
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning and controlling the morphology of spherical molecularly imprinted polymers
Hongzhi
Lu
a,
Hao
Tian
bc,
Changzheng
Wang
 b and
Shoufang
Xu
*c
b and
Shoufang
Xu
*c
aSchool of Chemistry and Chemical Engineering, Linyi University, Linyi 276005, China
bBeijing Key Laboratory of Functional Materilas for Building Structure and Environment Remediation, Being University of Civil Engineering and Architecture, Beijing, 100044, China
cLaboratory of Functional Polymers, School of Materials Science and Engineering, Linyi University, Linyi 276005, China. E-mail: shfxu1981@163.com
First published on 17th August 2020
Abstract
Molecularly imprinted polymers (MIPs) display specific recognition ability for their template in shape, size and functional monomers. The morphology has an important influence on the binding capacity of MIPs, thus affecting the enrichment efficiency and detection sensitivity. In this critical review, we highlight the morphology design and control of spherical MIPs, mainly focusing on solid sphere, core–shell, hollow and mesoporous MIPs. The methods for preparation of MIPs of different morphologies are summarized, and typical TEM or SEM images are displayed. The influence of morphology on the application of MIPs is highlighted. Finally, we discuss the remaining challenges, and some significant attempts in further improving the morphology of MIPs are also proposed.
 Hongzhi Lu | Hongzhi Lu received his Master's degree from Shandong Normal University in 2007. He is currently a lecturer at the School of Chemistry & Chemical Engineering, Linyi University. His research interests focus on the synthesis and application of functional polymers. |
 Hao Tian | Hao Tian received his BE degree in water supply and drainage from Beijing University of Civil Engineering and Architecture in 2019. He is currently pursuing a Master's degree at the School of Environment and Engineering, Beijing University of Civil Engineering and Architecture under the supervision of Prof. Changzheng Wang and Prof. Shoufang Xu. His research is focused on the preparation of molecularly imprinted polymers (MIPs). |
 Changzheng Wang | Dr Changzheng Wang received his MS degree (2005) from Yunnan Normal University (YNU) and PhD degree (2008) from Beijing Normal University (BNU). Currently, he is a professor in the School of Environment and Engineering of Beijing University of Civil Engineering and Architecture (BUCEA). His research focuses on the synthesis of functional nanomaterials for environmental remediation and their applications in photocatalysis and electrocatalysis. |
 Shoufang Xu | Shoufang Xu received her PhD degree from the Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences in 2012. She is currently a professor at the School of Materials Science and Engineering, Linyi University. She is the academic leader of Youth Innovation team of Shandong province. Her research interests focus on the preparation of molecularly imprinted polymers (MIPs) and the application of MIPs in the separation and analysis of typical organic pollutants. |
1. Introduction
Molecularly imprinted polymers (MIPs) are a class of polymers displaying specific recognition to template molecules. MIPs possess some prominent advantages, like structure predictability, chemical stability, recognition specificity and application universality.1,2 MIPs are prepared by pre-polymerization, polymerization and template removal in three consecutive steps, as shown in Fig. 1.3 When MIPs are placed in a complex sample matrix, the molecules matching the recognition sites can be specifically adsorbed. Due to the high selectivity, MIPs were widely used for sample pretreatment or chemical sensors. According to the polymerization mechanism, MIPs can be divided into organic MIPs and inorganic MIPs. Organic MIPs are prepared by polymerization of vinyl functional monomers such as methacrylic acid (MAA), acrylamide (AAm), 4-vinylpyridine (4-VP) with cross-linkers like ethylene glycol dimethacrylate (EGDMA), trimethylolpropane trimethacrylate (TRIM), or divinylbenzene (DVB) initiated by thermally sensitive or UV light sensitive initiators. Due to its tolerance to reagent purity, flexibility in terms of experimental setups and the wide range of commercially available functional monomers, the free-radical polymerization technique is widely used to prepare organic MIPs. Inorganic MIPs are mainly prepared by condensation of tetraethoxysilane (TEOS) with functional monomers like 3-aminopropyltriethoxysilane (APTES) and 3-isocyanatopropylethoxysilane (IPTS) in alkaline conditions to get sol–gel silica. Inorganic MIPs display higher thermal and chemical stability, and recognition sites can be well maintained because silica exhibits minimal swelling in the presence of solvents. Recently, self-polymerization of dopamine in a weak alkaline aqueous solution has been developed to form MIPs for protein imprinting due to the hydrophilicity and biocompatibility of polydopamine (PDA). The polymerization mechanism of dopamine to form PDA was discussed in a review article.4 Based on the above polymerization methods, MIPs with different morphologies for different applications have been prepared.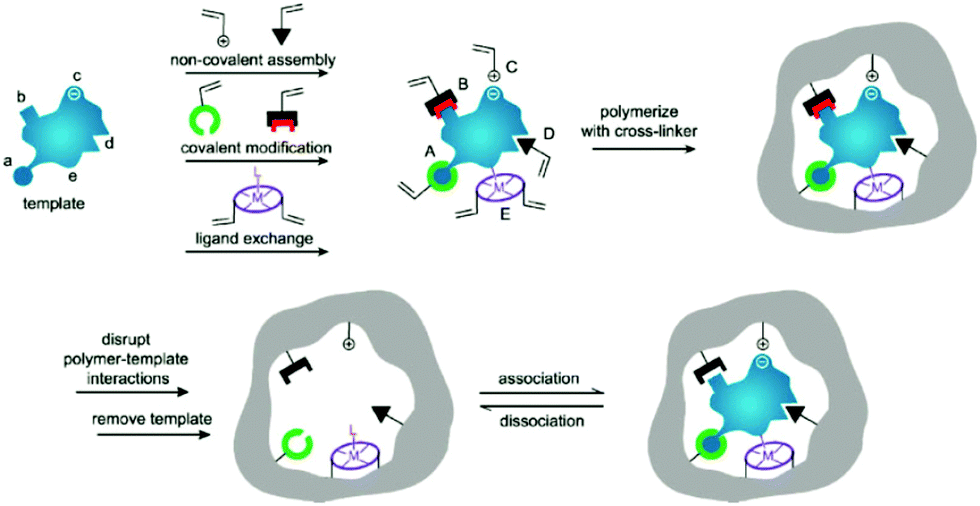 | ||
| Fig. 1 Preparation process of MIPs including pre-polymerization, polymerization, and elution template as three steps. During the pre-polymerization, functional monomers can bond with template by (i) noncovalent, (ii) electrostatic/ionic, (iii) covalent, (iv) semicovalent, and (v) metal centre coordination. Adapted from ref. 3. Copyright 2006 John Wiley and Sons. | ||
In the past three years, a series of excellent reviews focused on the preparation5–7 and application of MIPs8–18 have been published. In those review articles,2,6,7 the brief history of the molecular imprinting technique, the type of molecular imprinting (including noncovalent imprinting, covalent imprinting, semi-covalent imprinting), and the essential elements of molecular imprinting like functional monomer, cross-linker, target template, initiators and porogens have been introduced in detail many times. However, to the best of our knowledge, there are few review articles on the morphology control of MIPs.19–21 The morphology has an important influence on the binding capacity of MIPs, thus affecting the enrichment efficiency and detection sensitivity. Therefore, design and control of morphology are important in the field of molecular imprinting. The morphology of MIPs has evolved from irregular particles to regular spheres, hierarchical structures, and films. As shown in Fig. 2, MIP spheres are changeable and controllable, and widely used in the fields of filling columns, adsorption separation and sensor construction. So in this review, instead of providing a comprehensive review of MIPs, we focus on the preparation of MIPs with controlled spherical morphologies taking a small fraction of the literature, and the effect of morphology on MIP performance is discussed. We hope that this review article will be of guiding significance to researchers engaged in the design of MIPs and bridge the gap between the preparation method and application of MIPs. It should be noted that one type of MIP particles may have multiple characteristic morphologies at the same time, such as core–shell mesoporous MIPs can be classified as core–shell MIPs or mesoporous MIPs. We discuss them under the most representative mesoporous morphologies.
 | ||
| Fig. 2 Typical morphologies of spherical MIPs. | ||
2. Spherical MIPs
Initially, MIPs were simply prepared by bulk polymerization. Monomers, template molecules, cross-linker and a small amount of solvents were polymerized to obtain bulk polymers.22–24 The polymers needed to be ground and sieved to get irregular polymer particles before being used to pack a chromatography column or SPE column. Bulk polymers displayed obvious advantages like simple preparation and high polymer yield. At the same time, there were several obvious shortcomings, like time-consuming post-treatment procedure, low-affinity binding sites, poor site accessibility and low binding capacity stemming from big and irregular particle size. It was necessary to prepare monodisperse spherical MIPs. Inorganic spherical MIPs can be obtained by hydrolysis of TEOS in alkaline condition by the classical Stöber method. The preparation process is simple and repeatable, which is not discussed here. Organic spherical MIPs are discussed in this section.2.1 Spherical MIPs by precipitation polymerization
Spherical MIPs prepared by precipitation polymerization exhibit highly uniform particle size and clean surface because dispersants and surfactants are not needed to stabilize polymer droplets. During precipitation polymerization, monomers, templates and cross-linkers are dispersed in a large amount of solvent (generally, solvent accounts for 95% of the polymerization system). The resulting polymer is insoluble in the solvent and precipitates from the solvent to form regular spherical MIPs. For example, as early as 2003, Row25 prepared caffeine-imprinted uniform spherical MIPs by precipitation polymerization using acetonitrile as the dilute solvent with a concentration of 96%, MAA as the monomer and EGDMA as the crosslinker. The average particle size of MIPs was about 400 nm while the average particle size of spherical NIPs was 800 nm, which indicated that the template could affect the particle size. Subsequently, precipitation polymerization was employed to prepare many spherical MIPs for selective recognition of tetracycline,26 vitamin E,27etc. The key point of this method was the solubility and amount of solvent. Low amount of solvent would result in the adhesion and agglomeration of polymer particles, and it was hard to get monodispersed spherical MIPs. For some hydrophilic templates, modified precipitation polymerization can be adopted.28,29 In this method, the hydrophilic template is first dissolved in a small portion of water, and then MIPs are prepared by precipitation polymerization using acetonitrile or toluene as solvent.Spherical MIPs prepared by precipitation polymerization often display a broad size distribution due to uncontrollable chain transfer and termination reactions of free radical polymerization. To get monodisperse MIPs, living/controlled radical polymerization techniques30–35 were adopted, in which reversible addition–fragmentation chain transfer (RAFT) polymerization was the most employed. RAFT precipitation polymerization (RAFTPP) can be carried out by introducing a suitable chain transfer agent (RAFT agent, normally a thiocarbonylthio or dithiobenzoate compound) into a conventional precipitation polymerization system. In our previous work,30 we prepared atrazine-imprinted polymers by RAFTPP and the resultant RAFT-MIPs displayed uniform spherical shape with a rough surface, leading to an improvement in imprinting efficiency. With this advantage, RAFTPP was employed to prepare spherical MIPs for nicotine,31 glutathione,32etc. Atom transfer radical precipitation polymerization (ATRPP) also was employed for preparation spherical MIPs.33 The monodispersity was improved by living polymerization.
The process of traditional precipitation polymerization often needed 24 h at approximately 60 °C. To shorten the polymerization time, distillation precipitation polymerization (DPP) was developed, in which some of the solvent was distilled off during the polymerization process to accelerate the precipitation of MIPs.36–38 Li37 prepared MIPs by DPP for detection of BPA with a diameter of about 2 μm. Higher reaction temperature used in DPP resulted in decreased particle diameter. However, DPP could cause unstable polymerization because of the loss of solvent. To overcome the limitations, Sellergren39 proposed reflux precipitation polymerization (RPP) at 90 °C with constant volume of the solvent during the polymerization to generate uniform MIPs. After 2 h of reaction at high temperature, the MIPs were quite uniform with a diameter of 2.0 ± 0.2 μm. When comparing precipitation polymerization at 60 °C for 24 h and DPP under reflux condition for 3 h, the yields were comparable.
For the above mentioned several kinds of precipitation polymerization methods, living precipitation polymerization can improve the dispersion of MIPs, but additional polymerization reagents like RAFT agents are needed. DPP and RPP can shorten the polymerization time, but higher polymerization temperature is needed. Furthermore, more complicated reaction devices are needed. So DPP and RPP have not been widely used. Traditional precipitation polymerization is still the most widely used method because of its simple operation. It should be noted that precipitation polymerization needs a large amount of organic solvents, which brings about environmental pollution and increases the cost. The polymer yield is low due to the low concentration of monomers.
2.2 Spherical MIPs by emulsion polymerization
Typically, SPE packing materials prefer particle sizes of several micrometers. The size of MIPs prepared by precipitation polymerization is below 5 μm, which is generally not suitable for filling SPE columns. Suspension polymerization40–42 can be used to prepare spherical MIPs with suitable large size for SPE. Oil-soluble polymerization system was dispersed in the water phase for polymerization. In order to stabilize the polymer droplets, dispersants were added. For example, Lenain40 prepared MIPs by suspension polymerization for SPE procedure. 80% of the MIP particles were situated in the 12–58 μm range with an average diameter of 30 μm. In recent years, suspension polymerization has been less used because the surfactant would behave as an additional pollutant in the preparation process.Pickering emulsion polymerization can be employed to prepare spherical MIPs with diameters of about 100 μM, which are suitable as SPE sorbents. Pickering emulsion is stabilized by solid particles, which are located at the interface between the two immiscible liquids, preventing coalescence of the emulsion droplets. The important step for Pickering emulsion polymerization is to form stable Pickering emulsions. So the choice of nanoparticles is crucial. Hydrophilic particles tend to form oil-in-water emulsions while hydrophobic particles lead to the opposite situation. Until now, silica,43–53 Fe3O4 particles,54–59 chitosan,60–62 halloysite nanotubes,54–56,63 Janus hydroxyapatite NPs,64,65 BHb,66 yeast,67 lignin,68 fluorescent NPs,69etc., were adopted to stabilize Pickering emulsions for preparation of MIPs. Different particles enjoy different advantages. For example, silica nanoparticles are commonly used due to the stability, economy, and easy removal by HF etching. Pickering emulsion based on Fe3O4 particles can fabricate magnetic MIPs, which could be easily collected and separated. Chitosan is suitable as a stabilizer for Pickering emulsion polymerization due to its biocompatibility, environmental friendliness and low cost. Due to high mechanical strength, thermal stability, biocompatibility and abundance, halloysite nanotubes have been used for constructing polymer nanocomposites.
After formation of a stable Pickering emulsion, polymerization can be initiated to form polymers. Generally speaking, the polymerization was carried out in the dispersed phase, and finally spherical polymers were formed.43–51 The solid particles which are stable in the Pickering emulsion can be removed49,50 or remain54,55 on the polymer surface. The template molecules inside the polymer are removed to form MIPs. Generally, MIP microspheres obtained by Pickering emulsion polymerization are spherical with good dispersion, and particle size is generally about tens of microns. Frankly speaking, the polymer particle size is too large, and the recognition sites in the interior are difficult to use. In order to make the recognition sites on the MIP surface, Ye and his group49,50 proposed new Pickering polymerization methods using template molecules bonded to silica nanoparticles as stabilizer. After the silica nanoparticles were removed by HF solution, a permeable layer was obtained. All binding sites were located on the surface of MIPs, which is convenient for fast template binding.
