
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis of hierarchical carbon coupled with Fe3O4/Fe2C as an efficient catalyst for the oxygen reduction reaction†
Ling
Zhang‡
a,
Lili
Fan‡
 *a,
Pu
Yang
a,
Mengfei
Li
a,
Haobing
Zhang
a,
Yucheng
Tang
a,
Zixi
Kang
a,
Hailing
Guo
b,
Rongming
Wang
*a and
Daofeng
Sun
a
*a,
Pu
Yang
a,
Mengfei
Li
a,
Haobing
Zhang
a,
Yucheng
Tang
a,
Zixi
Kang
a,
Hailing
Guo
b,
Rongming
Wang
*a and
Daofeng
Sun
a
aSchool of Materials Science and Engineering, College of Science, China University of Petroleum (East China), Qingdao 266580, China. E-mail: lilifan@upc.edu.cn; rmwang@upc.edu.cn
bState Key Laboratory of Heavy Oil Processing, Key Laboratory of Catalysis, China National Petroleum Corp. (CNPC), China University of Petroleum (East China), Qingdao 266580, China
First published on 11th August 2020
Abstract
A green synthesis strategy is reported here for the fabrication of a hierarchical Fe-based carbon electrocatalyst for catalyzing the oxygen reduction reaction. This strategy involves the grinding of the organic ligand (1,4-benzenedicarboxylic acid) and metal salt (iron chloride hexahydrate) with a minute quantity of solvent at room temperature and thermal annealing under a N2 atmosphere. The metal salt introduced plays dual roles. One is as a hard template for creating a hierarchical porous structure, which is important for controlling the overall mechanism and kinetics of the catalysis process by affecting the mass transport and exposure of active sites. The other one is as a metal source for building the MOF structure that can drive Fe3O4 and Fe2C active sites for catalyzing the oxygen reduction reaction. Combining the merits of porosity and MOFs, the catalyst synthesized by 120 min grinding (BM-Fe@NC-120) possesses good ORR performance in 0.1 M KOH with a half-wave potential of 0.80 V, a limiting current density of −5.08 mA cm−2, excellent long-term durability and resistance to methanol, indicating the validity of this eco-friendly route for scalable synthesis of catalysts for future practical fuel-cell applications.
Introduction
Increasing demand for solving the worldwide energy crisis and environmental issues has triggered extensive research interests on sustainable energy conversion and storage systems, such as fuel cells and metal–air batteries, the commercialization of which, nevertheless, depends significantly on the development of electrocatalysts for the oxygen reduction reaction (ORR).1–4 Platinum (Pt) and Pt-based materials exhibit superior ORR activity and have been recognized as the bench mark for ORR catalysts so far. However, the prohibitive cost, low reserve, and limited durability of these materials hinder their large scale applications.5,6 Thus, it is of vital importance to explore alternative low-cost electrocatalysts with competitive performance and improved durability.To date, great efforts have been devoted and a wide range of catalysts have been developed, including the metal-free heteroatom (nitrogen (N), boron (B), sulfur (S), phosphorus (P), etc.)-doped carbon materials.7–10 and their composites with transition-metals (iron (Fe), cobalt (Co), nickel (Ni), copper (Cu)) or transition-metal oxides, carbides, chalcogenides and phosphides.11–20 Among these materials, Fe-based carbon composites are expected to exhibit good activity towards the ORR due to the synergy between the ORR-effective Fe (Fe3O4, Fe2C, etc.) and N (pyridinic N, graphitic N) species, as well as the carbon substrates with good conductivity and moderate defects, which has been confirmed by recently reported research studies.21–25
To fabricate such catalysts, the precursor-carbonization method has been considered as one of the best methods.26–30 Although several methods have been utilized to prepare precursors, the physical blending of the metal, carbon and nitrogen sources and using metal–organic frameworks (MOFs) as the self-sacrificing template are the most favorable and effective methods.31–35 The physical blending is simple and efficient for acquiring a precursor, but there is no effective way to control the final distribution of active sites and the structure. In respect of inheriting structural features of the precursor, MOFs that are built from periodically linked organic ligands and metal centers are attractive candidates owing to their structural diversity and metal centers uniformity.36–43 Although many MOFs have been explored as precursors for deriving non-precious metal/carbon-based materials,44–49 it is still challenging to introduce meso- and/or macro-porosity into the final carbon structure since the architecture plays an important role in controlling the overall mechanism and kinetics of the catalysis process by affecting the reactive site exposure, reactant accessibility and conductivity of the materials. Furthermore, conventional synthesis of MOFs is heat and organic-solvent consuming, which is not beneficial for large scale synthesis. Therefore, it would be favorable and challenging to combine the advantages of physical blending to develop more environmental-friendly synthetic methods for MOFs and introduce porosity into the final carbon structure as well.
Herein, a green synthesis strategy is reported for the fabrication of hierarchical Fe-based carbon electrocatalysts for catalysing the ORR. This strategy involves simple grinding of the organic ligand (1,4-benzenedicarboxylic acid) and metal salt (iron chloride hexahydrate) with a minute quantity of solvent at room temperature and thermal annealing under a N2 atmosphere. The metal salt used in this strategy performs two major roles: first, excess metal salt was added as a hard template to introduce meso- and/or macro-porosity into the final carbon structure, which is favorable for improving the mass transport and providing sufficient exposure of active sites; second, it provides the metal source needed for the construction of the MOF structure, which serves as a self-sacrificing template for deriving Fe3O4 and Fe2C active sites to catalyze the ORR process. By optimizing the grinding time to 120 min, the best catalytic performance was obtained in 0.1 M KOH with a half-wave potential of 0.80 V, a limiting current density of −5.08 mA cm−2, excellent long-term durability and resistance to methanol. This indicates the effectiveness of this eco-friendly route, which will provide new insight into the synthesis of hierarchical carbon catalysts for future practical fuel-cell applications.
