
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of ZnO@rGO nanocomposites for the enzyme free electrochemical detection of urea and glucose†
K. B.
Babitha
a,
P. S.
Soorya
a,
A.
Peer Mohamed
a,
R. B.
Rakhi
 *ab and
S.
Ananthakumar
*a
*ab and
S.
Ananthakumar
*a
aFunctional Materials Section, Materials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Industrial Estate P.O., Thiruvananthapuram 695019, Kerala, India. E-mail: ananthakumar70@gmail.com; ananthakumars@niist.res.in; rakhisarath@gmail.com; rakhiraghavanbaby@niist.res.in; Fax: +91 471 2491712; Tel: +91 471 2515289
bDepartment of Physics, University of Kerala, Kariavattom P.O., Thiruvananthapuram 695581, Kerala, India
First published on 7th August 2020
Abstract
Herein, we report a green synthetic strategy for the preparation of ZnO@rGO nanocomposites. Bioinspired synthesis of ZnO nanoarchitectures has been done using a series of biotemplates such as dextrose, sucrose, soluble starch, and carboxy methyl cellulose. The as-prepared morphologically tuned ZnO nanoarchitectures are homogeneously distributed over the surface of rGO sheets, resulting in the formation of ZnO@rGO nanocomposites. For the first time, zinc (Zn) dust, a waste byproduct from industry (Binani Zinc Limited, Kerala, India), is employed for the chemical reduction of GO. ZnO@rGO nanocomposite based nonenzymatic urea and glucose sensors have been developed over a glassy carbon electrode, and the electrochemical performance of the nanocomposite for the detection of urea and glucose are investigated. The biosensor electrode is sensitive to urea in the concentration range of 0.02 × 10−3 mM to 7.2 × 10−3 mM with a detection limit of 0.012 μM. When used for glucose detection, the linear range is 0.02 × 10−3 mM to 18 × 10−3 mM, with a detection limit of 0.008 μM. The developed sensor exhibits an ultra-high sensitivity of 682.8 μA mM−1 cm−2 towards urea and 481 μA mM−1 cm−2 towards glucose.
1. Introduction
Urea is the most widely used fertilizer, and its production and consumption in India are about 23 and 31 million tonnes. Due to its higher solubility in water, a large amount of the applied urea may get dissolved in groundwater and leads to waterpollution.1 From a biological point of view, urea is the final product of protein degradation and nitrogen metabolism. The normal concentration of urea in blood serum is 2.5–7.5 mM.2 The higher or lower amount of urea in the body leads to several diseases such as renal failure, urinary tract obstruction, dehydration, shock burns, gastrointestinal bleeding, hepatic failure, nephritic syndrome, and cachexia.3,4 So the monitoring of the urea level has great significance in both environmental and clinical samples.Glucose is the primary source of energy in the human body.5 The acceptable range of the blood glucose level in the human body is ∼3.9–6.2 (empty stomach) or ∼3.9–7.8 (2 h after food) mM.6 If the human body is unable to control the amount of glucose due to decreased insulin secretion, the condition results in diabetes mellitus.7 Furthermore, it leads to different health problems like cardiovascular, nervous, ocular, cerebral and peripheral vascular diseases, kidney failure, tissue damage, blindness, etc.5,8 Hence, quantitative monitoring of the blood glucose level is essential, to avoid the adverse effects of diabetes.5
A quantum of research studies has been conducted to develop an efficient and reliable method for urea and glucose sensing. The electrochemical sensing strategy has been considered as the most promising tool for urea and glucose detection.9,10 Metal oxides such as ZnO, Fe2O3, CeO2, SnO2, Ag2O, ZrO2, TiO2, Co3O4, NiO, Cu2O, MnO2, MgO, RuO2, and Mn3O4 have been extensively studied for electrochemical sensors. Among these, ZnO nanoparticles were most widely studied for electrochemical biosensing applications. In electrochemical sensors, nonenzymatic sensors are most popular because they can overcome the disadvantages of enzymatic sensors.11
Graphene-based nanomaterials are widely used in electrocatalytic sensing applications. rGO is found to be the most preferred sensor platform due to the following reasons: (1) rGO is electrically conductive compared to the non-conductive GO, (2) a large number of edges and defects facilitate electron transfer and (3) conductivity and surface functional groups could be tuned for the detection of specific chemical species. Different methods are reported for the preparation of rGO from GO. In 2010, X. Gao et al. showed that the deoxygenation of GO with hydrazine or heat treatment results in rGO.12 In 2011, P. Cui et al. reported a new reducing system i.e., hydriodic acid with trifluoroacetic acid, which can chemically convert GO into rGO at subzero temperature (below 0 °C) with mass production.13 In 2012, V. H. Pham et al. reported a simple and effective method to reduce the aqueous suspension of GO using nascent hydrogen generated in situ by the reaction between Al foil and HCl, Al foil and NaOH, and Zn powder and NaOH.14 In 2012, O. Akhavan et al. reported a single-step green method for the reduction and functionalization of GO by glucose.15 In 2012, R. S. Dey et al. demonstrated a facile route for the synthesis of rGO sheets by the chemical reduction of GO using Zn/acid solution at room temperature.16 To the best of our knowledge, no attempts were made to conduct the reduction of GO using waste materials.
The use of biomaterials as templates has become significant in the green synthesis of nanomaterials with controllable morphology. Biomaterials are cheap, economical, environmentally begin, and renewable. In the present work, ZnO nanoarchitectures with varied morphology were prepared via the biotemplate assisted microwave method. Here dextrose, sucrose, soluble starch, and carboxy methyl cellulose were used as biotemplates. At the same time, rGO was prepared by the chemical reduction of GO using nascent hydrogen. Zn dust, a waste material obtained from industry, was used for the generation of nascent hydrogen. It acts as a strong reducing agent. ZnO nanoarchitectures prepared with different biotemplates are attached on rGO via the microwave assisted ex situ technique, which results in ZnO@rGO nanocomposites. Structural and morphological studies were carried out, showing the homogeneous distribution of ZnO nanoarchitectures on rGO sheets. The as-prepared ZnO@rGO has been used for the nonenzymatic sensing of urea and glucose. Since the studies on nonenzymatic ZnO based glucose and urea sensors are rarely reported, the fabrication and performance of ZnO@rGO based sensors are important. Similarly, the synergistic effect of both ZnO and rGO provides excellent sensing characteristics in the as-prepared nonenzymatic sensor.
2. Experimental section
2.1 Materials
Graphite (99.9%), Nafion solution (5 wt% in lower aliphatic alcohols and water contains 15–20% water) and potassium ferri cyanide (C6N6FeK3, 99.9%) were procured from Sigma Aldrich, Germany. Sodium nitrate (NaNO3, 99.5%), potassium permanganate (KMnO4, 99%), hydrogen peroxide (H2O2, 99%), dextrose anhydrous (D-glucose anhydrous, C6H12O6, 99%), sucrose (saccharose, C12H22O11, 99%), and carboxy methyl cellulose sodium salt were obtained from S. D. Fine Chemicals limited, India. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%), conc. sulphuric acid (H2SO4, 99%) and soluble starch ((C6H10O5)n, 99%) were supplied by Merck, India. Lithium hydroxide (LiOH, 99%) was purchased from SRL, India. Zn dust obtained from Binani Zinc Limited was used for the reduction of GO. All the reagents were used as received without any further purification.2.2 Preparation of GO
GO has been prepared via Hummers’ method. In a typical procedure 1 g graphite and 1 g NaNO3 were dispersed in 50 mL conc. H2SO4 while keeping in an ice bath. 3 g of KMnO4 was added slowly to the reacting mixture and the reaction temperature was not allowed to increase to 20 °C. The reaction mixture was then transferred to a water bath kept at a temperature of 35 °C. 50 mL of distilled water was added to the system and the temperature raised to 98 °C. It was followed with the addition of 150 mL of distilled water and 10 mL of 30% H2O2. The oxidized graphite was then centrifuged and washed with double distilled water until the pH becomes 7. Finally, on freeze drying graphene oxide with a fluffy nature was obtained.17,182.3 Preparation of rGO via nascent hydrogen chemical reduction
In a typical procedure, 20 mg of GO was dispersed in a 200 mL double distilled water using a probe sonicator. It was further treated with conc. HCl (1 M) followed by the slow addition of Zn dust (2 g). The whole mixture was stirred overnight using a magnetic stirrer. The gradual change in the colour from orange-brown to black indicates the reduction of GO to rGO. The obtained rGO was then collected by centrifugation. Furthermore, it was washed three times with 0.1 M HCl, followed by washing with double distilled water. After removing all impurities, the rGO nanosheets were freeze-dried. Illustration for the nascent hydrogen chemical reduction is shown in Fig. 1. | ||
| Fig. 1 Illustration of the Zn mediated nascent hydrogen chemical reduction of GO in the presence of conc. HCl. | ||
2.4 Synthesis of ZnO nanoarchitectures via the biotemplate method
ZnO nanoarchitectures were prepared via the biotemplate assisted microwave technique (Fig. 2, step 1). In a typical procedure, 0.3 M Zn(NO3)2·9H2O was dissolved in 500 mL double-distilled water, and an appropriate amount of carbohydrate/cellulose was dispersed eventually by keeping the wt ratio of Zn(NO3)2·9H2O and carbohydrate/cellulose as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Dextrose, sucrose, soluble starch, and carboxy methyl cellulose were used as structure-directing agents. The pH of the solution was adjusted with 10 wt% LiOH solution. The obtained solution was kept in a microwave synthesis work station (Sineo MAS II) at 80 °C for 30 min (P = 300 W). The obtained powder was washed repeatedly with double distilled water to remove the impurities. The final product was dried in an oven at 50 °C. The obtained ZnO samples were denoted as ZD, ZS, ZSs, and ZC, respectively.