Ye and his group43 also explored the preparation of water-compatible MIPs by oil-in-water Pickering emulsion polymerization using propranolol as a model template, MAA as functional monomer and EGDMA as cross-linker, denoted as MIPs-P. For comparison, MIP particles were also synthesized using traditional bulk polymerization, denoted as MIPs-B. The amount of water adsorbed by MIPs-P was 1.40 times higher than that by MIPs-B, whereas the amount of toluene adsorbed by MIPs-P was only 15.6% of that by MIPs-B. They attributed this phenomenon to the functional monomer of MAA. Because of the solubility of MAA both in the oil and in the water phase, MIPs-P have abundant surface carboxyl groups and therefore a hydrophilic surface. This new surface characteristic allowed the MIPs to recognize the template molecule with high selectivity in pure water. Water-in-oil Pickering emulsion also was adopted to prepare water-compatible MIP hydrogel.53,55 The water phase contained the template and the hydrophilic monomers polymerized to form MIPs. The key step was to obtain a stable pre-polymerization complex, as water may disrupt the interactions between template and monomer molecules through competitive hydrogen bonding. For example, Agrofoglio52 synthesized MIP hydrogel by oil-in-water Pickering emulsion for controlled release of adenosine 5-monophosphate. To obtain a stable pre-polymerization complex, 2-(dimethylamino)ethyl methacrylate and N-isopropylacrylamide were selected as monomers providing ionic interactions with anionic phosphate moiety of 5-monophosphate. From the above examples we can see that the research work concerning MIPs obtained by Pickering emulsion polymerization mainly focused on Ye's group. This may be due to the following reasons. First, a stable Pickering emulsion polymerization system is difficult to control, which requires rich experience. Second, the larger particle size leads to low adsorption capacity, which means that MIPs obtained by Pickering emulsion polymerization do not display obvious advantages in sample pretreatment or sensor construction.
2.3 Spherical MIPs with polymer brush
It is often necessary to detect a target in aqueous media, such as environmental water samples, beverages, and biological fluids. To achieve aqueous media recognition, inorganic MIPs based on the sol–gel process or PDA can be employed. For organic MIPs, surface hydrophilicity modification is necessary. Zhang70–79 proposed a strategy to graft hydrophilic polymer brush on the surface of spherical MIPs. Generally, spherical MIPs were first prepared by living precipitation polymerization (RAFTPP71,73,74 or ARTPP72), then hydrophilic polymer brushes were grown in situ on their surfaces by surface initiated living polymerization. For example, Zhang prepared MIPs with PHEMA brush by ATRPP under ambient temperature with UV light, as shown in Fig. 3A.72Fig. 3Ha shows the obtained spherical MIPs were narrow or monodisperse. It should be noted that from the SEM images, the differences between MIPs with or without polymer brush could not be observed. Grafting hydrophilic polymer brushes has been widely used in many situations, such as grafting hydrophilic polymer brushes on the surface of fluorescent MIPs (Fig. 3B),76,78 grafting hydrophilic temperature-responsive PNIPAAm brushes on the surface of photo-responsive MIPs to produce multi-responsive MIPs (Fig. 3C),71,75in situ growth of magnetic microspheres on hydrophilic polymer brushes to prepare monodispersed hydrophilic and magnetic MIPs (Fig. 3D) (the TEM image shown in Fig. 3Hb confirmed the successful anchor of magnetic microspheres),74 or the preparation of biological sample-compatible MIPs by combining RAFT polymerization and thiol–epoxy coupling chemistry (Fig. 3E).79 In our previous work,80 water-compatible MIPs with PHEMA brush were prepared and employed as sorbent for extraction of four triazines from aqueous media. Surface grafting of polymer brush is a general approach for hydrophilic modification of MIPs.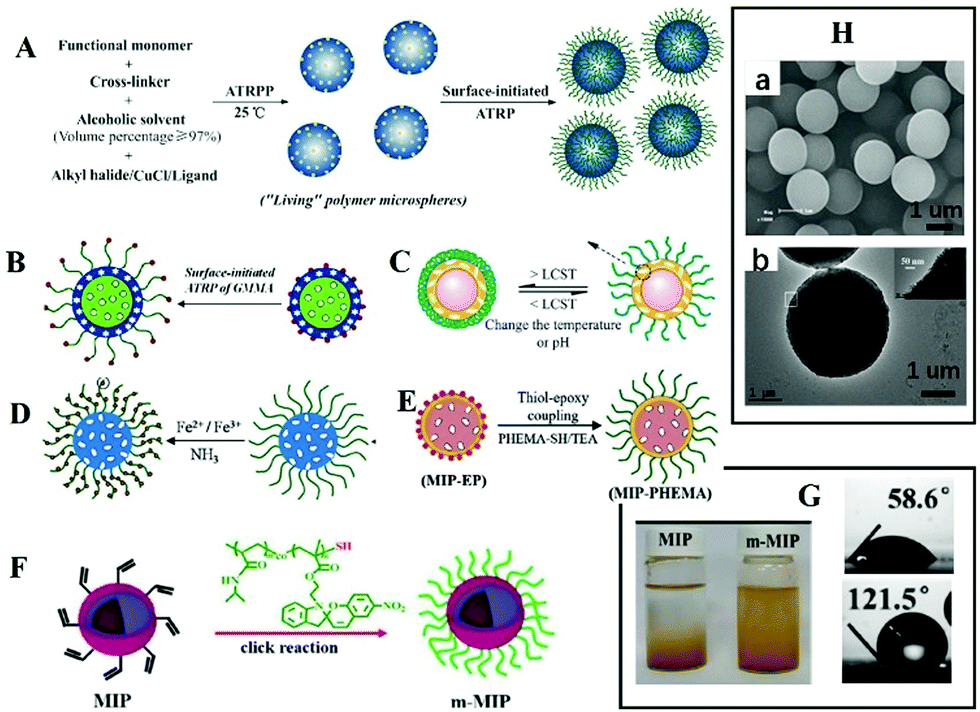 | ||
| Fig. 3 Representation of MIPs with polymer brush. (A) A scheme for preparation of spherical MIPs with polymer brushes by living polymerization, adapted from ref. 72, Copyright 2012. (B) Fluorescent MIPs with brush, adapted from ref. 77, Copyright 2016 American Chemical Society. (C) Multi-responsive MIPs, adapted from ref. 71, Copyright 2012. (D) Magnetic MIPs with brush, adapted from ref. 74, Copyright 2014. (E) Preparation of MIPs with brush by thiol epoxy coupling, adapted from ref. 79, Copyright 2019. (F) Polymer brush by click reaction, adapted from ref. 81, Copyright 2020 American Chemical Society. (H) Images of MIPs with polymer brush ((a) adapted from ref. 78, Copyright 2012; and (b) adapted from ref. 74, Copyright 2014). (G) The water contact angle and the dispersity of MIPs in water, adapted from ref. 81, Copyright 2020 American Chemical Society. | ||
Polymer brushes also can be grafted to MIPs by click reaction. Wang's group81 prepared magnetic core–shell MIPs, then grafted them with macromolecular chains by a “click reaction” to obtain hydrophilic MIPs. The obtained MIPs displayed excellent binding capacity and selectivity in aqueous solution (Fig. 3F). The static water contact angle decreased from 121.5° to 58.6°, and the modified MIPs could be dispersed in water, as shown in Fig. 3G. Polymer brushes “grafted from” the surface of MIPs can be completed by a one-pot method, which is simple. Grafting polymer brushes to the surface of MIPs requires three independent steps involving the preparation of polymer brushes and MIPs, which is relatively cumbersome. However, the structure of polymer brushes can be changeable.
2.4 Restricted access material MIPs (RAM-MIPs)
Macromolecules existing in complicated sample matrices would adsorb on the surface of MIPs and hinder the specific recognition of targets. Restricted access materials (RAM) are a kind of sorbent containing hydrophilic functional groups and/or proteins on their external surface, which could exclude macromolecules. As shown in Fig. 4A, there are three kinds of RAM, namely internal surface phase, semipermeable surface phase and MIP RAM, and macromolecules could be excluded by a physical or chemical diffusion barrier. The physical diffusion barrier is based on the size of the pore and the chemical diffusion barrier depends on functional groups with selective retention.82 Combining these two types of RAM with MIPs, RAM-MIPs could effectively avoid the interference of macromolecules in purification or detection. Modification of hydrophilic hydroxyl groups on MIP surfaces is a common method to prepare RAM-MIPs, and glycidyl methacrylate (GMA) or KH-560 containing active epoxy groups is a commonly used copolymerization functional monomer.83–88 When the epoxy groups were hydrolyzed to produce a large amount of hydroxyl groups, the surface of MIPs were hydrophilic and the interference of biological macromolecules could be eliminated, as shown in Fig. 4B. For example, Gong prepared RAM-M-MIPs using GMA as co-monomer for magnetic-solid phase extraction (MSPE) of tetracyclines from untreated milk and egg samples.88 The obtained RAM-M-MIPs can effectively eliminate the interference of protein macromolecules with an exclusion rate of 99.4%.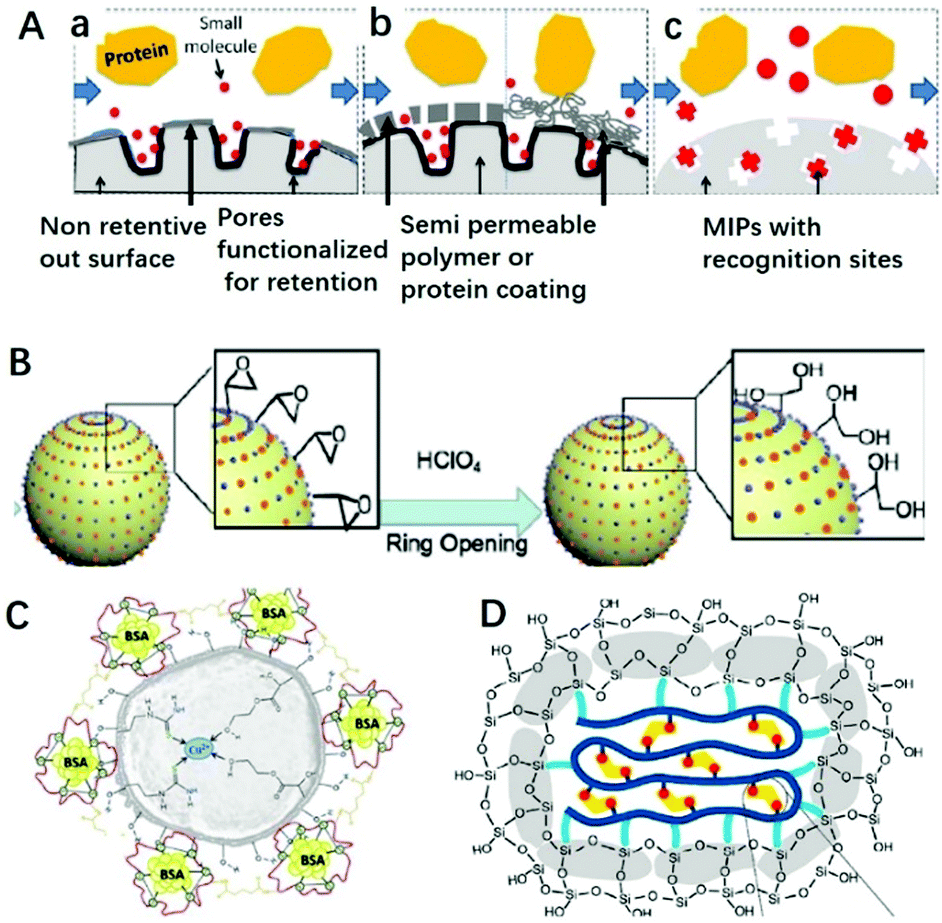 | ||
| Fig. 4 Different classifications of RAM (A) include (a) internal surface, (b) semi-permeable surface (including both polymeric and protein-based hybrids), and (c) MIP phases, adapted from ref. 82, Copyright 2013 John Wiley and Sons. (B) RAM based on GMA ring opening, adapted from ref. 85, Copyright 2020 Elsevier. (C) RAM based on protein BSA, adapted from ref. 89, Copyright 2018 Elsevier. (D) RAM based on SiO2 layer, adapted from ref. 93, Copyright 2019 American Chemical Society. | ||
A protein like bovine serum albumin (BSA) also can be coated onto MIPs using glutaraldehyde as a cross-linker to prepare RAM-MIPs-BSA. Due to the electrostatic repulsion between the proteins in samples and the BSA layer fixed on the MIP surface (both negatively charged), 99% of the proteins from samples could be eliminated.89–92 For example, BSA was anchored on the surface of Cu(II) imprinted MIPs in order to provides better performance towards application in aqueous medium,89 as shown in Fig. 4C. In order to anchor BSA, HEMA was used as co-functional monomer to provide hydroxyl groups. When comparing IIPs, IIPs-HEMA, IIPs-HEMA-BSA was the most efficient polymer for simultaneous Cu2+ preconcentration and BSA protein exclusion in aqueous medium. Gong90 prepared core–shell RAM-MIPs-BSA for SPE of ofloxacin and enrofloxacin from bovine serum. The maximum adsorption capacity of RAM-MIPs-BSA was 50.55 mg g−1 for ofloxacin, with a 99.4% protein exclusion rate.
Chang93 prepared SiO2-coated MIPs for the adsorption of BPA, as shown in Fig. 4D. The anchored SiO2 clusters confined the conformation of the copolymer, suppressed the nonspecific hydrophobic interactions and enhanced the water compatibility of MIPs. With the help of the SiO2 layer, the hydrophobic MIP particles could well disperse in aqueous solution. Furthermore, nonspecific hydrophobic interaction was reduced resulting in high adsorption kinetics and an outstanding imprinting effect, including an imprinting factor of 14, adsorption capacity of 40.3 mg g−1, and selectivity factor of 24.
