
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent molecular switches based on dicationic P-doped polycyclic aromatic hydrocarbons†
Thomas
Delouche
a,
Antoine
Vacher
a,
Thierry
Roisnel
 a,
Marie
Cordier
a,
Jean-Frederic
Audibert
b,
Boris
Le Guennic
a,
Fabien
Miomandre
*b,
Denis
Jacquemin
*c,
Muriel
Hissler
a and
Pierre-Antoine
Bouit
*a
a,
Marie
Cordier
a,
Jean-Frederic
Audibert
b,
Boris
Le Guennic
a,
Fabien
Miomandre
*b,
Denis
Jacquemin
*c,
Muriel
Hissler
a and
Pierre-Antoine
Bouit
*a
aUniv Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France. E-mail: pierre-antoine.bouit@univ-rennes1.fr
bPPSM, ENS Paris-Saclay, CNRS, Université Paris-Saclay, 94235 Cachan, France. E-mail: mioman@ppsm.ens-cachan.fr
cCEISAM, UMR CNRS 6230, University of Nantes, 44322 Nantes, France. E-mail: denis.jacquemin@univ-nantes.fr
First published on 19th October 2020
Abstract
We report the efficient synthesis of electron-acceptors based on cationic P-doped polycyclic aromatic hydrocarbons. In particular, exo-skeletal modifications of the π-core were performed and their effect on the optical and redox properties was studied and compared to the effect of endo-skeletal modifications. Combined experimental (spectroelectrochemical and electron paramagnetic resonance) and theoretical studies performed on this entire family provide in-depth characterization of the reduced species and specifically of the stable radical cations formed and afford a guideline for the property tuning of such derivatives. Finally, electrochemical modulation of fluorescence performed on two compounds highlights the potential of these intrinsically switchable electroactive fluorophores.
Polycyclic aromatic hydrocarbons (PAHs, also called nanographene when their size exceeds 1 nm) are bidimensional organic molecules composed of sp2 C atoms.1 This particular chemical composition endows them with unique properties such as strong light absorption and emission, charge transport in the solid state, etc. Consequently they are excellent candidates for building semi-conductors or active materials in a wide range of applications linked to molecular electronics. The modification of their carbon framework (ring size, edge configuration etc.) endows them with chiroptical or magnetic properties which even enlarge the scope of their potential applications in materials sciences.2 Another strategy to tune both the structure and properties is to replace C-atoms by heteroatoms.3 During the last decade, various synthetic approaches allowed introducing single heteroatoms (B, N, O, P, S etc.) into the C-framework, therefore endowing it with unique reactivity and electronic properties.4 Introducing a second identical heteroatom into the framework has been recently attracting significant interest since this allows the investigation of the electronic communication between the main group elements through a π-conjugated PAH backbone. As a representative example, Würthner et al. described electron-acceptors based on PAHs featuring two σ3,λ3 B atoms such as A (Fig. 1).5 These authors studied the complex structure–electronic property relationships (based on the position of the B atoms and the size of the PAH) and could also use one of their compounds as an electron acceptor in bulk heterojunction solar cells showing promising power conversion efficiencies of up to 3%. PAHs embedded with two σ3,λ4 N+ (B in Fig. 1) have also been described by Feng et al.6 They display viologen-like behavior (e.g., two easily accessible reduction waves including stable radical cations). Among the heteroatoms, P is also of particular interest due to its high variety of valence and coordinance. In our effort to prepare P-containing PAHs,7 we recently developed a synthetic approach toward 5 dicationic P-containing PAHs.8,9 Various π-backbones could be used and their viologen-like behavior was exemplified on one prototype derivative (C, Fig. 1). This particular behavior was attributed to the presence of two σ4,λ4-P+ connected through an even number of sp2 C-atoms, by analogy with the Hünig strategy.10 In the present article, we describe the molecular engineering of the C-type PAH platform by using exoskeletal modifications (modification of R1 and R2, Fig. 1). The effect on the optical and redox properties is studied and compared to the effect of endoskeletal modifications (π, Fig. 1) based on a joint experimental and theoretical approach. Spectro-electrochemical and EPR studies were carried out on all compounds to investigate in the entire series the “viologen-like” behaviour previously observed for C (Fig. 1). In addition, by taking advantage of the luminescence associated with the redox properties, we demonstrate the electrofluorochromic properties of two selected compounds. The development of intrinsically switchable electroactive fluorophores remains a challenge for the scientific community.11
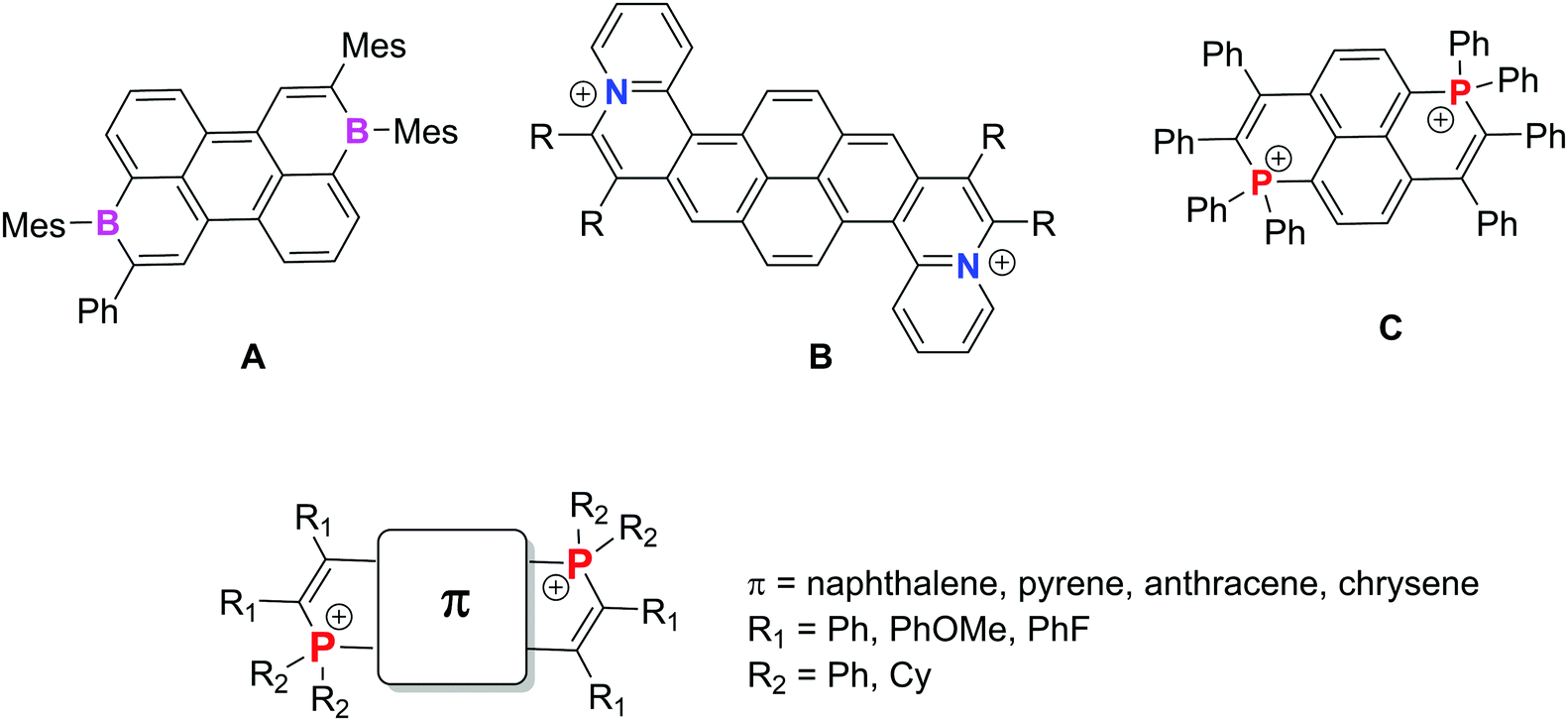 | ||
| Fig. 1 Reported examples of PAHs featuring two identical heteroatoms A–C (top) and illustration of our molecular engineering strategy (bottom) (counterions of B–C were omitted for clarity). | ||
Synthesis
To prepare the targeted dicationic P-containing PAHs, we relied on a copper mediated radical approach recently developed by Wang et al.12 In order to have in hand a reference compound, we first reproduced the synthesis of 2[OTf] (Scheme 1) (see Fig. S16, ESI,† for X-ray structure).12a Then, the phosphacyclization of bisphosphane 3a with phenylacetylene afforded 1,8-bisphosphapyrenium 4a[OTf]2 (87% yield, Scheme 1). The synthetic approach is highly versatile as it is compatible with a wide scope of phosphines and alkynes. In order to further tune the optical and redox properties of the bisphosphapyrenium, we prepared in good yields 4b[OTf]2 featuring electron rich exocyclic P-cyclohexyl groups. This compound was characterized by X-ray diffraction and the overall structure displays no important modifications compared to 4a[OTf]2 (mostly a planar π-framework and classical P–C bond lengths for phosphonium, Fig. S17, ESI†). We then modified the exocyclic C-core by introducing electron-donating p-methoxyphenyl groups (4c[OTf]2) and electron poor fluorophenyl (4d[OTf]2) by modification of the alkyne engaged in the phosphacyclization. Introduction of more electron rich (such as aminophenyl) or poor (cyanophenyl) substituents could not be achieved. However, these structural modifications highlight that the bisphosphapyrenium can be chemically tuned through straightforward exoskeletal modifications of various chemical functions (P atoms, C-frameworks).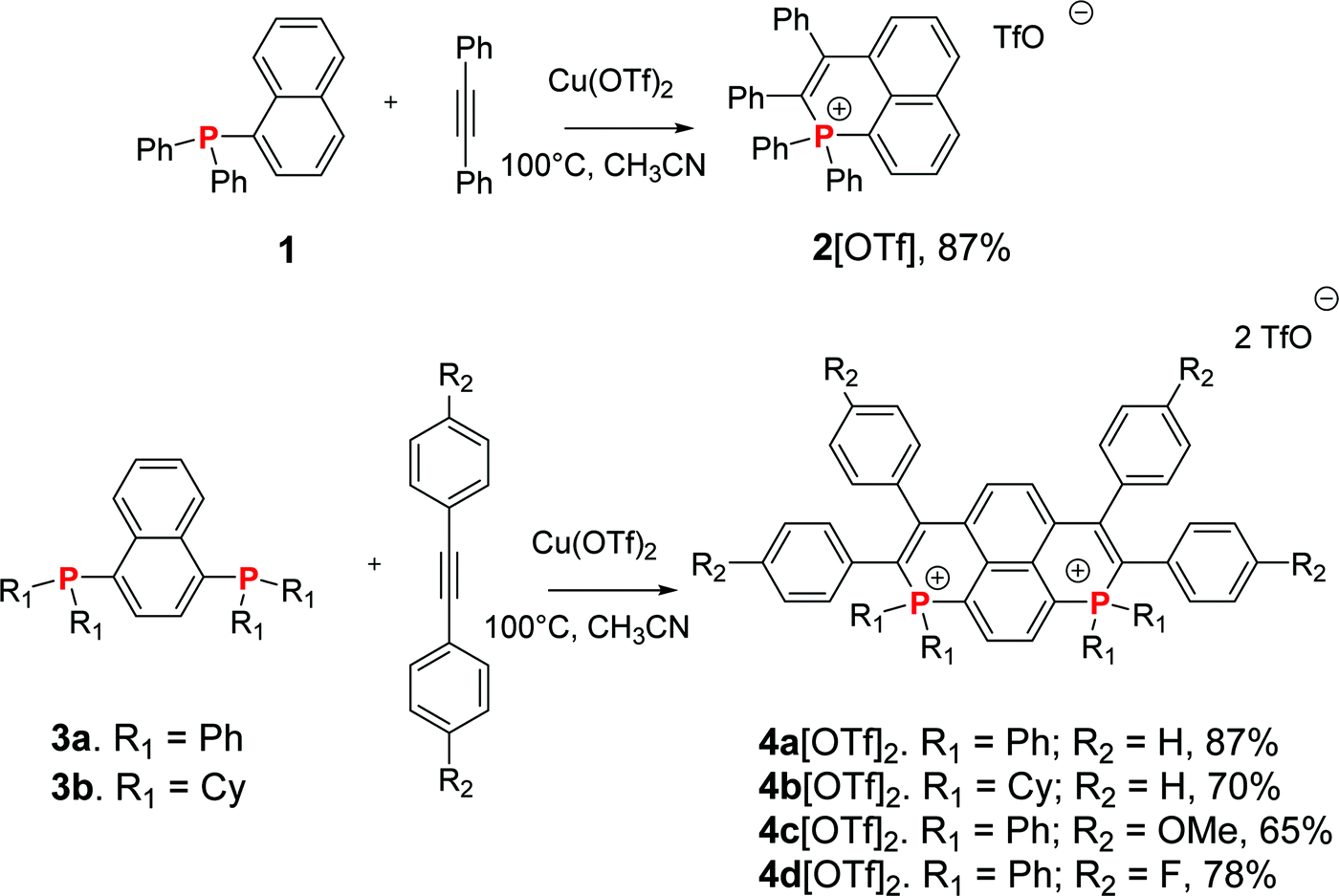
In short, we prepared three new P-containing PAHs with variable exocyclic P-substituents and C-substituents and a reference compound (2[OTf]). We thus have in hand an entire family of eight dicationic featuring variable π-cores (5–8[OTf]2, Fig. 2)8 or exocyclic substituents (4a–d[OTf]2, Scheme 1).
 | ||
| Fig. 2 Molecular structures of previously reported 5–82+.8 | ||
Optical properties
The spectroscopic properties of 2[OTf] and 4b–d[OTf]2 are investigated in diluted CH2Cl2 solutions (c = 5 × 10−6 mol L−1, Fig. 3 and Table 1) and compared to the previously reported 4a[OTf]2.8,134a[OTf]2 displays large absorption bands in the visible region with λabs at 420 nm.8 This transition can be assigned to a delocalized π–π* transition according to TD-DFT calculations (vide infra and Fig. 7). As expected, this absorption is considerably red-shifted and hyperchromically shifted compared to 2[OTf] that displays only one P-heterocycle (Fig. 3).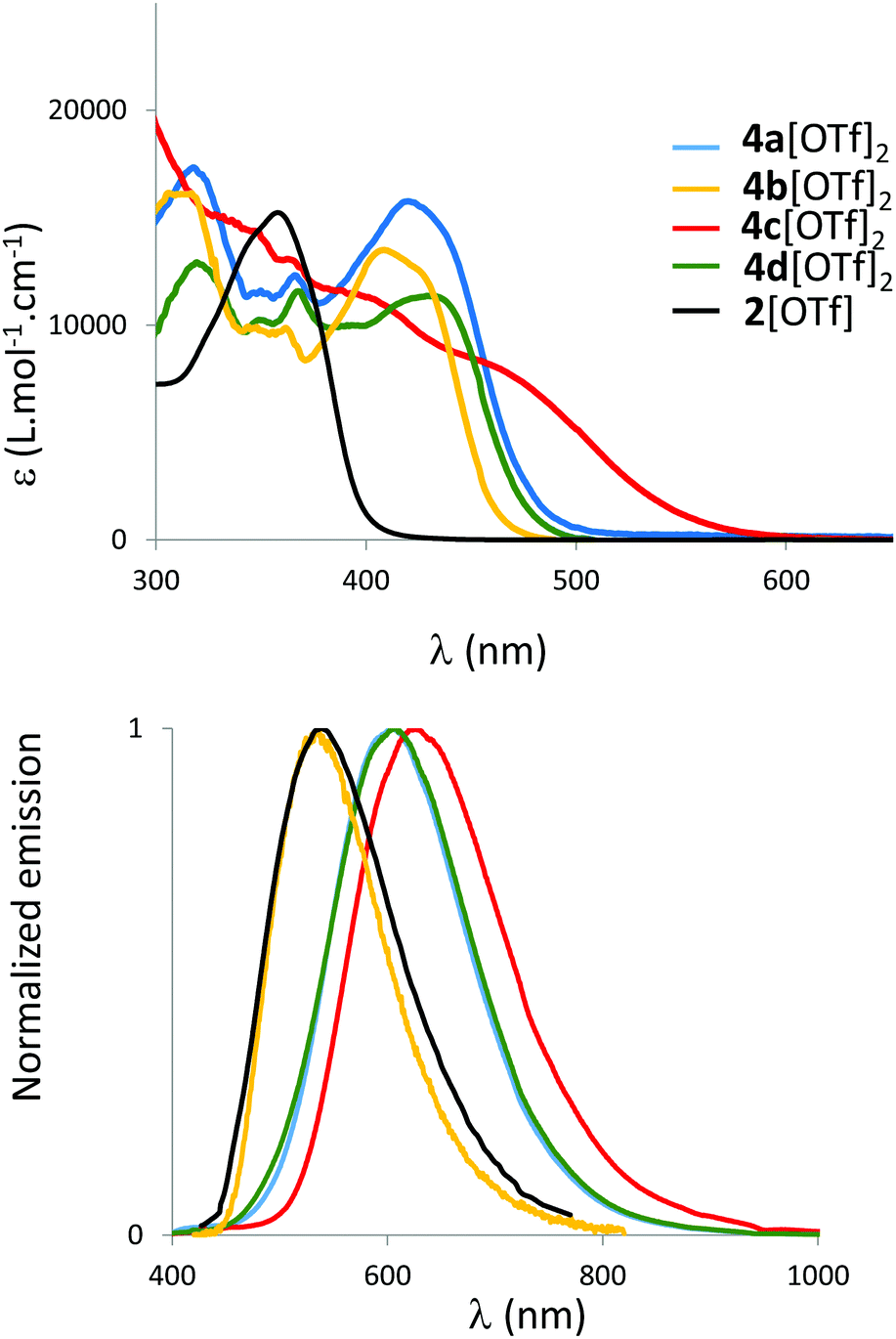 | ||
| Fig. 3 UV-Vis absorption (top) and normalized emission spectra (bottom) of 2[OTf] and 4ad[OTf]2 in CH2Cl2 (c = 5 × 10−6 M). | ||
| λ abs [nm] | ε [M−1 cm−1] | λ em [nm] | Φ (%) | σ (cm−1) |
λ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) solid
[nm] solid
[nm] |
Φ (%) |
E
red1d [V] |
E
red2d [V] |
|
|---|---|---|---|---|---|---|---|---|---|
| a In CH2Cl2 (10−5 M). b Measured in a calibrated integrated sphere. c Measured in powder. d In DCM with Bu4N+PF6− (0.2 M) at a scan rate of 200 mV s−1. Potentials vs. Fc+/Fc. e Reversible process (ΔEp < 60 mV). f Quasi-reversible process. | |||||||||
| 2[OTf] | 359 | 15000 |
539 | 14 | 9300 | 507 | 48 | −1.61e | — |
| 4a[OTf]2 | 420 | 16000 |
606 | 19 | 7300 | 552 | 39 | −0.84e | −1.23e |
| 4b[OTf]2 | 409 | 13500 |
538 | 75 | 5900 | 542 | 52 | −0.93e | −1.40e |
| 4c[OTf]2 | 470 | 8500 | 625 | 2 | 5700 | 640 | 10 | −0.89f | −1.29f |
| 4d[OTf]2 | 429 | 11500 |
607 | 13 | 6800 | 588 | 37 | −0.82e | −1.18e |
| 5[OTf]2 | 417 | 23000 |
608 | 14 | 7600 | 554 | 37e | −0.81e | −1.24e |
| 6[OTf]2 | 430 | 55000 |
575 | 13 | 6200 | 555 | 11e | −0.98e | −1.36e |
| 7[OTf]2 | 432 | 19000 |
560 | 6 | 5600 | 566 | 19 | −1.10e | −1.44f |
| 8[OTf]2 | 522 | 19000 |
672 | 4 | 4700 | 670 | 16 | −0.64e | −1.01e |
We then focused our attention on the effect of exoskeletal modifications with the study of new derivatives 4b–d[OTf]2. Modification of the exocyclic P-substituent (cyclohexyl instead of phenyl) leads to a moderate blue-shift of the absorption (λabs(4b[OTf]2) = 409 nm). While the introduction of a (weak) electron-withdrawing group on the C-framework leaves the absorption spectrum almost unaffected (λabs(4d[OTf]2) = 429 nm), insertion of electron-donating groups strongly affects the absorption with a tail spanning to λ = 518 nm and a smaller extinction coefficient. In this compound intramolecular charge transfer from the rich methoxy groups toward the electron-deficient polyaromatic platform is envisaged as the cause of these modifications (vide infra for TD-DFT rationalization). All derivatives also display fluorescence in solution and a similar trend regarding the effect of donors/acceptors on the emission wavelength can be observed with a gradual red-shift in the series 2–4b–4ad–4c[OTf]2. Surprisingly, 4b[OTf]2 displays high photoluminescence quantum yield in solution (75%) compared to the other compounds. All derivatives display a large Stokes shift (Table 1), despite their rigid polyaromatic backbone. TD-DFT nevertheless reveals a significant change in geometry between the ground and the excited states (see Fig. S35, ESI†). This feature seems general for π-conjugated phosphonium and bisphosphonium and is clearly evidenced when compared with pyrene (σ = 2800 cm−1) which displays a similar polyaromatic platform to 4a–d[OTf]2 and 5[OTf]2.12b,14,15 All dyes also show solid-state photoluminescence (in powders, Fig. S19, ESI†). No clear trend can be drawn on the impact of aggregation on the luminescence wavelength. Except for 4b[OTf]2, the luminescence in powder form is higher than in diluted solution. This increase nicely correlates with an emission enhancement induced by the restriction of the rotations of the 8 phenyl rotors and by the global bulkiness of the compounds that prevents π-stacking.
In conclusion, 4a–d[OTf]2 display strong absorption in the visible range as well as fluorescence both in solution and in the solid state. We show that exoskeletal modifications of the platforms also allow the tuning of the absorption/emission properties in the visible range.
Redox properties
The electrochemical properties of 2[OTf] and 4b–d[OTf]2 have been investigated by cyclic voltammetry (CV) in dichloromethane solution (see Fig. 4 and Fig. S19, ESI†) and compared to the previously reported 4a (5–8[OTf]2 can be found elsewhere).8 No oxidation processes are observed with any of the compounds under these experimental conditions. 4a[OTf]2 possesses two separated reversible reduction waves at low potential (Ered1(4a[OTf]2) = −0.84 V vs. Fc+/Fc; Ered2(4a[OTf]2) = −1.23 V vs. Fc+/Fc, Fig. 4 and Table 1). Comparison with 2[OTf] (a single reduction wave with Ered = −1.61 V vs. Fc+/Fc) clearly highlights the crucial role of the two cationic σ4,λ4-P+ connected through an even number of sp2 C-atoms in the viologen-like behavior of 4a[OTf]2.16 We underline that the first reduction potential of 4a matches with that of methyl-viologen triflate recorded under the same conditions. Then the properties of the novel compounds 4b–d[OTf]2 were examined. As expected, replacing the P–Ph of 4a[OTf]2 by a more electron-rich P–Cy leads to a cathodic shift of 0.1 V (Ered1(4b[OTf]2) = −0.93 V vs. Fc+/Fc). Such a phenomenon is also observed with the insertion of methoxy groups on the C-platform (Ered1(4c[OTf]2) = −0.89 V vs. Fc+/Fc). For this compound, the two reduction processes are quasi-reversible. The presence of fluorophenyl leaves the reduction almost unaffected (Ered1(4d[OTf]2) = −0.82 V vs. Fc+/Fc). These observations are consistent with the fact that reinforcing the electronic density on the platform makes the reduction more difficult and has an impact on the reversibility. However, the fluorophenyl group seems to be a too “weak” acceptor to induce significant changes, in agreement with the optical data. The effect of the exoskeletal modification is thus rather easy to rationalize. As we previously reported, such rationalization is more challenging with the endoskeletal modifications: no clear trend can be drawn regarding the effect of π-extension.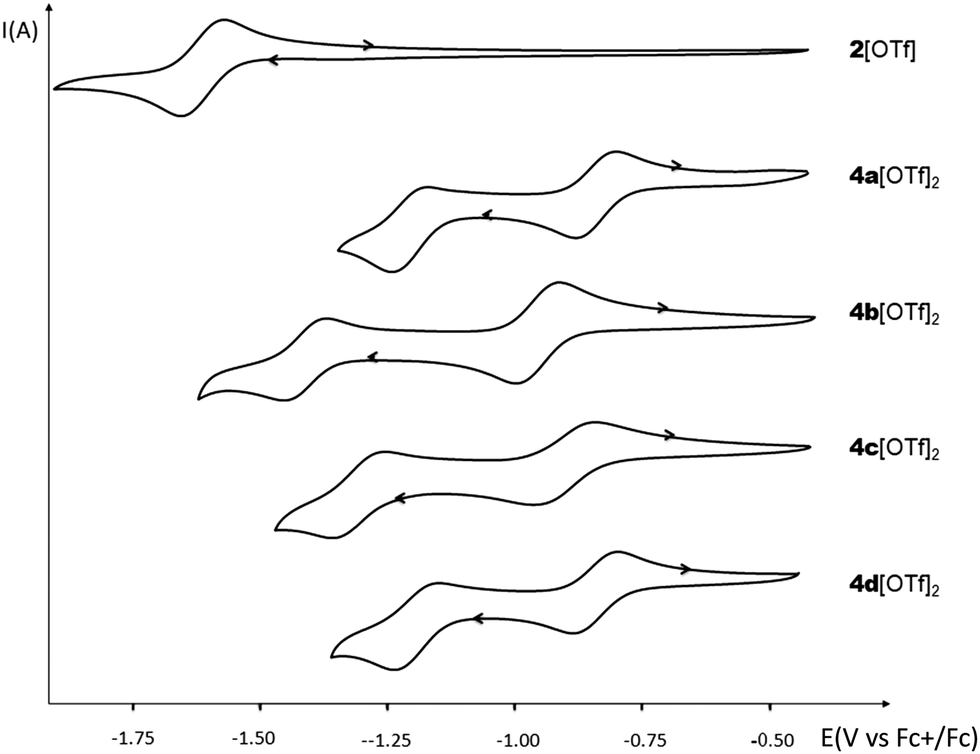 | ||
| Fig. 4 Cyclic voltammograms of 2[OTf] and 4a–d[OTf]2 (c = 10−3 M) in DCM (Bu4NPF6 (0.2 M), 200 mV s−1, potentials vs. Fc+/Fc). | ||
In short, the combined optical and redox study reveals that 4a–d[OTf]2 possess all the characteristics of intrinsically switchable electroactive fluorophores.11
Spectroelectrochemistry and EPR
In order to investigate the optical properties of the reduced forms of the dicationic P-containing PAHs, spectroelectrochemical and EPR analyses were performed on all derivatives. We previously reported the properties of 4a[OTf]2 for which the 3 redox states including a stable organic radical could be optically characterized.8 By studying the 7 other compounds, we show here that the entire family follows the same general trend (Fig. 5 and Fig. S19–S30, ESI†). In spectroelectrochemistry, the first reduced state is characterized by the appearance of new bands in the visible-NIR domains, which are characteristic of π-delocalized radicals. Upon further reduction toward the neutral state, these transitions disappear.17 The persistence in some cases of weak NIR absorption bands in the doubly reduced state probably comes from the presence of traces of the radical cations. The introduction of electron-rich substituents on the platform strongly impacts the stability of the reduced form as no reduced species of 4c2+ could be clearly identified and the redox-triggered modifications appeared irreversible (Fig. S21 and S22, ESI†). Regarding the impact of the endoskeletal modifications, it appears that the spectral shapes and maximal wavelength of the absorption of the radical cations both depend on the nature of the platform, with absorption maxima from 662 nm for 7+˙ to 1218 nm in the case of 6+˙. The return to the initial state is globally satisfying after the first reduction (see the ESI†). The return to the initial state is observed after the second reduction for 4a2+, 4b2+, 4d2+, 62+ and 82+ (see Fig. S19–S30, ESI†).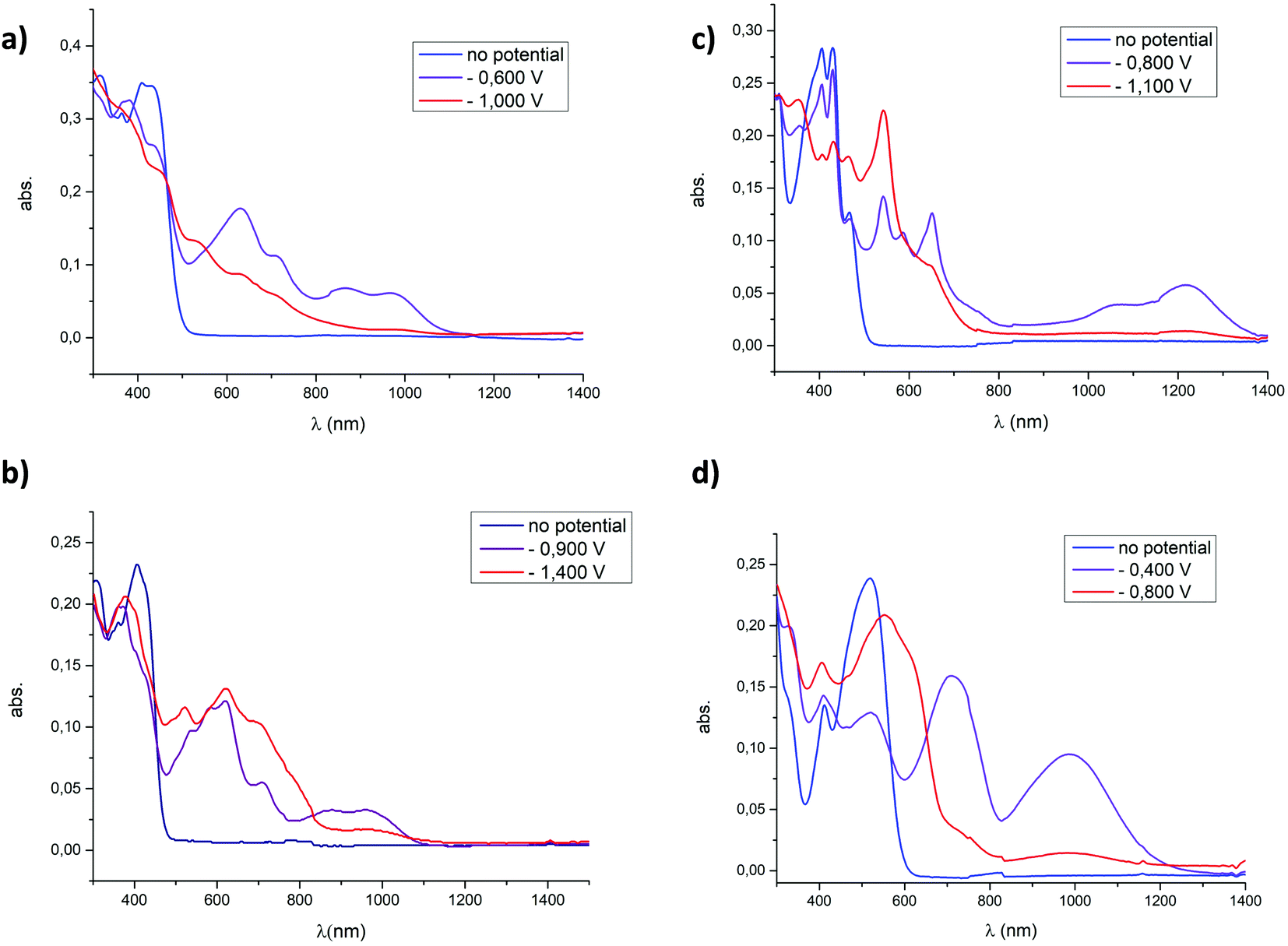 | ||
| Fig. 5 Absorption spectra measured upon electrochemical reduction of (a) 4a[OTf]2, (b) 4a[OTf]2 (c) 6[OTf]2, and (d) 8[OTf]2 (c = 5 × 10−4 M) in DCM (0.2 M Bu4NPF6). | ||
Electron paramagnetic resonance of all 8 chemically formed radicals was then investigated. In all cases but 4c+˙ and 7+˙, which appear too unstable to be recorded under these conditions, the organic nature of these radicals is confirmed by g-factor values in the 2.0032–2.0034 range (Fig. 6 and Fig. S32 and S33, ESI†). The hyperfine structure of all compounds is also fully consistent with the presence of a radical fully delocalized on the entire planar π-skeleton, without contribution of the exocyclic moieties. Hence, the hyperfine couplings are well reproduced by considering the coupling of the unpaired electron with the two P and the different sets of H atoms. Consequently, the modifications of exocyclic substituents in 4b+˙and 4d+˙ do not affect the hyperfine structure compared to 4a+˙ (Fig. S32, ESI†). The computed spin densities of all radical cations also support these findings (vide infra, Fig. 8).
 | ||
| Fig. 6 EPR spectra of the chemically generated 4a+˙ (a), 4b+˙ (b), 6+˙ (c) and 8+˙ (d) in DCM. | ||
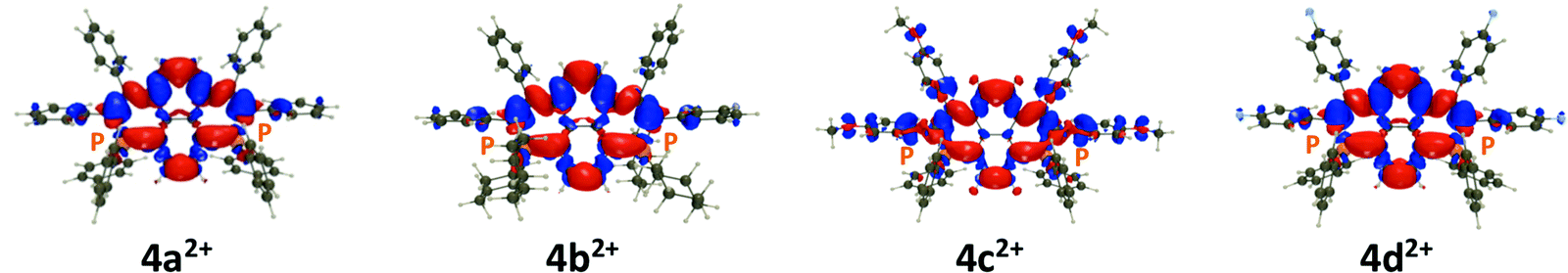 | ||
| Fig. 7 Electron density difference determined for 4a–d2+. The blue (red) lobes correspond to a decrease (increase) of density upon photon absorption, respectively. A contour threshold of 8 × 10−4 a.u. was used. | ||
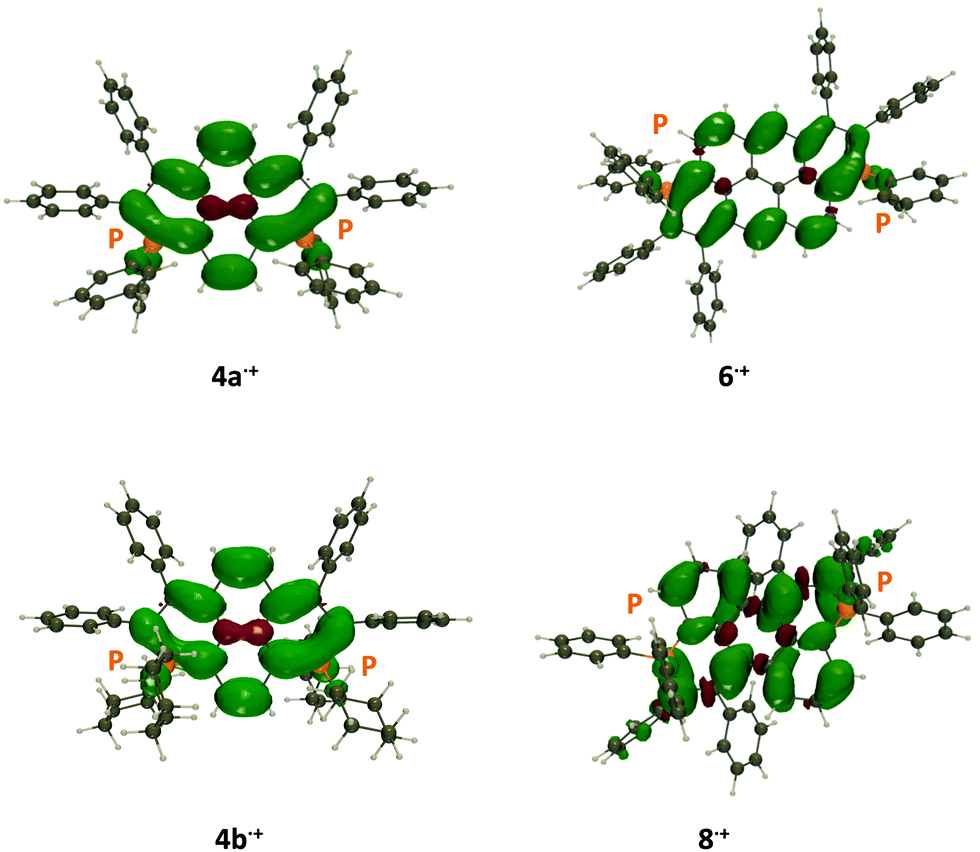 | ||
| Fig. 8 Representation of the spin density difference in the radical cation of 4a+˙, 4b+, 6+˙ and 8+˙ (at their minimal radical cation geometry). The contour threshold is 1 × 10−3 a.u. | ||
In conclusion, this spectroelectrochemical and EPR study allows generalizing to the entire family of compounds the “viologen-like” behaviour previously observed on 4a[OTf]2.8
Theoretical calculations
To gain more insights into the properties of the investigated systems, we have carried out DFT and TD-DFT calculations using a protocol described in the ESI.† The TD-DFT calculations return bright S0–S1 transitions with a clear π–π* character centered on the core of the dyes. The Electron Density Difference (EDD) plots are displayed for all compounds in Fig. 7. One notices that the patterns are very similar for all compounds differing by their exoskeletal substituents that play not a major role in the transition, with the exception of 4c2+ in which the donor methoxy group loses density upon transition, which is consistent with a slight CT character from the periphery to the core and hence a redshift of the absorption (see Fig. S36, ESI,† for the representation of the frontier orbitals of the four compounds). Consistent with the experimental trend (Fig. 3), the oscillator strength determined by TD-DFT is also significantly smaller in 4c2+ (0.40) than in 4a,b,d2+ (0.60–0.66). Regarding the various endoskeletal structures, one notices similar delocalization along all the π-core (Fig. S34, ESI†).8The vertical transition energies determined with TD-DFT using a refined solvation model correspond to absorption wavelengths of 394, 389, 425, 392, 374, 376, 370, and 487 nm, for 4a2+, 4b2+, 4c2+, 4d2+, 52+, 62+, 72+, and 82+, respectively. In the exoskeletal series, one clearly notices the significant redshift of 4c2+, again consistent with the measurements, whereas in the endoskeletal series, the strong bathochromic shift of 82+ is noticeable. We have also determined the vertical emission wavelengths: 599, 513, 722, 610, 622, 450, 462, and 763 nm, for 4a2+, 4b2+, 4c2+, 4d2+, 52+, 62+, 72+, and 82+, respectively. Although TD-DFT exaggerates the difference between the systems, the experimental ranking of the various compounds is reasonably reproduced.
The spin density plots are displayed in Fig. 8. As can be seen, the radical is highly delocalized on the π-conjugated core, in all cases, with no significant contributions from the exoskeletal substituents. Therefore, in agreement with the experimental results, 4a+˙ and 4b+˙ possess similar spin densities. Interestingly, the two central carbon atoms in 4a+˙ show the opposite spin excess, whereas the radical is only delocalized on the rim of the dye in 6+˙, a pattern not found in 8+˙, confirming the specificities of this particular core.
Electrofluorochromism
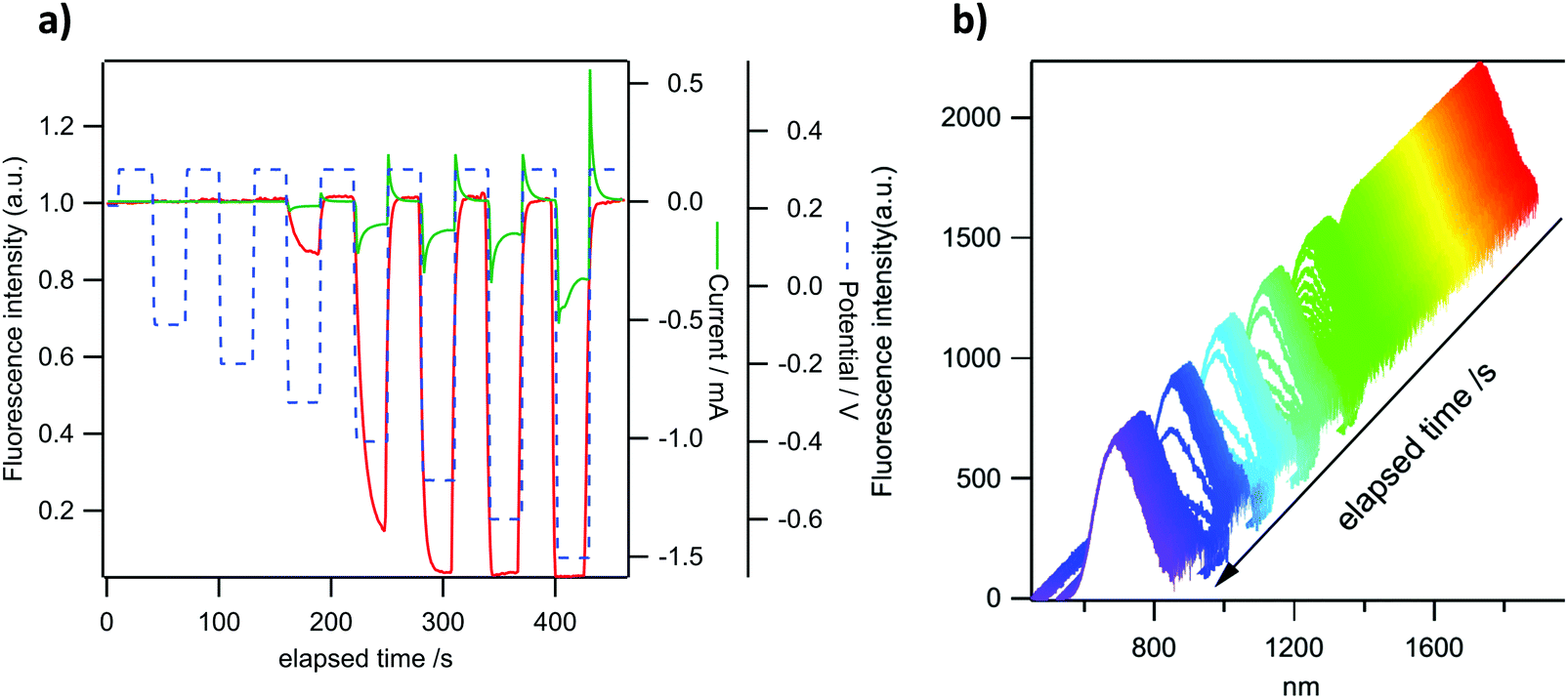
P-Doped PAHs 4–82+ display “viologen-like” redox behavior as illustrated by CV, spectroelectrochemistry and EPR. However, in contrast to most of the viologen derivatives, which fluoresce under very specific conditions only or upon complex molecular engineering,18 they are intrinsically fluorescent. In order to take advantage of this specific property, the electrochemical modulation of the luminescence of 4a2+ and 82+ has been investigated. These two derivatives have been chosen because they provide satisfactory electrochemical reversibility and they emit in different regions of the visible spectrum. Compounds with exoskeletal modifications were not tested as we showed that the effect of electron-withdrawing/electron-rich groups is either too weak to induce significant changes or detrimental for the stability of the reduced species. Emission spectroelectrochemistry and simultaneous variations of fluorescence intensity and redox current have been recorded under potential control for 4a2+ and 82+ in DCM solutions. The ITO substrate is polarized successively at +0.3 V (dicationic state) and −0.6 V for 4a2+ or −0.5 V for 82+ (radical cation state in each case). The results are shown in Fig. 9a and b. A clear modulation of fluorescence intensity can be seen for both compounds that are synchronous with the electrochemical current variations. One can observe a fluorescence intensity drop upon reduction and a full restoration upon reoxidation to the initial state. These results are consistent with the absorption spectroelectrochemical results in Fig. 5 where a clear increase of absorption can be seen in the wavelength range of emission upon reduction. In the case where the reduced form would be emissive, this impedes observing any emission in this spectral range. The fluorescence intensity modulation remains perfectly stable in three successive cycles for both compounds, although a slight drift can be observed for the fluorescence intensity of 4a2+. The relative fluorescence intensity drop is larger in 4a2+ (ca. 30%) than in 82+ (ca. 15%), as a result of the higher electrochemical current for the former compared to the latter. In the potential range explored, the electrochemical conversion is more complete for 4a2+ leading to a higher emission contrast. This phenomenon can also be seen in the variations of emission spectra with time for the same potential modulation (Fig. 9c and d). This allows us to check that the intensity recorded actually corresponds to the emission of both compounds (see Fig. 3 for comparison of the emission spectra in the absence of an electrochemical signal). The fluorescence intensity collected in Fig. 9a and b corresponds to the integration of the whole emission spectrum in Fig. 9c and d.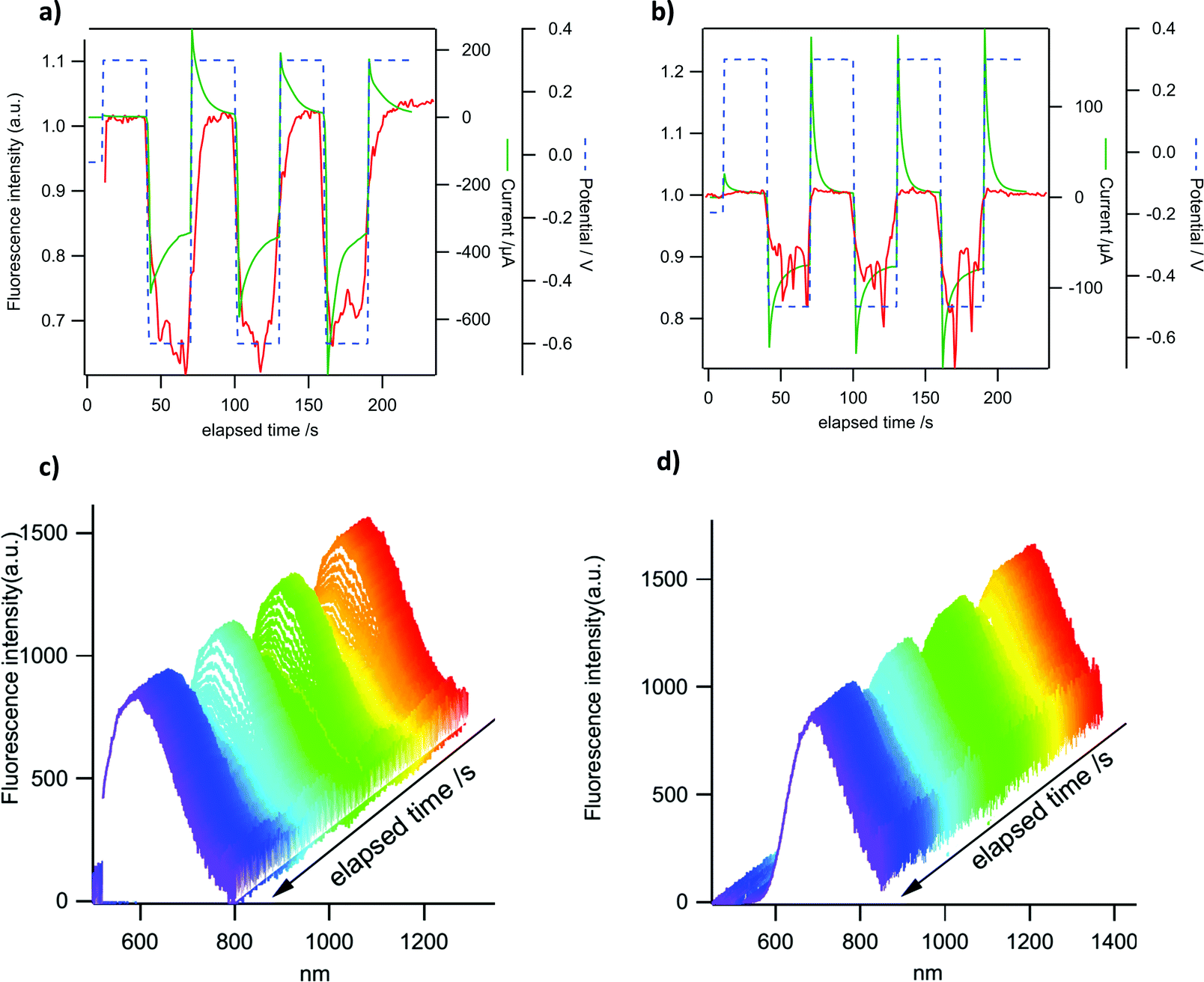 | ||
| Fig. 9 Top (a and b): modulation of fluorescence intensity (red trace) and electrochemical current (green trace) under potential steps (dashed blue trace) for 4a2+ (a) and 82+ (b). Bottom (c and d): corresponding variations of emission spectra of 4a2+ (c) and 82+ (d) with time for the same potential signal as above. Excitation wavelength: 470 nm. The emission is integrated from 520 to 800 nm to give the intensity recorded in (a) and from 600 to 820 nm for the intensity in (b). Concentration of 4a2+ and 82+: 1 mM. | ||
To check that the fluorescence intensity drop can be fully controlled by the applied potential, another potential signal was applied on 82+ (Fig. 10). To enhance the sensitivity, the fluorescence is measured in a microvolume delimited by the substrate and a microelectrode tip (25 μm) positioned 60 μm above the ITO substrate. The latter is polarized at more and more negative potentials at each cycle. The results indicate that the fluorescence starts to drop at −0.3 V for the reduction of 82+ (Fig. 10a) exactly when the electrochemical current starts to be significant. The modulation rapidly increases as the potential limit is pushed toward more negative values (from −0.3 to −0.5 V). From −0.5 V to −0.6 V the modulation remains nearly constant because at these potentials the electrochemical conversion remains fairly constant as well19 and this is confirmed by the amplitude of the electrochemical current. At −0.7 V, the current amplitude starts to increase again due to the start of the second reduction process but the fluorescence intensity does not change much. This gradual increase of the fluorescence intensity modulation can also be seen in Fig. 10b where deeper and deeper variations of the intensity can be seen but without modification of the shape and location of the emission spectra. In conclusion, a nearly full contrast between the ‘on’ and ‘off’ states can be obtained at a very moderately negative potential, which endows 82+ with very good electrofluorochromic properties if compared with already well known compounds in this field like e.g. tetrazines.20 When considering their emission in the red part of the spectrum, and their initial charged state, they can also be advantageously compared to ruthenium complexes21 by offering similar properties but with a reductive quenching. These experiments illustrate that the luminescence of these P-doped PAHs can be electrochemically modulated in the entire visible range, extending the scope of molecules exhibiting efficient luminochromism.22
 | ||
| Fig. 10 Modulation of fluorescence intensity (red trace) and electrochemical current (green trace) of compounds 82+ (a) for potential steps of increasing amplitude (dashed blue trace). (b) Corresponding variation of emission spectra with time of compound 82+ for the same potential signal as in (a). | ||
In conclusion, synthetic access to three novel dicationic P-doped PAHs and a reference compound is described allowing us to have in hand an entire family of 8 derivatives. Exo-skeletal modifications allow the fine-tuning of their optical and redox properties. 4–8[OTf]2 are electron acceptors with “viologen-like” redox behaviour. However, in contrast to most of the viologen derivatives, 4–8[OTf]2 are fluorescent both in solution and in the solid-state. Combined experimental (spectroelectrochemical and EPR) and theoretical studies allowed the more in depth investigation of the reduced species and in particular the stable radical cations formed. This extensive study thus allows generalizing the behaviour previously observed on 4a[OTf]2 and affords a guideline for the property tuning of such derivatives. Finally, the fluorescence properties of 4[OTf]2 and 8[OTf]2 were electrochemically modulated. These first results of electrofluorochromism illustrate the potential of these derivatives to be used as novel luminescent switches or in other relevant applications for multi-stage redox systems such as photoredox catalysis,23 artificial photosynthesis,24 spintronics25 or energy storage.26
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Ministère de la Recherche et de l’Enseignement Supérieur, the CNRS, the Région Bretagne, and the French National Research Agency (ANR Heterographene ANR-16-CE05-0003-01). Y. Molard and G. Taupier (Scanmat-UMS 2001) are thanked for PLQY measurements. This work used the computational resources of the CCIPL supercomputing center installed in Nantes. T. Guizouarn (ISCR) is thanked for assistance with the EPR measurements.References
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS; (b) A. Narita, X.-Y. Wang, X. Feng and K. Mullen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- (a) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS; (b) W. Zeng, T. Y. Gopalakrishna, H. Phan, T. Tanaka, T. S. Herng, J. Ding, A. Osuka and J. Wu, J. Am. Chem. Soc., 2018, 140, 14054–14058 CrossRef CAS; (c) Y. Nakakuki, T. Hirose, H. Sotome, H. Miyasaka and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 4317–4326 CrossRef CAS.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2016, 117, 3479–3716 CrossRef; (b) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS.
- (a) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS; (b) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS; (c) S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486–3487 CrossRef CAS; (d) D. Stassen, N. Demitri and D. Bonifazi, Angew. Chem., Int. Ed., 2016, 55, 5947–5951 CrossRef CAS; (e) P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS; (f) L. Chen, S. R. Puniredd, Y.-Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869–17872 CrossRef CAS.
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS.
- K. Xu, Y. Fu, Y. Zhou, F. Hennersdorf, P. Machata, I. Vincon, J. J. Weigand, A. A. Popov, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 15876–15881 CrossRef CAS.
- R. Szűcs, P.-A. Bouit, L. Nyulászi and M. Hissler, ChemPhysChem, 2017, 18, 2618–2630 CrossRef.
- T. Delouche, A. Vacher, E. Caytan, T. Roisnel, B. Le Guennic, D. Jacquemin, M. Hissler and P.-A. Bouit, Chem. – Eur. J., 2020, 26, 8226–8229 CrossRef CAS.
- Other polyaromatic bisphosphoniums described: (a) P. Hindenberg, F. Rominger and C. Romero-Nieto, Chem. – Eur. J., 2019, 25, 13146–13151 CrossRef CAS; (b) P. Federmann, H. K. Wagner, P. W. Antoni, J.-M. Mörsdorf, J. L. Pérez Lustres, H. Wadepohl, M. Motzkus and J. Ballmann, Org. Lett., 2019, 21, 2033–2038 CrossRef CAS; (c) S. Nieto, P. Metola, V. M. Lynch and E. V. Anslyn, Organometallics, 2008, 27, 3608–3610 CrossRef CAS.
- K. Deuchert and S. Hünig, Angew. Chem., Int. Ed. Engl., 1978, 17, 875–886 CrossRef.
- (a) P. Audebert and F. Miomandre, Chem. Sci., 2013, 575–584 RSC; (b) H. Al-Kutubi, H. R. Zafarani, L. Rassaei and K. Mathwig, Eur. Polym. J., 2016, 83, 478–498 CrossRef CAS; (c) A. Beneduci, S. Cospito, M. La Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240–1247 CrossRef CAS; (d) F. Miomandre, Curr. Opin. Electrochem., 2020, 24, 56–62 CrossRef; (e) C. Romero-Nieto, M. Marcos, S. Merino, J. Barbera, T. Baumgartner and J. Rodriguez-Lopez, Adv. Funct. Mater., 2011, 21, 4088 CrossRef CAS.
- (a) Q. Ge, J. Zong, B. Li and B. Wang, Org. Lett., 2017, 19, 6670–6673 CrossRef CAS; (b) A. Belyaev, Y.-T. Chen, Z.-Y. Liu, P. Hindenberg, C.-H. Wu, P.-T. Chou, C. Romero-Nieto and I. O. Koshevoy, Chem. – Eur. J., 2019, 25, 6332–6341 CrossRef CAS; (c) L. Qiu, W. Hu, D. Wu, Z. Duan and F. Mathey, Org. Lett., 2018, 20, 7821–7824 CrossRef CAS.
- Spectroscopic and redox properties of 5–8[OTf]2 are also found in ref. 8.
- A. Mocanu, R. Szűcs, E. Caytan, T. Roisnel, V. Dorcet, P.-A. Bouit, L. Nyulászi and M. Hissler, J. Org. Chem., 2019, 84, 957–962 CrossRef CAS.
- (a) I. O. Koshevoy, A. Belyaev and P.-T. Chou, Chem. – Eur. J., 2020 DOI:10.1002/chem.202001853; (b) S. Arndt, M. M. Hansmann, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2017, 23, 5429–5433 CrossRef CAS.
- L. Striepe and T. Baumgartner, Chem. – Eur. J., 2017, 23, 16924–16940 CrossRef CAS.
- This trend was previously corroborated by TD-DFT, see ref. 8.
- (a) M. Freitag, L. Gundlach, P. Piotrowiak and E. Galoppini, J. Am. Chem. Soc., 2012, 134, 3358–3366 CrossRef CAS; (b) K. Takahashi, T. Nihira, K. Akiyama, Y. Ikegami and E. Fukuyo, J. Chem. Soc., Chem. Commun., 1992, 620–622 RSC; (c) I. Roy, S. Bobbala, J. Zhou, M. T. Nguyen, S. K. M. Nalluri, Y. Wu, D. P. Ferris, E. A. Scott, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 7206–7212 CrossRef CAS; (d) A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467–8473 CrossRef CAS.
- Indeed the concentration ratio between oxidized and reduced forms follows a S-shape curve with the applied potential, as a result of the Nernst law.
- F. Miomandre, C. Allain, G. Clavier, J.-F. Audibert, R. B. Pansu, P. Audebert and F. Hartl, Electrochem. Commun., 2011, 13, 574–577 CrossRef CAS.
- F. Miomandre, R. B. Pansu, J.-F. Audibert, A. Guerlin and C. R. Mayer, Electrochem. Commun., 2012, 20, 83–87 CrossRef CAS.
- J. W. Sun, Y. N. Chen and Z. Q. Liang, Adv. Funct. Mater., 2016, 26, 2783–2799 CrossRef CAS.
- (a) H. Cheng, X. Wang, L. Chang, Y. Chen, L. Chu and Z. Zuo, Sci. Bull., 2019, 64, 1896–1901 CrossRef CAS; (b) V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS; (c) P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef.
- G. Li, L. Xu, W. Zhang, K. Zhou, Y. Ding, F. Liu, X. He and G. He, Angew. Chem., Int. Ed., 2018, 57, 4897–4901 CrossRef CAS.
- (a) Y. Huang and E. Egap, Polym. J., 2018, 50, 603–614 CrossRef CAS; (b) J. E. Barker, J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2020, 142, 1548–1555 CrossRef CAS; (c) T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- (a) G. Li, B. Zhang, J. Wang, H. Zhao, W. Ma, L. Xu, W. Zhang, K. Zhou, Y. Du and G. He, Angew. Chem., Int. Ed., 2019, 58, 8468–8473 CrossRef CAS; (b) M. Stolar, C. Reus and T. Baumgartner, Adv. Energy Mater., 2016, 6, 1600944 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2001023 and 2001025. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00711k |
| This journal is © The Royal Society of Chemistry 2020 |