
Nanocomposites have attracted considerable attention in recent years and have become key materials in modern synthetic chemistry due to their unique catalytic activities. Wide applications of nanocatalysts (NCs) in the chemical and pharmaceutical industries have been witnessed, which results from their high surface to volume ratio and easy recovery from the medium after completion of the reactions. Additional benefits are enhancements in energy efficiency with the involvement of green technology, which implies minimization of chemical waste generation.1
Among the numerous nanoparticles, silver nanoparticles represent the most sought-after candidate, mainly due to their unique electrical,2 optical,2–4 catalytic,5 and particularly antimicrobial properties,6,7 which are well established and have been extensively investigated. From a catalysis standpoint, as reported in the literature, the activity of silver is comparable to that of gold but the former one is cheaper to handle. Catalysis by silver is well known for C–H activation, C–C activation, and asymmetric aldol reactions with very high regioselectivity and excellent enantioselectivity towards product formation. The π-Lewis acidity of Ag(i) complexes is utilized for C–C activation in nucleophilic reactions.8
Palladium-based catalysts have become significant and interesting to the modern industrial world because of their superior activity, capacity to inhibit CO formation, high surface to volume ratio, maximum number of active centres, and comparatively lower cost than that of platinum-based catalysts. To enhance the catalytic activity of Pd-based NCs, the dispersed Pd nanoparticles are loaded on a supporting material with a developed specific surface area.9–13
Flavonoid compounds are secondary metabolites in plants that have no direct association with their growth or development. From a green perspective, flavonoid compounds possess good reducing and capping abilities in the synthesis of nanomaterials. Besides their role in nanotechnology, flavonoids have found applications in several other fields.14–17 One such flavonoid is rutin (2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one), the glycoside of quercetin, which is abundantly found in plants, such as in buckwheat seed, fruits, and fruit rinds, especially that of citrus fruits (like orange, grapefruit, lemon, etc.). Rutin can also possess significant scavenging properties towards a few oxidizing species like OH˙, O2˙− and O2˙2− radicals in human systems, as observed in several in vivo and in vitro experiments.18–21
The need for developing greener and biogenic routes in nanomaterial synthesis arises to avoid the use of toxic chemicals, reduce environmental loads, and make the process economically viable.22,23 The choice of solvent, and reducing and/or capping agent for the synthesis of metal nanocomposites plays an important role in such a greener approach and makes the methodology an important finding to the scientific world.24–28 Table S1 (ESI†) summarizes some literature data on the particle size of Ag and Pd nanoparticles depending on the green synthesis conditions and the biomaterials used and finally compares them with our work.
Allyl ethers are considered as one of the useful precursors for several organic reactions including [3,3]-sigmatropic shift, polymerization reactions, 1,3-hydrogen shift, photocatalytic reactions, etc.29,30 They are frequently used as protecting groups for alcoholic compounds.31 These ethers may be present as a core unit in several natural products and also in different biologically active molecules.32 The traditional allyl ether synthesis methods usually involve the use of drastic conditions and highly active equivalents of the allylic group.33 But allyl acetates are considered as a better candidate for allylation as compared to highly activated, unstable allyl halides or tosylates because of their high configurational stability and easy accessibility.34 Usually, allyl ethers can be synthesized using allyl halides and phenols in the presence of strong bases, like metal alkoxides.35 In contrast, the use of soft phenolic nucleophiles for allylation has received less attention. In this regard, complexes of some transition metals, viz. Ru,36 Ni,37 Pd,38 Ir,39etc., have been developed as catalysts to prepare allyl ethers. However, most of the existing methods involve costly non-reusable homogeneous catalysts as well as toxic solvent systems. Hence, the development of an efficient, cost-effective, recyclable and heterogeneous catalytic system is necessary to prepare allyl ethers. Incidentally, Saha et al., and Baig et al., have reported a magnetically separable heterogeneous Pd catalyst for the same.40 In parallel, a Pd metal anchored polystyrenal β-alanine-imine network has been developed by our group for the synthesis of allyl ethers.29 Nevertheless, heterogeneous transition metal nanoparticle-induced syntheses of allyl ethers are one of the most attractive areas. With increasing awareness and the growing importance of developing sustainable, eco-friendly, and economically sound synthetic routes, both Pd- and Ag-based NPs have potential to provide alternative promising solutions for researchers.
In the present study, we chose rutin as a capping agent for the synthesis of Ag nanostructures and both as a reducing and capping agent for Pd nanostructures. The nanocomposites so obtained were characterized using different analytical tools like absorption spectroscopy, N2 adsorption–desorption, field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and so on. Then, these nanocomposites were used as catalysts for the etherification reaction of different substituted cinnamyl acetates and phenolic compounds to find the best conditions of synthesis in water medium under reflux for 18 h (Scheme 1).
Rutin hydrate (2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one) (≥94%) was obtained from Sigma Aldrich, USA. Silver nitrate (99.9%) was obtained from Glaxo Laboratories Ltd, India. Palladium chloride (59 to 60%) was obtained from Loba Chemie, Mumbai, India. Cinnamyl acetate (99%) was obtained from Sigma Aldrich, USA. p-Cresol (≥99%) and K2CO3 (≥99%) were obtained from Merck Specialities Private Limited, India. CDCl3 was purchased from Sigma Aldrich, USA. Ethyl acetate (≤99.5%) was purchased from Merck Specialities Private Limited, India. Silica Gel G (∼13%) and Silica Gel GF (∼13%) were obtained from Spectrochem Pvt. Ltd, India. The pH of rutin solutions was adjusted using dilute HCl (37%) (Merck) or NaOH (≥97%) (Merck) solutions. All other chemicals were of analytical grade (AR) and used as obtained. All the solvents used were distilled and dried before actual use. Triple distilled water was used throughout the experiments.
Absorption spectra were recorded on a PerkinElmer Lambda 25 UV-Vis spectrophotometer. The temperature-controlled absorbance data were measured using a Varian Cary 50 Bio UV-Vis spectrophotometer. Nitrogen adsorption–desorption isotherms were measured at liquid nitrogen temperature (77 K) using a Quantachrome surface area analyzer. TEM images were obtained using a JEOL JEM 2100 HR with EELS transmission electron microscopy. The Fourier transform infrared (FTIR) spectra of the samples were recorded from 400 to 4000 cm−1 on a PerkinElmer FT-IR 783 spectrophotometer having a resolution of 1 cm−1 using KBr pellets. Powder X-ray diffraction (PXRD) was measured using an X-PERT-PRO Panalytical diffractometer to confirm the actual phase of the prepared material. A centrifuge machine, Remi Elektrotechnik Ltd R-4C, was used to separate the nanocomposites from the aqueous phase. The thermogravimetric analysis of the NCs was carried out using a Diamond TG/DTA Thermogravimetric/Differential Thermal Analyzer of PerkinElmer Instruments in the temperature range from 28 to 439 °C. The concentrations (mol%) of silver and palladium in the nanocomposites were estimated using an inductively coupled plasma-optical emission spectrometry (ICP-OES) instrument (ICAP duo 6500, Thermofisher-Scientific, RF power 1150 Watt, flush pump rate 50 rpm, analysis pump rate 50 rpm, auxiliary gas flow 1.0 L min−1, nebulizer gas flow 0.60 L min−1 and coolant gas flow 12 L min−1, wavelength 224.64 nm, 328.068 nm, and 339.289 nm for silver and 324.27 nm and 340.458 nm for palladium). X-ray photoelectron spectroscopy (XPS) measurement was carried out using a Thermo Scientific ESCALAB 250 Xi system. A Mettler Toledo digital balance correct up to the fourth decimal place was used for measuring the weights of samples. A constant temperature incubator, NOVA Model: SHCI 10(D), was used for obtaining the palladium and silver–palladium nanocomposites. A digital Mettler Toledo Seven Compact pH/ion meter was used to measure and adjust the pH of the rutin solution. Thin-layer chromatography (TLC) was performed on TLC silica gel 60 F254. The products were purified using silica gel (60–120 mesh) column chromatography. Nuclear magnetic resonance (NMR) spectra (1H and 13C NMR) were obtained on a 300 MHz Bruker spectrometer using CDCl3 as a solvent. The 1H and 13C chemical shifts are reported in ppm relative to tetramethylsilane (TMS).