3. Core–shell structured MIPs
The recognition sites in irregular or solid spherical MIPs are difficult to utilize due to the high mass transfer resistance. In order to reduce the resistance of mass transfer and improve the utilization rate of recognition sites, surface imprinting was developed to prepare core–shell structure MIPs. Surface imprinting usually involves the following three steps: first, preparation of a supporting matrix, then surface modification of the supporting matrix, and finally coating an organic or inorganic MIP layer on the surface of the supporting matrix. Surface modification is the key step for preparation of core–shell MIPs. The diverse surface modification methods are emphasized in the following section. The core–shell structure MIPs can be divided into solid core with solid shell, solid core with mesoporous shell, hollow core with solid shell, hollow core with mesoporous shell etc. In this section, only MIPs with solid core and solid shell structure are discussed, and the others will be discussed with hollow MIPs or mesoporous MIPs.3.1 The cores for core–shell MIPs
Many solid particles, like SiO2 nanoparticles,94–97 mesoporous SiO2,98–100 chitosan,101 metal–organic frameworks (MOFs),102–107 PtPd nanoflowers,108 TiO2,109,110 polymers like PS111 and PMMA,112 fluorescent nanoparticles (FNPs),113–115 Fe3O4 magnetic nanoparticles (MNPs),116–149etc., have been used as a supporting matrix. Silica nanoparticles are widely used to prepare core–shell structure MIPs because of their simple preparation and good stability. Mesoporous silica, like FDU-1298 and SBA-15,100 with large and ordered pore sizes, higher surface area and well-modified surface properties, also has been widely used. MNPs also have been widely used due to their magnetic separation ability. The advantages of hydrophilicity, biocompatibility and non-toxicity make chitosan suitable for imprinting. MOFs such as MOF-5,103 UiO-66-NH2,104 MOF-177,105 and MIL-101106 with tunable pore structure are often employed to prepare MOF@MIPs for separation, biosensors and drug delivery. Using fluorescent nanoparticles such as quantum dots (QDs)113 and carbon dots (CDs)115 as cores to prepare core–shell structured MIPs can produce fluorescence sensors. The core–shell MIPs using TiO2 as core can achieve highly efficient photocatalytic degradation of target pollutants under visible light irradiation. According to the required application, the appropriate core materials can be selected.3.2 Surface modification of cores
The key point for the preparation of core–shell MIPs lies in the growth of the MIP layer on the surface of the solid core, rather than nucleation in solution. Therefore, surface modification is necessary. Taking magnetic microspheres as an example, several surface modification methods are summarized in Fig. 5. Before introducing the surface modification methods of MNPs, it is necessary to introduce the preparation methods of MNPs. Fe3O4 MNPs can be prepared by the coprecipitation method116,117 or the solvothermal method.124–128 The MNPs prepared by coprecipitation were hydrophilic with size of about 10 nm and relatively weak magnetic properties. The MNPs prepared by the solvothermal method were about 500 nm with stronger magnetic property.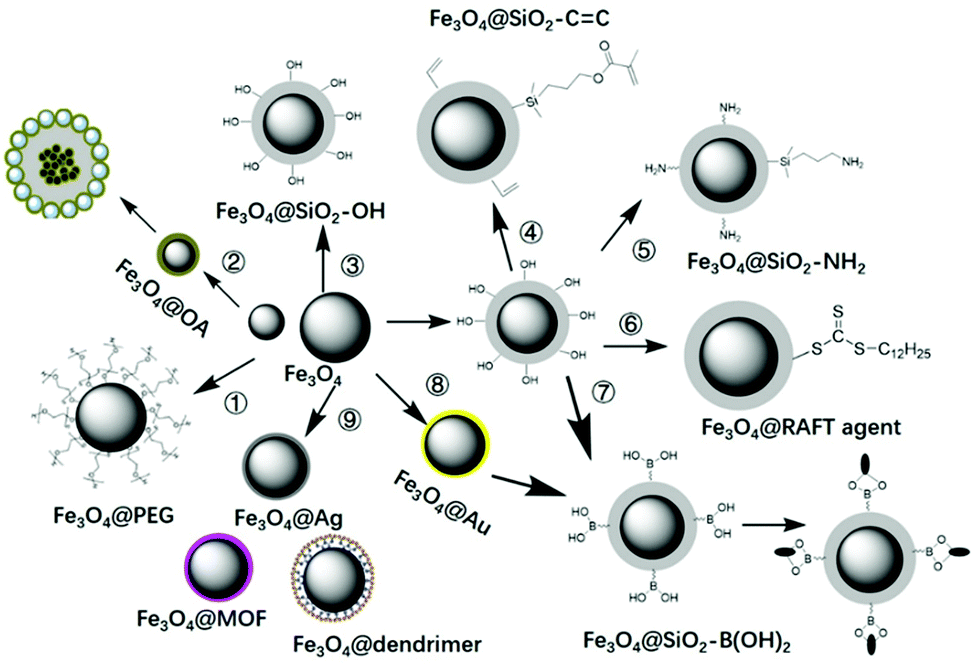 | ||
| Fig. 5 Surface modification of MNPs with PEG, OA, TEOS, vinyl double bond, amino groups, RAFT agents, boronic acid, gold or silver NPs, MOF, etc. | ||
In order to transform hydrophilic MNPs into hydrophobic MNPs, they were modified with polyethylene glycol (PEG)116,117 or oleic acid (OA),118–122 then were coated into MIPs by suspension polymerization118,122 or Pickering emulsion polymerization.60 For this kind of magnetic MIPs (M-MIPs), serval magnetic microspheres were coated into one MIP particle, as multiple core in one shell. The third was to introduce a thin SiO2 layer on the surface of the MNPs, and then an inorganic MIP layer was coated by the sol–gel process.123–125 The formed MIP layer may be a solid SiO2 layer123 or mesoporous SiO2 layer.125 The fourth was to coat a thin SiO2 layer and then introduce vinyl double bond on the surface of the MNPs, followed by coating an organic MIP layer by free radical polymerization.126–137 In order to fix more functional groups, the SiO2 layer on the surface of MNPs can be designed as a mesoporous silica layer.126–128 In order to make the recognition sites evenly distributed in the shell, the template molecules were fixed on the surface of cores. So amino groups138–142 or boronic acid137–150 were introduced into the surface of MNPs through APTES or phenylboronic acid, then templates were immobilized on the surface of MNPs, followed by the imprinted layer coating on the surface of magnetic microspheres. In order to facilitate the coating of the MIP layer on the core surface and effectively controlling the thickness of the shell, RAFT agents143–146 were introduced on the surface of MNPs, and the shell MIPs with controllable thickness were obtained by living polymerization. The above are general surface modification methods, which also can be used for surface modification of other solid particles.
In order to meet the requirements of different application fields, MNPs can be modified by other functional groups. For example, Zhang151 modified MNPs with poly(glycidyl methacrylate) for introduce boron–nitrogen (B–N) coordination, then further immobilized the template glycoproteins (ovalbumin, OVA). Finally, a redox-polymerized aniline imprinted layer was deposited onto the MNP surface. Xie152 used TiO2-functionalized MNPs as core to prepare M-MIPs. First, a TiO2 layer was deposited directly on the surface of the MNPs using a sol–gel method for immobilizing template phenylphosphonic acid by Lewis acid–base interaction. The noble metal gold (Au) was used as a coating material to prepare M-MIPs to allow immobilizing the template with other useful species based on the Au–S bond. For example, Ma et al.149 coated an Au layer on the surface of MNPs in order to anchor 4-mercaptophenylboronic acid, then with further immobilization of glycoprotein before the imprinting process. To achieve surface-enhanced Raman spectroscopy detection of target, silver NPs were employed.153,154 Yu and co-workers coated Ag NPs on the surface of MNPs, and then coated MIPs on the surface of Fe3O4@Ag.154 In this work, MNPs were prepared by a solvothermal approach, and functionalized with amino groups. Then Ag NPs were located on the Fe3O4@SiO2–NH2 surface via electrostatic interaction. Vinyl double bonds were modified on Fe3O4@Ag to facilitate imprinting by free radical polymerization. Zhou first coated MOF ZIF-8 on the surface of MNPs, then prepared Fe3O4@ZIF-8@MIPs for selective adsorption of tetrabromobisphenol A from water samples.155 The MOF layer provided a large specific surface area and abundant pores, enabling a better adsorption effect.
The properties and morphology of M-MIPs can be affected by surface modification. He's group156 modified MNPs with APTES and KH-570 to get Fe3O4–NH2 and Fe3O4–CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2H4, and then prepared M-MIPs. Fe3O4–CH2C2H4@MIPs exhibited more regular shapes and better dispersibility compared to Fe3O4–NH2@MIPs. The saturation magnetization of Fe3O4–NH2@MIPs (23.25 emu g−1) was higher than that of Fe3O4–CH2C2H4@MIPs (18.63 emu g−1), which may be attributed the poor dispersibility of Fe3O4–NH2. Therefore, it is very important to choose a suitable surface modifier. Armenta and co-workers157 discussed the effect of the MNP modifier used on the extraction performance of M-MIPs. They employed PEG and 3-(trimethoxysilyl)propyl methacrylate(V) to modify MNPs prepared by a co-precipitation method, then coated MIPs by bulk polymerization. The resulting V-MNPs were less prone to aggregation than PEG-MNPs and more V-MNPs were embedded into MIPs due to the presence of vinyl groups, which were able to participate in free radical copolymerization with other monomers. In the same way, the choice of MNPs also affects the morphology of M-MIPs. MNPs prepared by a coprecipitation method are easy to agglomerate, and the prepared M-MIPs prefer to be agglomerated. MNPs prepared by a solvothermal method have large particle size and good dispersion, and the prepared M-MIPs also have good dispersion, and core–shell structure can be easily observed.
C2H4, and then prepared M-MIPs. Fe3O4–CH2C2H4@MIPs exhibited more regular shapes and better dispersibility compared to Fe3O4–NH2@MIPs. The saturation magnetization of Fe3O4–NH2@MIPs (23.25 emu g−1) was higher than that of Fe3O4–CH2C2H4@MIPs (18.63 emu g−1), which may be attributed the poor dispersibility of Fe3O4–NH2. Therefore, it is very important to choose a suitable surface modifier. Armenta and co-workers157 discussed the effect of the MNP modifier used on the extraction performance of M-MIPs. They employed PEG and 3-(trimethoxysilyl)propyl methacrylate(V) to modify MNPs prepared by a co-precipitation method, then coated MIPs by bulk polymerization. The resulting V-MNPs were less prone to aggregation than PEG-MNPs and more V-MNPs were embedded into MIPs due to the presence of vinyl groups, which were able to participate in free radical copolymerization with other monomers. In the same way, the choice of MNPs also affects the morphology of M-MIPs. MNPs prepared by a coprecipitation method are easy to agglomerate, and the prepared M-MIPs prefer to be agglomerated. MNPs prepared by a solvothermal method have large particle size and good dispersion, and the prepared M-MIPs also have good dispersion, and core–shell structure can be easily observed.
The surface imprinting process after surface modification is still based on free radical polymerization, sol–gel process or self-polymerization of dopamine. Those methods are relatively well known, so the process is not repeated here. It should be pointed out that the adhesion between core–shell MIP particles is a common phenomenon, so the preparation of monodisperse ultra-thin shells is a development direction in the future.
4. Hollow MIPs (H-MIPs)
A core–shell structure can improve the utilization of recognition sites of MIPs, but a target molecule can only enter the recognition site from the outer surface. If MIPs with a hollow structure (H-MIPs) were to be prepared, the target molecules can enter the recognition site from the inner and outer double surfaces, which is more efficient. Therefore, preparation of H-MIPs has become an important research direction in this field. As shown in Fig. 6, H-MIPs can be prepared by the sacrificial solid cores method, seed swelling polymerization, Pickering emulsion polymerization, and surface imprinting on hollow core.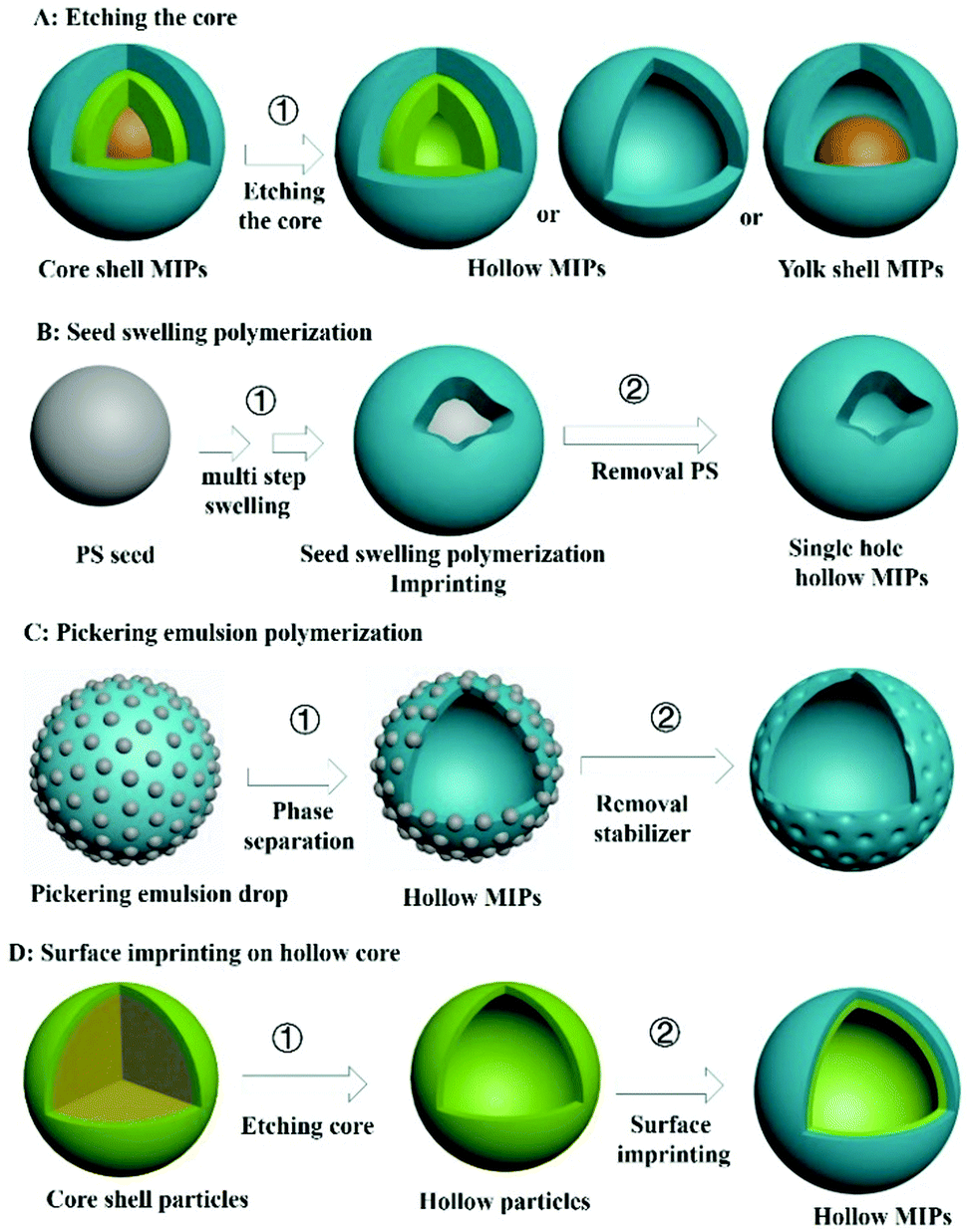 | ||
| Fig. 6 The methods for preparation of hollow MIPs. | ||
4.1 Sacrificial solid cores
Inspired by core–shell MIPs, H-MIPs can be obtained by removing core materials. In this method, the step of removing cores is added on the basis of preparing core–shell MIPs, and selective etching is used to remove cores. Usually, PS,158,159 SiO2,160–169 mesoporous silica,170–173 K2Ti4O9,174 CaCO3175 and Fe3O4@SiO2176–179 are used as the sacrificial material to prepare H-MIPs. This method is widely used because both organic and inorganic H-MIPs can be prepared. SiO2 is commonly used as the sacrificial solid core because of its chemical stability and easy removal by HF etching. Fig. 7A and C show TEM and SEM images of H-MIPs after removal of SiO2 core. The shell is well preserved. When using PS as the core, the hollow MIP shell often collapses because the dissolution and removal of the PS core are accompanied by its swelling. In order to get an intact shell, the MIP shell has to be thick. He158 proposed an approach to prepare H-MIPs with thin and solid MIP shells. PS@SiO2 particles were used as the core, and only the PS part was sacrificed. The SiO2 part kept in the H-MIPs made it possible to get a thin but solid MIP shell. The imprinted shell was estimated to be about 50 nm. As shown in Fig. 7B, the H-MIPs were rigid and kept a perfect spherical shape without breakage and deformation. Fe3O4@SiO2 also was used as a sacrificial support to prepare yolk–shell magnetic hollow MIPs by etching the SiO2 layer with HF to form Fe3O4@void@MIPs, as shown in Fig. 7D.176 The yolk–shell structure is of great interest because it retains the magnetic separation ability and has hollow characteristics like high surface area, suitable pore size and low density. The exposed inner surface can overcome the defects caused by the high cross-linking of the molecular imprinting. | ||
| Fig. 7 Images of different H-MIPs. (A) TEM image of H-MIPs by etching SiO2 core, adapted from ref. 161, Copyright 2017. (B) TEM image of H-MIPs with thin shell by etching PS core with the protection of SiO2 layer, adapted from ref. 158, Copyright 2014. (C) SEM image of H-MIPs by etching mesoporous SiO2 core, adapted from ref. 171, Copyright 2012. (D) TEM image of Fe3O4@void@MIPs by etching SiO2 layer, adapted from ref. 176, Copyright 2018, Elsevier. TEM (E) and SEM (F) images of H-MIPs prepared by seed swelling polymerization, adapted from ref. 180, Copyright 2011. (G) SEM image of H-MIPs prepared by Pickering emulsion polymerization, adapted from ref. 58, Copyright 2013, American Chemical Society. (H) SEM image of multi-hollow MIPs prepared by oil-in-water Pickering emulsion polymerization, adapted from ref. 185, Copyright 2015, John Wiley and Sons. (I) W-o-w Pickering emulsion polymerization, adapted from ref. 59, Copyright 2015 Elsevier. (J) SEM image of multi-core H-MIPs prepared by oil-in-water Pickering emulsion polymerization, adapted from ref. 51, Copyright 2018, Elsevier. TEM images of hollow SnO2 (K) and hollow SnO2 based H-MIPs (L), adapted from ref. 186, Copyright 2018, Elsevier. | ||
4.2 Seed swelling polymerization
H-MIPs also can be prepared by seed swelling polymerization,180–183 which was pioneered by our group in 2011.180 Monodispersed PS seed particles were first prepared by dispersion polymerization. Then PS was swelled, followed by polymerization on the surface of PS. Finally, the PS was washed to form the hollow polymers. In a typical multistep seed swelling polymerization process, PS particles were first swelled by dibutyl phthalate for 24 h, followed by the second step of swelling by toluene, functional monomer, crosslinker, and template for another 24 h. After the two swelling steps, the mixture was polymerized to get the single-hole hollow polymers, as shown in Fig. 7E and F. The formation mechanism of holes was based on the concomitant microphase separation and symmetrical volume shrinkage of shell materials, which can be broken down into five consecutive steps: (i) the formation of a prepolymer layer, (ii) the microphase separation of two polymer shell layers, (iii) the development of a small interior void in the shell, (iv) formation of a film-covered hole, and (v) formation of an open hole in the polymer shells. Using this seed swelling polymerization method, H-MIPs have been prepared for selective recognition of N-nitrosamine,181 triazines,182 and tetracycline.183Multistep swelling before polymerization is complex and time-consuming. Li184 proposed miniemulsion polymerization to prepare single-hole H-MIPs for recognition of BPA. MAA was chosen as the hydrophilic functional monomer and EGDMA as the cross-linker. In the swelling process, the oil phase, including template, functional monomer, crosslinker and dibutyl phthalate, was added dropwise into the water phase composed of sodium lauryl diphenyl ether disulfonate under vigorous stirring to form monomer miniemulsion. The monomer miniemulsion was added dropwise to dispersion liquid of PS seed, followed by 12 h of swelling. During the polymerization process, an eccentric sphere structure with internal void was formed firstly, the surface pores and internal pores were increased by eluting PS seeds, and finally single-pore H-MIP particles were formed. The swelling process was observed by dynamic light scattering. The particle size of mono-disperse PS seed prepared by emulsion polymerization was 145 nm and the droplet sizes of monomer miniemulsion were 287 nm. The size of PS seed after the miniemulsion swelling increased to 275 nm. After polymerization, the size of the product was 269 nm, which was a little smaller than the swelled seed due to the volume shrinkage during the polymerization. These microcapsules collapse into “bowl-like” shape with relatively low modulus of the shell polymer. The average thickness of the shell layer was about 28 nm.
4.3 Pickering emulsion
The third method for preparing H-MIPs is Pickering emulsion polymerization. In Section 2 we discussed that solid-sphere MIPs could be prepared by Pickering emulsion polymerization. More solvent is used to prepare H-MIPs than solid-sphere MIPs, and the hollow structure stems from phase separation. Furthermore, the shell of the H-MIPs is semipermeable, arising from the particle interstices, and the permeability can be modulated by changing the particle size and morphology. For example, Pan59 prepared M-H-MIPs for adsorption of l-cyhalothrin using Pickering emulsion polymerization stabilized by halloysite nanotubes and a few hydrophilic Fe3O4 nanoparticles. The Pickering emulsion polymerization was carried out between St and functional monomer 4-VP in the presence of toluene solvent. The cross-linked polymer network tended towards phase separation in the poor solvent, toluene, driven by the interfacial tension and precipitation at the interface. By elution with water and ethanol, hollow structures were readily formed by the removal of the residual toluene and unreacted 4-VP. The hollow structure can be observed and confirmed by optical micrographs of the MIPs. The emulsion droplets exhibited a spherical hollow structure without collapse, and the hollow structures existed in the MIPs after elution with asymmetric and nonuniform capsule shells with a thickness range of 7.0–20 μm. Because solid particles were used as stabilizer, the surface of H-MIPs was rough and those solid particles can be observed under high-resolution SEM, as shown in Fig. 7G.Using a water in oil in water (w1/o/w2) Pickering emulsion, multi-hollow MIPs also can be prepared. For example, Pan and co-workers62 synthesized multi-hollow M-MIPs using w1/o/w2 Pickering emulsion. The inner water in oil (w1/o) droplets were stabilized by hydrophobic Fe3O4 NPs, and the outer oil in water (o/w2) phase was stabilized by cellulose nanocrystals. The template and functional monomer were contained in the oil phase. After polymerization in the oil phase, multi-hollow holes stemming from the inner water phase were distributed uniformly inside the MIPs, as shown in Fig. 7I. Feng185 prepared multi-hollow M-MIPs by incorporating 3-indolebutyric acid and ferroferric oxide nanoparticles simultaneously into a poly(styrene-co-methacrylic acid) copolymer (poly(St-co-MAA)) matrix for the selective enrichment of indolebutyric acid. Firstly, MNPs and poly(St-co-MAA) were each prepared. Then MNPs, template 3-indolebutyric acid, and poly(St-co-MAA) were added into chloroform solvent. And then the mixture was transferred into 10 mL NaOH solution under vigorous stirring at room temperature. After 1 h, the chloroform was removed by heating under stirring. After cooling to room temperature, the products were collected, purified and dried. Lots of pores with sizes of 0.2–4 μm can be observed on the surface of the obtained MIPs with size around 10 μm, as shown in Fig. 7H. However, the preparation and hole formation mechanism was not discussed in that work. The multi-hollow holes may stem from the removal of chloroform.
Using Pickering emulsion polymerization, Li51 prepared H-MIPs with multiple cores for recognition of BPA, as displayed in Fig. 7J. The formation of the hollow structure was attributed to the polymerization-related phase separation and interfacial tensions. The formation of multicore structure could be due to BPA precipitation nucleation. In this polymerization system, 4-VP and DVB were functional monomer and crosslinker, respectively, while hexadecane was employed as solvent. Because of the poor solubility of BPA in hexadecane, BPA precipitated from the oil phase, and played the part of nucleating agent in the phase separation process of the propagating copolymers. The gradual deposition of polymer on the nuclei finally resulted in the formation of multicore structured hollow spheres. This method is not universal because it has high requirements for the solubility of template molecules and solvents.
4.4 Surface imprinting on hollow cores
H-MIPs also can be prepared by surface imprinting on the hollow cores, like hollow SnO2,186 hollow mesoporous silica,187,188 hollow Fe3O4,130 hollow Ag NPs,189etc. For the sacrificial solid cores method, the recognition sites would be destroyed in the long-term etching process. This process can be avoided by using surface imprinting on the hollow cores. For example, Tan130 prepared M-H-MIPs by wrapping MIP layer on the surface of hollow magnetic Fe3O4, which was prepared via a one-pot hydrothermal method. When compared with solid Fe3O4 microspheres, hollow Fe3O4 displays many advantages, like uniform particle size, lower density, higher specific surface area, and excellent dispersibility in aqueous phase, making it well dispersed in the next polymer-coating processes. Jia184 prepared H-MIPs by a surface imprinting method using hollow SnO2 as support for selective recognition and separation of luteolin. First SiO2@SnO2 was prepared, and the SiO2 was etched to form hollow SnO2 (Fig. 7K). Then H-MIPs (Fig. 7L) were prepared by living ATRP on the surface of hollow SnO2. Yu185 prepared H-MIPs by surface imprinting on hollow silica mesoporous microspheres. First hollow silica mesoporous microspheres were prepared and modified with APTES and acryloyl chloride. Then MIP layer was coated on the surface of the hollow silica mesoporous microspheres using AA as functional monomer, EGDMA as crosslinker and tetradifon as template. After the original hollow silica mesoporous microspheres were removed by HF, the shell morphology still remained intact.Among the four kinds of method, the method of etching cores is widely used because both organic and inorganic H-MIPs could be prepared and the particle size could be adjusted by adjusting the size of cores. Surface imprinting on hollow cores is similar to the etching of cores. Big hollow MIPs with particle size suitable for SPE could be prepared by the seed swelling polymerization method. However, hollow MIPs with thin shell are hard to control. Moreover, multi-swelling is time consuming. Only few H-MIPs have been prepared by Pickering emulsion polymerization, maybe because of the big particle size and relatively low binding capacity.
4.5 M-H-MIPs
M-MIPs often use MNPs as core, and hollow MIPs often sacrifice the cores. Fortunately, magnetic hollow MIPs (M-H-MIPs) can be prepared by in situ growth on the surface of H-MIPs.190–192 In our previous work,190 we prepared M-H-MIPs for selective enrichment of atrazine by in situ growth of MNPs on the surface of H-MIPs. First, H-MIPs were prepared by multi-step swelling polymerization (which was discussed in Section 4.2). To facilitate the growth of MNPs on the surface of H-MIPs, GMA was used as a co-functional monomer, which offers potential hydroxyl groups on the surface of H-MIPs for in situ growth of Fe3O4 NPs. Epoxy bond opening provided abundant hydroxyl groups on the surface of H-MIPs, on which Fe3O4 was prior grown. Then, under base medium in a N2 atmosphere, Fe3O4 was grown in situ based on the cooperation of Fe3+ and Fe2+. Chen191 prepared M-H-MIPs for specific extraction of protocatechuic acid using a similar method. The difference is that H-MIPs were prepared using mesoporous silica spheres (MCM-48) as sacrificial support. From the above reference we can see that to achieve successful in situ growth of Fe3O4 NPs, GMA was necessary as co-functional monomer because Fe3O4 NPs can be chemisorbed onto PGMA by forming five-membered chelate rings with the Fe atoms.1925. Mesoporous MIPs
Mesoporous MIPs were developed from mesoporous silica. Mesopores have diameters between 2 and 50 nm according to the IUPAC classification. Due to the large specific surface area and high adsorption capacity of mesoporous MIPs, they are widely used in separation, adsorption and chemical sensors. Mesoporous MIPs can be divided into homogeneous mesoporous, core–shell mesoporous, hollow mesoporous and multi-level mesoporous MIPs depending on the morphology.5.1 Homogeneous mesoporous MIPs
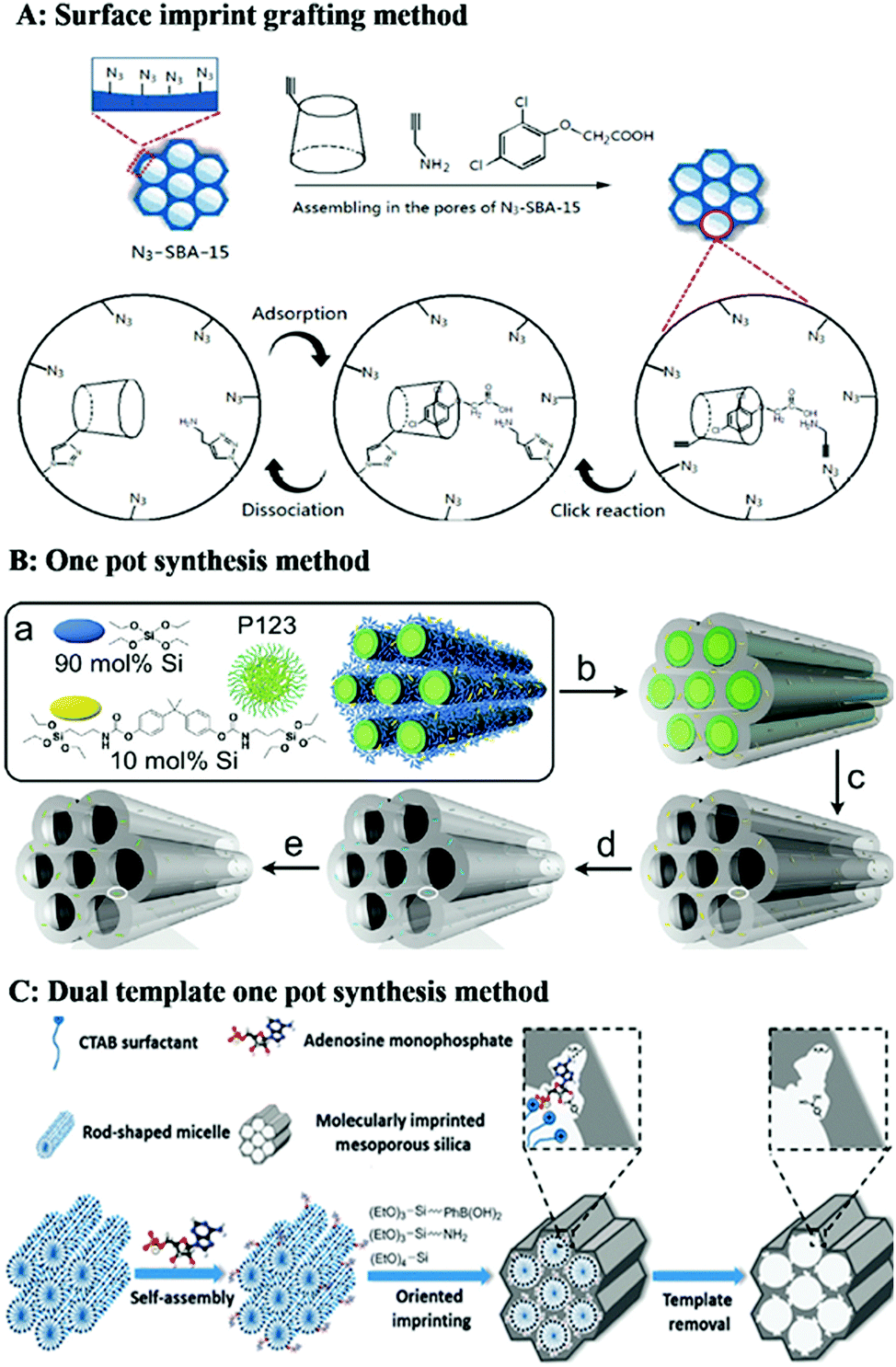 | ||
| Fig. 8 Representation of preparation of homogeneous mesoporous MIPs. (A) 2D Imprinting in the pores of mesoporous silica by click reaction, adapted from ref. 194, Copyright 2019, Elsevier. (B) One-pot synthesis method for formation of mesoporous MIPs using P123 as soft template for mesopores, adapted from ref. 195, Copyright 2011, American Chemical Society. (C) Dual-template docking oriented molecular imprinting method, adapted from ref. 204, Copyright 2015. | ||
 | ||
| Fig. 9 TEM and SEM images of mesoporous MIPs. TEM images of SBA before (A) and after (B) anchoring of MIP layer, adapted from ref. 193, Copyright 2014. SBA-15 (C) and MCM-41 (D) based Ni(II) ion imprinted polymers by one-pot synthesis method, adapted from ref. 201, Copyright 2018, Elsevier. (E) Phosphate-imprinted mesoporous silica using dual-template method, adapted from ref. 204, Copyright 2015. (F and G) TEM images of magnetic mesoporous silica-based mesoporous core–solid shell MIPs, adapted from ref. 215, Copyright 2016, Elsevier. (H and I) TEM images of solid core–mesoporous shell MIPs, adapted from ref. 222, Copyright 2015. (J) TEM image of hollow mesoporous MIPs, adapted from ref. 227 Copyright 2017, Elsevier. (K and L) SEM and TEM images of multilevel mesoporous silica, adapted from ref. 230, Copyright 2019. (M) TEM image of Fe3O4@fibrous SiO2 after anchoring of MIP layer, adapted from ref. 233, Copyright 2015, American Chemical Society. | ||
Mesoporous MIPs are widely used for removal or enrichment of targets due to the high surface area. For example, mesoporous IIPs were prepared for rapid and specific adsorption of Cr(III) ions in effluents. Due to the mesoporous structure, the adsorption reached equilibrium within 5 min.196 Mesoporous MIPs were prepared for SPE of chloramphenicol from milk. The maximum imprinting factor was 9.7, whereas the highest capacity was 23 mg g−1.197 Ni ion imprinted mesoporous IIPs were employed for removal of Ni ions from wastewaters with adsorption efficiency above 92.5% even after seven extraction-stripping cycles.201
For the mesoporous structured MIPs prepared by the above method, it cannot be guaranteed that all the recognition sites are located on the surface of the mesoporous MIPs. Most recognition sites are still located in the MIPs. Therefore, it is highly desirable to develop straightforward and efficient imprinting strategies to generate recognition sites on the surface of mesoporous silica. A dual-template (molecular imprinting template and mesoporous template) docking oriented molecular imprinting method for one-pot synthesis of mesoporous MIPs with recognition sites located on the surface of mesoporous silica was proposed.202–206 As shown in Fig. 8C, rod-like positively charged CTAB micelles were mixed with negatively charged template to form dual-template complexes based on electrostatic attraction. Then an appropriate Si source precursor co-condensation occurred around the dual-template complexes. After removal of the soft template, recognition sites were located on the surface of mesoporous silica materials. This method was easy to control. Fig. 9E shows a TEM image of phosphate-imprinted mesoporous silica using the dual-template method, with mesopore diameter of 2.6 nm and specific surface area of 792 m2 g−1.204 Similarly, Hu205 prepared mesoporous MIPs to recognize phosphorylated tyrosine residue using this dual-template docking method. Template phenylphosphonic acid is negatively charged in alkaline solution, which can be docked onto CTAB micelles to form a dual-template due to electrostatic attraction. However, this imprinting strategy was designed only for charged targets.
The pore size of mesoporous silica using CTAB as the soft template is less than the mean size of proteins, which is not suitable for protein imprinting. Therefore, suitable soft templates, which can generate suitable sizes of mesopores and channels for the diffusion of proteins, are in high demand. Researchers have found that amphiphilic ionic liquids (ILs) consisting of an imidazolium cation and a hydrophobic long alkyl chain along with a kosmotropic anion could be used as soft templates to produce well-ordered mesoporous materials. More importantly, the mesopore size can be tuned by varying the length of the alkyl chain and the anion. So, using the amphiphilic IL 1-octadecyl-3-methylimidazolium chloride (C18MIMCl) as surfactant, Qian206 prepared mesoporous MIPs for protein recognition. The imidazolium cation of the IL provided multiple interactions with nonapeptide template, so nonapeptide template was anchored on the surface of C18MIMCl micelle rods to generate surfactant–template complexes. MIPs displayed a fibrous porous morphology that was directed by C18MIMCl. The diameter of spherical MIPs was about 200 nm and the pore diameter was 3.62 nm. The dimensions of cytochrome c are about 2.6 nm × 3.2 nm × 3.3 nm, so the size of the pores produced by ILs is suitable for Cyc imprinting.
5.2 Core–shell mesoporous MIPs
These kinds of mesoporous core–solid shell MIPs are often used for selective extraction and enrichment of targets from complicated sample matrices. However, separation of those mesoporous core–shell MIPs is difficult because of the small particle size. So Yan215 prepared core–shell magnetic mesoporous MIPs for specific magnetic separation of BPA. The novelty of this work was using magnetic mesoporous silica as supporting matrix. Mesoporous silica was first prepared, followed by embedding of MNPs into the pores of mesoporous silica. The as-prepared MIPs combined the advantages of magnetic separation and faster mass transfer and higher adsorption capacity stemming from the mesoporous support. Fig. 9F and G display the TEM images of magnetic mesoporous silica and the core–shell MIPs. An imprinted MIP layer coated on the magnetic mesoporous silica could be observed clearly. Magnetic mesoporous silica also can be prepared by pyrolysis method. For example, Liu216 prepared magnetic mesoporous MIPs for Sr(II) by RAFT polymerization. First, Fe3O4@SBA-15 was obtained by calcining SBA-15 and Fe(NO3)3 in N2 flowing atmosphere. Then Fe3O4@SBA-15 was modified with APTES and RAFT agent. The imprinted polymers were obtained on the external and internal surfaces of Fe3O4@SBA-15 through RAFT polymerization.
Many scholars have pointed out that binding capacity is increased by using a mesoporous core. However, we think the effect of mesoporous cores on the binding capacity is not obvious. This is because the surface of a mesoporous core is covered with solid imprinted shell, and the effective recognition sites are only located within a dozen nanometers of the MIP surface. Unless ultra-thin imprinted shell of several nanometers is covered on the surface of the mesoporous core, the advantages of the mesoporous core cannot be realized. From the viewpoint of increasing adsorption capacity, it is more effective to prepare MIPs with mesoporous shells.
Solid Fe3O4 core–mesoporous shell MIPs are usually used for extraction of target from complicated samples. Magnetic mesoporous yolk–shell MIPs also were prepared for extraction of 17β-estradiol (E2).226 First, Fe3O4 was coated with an inorganic SiO2 layer. Then mesoporous MIP layer was coated on the surface of Fe3O4@SiO2 using APTES as functional monomer, bis(triethoxysilyl)benzene (BTESE) as organic crosslinker, and CTAB as soft template for mesopores. To form the magnetic mesoporous yolk–shell structure, the inorganic SiO2 was etched by NaCO3 solution. The magnetic mesoporous yolk–shell structure MIPs were applied to magnetic solid-phase extraction (MSPE) of E2. Recoveries ranged from 88.3 to 102.4% and the relative standard deviations (RSDs) were lower than 5.5%.
5.3 Hollow mesoporous MIPs
Hollow mesoporous MIPs (HM-MIPs) can be prepared by etching of the core as discussed in Section 4. First, solid core–mesoporous shell structure MIPs are prepared, and then the cores are etched to form hollow mesoporous MIPs. For example, in our previous work,226 HM-MIPs were prepared by a one-pot surface imprinting method followed by chemical selective etching to remove the solid silica core. First, solid SiO2 was synthesized using inorganic TEOS as precursor. Then mesoporous E2-imprinted layer was coated onto the surface of inorganic SiO2 using organosilica precursor bis(triethoxysilyl)benzene instead of TEOS. Solid inorganic SiO2 was etched by hot Na2CO3 solution to form hollow voids. It should be pointed out that the organosilica is much more stable than inorganic silica, so the organosilica mesoporous shell could be well preserved after etching. Fig. 9J shows the E2-imprinted hollow mesoporous MIPs. The diameter of HM-MIPs was about 460 nm while the thickness of the shell was about 55 nm. HM-MIPs displayed a higher binding capacity (18.3 mg g−1) when compared with hollow MIPs (12.15 mg g−1)227 and core–shell mesoporous MIPs (2.93 mg g−1).125HM-MIPs also can be prepared by grafting MIPs onto hollow mesoporous silica. For example, Li228 prepared Pb(II) ion imprinted hollow mesoporous MIPs by imprinting on the surface of hollow mesoporous silica. The hollow mesoporous silica was prepared by etching SiO2@mSiO2 by Na2CO3 solution. Then a suspension polymerization procedure was adopted for the preparation of HM-MIPs. Using hollow mesoporous silica as solid matrix, Li also prepared HM-MIPs for extraction of trace BPA in real water samples.229 Comparing the above two methods, the first method involved in the one pot preparation of core–mesoporous shell MIPs and then etching of the core layer was simpler.
5.4 Multilevel mesoporous MIPs
Multilevel mesoporous silica has many kinds of channels with different sizes, from mesoporous channels to macropores. MIPs employing multilevel mesoporous silica combine the advantages of mesoporous and macroporous structures. The coexistence of multiple-scale pores may enhance and harmonize the diffusion of guest molecules. For example, in our previous work,230 BPA-imprinted multilevel mesoporous silica was prepared. During the preparation process, water, ethanol and ethyl ether were employed as co-solvents. CTAB micelles were employed to form mesopores, while the heterogeneous gasification of ethyl ether formed large mesopores. Fig. 9K and J show SEM and TEM images of multilevel mesoporous silica, shaped like a hydrangea flower. These multilevel mesoporous MIPs were employed to produce a fluorescent sensor by post-imprinting strategy by anchoring FNPs on the large mesopores. The small mesopores led to increased surface area, and facilitated the diffusion of BPA. The advantage of multilevel mesoporous silica is that QDs can be uniformly bonded in MIPs due to the presence of large pores, and the bound QDs do not hinder the adsorption of BPA. The sensitivity of the p-MIFPs was two orders of magnitude higher than that of the d-MIFPs.Liu231 prepared Ni(II)-IIP by surface imprinting on microporous–mesoporous silica. P123 was used as soft template to form mesopores while solid PS particles were used to form macropores. After modification with MPA, Ni(II) imprinting was carried out using AM as functional monomer and EGDMA as crosslinker. The results proved that the introduction of macropores facilitated mass transfer and reduced transport limitations.
The pore size of mesoporous silica using CTAB as a soft template was less than 3 nm, which was not suitable for protein imprinting. Fibrous silica was synthesized using a biphase reaction. CTAB and urea were dissolved in H2O as the aqueous phase, and a mixture of cyclohexane, isopropanol, and TEOS was the oil phase. The oil phase and water phase were mixed and stirred at a fixed rate. By simply controlling the stirring rate, the pore size can be tuned. The pore size of fibrous silica can be tuned from a few nanometers to tens of nanometers. Multilevel mesoporous silica like fibrous silica also could be used as a solid matrix to prepare MIPs by surface imprinting.232,233 For example, Ge's group233 prepared lysozyme-imprinted PDA layer on Fe3O4@fibrous SiO2 microspheres. Fibrous SiO2 was coated on the surface of Fe3O4. The size of fibrous channel was about 12 nm and the specific surface area of Fe3O4@fibrous SiO2 support was 570 m2 g−1. After lysozyme imprinting, the surface area of MIP-lysozyme was 309 m2 g−1, and the intensity of the pores with diameter of 12 nm decreases only 12%, which indicates PDA only attached to the surface of the pores of Fe3O4@F-SiO2, rather than completely blocking those pores. Due to the high surface area of Fe3O4@fibrous SiO2 support, the obtained Fe3O4@fibrous SiO2@PDA microspheres have a high saturation adsorption capacity of lysozyme of 700 mg g−1 within 30 min. Fig. 9M shows a TEM image of lysozyme-imprinted Fe3O4@F-SiO2@PDA, which displays the clear core–shell structure and radial fibers.
6. Conclusions and outlook
In the past two decades, great progress has been made in the development of MIPs with different morphologies. Various methods have been developed to generate MIPs with the desired size, shape and particle size distribution. Many kinds of spherical, core–shell, hollow, mesoporous and porous MIPs have been successfully prepared for packed columns, enrichment of targets, and chemical sensors. The diameter and size of MIP particles are affected by the preparation method and morphology. As shown in Fig. 10, we analyzed the size of MIPs from about 200 references listed in this article, and arrived at the following conclusions. The particle size of mesoporous MIPs prepared by one-pot methods is generally below 100 nm with good dispersivity. The size of core–shell structured MIPs (including solid core–solid shell, solid core–mesoporous shell and mesoporous core–solid shell) is slightly larger, being distributed between 100 nm and 1000 nm, depending on the size of the core material. The size of hollow MIPs obtained by etching of cores is similar to that of core–shell structures, while the size of hollow MIPs prepared by the seed swelling method is larger, about 3–5 μm. The particle size of solid spherical MIPs is greatly affected by the polymerization method. The spherical MIPs prepared by precipitation polymerization are mostly in the range of 0.5–5 μm, while the particle size obtained by Pickering polymerization and suspension polymerization is larger, in the range of tens to hundreds of micrometers.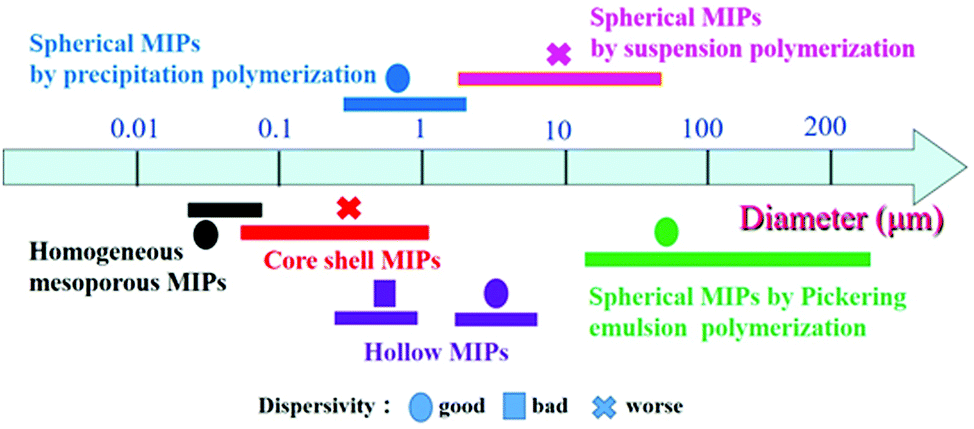 | ||
| Fig. 10 The diameter distribution of MIPs with different morphologies obtained using different preparation methods. | ||
At the same time, we can choose the appropriate morphology and preparation method for the application field. For example, for SPE adsorbents, Pickering emulsion polymerization and suspension polymerization can be used to prepare large size MIPs. In order to get improved adsorption capacity, hollow or mesoporous MIPs can be selected. If one wants to endow MIPs with performances like photodegradation and magnetic separation, core–shell structured MIPs are a good choice. The spherical MIPs prepared by precipitation polymerization have a wide particle size distribution and can be used in many fields. The main advantages and disadvantages of different morphologies are listed in Table 1.
| Morphology | Preparation method | Main advantages | Main disadvantages |
|---|---|---|---|
| Sphere | Precipitation polymerization | Uniform and controllable particle size; | Large amount of solvent; |
| simplicity in preparation | relatively low yield | ||
| Suspension polymerization | One-step simple polymerization process; | Poor dispersivity; | |
| big particle size suitable for SPE | surfactant contamination; | ||
| low binding capacity | |||
| Pickering emulsion | Big particle size suitable for SPE; | Low binding capacity | |
| good dispersivity; | |||
| surfactant free | |||
| Sol–gel process | Ease of fabrication at room temperature; | Lack of functional monomer | |
| green reaction solvent; | |||
| high stability | |||
| Core–shell | Surface imprinting | High utilization of recognition sites; | Aggregation; |
| multiple cores endow multiple functions | multiple steps | ||
| Hollow | Etching core | Method with wide application; | Aggregation; |
| high binding capacity | multiple steps | ||
| Seed swelling | Big particle size suitable for SPE | Time consuming; thick layer | |
| Mesoporous | Sol–gel process | Good dispersivity; | Small particle size hard to separate |
| high binding capacity |
There is a lot of work that needs to be done in the future to improve the morphology of MIPs.
(i) Many of the existing morphologies are unsatisfactory. For example, although living polymerization has been employed to prepare MIPs, the dispersion of spherical MIPs is not ideal. The phenomena of adhesion and agglomeration are commonly observed for many core–shell MIPs, and the recognition sites are still difficult to use due to the thick shell. Many hollow MIPs collapse easily and fracture, and imprinting in the pores of mesoporous silica is still difficult.
(ii) The kinds of morphology of MIPs are relatively rare, and most of the morphologies are relatively simple. Many new morphologies, such as dumbbell and Janus, have not been used in MIPs. This may be because the ideal application areas of these special structures have not been found.
(iii) Attention for most MIPs has focused on separation/purification/detection in environmental, food and biological matrices. Fewer MIPs have been employed in vivo and in cells. The good news is that fluorescent MIPs have been employed for drug delivery and cell imaging in cells using a dual-template imprinting strategy.234,235 In the future, the application of spherical MIPs can be extended to in vivo and cells.
(iv) Due to the small space of recognition sites, the existing recognition sites in MIPs cannot be characterized. Many imprinting strategies, like solid-phase imprinting and dual-template oriented imprinting, cannot be strongly supported by the existing morphology characterization methods.
(v) There are many factors affecting the adsorption capacity of MIPs, such as morphology, particle size, functional monomer, solvent and so on. The adsorption capacity of MIPs and detection sensitivity of MIP-based chemical sensors should be improved by many aspects together.
List of abbreviations and acronyms
| AAm | Acrylamide |
| APTES | 3-Aminopropyltriethoxysilane |
| ATRP | Atom transfer radical polymerization |
| AuNCs | Gold nanoclusters |
| BET | Brunauer–Emmett–Teller |
| BHb | Bovine hemoglobin |
| BPA | Bisphenol A |
| BSA | Bovine serum albumin |
| BTESE | Bis(triethoxysilyl)benzene |
| CDs | Carbon dots |
| CTAB | Cetyltrimethylammonium bromide |
| DA | Dopamine |
| DPP | Distillation precipitation polymerization |
| DVB | Divinylbenzene |
| E2 | 17β-Estradiol |
| EGDMA | Ethylene glycol dimethacrylate |
| FNPs | Fluorescent nanoparticles |
| GMA | Glycidyl methacrylate |
| HEMA | Hydroxyethyl methacrylate |
| HM-MIPs | Hollow mesoporous MIPs |
| HMS | Hollow mesoporous silica |
| IIPs | Ion imprinted polymers |
| ILs | Ionic liquids |
| IPTS | 3-Isocyanatopropylethoxysilane |
| MAA | Methacrylic acid |
| MAPS | Methacryloxypropyltrimethoxysilane |
| M-H-MIPs | Magnetic hollow MIPs |
| MIPs | Molecularly imprinted polymers |
| M-MIPs | Magnetic MIPs |
| MNPs | Magnetic nanoparticles |
| MOF | Metal–organic framework |
| MSPE | Magnetic solid-phase extraction |
| NIPAm | N-Isopropylacrylamide |
| NPs | Nanoparticles |
| OVA | Ovalbumin |
| P123 | EO20PO70EO20 |
| PDA | Polydopamine |
| PEG | Polyethylene glycol |
| PHEMA | Polyhydroxyethyl methacrylate |
| PMMA | Poly(methyl methacrylate) |
| PNIPAAm | Poly(N-isopropylacrylamide) |
| PS | Polystyrene |
| PVA | Poly(vinyl alcohol) |
| QDs | Quantum dots |
| RAFT | Reversible addition–fragmentation chain transfer |
| RAM-MIPs | Restricted access material MIPs |
| RPP | Reflux precipitation polymerization |
| SDS | Sodium dodecyl sulfate |
| SEM | Scanning electron microscope |
| SPE | Solid-phase extraction |
| TBBPA | Tetrabromobisphenol A |
| TEM | Transmission electron microscope |
| TEOS | Tetraethoxysilane |
| TRIM | Trimethylolpropane trimethacrylate |
| 2D | Two dimensional |
| 2,4-D | 2,4-Dichlorophenoxyacetic acid |
| 3D | Three dimensional |
| 4-VP | 4-Vinylpyridine |
| β-CDs | β-Cyclodextrins |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21777065) and Youth Innovation Project for Colleges of Shandong Province (2019KJA021).Notes and references
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O’Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS PubMed.
- 16 W. Cheng, X. Zeng, H. Chen, Z. Li, W. Zeng, L. Mei and Y. Zhao, ACS Nano, 2019, 13, 8537–8565 CrossRef CAS PubMed.
- T. Zhou, L. Ding, G. Che, W. Jiang and L. Sang, TrAC, Trends Anal. Chem., 2019, 114, 11–28 CrossRef CAS.
- S. Xu, H. Lu, X. Zheng and L. Chen, J. Mater. Chem. C, 2013, 1, 4406–4422 RSC.
- J. Fu, L. Chen, J. Li and Z. Zhang, J. Mater. Chem. A, 2015, 3, 13598–13627 RSC.
- Q. Yang, J. Li, X. Wang, H. Peng, H. Xiong and L. Chen, Biosens. Bioelectron., 2018, 112, 54–71 CrossRef CAS PubMed.
- M. Sobiech, P. Bujak, P. Luliński and A. Pron, Nanoscale, 2019, 11, 12030–12074 RSC.
- X. Guo, J. Li, M. Arabi, X. Wang, Y. Wang and L. Chen, ACS Sens., 2020, 5, 601–619 CrossRef CAS PubMed.
- P. Wang, X. Sun, X. Su and T. Wang, Analyst, 2016, 141, 3540–3553 RSC.
- S. Farooq, J. Nie, Y. Cheng, Z. Yan and J. Li, Analyst, 2018, 143, 3971–3989 RSC.
- R. Gui, H. Guo and H. Jin, Nanoscale Adv., 2019, 1, 3325–3363 RSC.
- J. Pan, W. Chen, Y. Ma and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- R. Xing, Y. Wen, H. He, Z. Guo and Z. Liu, TrAC, Trends Anal. Chem., 2019, 110, 417–428 CrossRef CAS.
- R. Schirhagl, Anal. Chem., 2014, 86, 250–261 CrossRef CAS PubMed.
- G. Sharma and B. Kandasubramanian, J. Chem. Eng. Data, 2020, 65, 396–418 CrossRef CAS.
- V. Pichon, N. Delaunay and A. Combès, Anal. Chem., 2020, 92, 16–33 CrossRef CAS PubMed.
- L. Wan, Z. Chen, C. Huang and X. Shen, TrAC, Trends Anal. Chem., 2017, 95, 110–121 CrossRef CAS.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911–933 RSC.
- M. Dinc, C. Esen and B. Mizaikoff, TrAC, Trends Anal. Chem., 2019, 114, 202–217 CrossRef CAS.
- T. Alizadeh and N. Memarbashi, Sep. Purif. Technol., 2012, 90, 83–91 CrossRef CAS.
- C. Gonzato, P. Pasetto, F. Bedoui, P. Mazeranc and K. Haupt, Polym. Chem., 2014, 5, 1313–1322 RSC.
- M. R. Halhalli and B. Sellergren, Polym. Chem., 2015, 6, 7320–7332 RSC.
- D. Wang, S. Hong, G. Yang and K. Row, Korean J. Chem. Eng., 2003, 20, 1073–1076 CrossRef CAS.
- J. Sun, J. Dai, W. Ma, L. Gao, A. Xie, J. He, X. Wei, Z. Zhou, C. Li and Y. Yan, J. Iran. Chem. Soc., 2016, 13, 489–497 CrossRef CAS.
- Y. Lu, Y. Zhu, Y. Zhang and K. Wang, J. Chem. Eng. Data, 2019, 64, 1045–1050 CrossRef CAS.
- J. Haginaka, C. Miura, N. Funaya and H. Matsunaga, Anal. Sci., 2012, 28, 315–317 CrossRef CAS PubMed.
- C. Miura, H. Li, H. Matsunaga and J. Haginaka, J. Pharm. Biomed. Anal., 2015, 114, 139–144 CrossRef CAS.
- S. Xu, J. Li and L. Chen, Talanta, 2011, 85, 282–289 CrossRef CAS PubMed.
- T. Zhou, L. Jørgensen, M. Mattebjerg, I. Chronakis and L. Ye, RSC Adv., 2014, 4, 30292–30299 RSC.
- R. Song, X. Hu, P. Guan, J. Li, L. Qian and Q. Wang, Chin. J. Polym. Sci., 2015, 33, 404–415 CrossRef CAS.
- W. Han, L. Gao, X. Li, L. Wang, Y. Yan, G. Chen, B. Hu, X. Lin and M. Song, RSC Adv., 2016, 6, 81346–81353 RSC.
- M. J. Garcia-Soto, K. Haupt and C. Gonzato, Polym. Chem., 2017, 8, 4830–4834 RSC.
- F. Bai, X. Yang and W. Huang, Macromolecules, 2004, 37, 9746–9752 CrossRef CAS.
- K. Yang, M. M. Berg, C. Zhao and L. Ye, Macromolecules, 2009, 42, 8739–8746 CrossRef CAS.
- Y. Liu, L. Zhang, N. Zhao, Y. Han, F. Zhao, Z. Penge and Y. Li, Analyst, 2017, 142, 1091–1098 RSC.
- M. Safdarian and Z. Ramezani, New J. Chem., 2019, 43, 48–57 RSC.
- X. Shen, C. Huang, S. Shinde, M. SwitnickaPlak, P. Cormack and B. Sellergren, RSC Adv., 2016, 6, 81491–81499 RSC.
- P. Lenain, J. D. D. Mavungu, P. Dubruel, J. Robbens and S. Saeger, Anal. Chem., 2012, 84, 10411–10418 CrossRef CAS PubMed.
- N. Shah, J. H. Ha, M. Ul-Islam and J. K. Park, Korean J. Chem. Eng., 2011, 28, 1936–1944 CrossRef CAS.
- X. Shen and L. Ye, Chem. Commun., 2011, 47, 10359–10361 RSC.
- X. Shen, C. Xu and L. Ye, Soft Matter, 2012, 8, 7169–7176 RSC.
- T. Fan, W. Yang, N. Wang, X. Ni, J. Wen and W. Xu, J. Appl. Polym. Sci., 2016, 133, 43484 Search PubMed.
- M. Kujawska, T. Zhou, A. W. Trochimczuk and L. Ye, J. Mol. Recognit., 2017, e2626 Search PubMed.
- J. Li, X. Hu, P. Guan, X. Zhang, L. Qian, N. Zhang, C. Du and R. Song, J. Sep. Sci., 2016, 39, 1863–1872 CrossRef CAS PubMed.
- H. Sun, Y. Li, J. Yang, X. Sun, C. Huang, X. Zhang and J. Chen, J. Sep. Sci., 2016, 39, 2188–2195 CrossRef CAS PubMed.
- T. Zhou, X. Shen, S. Chaudhary and L. Ye, J. Appl. Polym. Sci., 2014, 131, 39606 CrossRef.
- X. Shen and L. Ye, Macromolecules, 2011, 44, 5631–5637 CrossRef CAS PubMed.
- C. Passini, L. Anfossi, F. Nardo, G. Spano, V. Maurino and C. Baggiani, J. Sep. Sci., 2015, 38, 3661–3668 CrossRef PubMed.
- Z. Wang, T. Qiu, L. Guo, J. Ye, L. He and X. Li, Chem. Eng. J., 2018, 332, 409–418 CrossRef CAS.
- M. G. Ayari, P. Kadhirvel, P. Favetta, B. Plano, C. Dejous, B. Carbonnier and L. A. Agrofoglio, Mater. Sci. Eng., C, 2019, 101, 254–263 CrossRef CAS PubMed.
- X. Shen, T. Zhou and L. Ye, Chem. Commun., 2012, 48, 8198–8200 RSC.
- J. Pan, W. Zhu, X. Dai, X. Yan, M. Gan, L. Li, H. Hang and Y. Yan, RSC Adv., 2014, 4, 4435–4443 RSC.
- Y. Sun, J. Chen, Y. Li, H. Li, X. Zhu, Y. Hu, S. Huang, J. Li and S. Zhong, New J. Chem., 2016, 40, 8745–8752 RSC.
- H. Hang, C. Li, J. Pan, L. Li, J. Dai, X. Dai, P. Yu and Y. Feng, J. Sep. Sci., 2013, 36, 3285–3294 CrossRef CAS PubMed.
- Y. Zhu, D. Jiang, D. Sun, Y. Yan and C. Li, J. Environ. Chem. Eng., 2016, 4, 3570–3579 CrossRef CAS.
- J. Pan, L. Li, H. Hang, R. Wu, X. Dai, W. Shi and Y. Yan, Langmuir, 2013, 29, 8170–8178 CrossRef CAS PubMed.
- W. Zhu, W. Ma, C. Li, J. Pan and X. Dai, Chem. Eng. J., 2015, 276, 249–260 CrossRef CAS.
- R. Jalilian, M. Shahmari, A. Taheria and K. Gholami, Ultrason. Sonochem., 2020, 61, 104802 CrossRef CAS PubMed.
- H. Ou, Q. Chen, J. Pan, Y. Zhang, Y. Huang and X. Qi, J. Hazard. Mater., 2015, 289, 28–37 CrossRef CAS PubMed.
- X. Shen, J. S. Bonde, T. Kamra, L. Bülow, J. C. Leo, D. Linke and L. Ye, Angew. Chem., Int. Ed., 2014, 53, 10687–10690 CrossRef CAS PubMed.
- H. Li, H. Zhou, H. Wang, P. Yang and S. Zhong, J. Sep. Sci., 2015, 38, 1365–1371 CrossRef PubMed.
- Y. Sun, T. Ren, Z. Deng, Y. Yang and S. Zhong, New J. Chem., 2018, 42, 7355–7363 RSC.
- Y. Sun, Y. Li, J. Xu, L. Huang, T. Qiu and S. Zhong, Colloids Surf., B, 2017, 155, 142–149 CrossRef CAS PubMed.
- Y. Sun and S. Zhong, Colloids Surf., B, 2017, 159, 131–138 CrossRef CAS PubMed.
- W. Zhu, W. Ma, C. Li, J. Pan, X. Dai, M. Gan, Q. Qu and Y. Zhang, Eng. Aspects, 2014, 453, 27–36 CrossRef CAS.
- R. Yin, L. Chen and L. Ma, J. Sep. Sci., 2019, 42, 3563–3570 CrossRef CAS PubMed.
- C. Liu, Z. Song, J. Pan, X. Wei, L. Gao, Y. Yan, L. Li, J. Wang, R. Chen, J. Dai and P. Yu, J. Phys. Chem. C, 2013, 117, 10445–10453 CrossRef CAS.
- G. Pan, Y. Ma, Y. Zhang, X. Guo, C. Li and H. Zhang, Soft Matter, 2011, 7, 8428–8439 RSC.
- Y. Ma, Y. Zhang, M. Zhao, X. Guo and H. Zhang, Chem. Commun., 2012, 48, 6217–6219 RSC.
- J. Jiang, Y. Zhang, X. Guo and H. Zhang, RSC Adv., 2012, 2, 5651–5662 RSC.
- M. Zhao, X. Chen, H. Zhang, H. Yan and H. Zhang, Biomacromolecules, 2014, 15, 1663–1675 CrossRef CAS PubMed.
- M. Zhao, C. Zhang, Y. Zhang, X. Guo, H. Yan and H. Zhang, Chem. Commun., 2014, 50, 2208–2210 RSC.
- C. Li, Y. Ma, H. Niu and H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 27340–27350 CrossRef CAS PubMed.
- H. Niu, Y. Yang and H. Zhang, Biosens. Bioelectron., 2015, 74, 440–446 CrossRef CAS PubMed.
- Y. Yang, H. Niu and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 15741–15749 CrossRef CAS PubMed.
- Y. Yang, Z. Wang, H. Niu and H. Zhang, Biosens. Bioelectron., 2016, 86, 580–587 CrossRef CAS PubMed.
- Y. Ma, Ji. Gao, C. Zheng and H. Zhang, J. Mater. Chem. B, 2019, 7, 2474–2483 RSC.
- S. Xu, H. Lu and L. Chen, J. Chromatogr. A, 2014, 1350, 23–29 CrossRef CAS PubMed.
- F. Lin, J. Chen, M. Lee, B. Li and J. Wang, ACS Appl. Nano Mater., 2020, 3, 1147–1152 CrossRef CAS.
- S. H. Yang, H. Fan, R. J. Classon and K. A. Schug, J. Sep. Sci., 2013, 36, 2922–2938 CrossRef CAS PubMed.
- T. Liang, L. Chen and Y. Ma, J. Chromatogr. A, 2020, 1609, 460453 CrossRef CAS PubMed.
- Z. Sun, H. Liu, Y. Zhou, S. Zhao, J. Li, X. Wang and B. Gong, RSC Adv., 2019, 9, 27953–27960 RSC.
- H. Liu, J. Ding, K. Zhang and L. Ding, Talanta, 2020, 209, 120508 CrossRef CAS PubMed.
- F. Puoci, F. Iemma, G. Cirillo, M. Curcio, O. I. Parisi, U. G. Spizzirri and N. Picci, Eur. Poly. J., 2009, 45, 1634–1640 CrossRef CAS.
- J. He, L. Song, S. Chen, Y. Li, H. Wei, D. Zhao, K. Gu and S. Zhang, Food Chem., 2015, 187, 331–337 CrossRef CAS PubMed.
- Y. Zhou, H. Liu, J. Li, Z. Sun, T. Cai, X. Wang, S. Zhao and B. Gong, J. Chromatogr. A, 2020, 1613, 460684 CrossRef CAS PubMed.
- F. A. C. Suquila, L. L. G. de Oliveira and C. R. T. Tarley, Chem. Eng. J., 2018, 350, 714–728 CrossRef CAS.
- Z. Sun, H. Liu, Y. Zhou, S. Zhao, J. Li, X. Wang and B. Gong, RSC Adv., 2019, 9, 27953–27960 RSC.
- L. L. G. de Oliveira, F. A. C. Suquilaa, F. M. de Oliveiraa, G. L. Scheela and C. R. T. Tarley, React. Funct. Polym., 2019, 134, 93–103 CrossRef CAS.
- D. Wang, D. Gao, Y. Huang, W. Xu and Z. Xia, Talanta, 2019, 202, 392–401 CrossRef CAS PubMed.
- K. Chin and S. Chang, ACS Appl. Nano Mater., 2019, 2, 89–99 CrossRef CAS.
- R. Fernandes, M. Dinc, I. Raimundo Jr. and B. Mizaikoff, Microporous Mesoporous Mater., 2018, 264, 28–34 CrossRef CAS.
- X. Yang, Y. Sun, Y. Xiang, F. Qiu and G. Fu, Analyst, 2019, 144, 5439–5448 RSC.
- Q. Li, K. Yang, Y. Liang, B. Jiang, J. Liu, L. Zhang, Z. Liang and Y. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 21954–21960 CrossRef CAS PubMed.
- M. R. Halhalli, C. S. A. Aureliano, E. Schillinger, C. Sulitzky, M. M. Titirici and B. Sellergren, Polym. Chem., 2012, 3, 1033–1042 RSC.
- C. Rui, J. He, Y. Li, Y. Liang, L. You, L. He, K. Li and S. Zhang, Talanta, 2019, 201, 342–349 CrossRef CAS PubMed.
- J. Guo, H. Chen and X. Wei, J. Chem. Eng. Data, 2019, 64, 4005–4012 CrossRef CAS.
- H. Wang, Y. Liu, S. Yao and P. Zhu, Food Chem., 2018, 240, 1262–1267 CrossRef CAS PubMed.
- D. Rahangdale and A. Kumar, Carbohydr. Polym., 2018, 202, 334–344 CrossRef CAS PubMed.
- Z. Iskierko, P. Sharma, D. Prochowicz, K. Fronc, F. D’Souza, D. Toczydłowska, F. Stefaniak and K. Noworyta, ACS Appl. Mater. Interfaces, 2016, 8, 19860–19865 CrossRef CAS PubMed.
- K. Qian, G. Fang and S. Wang, Chem. Commun., 2011, 47, 10118–10120 RSC.
- Z. Sun, H. Liu, Y. Zhou, Sh. Zhao, J. Li, X. Wang and B. Gong, RSC Adv., 2019, 9, 27953–27960 RSC.
- Z. Wei, R. Zhang, L. Mu, Y. Huang and Z. Liu, Eur. Polym. J., 2019, 121, 109301 CrossRef.
- H. Liu, L. Mu, Xi. Chen, J. Wang, S. Wang and B. Sun, J. Agric. Food Chem., 2017, 65, 986–992 CrossRef CAS PubMed.
- Y. Huang and R. Wang, J. Mater. Chem. A, 2019, 7, 12105–12114 RSC.
- C. Fan, J. Liu, H. Zhao, L. Li, M. Liu, J. Gao and L. Ma, RSC Adv., 2019, 9, 33678–33683 RSC.
- T. S. Anirudhan and S. MadananAnju, J. Environ. Chem. Eng., 2019, 7, 103355 CrossRef CAS.
- R. Fiorenza, A. Di Mauro, M. Cantarella, A. Gulino, L. Spitaleri, V. Privitera and G. Impellizzeri, Mater. Sci. Semicond. Process., 2020, 112, 105019 CrossRef CAS.
- H. Basan, M. Dinca and B. Mizaikoff, Anal. Methods, 2018, 10, 997–1005 RSC.
- Q. Liu, J. Wan and X. Cao, Process Biochem., 2018, 70, 168–178 CrossRef CAS.
- X. Wang, J. Yu, J. Li, Q. Kang, D. Shen and L. Chen, Sens. Actuators, B, 2018, 255, 268–274 CrossRef CAS.
- W. Zhang, W. Liu, P. Li, H. Xiao, H. Wang and B. Tang, Angew. Chem., 2014, 126, 12697–12701 CrossRef.
- X. Wu, Z. Zhan, J. Li, H. You, Y. Li and L. Chen, Sens. Actuators, B, 2015, 211, 507–514 CrossRef CAS.
- M. Arabi, M. Ghaedi and A. Ostovan, ACS Sustainable Chem. Eng., 2017, 5, 3775–3785 CrossRef CAS.
- Y. Hu, Y. Li, R. Liu, W. Tan and G. Li, Talanta, 2011, 84, 462–470 CrossRef CAS PubMed.
- N. Kumar, N. Narayanan and S. Gupta, Food Chem., 2018, 255, 81–88 CrossRef CAS PubMed.
- M. Peyrovi, M. Hadjmohammadi and I. Saeidi, Biomed. Chromatogr., 2019, 33, e4404 CrossRef PubMed.
- R. Uzuriaga-Sánchez, A. Wong, S. Khan, M. Pividori, G. Picasso and M. Sotomayor, Mater. Sci. Eng., C, 2017, 74, 365–373 CrossRef PubMed.
- X. Wang, P. Huang, X. Ma, X. Du and X. Lu, J. Chromatogr. A, 2018, 1537, 35–42 CrossRef CAS PubMed.
- S. Xu, J. Li, X. Song, J. Liu, H. Lu and L. Chen, Anal. Methods, 2013, 5, 124–133 RSC.
- R. Gao, Y. Hao, S. Zhao, L. Zhang, X. Cui, D. Liu, Y. Tang and Y. Zheng, RSC Adv., 2014, 4, 56798–56808 RSC.
- R. Gao, Y. Hao, L. Zhang, X. Cui, D. Liu and Y. Tang, Anal. Methods, 2014, 6, 9791–9799 RSC.
- H. Lu and S. Xu, Talanta, 2015, 144, 303–311 CrossRef CAS PubMed.
- B. Wang, H. Deng, M. Wu, S. Xiang, Q. Ma, S. Shi, L. Xie and Y. Guo, Anal. Methods, 2018, 10, 3317–3324 RSC.
- J. Huang, C. Sun, D. Yao, C. Wang, L. Zhang, Y. Zhang, L. Chen and C. Yuan, J. Mater. Chem. B, 2018, 6, 1531–1542 RSC.
- L. Xie, J. Guo, Y. Zhang, Y. Hua, Q. You and S. Shi, Food Chem., 2015, 178, 18–25 CrossRef CAS PubMed.
- S. Miao, M. Wu, H. Zuo, C. Jiang, S. Jin, Y. Lu and H. Yang, J. Agric. Food Chem., 2015, 63, 3634–3645 CrossRef CAS PubMed.
- J. Zhou, Y. Wang, Y. Ma, B. Zhang and Q. Zhang, Appl. Surf. Sci., 2019, 486, 265–273 CrossRef CAS.
- J. Guo, M. Yu, X. Wei and L. Huang, J. Chem. Eng. Data, 2018, 63, 3068–3073 CrossRef CAS.
- Z. Lia, C. Lei, N. Wang, X. Jiang, Y. Zeng, Z. Fu, L. Zou, L. He, S. Liu, X. Ao, K. Zhou and S. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1100–1101, 113–121 CrossRef PubMed.
- X. He, Z. Lian, L. Tan and J. Wang, J. Chromatogr. A, 2018, 1469, 8–16 CrossRef PubMed.
- X. Yu, H. Liu, J. Diao, Y. Sun and Y. Wang, Sep. Purif. Technol., 2018, 204, 213–219 CrossRef CAS.
- D. Wang, D. Gao, W. Xu, F. Li, M. Yin, Q. Fu and Z. Xi, Talanta, 2018, 184, 307–315 CrossRef CAS PubMed.
- W. Xu, Q. Dai, Y. Wang, X. Hu, P. Xu, R. Ni and J. Meng, RSC Adv., 2018, 8, 21850–21856 RSC.
- K. Hemmati, R. Sahraei and M. Ghaemy, Polymers, 2016, 101, 257–268 CrossRef CAS.
- J. Zhou, Y. Wang, J. Bu, B. Zhang and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 25682–25690 CrossRef CAS PubMed.
- Z. Zhang, H. Wang, H. Wang, C. Wu, M. Li and L. Li, Analyst, 2018, 143, 5849–5856 RSC.
- H. Gholami, M. Arabi, M. Ghaedi, A. Ostovan and A. Bagheri, J. Chromatogr. A, 2019, 1594, 13–22 CrossRef CAS PubMed.
- D. Gao, D. Wang, Q. Fu, L. Wang, K. Zhang, F. Yang and Z. Xia, Talanta, 2018, 178, 299–307 CrossRef CAS PubMed.
- M. Liu, X. Li, J. Li, Z. Wu, F. Wang, L. Liu, X. Tan and F. Lei, J. Colloids Interface Sci., 2017, 504, 124–133 CrossRef CAS PubMed.
- Y. He, Y. Huang, Y. Jin, X. Liu, G. Liu and R. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 9634–9642 CrossRef CAS PubMed.
- J. Wang and J. Wei, J. Mater. Chem. A, 2017, 5, 4651–4659 RSC.
- Z. Liu, Z. Hu, Y. Liu, M. Meng, L. Ni, X. Meng, G. Zhong, F. Liu and Y. Gao, RSC Adv., 2015, 5, 52369–52381 RSC.
- J. Li, R. Dong, X. Wang, H. Xiong, S. Xu, D. Shen, X. Song and L. Chen, RSC Adv., 2015, 5, 10611–10618 RSC.
- H. Lu and S. Xu, Sens. Actuators B, 2020, 306, 127566 CrossRef.
- Y. Li, Y. Chen, L. Huang, B. Lou and G. Chen, Analyst, 2017, 142, 302–309 RSC.
- R. Ma, W. Ha, J. Chena and Y. Shi, J. Mater. Chem. B, 2016, 4, 2620–2627 RSC.
- Q. Han, X. Shen, W. Zhu, C. Zhu, X. Zhou and H. Jiang, Biosens. Bioelectron., 2016, 79, 180–186 CrossRef CAS.
- H. Zhu, H. Yao, K. Xia, J. Liu, X. Yin and W. Zhang, Chem. Eng. J., 2018, 346, 317–328 CrossRef CAS.
- Q. Liu, K. Zhang, Y. Jin, X. Wang, Y. Liu, H. Liu and M. Xie, Talanta, 2018, 186, 346–353 CrossRef CAS.
- M. Khajeh, M. Sharifirad, M. Bohlooli and M. Ghaffari-Moghaddam, RSC Adv., 2016, 6, 54702–54708 RSC.
- Z. Liu, Y. Gao, L. Jin, H. Jin, N. Xu, X. Yu and S. Yu, ACS Sustainable Chem. Eng., 2019, 7, 8168–8175 CrossRef CAS.
- M. Li, P. Sun, Q. Wu, D. Liu and L. Zhou, Environ. Sci.: Nano, 2018, 5, 2651–2662 RSC.
- Y. He, F. Zhao, C. Zhang, A. EI-Aty, D. Baranenko, A. Hacimüftüoğlu and Y. She, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1132, 121811 CrossRef CAS.
- A. Sorribes-Soriano, F. Esteve-Turrillas, S. Armenta, A. Montoya, J. Herrero-Martínez and M. Guardia, J. Chromatogr. A, 2018, 1545, 22–31 CrossRef CAS.
- D. Ren, J. He and H. Zhang, Anal. Methods, 2014, 6, 3079–3085 RSC.
- I. Mohiuddin, A. Grover, J. Singh, A. Sang-Soo Lee, A. K. Malik and K. Kim, Chem. Eng. J., 2020, 382, 123002 CrossRef.
- W. Chen, M. Fu, X. Zhu and Q. Liu, Biosens. Bioelectron., 2019, 142, 111492 CrossRef CAS PubMed.
- J. Wang, Z. Meng, M. Xue, L. Qiu, X. Dong, Z. Xu and X. He, New J. Chem., 2017, 41, 1129–1136 RSC.
- W. J. Tang, T. Zhao, C. H. Zhou, X. J. Guan and H. X. Zhang, Anal. Methods, 2014, 6, 3309–3315 RSC.
- Y. Guo and T. Guo, Chem. Commun., 2013, 49, 1073–1075 RSC.
- H. Fan, J. Wang, Q. Meng and Z. Jin, Food Chem., 2019, 281, 1–7 CrossRef CAS PubMed.
- C. Gong, Y. Wei, L. Liu, A. Zheng, Y. Yang, C. Chow and Q. Tang, Mater. Sci. Eng., C, 2017, 76, 568–578 CrossRef CAS PubMed.
- C. Gong, Y. Yang, Y. Yang, A. Zheng, S. Liu and Q. Tang, J. Colloid Interface Sci., 2016, 481, 236–244 CrossRef CAS PubMed.
- H. Lu and S. Xu, J. Chromatogr. A, 2017, 1051, 10–17 CrossRef PubMed.
- B. Bali and P. Kislay Singh, Sens. Actuators B, 2017, 244, 167–174 CrossRef.
- D. Fan, H. Li, S. Shi and X. Chen, J. Chromatogr. A, 2016, 1470, 27–32 CrossRef CAS PubMed.
- D. Fan, L. Jia, H. Xiang, M. Peng, H. Li and S. Shi, Food Chem., 2017, 24, 32–36 CrossRef PubMed.
- D. Zang, M. Yan, P. Zhao, L. Ge, S. Liu and J. Yu, Analyst, 2012, 137, 4247–4253 RSC.
- A. Ostovan, M. Ghaedi, M. Arabi and A. Asfaram, J. Chromatogr. A, 2017, 1520, 65–74 CrossRef CAS PubMed.
- J. Zhang, Y. Chen, W. Wu, Z. Wang, Y. Chu and X. Chen, Microchem. J., 2019, 145, 1176–1184 CrossRef CAS.
- S. Ansari, Anal. Methods, 2017, 9, 3200–3212 RSC.
- P. Wang, A. Zhang, Y. Jin, Q. Zhang, L. Zhang, Y. Peng and S. Du, RSC Adv., 2014, 4, 26063–26073 RSC.
- J. Fan, J. Yu, X. Yang, X. Zhang, T. Yuan and H. Peng, Chem. Eng. J., 2018, 337, 722–732 CrossRef CAS.
- A. Xie, J. Dai, X. Chen, T. Zou, J. He, Z. Chang, C. Li and Y. Yan, RSC Adv., 2016, 6, 51014–51023 RSC.
- R. Yang, Y. Liu, X. Yan and S. Liu, Talanta, 2016, 161, 114–121 CrossRef CAS PubMed.
- Y. Yang, Y. Sun, H. Chen, X. Dang, Y. Ai, X. Liu and H. Chen, Anal. Methods, 2020, 12, 507–513 RSC.
- S. Xu, L. Chen, J. Li, W. Qin and J. Ma, J. Mater. Chem., 2011, 21, 12047–12053 RSC.
- X. Zheng, T. Xu, R. Shi, N. Lu, J. Zhang, C. Jiang, C. Zhang and J. Zhou, Mater. Lett., 2018, 211, 21–23 CrossRef CAS.
- Q. Zhao, H. Li, Y. Xu, F. Zhang, J. Zhao, L. Wang, J. Hou, H. Ding, Y. Li, H. Jin and L. Ding, J. Chromatogr. A, 2015, 1376, 26–34 CrossRef CAS PubMed.
- H. Li, L. Zhao, Y. Xu, T. Zhou, H. Liu, N. Huang, J. Ding, Y. Li and L. Ding, Talanta, 2018, 185, 542–549 CrossRef CAS PubMed.
- Z. Wang, T. Qiu, L. Guo, J. Ye, L. He and X. Li, Chem. Eng. J., 2019, 357, 348–357 CrossRef CAS.
- S. Li, C. Yin, S. Ren, T. Yang, J. Wang and S. Feng, J. Sep. Sci., 2015, 38, 2573–2579 CrossRef CAS PubMed.
- Q. Jia, Y. Ma, Y. Peng, Y. Liu and W. Zhang, Chem. Eng. J., 2018, 342, 293–303 CrossRef CAS.
- L. Li, K. Yu, M. Tian, Y. Wang, Z. Zhang, G. Jiang and L. Li, Anal. Methods, 2018, 10, 3926–3932 RSC.
- X. Zhao, Y. He, Y. Wang, S. Wang and J. Wang, Food Chem., 2020, 309, 125787 CrossRef CAS PubMed.
- C. Chen, Y. Wang, S. Ding, C. Hong and Z. Wang, Microchem. J., 2019, 147, 191–197 CrossRef CAS.
- A. Wang, H. Lu and S. Xu, J. Agric. Food Chem., 2016, 64, 5110–5116 CrossRef CAS PubMed.
- W. Huang, X. Hou, Y. Tong and M. Tian, RSC Adv., 2019, 9, 5394–5401 RSC.
- H. Li, X. Hu, Y. Zhang, S. Shi, X. Jiang and X. Chen, J. Chromatogr. A, 2015, 1404, 21–27 CrossRef CAS PubMed.
- Z. Xu, P. Deng, S. Tang, D. Kuang, F. Zhang and J. Li, J. Mater. Chem. B, 2014, 2, 8418–8426 RSC.
- Z. Xu, P. Deng, J. Li, S. Tang and Y. Cui, Mater. Sci. Eng., C, 2019, 94, 684–693 CrossRef CAS PubMed.
- J. Lofgreen, I. Moudrakovski and G. Ozin, ACS Nano., 2011, 5(3), 2277–2287 CrossRef CAS PubMed.
- S. Cen, W. Li, R. He, J. Tan, H. Wang, C. Wei and Y. Tang, RSC Adv., 2017, 7, 37778–37786 RSC.
- S. Xu and H. Lu, Biosens. Bioelectron., 2016, 85, 950–956 CrossRef CAS PubMed.
- Y. Geng, M. Guo, J. Tan, S. Huang, Y. Tang, L. Tan and Y. Liang, Sens. Actuators B, 2019, 281, 821–829 CrossRef CAS.
- H. Lu, S. Xu and J. Liu, ACS Sens., 2019, 4, 1917–1924 CrossRef CAS PubMed.
- H. Lu, C. Yu and S. Xu, Sens. Actuators B, 2019, 288, 691–698 CrossRef CAS.
- R. He, Z. Wang, L. Tan, Y. Zhong, W. Li, D. Xing, C. Wei and Y. Tang, Microporous Mesoporous Mater., 2018, 257, 212–221 CrossRef CAS.
- Y. Chen, D. Li, Z. Bie, X. He and Z. Liu, Anal. Chem., 2016, 88, 1447–1454 CrossRef CAS PubMed.
- X. Pan, X. He and Z. Liu, Anal. Chim. Acta, 2018, 1019, 65–73 CrossRef CAS PubMed.
- Y. Chen, X. Li, D. Yin, D. Li, Z. Bie and Z. Liu, Chem. Commun., 2015, 51, 10929–10932 RSC.
- G. Zhang, L. Jiang, J. Zhou, L. Hu and S. Feng, Chem. Commun., 2019, 55, 9927–9930 RSC.
- Z. Li, P. Guan, X. Hu, S. Ding, Y. Tian, Y. Xu and L. Qian, Polymers, 2018, 10, 298 CrossRef PubMed.
- J. Yang, Y. Li, J. Wang, X. Sun, S. M. Shah, R. Cao and J. Chen, Anal. Chim. Acta, 2015, 853, 311–319 CrossRef CAS PubMed.
- S. Xu, H. Lu, J. Li, X. Song, A. Wang, L. Chen and S. Han, ACS Appl. Mater. Interfaces, 2013, 5, 8146–8154 CrossRef CAS PubMed.
- S. Xu and H. Lu, Biosens. Bioelectron., 2015, 73, 160–166 CrossRef CAS PubMed.
- L. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16248–16256 CrossRef CAS PubMed.
- Y. Liu, G. Zhong, Z. Liu, M. Meng, F. Liu and L. Ni, Chem. Eng. J., 2016, 296, 437–446 CrossRef CAS.
- X. Gu, H. He, C. Wang, Y. Gao, H. Zhang, J. Hong, S. Du, L. Chen and C. Yuan, RSC Adv., 2015, 5, 41377–41384 RSC.
- H. Wang, Y. Liu, S. Yao and P. Zhu, Food Chem., 2018, 240, 1262–1267 CrossRef CAS PubMed.
- F. Yang, D. Hu and X. Dong, Chromatographia, 2015, 78, 45–54 CrossRef CAS.
- J. Dai, J. He, A. Xie, L. Gao, J. Pan, X. Chen, Z. Zhou, X. Wei and Y. Yan, Chem. Eng. J., 2016, 284, 812–822 CrossRef CAS.
- Y. Liu, F. Liu, L. Ni, M. Meng, X. Meng, G. Zhong and J. Qiu, RSC Adv., 2016, 6, 54679–54692 RSC.
- X. Pan, X. He and Z. Liu, Anal. Chim. Acta, 2018, 1019, 65–73 CrossRef CAS PubMed.
- Z. Zhang, J. Li, X. Wang, D. Shen and L. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 9118–9127 CrossRef CAS PubMed.
- Y. Miao, X. Sun, J. Lv and G. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 2264–2272 CrossRef CAS PubMed.
- M. Jia, Z. Zhang, J. Li, H. Shao, L. Chen and X. Yang, Sens. Actuators B, 2017, 252, 934–943 CrossRef CAS.
- S. Xu and H. Lu, Chem. Commun., 2015, 51, 3200–3203 RSC.
- W. Gui, H. Wang, Y. Liu and Q. Ma, Sens. Actuators B, 2018, 266, 685–691 CrossRef CAS.
- M. Li, H. Liu and X. Ren, Biosens. Bioelectron., 2017, 89, 899–905 CrossRef CAS PubMed.
- M. Amjadi and R. Jalili, Biosens. Bioelectron., 2017, 96, 121–126 CrossRef CAS PubMed.
- R. Yang, Y. Liu, X. Yan, S. Liu and H. Zheng, J. Mater. Chem. A, 2016, 4, 9807–9815 RSC.
- H. Lu and S. Xu, J. Chromatogr. A, 2017, 1501, 10–17 CrossRef CAS PubMed.
- W. Chen, M. Xue, F. Xue, X. R. Mu, Z. B. Xu, Z. H. Meng, G. X. Zhu and K. J. Shea, Talanta, 2015, 140, 68–72 CrossRef CAS PubMed.
- Z. Zhang, X. Zhang, D. Niu, Y. Li and J. Shi, J. Hazard. Mater., 2017, 328, 160–169 CrossRef CAS PubMed.
- L. Li, K. Yu, M. Tian, Y. Wang, Z. Zhang, G. Jiang and L. Li, Anal. Methods, 2018, 10, 3926–3932 RSC.
- H. Lu and D. Wei, Analyst, 2019, 144, 6283–6290 RSC.
- Y. Liu, F. Liu, M. Meng, Z. Liu and L. Ni, New J. Chem., 2016, 40, 3821–3832 RSC.
- Z. Feng, Y. Wang, L. Yang and T. Sun, Sci. Total Environ., 2019, 685, 442–450 CrossRef CAS PubMed.
- J. Chen, S. Lei, Y. Xie, M. Wang, J. Yang and X. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 28606–28615 CrossRef CAS PubMed.
- Y. Qin, H. Peng, X. He, W. Li and Y. Zhang, Anal. Chem., 2019, 91, 12696–12703 CrossRef CAS PubMed.
- C. Jia, M. Zhang, Y. Zhang, Z. Ma, N. Xiao, X. He, W. Li and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 32431–32440 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |