mmol) in place of compound (2a). The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 247 mg. Colour: Brown. MALDI-TOF-MS (m/z): calculated: 1140. Found: 1105 [M − Cl]+.
(6d) 4β-(3-(3,4-dicyanophenoxy) benzoic acid) manganese(III) chloride phthalocyanine. Complex (6d) was prepared according to the same procedure described for compound (3d) by using manganese(II) chloride (0.10 g, 0.618 mmol) in place of cobalt(II) acetate. The solid product was subjected to Soxhlet extraction with MeOH for 24 hours to remove unreacted starting materials. A final purification step was performed using column chromatography with CH3Cl as the eluting solvent. Yield: 209 mg. Colour: Brown. MALDI-TOF-MS (m/z): calculated: 1149. Found: 1114 [M − Cl]+.
Conclusions
This work demonstrated the synthesis of traditional and novel MPcs with four metal centres, i.e. Co, Ni, Ti and Mn that were tetra substituted with four functional groups displaying slight structural differences. Complete molecular energy diagrams indicated that the MPcs are suitable candidates as ternary materials for a P3HT:PC70BM blended BHJ active layer. The inclusion of the MPc ternary layer (P3HT:MPc:PC70BM) led to the proposed improvement in OSC efficiencies by driving efficient electron transfer and absorption of a larger fraction of the solar spectrum. The performance of the ternary BHJ OSCs showed variability amongst the MPcs with regard to the valency of the central metal and associated properties, especially with their molecular energy levels. Although the CoPcs, TiPcs and MnPcs displayed experimentally favourable HOMO energy alignments between P3HT and PC70BM, the axially coordinated MnPcs proved to be the most efficient additive as they exhibited the best BHJ OSC performance. The OSC performances did not show great variability between substituents. The MnPcs presented with the lowest HOMOs which may have facilitated a better electron transfer and cascading effect between the P3HT and PC70BM, thus marginally outperforming the TiPcs. The EQE assessment showed that the MPcs were unable to contribute to greater charge generation despite being strong absorbers within the near-infrared region, therefore the improvement of the OSC efficiencies relative to the binary P3HT:PC70BM reference system was likely a result of either improved charge transfer between P3HT and PC70BM or reduced charge recombination rather than the generation of more photoinduced charge carriers. It is anticipated that physically attaching the MPc molecules to the P3HT donor molecules will achieve greater absorption and obtain enhanced OSC performance.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
DG thanks Prof. Dr Lukas Schmidt-Mende (University of Konstanz) for hosting her, and the Centre for High Performance Computing's (CHPC), Cape Town, South Africa. This work was financially supported by the National Research Foundation (NRF) (Grant number: 108424) and the Zukunftskolleg of the University of Konstanz.References
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633–17648 RSC.
- B. Azzopardi, C. J. M. Emmott, A. Urbina, F. C. Krebs, J. Mutale and J. Nelson, Energy Environ. Sci., 2011, 4, 3741–3753 RSC.
- L. Zhu, M. Zhang, G. Zhou, T. Hao, J. Xu, J. Wang, C. Qiu, N. Prine, J. Ali, W. Feng, X. Gu, Z. Ma, Z. Tang, H. Zhu, L. Ying, Y. Zhang and F. Liu, Adv. Energy Mater., 2020, 10, 1904234 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- Z. Guo, D. Lee, R. D. Schaller, X. Zuo, B. Lee, T. Luo, H. Gao and L. Huang, J. Am. Chem. Soc., 2014, 136, 10024–10032 CrossRef CAS.
- N. K. Elumalai, C. Vijila, R. Jose, K. Zhi Ming, A. Saha and S. Ramakrishna, Phys. Chem. Chem. Phys., 2013, 15, 19057–19064 RSC.
- T. M. Grant, T. Gorisse, O. Dautel, G. Wantz and B. H. Lessard, J. Mater. Chem. A, 2017, 5, 1581–1587 RSC.
- G. Zhang, K. Zhang, Q. Yin, X.-F. Jiang, Z. Wang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS.
- N. Gasparini, L. Lucera, M. Salvador, M. Prosa, G. D. Spyropoulos, P. Kubis, H.-J. Egelhaaf, C. J. Brabec and T. Ameri, Energy Environ. Sci., 2017, 10, 885–892 RSC.