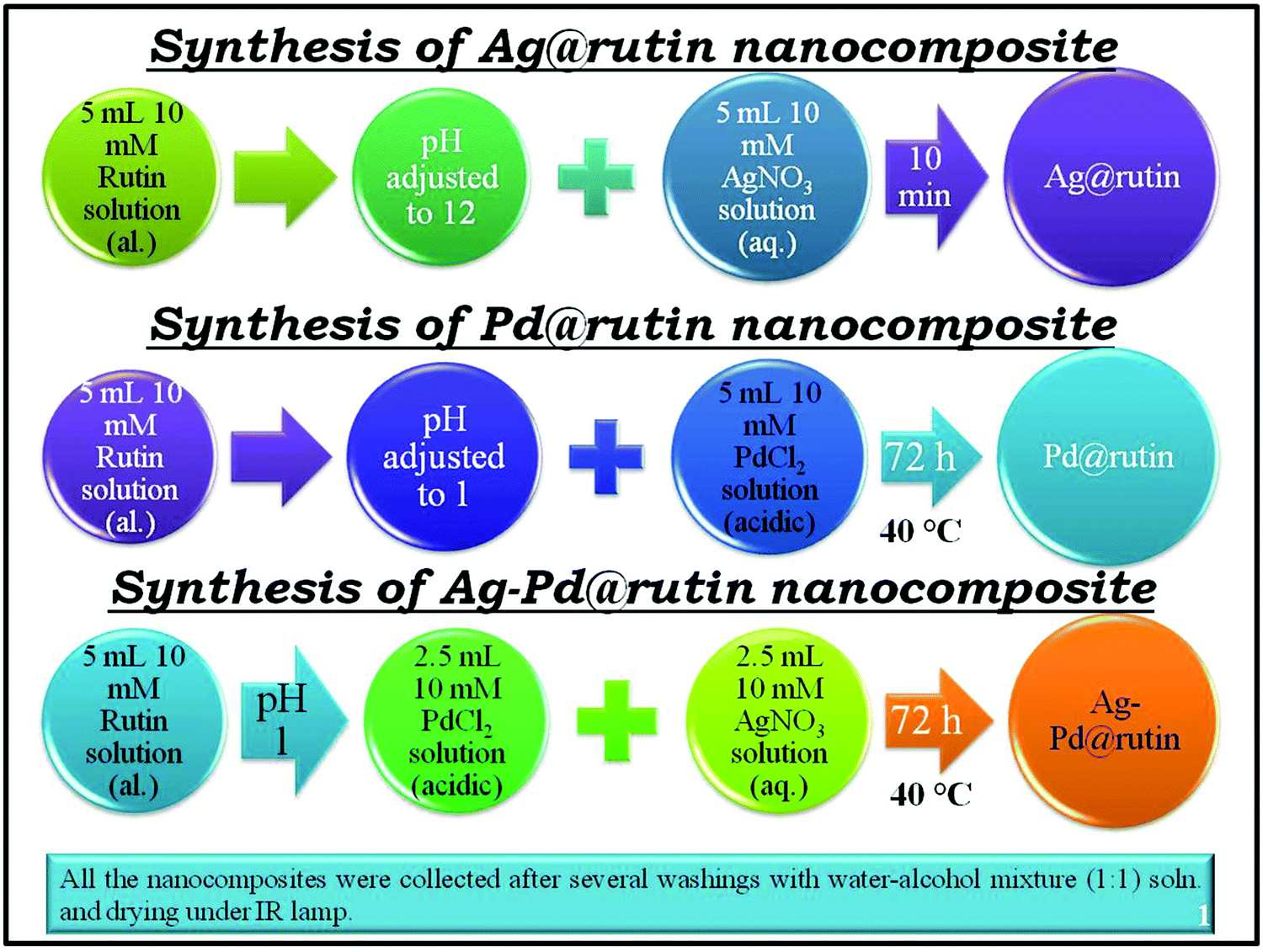
5 mL of a 10 mM ethanolic solution of rutin adjusted to pH 12 was treated with 5 mL of a 10 mM aqueous solution of AgNO3. As soon as the addition was complete, the color of the rutin solution changed from faint red to deep brown, indicating the formation of the nanocomposite. To complete the reaction, the solution was kept undisturbed in the dark for 5 to 10 min. A small portion of it was taken out for characterization. The supernatant solution was removed by centrifugation. The obtained solid was then washed two to three times with a water–ethanol (1 : 1) mixture to remove the unreacted rutin and AgNO3. The final product was obtained by drying under an IR lamp.
5 mL of a 10 mM rutin solution adjusted to pH 1 was treated with 5 mL of a 10 mM acidic solution of PdCl2 (prepared in 1 M HCl). Then, the solution was stirred for 5 min. Finally, the mixture was kept at 40 °C for 72 h. After completion of the reaction, a small portion of the colloidal solution was taken out for characterization. The solution was then centrifuged and the supernatant was discarded. The nanocomposite in the centrifugate was washed with a water–ethanol (1 : 1) mixture two to three times and finally dried under an IR lamp.
5 mL of a 10 mM rutin solution adjusted to pH 1 was treated with 2.5 mL of a 10 mM acidic solution of PdCl2 and 2.5 mL of a 10 mM aqueous solution of AgNO3. Then, the solution was stirred for 5 min. Finally, the mixture was kept at 40 °C for 72 h. A small portion was taken out for characterization. The solution was then centrifuged and the supernatant was discarded. The final product was collected as described above. Scheme 2 gives a clear understanding of the synthesis approach.
The metal–rutin complexation was determined using UV-Vis spectrometry at different pHs between 1 and 12. A spectacular color change was observed at pH 1 and 12 (Fig. S1, ESI†), indicating that the most prominent changes occurred at these two pH values. Absorption spectroscopy was therefore used to determine the association constants of metal–rutin complexation at pH 1 and pH 12. Silver–rutin complexation was studied at pH 12 and palladium–rutin complexation and silver–palladium bimetallic complexation with rutin were studied at pH 1 as indicated by the visual color changes. In 2.5 mL of pH adjusted water, 7 μL of a 10 mM ethanolic solution of rutin was added, and then the mixture was treated with the gradual addition (6 μL aliquots) of 1 mM metal ion solutions. The change in absorption data was taken as a function of the concentration of metal ions. Finally, 1/(A − A0) was plotted against 1/[metal], which gave a straight line maintaining the general relationship (Benesi–Hildebrand equation):
 (1)
where A0 = the absorbance of the rutin solution in the absence of metal, A1 = absorbance of rutin when it is completely bound with the metal, A = absorbance of rutin with the gradual addition of metal, [M] = concentration of metal, and Ka = binding or association constant.41
(1)
where A0 = the absorbance of the rutin solution in the absence of metal, A1 = absorbance of rutin when it is completely bound with the metal, A = absorbance of rutin with the gradual addition of metal, [M] = concentration of metal, and Ka = binding or association constant.41
The time scan profile obtained using absorption spectroscopy helped to estimate the possible mechanism behind the formation of the nanocomposites.
Before the experiment, the solid material was degassed at ∼300 °C in a surface area analyzer instrument for ∼3 h.
The nano dimensions of the materials were confirmed by the TEM images. To perform this experiment, a colloidal solution containing each of the nanocomposites was drop cast on a carbon-coated Cu grid. Then, the grid was dried under an IR lamp before experimenting.
FTIR analysis was utilized to get a clear idea about the different types of bonds that are present in the nanocomposites. We also extended our study to the change of bond frequencies upon nanocomposite formation from that of a pristine rutin molecule. A little amount of the solid material was mixed thoroughly with dried KBr in a mortar and pestle. Then, the solid mixture was introduced into a steel arrangement followed by application of about 5 tons of pressure on the solid mass through a hydraulic pressure instrument to make a transparent pellet. Finally, the pellet was loaded into a sample holder inside the FTIR instrument analyzer to obtain the spectrum.
The powder sample was further characterized using X-ray diffraction analysis to ascertain the crystalline nature of the nanocomposites. The powder sample was first taken in a rectangular metal holder and then lightly pressed for PXRD analysis.
Thermogravimetric analyses of the three samples were performed to check the thermal properties of the NCs. The experiment was carried out under a N2 atmosphere with a flow rate of 10 mL min−1. Around 6 mg of solid material was taken on a platinum pan and then gradually heated to ∼439 °C with a heating rate of 10 °C min−1 to record the change of weight percentage of the material with the change of temperature.
XPS is a surface-sensitive technique with a sampling depth of 7.5 nm to 10 nm of the top surface under analysis in the case of polymers.42 XPS was used to confirm the composition and the respective oxidation states of each of the elements present in the nanocomposites. The XPS instrument is equipped with a monochromated Al Kα X-ray source with an X-ray spot size of 900 × 900 μm2. Data were analyzed using Thermo Avantage Software (Version 5.952) using a smart background.
As described earlier, we have prepared three nanocomposites, namely, Ag@rutin, Pd@rutin, and Ag–Pd@rutin NCs. All of these nanocomposites were used as catalysts for the etherification reaction between cinnamyl acetate (1a) and p-cresol (2a). The general procedures for the reaction are described in the ESI.† The identity of the product was confirmed by 1H and 13C-NMR spectroscopy.
The leaching experiment was carried out with all three NCs. After the completion, the reaction mixture was centrifuged. The NC was separated from the reaction mixture and kept aside. Besides, the liquid part containing the reaction mixture was collected and then digested with hot concentrated nitric acid. After three consecutive addition and evaporation steps of the concentrated acid, a solid mass was obtained and it was cooled to room temperature. Thereafter, the solid mass was mixed with a requisite amount of Milli-Q water and then filtered using a microsyringe filter (pore size 0.22 μm). Finally, the filtrate was diluted with water to a specific volume and the resulting solution was analyzed using an ICP-OES instrument.
The reusability study was only performed with the Pd@rutin NC as it produced the maximum product yield among the examined NCs. To examine the reusability of the Pd@rutin nanocomposite, an O-allylation reaction was performed using cinnamyl acetate (1a) and p-cresol (2a). After completion of the reaction, the NC was separated from the reaction mixture through centrifugation. Then, the NC was repetitively washed with water and ethyl acetate followed by dichloromethane to remove any traces of the organic part. Finally, the NC was dried at 75 °C for 2 h under vacuum for reactivation. The NC was found to be recyclable for 4 cycles without significant loss in its activity.
The feed compositions of the nanocomposites of Ag@rutin, Pd@rutin, and Ag–Pd@rutin prepared in different pH media are described with their yields in Table S2 (ESI†).
The rutin solution has three positions of λmax (viz. 225 (π → π* transition), 270 (π → π* transition) and 411 nm (n → π* transition)) in pH 12 solution. Upon treatment with the silver solution, absorbance increased at all three positions (Fig. 1). The increase at λ = 225 and 270 nm was due to silver itself (Fig. S2, ESI†). The peaks for silver in pH 12 at λ = 217 and 255 nm were shifted to 225 and 270 nm, respectively, in the presence of the rutin solution. The increase at λ = 411 nm was only due to the interaction between silver and rutin molecules. So, we tried to obtain the association constant for interaction between silver and rutin at this particular λmax using the Benesi–Hildebrand (B–H) eqn (1). The plot so obtained (Fig. 1 inset) using the B–H equation suggested that the association constant (Ka) for the metal with the ligand is 3833.33 M−1 with the stoichiometry of 1 : 1. The results are tabulated in detail in Table 1.

| Nanocomposite | λmax (nm) | Stability constant (M−1) | Stoichiometric ratio |
|---|---|---|---|
| Ag@rutin | 411 | 3833.33 | Ag : rutin = 1 : 1 |
| Pd@rutin | 350 | 606.48 | Pd : rutin = 1 : 1 |
| Ag–Pd@rutin | 350 | 492.36 | Ag : Pd : rutin = 0.5 : 0.5 : 1 |
At pH 1 and 40 °C, rutin showed characteristic absorption maxima at three wavelengths (viz. ∼205 (π → π* transition), 250 (π → π* transition) and 350 nm (n → π* transition)). Upon addition of palladium, absorption at 205 and 250 nm increased, and a new shoulder appeared at ∼287 nm. The peak at 250 nm and the shoulder at 287 nm were due to the palladium solution itself under these conditions (Fig. S3, ESI†). The slight change in absorbance at ∼350 nm with the gradual addition of palladium solution to rutin was then the main focus of interaction (Fig. 2). A B–H graph was then plotted at this particular wavelength (Fig. 2 inset) giving an association constant value (Ka) of 606.48 M−1 with a metal:rutin stoichiometry of 1 : 1. The detailed results are tabulated in Table 1.

In the presence of the silver–palladium bimetallic solution, the situation was similar to that of Pd@rutin (Fig. 3). The increase in absorbance at λ = 250 and 287 nm was due to the bimetallic solution at pH 1 and 40 °C (Fig. S4, ESI†). The slight change of absorbance at 350 nm wavelength was due to the interaction between the bimetallic solution and rutin molecules. The B–H plot (Fig. 3 inset (for λ = 350 nm)) was then obtained that suggested a 1 : 1 coordination of metal:rutin with the association constant (Ka) = 492.36 M−1. The detailed results are tabulated in Table 1.

The mechanism of nanocomposite formation was studied using an absorption time scan at a particular wavelength taking equal volumes and the same concentration of rutin and metal in the respective pH medium. For the Ag@rutin nanocomposite, equal volumes and the same concentration of AgNO3 and rutin were taken in pH 12 medium and then the absorption time scan was measured at 411 nm up to 45 minutes (2700 s) (Fig. 4). Whereas, for Pd@rutin and Ag–Pd@rutin, equal volumes and the same concentration of rutin and the metal solution were taken in pH 1 medium and then the adsorption time scan was measured at 350 nm up to 45 min (2700 s) (Fig. 4).

For Ag@rutin, a gradual decrease in absorption at 411 nm with time may be due to the rapid reaction between Ag and the rutin moiety, which resulted in the precipitation of the Ag@rutin nanocomposite from the solution.43 Due to such precipitation of the nanocomposite, the amount of free rutin or Ag@rutin in solution decreased, which resulted in a gradual decrease in the absorption of the solution.
For the Pd@rutin nanocomposite, there was a gradual increase in the absorption at 350 nm with time. The above synthesis procedure suggested that about 72 h was required to synthesize the Pd@rutin nanocomposite. So, we may say that this gradual increase of the absorption suggested slow and steady nucleation and growth of the nanocomposite, as described in the classical approach of nucleation and growth.44
For the bimetallic nanocomposite, the situation is quite similar and resultant of the mechanistic pathway of both Ag and Pd. The rapid decrease in the absorption in the first few minutes was due to the interaction between Ag and rutin, whereas the latter increase in the absorption was due to the interaction between Pd and rutin following the classical nucleation and growth mechanism.44
Fig. 5 shows the N2 adsorption–desorption isotherms of Ag@rutin, Pd@rutin, and Ag–Pd@rutin nanocomposites. The isotherm for Ag@rutin resembled type IV with the H4 hysteresis loop. This type of isotherm corresponds to a mesoporous adsorbent and the adsorption behavior can be determined from the adsorbent–adsorptive interaction. This type of adsorption was observed due to the simultaneous occurrence of monolayer and multilayer adsorption on the surface by a pore condensation mechanism. Particle aggregation in the nano-state developed this type of hysteresis (H4).45 For Pd@rutin and Ag–Pd@rutin, the isotherms looked like type II with a H3 hysteresis loop. This convex type of isotherm is generally observed for free monolayer-multilayer adsorption on the surface of the adsorbent at high P/P0 values with a tiny knee at lower P/P0 due to completion of monolayer coverage. The knee is characteristic of an overlap of monolayer adsorption and the beginning of multilayer adsorption. The H3 hysteresis is common for a particle having a plate-like assembly at the surface.45 The difference in the surface properties of Ag@rutin from those of Pd@rutin and Ag–Pd@rutin may have occurred due to the reaction time of the formation of the nanocomposites. For Ag@rutin, aggregated particles were mostly formed due to the rapid development of the nanocomposite. For Pd@rutin and Ag–Pd@rutin, they took sufficient time to form the nanocomposites and developed one layer on top of the other, giving a plate-like assembly.