:1. Dextrose, sucrose, soluble starch, and carboxy methyl cellulose were used as structure-directing agents. The pH of the solution was adjusted with 10 wt% LiOH solution. The obtained solution was kept in a microwave synthesis work station (Sineo MAS II) at 80 °C for 30 min (P = 300 W). The obtained powder was washed repeatedly with double distilled water to remove the impurities. The final product was dried in an oven at 50 °C. The obtained ZnO samples were denoted as ZD, ZS, ZSs, and ZC, respectively.
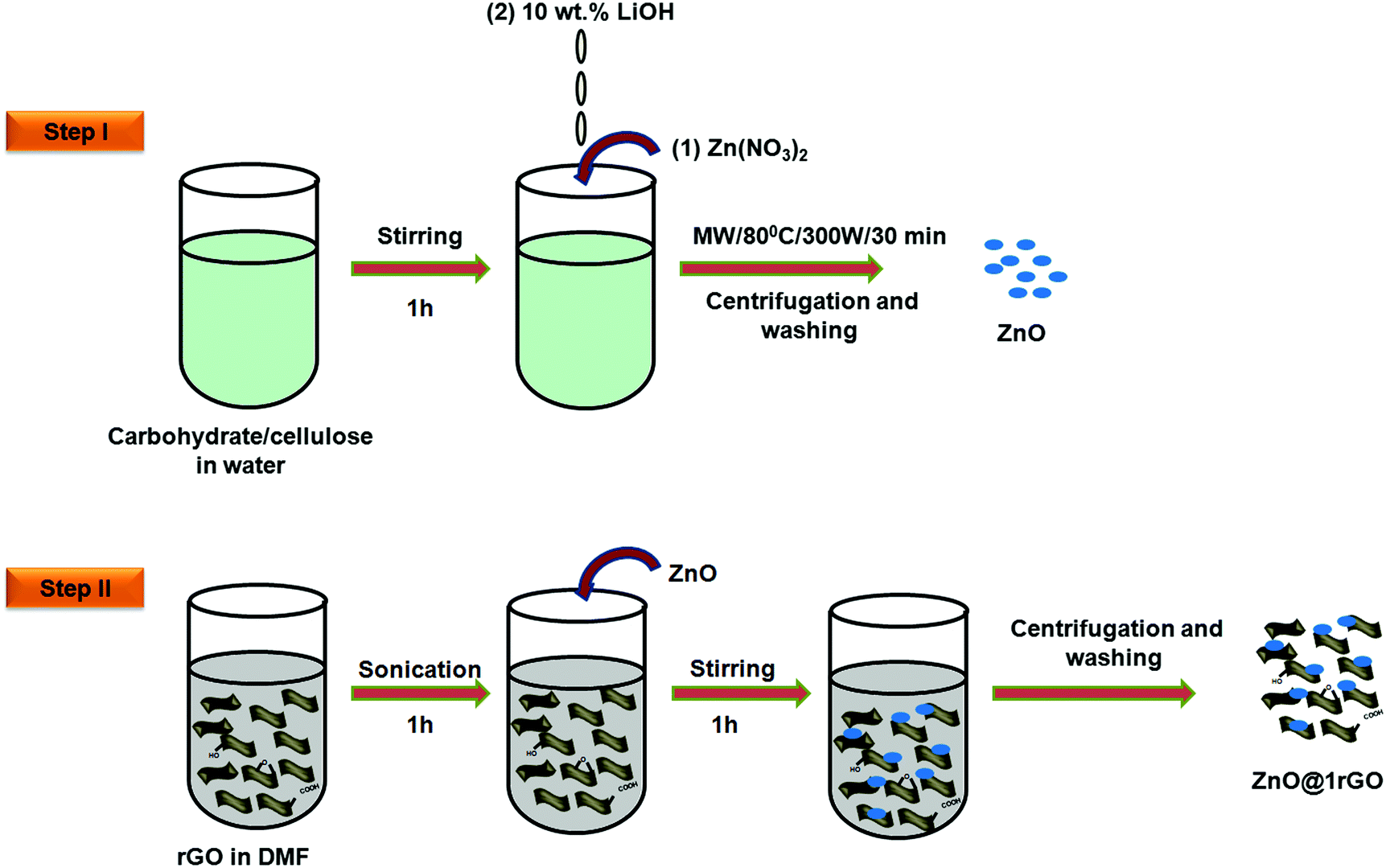 | ||
| Fig. 2 Schematic illustration showing the synthetic procedure of ZnO and ZnO@1rGO. | ||
2.5 Preparation of ZnO@1rGO nanocomposites
In a typical synthesis of the nanocomposites, 0.0025 g of rGO (1 wt%) was uniformly dispersed in 30 mL of DMF with the aid of ultrasonication for 1 h to obtain a uniform suspension of rGO. Then 0.2475 g of ZnO (ZD, ZSs, ZS, and ZC) was added. It was followed by magnetic stirring for 1 h to attain uniform dispersion. The product obtained was collected by centrifugation and repeatedly washed with ethanol to remove the impurities and dried at 85 °C. The illustration is provided in Fig. 2, step II.2.6 Characterization
The X-ray diffraction patterns of the samples were collected on an X-ray powder diffractometer X’Pert Pro Philips using Cu Kα radiation (λ = 1.5406 Å) in the diffraction angle (2θ) range between 10° and 60°. The crystallite size was calculated using the Debye–Scherrer formula DXRD = kλ/βcosθ, where DXRD is the average crystallite size in nm, k is the shape factor (0.9), λ is the X-ray wavelength in nm, β is the full width at half maximum of the (101) peak in radian and θ is the Bragg angle (degree).19 The lattice parameters of the samples were calculated using the equation 1/d2 = 4/3(1/a2) + 1/c2, where d is the interplanar distance, and a and c are the lattice parameters in nm (being hexagonal c/a = (8/3)1/2).20 The optical absorption spectra of the samples were recorded over a wavelength range of 200–800 nm using a UV-vis spectrophotometer UV 2401-PC, Shimadzu. Fourier transform infrared spectra (FTIR) of the samples were recorded using an IR Prestige 21 (Shimadzu) spectrophotometer using the KBr method in the scanning range of 500 to 4000 cm−1. The morphology of the samples was determined by using a scanning electron microscope (SEM, ZEISS EVO 18) applying an acceleration voltage of 20 kV. The elemental analysis of the samples was performed by the energy dispersive X-ray (EDX) technique equipped with a SEM instrument. Elemental mapping was performed by using an EDX spectroscopic technique associated with a ZEISS EVO 18 instrument. The transmission electron microscopy (TEM) images were collected by using a Tecnai G2 model FEI microscope operating with an accelerating voltage of 300 kV. The Brunner–Emmett–Teller (BET) surface area of the samples was evaluated on the basis of N2 adsorption–desorption isotherms with a Micromeritics Gemini 2375 Surface Area Analyzer. Prior to the surface area analysis, the samples were degassed for 2 h at 150 °C.
