
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobust, remeltable and remarkably simple to prepare biomass–sulfur composites†‡
Moira K.
Lauer
a,
Menisha S.
Karunarathna
a,
Andrew G.
Tennyson
 ab and
Rhett C.
Smith
*a
ab and
Rhett C.
Smith
*a
aDepartment of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: rhett@clemson.edu
bDepartment of Materials Science and Engineering, Clemson University, Clemson, South Carolina 29634, USA
First published on 30th September 2020
Abstract
Lignocellulosic biomass holds a tremendous opportunity for transformation into carbon-negative materials, yet the expense of separating biomass into its cellulose and lignin components remains a primary economic barrier to biomass utilization. Herein is reported a simple procedure to convert several biomass-derived materials into robust, recyclable composites through their reaction with elemental sulfur by inverse vulcanization, a process in which olefins are crosslinked by sulfur chains. In an effort to understand the chemistry and the parameters leading to the strength of these composites, sulfur was reacted with four biomass-derivative comonomers: (1) unmodified peanut shell powder, (2) allyl peanut shells, (3) ‘mock’ allyl peanut shells (a mixture containing independently-prepared allyl cellulose and allyl lignin), or (4) peanut shells that have been defatted by extraction of peanut oil. The reactions of these materials with sulfur produce the biomass–sulfur composites PSx, APSx, mAPSx and dfPSx, respectively, where x = wt% sulfur in the monomer feed. The influence of biomass![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sulfur ratio was assessed for PSx and APSx. Thermal/mechanical properties of composites were evaluated for comparison to commercial materials. Remarkably, unmodified peanut shell flour can simply be heated with elemental sulfur to produce composites having flexural/compressive strengths exceeding those of Portland cement, an effect traced to the presence of olefin-bearing peanut oil in the peanut shells. When allylated peanut shells are used in this process, a composite having twice the compressive strength of Portland cement is attained.
:sulfur ratio was assessed for PSx and APSx. Thermal/mechanical properties of composites were evaluated for comparison to commercial materials. Remarkably, unmodified peanut shell flour can simply be heated with elemental sulfur to produce composites having flexural/compressive strengths exceeding those of Portland cement, an effect traced to the presence of olefin-bearing peanut oil in the peanut shells. When allylated peanut shells are used in this process, a composite having twice the compressive strength of Portland cement is attained.
Introduction
Lignocellulosic biomass constitutes the most abundant and underutilized by-product of human industry, yet the economic conversion of biomass into fuels and feedstocks for chemical processes remains one of the great challenges in science. There is little doubt that harnessing biomass is the key to the future green economy needed to ensure the sustainability and resilience of civilization, and while great strides have been made in developing efficient processes for the conversion of cellulose to fuel ethanol, the need to separate lignin from cellulose prior to that conversion greatly detracts from the economy of the process.1–5 Even once separated, the lignin is a high-volume, low-value commodity whose full potential has yet to be leveraged.6As part of our ongoing effort to explore the viability of lignocellulosic biomass as a precursor to durable composites, we recently reported a process by which agricultural by-product peanut shells can be converted to a composite having mechanical properties that rival those of familiar structural materials like Portland cement.7 This process involved a low-temperature aqueous olefination of finely-ground peanut shell powder (particle size ≤300 μm, Fig. S1, ESI‡) and subsequent polymerization by the ∼100% atom-economical inverse vulcanization upon reaction with elemental sulfur to yield APS95 (Scheme 1, x = wt% sulfur in the monomer feed). Inverse vulcanization is a process in which olefins are crosslinked upon their reaction with thermally-generated sulfur radicals (Scheme 2) and has proven quite versatile to prepare high sulfur-content materials (HSMs) from both naturally-occurring and synthetic olefin crosslinkers.8–11 Such high sulfur-content materials can be used in a variety of applications including thermal imaging, batteries, structural materials and water purification, driving the development of innovative synthetic and processing approaches.12–36 Significant advances have been made in lowering the reaction temperature and controlling curing of HSMs through the use of prepolymers,23 ternary systems15 or catalysts.14,37 Pioneering insight into facile processing techniques relying on a clever S–S metathesis process have made HSMs even more attractive as candidates for commercial applications by opening up the possibility for their use in additive manufacturing and compression moulding.38,39