The TEM micrographs of the nanocomposites reflect their nano-dimensional properties (Fig. 6). For Ag@rutin, the figure itself and the inset suggest the aggregated nature of the nanocomposite along with the lattice fringes. The porous nature of the Ag@rutin nanocomposite can also be confirmed from these images. For Pd@rutin, the TEM image (Fig. 6) showed discrete smaller particles with clear lattice fringes. A layered structure with a porous nature of the Pd@rutin nanocomposite can also be viewed from the image itself and the inset picture. For Ag–Pd@rutin, the TEM image (Fig. 6) shows aggregated particles with very insignificant lattice fringes. The porous nature cannot be confirmed from this image, whereas the corresponding inset picture indicates a plate-like assembly of the Ag–Pd@rutin nanocomposite. This was the combined result of both Ag@ruitn and Pd@rutin. The small reaction time of the Ag@rutin nanocomposite allowed aggregated particles to form that deposited readily within 5 to 10 min. For the Pd@rutin nanocomposite, the comparatively long reaction time with a very small deposition of the nanocomposite at the bottom of the container after the completion of the reaction suggested that discrete smaller particles were formed with layers of the nanocomposite depositing one after the other. For Ag–Pd@rutin, although the reaction time and type of deposition were similar to that of the Pd@rutin nanocomposite, the overall particle nature was the result of the conjugate effect of both the metals. The particle agglomeration mainly occurred due to silver and the layer like assemblies were observed due to palladium.

The elemental mapping of the NCs is shown in Fig. 7. The red color indicates the Ag metal in Ag@rutin NC, whereas green indicates the Pd metal in Pd@rutin NC. In the bimetallic nanocomposite, the efficient mixing of these two colors suggests alloy formation between the two metals, Ag and Pd, in the Ag–Pd@rutin nanocomposite. The alloy formation is also supported by results obtained from PXRD and XPS analyses, discussed later in the XPS section.

The histograms of the nanocomposites (Fig. 8) suggested that the average particle size of Ag@rutin, Pd@rutin, and Ag–Pd@rutin was ∼50 nm, ∼10 nm, and ∼80 nm, respectively.

Fourier transform infrared spectra of the nanocomposites along with the pure rutin compounds are shown in Fig. 9. Although there were several peaks, we were mainly interested in the zone of 500 to 1650 cm−1, which was modified to a good extent upon the addition of different metal solutions. For the Ag@rutin nanocomposite, there was a peak shift from 1362 to 1376 cm−1 (stretching of the C–O–C bond)46 due to the modification of silver on the rutin molecule. At the same time, the multiplet like feature of the pure rutin also disappeared upon modification by silver. For the Pd@rutin nanocomposite, again, the appearance of the multiplet was observed but its peak intensity was quite low. At the same time, there was a new peak at 590 cm−1 due to the formation of the Pd–O bond.47 For the Ag–Pd@rutin nanocomposite, the FTIR spectrum was quite similar to that of the Pd@rutin nanocomposite, indicating that upon bimetallic modification, there was a greater conjugation with palladium than silver metal due to the long reaction time. A similar observation was also obtained from XPS analysis as discussed later. Table 2 describes the different frequencies with possible functionalities obtained from the FTIR analysis.

| Sample | Peak position (cm−1) | Functionality | Ref. |
|---|---|---|---|
| Pure rutin | 3426 | Aromatic –OH stretching | 46 |
| 2936 | Asymmetric –CH2 + asymmetric –CH3 | ||
| 1653 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching O stretching |
||
| 1600–1456 | C–C stretching | ||
| 1363 | C–O–C stretching | ||
| 1294 | |||
| 1211 | |||
| 1149 | Stretching and bending of the keto group | ||
| 1066 | C–O–C stretching | ||
| 1018 | |||
| 811 | C–H stretching | ||
| Ag@rutin NC | 3440 | Aromatic –OH stretching | 46 |
| 2922 | C–H stretching | ||
| 1653 |
CO stretching |
||
| 1377 | C–O–C stretching | ||
| 1204 | |||
| 1060 | |||
| 804 | C–H stretching | ||
| 659 | O–H deformation | 47 | |
| Pd@rutin NC | 3426 | Aromatic –OH stretching | 46 |
| 2964 | C–H stretching | ||
| 1701 |
CO stretching |
47 | |
| 1605 | C–C stretching | 46 | |
| 1515 | |||
| 1445 | |||
| 1370 | C–O–C stretching | ||
| 1287 | |||
| 1176 | Stretching and bending of the keto group | ||
| 790 | C–H vibration | 47 | |
| 590 | Pd–O bond vibration | 46 | |
| Ag–Pd@rutin NC | 3426 | Aromatic –OH stretching | 46 |
| 2929 | Asymmetric –CH2 + asymmetric –CH3 stretching | ||
| 1714 |
CO stretching |
47 | |
| 1605 | C–C stretching | 46 | |
| 1515 | |||
| 1439 | |||
| 1356 | C–O–C stretching | ||
| 1287 | |||
| 1190 | |||
| 997 | |||
| 756 | C–H vibration | 47 | |
| 590 | Pd–O bond vibration | 46 | |
The obtained nanocomposites were further characterized by powder X-ray diffraction analysis (Fig. 10). The PXRD data so obtained were then compared with the unique number obtained from the Joint Committee on Powder Diffraction Standards (JCPDS). For the Ag@rutin nanocomposites, the major diffractions located at 2θ values of 31.84°, 38.25°, 44.44°, and 45.57°, which correspond to the (060), (111), (200) and (100) planes. For the Pd@rutin nanocomposites, the most intense diffraction came from the (023) plane at a 2θ value of 40.24°. Whereas, the Ag–Pd@rutin nanocomposite showed diffraction mainly at four positions having 2θ values of 27.94°, 32.35°, 40.11°, and 46.34°. The PXRD data of the prepared samples are tabulated in Table 3 with possible (hkl) planes and JCPDS numbers.

| Sample | Ag@rutin NC | |||
|---|---|---|---|---|
| Obtained position [°2θ] | Actual position [°2θ] | Possible hkl plane | Possible lattice | JCPDS no. |
| 27.513 | 27.493 | 024 | Rhomb-centered | 87-0422 |
| 29.531 | 29.608 | 214 | Rhomb-centered | 87-0422 |
| 31.846 | 31.770 | 060 | Primitive | 79-2441 |
| 38.259 | 38.202 | 111 | Face-centered | 87-0720 |
| 44.441 | 44.402 | 200 | Face-centered | 87-0720 |
| 45.578 | 45.539 | 100 | Primitive | 41-1402 |
| 56.588 | 56.508 | 544 | Rhomb-centered | 87-0422 |
| 64.562 | 64.426 | 220 | Face-centered | 04-0783 |
| 66.329 | 66.315 | 113 | Primitive | 89-3081 |
| 75.392 | 75.323 |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .23 .23 |
Primitive | 89-3081 |
| 77.497 | 77.472 | 311 | Face-centered | 04-0783 |
| 81.679 | 81.606 | 1106 | Rhomb-centered | 87-0422 |
| 84.079 | 84.136 | 063 | Primitive | 84-0713 |
| Pd@rutin NC | ||||
| 40.243 | 40.263 | 023 | End-centered | 79-0322 |
| 46.723 | 46.733 |
18 |
Primitive | 45-0773 |
| 68.198 | 68.141 |
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 16 16 |
Primitive | 89-6629 |
| 82.234 | 82.245 | 006 | End-centered | 79-0264 |
| 86.692 | 86.619 | 260 | End-centered | 79-0264 |
| Ag–Pd@rutin NC | ||||
| 27.949 | 27.736 |
12 |
Primitive | 45-0773 |
| 32.357 | 32.467 | 102 | Primitive | 50-1058 |
| 40.111 | 40.116 | 130 | End-centered | 79-0264 |
| 46.344 | 46.380 |
15 |
Primitive | 89-6629 |
| 54.959 | 54.909 | 214 | Primitive | 89-6629 |
| 57.568 | 57.499 |
![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 03 03 |
Primitive | 51-1440 |
| 67.852 | 67.899 | 150 | End-centered | 79-0264 |
| 76.814 | 76.876 | 044 | End-centered | 79-0264 |
| 82.058 | 82.175 | 461 | End-centered | 79-0322 |
| 85.806 | 85.812 | 171 | End-centered | 79-0322 |
The TGA graphs of the samples (Fig. 11) indicate the stability of the prepared NCs up to ∼200 °C. Only ∼15%, 10%, and 4% mass loss occurred at ∼100 °C for Ag@rutin, Pd@rutin, and Ag–Pd@rutin NCs, respectively, which may be due to the degradation of the polyphenol molecules present in the NCs. As our reaction proceeded at ∼100 °C, we can conclude that the NCs are stable enough at this temperature and are capable of catalyzing the reaction.

XPS is a surface-sensitive technique with a sampling depth of 7.5 nm to 10 nm of the top surface under analysis in the case of polymers.42 Understandably, the analysis carried out using XPS would certainly provide details of up to 10 nm depth of the top surface layer, rather than the bulk material. Also, the surface free energy of rutin is a lot lower than those of Ag and Pd.48 Pd has a much higher surface free energy (1950 mJ m−2)48 than that of Ag (1300 mJ m−2).48 For that reason, when in a composite, it is expected that Pd would locate underneath the top surface layer to decrease surface free energy for stable equilibrium. With silver having a lower surface free energy and higher sensitivity to XPS, when a composite made up of Ag and Pd and biomaterials is under investigation by XPS, the detected amount of Ag is expected to be higher. To extract specific information from the XPS core-level spectra, deconvolution into several component peaks was carried out, revealing oxidation states along with the bonding environment of the element. However, without cross-matching with each element (for example, if Ag2p spectra are analyzed to find oxidation states, O1s spectra should also be analyzed and referenced appropriately to complete the set), peak assignment solely based on comparisons to the reference binding energy values was incomplete. Binding energy cross-matching with references can only provide some indication and can only be used as a necessary but not sufficient condition to claim bond formation.49 In our present set of samples, the O1s binding energy region (∼531 eV) was overlapped with Pd3p3/2 (∼530 eV). The amount of oxygen (O1s) quantified does include contributions from Pd3p regions and for that reason, the amount reported here from the survey spectra is just for indication and trends only. For the same reason, O1s narrow scan data has not been analyzed in this report either.
The survey scan spectra of these Ag@rutin, Pd@rutin, and Ag–Pd@rutin nanocomposites are shown in the ESI,† Fig. S5. As Ag has a lower surface free energy and higher sensitivity towards XPS, a lot more amount of Ag can be detected than Pd, as reflected in Table 4.
| Sample | Atomic % of | ||||
|---|---|---|---|---|---|
| Ag3d5/2 | Pd3d5/2 | O1s | C1s | Cl2p | |
| Ag–Pd@rutin | 0.3 | 0.3 | 29.8 | 67.1 | 2.6 |
| Ag@rutin | 25.6 | 0.0 | 29.9 | 42.7 | 1.9 |
| Pd@rutin | 0.0 | 0.7 | 32.2 | 66.5 | 0.7 |
In Fig. 12a–d, the narrow scan spectra of silver and palladium are shown. All narrow scan spectral positions were referenced by fixing the C1s sp2–sp3 mixed hybridized position at 284.7 eV. Rutin has both sp2 and sp3 hybridized carbons and differentiating sp2 and sp3 carbons requires further analysis. A combined peak position was the reference position and it was fixed to calculate charge shifting of the narrow scan peaks. C1s narrow scan spectra of all the samples are shown in the ESI,† Fig. S6.

In the case of the Pd@rutin nanocomposite (Fig. 12a), Pd showed the presence of metallic palladium50 and palladium(ii) oxide.51 There was no silver present in this sample. Silver in the Ag@rutin nanocomposite (Fig. 12b) was present as silver(i) oxide (Ag2O). However, things were different in the case of the Ag–Pd@rutin nanocomposite. XPS revealed some very interesting phenomena happening in these synthesized nanocomposites. As shown in Fig. 12c and d, the Pd3d5/2 peak was positioned at 336.4 eV and the Ag3d5/2 peak was positioned at 368.6 eV. There might have been the formation of Pd–Ag alloy and as per the NIST XPS database, palladium–silver alloy (90–10)/carbon is the compound.52 Using density functional theory (DFT) and by calculating the segregation energies for binary alloy nanoparticles, it has previously been reported that the formation of a Pd core and Ag shell is energetically favorable.53 To cross-check the alloy formation, Ag–Pd alloy should have XRD peaks at 2θ = 39.92° and 2θ = 46.29° corresponding to the 111 and 200 planes, respectively, of Ag–Pd alloy.54 As shown in the PXRD table (Table 3), the Ag–Pd@rutin nanocomposite has a couple of major peaks at 2θ = 40.11° and 2θ = 46.34°. So, these results matched very closely with the palladium silver alloy formation theory, supported by both PXRD and XPS techniques. There was another explanation that could also be investigated. It has been previously reported that Ag3d5/2 and Pd3d5/2 peaks are shifted by 0.3 eV towards a higher BE due to the Ag–Pd interaction55 due to the charge transfer between Ag and Pd.56,57 Considering the extra 0.3 eV charge shifting towards a higher BE, the Ag3d5/2 peak corresponded to silver(i) oxide (Ag2O)58 and the Pd3d5/2 peak corresponded to either palladium(vi) oxide (PdO3)59 or palladium(ii) oxide (PdO). The charge shift happened due to charge transfer between Ag and Pd. These results provided evidence of bimetallic interfaces between Ag and Pd, which are partially responsible for the high catalytic activity.
Catalytic activity of the prepared nanocomposites was examined for the O-allylation reaction of phenolic compounds. Our preliminary study aimed to screen the catalyst using cinnamyl acetate (1a) and p-cresol (2a) as the model substrates (Table 5). The as-synthesized three NCs, Ag@rutin, Pd@rutin, and Ag–Pd@rutin nanocomposites, were used for this study. It was found that both Ag@rutin and Pd@rutin nanocomposites were successful in furnishing the corresponding ether (Table 5, entries 1 and 2). The reactivity of the Pd@rutin nanocomposite was much greater than that of the Ag@rutin system. The activity of the Ag–Pd@rutin nanocomposite was also examined and was found to have nearly the same reactivity as that of the Ag@rutin nanocomposite (Table 5, entry 3). The reaction optimization using Ag@rutin and Ag–Pd@rutin NCs using the same reaction was performed and the results are tabulated in Tables S3 and S4, respectively, in the ESI.† From the TEM data, we found that the average particle size of Ag@rutin was much larger than that of Pd@rutin. Therefore, Pd@rutin having a smaller particle size can provide a higher surface area for the substrates and thus furnished higher product yields than Ag@rutin. Similarly, the Ag–Pd@rutin bimetallic nanocomposite furnished comparatively lower product yields due to the large size of the Ag@rutin NC in the nanocomposite that decreased the availability of the reactive but small Pd NC. However, this result is quite interesting as no report is available in the literature regarding a Ag NC-based catalyst induced O-allylation reaction of phenolic compounds to the best of our knowledge. Finally, the activity of rutin was also examined and it was found that rutin has no catalytic activity in this reaction system (Table 5, entry 4). The most probable reaction mechanism is presented in Scheme 3. The reaction proceeds through an interaction of Pd(ii) or Ag (i) with cinnamyl acetate followed by oxidative addition with p-cresol resulting in the formation of a η3-π-allyl complex that undergoes reductive elimination to give the final product,29 which was further characterized by 1H and 13C-NMR spectroscopy (Fig. S5–S9 in the ESI†).
| Entry | Catalyst | Time (h) | Yieldb (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ag@rutin | 18 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Pd@rutin | 18 | 88 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Ag–Pd@rutin | 18 | 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4c | Rutin | 25 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Conditions: cinnamyl acetate (1 mmol), p-cresol (1 mmol), K2CO3 (2 mmol), catalyst (0.00027 mol%), and water, reflux. b Yields refer to those of isolated products obtained by column chromatography. c 6 mg of catalyst. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

After screening the catalysts, a series of reactions was performed to get the optimized reaction conditions using the Pd@rutin NC. Among various solvents (DMF, toluene, acetonitrile, xylene), water gave the best result in terms of yields (Table 6, entries 1–5). Besides, K2CO3 was found to be suitable as a base as compared to NaHCO3, Cs2CO3, K3PO4 (Table 6, entries 5–8). All the reactions were performed under refluxing conditions as a high temperature was required for the activation of the catalyst (Table 6, entries 9, and 10). During the experiments on the reaction, no product was formed without any catalyst even after 30 h. This observation indicates the necessity of the catalyst (Table 6, entry 11). The best product yield was obtained using water as a green solvent and K2CO3 as a base under refluxing conditions for 18 h with stirring using 0.00027 mol% Pd@rutin NC.

|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Temp. (°C) | Time (h) | Yieldb (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | DMF | K2CO3 | 120 | 18 | 68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Toluene | K2CO3 | 110 | 18 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Acetonitrile | K2CO3 | 78 | 18 | 31 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Xylene | K2CO3 | 120 | 18 | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Water | K2CO3 | 100 | 18 | 88 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Water | NaHCO3 | 100 | 18 | 82 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Water | Cs2CO3 | 100 | 18 | 41 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Water | K3PO4 | 100 | 18 | 41 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Water | K2CO3 | 60 | 18 | 59 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Water | K2CO3 | rt | 30 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11c | Water | K2CO3 | 100 | 30 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Conditions: cinnamyl acetate (1 mmol), p-cresol (1 mmol), base (2 mmol), Pd@rutin (6 mg, 0.00027 mol%), solvent, temp., and time. b Yields refer to those of isolated products obtained by column chromatography. c Without any catalyst. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
After optimization of the reaction conditions, we started to explore the versatility of the Pd@rutin nanocomposites. Several substituted cinnamyl acetates and phenolic compounds were studied through this reaction to produce the corresponding product ethers (Table 7). Phenols containing an electron-donating group (–Me) and withdrawing group (–Br) in their aromatic ring underwent this etherification reaction smoothly. Similarly, cinnamyl acetate with –Me and –OMe substitution in its aromatic ring also reacted to give the corresponding ethers with good yields (entries 3c–e, Table 7).

|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TOF = TON/time, [TON = moles of substrate converted per mole of active site] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
The Pd@rutin NC can be reused for 4 consecutive cycles. The reusability chart is presented in Fig. 13. After the recycling experiment, the NC was analyzed thoroughly by FTIR spectroscopic analysis (after the 4th run) (Fig. 14). A table (Table 8) was also made to compare the FTIR peak position of the fresh Pd@rutin catalyst to that of the catalyst after the 4th cycle. The results of the analysis indicates that the nanocomposites were significantly stable up to 4 cycles. Moreover, the leaching experiment of the reaction mixture was also performed by ICP-OES analysis, and no Ag or Pd NPs were detected in the solution, or in the case where Pd was detected, it was below the limit of detection for any of the nanocomposites. Finally, the utility of our newly developed methodology is compared with literature reports and is demonstrated in Table S5 (ESI†). It can be seen that entry 1 in Table S5 (ESI†) suggests a lower time of reaction and higher reusability than our work, but the present work not only deals with the catalytic activity of the nanocomposites but also offers a green synthesis of the nanocatalysts. Both entry 1 and the present work show similar catalytic activity, but the synthesis of the catalyst in entry 1 neither used a biogenic source nor involved a green methodology.


| Initial peak position (cm−1) | Peak position (cm−1) after reuse |
|---|---|
| 3426 | 3426 |
| 2964 | 2923, 2852, 2357 |
| 1701, 1605, 1515, 1445 | 1633 |
| 1370 | 1399, 1383 |
| 1287, 1176 | 1150, 1033, 875 |
| 790 | 798, 753, 698 |
| 590 | 561 |
In this work, we have prepared Ag, Pd, and Ag–Pd bimetallic nanocomposites using a greener approach to reduce the environmental load and established interesting catalytic activity of the nanocomposites. We have characterized these metal nanocomposites using different analytical tools like – absorption spectroscopy, N2 adsorption–desorption, TEM, FTIR, PXRD, and XPS analyses. Finally, the catalytic activity of the prepared nanocomposites towards the etherification reaction was studied. From the study, we confirmed that the palladium nanocomposites showed higher catalytic activity than bimetallic and Ag@rutin nanocomposites towards this studied reaction. The Pd@rutin NC can also be reused up to four cycles without considerable loss in product yield. Additionally, the role of Ag in catalyzing such reactions has been demonstrated for the first time to the best of our knowledge with a fairly encouraging yield. It has been perceived that although the formation of Pd@rutin shows a slow formation rate, it exhibits a smaller particle size with dual oxidation states (Pd(ii) and Pd(0)) and a better catalytic activity than those of the other two materials. The most significant observation is the drastically reduced amount of the costly metal Pd that is required for the catalysis as compared to homogeneous catalysis. The amount of active catalyst required is only 0.00027 mol% of the material, which is again reusable after regeneration for at least 4 cycles. This is in coherence with the results of the ICP-OES analysis of the reaction media for the leached out metal. Additionally, the reaction proceeds in a water medium with 88% product yield. Therefore, in a nutshell, the work is in agreement with green chemistry perspectives and opens a vast field of organic synthesis with the easy synthesis and commendable use of such potential catalysts.
There are no conflicts to declare.
PS expresses sincere thanks to the University Grants Commission (UGC) (Ref. No. of PS 20/12/2015(ii)EU-V dated 24.08.2016) for providing the necessary fellowship. The authors would also like to acknowledge the funding received under the Global Challenge Research Fund (GCRF) provided by the University of Brighton, UK. We also express our sincere gratitude to Mr Nayan Ranjan Saha, Department of Chemical Technology, University of Calcutta, India for carrying out the PXRD analysis. We want to express our sincere gratitude to Prof. Susanta Lahiri and his fellow Dr Nabanita Naskar, Chemical Science Division, Saha Institution for Nuclear Physics, India for performing ICP-OES of our samples to determine the mol percentage of the NCs. We are also thankful to Prof. Abhijit Chakrabarti and his scholar Dr Dipayan Bose, Crystallography & Molecular Biology Division, Saha Institute of Nuclear Physics, India for helping to measure the absorbance data at 40 °C.
This journal is © The Royal Society of Chemistry 2020