2.7 Fabrication of the nonenzymatic electrochemical sensor electrodes
The nonenzymatic electrochemical sensor electrodes (glassy carbon electrode (GCE)-with ZnO or ZnO@1rGO) have been prepared by the following procedure. The GCE was first cleaned by polishing with 0.3 μm alumina powder. The polished GCE was further processed by ultrasonication for 5 min each in acetone and double-distilled water, respectively, and dried in ambient air. 1 mg mL−1 of ZnO or ZnO@1rGO was sonicated in Nafion for 10 min to get a uniform dispersion. The cleaned GCE was drop cast with 2 μL of ZnO or ZnO@1rGO dispersed in Nafion. This thin film of the materials deposited on the surface of the electrodes was left overnight at room temperature for drying the deposited material, and the same was used as a working electrode (nonenzymatic electrochemical sensor electrode) for the detection of urea or glucose.2.8 Electrochemical studies
The electrochemical behavior of ZnO (ZD, ZS, ZSs, and ZC) and ZnO@1rGO nanocomposite (ZD@1rGO, ZS@1rGO, ZSs@1rGO, and ZC@1rGO) based electrochemical sensor electrodes has been determined with urea or glucose. Electrochemical experiments, such as cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) analysis, were conducted in a conventional three-electrode cell using an electrochemical work station (VMP3-Biologic). The modified glassy carbon electrode (GCE) was used as the working electrode (with a GC disc diameter of 3 mm) in combination with a platinum (Pt) wire and Ag/AgCl (in saturated KCl) as the counter electrode and reference electrode, respectively. 0.1 M potassium ferricyanide solution was used as the electrolyte. All measurements were conducted under ambient conditions. The EIS analysis was performed in 0.1 M potassium ferricyanide solution at a potential of 0.32 V in the frequency range of 10–106 Hz at a signal amplitude of 10 mV.3. Results and discussion
3.1 Characterization of rGO prepared via Zn mediated nascent hydrogen reduction
Zn dust obtained as a waste by-product from industry was used for the generation of nascent hydrogen for the chemical reduction of GO. The detailed characterization studies of freeze-dried GO prepared via Hummers’ method had been reported in our earlier work.18 The XRD pattern of the Zn dust is given in Fig. S1 (ESI†). The characteristic diffraction peaks at 2θ positions of 36.5°, 39.2°, 43.4°, 54.36°, 70.54°, and 70.7° corresponding to the (002), (100), (101), (102), (103), and (110) lattice planes of pure Zn metal, respectively (JCPDS file no. 00-004-0831),21 confirm the presence of pure Zn metal in the waste by-product.The XRD analysis of rGO sheets prepared using nascent hydrogen reduction with Zn dust is given in Fig. 3A. The XRD pattern of GO is given in the inset of Fig. 3A. The main intensity peak of GO is at 10.8° corresponding to the (001) reflection plane. After reduction with Zn dust, this peak completely disappeared, and a new broad diffraction peak centred at ∼24.5° appeared indicating the complete conversion of GO to rGO. This peak corresponds to the (002) plane of rGO nanosheets.22 The appearance of a low-intensity peak exhibited by rGO nanosheets at ∼44° corresponds to the (100) reflection plane of rGO.23
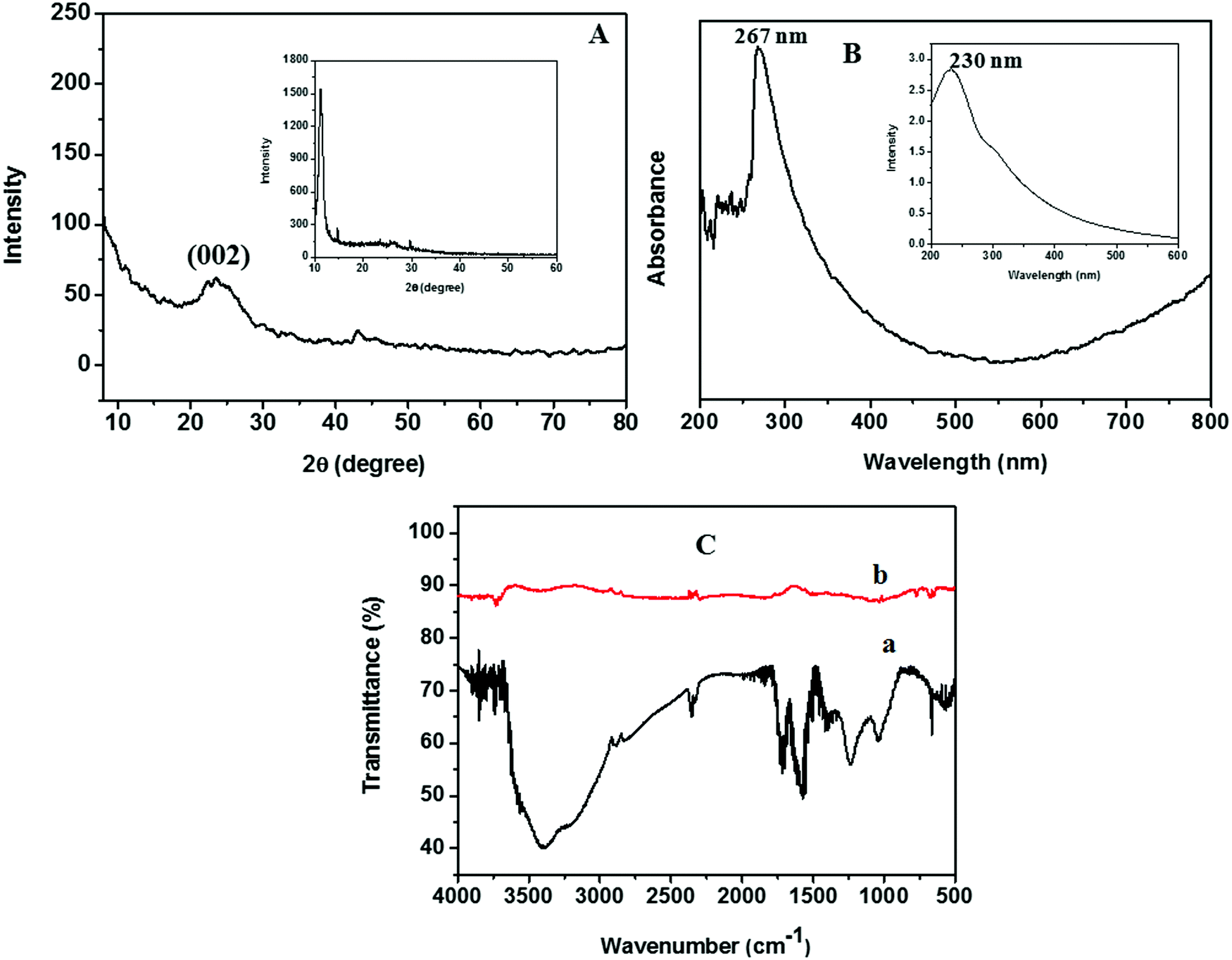 | ||
| Fig. 3 (A) XRD pattern of rGO prepared using Zn dust. (B) UV-vis spectra of rGO dispersed in DMF. (C) FTIR spectra of (a) GO and (b) rGO. The inset of (A) shows the XRD pattern of GO and the inset of (B) shows the UV-vis spectra of GO. | ||
The UV-vis absorption spectrum of rGO, studied by dispersing rGO nanosheets in DMF, is given in Fig. 3B. The optical properties of GO are also given in the inset of Fig. 3B. The maximum absorption peak of rGO is found to be at 267 nm. During the reduction of GO to rGO, the absorption band corresponding to the aromatic C–C bond (π–π* transition) is red-shifted from 230 to 267 nm. Hence, the restoration of electronic conjugation and structural ordering in the carbon framework after Zn mediated hydrogen reduction provides clear evidence for the complete reduction of GO to rGO.16
The FTIR spectra obtained for GO and rGO are given in Fig. 3C. In the FTIR spectra of GO we could see a broad absorption band in between 3000 and 3400 cm−1, which corresponds to the –OH stretching. This peak completely vanishes in the FTIR spectra of rGO. Similarly, a remarkable decrease in the band intensity was observed in the bands corresponding to the stretching of oxygen-containing functionalities such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl (1720 cm−1), epoxy C–O (1231 cm−1), –OH deformation vibrations (1389 cm−1), and alkoxy C–O (1043 cm−1) after the nascent hydrogen reduction of GO.16
O carbonyl (1720 cm−1), epoxy C–O (1231 cm−1), –OH deformation vibrations (1389 cm−1), and alkoxy C–O (1043 cm−1) after the nascent hydrogen reduction of GO.16
Typical low and high magnification SEM images of rGO are presented in Fig. 4A and B, respectively. The fluffy and thin layered nature of rGO nanosheets was confirmed from the SEM images. The microstructure of the rGO nanosheets was again confirmed with the TEM image of rGO, as shown in Fig. 4C.
 | ||
| Fig. 4 (A and B) Typical low and high magnification SEM images of rGO prepared via Zn dust mediated nascent hydrogen reduction of GO. (C) TEM image of rGO prepared via chemical reduction with Zn dust. | ||
The probable mechanism for the nascent hydrogen chemical reduction of GO with Zn dust can be explained as follows: epoxy, carbonyl, and hydroxyl groups are the main oxygen-containing functional groups presented on the GO surface. The homogeneous dispersion of GO in water facilitates the intimate contact between Zn dust and conc. HCl thus leading to the in situ generation of nascent hydrogen. This highly reactive nascent hydrogen acts as a reducing agent for the chemical reduction of GO.
| Zn dust + 2 conc. HCl → ZnCl2 + 2[H] | (1) |
The chemical reduction of GO involves the removal of the above mentioned functional groups. It involves the deoxygenation of epoxide groups through the ring-opening mechanism and decarboxylation of carboxylic acid groups.16 Both reactions led to the restoration of π conjugation14 and resulted in olefins. Similarly, carbonyl groups are reduced to the corresponding alcohols, which further give olefins. The conversion of GO to rGO is again clear from the colour change of the reactant mixture from dark brown to black.24
The BET surface areas of GO and rGO were measured using N2 adsorption desorption isotherms and are presented in ESI,† Fig. S2. The BET surface areas of GO and rGO are 161 m2 g−1 and 153 m2 g−1. During the reduction process there is a chance of agglomeration of the nanosheets and it will result in the decreased surface area of rGO compared to GO.
Electrochemical impedance spectroscopy is a powerful tool for identifying the electrical conductivity of the sensor electrode material. The Nyquist plot of the electrochemical impedance spectra (EIS) of the rGO modified GCE in 0.1 M K3[Fe(CN)6] electrolyte solution is shown in Fig. S3 (ESI†). An extremely low value of the charge transfer resistance (250 mΩ) of rGO clearly indicates that the material has high electrical conductivity.
3.2 Characterization of ZnO nanoparticles prepared via the biotemplate assisted method
Fig. 5A shows the XRD patterns of pure ZnO synthesized with carbohydrates/cellulose. The XRD pattern indicates that all samples are highly crystalline. All of the diffraction peaks of the as-prepared ZnO nanoarchitectures match well with those of the standard XRD file of ZnO (JCPDS standard card no. 79-0205) and can be indexed as the wurtzite phase of ZnO. The diffraction peaks at 31.7°, 34.57°, 36.09°, 47.53°, 56.67°, 62.78°, and 67.86° correspond to the (100), (002) (101), (110), (103), and (112) lattice planes of wurtzite ZnO. No other impurity peaks arising due to carbohydrate/cellulose as well as no remarkable shift in all diffraction peaks were detected in the XRD patterns of the as-prepared samples, which confirm the phase purity of the products. The lattice parameters of the as-prepared samples were calculated and are given in Table 1. The calculated values of lattice parameters a = b = 0.32379–0.32406 nm and c = 0.52865–0.5291 nm are consistent with the reported values of ZnO.25 The crystallite size was evaluated using the Scherrer formula and is presented in Table 1. From the Scherrer formula, the crystallite size was estimated to be ∼43.99, 27.77, 26.62, and 33.75 nm, respectively, for ZD, ZS, ZSs, and ZC.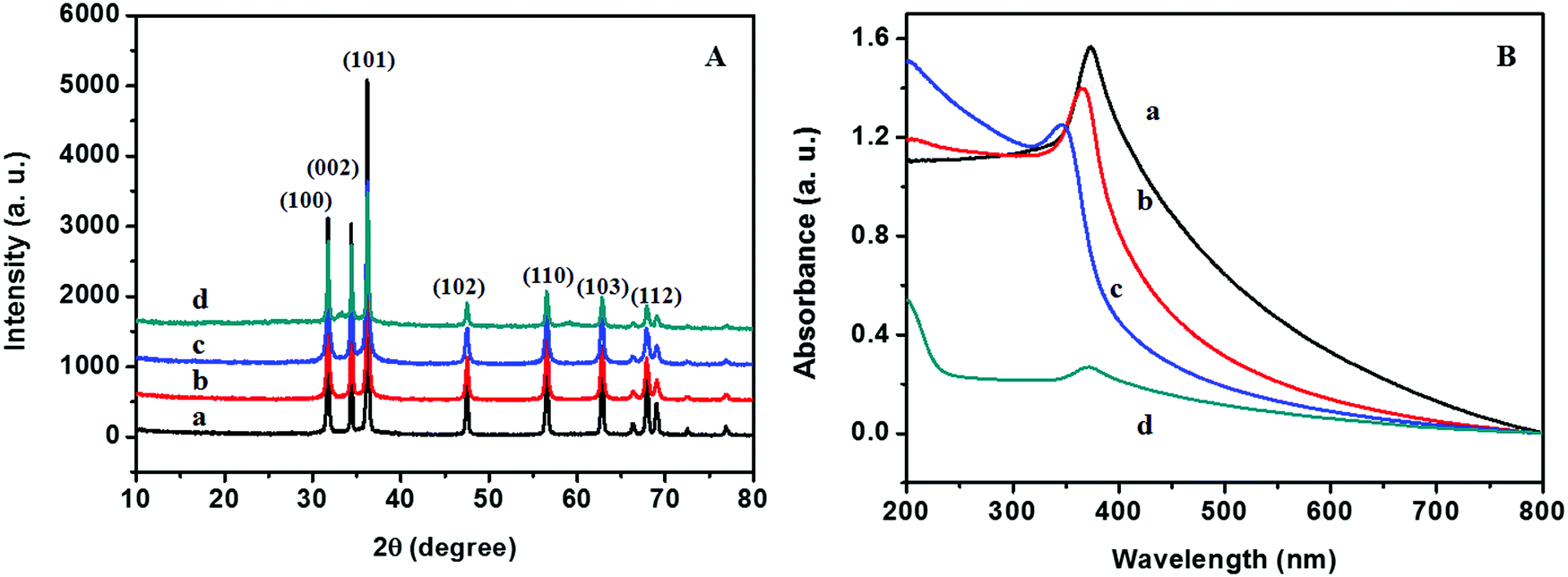 | ||
| Fig. 5 (A) XRD patterns and (B) UV-vis absorption spectra of (a) ZD, (b) ZS, (c) ZSs, and (d) ZC. | ||
| Samples | D XRD (nm) | Lattice parameter (nm) | λ max | Morphology | D TEM | ||
|---|---|---|---|---|---|---|---|
| a | c | c/a | |||||
| ZD | 43.99 | 0.32406 | 0.5291 | 1.6327 | 372.95 | Rods | L = 800.8, W = 140.5 |
| ZS | 27.77 | 0.32385 | 0.52875 | 1.6327 | 367.84 | Spheres | 109.72 |
| ZSs | 26.62 | 0.32379 | 0.52865 | 1.6326 | 344.9 | Aggregates | 50 |
| ZC | 33.75 | 0.32386 | 0.52877 | 1.6327 | 370.4 | Square | 188.19 |
The optical properties of ZnO nanoarchitectures synthesized with different carbohydrates/cellulose are studied using UV-vis absorption spectroscopy and are given in Fig. 5B. The maximum absorption peak of ZD is at 372.95 nm. A blueshift in the UV-vis absorption spectra was observed from ZD to ZSs (372.95–344.99 nm), which is mainly due to the crystallite size reduction from 43.99 to 26.62 nm. From ZSs to ZC a redshift was observed from 344.9 to 370.4 nm and is mainly attributed to the increase in the crystallite size from 26.62 to 33.75 nm.
Fig. 6 displays the SEM images of ZnO nanoarchitectures prepared with different carbohydrates/cellulose. ZnO nanoarchitectures (ZD) synthesized with dextrose as a structure-directing agent were found to have a rod shape with ∼800 nm length and ∼140 nm width (Fig. 6A). From Fig. 6B, it can be seen that sucrose leads to the formation of ZnO nanoarchitectures with a diameter of ∼91.5 nm. The synthesized ZnO nanoarchitectures (ZSs) using soluble starch as a structure-directing agent were found to be spherical in shape with smaller particle size (Fig. 6C). But some agglomeration was also observed in this case. Carboxy methyl cellulose leads to the formation of ZnO nanoarchitectures (ZC) with agglomerates of spherical particles of relatively larger size compared to ZS and ZSs nanoarchitectures (Fig. 6D). Similar observations are obtained in the TEM imaging also. The synthesized ZD nanoarchitectures having a rod shape with ∼800.8 nm length and ∼140.5 nm diameter (Fig. 7A). The spherical shape of the ZS nanoarchitectures is more clearly seen in the TEM image (Fig. 7B) with a diameter of ∼109.72 nm. Soluble starch leads to the formation of ZnO nanoarchitectures (ZSs) with a relatively small size of ∼50 nm. As seen in the SEM image, Fig. 7C also shows uniformly sized agglomerated particles of ZSs nanoarchitectures. Relatively square-shaped particles are observed in the TEM image of ZC with a particle size of 188.19 nm (Fig. 7D).
 | ||
| Fig. 6 SEM images of (A) ZD, (B) ZS, (C) ZSs, and (D) ZC. | ||
 | ||
| Fig. 7 TEM images of (A) ZD, (B) ZS, (C) ZSs, and (D) ZC. | ||
3.3 Characterization of ZnO@1rGO nanocomposites
The XRD patterns of ZnO@1rGO prepared with different carbohydrates/cellulose are presented in Fig. 8A. All samples show the diffraction patterns of wurtzite ZnO even in the presence of rGO. The XRD results of all samples show diffraction peaks at 31.77°, 34.31°, 36.09°, 47.53°, 56.67°, 62.78°, and 67.6° corresponding to the lattice planes of (100), (101), (102), (110), (103), and (112). No remarkable shift in the diffraction peaks was observed in the presence of rGO in the nanocomposite. Since the amount of rGO in the nanocomposite is very small, no peaks of rGO were observed in the XRD patterns of nanocomposites. The lattice parameter calculated is listed in Table 2. The lattice parameter values (a = 0.3232–0.3236 nm, and c = 0.52765–0.52834 nm) are consistent with the reported lattice parameter values of ZnO. The crystallite size evaluated using the Scherrer equation is given in Table 2. The results indicate that the presence of rGO in the nanocomposite did not remarkably affect the crystallite size and lattice parameter of ZnO nanoarchitectures.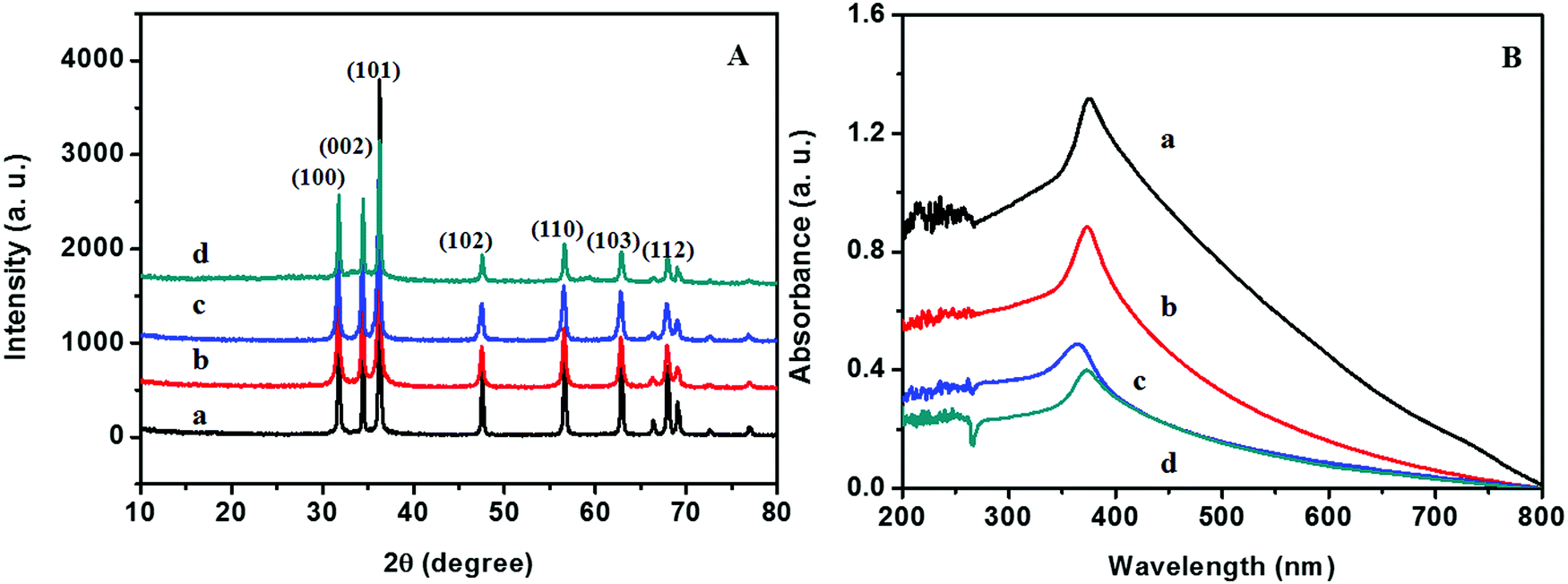 | ||
| Fig. 8 (A) XRD patterns and (B) UV-vis absorption spectra of (a) ZD@1rGO, (b) ZS@1rGO, (c) ZSs@1rGO, and (d) ZC@1rGO. | ||
| Samples | D XRD (nm) | Lattice parameter (nm) | λ max | Morphology | D TEM | ||
|---|---|---|---|---|---|---|---|
| a | c | c/a | |||||
| ZD@1rGO | 38.17 | 0.3236 | 0.52834 | 1.6326 | 375.77 | Rods | L = 780, W = 140 |
| ZS@1rGO | 25.32 | 0.3236 | 0.52829 | 1.6325 | 373.71 | Spheres | 102.5 |
| ZSs@1rGO | 23.61 | 0.3232 | 0.52765 | 1.6325 | 362.75 | Aggregates | 50 |
| ZC@1rGO | 30.12 | 0.3236 | 0.52833 | 1.6326 | 373.71 | Square | 187.8 |
The optical properties of the as-prepared ZnO@1rGO nanocomposites are given in Fig. 8B. We could see a redshift in the absorption spectra of all nanocomposites compared to the spectra of ZnO nanoarchitectures. The redshift in the λmax values is attributed to the incorporation of rGO in the nanocomposites, and the λmax values are presented in Table 2.
Fig. 9 shows the SEM images of ZnO@1rGO nanocomposites synthesized with different carbohydrate/cellulose. The insets of Fig. 9B and D show the SEM images at high resolution. The presence of both rGO nanosheets and ZnO are clearly seen in the SEM images of all samples. It is clear that ZnO nanoarchitectures are deposited on the surface of rGO sheets. The same observations were again found in the TEM images of ZnO@1rGO (Fig. 10). The SEM and TEM images confirm that the morphology of pure ZnO is not altered after the preparation of the composite with rGO.
 | ||
| Fig. 9 SEM images of (A) ZD@1rGO, (B) ZS@1rGO, (C) ZSs@1rGO, and (D) ZC@1rGO. | ||
 | ||
| Fig. 10 TEM images of (A) ZD@1rGO, (B) ZS@1rGO, (C) ZSs@1rGO, and (D) ZC@1rGO. | ||
The N2 adsorption–desorption isotherms of ZnO@1rGO nanocomposites are shown in Fig. 11. All samples show type IV isotherms. The BET surface areas of ZD@1rGO, ZS@1rGO, ZSs@1rGO, and ZC@1rGO are 6.47, 18.33, 20.03, and 13.21 m2 g−1. Based on the BET surface area analysis, it is clear that a higher surface area is obtained for the ZSs@1rGO nanocomposite, which is in agreement with the XRD and TEM/SEM results. It is due to the low crystallite size or low particle size shown by the ZSs@1rGO nanocomposite.
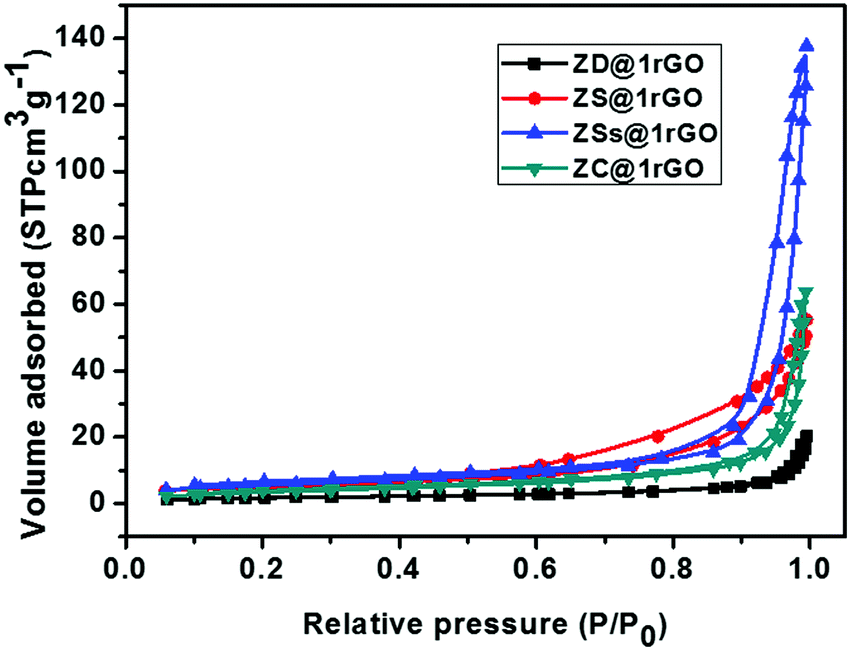 | ||
| Fig. 11 N2 adsorption–desorption isotherms of ZnO@1rGO. | ||
The elemental analysis (EDS) spectra of rGO and ZSs@1rGO are presented in Fig. S4 (ESI†). The EDS spectrum of rGO (Fig. S4A, ESI†) shows the peaks of carbon and oxygen. However, the intensity of oxygen is less compared to carbon in rGO. The presence of Zn in the nanocomposite is confirmed by the EDS spectrum of ZSs@1rGO. Additional peaks of C and O are also seen in the composite. The elemental mapping of ZSs@1rGO is shown in Fig. S5 (ESI†). The presence of elements such as C, O, and Zn is well mapped in the images.
3.4 Electrochemical studies
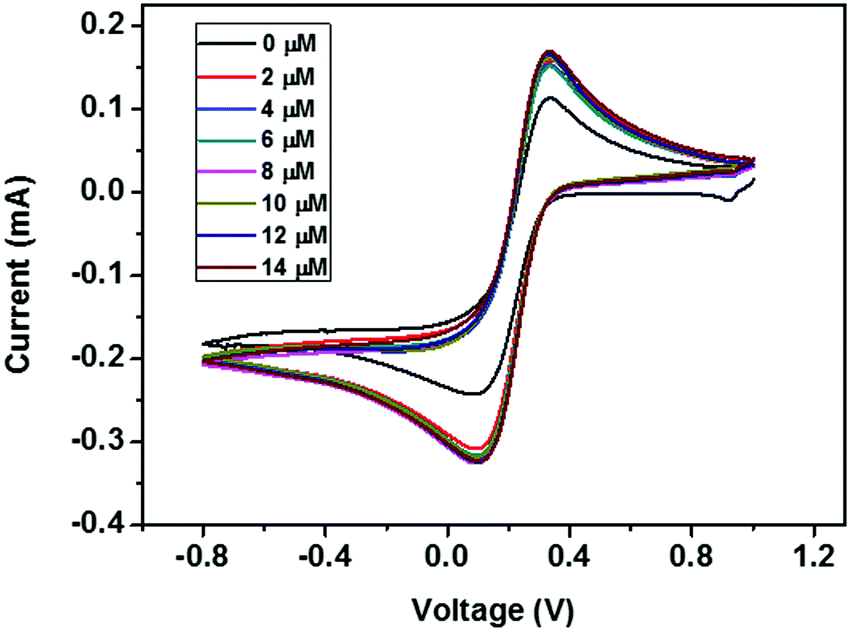 | ||
| Fig. 12 CV curves of the ZD modified GCE with addition of various concentrations of urea from 0 to 14 μM. | ||
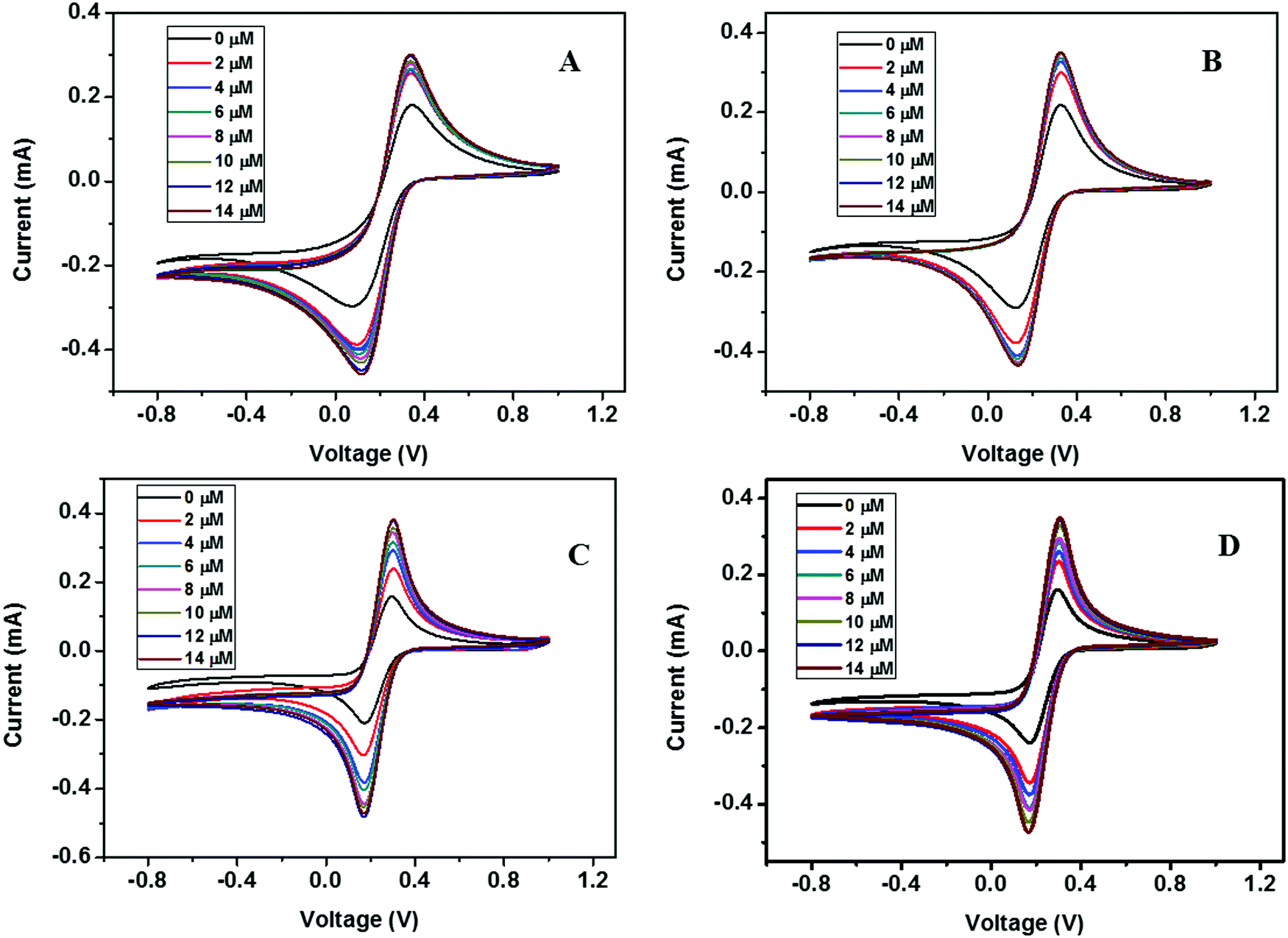 | ||
| Fig. 13 CV curves of (A) ZD@1rGO, (B) ZS@1rGO, (C) ZSs@1rGO, and (D) ZC@1rGO modified GCEs with the addition of various concentrations of urea from 0 to 14 μM. | ||
The presence of both ZnO nanoarchitectures and rGO nanosheets in the nanocomposite results in different architectures on the electrode surface. This will provide a greater active surface, improved immobilization of the target molecules, and a more efficient electron transfer. A comparative study of the CV measurements has been carried out using different nanocomposite modified GC electrodes (ZD@1rGO, ZS@1rGO, ZSs@1rGO, and ZC@1rGO) after the addition of 14 μM urea into the electrolyte solution and the results are shown in Fig. 14. The best performance was exhibited by ZSs@1rGO. The higher electrochemical activity of ZSs@1rGO is due to the smaller crystallite size or particle size, as well as, the higher BET surface area of ZSs@1rGO compared to other nanocomposites.
 | ||
| Fig. 14 CV measurements of ZnO (ZD) and ZnO@1rGO nanocomposites (ZD@1rGO, ZS@1rGO, ZSs@1rGO, and ZC@1rGO) modified GCEs after the addition of 14 μM urea. | ||
EIS is a commonly used characterization technique to study the impedance changes of the electrode surface, mainly due to the electron transfer between electrolyte and the electrode surface. The Nyquist plot contains a semicircle region and a linear region. The semicircle portion corresponds to the electron transfer limited process, and the linear part corresponds to the diffusion process. The diameter of the semicircle arc is equivalent to the electron transfer resistance (Rct), which reflects the conductivity and the electron transfer process.26 A large semicircle arc with high Rct indicates that the system has a higher resistance to the flow of electrons. In Fig. 15A and B the ZnO modified GCE showed a higher Rct with a larger semicircle diameter (Rct = 169 Ω), indicating that ZnO nanoarchitectures were successfully immobilized on the GCE surface, which hindered the electron transfer of the electrochemical probe. This hindrance was decreased (Rct = 74 Ω) after the incorporation of rGO in nanocomposites. The small semicircular diameter of the ZSs@1rGO modified GCE implies that it has low resistance towards the electron transfer process. These results showed the efficiency of the ZSs@1rGO modified GCE compared to the ZnO modified GCE.9,27
 | ||
| Fig. 15 (A and B) Nyquist diagram of electrochemical impedance spectra of ZD and ZSs@1rGO modified GCEs in 0.1 M K3[Fe(CN)6] electrolyte solution. (C) Randles equivalent circuit. | ||
The calibration study was conducted using CV by the addition of 0.02 μM to 32 μM urea to the electrolyte solution (Fig. 16A). From the plot of IPversus concentration of urea given in Fig. 16B, the linear range was calculated and is between 0.02 × 10−3 and 7.2 × 10−3 mM. The effect of the scan rate for 50, 100, 150, 200, 300, 400, and 500 mV s−1 towards the electrochemical oxidation of urea using cyclic voltammetry as the determining mode is shown in Fig. 16C. The oxidation/reduction peak current increases with increasing scan rate. It is evident that the oxidation peak current has a linear dependence on the square root of scan rate (Fig. 16D) compared to IPversus scan rate. It means that the electrochemical oxidation process is purely diffusion-controlled on the surface of the ZSs@1rGO modified GCE. The lowest detection limit was calculated to be 0.012 μM, and the sensitivity was found to be 682.8 μA mM−1 cm−2. The proposed sensing mechanism for the fabricated urea sensor is described as
| CO(NH2)2 + 8OH− → 6H2O + CO32− + N2 + e− | (2) |
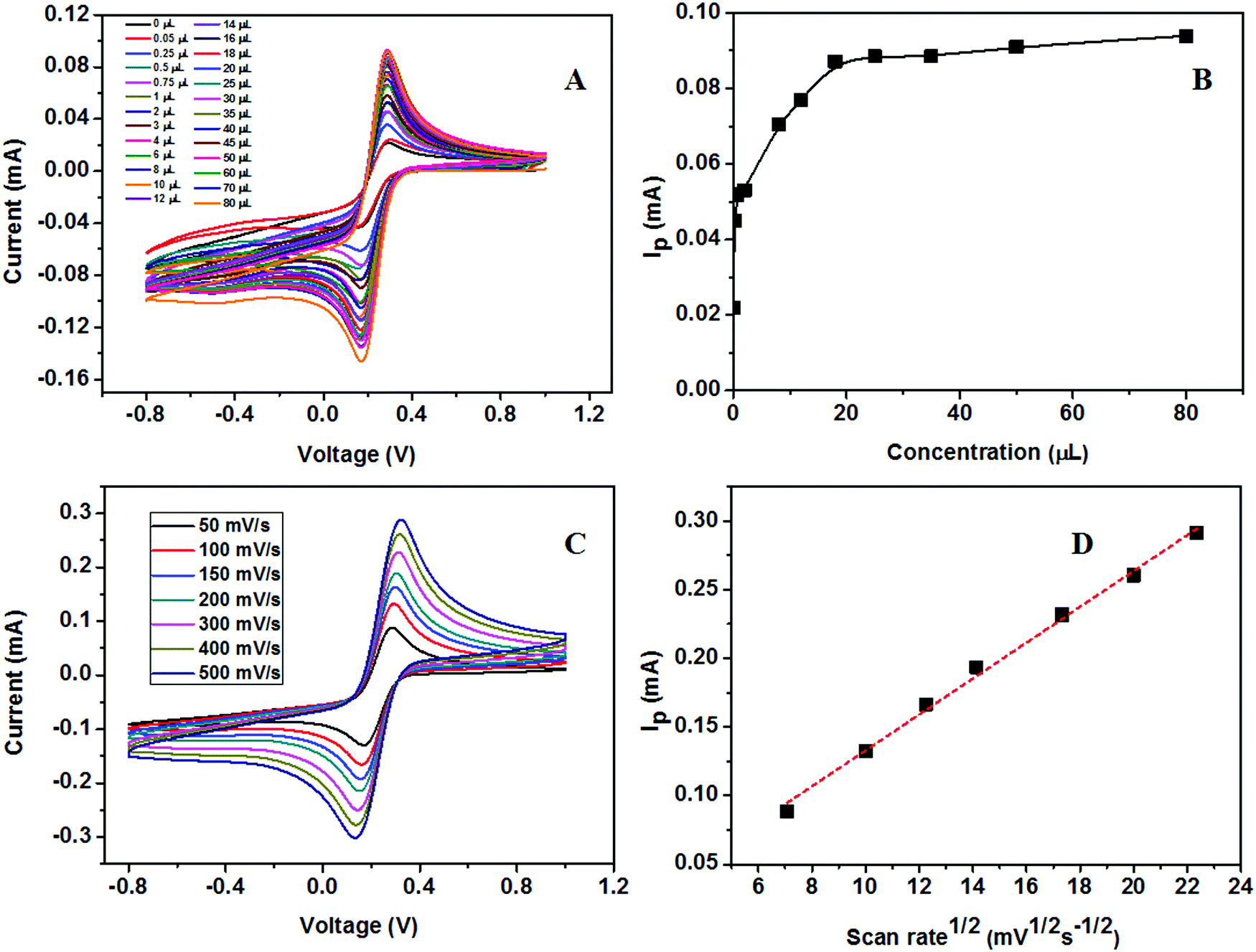 | ||
| Fig. 16 (A) Calibration study of the ZSs@1rGO modified electrode towards 4 mM urea using CV. (B) Plot of IPversus concentration. (C) Scan rate study using the ZSs@1rGO modified electrode with different scan rates. (D) Plot of IPversus square root of scan rate. | ||
 | ||
| Fig. 17 (A) Calibration study of the ZSs@1rGO modified electrode towards 4 mM glucose. (B) Plot of IPversus concentration. (C) Scan rate study using the ZSs@1rGO modified electrode with different scan rates. (D) Plot of IPversus square root of scan rate. | ||
The proposed sensing mechanism for the fabricated glucose sensor is described as
| O2 g (air/liquid interface) ↔ O2 ads (ZnO@rGO) | (3) |
| O2 ads (ZnO) + 2e− (ZnO@rGO) ↔ 2O− ads (O−/O2−) | (4) |
| Glucose + O−glucono- → δ-lactone + 2e− | (5) |
| Glucono-δ-lacone → gluconic acid | (6) |
The present study confirms that the ZnO nanoarchitectures act an efficient electron mediator for the fabrication of efficient nonenzymatic sensor.
The results obtained in the present study are compared with the literature data on similar types of nonenzymatic sensors for detecting urea or glucose. The comparison shown in Table 3 reveals that the fabricated urea and glucose sensor based on the ZSs@1rGO modified GCE is highly effective in the accurate detection of urea and glucose. The developed ZSs@1rGO modified GCE has shown the lowest detection (0.008 μM for glucose and 0.012 μM for urea) and excellent sensitivity (481 mA mM−1 cm−2 for glucose and 682.8 mA mM−1 cm−2 for urea) in the concentration range of 0.02 × 10−3–18 × 10−3 μM for glucose and of 0.02 × 10−3–7.2 × 10−3 μM for urea.
| Electrode material | Analyte | Linear range (mM) | Lowest detection limit (μM) | Sensitivity (mA mM−1 cm−2) | Ref. |
|---|---|---|---|---|---|
| ZnO NRs | Glucose | — | 0.5 | 5.601 | 5 |
| Cu/CuO/ZnO | Glucose | 0.1–1 | 18 | 408 | 28 |
| Ni0.31Co0.69S2/rGO | Glucose | 0.001–5, 5–16 | 0.078 | 1753, 954.7 | 29 |
| Ni(OH)2–rGO | Glucose | 15–30 | 15 | 11.4 | 30 |
| Cu@Cu2O/rGO | Glucose | 0.005–7 | 0.5 | 0.1452 | 31 |
| ZnO/rGO | Glucose | 0–33.5 × 10−3 | 0.0002 | 39.78 | 32 |
| ZSs@1rGO | Glucose | 0.02 × 10−3–18 × 10−3 | 0.008 | 481 | Present work |
| NanoZnO | Urea | 0.6–4.4 | 0.1527 | 0.0259 | 33 |
| NiO | Urea | 0.1–1.1 | 10 | — | 4 |
| ZSs@1rGO | Urea | 0.02 × 10−3–7.2 × 10−3 | 0.012 | 682.8 | Present work |
Interference study of rGO based metal oxides is well established. From the literature it was observed that in the case of CeO2/rGO or AgFe2O3/rGO modified GCEs, the presence of biological interfering species show no significant effect on the peak current.34,35 The reported studies say that a similar observation will be obtained with AA, UA, H2O2 and DA.
4. Conclusions
In the present work, the as-prepared GO was reduced with a nascent hydrogen reduction mechanism using metallic zinc obtained as a waste by-product from industry. The formed rGO was then analyzed with XRD, UV-vis, SEM, and TEM. The peak shift from 10.8 to 24.5° in the XRD pattern and the redshift in the UV-vis absorption spectra from 230 to 267 nm confirm the complete reduction of GO to rGO. In addition to this, ZnO nanoarchitectures with various morphologies were prepared using carbohydrate/cellulose as a bio-template. The as-prepared ZnO nanoarchitectures were used to decorate rGO sheets, which results in a ZnO@rGO nanocomposite. Due to the synergistic effect of ZnO and rGO, the prepared ZnO@rGO nanocomposite was found to be an excellent probe for the nonenzymatic electrochemical sensing of urea and glucose. The electrochemical studies revealed that the fabricated electrode was sensitive to urea in the concentration range of 0.02 × 10−3–7.2 × 10−3 mM with a detection limit of 0.012 μM. The developed nonenzymatic sensor electrode was sensitive to glucose in the concentration range of 0.02 × 10−3–18 × 10−3 mM with a detection limit of 0.008 μM. The developed sensor exhibited an ultra-high sensitivity of 682.8 μA mM−1 cm−2 towards urea and 481 μA mM−1 cm−2 towards glucose.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Director, CSIR-National Institute of Science and technology (NIIST), for providing the laboratory facilities to carry out the experiments. K. B. Babitha is thankful to CSIR, India, for providing the Senior Research Fellowship (SRF). R. B. Rakhi is thankful to DST-SERB for the Ramanujan Fellowship (SB/S2/RJN-098/2015). The authors also thank M/s Binani Zinc Limited for the zinc dust which was used in the reduction of GO. The authors also thank Mr Prithviraj for XRD, Mr Kiran Mohan for TEM, Mrs Soumya and Mr V. Harish Raj for SEM, and Mr George for FTIR analysis. All the members of the Materials Science and Technology Division are acknowledged for providing general support.References
- D. Dutta, S. Chandra, A. K. Swain and D. Bahadur, Anal. Chem., 2014, 86, 5914–5921 CrossRef CAS PubMed.
- R. C. Bianchi, E. R. D. Silva, L. H. Dall'Antonia, F. F. Ferreira and W. A. Alves, Langmuir, 2014, 30, 11464–11473 CrossRef CAS PubMed.
- O. O. Soldatkin, I. S. Kucherenko, S. V. Marchenko, K. B. Ozansoy, B. Akata, A. P. Soldatkin and S. V. Dzyadevych, Mater. Sci. Eng., C, 2014, 42, 155–160 CrossRef CAS PubMed.
- M. Arain, A. Nafady, Sirajuddin, Z. H. Ibupoto, S. T. H. Sherazi, T. Shaikh, H. Khan, A. Alsalme, A. Niaz and M. Willander, RSC Adv., 2016, 6, 39001–39006 RSC.
- G. N. Dar, A. Umar, S. A. Zaidi, S. Baskoutas, S. H. Kim, M. Abaker, A. Al-Hajry and S. A. Al-Sayari, Sci. Adv. Mater., 2011, 3, 901–906 CrossRef CAS.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- V. K. L. Shanbhag and K. S. Prasad, Anal. Methods, 2016, 8, 6255–6259 RSC.
- T. R. Kumar, K. J. Babu, D. J. Yoo, A. R. Kim and G. G. Kumar, RSC Adv., 2015, 5, 41457–41467 RSC.
- J. Jiang, P. Zhang, Y. Liua and H. Luo, Anal. Methods, 2017, 9, 2205–2210 RSC.
- P. T. Yin, T. H. Kim, J. W. Choi and K. B. Lee, Phys. Chem. Chem. Phys., 2013, 15, 12785–12799 RSC.
- Y. Zhao, L. Fan, B. Hong, J. Ren, M. Zhanga, Q. Queb and J. Jib, Sens. Actuators, B, 2016, 231, 800–810 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CrossRef CAS.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- V. H. Pham, H. D. Pham, T. T. Dang, S. H. Hur, E. J. Kim, B. S. Kong, S. Kim and J. S. Chung, J. Mater. Chem., 2012, 22, 10530–10536 RSC.
- O. Akhavan, E. Ghaderi, S. Aghayee, Y. Fereydoonia and A. Talebia, J. Mater. Chem., 2012, 22, 13773–13781 RSC.
- R. S. Dey, S. Hajra, R. K. Sahu, C. R. Raj and M. K. Panigrahic, Chem. Commun., 2012, 48, 1787–1789 RSC.
- M. B. M. Krishna, N. Venkatramaiah, R. Venkatesan and D. Narayana Rao, J. Mater. Chem., 2012, 22, 3059–3068 RSC.
- K. B. Babitha, S. Nishanth Kumar, V. Pooja, B. S. Dileep Kumar, A. Peer Mohamed and S. Ananthakumar, Chem. Eng. J., 2017, 324, 154–167 CrossRef CAS.
- K. B. Jaimy, K. V. Baiju, S. K. Ghosh and K. G. K. Warrier, J. Solid State Chem., 2012, 186, 149–157 CrossRef CAS.
- K. Jayanthi, S. Chawla, A. G. Joshi, Z. H. Khan and R. K. Kotnala, J. Phys. Chem. C, 2010, 114, 18429–18434 CrossRef CAS.
- S. Balanand, K. B. Babitha, M. Jeen Maria, A. A. Peer-Mohamed and S. Ananthakumar, ACS Sustainable Chem. Eng., 2018, 6, 143–154 CrossRef CAS.
- M. S. A. S. Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 CrossRef PubMed.
- T. Lv, L. Pan, X. Liu and Z. Sun, Catal. Sci. Technol., 2012, 2, 2297–2301 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- D. Pradhan and K. T. Leung, Langmuir, 2008, 24, 9707–9716 CrossRef CAS PubMed.
- K. S. Bhat and H. S. Nagaraja, Mater. Res. Innovations, 2019, 1–24 CAS.
- R. M. Shereema, S. R. Nambiar, S. S. Shankar and T. P. Rao, Anal. Methods, 2015, 7, 4912–4918 RSC.
- S. SoYoon, A. Ramadoss, B. Saravanakumar and S. J. Kim, J. Electroanal. Chem., 2014, 717–718, 90–95 CrossRef CAS.
- G. Li, H. Huo and C. Xu, J. Mater. Chem. A, 2015, 3, 4922–4930 RSC.
- P. Subramanian, J. Niedziolka-Jonsson, A. Lesniewski, Q. Wang, M. Li, R. Boukherroub and S. Szunerits, J. Mater. Chem. A, 2014, 2, 5525–5533 RSC.
- H. Huo, C. Guo, G. Li, X. Han and C. Xu, RSC Adv., 2014, 4, 20459–20465 RSC.
- M. Sreejesh, S. Dhanush, F. Rossignol and H. S. Nagaraja, Ceram. Int., 2017, 43, 4895–4903 CrossRef CAS.
- A. Ali, A. A. Ansari, A. Kaushik, P. R. Solanki, A. Barik, M. K. Pandey and B. D. Malhotra, Mater. Lett., 2009, 63, 2473–2475 CrossRef CAS.
- A. A. Ensafi, R. Noroozi, N. Z. Atashbar and B. Rezaei, Sens. Actuators, B, 2017, 245, 980–987 CrossRef CAS.
- N. Zhang and J. Zheng, J. Mater. Sci.: Mater. Electron., 2017, 28, 11209–11216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00445f |
| This journal is © The Royal Society of Chemistry 2020 |