Experimental
Materials
All chemicals were used directly without further refinement. Iron chloride hexahydrate (FeCl3·6H2O), hydrochloric acid (HCl, 12 mol L−1) and potassium hydroxide (KOH) were supplied by Sinopharm Chemical Reagent Co., Ltd. 1,4-Benzenedicarboxylic acid (H2BDC) was purchased from Energy Chemical Co., Ltd. N,N-Dimethylformamide (DMF), methanol, ethanol and isopropanol were received from Tianjin Fuyu Fine Chemical Co. Ltd. The commercial 20 wt% Pt/C and 5 wt% Nafion solution were obtained from Sigma-Aldrich.Preparation of the MOF precursor
Typically, a certain amount of FeCl3·6H2O (1.1474 g, 4.2 mmol) and H2BDC (0.3526 g, 2.1 mmol) were added into an agate grinding jar. Then, 1 mL of DMF were injected into the jar, and the mixture was ground at a rotating speed of 700 rpm for different times (30, 60, 90, 120 and 150 min). Finally, the products were preserved at room temperature for 5 h.Preparation of BM-IM-T
The as-prepared precursor was washed with ethanol to remove the unreacted metal salt and the collected samples were denoted as BM-IM-T (T represents the grinding time from 30–150 min).Preparation of BM-Fe@NC-T catalyst
The as-prepared precursor was transferred to a crucible, which was placed in a tubular furnace and heated at 800 °C under a N2 atmosphere for 3 h with a heating rate of 5 °C min−1. After cooling to room temperature, the obtained sample was immersed in HCl (12 mol L−1) for 30 min, collected by centrifugation, washed with deionized water to neutral and dried at 80 °C overnight, respectively. The samples were denoted as BM-Fe@NC-T (T represents the grinding time from 30–150 min).Characterization
The phase purity and crystallinity were tested on a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 0.15418 nm). A scanning electron microscope (SEM, JEOL-7900) and a transmission electron microscope (TEM, JEM-2100F) were employed to investigate the detailed structure and morphology of the synthesized samples. The structure information of the carbon matrix was acquired from a LABRAM HR EVO Raman microscope spectrometer with a 532 nm excitation wavelength. X-ray photoelectron spectra were recorded on an ESCA250Xi spectrometer and the binding energies were calibrated based on the graphite C 1s peak. The content of Fe was determined by using an Agilent 710 inductively coupled plasma optical emission spectrometer (ICP-OES). The BET surface areas and pore size distributions were characterized on a JW-BK200 surface area analyzer.Electrochemical measurements
All electrochemical measurements were obtained using a Gamry electrochemical work station (INTERFACE 1000 T) under a three-electrode cell configuration, using a glassy carbon rotating disk electrode (RDE, 5 mm in diameter) as the working electrode, a platinum plate as the counter electrode and an Ag/AgCl (KCl saturated solution) electrode as the reference electrode. The measured potentials were referenced to reversible hydrogen electrode (RHE) on the basis of the formula ERHE = EAg/AgCl + 0.059pH + EθAg/AgCl. For preparation of the catalyst ink, 4 mg of samples was added to a solution of 970 μL isopropanol and 30 μL Nafion (5 wt%). After ultrasonic dispersion for 1 h, the homogeneous catalyst ink was obtained. The working electrode was prepared by pipetting 15 μL of catalyst ink onto the GC electrode with a loading equivalent to 0.300 mg cm−2. Linear sweep voltammetry (LSVs) and cyclic voltammetry (CV) were performed in a N2/O2 saturated 0.1 M KOH solution in the potential range from 0.2 to 1.2 V with a scan rate of 10 mV cm−1. The long time stability experimented during CV cycling and current vs. time (i–t) chronoamperometry responses were compared with those of 20% Pt/C.The electron transfer number was evaluated by two methods. One is by using the Koutecky–Levich (K–L) equation:
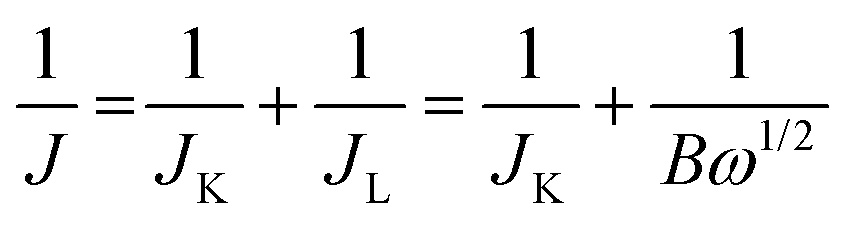
| B = 0.2nFC0D02/3V−1/6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (C0 = 1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm2 s−1 in 0.1 M KOH), and V is the kinematic viscosity of the electrolyte (V = 0.01 cm2 s−1 in 0.1 M KOH).50
485 C mol−1), C0 is the bulk concentration of O2 (C0 = 1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm2 s−1 in 0.1 M KOH), and V is the kinematic viscosity of the electrolyte (V = 0.01 cm2 s−1 in 0.1 M KOH).50
According to the rotating ring disk electrode (RRDE) test, the yield of hydrogen peroxide (H2O2%) and electron transfer number (n) in the ORR process can be calculated using the following equations:
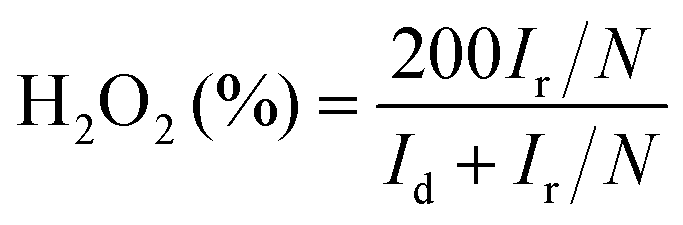
where Id and Ir are the disk and ring current densities, respectively, and N is the ring collection efficiency (0.37).

Results and discussion
A typical synthesis route of the BM-Fe@NC-T composites is illustrated in Fig. 1. Metal salts and organic ligands were added into the grinding jar with 1 mL of DMF as the solvent, the mixture of which was ground for a certain time and further heated at 800 °C under an inert atmosphere to construct hierarchical porous N doped carbon flakes with enchased Fe3O4/Fe2C active sites (see the Experimental section for more details) for catalyzing the ORR.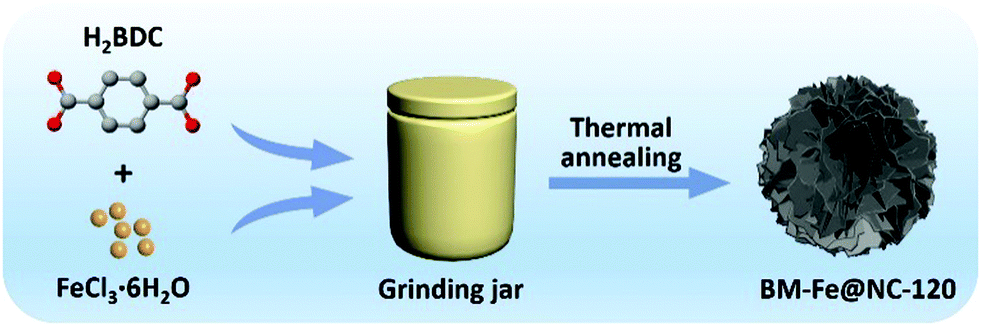 | ||
| Fig. 1 Schematic illustration of the synthesis procedures for BM-Fe@NC-120. | ||