- F. Bonaccorso, N. Balis, M. M. Stylianakis, M. Savarese, C. Adamo, M. Gemmi, V. Pellegrini, E. Stratakis and E. Kymakis, Adv. Funct. Mater., 2015, 25, 3870–3880 CrossRef CAS.
- H. Cha, D. S. Chung, S. Y. Bae, M.-J. Lee, T. K. An, J. Hwang, K. H. Kim, Y.-H. Kim, D. H. Choi and C. E. Park, Adv. Funct. Mater., 2013, 23, 1556–1565 CrossRef CAS.
- Y. Zhang, D. Deng, K. Lu, J. Zhang, B. Xia, Y. Zhao, J. Fang and Z. Wei, Adv. Mater., 2015, 27, 1071–1076 CrossRef CAS.
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158–162 CrossRef CAS.
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 CrossRef.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- M.-T. Dang, T. M. Grant, H. Yan, D. S. Seferos, B. H. Lessard and T. P. Bender, J. Mater. Chem. A, 2017, 5, 12168–12182 RSC.
- M. Koçak, A. Cihan, A. I. Okur, A. Gül and Ö. Bekaroğlu, Dyes Pigm., 2000, 45, 9–14 CrossRef.
- M. Özçeşmeci and E. Hamuryudan, Dyes Pigm., 2008, 77, 457–461 CrossRef.
- J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281–321 CrossRef CAS.
- L. Edwards and M. Gouterman, J. Mol. Spectrosc., 1970, 33, 292–310 CrossRef CAS.
- A. Harriman and M.-C. Richoux, J. Chem. Soc., Faraday Trans. 2, 1980, 76, 1618–1626 RSC.
- D. Dini and M. Hanack, Physical Properties of Phthalocyanine-based Materials, Academic Press, 2003 Search PubMed.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- J. M. Birchall, R. N. Haszeldine and J. O. Morley, J. Chem. Soc. C, 1970, 2667–2672 RSC.
- P. Bhyrappa and V. Krishnan, Inorg. Chem., 1991, 30, 239–245 CrossRef CAS.
- F. D’Souza, A. Villard, E. Van Caemelbecke, M. Franzen, T. Boschi, P. Tagliatesta and K. M. Kadish, Inorg. Chem., 1993, 32, 4042–4048 CrossRef.
- B. N. Achar, T. M. Mohan Kumar and K. S. Lokesh, J. Coord. Chem., 2007, 60, 1833–1846 CrossRef CAS.
- N. Ceylan, G. Gümrükçü, G. K. Karaoğlan and A. Gül, Synth. Met., 2015, 206, 55–60 CrossRef CAS.
- I. Yilmaz, New J. Chem., 2008, 32, 37–46 RSC.
- J. Obirai and T. Nyokong, Electrochim. Acta, 2005, 50, 3296–3304 CrossRef CAS.
- S. Altun, Z. Odabaş, A. Altındal and A. R. Özkaya, Dalton Trans., 2014, 43, 7987–7997 RSC.
- R. Seoudi, G. S. El-Bahy and Z. A. El Sayed, Opt. Mater., 2006, 29, 304–312 CrossRef CAS.
- S. Park, B. T. Lim, B. Kim, H. J. Son and D. S. Chung, Sci. Rep., 2014, 4, 5482 CrossRef CAS.
- A. J. Thomson, Biochem. Educ., 1981, 9, 35 CrossRef.
- K. Lian, R. Li, H. Wang, J. Zhang and D. Gamota, Mater. Sci. Eng., B, 2010, 167, 12–16 CrossRef CAS.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS.
- M. Cheng, M. Safdari, P. Liu, L. Kloo, L. Sun, C. Chen and Y. Li, Adv. Energy Mater., 2017, 7, 1602556 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- E. A. Davis and N. F. Mott, Philos. Mag. A, 1970, 22, 903–922 CrossRef CAS.
- L. N. Ramavathu, K. K. Maniam, K. Gopalram and R. Chetty, J. Appl. Electrochem., 2012, 42, 945–951 CrossRef CAS.
- H. Kalvelage, A. Mecklenburg, U. Kunz and U. Hoffmann, Chem. Eng. Technol., 2000, 23, 803–807 CrossRef CAS.
- Y. Lu and R. G. Reddy, Electrochim. Acta, 2007, 52, 2562–2569 CrossRef CAS.
- R. Seoudi, G. S. El-Bahy and Z. A. El Sayed, J. Mol. Struct., 2005, 753, 119–126 CrossRef CAS.
- G. K. Mor, S. Kim, M. Paulose, O. K. Varghese, K. Shankar, J. Basham and C. A. Grimes, Nano Lett., 2009, 9, 4250–4257 CrossRef CAS.
- Y. Park, V. Choong, Y. Gao, B. R. Hsieh and C. W. Tang, Appl. Phys. Lett., 1996, 68, 2699–2701 CrossRef CAS.
- M. Fang, C. Zhang and Q. Chen, Appl. Surf. Sci., 2016, 385, 28–33 CrossRef CAS.
- J. Y. Kim, S. H. Kim, H.-H. Lee, K. Lee, W. Ma, X. Gong and A. J. Heeger, Adv. Mater., 2006, 18, 572–576 CrossRef CAS.
- S. Kashiwaya, J. Morasch, V. Streibel, T. Toupance, W. Jaegermann and A. Klein, Surfaces, 2018, 1, 73–89 CrossRef.
- C. S. Beleznai, D. Vouagner and J. P. Girardeau-Montaut, Appl. Surf. Sci., 1999, 138–139, 6–11 CrossRef CAS.
- D. R. Lide, J. Am. Chem. Soc., 2009, 131, 12862 Search PubMed.
- Z.-X. Xu, V. A. L. Roy, K.-H. Low and C.-M. Che, Chem. Commun., 2011, 47, 9654–9656 RSC.
- B. Kadem, A. Hassan, M. Göksel, T. Basova, A. Şenocak, E. Demirbaş and M. Durmuş, RSC Adv., 2016, 6, 93453–93462 RSC.
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC.
- R. A. Street, Adv. Mater., 2016, 28, 3814–3830 CrossRef CAS.
- J.-C. Ke, Y.-H. Wang, K.-L. Chen and C.-J. Huang, Sol. Energy Mater. Solar Cells, 2015, 133, 248–254 CrossRef CAS.
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941–1960 CrossRef CAS.
- M. J. Trimpl, A. D. Wright, K. Schutt, L. R. V. Buizza, Z. Wang, M. B. Johnston, H. J. Snaith, P. Müller-Buschbaum and L. M. Herz, Adv. Funct. Mater., 2020, 2004312 CrossRef CAS.
- R. Zhu, C.-Y. Jiang, B. Liu and S. Ramakrishna, Adv. Mater., 2009, 21, 994–1000 CrossRef CAS.
- J. U. Lee, Y. Do Kim, J. W. Jo, J. P. Kim and W. H. Jo, J. Mater. Chem., 2011, 21, 17209–17218 RSC.
- A. J. Parnell, A. J. Cadby, A. D. F. Dunbar, G. L. Roberts, A. Plumridge, R. M. Dalgliesh, M. W. A. Skoda and R. A. L. Jones, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 141–146 CrossRef CAS.
- T. S. Glen, N. W. Scarratt, H. Yi, A. Iraqi, T. Wang, J. Kingsley, A. R. Buckley, D. G. Lidzey and A. M. Donald, Sol. Energy Mater. Sol. Cells, 2015, 140, 25–32 CrossRef CAS.
- S. Alam, A. Gavrik, R. Meitzner, S. Hoeppener, V. Dyakonov, A. Baumann, U. S. Schubert and H. Hoppe, J. Phys. D: Appl. Phys., 2019, 52, 475501 CrossRef CAS.
- D. Ghosh, M. Y. Ali, D. K. Chaudhary and S. Bhattacharyya, Sol. Energy Mater. Solar Cells, 2018, 185, 28–35 CrossRef CAS.
- Y. Jiang, M. Remeika, Z. Hu, E. J. Juarez-Perez, L. Qiu, Z. Liu, T. Kim, L. K. Ono, D.-Y. Son, Z. Hawash, M. R. Leyden, Z. Wu, L. Meng, J. Hu and Y. Qi, Adv. Energy Mater., 2019, 9, 1803047 CrossRef.
- E. Zimmermann, Matlab Progr., University of Konstanz, 2019 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Starov, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. N. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView - Version 6.1, Semichem Inc., Shawnee Mission, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00725k |
| This journal is © The Royal Society of Chemistry 2020 |