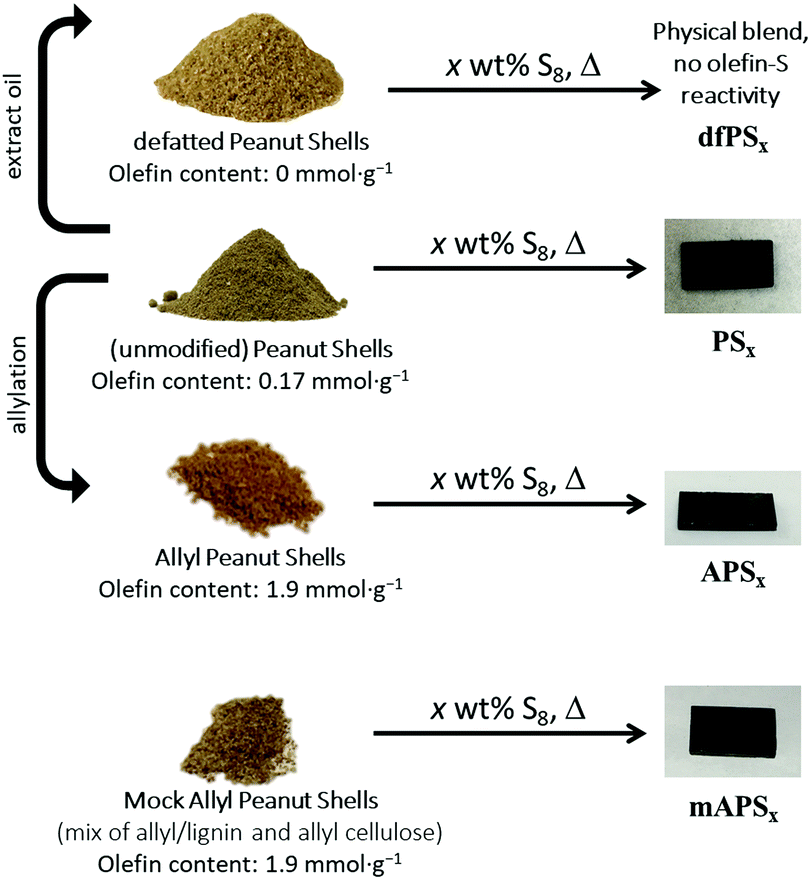 | ||
| Scheme 1 Preparation of biomass–sulfur composites, where x denotes wt% sulfur in the monomer feed. | ||
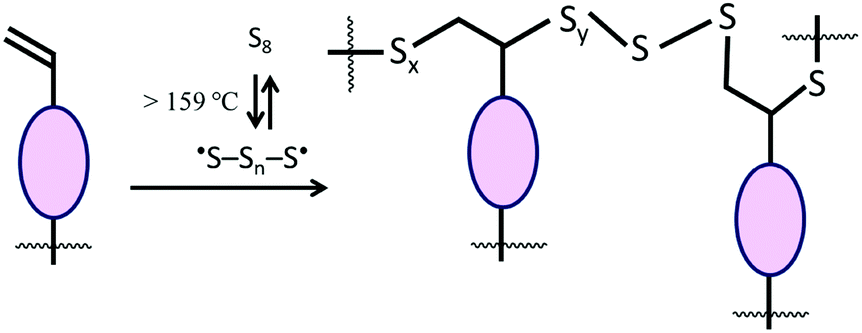 | ||
| Scheme 2 Schematic for the general inverse vulcanization process. | ||
Encouraged by advances in HSM processing and the high mechanical strength of APS95, we hypothesized that the properties of durable biomass–sulfur composites could be tuned by subtly changing the biomass:sulfur ratio or biomass composition in monomer feeds. We also hypothesized that unsaturated triglycerides that make up naturally occurring plant oils – present to varying extent in lignocellulosic waste streams – could contribute to crosslinking in biomass–sulfur composites derived from less-processed sources. The inverse vulcanization of triglycerides from a variety of plant oils has been reported16,19,20,37,40–43 and high mechanical strength can be achieved with relatively low (≤5 wt%) triglyceride content.44 The presence of plant oils could thus be an important factor in predicting how predominantly lignocellulosic biomass sources would perform as comonomers with sulfur. To test these hypotheses, a range of biomass–sulfur composites were prepared (Scheme 1). To prepare the HSMs, elemental sulfur was polymerized with one of several biomass-derived comonomers: (1) peanut shells (which contain some peanut oil) to give PSx; (2) allylated peanut shells to give APSx, (3) ‘mock’ allylated peanut shells to give mAPSx, and defatted peanut shells to give dfPSx. For all the materials prepared, x = wt% S in the monomer feed, and each biopolymer–sulfur composite had either 5 or 10 wt% of biomass crosslinker in the monomer feed.
The mock allylated peanut shell material was comprised of lignin and independently-synthesized allyl lignin and allyl cellulose mixed in such proportion as to accomplish the same lignin:cellulose ratio and to have the same concentration of olefins per unit mass as the authentic allylated peanut shells have. A comparison of properties for APSxversus those for mAPSx was conceived as a way to assess the extent to which properties are affected by the presence of lignin–cellulose crosslinks in APSx but not in the cellulose–lignin physical blend used to prepare mAPSx. The defatted peanut shells have had the peanut oil extracted prior to their reaction with sulfur to preclude the involvement of unsaturated triglycerides in the attempted crosslinking reaction. A comparison of the properties of PSx to dfPSx should thus inform on the extent to which triglyceride crosslinking contributes to material properties. The thermal and mechanical properties of the biomass–sulfur materials were characterized for comparison to familiar structural materials. In some cases, flexural and compressive strengths twice those of portland cement are accomplished in the biomass–sulfur composites.
Results and discussion
Synthesis of biomass–sulfur composites
We recently reported a method to allylate finely-ground peanut shells as a representative sample of lignocellulosic biomass (Fig. S1, ESI‡).7 This simple aqueous allylation process gave allylated peanut shells containing 1.9 mmol olefin per g material (determined by iodometric titration, the details of which can be found in the ESI‡). The allylated peanut shells were used directly as the organic monomer in inverse vulcanization with 95 wt% sulfur to give APS95 (Scheme 1). An expanded set of biomass–sulfur composites was targeted for the current study in an effort to better understand the process and delineate the factors that influence mechanical properties. Compositions of the four biomass-derived monomers selected for this purpose are summarized in Scheme 1 and Table 1.| Biomass derivativea | Cellulose:lignin ratio |
Oil content (wt%) | Olefin content (mmol g−1) | Resultant compositee |
|---|---|---|---|---|
| a Each biomass derivative was filtered to pass a 300 μm sieve prior to polymerization. b Determined by iodometric titration. c Determined by mass loss upon hexanes extraction. d Determined by iodometric titration and 31P NMR spectrometry of phosphidylated derivative. e The subscript x in the names indicates the wt% of sulfur in the monomer feed. f A mixture of lignin with independently-prepared allyl cellulose and allyl lignin. g Peanut shells from which the peanut oil has been extracted with hexanes. | ||||
| Allyl peanut shells | 3:2 |
0 | 1.9 ± 0.04b | APSx |
| Unmodified peanut shells | 3:2 |
1c | 0.17 ± 0.05b | PSx |
| Mock allyl peanut shellsf | 3:2 |
0 | 1.9d | mAPSx |
| Defatted peanut shellsg | 3:2 |
0 | 0 | dfPSx |
One goal of the current study was to assess the influence of biomass content on composite properties. APS90 was thus prepared with twice the biomass content in the monomer feed as compared to APS95. Both APS90 and APS95 are dark-coloured, readily-remeltable materials that are readily re-shaped by simply melting and pouring into moulds, where they are allowed to cure under ambient conditions (Scheme 1). Composites PSx, made from sulfur and unmodified peanut shells were also prepared having 5 or 10 wt% of biomass for comparison.
Another goal of the current study was to assess the extent to which potential cellulose–lignin crosslinking present in allylated peanut shells contributes to the strength of the resulting biomass–sulfur composites. Towards this end a ‘mock’ allylated peanut shell powder was prepared by mixing independently-prepared samples of lignin, allyl lignin and allyl cellulose in such a ratio as to accomplish the same lignin:cellulose ratio and olefin content as in the allyl peanut shells that were prepared directly from the raw peanut shell powder. Composite mAPS95, prepared by the reaction of mock peanut shells and sulfur, led to a remeltable solids that was quite similar in appearance to APSx (Scheme 1).
A third goal of the current study was to assess whether plant oils in biomass waste might contribute to material properties of the biomass–sulfur composites. Plant oils are comprised primarily of unsaturated triglycerides, which are known substrates for inverse vulcanization.16,17,20,40–42 Peanut shells generally contain about 1 wt% peanut oil.45 The peanut oil triglycerides are comprised of 18% saturated, 50% monounsaturated (48% oleate) and 31% polyunsaturated (30% linoleate) fatty acid chains.46 Although the peanut oil content of peanut shells is only 1 wt%, the storage modulus of elemental sulfur is known to increase by nearly an order of magnitude when it undergoes inverse vulcanization with just 1 wt% of free fatty acids linoleic acid26 or oleic acid.30 Furthermore, each triglyceride molecule already links three fatty acid molecules, comprising up to six olefins in the case of a triglyceride having three linoleate chains, effectively increasing the crosslink density compared to that afforded by the free fatty acids. If crosslinking of sulfur by the peanut oil present in the peanut shells provides sufficient strength to the composite materials, then peanut shells would not need to be allylated to produce durable materials, which would clearly be a significant improvement in terms of the sustainability and affordability of the process. In a broader context, understanding how the presence of plant oils influences properties could be an important factor in identifying other privileged biomass sources for production of durable materials by the current route.
To first confirm the presence of unsaturated peanut oil triglycerides in the peanut shell product used for the current study, a sample of the ground peanut shell powder was extracted with hexanes, in which peanut oil is soluble but proteins, lignin, cellulose and hemicellulose are quite insoluble. The hexanes solvent was removed under reduced pressure, and the hexanes-soluble material was found to account for 1% of the peanut shell mass.
Analysis of the hexanes-extractable material by 1H NMR spectrometry and comparison to a literature spectrum for peanut oil (Fig. 1) confirmed that the hexanes-soluble extract consists primarily of peanut oil, though there are also some other less intense resonances present that cannot be definitively assigned to peanut oil. Additional confirmation and quantification of unsaturation content in the peanut shells was obtained by iodometric titration of the peanut shell power, from which it was determined that the raw shells have an olefin content of 0.17 mmol g−1.
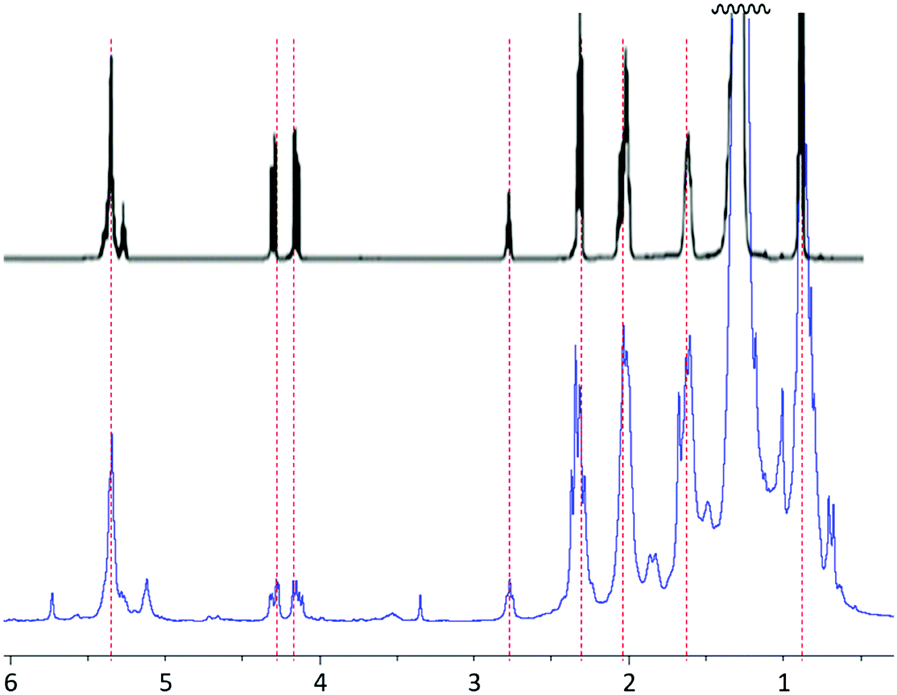 | ||
| Fig. 1 Proton NMR spectra in CDCl3 for isolated peanut oil (upper spectrum) and hexanes-extractable material (lower spectrum) from peanut shells used to prepare PSx. The spectrum for isolated peanut oil is reprinted from E. G. Silveira Jr, et al., Potential of Virginia-type peanut (Arachis hypogaea L.) as feedstock for biodiesel production, Ind. Crops Prod., 89, 451, © 2016, with permission from Elsevier. | ||
Having confirmed that the unmodified peanut shells contained the olefin moieties needed for crosslinking by sulfur, the shells were tested by their reaction with 90 or 95 wt% elemental sulfur to form composites PSx (x = wt% sulfur in monomer feed). When raw peanut shell powder is heated with elemental sulfur at 180 °C, durable composites are in fact produced that are remeltable and similar in appearance to APSx (Scheme 1).
If the presence of peanut oil is the critical feature that allows raw peanut shell powder to serve as a substrate for inverse vulcanization, then peanut shells from which peanut oil has been extracted should not produce a homogenous material, but rather a physical blend of peanut shell powder and sulfur. As a control experiment, defatted peanut shells (resulting from the hexanes extraction study described above) were thus heated with sulfur to yield composite materials dfPSx comprising 90 or 95 wt% sulfur (Scheme 1).
Scanning electron microscopy with element mapping by energy-dispersive X-ray analysis (SEM-EDX) confirmed that dfPS95 and dfPS90 are physical blends in which unreacted peanut shell particles are clearly visible (Fig. 2). These results are consistent with our previously reported findings that a physical blend of sulfur and either unmodified cellulose or unmodified lignin is likewise heterogeneous and phase-separates.27,32 In contrast to what was observed for dfPSx, the SEM-EDX analyses shown in Fig. 2 provides evidence that olefin-containing APSx, PSx and mAPSx all form microscopically homogeneous materials. All of these materials show a uniform distribution of sulfur, carbon and oxygen (carbon and sulfur maps are provided in Fig. 2, while oxygen elemental maps are included in the ESI‡ file, Fig. S17–S22). These data provide evidence that chemical reaction between biomass-bound olefins and sulfur is a requirement for homogenization. Further evidence that the strength of PSx is derived from the presence of the peanut oil is that a sample of pure sulfur is too brittle to survive clamping even at the minimum clamping force possible with the test instruments.
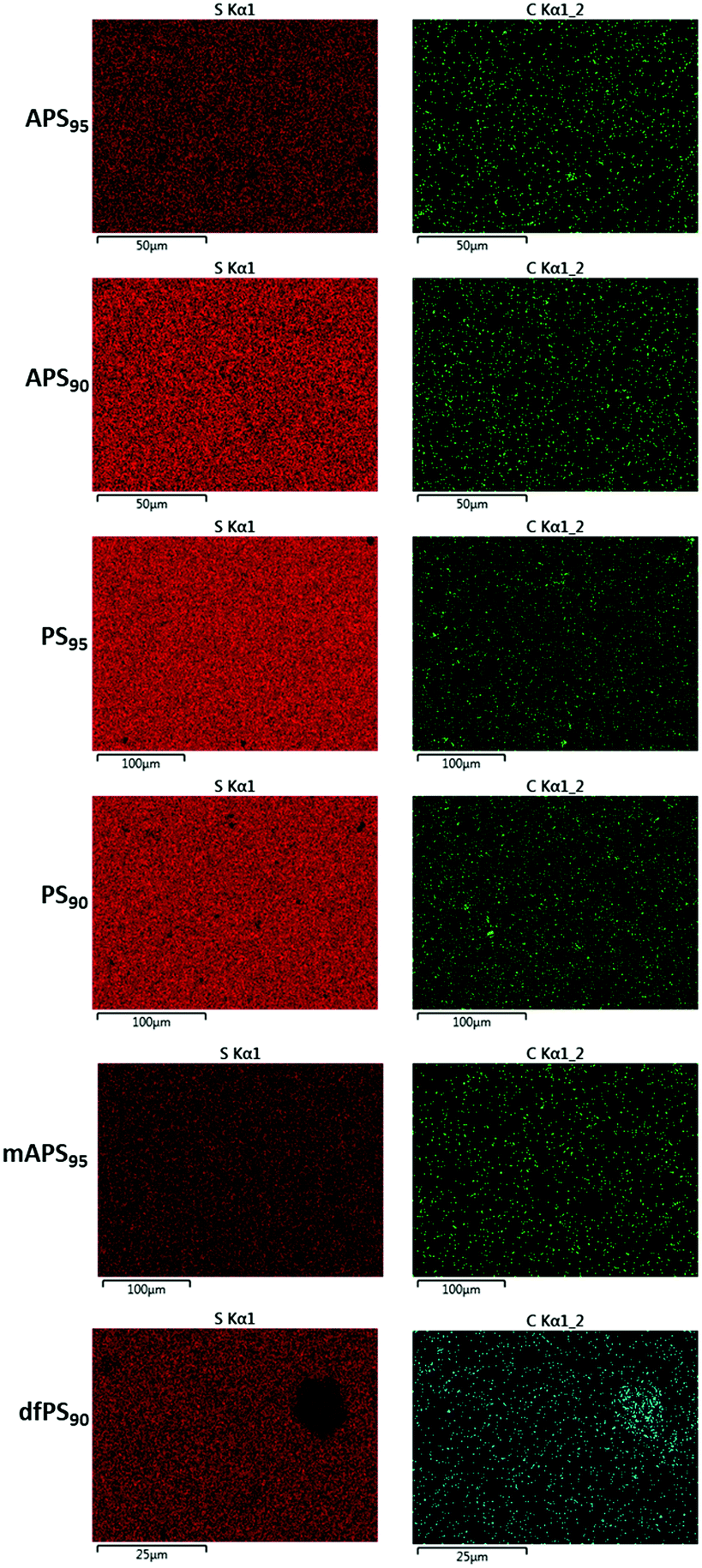 | ||
| Fig. 2 Surface analysis of biomass–sulfur composites by EDX revealed the homogeneous distribution of sulfur (left) and carbon (right) for biomass–sulfur composites. Voids in the sulfur map where high carbon content is observed are identified as unreacted peanut shell particles in the physical blend dfPS90. | ||
Many microscopically homogeneous HSMs are composites characterized by a sulfur-crosslinked organic network and some elemental sulfur that is not covalently linked to the organic component. Sulfur that is not covalently attached to the crosslinked network is readily extracted into CS2. Gravimetric and elemental analysis of the extractable sulfur and insoluble fractions were thus undertaken as a means to quantify the extent to which sulfur was covalently incorporated into the materials. Fractionation studies paired with evidence of quantitative consumption of olefins by infrared spectroscopy (Fig. S8 and S9, ESI‡) allow for the calculation of average number of sulfur atoms per crosslink, known as the sulfur rank. The results of these fractionation studies are summarized in Table 2. From these data, it can be observed that increasing the wt% of organic in the material led to a decrease in CS2 solubility in all cases, as would be anticipated on the basis of available sites for covalent tethering of sulfur chains. The results observed for APS95 and APS90 were similar to what was observed with sulfur composites prepared from allyl cellulose alone, which had sulfur ranks of ∼20.31 Surprisingly, composites PS90 and PS95 exhibited a lower fraction of CS2-extractable sulfur compared to their APSx analogues. This observation could be due to longer polysulfur crosslinks or occurrence of crosslinking by reactions other than the typical inverse vulcanization. Based on the titratable alkenes in peanut shells, sulfur ranks were calculated to be as large as 330 and 257 for PS95 and PS90, respectively. Sulfur rank in mAPS95 was somewhat larger than for corresponding APSx but much smaller than PSx and was similar to that observed in composites made from allyl lignin and elemental sulfur.32
| Material | CS2 insolublea (%) | Sulfur rankb | T d (°C) | T cc (°C) | Char yielde (%) |
|---|---|---|---|---|---|
| a The percentage of material that was insoluble in CS2 after recursive washings. b The average length of sulfur atoms per crosslink as determined by CS2 extraction. c The temperature at which 5% of the material had decomposed by TGA. d The range of cold crystallization temperatures observed in cycles 2–5 of DSC scans. e The residual mass after heating to 800 °C at 10 °C min−1 under N2 as an average of duplicate runs with standard deviations. | |||||
| APS95 | 11 | 20 | 217 ± 1 | 28–33 | 1.7 ± 0.4 |
| APS90 | 23 | 21 | 215 ± 2 | 25–28 | 3.7 ± 0.2 |
| PS95 | 14 | 330 | 223 ± 4 | 22–24 | 1.3 ± 0.5 |
| PS90 | 24 | 257 | 217 ± 1 | 33–34 | 5.3 ± 0.0 |
| mAPS95 | 20 | 33 | 219 ± 7 | 24–28 | 2.1 ± 0.0 |
All of the materials had similar Td values attributable to decomposition of S8 (Table 2). Both APS95 and PS95 exhibited a second mass loss step at just above 300 °C that was not observed in the other materials (Fig. S10, ESI‡), likely a result of the more extensive crosslinking in these materials (vide infra). Char yields scaled predictably with the amount of organic content, with the exception of PS90, which showed a larger than anticipated char yield, potentially due to attendant thermal crosslinking of hemicelluloses constituents that are not present in either the allylated peanut shells used to prepare APSx or the constituents used to prepare mAPSx. That lignin and hemicellulose improve one another's thermal stability in such a way has been established for synthetic hydrogels47 as well as for thermally-treated wood products.48–51
The differential scanning calorimetry (DSC) data revealed that all of the materials exhibited cold crystallization peaks but no observable Tg (full traces can be found in ESI,‡ Fig. S12–S16). The presence of cold crystallization peaks can be attributed to the confinement of S8 domains by crosslinked networks resulting in limited mobility and subsequent delayed recrystallization. Although this phenomenon has been reported for allyl lignin–sulfur composites containing 80–85 wt% sulfur,32 the confinement in cellulose–sulfur networks was not sufficient to facilitate such cold crystallization.31
Mechanical properties of biomass–sulfur composites
The influence of monomer composition on the mechanical strength of HSMs has been surveyed in some detail by Chalker and Hasell.16 It has also recently been demonstrated that introduction of post-vulcanization urethane crosslinks vastly improves the mechanical properties of HSMs.52 With respect to lignocellulosic materials, we previously demonstrated methylpropenyl cellulose–sulfur composites having flexural strengths similar to those of familiar building materials like ordinary Portland cement (OPC), which has a flexural strength of 3–5 MPa, but those materials had apparently poor compressive strengths. Allyl lignin–sulfur composites exhibited poor flexural strength and the amount of allyl lignin that could be incorporated as a crosslinker was limited to under 5 wt% due at least in part to phase separation of lignin and sulfur phases and precipitation of allyl lignin when it was added in higher quantities.32 Lignin–sulfur HSMs having improved monomer mixing at higher lignin loadings can be accomplished by RASP (radical aryl halide–sulfur polymerization) of chlorolignin and sulfur, leading to materials whose flexural strengths are also in the range of OPC.27 For the current context, each of the biomass crosslinking agents is comprised of both lignin and cellulose (Fig. 3).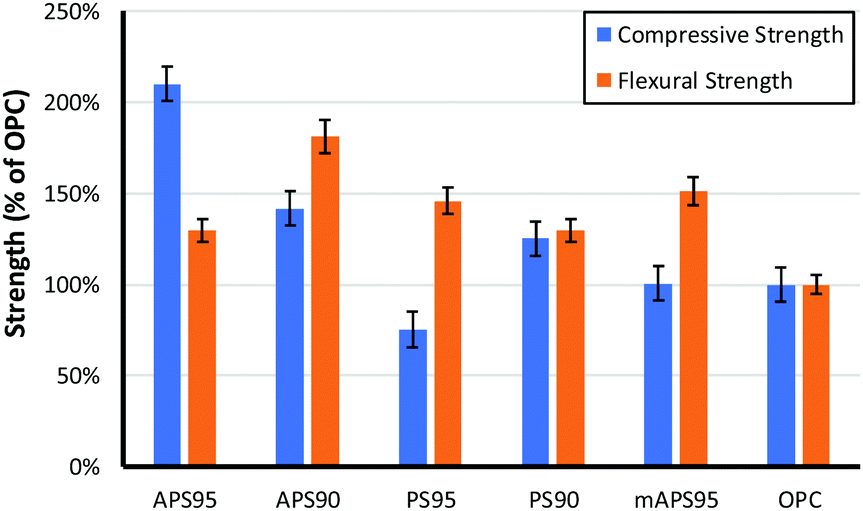 | ||
| Fig. 3 Comparison of the compressive and flexural strength of biomass–sulfur polymers to that of ordinary portland cement (OPC). | ||
The composites crosslinked by 5 wt% of biomass derivative exhibit similar flexural strengths, in the range of 130–150% that of OPC. The compressive strength, however, is significantly higher for APS95 than for the other biomass–sulfur composites and nearly twice that of OPC. Notably, the mAPS95 has only 49% the compressive strength of APS95 despite the fact that both of these composites were made using biomass crosslinkers consisting of the same cellulose–lignin ratio and olefin content in the material. While it is possible that the notably higher compressive strength of APS95 is attributable to cellulose–lignin crosslinking retained from those in the peanut shell precursor, it is also possible that the alkaline conditions to which the peanut shells were exposed for the allylation step catalysed some esterification of the carboxylate groups in lignin with cellulose-bound alcohol units. Phosphitylation of the lignin and quantification of carboxylate groups by 31P NMR spectrometry with an internal standard53,54 confirmed that the lignin contains 0.36 mmol g−1 of carboxylate units that could participate in esterification. There is also the possibility for thermal transesterification during heating, but this should also take place during formation of mAPS95, so this possibility does not adequately explain the notable strength difference between the two composites.
As may be anticipated, the compressive strength of PS95, in which crosslinking is provided only by peanut oil present in the peanut shells, had by far the lowest compressive strength, only 67% that of OPC and 36% that of APS95. If the amount of unmodified peanut shells (and thus crosslinkable olefins) is doubled, however, resulting composite PS90 had flexural and compressive strengths exceeding those of Portland cement. The striking observation that unmodified peanut shells and sulfur can simply be heated together to produce a material that is stronger than cement was initially surprising. Such strength may initially seem higher than what might be expected on the basis of crosslinking by a small percentage of peanut oil in the absence of any covalent contribution by the lignin or cellulose. In contrast, the dfPSx samples proved to be primarily physical blends. Triglycerides, however, undergo transesterification in the absence of catalyst at temperatures above 170 °C,55 below the polymerization temperature used in the current study.56–59 Increased strength is thus expected to arise from ester functionalities in the peanut shell triglycerides undergoing facile transesterification with alcohol functionalities in lignin and cellulose during heating to 180 °C. Such thermal transesterification would provide a mechanism for covalent bonding of the cellulose and lignin backbones to the network structure, accounting for the improved strength of the materials.
The compressive strengths of materials having 10 wt% biomass crosslinker are relatively similar to one another and are higher than those in OPC spanning 112–138% that of OPC. Flexural strengths differed more significantly with APS90 having a flexural strength almost twice that of OPC while PS90 exhibited a flexural strength only 30% larger. Perhaps most interestingly, increasing the ratio of organic for APSx lead to improvements in flexural strengths while decreasing compressive strength whereas PSx showed the opposite trend. Although both PS and APS can act as crosslinkers and fillers, the ratio of these influences may be the culprit for the differences in mechanical properties with increasing organic content. Increases in crosslink density which occur more readily with APS provides significant improvements to flexural properties while compressive properties can be improved by the addition of more filler (as was the case for PSx). The limits of strength of both materials may also represent an upper limit to the influence of organic additive.
Given the generally quite low flexural strength but relatively high compressive strength of the biomass–sulfur composites, potential application spaces may be limited to static installations that do not require high flexibility. A few commercial products used in such applications include Portland cement, glass fibre-polypropylene composites (Azdel SuperLite®) used in nonstructural automotive door panel components,60 and an ETFE product (Tefzel™ ETFE HT-2188, manufactured by Chemours) used commercially in electrical outlets/switches.61 The mechanical properties of these three commercial materials are provided in Table 3. The compressive strength and flexural modulus of APS95 are quite comparable to those of the ETFE product, while composites having lower compressive strengths but higher flexural moduli (APS90, PS95 and mAPS95) are more comparable to the glass fibre–polypropylene composites. The trade off of flexural and compressional metrics accessible to the biomass–sulfur composites based on the composition and biomass source thus hold potential to serve in variable application spaces.
| Material | Compressive strengtha (MPa) | Flexural strength (MPa) | Flexural modulus (MPa) | Modulus of resilience (Pa) |
|---|---|---|---|---|
| a All measured values based on the average of at least three trials. b OPC is ordinary Portland cement. c Minimum value required for residential building by building standard ACI 332.1R-06. d Azdel SuperLite® SL550600-100. e Poly(ethylene-co-tetrafluoroethylene), Tefzel™ ETFE HT-2188, manufactured by Chemours. | ||||
| APS95 | 35.7 ± 1.8 | 4.8 | 690 | 180 |
| APS90 | 24.1 ± 5.7 | 6.7 | 1490 | 1180 |
| PS95 | 12.8 ± 0.0 | 5.4 | 1360 | 860 |
| PS90 | 21.3 ± 1.2 | 4.8 | 950 | 1020 |
| mAPS95 | 17.1 ± 0.0 | 5.6 | 1410 | 1550 |
| OPC | 17c | 3.7 | 580 | 560 |
| Polypropylene–glass fibre composited | — | 3.5 | 1350 | — |
| ETFEe | 38 | — | 860 | — |
Conclusions
In conclusion we report a facile strategy for the modification of waste biomass and its subsequent reaction with sulfur by inverse vulcanization to generate materials with mechanical properties significantly exceeding those of Portland cement. A remarkably simple route to robust, remeltable composites was revealed by heating native biomass waste with sulfur. In this case, control experiments suggest that the peanut oil triglycerides in the biomass are required for producing such composites. These strategies reported herein should be readily extendable to a near-unlimited array of waste biomass, yet the influence of even minute constituents such as plant oils must be considered in all cases to explain and predict the sources of material properties. Although no change in mechanical properties after the first seven days of curing was noted, the long-term curing behaviour will need to be characterized before commercialization of biomass–sulfur construction materials. Given that heating organics and sulfur to elevated temperatures can produce toxic H2S gas, careful analysis of combustion products should also be undertaken for any organosulfur building material. In terms of developing mechanically durable materials the potential of biomass–sulfur composites as green surrogates seems to be limited predominantly by the immiscibility of biomass with sulfur, dictating further exploration of strategies to improve monomer compatibility. Increased organic incorporation, expansion of substrate scope, and increasingly green modification strategies may allow such biomass–sulfur materials to expand into commercial use.Experimental
General considerations
Fourier transform infrared spectra were obtained using an IR instrument (Shimadzu IRAffinity-1S) with an ATR attachment. Scans were collected over the range 400–4000 cm−1 at ambient temperature with a resolution of 8. TGA was recorded (Mettler Toledo TGA 2 STARe System) over the range 20–800 °C with a heating rate of 10 °C min−1 under a flow of N2 (100 mL min−1). Each measurement was acquired in duplicate and presented results represent an average value. DSC was acquired (Mettler Toledo DSC 3 STARe System) over the range −60 to 150 °C with a heating rate of 5 °C min−1 under a flow of N2 (200 mL−1 min−1). Each DSC measurement was carried out over five heat–cool cycles. Each measurement was acquired in duplicate to ensure consistent results were obtained.DMA was performed (Mettler Toledo DMA 1 STARe System) in single cantilever mode. DMA samples were cast from silicone resin moulds (Smooth-On Oomoo® 30 tin-cure). Samples were manually sanded to ensure uniform dimensions. The sample dimensions were approximately 15 × 8 × 1.5 mm but due to instrumental limitations (maximum force of 10 N), each sample differed slightly in thickness in order to obtain a stress at break. Sample dimensions were measured with a digital calliper with ±0.01 mm resolution. The clamping force was 5 cN m for all materials. The force was varied from 0 to 10 N with a ramp rate of 0.2 N min−1 measured isothermally at 25 °C.
Carbon disulfide extractions were performed by suspending 0.3 g of finely ground material (measured to 0.0001 g) in 20 mL of CS2, allowing the solid to settle for 30 minutes, pipetting off the supernatant into a separate vial, and adding another 20 mL of CS2. This process was repeated an additional 3 times so that a total of 5 washes was performed. The residual CS2 was evaporated under a flow of N2 and each vial was weighed to determine the fraction that was soluble (collected as supernatant) or insoluble (remained in the initial vial).
SEM was acquired on a Schottky Field Emission Scanning Electron Microscope SU5000 operating in variable pressure mode with an accelerating voltage of 15 keV. Compressional analysis was performed on a Mark-10 ES30 test stand equipped with a M3-200 force gauge (1 kN maximum force with ±1 N resolution). Compression cylinders were cast from silicone resin moulds (Smooth-On Oomoo® 30 tin-cure) with diameters of approximately 6 mm and heights of approximately 10 mm. Samples were manually sanded to ensure uniform dimensions and measured with a digital calliper with ±0.01 mm resolution. Compressional analysis was performed in triplicate and results were averaged.
Materials and methods
Peanut shell powder (Golden Peanut and Tree Nuts, Product ES) elemental sulfur (99.5%, Alfa Aesar), allyl bromide (99% Alfa Aesar), sodium hydroxide (≥97% VWR Chemicals), acetic acid (glacial, XX) hexanes (certified ACS, Fisher), cellulose (Alfa Aesar), alkali lignin (Sigma Aldrich), allyl lignin (prepared and characterized in Karunarathna et al.32) were all used as received unless otherwise specified. Details on the synthesis and characterization of allyl cellulose and APS as well as the preparation of mAPS and dfPS can be found in the ESI‡ (pages 2–7, Fig. S2–S7, Table S1, ESI‡).General composite synthesis
A quantity of organic material (compensating for moisture as determined by duplicate TGA experiments) and sulfur totalling 10 g were weighed out and thoroughly mixed into a 20 mL scintillation vial equipped with a Teflon coated stir bar. The vials were sealed with polypropylene lids pierced with a needle to allow moisture to escape during reaction. The reactions were allowed to continue until visibly homogeneous (variable depending on the wt% and identity of the organic species). Samples were allowed to cure for at least 14 days before any further analysis. Specific material synthesis can be found in the ESI‡ (pages 8–9).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Jake Harrell of Golden Peanut and Tree Nuts (Alpharetta, Georgia, USA) for supplying peanut shell product ES used in this study. Funding for this project from the National Science Foundation (CHE-1708844) is gratefully acknowledged. R. C. S. thanks John D. Protasiewicz of Case Western Reserve University for inspiring this work.Notes and references
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 Search PubMed.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 Search PubMed.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 Search PubMed.
- A. Gupta and J. P. Verma, Renewable Sustainable Energy Rev., 2015, 41, 550–567 Search PubMed.
- M. K. Lauer and R. C. Smith, Compr. Rev. Food Sci. Food Saf., 2020, 1–53, DOI:10.1111/1541-4337.12627.
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734–748 Search PubMed.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 590–594 Search PubMed.
- T. Thiounn, M. S. Karunarathna, L. M. Slann, M. K. Lauer and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 Search PubMed.
- C. P. Maladeniya, M. S. Karunarathna, M. K. Lauer, C. V. Lopez, T. Thiounn and R. C. Smith, Mater. Adv., 2020, 1, 1665–1674 Search PubMed.
- C. V. Lopez, C. P. Maladeniya and R. C. Smith, Electrochem, 2020, 1, 226–259 Search PubMed.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 Search PubMed.
- S. Park, D. Lee, H. Cho, J. Lim and K. Char, ACS Macro Lett., 2019, 8, 1670–1675 Search PubMed.
- T. Hasell, P. Yan, W. Zhao, B. Zhang, S. Petcher, A. Smith Jessica, J. Parker Douglas, I. Cooper Andrew, L. Jiang and J. Lei, Angew. Chem., Int. Ed., 2020, 59, 2–10 Search PubMed.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675 Search PubMed.
- B. Zhang, S. Petcher and T. Hasell, Chem. Commun., 2019, 55, 10681–10684 Search PubMed.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440 Search PubMed.
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Adv. Sustainable Syst., 2020, 4, 1900111 Search PubMed.
- N. A. Lundquist, M. J. Sweetman, K. R. Scroggie, M. J. H. Worthington, L. J. Esdaile, S. F. K. Alboaiji, S. E. Plush, J. D. Hayball and J. M. Chalker, ACS Sustainable Chem. Eng., 2019, 7, 11044–11049 Search PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 Search PubMed.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhargava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 23, 16106 Search PubMed.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 Search PubMed.
- C. Herrera, K. J. Ysinga and C. L. Jenkins, ACS Appl. Mater. Interfaces, 2019, 11, 35312–35318 Search PubMed.
- C. R. Westerman and C. L. Jenkins, Macromolecules, 2018, 51, 7233–7238 Search PubMed.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 Search PubMed.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustainable Chem. Pharm., 2020, 16, 100249 Search PubMed.
- A. D. Smith, C. D. McMillin, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 Search PubMed.
- M. S. Karunarathna, A. G. Tennyson and R. C. Smith, J. Mater. Chem. A, 2020, 8, 548–553 Search PubMed.
- M. S. Karunarathna, M. K. Lauer, A. G. Tennyson and R. C. Smith, Polym. Chem., 2020, 11, 1621–1628 Search PubMed.
- T. Thiounn, A. G. Tennyson and R. C. Smith, RSC Adv., 2019, 9, 31460–31465 Search PubMed.
- A. D. Smith, T. Thiounn, E. W. Lyles, E. K. Kibler, R. C. Smith and A. G. Tennyson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1704–1710 Search PubMed.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 Search PubMed.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 Search PubMed.
- T. Thiounn, M. K. Lauer, M. S. Bedford, R. C. Smith and A. G. Tennyson, RSC Adv, 2018, 8, 39074–39082 Search PubMed.
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303 Search PubMed.
- V. S. Wadi, K. K. Jena, K. Halique and S. M. Alhassan, ACS Appl. Polym. Mater., 2020, 2, 198–208 Search PubMed.
- V. S. Wadi, K. Halique and S. M. Alhassan, Ind. Eng. Chem. Res., 2020, 59, 13079–13087 Search PubMed.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 10035–10044 Search PubMed.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 Search PubMed.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044 Search PubMed.
- M. Mann, J. E. Kruger, F. Andari, J. McErlean, J. R. Gascooke, J. A. Smith, M. J. H. Worthington, C. C. C. McKinley, J. A. Campbell, D. A. Lewis, T. Hasell, M. V. Perkins and J. M. Chalker, Org. Biomol. Chem., 2019, 17, 1929–1936 Search PubMed.
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 Search PubMed.
- N. A. Lundquist, M. J. H. Worthington, N. Adamson, C. T. Gibson, M. R. Johnston, A. V. Ellis and J. M. Chalker, RSC Adv., 2018, 8, 1232–1236 Search PubMed.
- L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2018, 24, 6905–6916 Search PubMed.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 Search PubMed.
- P. Brown, I. Atly Jefcoat, D. Parrish, S. Gill and E. Graham, Adv. Environ. Res., 2000, 4, 19–29 Search PubMed.
- V. Dubois, S. Breton, M. Linder, J. Fanni and M. Parmentier, Eur. J. Lipid Sci. Technol., 2007, 109, 710–732 Search PubMed.
- B. Al-Rudainy, M. Galbe, O. Wallberg, M. Arcos Hernandez and P. Jannasch, Polymers, 2018, 11, 35 Search PubMed.
- R. Rowell, Cellul. Chem. Technol., 2012, 46, 443–448 Search PubMed.
- A. Ishikawa and T. Ohira, Mokuzai Kogyo, 2009, 64, 258–262 Search PubMed.
- J.-P. Haluk and M. Irmouli, J. Sci. Tech. Tonnellerie, 1998, 4, 1–82 Search PubMed.
- P. Topf, Holz Roh- Werkst., 1971, 29, 269–275 Search PubMed.
- T. Hasell, P. Yan, W. Zhao, B. Zhang, S. Petcher, A. Smith Jessica, J. Parker Douglas, I. Cooper Andrew, L. Jiang and J. Lei, Angew. Chem., Int. Ed., 2020, 59, 2–10 Search PubMed.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 Search PubMed.
- D. S. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 45–63 Search PubMed.
- M. Diasakou, A. Louloudi and N. Papayannakos, Fuel, 1998, 77, 1297–1302 Search PubMed.
- A. S. Reshad, P. Tiwari and V. V. Goud, Energy Fuels, 2017, 31, 9642–9651 Search PubMed.
- J.-J. Lin and Y.-W. Chen, J. Taiwan Inst. Chem. Eng., 2017, 75, 43–50 Search PubMed.
- J. Geuens, S. Sergeyev, B. U. W. Maes and S. M. F. Tavernier, Energy Fuels, 2013, 27, 2637–2642 Search PubMed.
- S. A. Pasias, N. K. Barakos and N. G. Papayannakos, Ind. Eng. Chem. Res., 2009, 48, 4266–4273 Search PubMed.
- Technical data sheet “Azdel SuperLite® SL550600-100” Supplied by Azdel April 30, 2020.
- Technical data sheet “Tefzel™ ETFE HT-2188” Supplied by Chemours May 27, 2020.
Footnotes |
| † Dedicated to John D. Protasiewicz for his contributions to using main group elements in polymer science. |
| ‡ Electronic supplementary information (ESI) available: Proton NMR spectral data, FTIR spectra, TGA curves, analysis of char yield versus composition; DSC curves. See DOI: 10.1039/d0ma00538j |
| This journal is © The Royal Society of Chemistry 2020 |