Before the thermal annealing treatment, the mixtures after different grinding times were first washed with ethanol to remove the unreacted metal salt, and then, characterized by powder X-ray diffraction (PXRD) measurements. As shown in Fig. 2a, the peaks in the 2θ range of 8–10° gradually appear with the increase of grinding time. The highest peak intensity can be acquired when the grinding time reaches 120 min, which is well consistent with the simulated pattern of MOF MIL-101, proving the successful synthesis of the MIL-101 structure in BM-IM-120. As illustrated in digital photographs (Fig. S1, ESI†), the BM-IM-120 sample shows a light brown color instead of the white color of the organic ligand, which further proves the formation of the MOF stucture. However, a longer grinding time of 150 min would result in the destruction of the formed MOF structure, which can be attributed to the breakage of the coordination bond under higher energy delivered from the mechanical force. All the other diffraction peaks of BM-IM-T observed in the range of 17–70° are in accordance with the characteristic peaks of H2BDC, indicating that a large part of the ligand was not involved in the construction of the MOF structure. Detailed morphology of BM-IM-T was analyzed by scanning electron microscopy (SEM). As shown in Fig. 2b–d and Fig. S2 (ESI†), the unreacted ligands present a similar stacking structure under the mechanical force, and spaces are observed between the bricks. These spaces were filled with metal salt and appear when the metal salt was removed, which would be preserved after thermal annealing, facilitating the fabrication of hierarchical porosity. That is where the metal salt plays the template role. Furthermore, a growing number of a new honeycomb-shaped phase that is associated with the MOF structure can also be recognized with the increase of grinding time from 30 to 120 min. Unlike its counterparts, this honeycomb structure no longer exists in the sample of BM-IM-150, and only an amorphous phase can be found on the surface of ligand blocks, which is consistent with the PXRD results. A control experiment was carried out by using BM-IM-120 as the precursor. After the same heating procedure, there was little carbon left due to the decomposition of H2BDC. This suggests the protective role of metal salt on the carbon source by reacting with H2BDC to form metal terephthalate, which possesses much higher decomposition temperature and can be retained after the annealing process.51
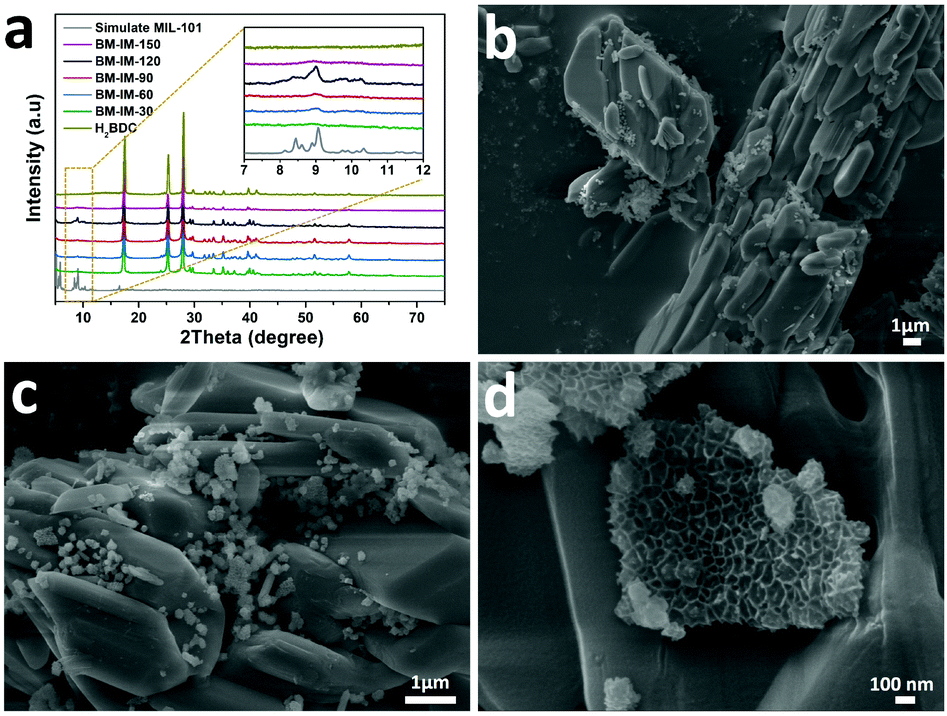 | ||
| Fig. 2 (a) PXRD patterns of BM-IM-30, 60, 90, 120 and 150; (b–d) SEM pictures of BM-IM-120 with different magnifications. | ||
After thermal annealing under an inert atmosphere, the ground mixtures were successfully transformed into BM-Fe@NC-T composites, the detailed morphology of which was characterized by SEM and is shown in Fig. 3a, b and Fig. S3 (ESI†). As revealed by the SEM images (Fig. 3a and b), the built MOF structure and the unreacted organic ligand in BM-Fe@NC-120 had been converted to the carbon matrix in the form of carbon flakes with hierarchical porosity. Such hierarchical porosity is desirable for the ORR since it is beneficial for mass transport during the catalytic process. The formation of this hierarchical structure can be attributed to the following three reasons: (1) the oxygen atoms in the organic ligand carried away a part of the carbon element during the thermal annealing process, turning the stacking structure of the unreacted organic ligand into thinner carbon flakes; (2) the unreacted metal salt embedded in the stacking ligand by the grinding force had acted as a template for generating pores; (3) part of the solvent DMF preserved in the precursor escaped at high temperatures, leaving pores in the final structure. Similar carbon flakes were also synthesized for BM-Fe@NC-30, 60 and 90 (Fig. S3a–f, ESI†). For the sample of BM-Fe@NC-150, the carbon flakes became much thicker due to the longer grinding time (Fig. S3g and h, ESI†). Additionally, it can be noted that the BM-Fe@NC-120 shows more compact hierarchical porosity than its counterparts. The distance between carbon flakes is narrowed because the honeycomb-shaped MOF fabricated by grinding has played the “binder” role in the thermal annealing process since the MOF itself can also derive carbon species at high temperatures and under an inert atmosphere. To reveal the detailed structure of BM-Fe@NC-120, transmission electron microscopy (TEM) was used and the results are presented in Fig. 3c–e. This indicates that the BM-Fe@NC-120 is composed of Fe3O4, Fe2C and the carbon matrix. The high-resolution TEM (HRTEM) images (Fig. 3d and e) present the lattice fringes with interplanar distances of 0.252 and 0.297 nm, which are in accordance with the (311) and (220) crystal planes of Fe3O4, respectively. A lattice spacing of 0.201 nm can be ascribed to the lattice distance of the (111) plane of Fe2C. As shown in Fig. 3e, the derived Fe2C phase is covered by the Fe3O4 phase, which is probably due to the contact with the ligand–O of its outer surface. This might protect the Fe2C from the chemical etching in the ORR environment and extending its service life in the BM-Fe@NC-120. EDS elemental mapping was further performed to confirm the distribution of different species in BM-Fe@NC-120. As revealed in Fig. 3f–j, the participating elements (C, O, N, and Fe) are dispersed homogeneously throughout the carbon matrix. As no other N source was added to the synthesis system, it can be concluded that the small amount of doped N comes from the solvent DMF wrapped in the precursor. That is one of the three reasons that the as-prepared precursor was not dried thoroughly. The other two reasons to retain the solvent DMF in the precursor are to provide a better growth environment for the MOF and becuase it probably generates pores when removed from the solvent during thermal annealing as mentioned above.
 | ||
| Fig. 3 (a and b) SEM images, (c) TEM image, (d and e) high-resolution TEM images, (f) HAADF-STEM image and the corresponding EDS elemental mappings of (g) C, (h) O, (i) N and (j) Fe of BM-Fe@NC-120, respectively. | ||
The structure and composition of the as-synthesized BM-Fe@NC-T samples were further confirmed by PXRD characterization. As shown in Fig. 4a, the formation of the carbon matrix can be illustrated by the broad diffraction peak centered around 26°, which is assigned to the (002) plane of the hexagonal graphite structure. Other diffraction peaks observed at 35.4°, 43.2°, 57.3° and 68.1° correspond to the (311) plane of Fe3O4 (JCPDS-19-0629), and the (101), (102) and (110) planes of Fe2C (JCPDS-36-1249), respectively. These Fe-based diffraction peaks gradually show up with grinding time and the intensities reach the maximum in BM-Fe@NC-120, indicating that they are related to the building of the MOF structure and not involved with the unreacted metal salt. Thus, it is not surprising to find attenuation of these peaks for BM-Fe@NC-150. The structural characteristics of the carbon in BM-Fe@NC-T samples were further investigated using Raman spectra (Fig. 4b). In all the examined samples, two sharp peaks centered at 1343 and 1584 cm−1 are observed, which correspond to the featured D-band (disordered, sp3 hybridized carbon) and G-band (ordered, sp2 hybridized carbon) of carbon-based nanostructures. Also, the relative intensity ratio of D-band to G-band (ID/IG) was calculated to reflect the defects or graphitization degree of carbon for each sample. The ID/IG value for BM-Fe@NC-120 (0.95) is slightly lower than BM-Fe@NC-30 (1.02), BM-Fe@NC-60 (0.99), BM-Fe@NC-90 (0.97) and BM-Fe@NC-150 (0.99). This lower ID/IG value indicates that the graphitization degree of BM-Fe@NC-120 is slightly higher than its counterparts, which is favorable for improving the electrical conductivity and ORR catalytic performance. The N2 adsorption–desorption isotherms were tested to characterize the hierarchical structure of BM-Fe@NC-T. As observed in Fig. 4c, except for BM-Fe@NC-30, all the other samples exhibit type-IV isotherms and H3-type hysteresis loops, revealing their mesoporous structure characteristics. From the inset pore size distribution plot, two main pore systems are identified to be centered around 2.5–3.5 nm and 10–11 nm, confirming the formed hierarchical structure of these samples. The total BET surface areas of BM-Fe@NC-30, 60, 90, 120 and 150 were calculated to be 112.1, 365.9, 502.4, 964.8 and 646.9 m2 g−1. The built hierarchical structure and higher BET surface area of BM-Fe@NC-120 are beneficial for producing more active sites and rapid mass transfer during the ORR process.
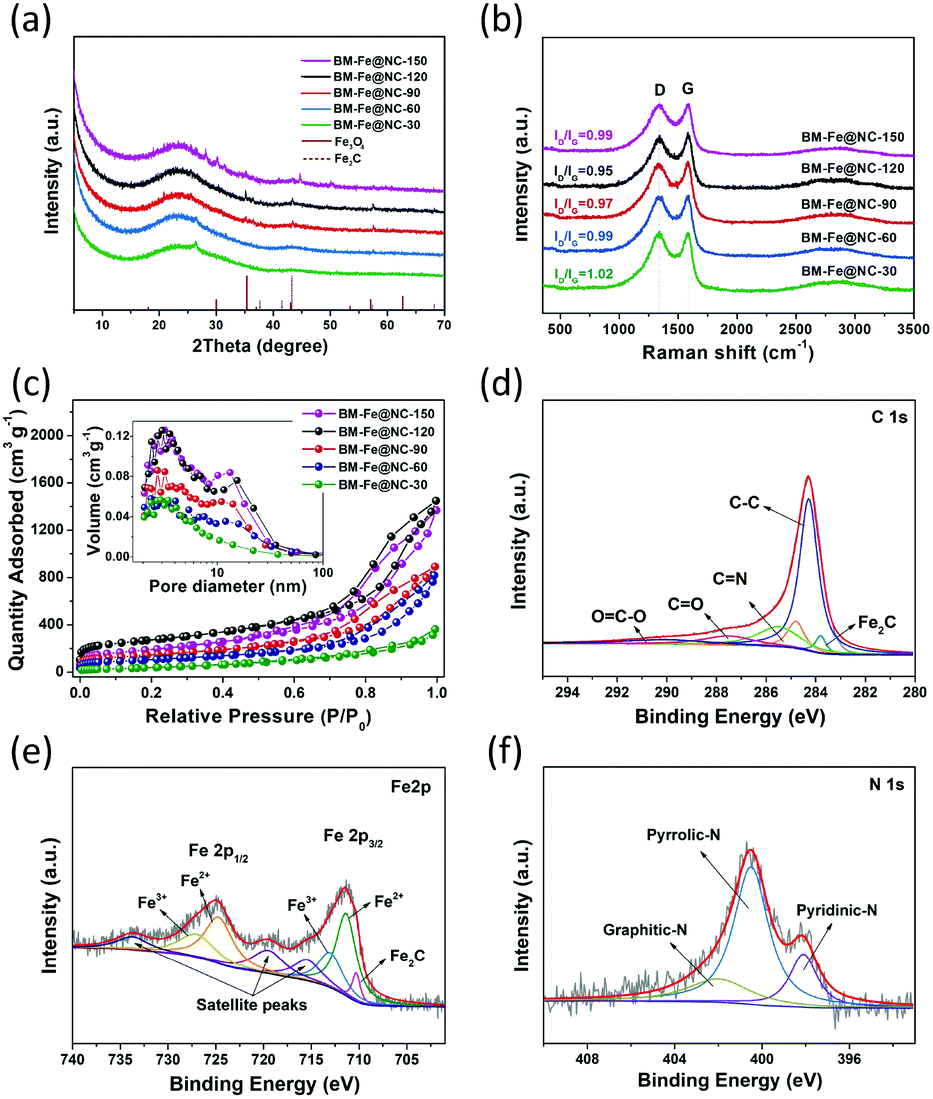 | ||
| Fig. 4 (a) PXRD patterns, (b) Raman spectra and (c) N2 adsorption–desorption isotherm of the BM-Fe@NC-T samples (inset: plots of pore-size distribution); (d) C 1s, (e) Fe 2p and (f) N 1s XPS spectrum of BM-Fe@NC-120. | ||
X-ray photoelectron spectroscopy (XPS) was further used to provide insights into the surface elemental chemical states and electronic structures of the BM-Fe@NC-120 sample. Elemental C, Fe, N and O were detected in the survey spectrum (Fig. S4, ESI†) and the XPS results of surface elemental contents are summarized in Table S1 (ESI†). In the high-resolution C 1s spectrum (Fig. 4d), five peaks at 283.8, 284.3, 285.5, 287.6 and 290.1 eV can be deconvoluted that correspond to the formation of C–Fe,52 C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CO and O–CO bonding respectively and the presence of the CN bond also confirms the doping of N atoms into the carbon matrix. Fittings of the Fe 2p spectrum show eight peaks at 710.3, 711.4, 713.2, 715.5, 719.6, 724.8, 727.2 and 733.8 eV (Fig. 4e). The peak appearing at 710.3 eV can be ascribed to Fe2C species, further confirming the existence of Fe–C bonding in BM-Fe@NC-120.53 Two pairs of 2p3/2/2p1/2 doublets are also observed in the Fe 2p spectrum. The peaks at 711.4 and 713.2 eV can be respectively assigned to the 2p3/2 binding energy of Fe2+ and Fe3+, while the peaks at 724.8 and 727.2 eV are respectively related to the 2p1/2 binding energy of Fe2+ and Fe3+. Three fitted peaks at 715.5, 719.6 and 733.8 eV are satellite peaks.54 This coexistence of Fe2+ and Fe3+ matches well with the above discussed TEM and XRD results. As for the N 1s spectrum illustrated in Fig. 4f, the deconvoluted three peaks at 398.1, 400.5 and 402.1 eV are assigned to pyridinic N, pyrrolic N and graphitic N, respectively. Among these doped N species, the pyridinic N and graphitic N are considered as contributors for boosting the ORR catalytic activity. Since these two kinds of N species account for the minority, the catalytic ability of BM-Fe@NC-120 for the ORR can be affirmed to mainly come from the formed Fe3O4 and Fe2C sites. The accurate content of Fe in the BM-Fe@NC-120 sample was determined using an inductively coupled plasma optical emission spectrometer (ICP) to be 0.96 wt%. By dissolving the ligand in BM-IM-120 with DMF, the amount of the formed MOF-related structure was calculated around 4.64 wt%, based on which it can be estimated that the Fe content in BM-Fe@NC-120 would be in the range of 1.17–1.85 wt% with or without the consideration of the O atoms of the unreacted ligand in BM-IM-120. Since the HCl treatment after thermal annealing may also take away a small amount of Fe metal, it is reasonable to have a low Fe content of 0.96 wt% in BM-Fe@NC-120. These results confirm an important point that the Fe3O4 and Fe2C presented in BM-Fe@NC-120 comes totally from the synthesized MOF structure, and not from the unreacted metal salt. Thus, the three roles that are played by the metal salts can be concluded as follows: (1) as a metal source for growth of the MOF; (2) as a template for producing hierarchical porosity; (3) as a protector of carbon against carbon loss during thermal annealing.
N, CO and O–CO bonding respectively and the presence of the CN bond also confirms the doping of N atoms into the carbon matrix. Fittings of the Fe 2p spectrum show eight peaks at 710.3, 711.4, 713.2, 715.5, 719.6, 724.8, 727.2 and 733.8 eV (Fig. 4e). The peak appearing at 710.3 eV can be ascribed to Fe2C species, further confirming the existence of Fe–C bonding in BM-Fe@NC-120.53 Two pairs of 2p3/2/2p1/2 doublets are also observed in the Fe 2p spectrum. The peaks at 711.4 and 713.2 eV can be respectively assigned to the 2p3/2 binding energy of Fe2+ and Fe3+, while the peaks at 724.8 and 727.2 eV are respectively related to the 2p1/2 binding energy of Fe2+ and Fe3+. Three fitted peaks at 715.5, 719.6 and 733.8 eV are satellite peaks.54 This coexistence of Fe2+ and Fe3+ matches well with the above discussed TEM and XRD results. As for the N 1s spectrum illustrated in Fig. 4f, the deconvoluted three peaks at 398.1, 400.5 and 402.1 eV are assigned to pyridinic N, pyrrolic N and graphitic N, respectively. Among these doped N species, the pyridinic N and graphitic N are considered as contributors for boosting the ORR catalytic activity. Since these two kinds of N species account for the minority, the catalytic ability of BM-Fe@NC-120 for the ORR can be affirmed to mainly come from the formed Fe3O4 and Fe2C sites. The accurate content of Fe in the BM-Fe@NC-120 sample was determined using an inductively coupled plasma optical emission spectrometer (ICP) to be 0.96 wt%. By dissolving the ligand in BM-IM-120 with DMF, the amount of the formed MOF-related structure was calculated around 4.64 wt%, based on which it can be estimated that the Fe content in BM-Fe@NC-120 would be in the range of 1.17–1.85 wt% with or without the consideration of the O atoms of the unreacted ligand in BM-IM-120. Since the HCl treatment after thermal annealing may also take away a small amount of Fe metal, it is reasonable to have a low Fe content of 0.96 wt% in BM-Fe@NC-120. These results confirm an important point that the Fe3O4 and Fe2C presented in BM-Fe@NC-120 comes totally from the synthesized MOF structure, and not from the unreacted metal salt. Thus, the three roles that are played by the metal salts can be concluded as follows: (1) as a metal source for growth of the MOF; (2) as a template for producing hierarchical porosity; (3) as a protector of carbon against carbon loss during thermal annealing.
The electrocatalytic properties of the BM-Fe@NC-T samples toward the ORR were first evaluated by cyclic voltammetry (CV) in N2- or O2-saturated 0.1 M KOH aqueous solution. As presented in Fig. 5a, well-defined cathodic peaks appear for all the samples in O2-saturated 0.1 M KOH aqueous solution, while no obvious peaks can be observed when the electrolyte is saturated with N2. The most positive reduction current peak occurred at 0.830 V vs. the reversible hydrogen electrode (RHE) for BM-Fe@NC-120, indicating its good catalytic activity toward the ORR. Further evidence was provided by the linear sweep voltammetry (LSV) curves tested at a rotating rate of 1600 rpm (Fig. 5b). For comparison, the commercial 20% Pt/C was also examined under the same conditions. Similar to the CV results, the BM-Fe@NC-120 exhibits the best ORR performance among its counterparts with the onset reduction potential, half-wave potential and diffusion-limited current density of 0.89 V, 0.80 V and −5.08 mA cm−2, respectively, which is comparable to that of 20% Pt/C (0.94 V, 0.85 V and 5.33 mA cm−2) and other reported carbon-based materials (Table S2, ESI†). In this work, HCl treatment was applied before obtaining the BM-Fe@NC-T catalysts, during which, the species that can be removed have been washed away. The Fe2C phase retained in BM-Fe@NC-120 was due to the protection of the Fe3O4 shell formed on its outer surface (Fig. 3e). Thus, it is believed that the Fe3O4 phase plays a much more important role in catalyzing the ORR process than the Fe2C phase.
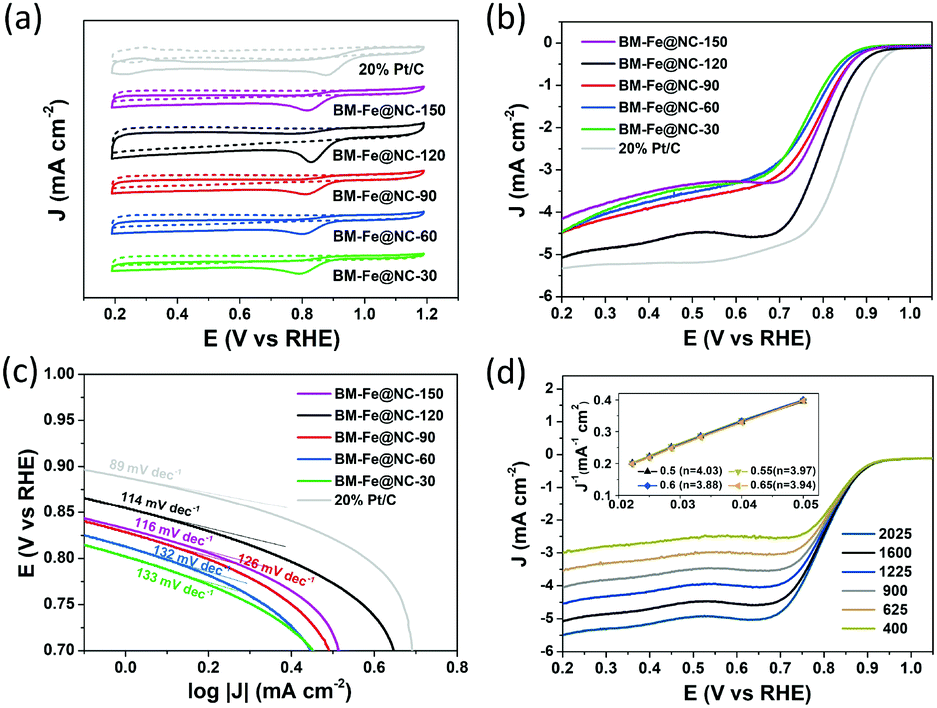 | ||
| Fig. 5 (a) CV curves of the BM-Fe@NC-T samples and 20% Pt/C in both N2- (dotted line) and O2- (solid line) saturated 0.1 M KOH solution; (b) LSV curves and (c) the corresponding Tafel plots of the BM-Fe@NC-T samples in comparison with 20% Pt/C in O2-saturated 0.1 M KOH solution; (d) LSV curves of BM-Fe@NC-120 recorded at different rotation rates in O2-saturated 0.1 M KOH solution (inset: the K–L plots). | ||
Fig. 5c presents the corresponding Tafel plots of the as-prepared BM-Fe@NC-T catalysts, in which, BM-Fe@NC-120 shows a smaller Tafel slope (114 mV dec−1) relative to BM-Fe@NC-30 (133 mV dec−1), BM-Fe@NC-60 (132 mV dec−1), BM-Fe@NC-90 (126 mV dec−1) and BM-Fe@NC-150 (116 mV dec−1), and larger Tafel slope than that of Pt/C (89 mV dec−1), indicating the better ORR activity of BM-Fe@NC-120 than its counterparts and slower catalytic kinetics than Pt/C. The Nyquist plots of these samples further confirm the smallest charge-transfer resistance of BM-Fe@NC-120, revealing its better electron conductivity towards the ORR process (Fig. S6, ESI†).
To gain detailed insight into the catalytic behaviors and ORR mechanism of BM-Fe@NC-T samples, electrocatalytic kinetics measurements were also carried out and the LSVs at electrode rotating rates of 400, 625, 900, 1225, 1600 and 2025 rpm were recorded (Fig. 5d and Fig. S5, ESI†). Enhancement in the current density with an increase of rotation speed is observed for all these samples, based on which, the Koutecky–Levich (K–L) plots of BM-Fe@NC-120 were acquired with parallel slopes and good linearity, and the average electron transfer number (n) is calculated to be 3.95. This indicates a dominant four-electron pathway, reducing the O2 directly into H2O (O2 + 2H2O + 4e− = 4OH−). The ORR catalytic pathway of BM-Fe@NC-120 was further explored using the rotating ring-disk electrode (RRDE) measurements by monitoring the formation of peroxide species (H2O2) during the ORR process. As shown in Fig. 6a, a less than 13% H2O2 yield was detected over the potential range of 0.20–0.70 V and the related electron transfer number is ∼3.85, which is consistent with the results of K–L plots. Thus, the ORR process catalyzed by the BM-Fe@NC-120 is proved to be through a four-electron reduction.
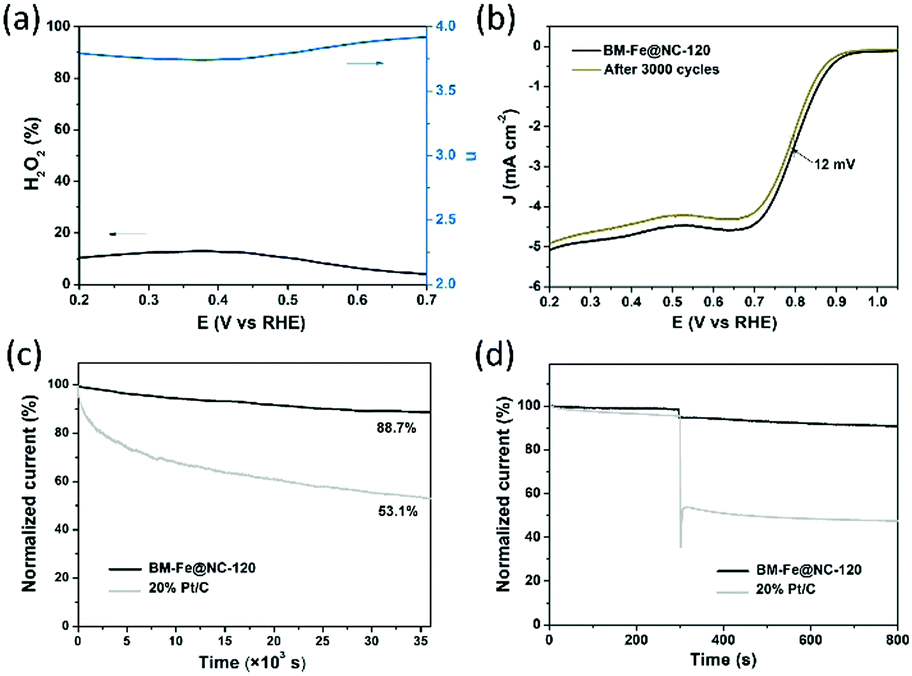 | ||
| Fig. 6 (a) Plots of H2O2 yield (black line) and electron-transfer number (blue line) of BM-Fe@NC-120 in 0.1 M KOH solution; (b) LSV curves of BM-Fe@NC-120 before (black) and after (dark yellow) 3000 CV cycles in 0.1 M KOH solution; (c) chronoamperometric responses of BM-Fe@NC-120 and 20% Pt/C at 0.80 V (vs. RHE) for 10 h; (d) chronoamperometric responses of BM-Fe@NC-120 and 20% Pt/C at 0.80 V (vs. RHE) by adding 3% volume methanol into the 0.1 M KOH solution after 300 s. | ||
For application, the durability of the catalyst and its resistance to the methanol crossover effect are two important aspects to be considered. To probe the durability of BM-Fe@NC-120 in a constant ORR process, CV cycling and chronoamperometry measurements were carried out in comparison with 20% Pt/C. Fig. 6b presents the LSVs of BM-Fe@NC-120 before and after long-term CV cycles. As illustrated, only a slight decrease of half-wave potential (12 mV) and diffusion-limited current density (3.1%) was observed after 3000 CV cycles, indicating the good durability of BM-Fe@NC-120 for the ORR. The chronoamperometry response was also recorded at 0.80 V vs. RHE and the time-dependent current density curve is shown in Fig. 6c. The BM-Fe@NC-120 can reserve 88.7% of its initial current after continuously working for 10 h in an O2-saturated 0.1 M KOH aqueous solution, while the current of the 20% Pt/C gradually decreased by 46.1% within the same time frame, revealing the superior catalytic stability of BM-Fe@NC-120. The small reduction of catalytic activity after the CV and chronoamperometry measurement is probably caused by the gradual electrooxidation of Fe2C during the ORR process. To further investigate the influence of the crossover effect on BM-Fe@NC-120, about 3% volume methanol was injected into the 0.1 M KOH solution after 300 s (Fig. 6d). Compared with the dramatic current decrease of 20% Pt/C (∼50%) due to the catalytic oxidation of methanol, the BM-Fe@NC-120 only presents a small current reduction of ∼3.8%, showing great promise as a methanol-tolerant cathode catalyst to be applied in alkaline direct-methanol fuel cells.
By using the model proposed by Damjanovic et al. and Hsueh et al. (Scheme S1 in the ESI†),55–57 the kinetics of electron transfer in the ORR can be revealed by RRDE voltammetry tests. On the basis of eqn (S1)–(S9) (ESI†), the rate constants (k1, k2 and k3) were calculated by the slope and intercept of Id/Irversus ω−1/2 and Idl/(Idl − Id) versus ω−1/2 within the potential range from 0.20 to 0.80 V vs. RHE (Fig. 7a–c). In Fig. 7b, the k1 ≫ k2 ≈ k3 reveals that the reduction of O2 to OH− catalyzed by BM-Fe@NC-120 is predominantly driven through the k1 pathway with four-electron kinetics. In the cases of BM-Fe@NC-90 and BM-Fe@NC-150, the rate constant k2 is close to k1 although k1 is larger than k2, indicating that the formation of HO2− is a rapid process with a sluggish reduction to OH−. The ratio of k1/k2 of these samples is also calculated and the results are shown in Fig. 7d. It is noteworthy that the BM-Fe@NC-120 presents a much higher value of k1/k2 compared with its counterparts, whose k1/k2 values are only a little larger than 1, further confirming the four-electron k1 pathway kinetics on BM-Fe@NC-120 for catalyzing the ORR process.58,59 As the difference between BM-Fe@NC-T only lies in the mechanical grinding time, the best ORR performance of BM-Fe@NC-120 is due to the optimized 120 min that is crucial for maximum formation of the MOF structure, deriving more catalytic active sites and generating more efficient hierarchical structures for rapid mass transfer.
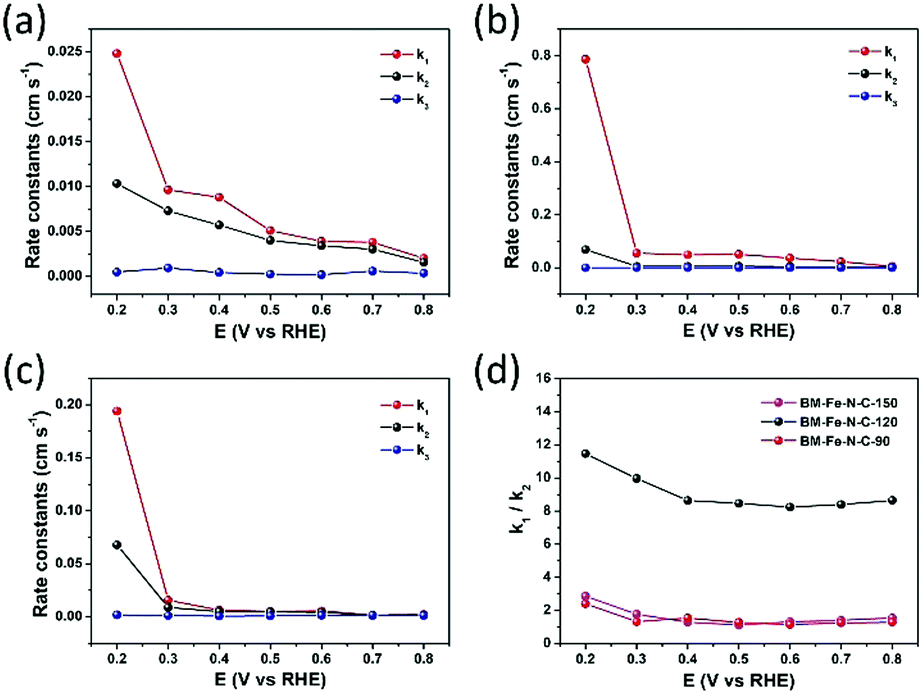 | ||
| Fig. 7 Rate constants of (a) BM-Fe@NC-90, (b) BM-Fe@NC-120 and (c) BM-Fe@NC-150; (d) the ratio of rate constants k1/k2 for BM-Fe@NC-90, BM-Fe@NC-120 and BM-Fe@NC-150 in the potential range from 0.20 to 0.80 V. | ||
By combining all the discussions above, the BM-Fe@NC-120 shows a superior ORR performance, which is the result of a combination of factor synergies: (1) adequate Fe3O4/Fe2C active sites derived from the synthesized MOF structure and doped N species for catalyzing the ORR process; (2) hierarchical structure with a large surface area built using the metal source template for rapid mass transfer; (3) partially graphitized carbon matrix formed for better electron transportation. By utilizing the mechanical grinding method, the metal source can be used as the template for building the hierarchical structure, which is of crucial importance in this work for boosting the kinetic processes for the ORR of BM-Fe@NC-120. This work will provide new insight into the synthesis of hierarchical carbon catalysts from this simple and eco-friendly strategy for future practical fuel-cell applications.
Conclusions
In summary, ORR catalysts with hierarchical structures were developed by utilizing the simple and scalable mechanical grinding strategy. A metal salt, organic ligand and a minute quantity of solvent were used as starting materials and ground at room temperature for different time periods, followed by a subsequent calcination process. The metal salt played the roles of a metal source for the MOF and a template for hierarchical porosity, while the organic ligand participated in the building of the MOF and served as the carbon matrix. As for the minute quantity of solvent, it was favorable for MOF growth and N doping, and may generate pores from volatilization as well. A grinding time of 120 min was optimized for MOF synthesis and the corresponding catalyst (with Fe3O4/Fe2C active sites) showed the best ORR performance, which can be attributed to the synergy between the active sites, hierarchical structures, large surface area and good electron conductivity. This work provides a guideline for future synthesis of hierarchical catalysts from this simple and scalable mechanical grinding strategy.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21875285, 21771193), Taishan Scholar Foundation (ts201511019), and the Fundamental Research Funds for the Central Universities (18CX02047A, 18CX07001A, 18CX05020A).Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. D. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- Y. Xu, T. Chen, T. Wang, J. Yang, Y. Zhu and W. Ding, Nanoscale, 2018, 10, 21944–21950 RSC.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- B. Garlyyev, K. Kratzl, M. Rück, J. Michalička, J. Fichtner, J. M. Macak, T. Kratky, S. Günther, M. Cokoja, A. S. Bandarenka, A. Gagliardi and R. A. Fischer, Angew. Chem., Int. Ed., 2019, 58, 9596–9600 CrossRef CAS PubMed.
- Z. Kou, B. Guo, D. He, J. Zhang and S. Mu, ACS Energy Lett., 2017, 3, 184–190 CrossRef.
- A. Ilnicka, P. Kamedulski, M. Skorupska and J. P. Lukaszewicz, J. Mater. Sci., 2019, 54, 14859–14871 CrossRef CAS.
- Y. Ito, H. J. Qiu, T. Fujita, Y. Tanabe, K. Tanigaki and M. Chen, Adv. Mater., 2014, 26, 4145–4150 CrossRef CAS PubMed.
- W. Ai, Z. Luo, J. Jiang, J. Zhu, Z. Du, Z. Fan, L. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- Y. Dong, M. Zhou, W. Tu, E. Zhu, Y. Chen, Y. Zhao, S. Liao, Y. Huang, Q. Chen and Y. Li, Adv. Funct. Mater., 2019, 29, 1900015 CrossRef.
- J. Liang, R. F. Zhou, X. M. Chen, Y. H. Tang and S. Z. Qiao, Adv. Mater., 2014, 26, 6074–6079 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- S. Cai, Z. Meng, H. Tang, Y. Wang and P. Tsiakaras, Appl. Catal., B, 2017, 217, 477–484 CrossRef CAS.
- Z. Meng, S. Cai, R. Wang, H. Tang, S. Song and P. Tsiakaras, Appl. Catal., B, 2019, 244, 120–127 CrossRef CAS.
- S. Zhao, J. Yang, M. Han, X. Wang, Y. Lin, R. Yang, D. Xu, N. Shi, Q. Wang, M. Yang, Z. Dai and J. Bao, Appl. Catal., B, 2020, 260, 118207 CrossRef CAS.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W. C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- Z. Liang, X. Fan, H. Lei, J. Qi, Y. Li, J. Gao, M. Huo, H. Yuan, W. Zhang, H. Lin, H. Zheng and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 13187–13191 CrossRef CAS PubMed.
- L. Liu, X. Yang, N. Ma, H. Liu, Y. Xia, C. Chen, D. Yang and X. Yao, Small, 2016, 12, 1295–1301 CrossRef CAS PubMed.
- Z. Huang, H. Pan, W. Yang, H. Zhou, N. Gao, C. Fu, S. Li, H. Li and Y. Kuang, ACS Nano, 2018, 12, 208–216 CrossRef CAS PubMed.
- Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- M. Li, Z. Xiao, L. Fan, F. Wang, X. Du, Z. Kang, W. Fan, Z. Guo and D. Sun, Inorg. Chem. Front., 2019, 6, 2296–2303 RSC.
- S. Zeng, F. Lyu, H. Nie, Y. Zhan, H. Bian, Y. Tian, Z. Li, A. Wang, J. Lu and Y. Y. Li, J. Mater. Chem. A, 2017, 5, 13189–13195 RSC.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS.
- P. Song, M. Luo, X. Liu, W. Xing, W. Xu, Z. Jiang and L. Gu, Adv. Funct. Mater., 2017, 27, 1700802 CrossRef.
- W. Zhao, P. Yuan, X. She, Y. Xia, S. Komarneni, K. Xi, Y. Che, X. Yao and D. Yang, J. Mater. Chem. A, 2015, 3, 14188–14194 RSC.
- W. Wei, X. Shi, P. Gao, S. Wang, W. Hu, X. Zhao, Y. Ni, X. Xu, Y. Xu, W. Yan, H. Ji and M. Cao, Nano Energy, 2018, 52, 29–37 CrossRef CAS.
- T. Wang, R. Yang, N. Shi, J. Yang, H. Yan, J. Wang, Z. Ding, W. Huang, Q. Luo, Y. Lin, J. Gao and M. Han, Small, 2019, 15, 1902410 CrossRef CAS PubMed.
- X. Long, D. Li, B. Wang, Z. Jiang, W. Xu, B. Wang, D. Yang and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 11369–11373 CrossRef CAS PubMed.
- X. Chen, N. Wang, K. Shen, Y. Xie, Y. Tan and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 25976–25985 CrossRef CAS PubMed.
- Y. Qin, H. Wu, L. A. Zhang, X. Zhou, Y. Bu, W. Zhang, F. Chu, Y. Li, Y. Kong, Q. Zhang, D. Ding, Y. Tao, Y. Li, M. Liu and X. C. Zeng, ACS Catal., 2018, 9, 610–619 CrossRef.
- H. Wang, F. X. Yin, N. Liu, R. H. Kou, X. B. He, C. J. Sun, B. H. Chen, D. J. Liu and H. Q. Yin, Adv. Funct. Mater., 2019, 29, 1901531 CrossRef.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- H. Yang, F. Peng, C. Dang, Y. Wang, D. Hu, X. Zhao, P. Feng and X. Bu, J. Am. Chem. Soc., 2019, 141, 9808–9812 CrossRef CAS PubMed.
- H. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2011, 112, 673–674 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, 1870277 CrossRef.
- M. Liqing, C. Abney and L. Wenbin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- D. Hong, Y. K. Hwang, C. Serre, G. Férey and J. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- Y. Wang, X. Zhao, H. Yang, X. Bu, Y. Wang, X. Jia, J. Li and P. Feng, Angew. Chem., Int. Ed., 2019, 58, 6316–6320 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- Y. Wang, J. Wang, D. Wei and M. Li, ACS Appl. Mater. Interfaces, 2019, 11, 35755–35763 CrossRef CAS PubMed.
- S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660–12668 CrossRef CAS PubMed.
- B. Y. Guan, L. Yu and X. W. D. Lou, Adv. Sci., 2017, 4, 1700247 CrossRef PubMed.
- L. Chen, Y. Li, N. Xu and G. Zhang, Carbon, 2018, 132, 172–180 CrossRef CAS.
- L. Chai, L. Zhang, X. Wang, Z. Hu, Y. Xu, T. Li, Y. Hu, J. Qian and S. Huang, Carbon, 2020, 158, 719–727 CrossRef CAS.
- F. Xiao, G. Xu, C. Sun, M. Xu, W. Wen, Q. Wang, M. Gu, S. Zhu, Y. Li, Z. Wei, X. Pan, J. Wang, K. Amine and M. Shao, Nano Energy, 2019, 61, 60–68 CrossRef CAS.
- Y. Li, H. Huang, S. Chen, X. Yu, C. Wang and T. Ma, Nano Res., 2019, 12, 2774–2780 CrossRef CAS.
- J. Yu and J. Wang, China Pet. Process. Petrochem. Technol., 2017, 19, 1–8 Search PubMed.
- J. Li, H. Lan, H. Liu, G. Zhang, X. An, R. Liu and J. Qu, ACS Appl. Mater. Interfaces, 2019, 11, 15709–15717 CrossRef CAS PubMed.
- Z. Yang, T. Zhao, X. Huang, X. Chu, T. Tang, Y. Ju, Q. Wang, Y. Hou and S. Gao, Chem. Sci., 2017, 8, 473–481 RSC.
- S. Gao, B. Fan, R. Feng, C. Ye, X. Wei, J. Liu and X. Bu, Nano Energy, 2017, 40, 462–470 CrossRef CAS.
- K. L. Hsueh, D. T. Chin and S. Srinivasan, J. Electroanal. Chem., 1983, 153, 79–95 CrossRef CAS.
- A. Damjanovic, M. A. Genshaw and J. O. Bockris, J. Electrochem. Soc., 1967, 114, 466–472 CrossRef CAS.
- N. A. Anastasijevic, Z. M. Dimitrijevic and R. R. Adzic, J. Electroanal. Chem., 1986, 199, 351–364 CrossRef CAS.
- A. Muthukrishnan, Y. Nabae, T. Hayakawa, T. Okajima and T. Ohsaka, Catal. Sci. Technol., 2015, 5, 475–483 RSC.
- K. Lee, L. Zhang, H. Lui, R. Hui, Z. Shi and J. Zhang, Electrochim. Acta, 2009, 54, 4704–4711 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Photographs, SEM images, survey XPS spectra, and CV and LSV curves. See DOI: 10.1039/d0ma00454e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |