
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive hydrogel consisting of hydrazide-functionalized poly(oligo(ethylene glycol)methacrylate) and dialdehyde cellulose nanocrystals†
Rasool
Nasseri
ab and
Kam C.
Tam
 *ab
*ab
aDepartment of Chemical Engineering, University of Waterloo, 200 University Avenue West, Waterloo, Ontario N2L 3G1, Canada. E-mail: mkctam@uwaterloo.ca; Tel: +1 (519) 888-4567 ext38339
bWaterloo Institute for Nanotechnology, University of Waterloo, 200 University Avenue West, Waterloo, Ontario N2L 3G1, Canada
First published on 4th August 2020
Abstract
A self-healable thermo-responsive hydrogel was produced via the formation of acylhydrazone bond between dialdehyde-functionalized cellulose nanocrystals (DACNC) and hydrazide-functionalized poly(oligo(ethylene glycol) methacrylate) (POEGMA-H). POEGMA-H displayed a two-step phase transition upon increasing the temperature due to the coil to globule transition of the middle and end segments of the polymer chains. Rheological measurements revealed a significant increase in hydrogel stiffness (from 11 Pa to 1907 Pa) due to the coil to globule transition of POEGMA-H chains when the temperature was increased from 25 to 75 °C. With increasing concentration of DACNC, the stiffness of the hydrogel was enhanced. At low and medium pHs, stable hydrogels were formed, however, at high pH, gradual formation of acylhydrazone bonds led to a loose network. The hydrogel possessed self-healing capability with close to 100% recovery in the storage modulus at medium pH, and it has great potential in biomedical applications.
1. Introduction
Polymeric hydrogels with high water content and three-dimensional structure mimic natural matrices. They have attracted increasing attention in several applications, such as tissue engineering,1 delivery vehicles2–4etc. Stimuli-responsive hydrogels are increasingly being used in the biomedical science due to their sol–gel transition and reversible swelling–shrinking behavior in response to stimuli, such as pH, temperature, light, and magnetic fields.5–9In situ gelling or injectable hydrogels are of particular interest, because their delivery modality is via minimally invasive injection, contrary to pre-formed hydrogels that must be surgically implanted.10,11 The crosslinking mechanism plays an important role in the final properties of the hydrogels. Self-healing or the ability of self-repair is one of the properties that can be produced in a hydrogel with a rational crosslinking strategy. Two major crosslinking approaches can be employed to prepare self-healing hydrogel, namely dynamic noncovalent interactions (host–guest interactions,12 metal ion co-ordinations,13,14 electrostatic interactions15,16) and reversible covalent bonds (boroneoxygen bonds,17 sulfur–sulfur bonds (disulfide),18 carbon–nitrogen bonds (imine,19 enamine,20 and acylhydrazone21–23), cyclohexenes (reversible Diels–Alder cycloaddition24)). Some of the reversible covalent bonds, such as acylhydrazone and disulfide are degradable in physiological environment, making them particularly advantageous in biomedical applications.25 Conflicting needs for both high stiffness (necessary for practical application) and high molecular dynamics (a requirement for the healing process), are some of the challenges in designing self-healable hydrogels. To address this, cellulose nanocrystals (CNCs) can be employed in self-healable hydrogels due to their desirable mechanical characteristics, such as high surface area (∼250 m2 g−1), high tensile strength (7500 MPa), and high stiffness (Young's modulus up to 140 GPa).26 Additionally, the abundance of reactive surface hydroxyl groups on the surface of CNC, provides a platform for functionalization and integration to form self-healable hydrogels.27 By adding CNC, the stiffness of the hydrogel can be enhanced with minimal disruption of the molecular dynamics.28Poly(oligo(ethylene glycol) methacrylate) (POEGMA) based hydrogels have gained increasing attention in biomedical applications, owing to their high tissue compatibility, facile functionalization and thermo-responsive properties.29–31 A significant advantage of hydrogels based on oligo(ethylene glycol) methyl ether methacrylate copolymers is the ability to precisely tune the thermoresponsive properties using co-monomers of different hydrophilicity.31–34 For instance, Lutz and coworkers copolymerized di(ethylene glycol) methyl ether methacrylate (MEO2MA) and oligoethylene glycol methyl ether methacrylate (OEGMA300, Mn = 300 g mol−1) containing different molar ratios of the two co-monomers, yielding POEGMA copolymers with LCSTs ranging from 27 °C to 65 °C.33 Smeets et al. reported in situ gelling hydrogels based on POEGMA precursor functionalized with hydrazide/aldehyde functional groups. The resulting hydrogel possessed desirable biological properties of poly(ethylene glycol) (PEG) based hydrogels with tunable mechanical properties and degradation time.35 Hoare and coworkers extended their study by examining the effect of varying the degree of functionality of the precursor POEGMA polymers and length of PEG side chains on the stiffness, degradation rate, and gelation of injectable POEGMA hydrogels.10,36 They revealed that the properties of the gel could be tuned by changing the molecular weight of the precursor polymers.11 Hydrogels prepared with higher molecular weight precursor polymers possessed faster gelling times and higher compressive and shear moduli compared to gels prepared with lower molecular weight precursor polymers. Recently, they reported that the architecture of the precursor polymers (branching) could be used to tune the properties of injectable hydrogels.37 Hydrogels prepared using highly branched precursor polymers possessed faster gelation and higher stiffness than hydrogels prepared with linear precursors.
Weak mechanical properties of in situ gel forming hydrogels often limit their application. CNCs have been evaluated as reinforcing agents for a range of hydrogels38–44 due to their large aspect ratio, high stiffness, low cytotoxicity and ecotoxicological risk.45–47 Gicquel et al.48 investigated hydrogels produced from the adsorption of a block copolymer composed of poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) cationic polyelectrolyte and poly(di(ethylene glycol)methyl ether methacrylate) (PMEO2MA) on oxidized cellulose nanocrystals. Upon heating beyond the LCST of a solution containing CNC and thermo-responsive copolymer, a transition from “liquid” to “gel” state occurred due to the network formation of CNC aggregates and non-adsorbed copolymers. McKee and coworkers43 reported a thermo-reversible hydrogel with tunable stiffness based on thermo-responsive methylcellulose (LCST around 50–60 °C) and CNC. By increasing the temperature above the LCST of methylcellulose, a gel (G′ > G′′) with G′ an order of magnitude larger than the liquid mixture of CNC and methylcellulose (below LCST of methylcellulose) was observed. De France et al.49 prepared an in situ gelling hydrogel with tunable mechanical properties by physical incorporation of CNCs into the hydrazone crosslinked hydrazide/aldehyde functionalized POEGMA precursors. Strong interaction of the CNCs with POEGMA precursors led to the uniform dispersion of CNCs within the hydrogel, inducing physical crosslinking that significantly increased the stiffness of the gel. They reported up to 35-fold increase in G′ by adding 5 wt% of CNC to the hydrogel.
Acylhydrazone bond exhibits pH-responsive characteristics,23,50 where the bond forms slowly in neutral condition, and the kinetics of bond formation accelerate in the presence of catalytic amounts of acid.51 The pH of surrounding medium has a large influence on the thermo-mechanical properties and self-healing behavior of the hydrogels prepared with acylhydrazone bonds.21
In this study, we prepared an in situ gelling hydrogel via acylhydrazone bonds using a mixture of hydrazide–functionalized POEGMA (POEGMA-H) and dialdehyde-functionalized CNC (DACNC). The rheological properties of the hydrogel can be tuned by changing the concentration of DACNC. Since POEGMA is a thermo-responsive polymer with a defined LCST, the mechanical properties of the hydrogel depended on the phase transition induced by changes in the temperature, and the dynamic nature of the acylhydrazone bonds imparts self-healing properties to the hydrogel. We investigated the effect of pH on the self-healing characteristics and mechanical properties of the hydrogel, and these hydrogels are suitable for biomedical applications.
2. Experimental
2.1. Materials
Cellulose nanocrystals (CNCs) were provided by CelluForce Inc. Sodium periodate (NaIO4, ≥99.8%, Sigma-Aldrich), ethylene glycol (99.8%, Sigma-Aldrich), anhydrous tetrahydrofuran (>99.5%, TCI), thriethylamine (TEA, ≥99%, Sigma-Aldrich), α-bromoisobutyryl bromide (BiBB, 98%, Sigma-Aldrich), sodium carbonate (≥99.0%, Sigma-Aldrich), dichloromethane (DCM, ≥99.8%, Sigma-Aldrich), anhydrous magnesium sulfate (Fisher), hydroxylamine hydrochloride (NH2OH·HCl, 99%, Sigma-Aldrich), sodium hydroxide (NaOH, ≥98%, Sigma-Aldrich), di(ethylene glycol) methyl ether methacrylate (MEO2MA, 95%, Aldrich), oligoethylene glycol methyl ether methacrylate (OEGMA300, 95%, Aldrich), copper(II) bromide (CuBr2, 99%, Aldrich), 2,2′-bipyridyl (bPy, ≥99%, Sigma-Aldrich), L-ascorbic acid (99%, Sigma-Aldrich), methacrylic acid (MAA, 99%, Sigma-Aldrich), adipic acid dihydrazide (ADH, >99%, TCI), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC, ≥98%, Sigma-Aldrich), and dimethyl sulfoxide-d6 (C2D6OS, 99.9%, Aldrich) were used as received.2.2. Periodate oxidation of CNC
Dialdehyde groups were formed on the CNC surfaces via sodium periodate oxidation. Sodium periodate (8 g) was added to 400 mL of 0.25 wt% CNC suspension and the pH was adjusted to 3.5 using glacial acetic acid. The suspension was heated to 45 °C in a one-neck round-bottom flask covered with aluminum foil (to avoid exposure to light), and stirred for 2.5 h in a dark fume hood. In the next step, 20 mL ethylene glycol was added to decompose the remaining periodate to iodate, and after 10 min, the suspension was cooled to room temperature. After three cycles of centrifugation, the suspension was dialyzed against DI to pH = 6 and stored in a refrigerator at 4 °C.The aldehyde content of DACNCs was determined by the titration of generated acid from the reaction of NH2OH·HCl with aldehyde groups using sodium hydroxide.52 Briefly, 0.0125 g DACNC was dissolved in 10 mL DI water and the pH was adjusted to 5 using NaOH solution. A solution of NH2OH·HCl (10 mL, 0.05 g L−1) was then prepared and the pH was adjusted to 5 using NaOH solution. After mixing the DACNC suspension with NH2OH·HCl and incubated for 4 h at 40 °C, the titration was then performed using 0.01 M NaOH solution. The amount NaOH solution to achieve a pH of 5 was recorded, and this was repeated for CNC suspension. The aldehyde content was then calculated using eqn (1):
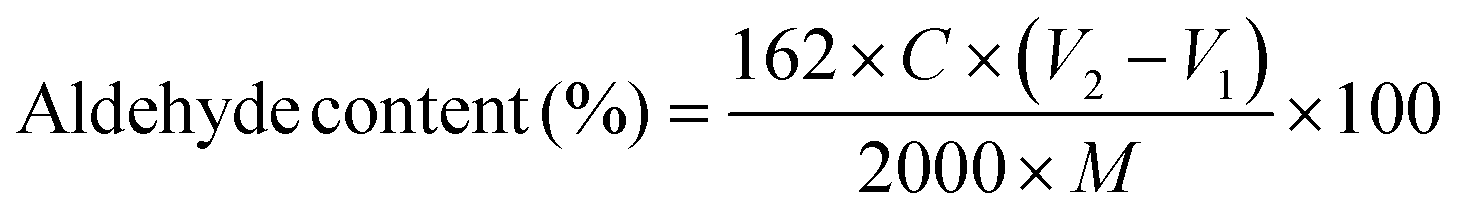 | (1) |
2.3. Synthesis of ethylene bis(2-bromoisobutyrate) (2f-BiB) bifunctional initiator.
Ethylene glycol (1.8 mL) and TEA (9.90 mL) were added to a round-bottom flask placed in an ice bath and purged with nitrogen for 15 min. Anhydrous THF (150 mL) was added to the flask and purged for 15 min, and cooled to 0 °C. Under a nitrogen atmosphere, 8.4 mL BiBB was added dropwise via a pressure funnel. After vigorous shaking, the solution was stirred overnight at ambient temperature. The reaction mixture was then filtered to remove the triethylammonium bromide salt formed during the reaction and then concentrated using a rotary evaporator. The bright brown solution was then mixed with 10 mL sodium carbonate solution (0.1 M) to hydrolyze any remaining BiBB. The product was then extracted with DCM and dried with anhydrous magnesium sulfate and filtered. The solvent was then removed by rotary evaporation. After three recrystallization steps in methanol, a yellowish white crystal was produced.2.4. Synthesis of poly(oligoethylene glycol methyl ether methacrylate-co-methacrylic acid) (POEGMA-MAA)
POEGMA-MAA was synthesized via atom transfer radical polymerization using activators generated by electron transfer (AGET ATRP). In a two-neck round-bottom flask connected to a pressure funnel, 79 mg of 2f-BiB (0.22 mmol) was dispersed in a mixture of methanol and water (20 mL methanol + 5 mL water). MEO2MA (8 mL, 44 mmol), OEGMA300 (12.4 mL 44 mmol), bPy (755.8 mg, 4.84 mmol), and L-ascorbic acid (38.7, 0.22 mmol) were then added to the flask and purged with nitrogen for 30 min. A solution of CuBr2 (4.9 mL of 100 mg mL−1) was then added to the pressure funnel and purged with nitrogen for 15 min before dropwise addition to the flask. The temperature was raised to 55 °C and after 4 h, 742 μL of MAA (8.8 mmol, purged with nitrogen for 15 min) was added to the flask through a pressure funnel and stirred overnight. The final molar ratio of ingredients was [MEO2MA]/[OEGMA300]/[MAA]/[2f-BiB]/[CuBr2]/[bpy]/[L-ascorbic acid] = 200/200/40/1/10/22/1. The polymer was purified in a dialysis tubing (MWCO 3500 Da, Fisher) against DI water/acetone mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 3 days, followed by DI for 3 days and freeze dried. POEGMA was synthesized with the similar procedure without adding MAA.
:1) for 3 days, followed by DI for 3 days and freeze dried. POEGMA was synthesized with the similar procedure without adding MAA.
2.5. Hydrazide functionalization of POEGMA-MAA (POEGMA-H)
Dried POEGMA-MAA (1 g) was dissolved in 10 mL DI water and 769.2 mg of ADH (10 fold molar excess) was added to the solution. The pH of solution was reduced to 4.75 using 1 M HCl. EDC (295.2 mg, 3.5 fold molar excess) was then added and the pH of solution was kept at 4.75 by adding 1 M HCl and stirred overnight. The functionalized-polymer was purified in a dialysis tubing (MWCO 3500 Da, Fisher) against DI water for 3 days and freeze dried.2.6. Formation of in situ hydrogels
Dry POEGMA-H polymer was dissolved in DI water to achieve a concentration of 20 wt%. DACNC suspension was concentrated to 3 wt% using a centrifuge filter tube (MWCO 10000 Da, Merck Millipore Amicon). In order to prepare gels of different pHs, equal volumes of POEGMA-H solution and DACNC suspension (200 μL) were thoroughly mixed and the pH was adjusted to low (∼3), medium (∼4) and high (∼11) values by adding one drop (∼10 μL) of 0.1 M HCl, 0.01 M HCl, and 0.1 M NaOH solutions, respectively. The solutions were then allowed to gel overnight in sealed containers. In studying the effect of DACNC concentration, DACNC suspensions with concentration of 1.5 wt% and 2.25 wt% were used in the hydrogel preparation and the pH was adjusted to a pH of 4 by adding one drop (∼10 μL) of 0.01 M HCl. Table 1 summarizes the preparation recipe of the hydrogels and the nomenclature, GelXY was used, where X is the concentration of DACNC suspension in wt% and Y describes the pH condition with L, M, and H for low, medium, and high respectively.
| Volume of POEGMA-H 20 wt% solution (μL) | Volume of DACNC suspension (μL) | Concentration of DACNC suspension (wt%) | pH | |
|---|---|---|---|---|
| Gel3M | 200 | 200 | 3 | Medium |
| Gel2.25M | 200 | 200 | 2.25 | Medium |
| Gel1.5M | 200 | 200 | 1.5 | Medium |
| Gel3L | 200 | 200 | 3 | Low |
| Gel3H | 200 | 200 | 3 | High |
2.7. Characterization
The zeta potential, size and distribution of DACNCs were determined using a Malvern Zetasizer Nano ZS90. 1H NMR was performed on a Bruker 300 MHz spectrometer at room temperature in DMSO-d6. A Nicolet 6300 Fourier-transform infrared (FTIR) spectrometer was employed to obtain the FTIR spectra. Samples were mixed with KBr powder and pressed to pellets for the measurements with a wave number range set at 4000–500 cm−1 and a resolution of 1 cm−1. A total of 32 scans were accumulated to reduce the spectra noise. The thermal responsive properties of the polymers were investigated using a Micro-Differential Scanning Calorimetry (μDSC) (Microcal VP-DSC) and a temperature-controlled UV-Visible spectrophotometer (UV-vis) (Cary 100 Bio). The polymer solutions with a concentration of 5 mg mL−1 were heated from 25 °C to 75 °C at 1 °C min−1 rate in the μDSC. A fixed wavelength (500 nm) was passed through the polymer solutions (5 mg mL−1) in UV-vis spectrophotometer as they were heated from 25 °C to 75 °C at 1 °C min−1 rate. The rheological properties of the hydrogel were characterized on a Malvern Kinexus ultra+ rheometer with a 25 mm parallel plate geometry and a solvent trap.3. Results and discussion
DACNCs were prepared via periodate oxidation of CNCs, where the emergence of a new peak at 1730 cm−1 (vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group)52 confirmed the formation of aldehyde groups on the CNCs (Fig. 1). The average zeta-potential and hydrodynamic diameter of pristine CNC were −52.6 ± 0.3 mV and 95.0 ± 1.8 nm respectively. After periodate oxidation the zeta-potential decreased to −41.1 ± 0.3 mV, while the hydrodynamic diameter remained constant (97.8 ± 3.5 nm), confirming the preservation of the morphology of CNCs. The aldehyde content as a percentage of hydroxyl groups on CNC was determined to be 12.8%.
O group)52 confirmed the formation of aldehyde groups on the CNCs (Fig. 1). The average zeta-potential and hydrodynamic diameter of pristine CNC were −52.6 ± 0.3 mV and 95.0 ± 1.8 nm respectively. After periodate oxidation the zeta-potential decreased to −41.1 ± 0.3 mV, while the hydrodynamic diameter remained constant (97.8 ± 3.5 nm), confirming the preservation of the morphology of CNCs. The aldehyde content as a percentage of hydroxyl groups on CNC was determined to be 12.8%.
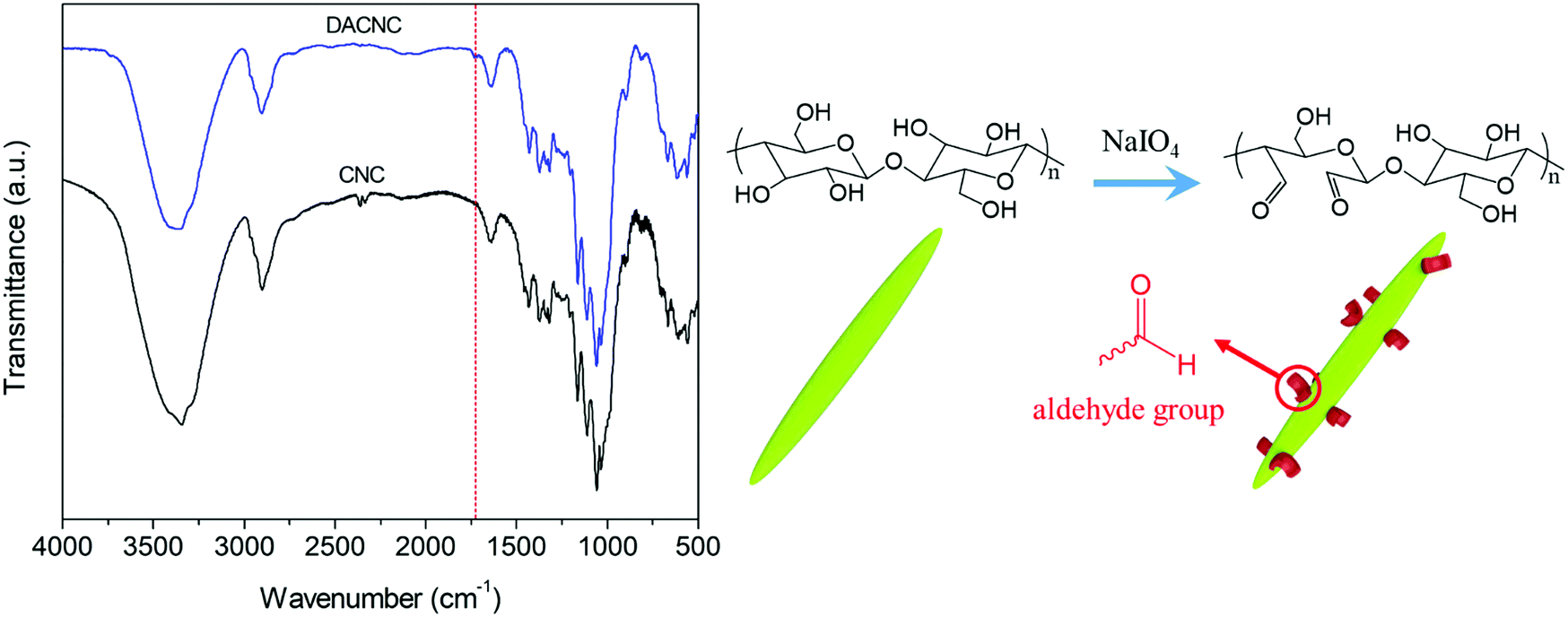 | ||
| Fig. 1 FTIR spectra of CNC and DACNC and the reaction route for periodate oxidation of CNC. | ||
The synthesis of 2f-BiB was confirmed by 1H NMR and FTIR, and Fig. 2a depicts the 1H NMR spectrum of 2f-BiB and its associated peaks (δ = 4.37 ppm, s, 4H, RO–(CH2)2–OR and δ = 1.87, s, 12H, ppm, –(CH3)2).53 The carbonyl stretching peak at 1730 cm−1 in the FTIR spectrum of 2f-BiB also confirmed the success of the synthesis53 (Fig. 2b). POEGMA-MAA was synthesized by a two-step AGET ATRP polymerization from 2f-BiB bifunctional initiator and subsequently functionalized with ADH through the carboxyl groups of MAA via the EDC chemistry11 as shown in Scheme 1. The copolymerization conversion was ∼10% and the molecular weight was ∼10 kDa (details in ESI†). The characteristic 1H NMR spectrum of POEGMA-H confirmed the presence of hydrazide groups in the copolymer (δ = 8.87 ppm, s, –NH–NH2).54 FTIR spectra of POEGMA, POEGMA-MAA and POEGMA-H displayed clear differences in the wavelength range of the –OH stretching (3250 cm−1 to 3750 cm−1) (inset in Fig. 2b). POEGMA showed a very small peak that is associated with the remaining –OH groups in 2f-BiB. After copolymerization with MAA, a broad peak emerged as a result of the carboxyl groups. By functionalizing POEGMA-MAA with hydrazide groups, the intensity and shape of this peak broadened and shifted to a higher wave number (Fig. 2b).
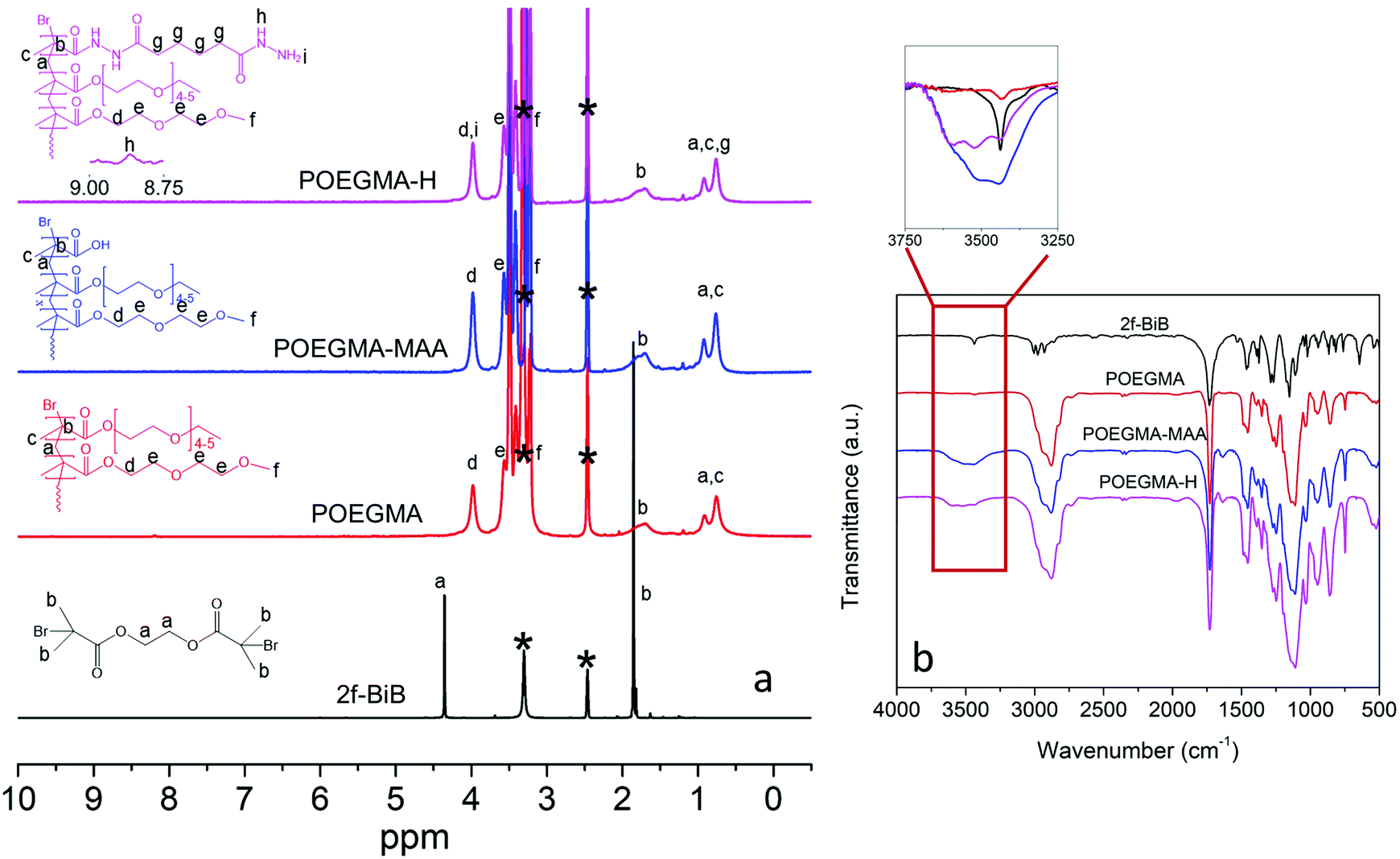 | ||
| Fig. 2 (a) 1H NMR spectra of 2f-BiB, POEGMA, POEGMA-MAA, and POEGMA-H in C2D6OS. Peak assignments are based on the literature.54,59 Solvent peaks are marked with *. (b) FTIR spectra of 2f-BiB, POEGMA, POEGMA-MAA, and POEGMA-H. Inset shows the –OH stretching range (3250 cm−1 to 3750 cm−1). | ||
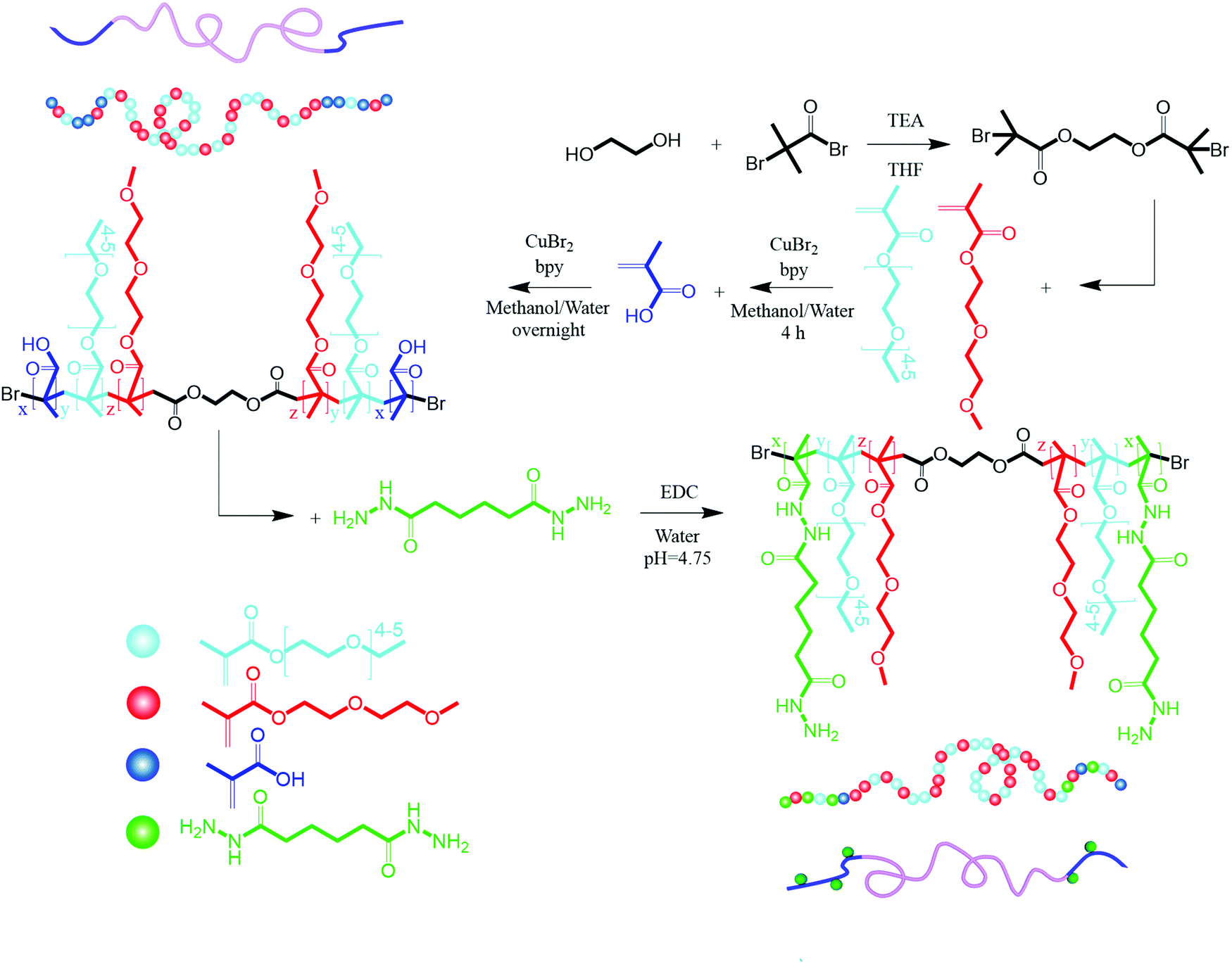 | ||
| Scheme 1 Synthesis of 2f-BiB, POEGMA-MAA, and POEGMA-H. | ||
The thermo-responsive behavior of the copolymers was studied by UV-vis spectrometry and μDSC. Due to the delicate balance between favorable and unfavorable interactions, POEGMA displayed a phase transition defined by its LCST in water. Upon increasing the temperature, the extensive hydrogen bonding between the ether groups on POEGMA and hydrogen atoms in water was disrupted, and the polyethylene glycol (PEG) segments on the POEGMA collapsed onto the methacrylate domains resulting in a coil to globule transition.32,55 Cloud point measurements by UV-vis spectrometry performed to determine the coil to globule transition temperature led to the higher scattering compared to the soluble chains. UV-vis spectrometry results indicated that POEGMA exhibited a single step phase transition with an onset temperature at 50 °C in accordance with reported value33 (Fig. 3a).
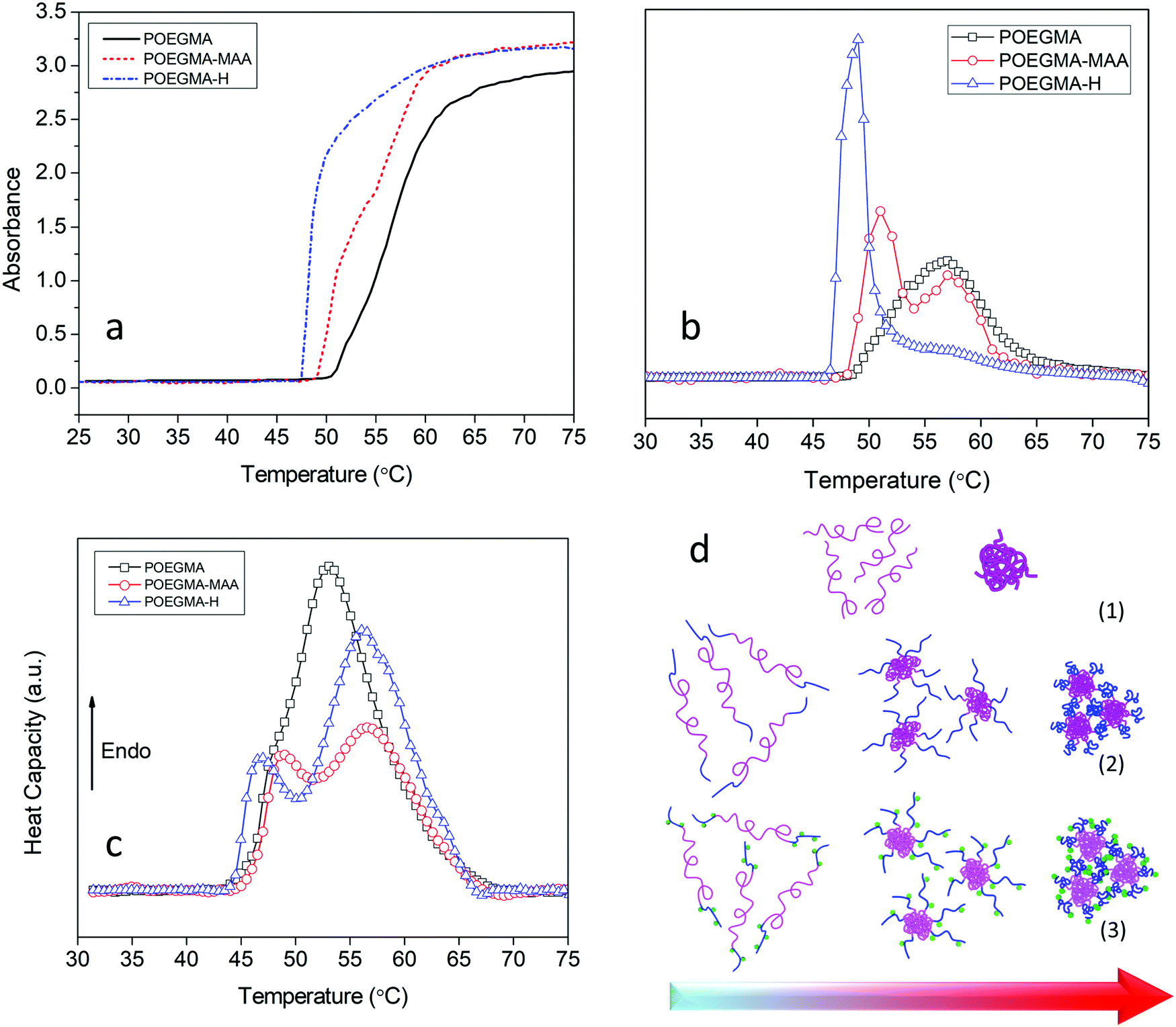 | ||
| Fig. 3 (a) UV-vis spectrum at 500 nm versus temperature of POEGMA, POEGMA-MAA, and POEGMA-H aqueous solutions (5 mg mL−1). (b) Derivative of UV-vis data for more convenient visualization of two-step phase transitions. (c) μDSC curves of aqueous solutions (5 mg mL−1) of POEGMA, POEGMA-MAA, and POEGMA-H. (d) Proposed mechanism for phase separation of (1) POEGMA, (2) POEGMA-MAA, and (3) POEGMA-H in aqueous solutions upon heating. | ||
By adding the MAA units to the POEGMA chains, the copolymer displayed a two-step phase transition. The onset temperature for the first transition shifted to a lower temperature (49 °C) while the second transition commenced at a higher temperature (55 °C) compared to only POEGMA. The lower phase transition temperature could be due to inter-molecular hydrogen bonding and complexation between carboxyl groups of MAA and ether groups on the PEG pendants that disrupts the hydrogen bonding between the PEGs and water molecules.56–58 The two-step phase transition reflects the presence of two different blocks in the copolymer with two different LCSTs.59,60 As discussed earlier, MAA monomer was added to the reaction after 4 h.
As MEO2MA and OEGMA300 monomers were present in the reaction medium, the end blocks consisted of MEO2MA and OEGMA300 and MAA random copolymers (with higher amount of MAA compare to other two monomers, Scheme 1). Due to the presence of a more hydrophilic monomer (MAA) in these blocks, the phase transition temperature became higher. The same two-step phase transition was observed for POEGMA-H with the first phase transition occurring at 46 °C and the second at 55 °C (Fig. 3a and b). The smaller second peak in derivative of UV-vis data of POEGMA-H compared to POEGMA-MAA (Fig. 3b) could be associated to the isolated smaller domains due to the higher hydrophilicity and affinity for water of the hydrazide groups compared to the carboxylic acid groups. The conformational changes due to the formation and breaking of hydrogen bonds between the copolymers and water involved a thermodynamic process that could be studied by μDSC. The results of μDSC also confirmed a single-step phase transition for POEGMA and two-step phase transition for POEGMA-MAA and POEGMA-H (Fig. 3c). The phase transition temperatures were observed at lower temperatures in all cases compared to the results from UV-vis spectroscopy. The reason for this difference is that DSC directly measures the fluctuations of heat flow associated with the phase separation, while, in UV-vis spectroscopy, the phase separation associated with the coil to globule transition and aggregation of the polymeric chains was determined from the turbidity, yielding a higher phase transition temperature. The proposed mechanism for the two-step phase transition of POEGMA-MAA and POEGMA-H is shown in Fig. 3d. At low temperatures, individual copolymer chains are present, and when the temperature was increased beyond the first phase transition temperature, core–shell micelles started to form and stabilize by the end-blocks. With further increase in the temperature beyond the second phase transition temperature, larger micelles were produced via the association of smaller micelles.
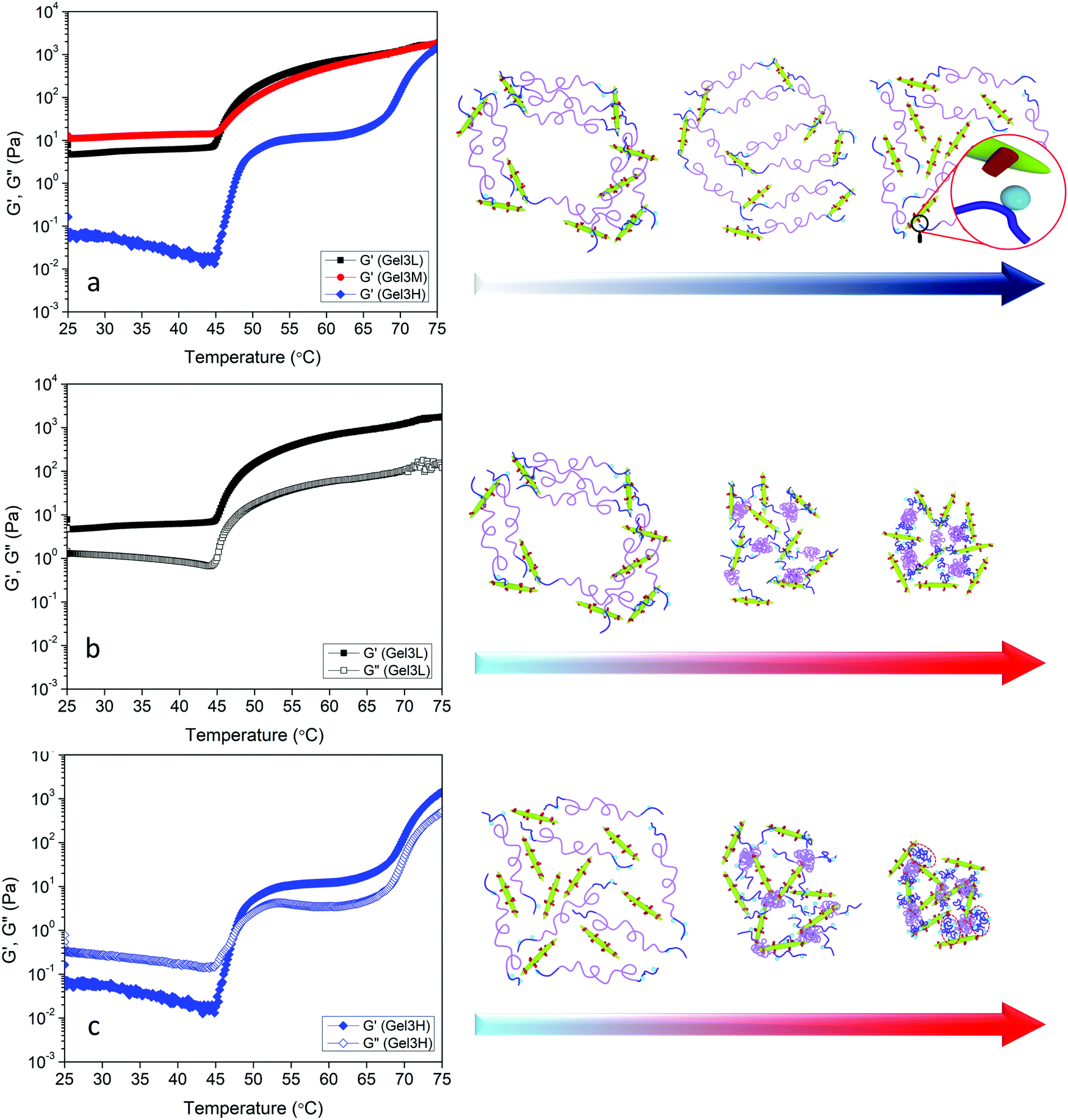
The effect of phase transition on the rheology of POEGMA-H aqueous solution was also evaluated. Fig. S1a (ESI†) shows the temperature sweep of 20 wt% POEGMA-H aqueous solution at 1 Hz and 1% strain (within the linear viscoelastic region). At the temperature below the first phase transition temperature, the moduli displayed a small decrease with increasing temperatures. Above the first phase transition temperature, the loss modulus G′′ was greater than storage modulus G′, where the POEGMA-H solution was a viscous liquid. At the first phase transition temperature, the coil to globule transition of the middle blocks on POEGMA-H chains produced a larger G′ and G′′. A further increase of G′ and G′′ was observed with increasing temperature that is associated to the coil to globule transition of the end-blocks of the polymer chains (Fig. S1b, ESI†). The hydrogels were formed by the bond formation between the hydrazide groups of POEGMA-H and aldehyde groups of DACNC (Fig. 4), which was confirmed by comparing the storage and loss modulus of POEGMA-H solution (20 wt%), DACNC suspension (3 wt%) and Gel3M measured at various temperatures at 1 Hz and 1% strain (Fig. 5a). The DACNC suspension displayed a monotonous decrease in the G′ and G′′ with temperature. However, in Gel3M, an abrupt increase in both G′ and G′′ was observed at the first phase transition temperature of POEGMA-H with G′ being much larger than G′′ over the entire temperature range, which confirmed the formation of a stable gel. The coil to globule transition of the middle and end-blocks of PEGMA-H is responsible for the increase in gel stiffness60 (Fig. 5b).
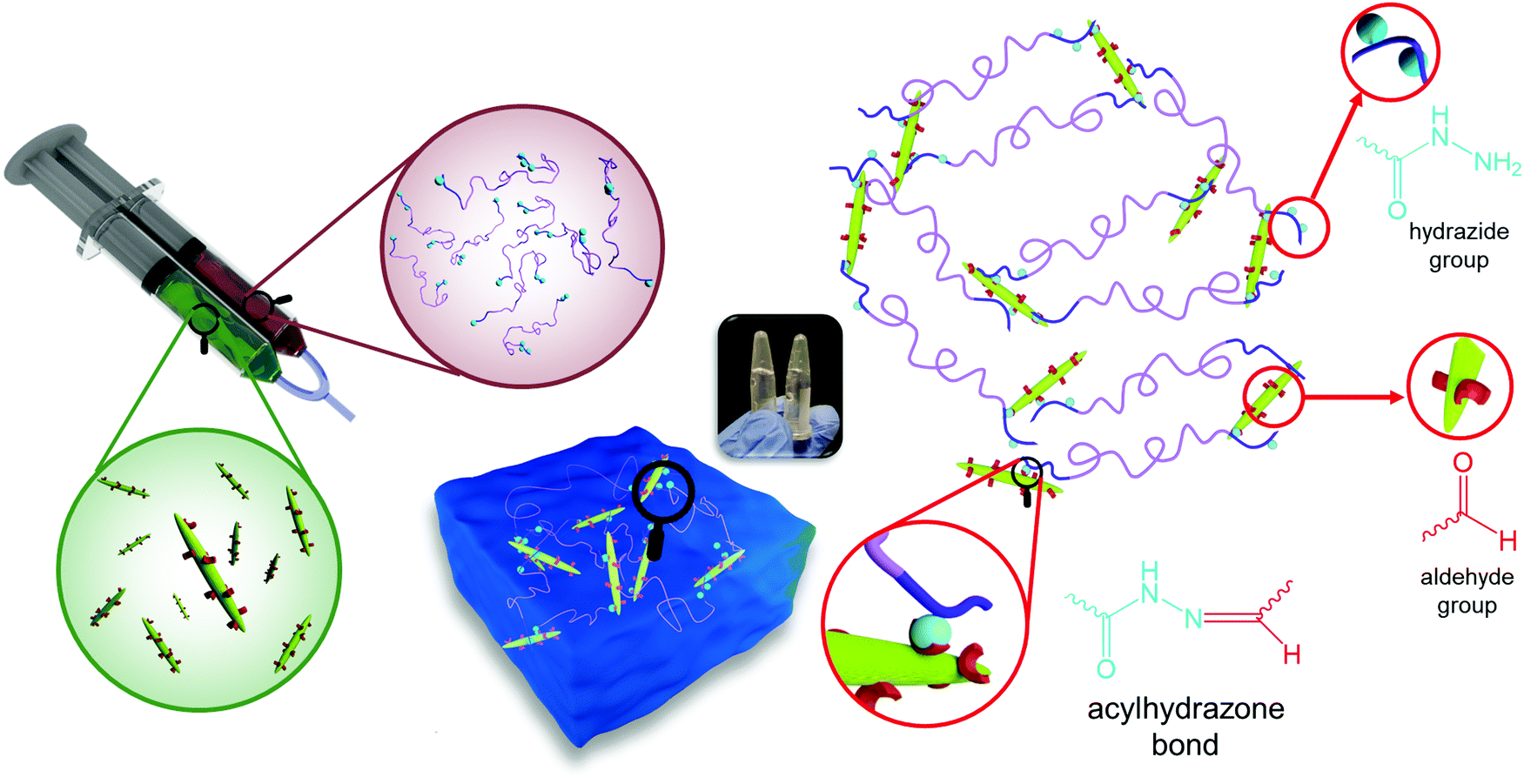 | ||
| Fig. 4 Formation of in situ gelling hydrogel by reaction of hydrazide groups of POEGMA-H and aldehyde groups of DACNC. | ||
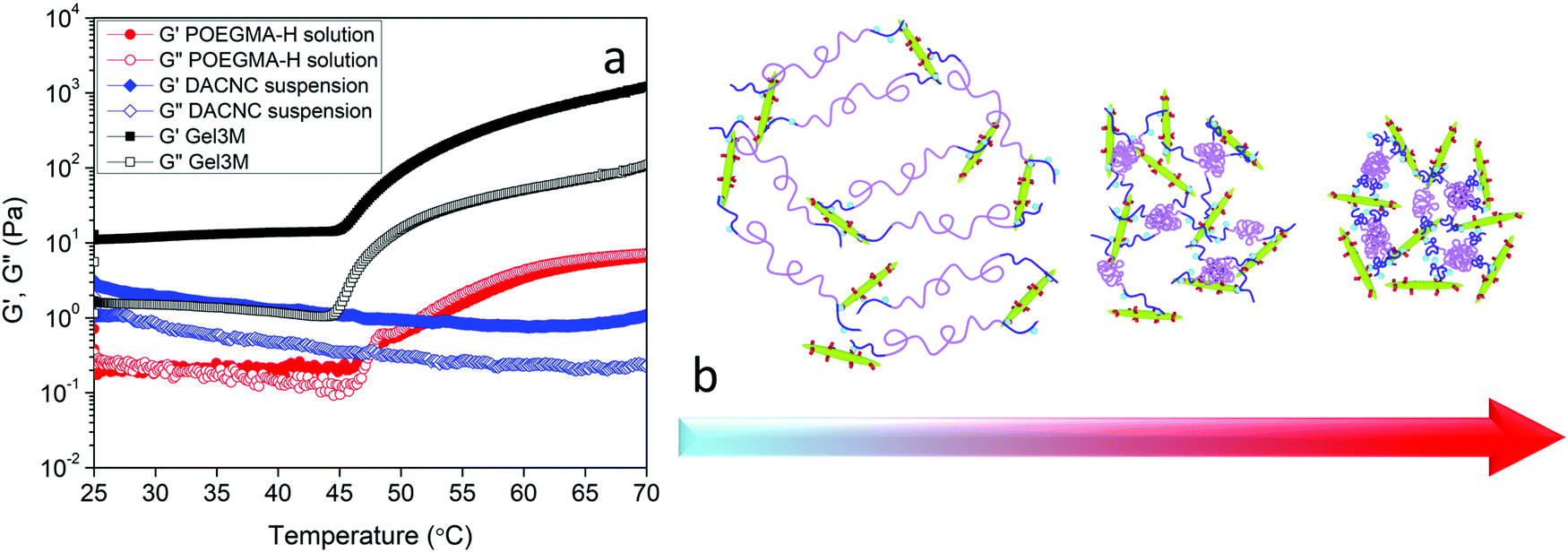 | ||
| Fig. 5 (a) Storage and loss modulus of POEGMA-H solution, DACNC suspension, and Gel3M versus temperature at 1 Hz and 1% strain. (b) Schematic representation of coil to globule transition of middle and end blocks of POEGMA-H in presence of DACNC. | ||
The effect of DACNC concentration on the rheological properties of hydrogel was examined. Fig. 6a shows the effect of DACNC concentration on the linear viscoelastic regime of the hydrogel at room temperature and 1 Hz. Gel1.5M displayed a very narrow linear viscoelastic range, where the G′ and G′′ had the same magnitude, indicating the formation of a weak gel. By increasing the concentration of DACNC, the linear viscoelastic regime broadened significantly to 400% and G′ was much larger than G′′. The deviation from linearity is a result of the breakdown of the dynamic cross-linked hydrogel.12 Temperature sweep experiments were also conducted to investigate the effect of temperature on the rheological properties of the hydrogels containing different amounts of DACNC (Fig. 6b). By decreasing the DACNC concentration to 2.25, a two-step increase in both G′ and G′′ was observed (Fig. 6b). The second step might be attributed to the coil to globule transition and association of the end blocks of POEGMA-H chains that were free due to insufficient DACNC nanoparticles. The increase in the amplitude for the second step (∼60–65 °C) of G′ from 180 Pa in Gel2.25M to 550 Pa in Gel1.5M confirmed this hypothesis, since there were more unassociated end-blocks in Gel1.5M. The reason for the single-step transition of Gel3M could then be attributed to the absence of unassociated end-blocks in this hydrogel. The temperature for the second step of G′ was greater than the second phase transition temperature of POEGMA-H (55 °C measured by UV-vis and μDSC) could be related to the association of the chains within the network structure or higher concentration of POEGMA-H in hydrogels compared to UV-vis and μDSC samples. The second step of G′ shifted to lower temperatures when the concentration of DACNC was reduced. Similar changes in the phase transition temperature arising from the addition of nanoparticle was reported previously.61 Gel2.25M existed as a gel over the entire temperature range (G′ > G′′) while, Gel1.5M experienced a sol–gel transition at the first phase transition temperature of the POEGMA-H. After the formation of micelles due to the coil to globule transition of the middle blocks, unlike POEGMA-H solution only, DACNCs bridged the micelles leading to the formation of a network (Fig. 6d). Increasing the amount of DACNC enhanced the gel stiffness both at low temperatures (below the phase transition) and high temperatures (above the phase transition) (Fig. S2, ESI†).
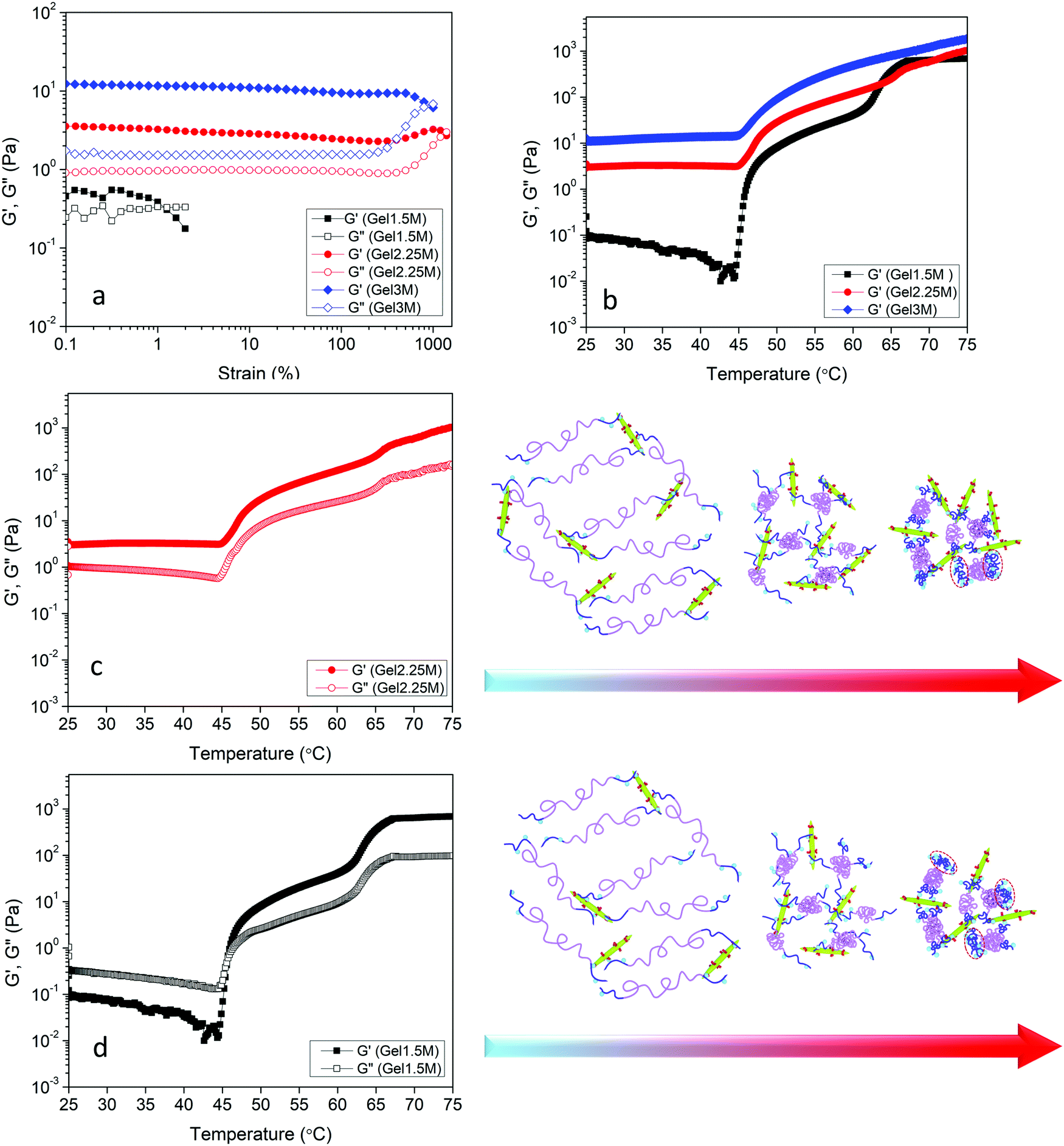 | ||
| Fig. 6 (a) Strain amplitude sweep results of Gel1.5M, Gel2.25M, and Gel3M at fixed angular frequency 1 Hz. (b) Storage modulus versus temperature of Gel1.5M, Gel2.25M, and Gel3M at 1 Hz and 1% strain. Storage and loss modulus at 1 Hz and 1% strain of (c) Gel2.25M and (d) Gel1.5M. Red dashed circled regions show aggregated globules of end blocks of PEGMA-H chains. | ||
A theoretical model based on the extensible worm-like model62,63 was employed to examine the rheological behavior of the hydrogels. This model considers the network as a collection of thermally fluctuating bundles of semi-flexible chains. Kouwer et al.64 modified the existing model to describe the thermal behavior of the storage modulus of their polyisocyanopeptide hydrogels which is similar to the present study. The plateau modulus G0 of a gel consisting of N polymer chains is dependent on the persistence length of the chains at the reference temperature lp,0 and the length lc between crosslinks:
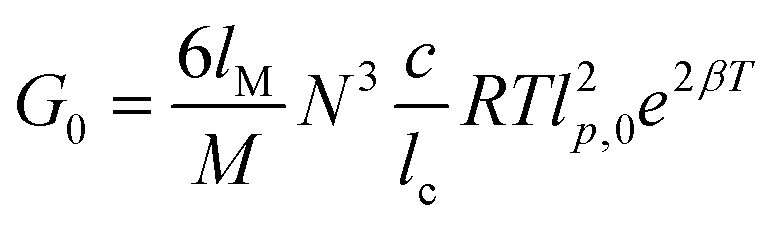 | (2) |
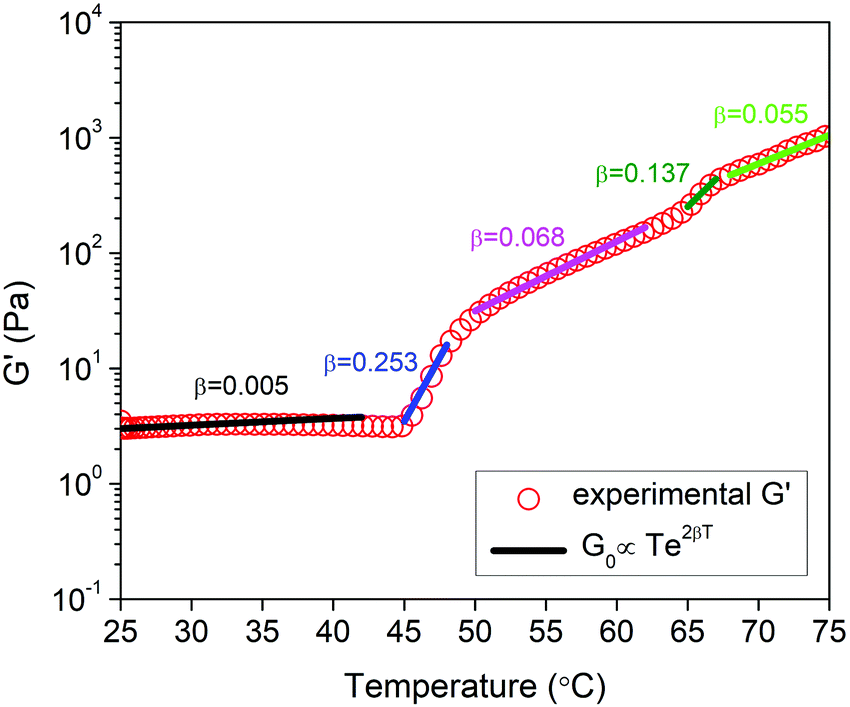 | ||
| Fig. 7 Exponential fits to experimental storage modulus data of Gel2.25M. | ||
The effect of pH on the rheological properties of hydrogels was also investigated. First, the effect of pH on the phase transition of POEGMA-H aqueous solution was investigated (Fig. S3, ESI†). Changing the pH appeared to have a minor effect on the temperature and shape of UV-vis spectra. Slight increase in the phase transition temperatures in acidic and basic conditions compared to neutral pH were observed for POEGMA-H. Similar increase of the phase transition temperature in acidic and basic pHs compared to pH 7 was reported for POEGMA and this was attributed to the slightly stronger interactions between PEG side chains of POEGMA and hydroxide ions or hydronium protons compared to water.65 The linear viscoelastic regime of the hydrogel containing the highest content of DACNC was evaluated with respect to pH at room temperature and 1 Hz (Fig. S4, ESI†). At low and medium pHs, the linear viscoelastic region of the hydrogels was broad (up to 400%) with G′ > G′′. The deviation from linearity is a result of the breakdown of the cross-links in the hydrogel.12 By increasing the pH, the linear viscoelastic range was dramatically reduced to below 1% and G′ and G′′ were close, indicating the formation of a weak gel (Fig. S4, ESI†). Temperature sweep experiments at 1 Hz and 1% strain were also conducted to investigate the effect of pH on G′ and G′′. The storage modulus of Gel3L showed a single-step increase at the first phase transition temperature of POEGMA-H, behavior which resembled that of the Gel3M (Fig. 8a). The storage modulus of Gel3M below the phase transition was greater than that reported for Gel3L. The rate of formation of acylhydrazone bonds increased significantly with decreasing pH values, thus, the gelation occurred much faster at low pH leading to the formation of an irregular transient network. The lower gelation rate at medium pH facilitated the development of a more regular network closer to the equilibrium (Fig. 8a). At high pH condition, very slow gelation rate was observed, leading to a loose network compared to the low and medium pH conditions (Fig. 7a and Fig. S5, ESI†). Unlike low and medium pHs, a sol–gel transition was observed at the first phase transition temperature of the system at high pH, where the formation of acylhydrazone bonds was inhibited, and only physical interactions were present in Gel3H. The micelle formation due to the coil to globule transition of the middle blocks of POEGMA-H chains at the first phase transition temperature and bridging of the micelles by DACNCs through physical interactions were responsible for the sol–gel transition. The second step in the moduli increase is associated with the coil to globule transition of the end blocks of the chains due to the lack of acylhydrazone bond formation. This increase for Gel3H occurred at higher temperature compared to Gel1.5M and Gel2.25M that could be related to the higher concentration of DACNC61 and higher pH of the system (Fig. S3, ESI†).
 | ||
| Fig. 8 (a) Storage modulus versus temperature of Gel3L, Gel3M, and Gel3H at 1 Hz and 1% strain. Storage and loss modulus at 1 Hz and 1% strain of (b) Gel3L and (c) Gel3H. Red dashed circled regions show aggregated globules of end blocks of PEGMA-H chains. | ||
Rheological recovery tests were employed to evaluate the self-healing capability of the hydrogels.17,19,50 Measurements of the linear viscoelastic regime of the hydrogels revealed the critical strain where the dynamic network of hydrogels was destroyed (Fig. S4, ESI†). In order to assess the restoration of the hydrogel network after destruction, step-strain tests were conducted at a fixed angular frequency 1 Hz (Fig. 9a). When the Gel3M was subjected to a small strain in the linear viscoelastic region (1%) for 100 s, G′ was greater than G′′, and they remained constant over the duration of the tests, indicating a stable hydrogel. By increasing the strain to a value greater than the linear viscoelastic threshold (2000%) for 100 s, G′′ became larger than G′ indicating that the hydrogel was liquefied. After 100 s rest, the strain was reduced to 1% again, and as a result, hydrogel was reformed (G′ > G′′) and storage modulus was completely recovered. This cycle was repeated 2 more times to ensure the reproducibility of the results (Fig. 8a). The effect of pH was also studied on the self-healing behavior of the hydrogel. Similar step-strain experiments were performed on Gel3L and Gel3H. For Gel3H, the second strain was 20% that was much greater than the linear viscoelastic threshold (Fig. S4, ESI†). Interestingly, the storage modulus of Gel3L was increased via reformation after the network was disrupted (Fig. 9b). This might be due to the attainment of a more regular network that was closer to equilibrium after each destruction and reformation of the network as schematically represented in Fig. 9b. However, for Gel3H, the storage modulus was not restored after the destruction of the network which suggested the lack of restoration capability due to a very slow rate of formation of acylhydrazone bonds at high pH (Fig. 9c).
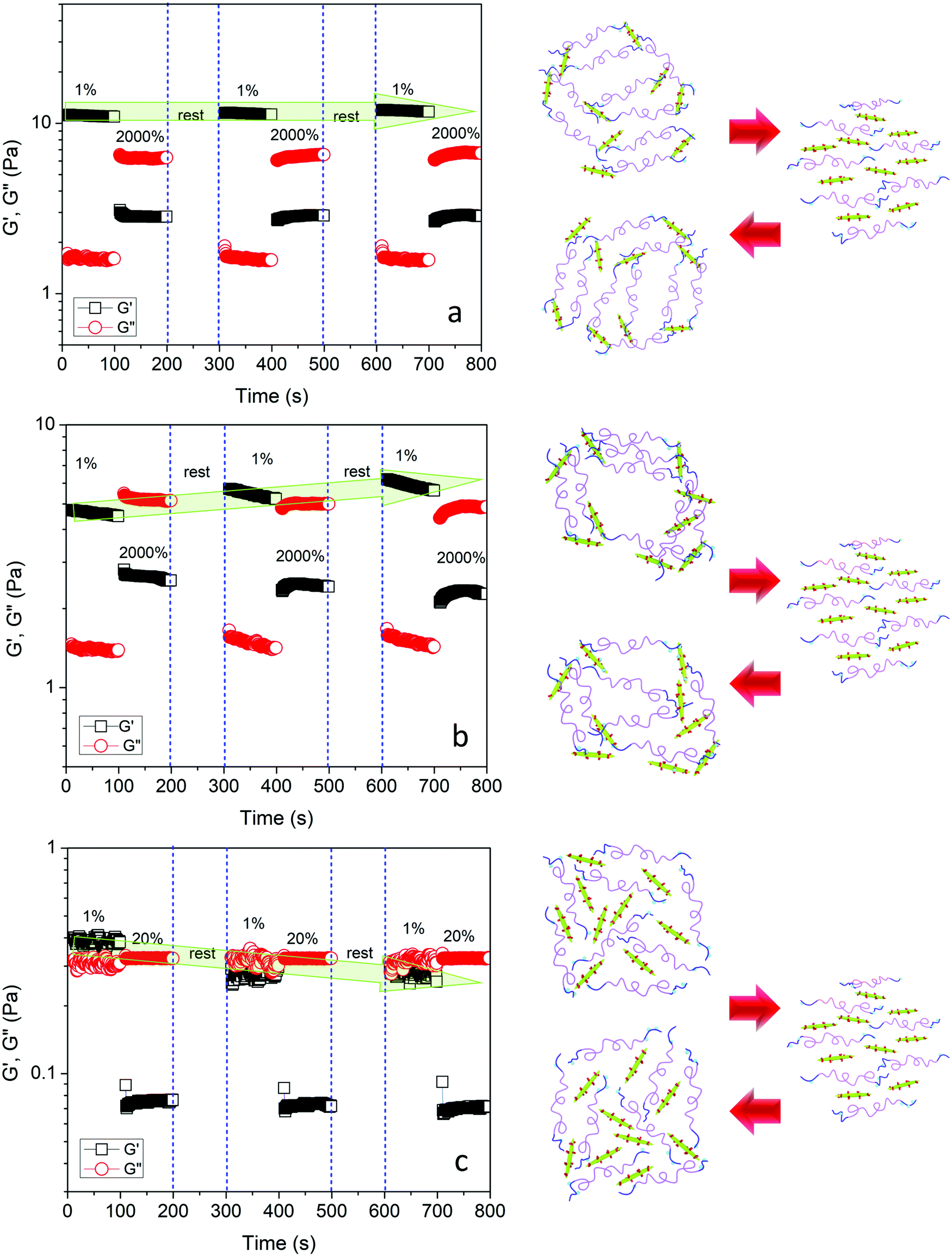 | ||
| Fig. 9 Step-strain experiments by switching from small strain (1%) to large strain (2000% or 20%) at a fixed angular frequency 1 Hz along with the schematic representation of the experiment for (a) Gel3M, (b) Gel3L, and (c) Gel3H. | ||
4. Conclusions
A thermo-responsive self-healable hydrogel was developed via the formation of acylhydrazone bonds between the dialdehyde CNC and hydrazide functionalized POEGMA. AGET ATRP was used to synthesize the POEEGMA-MAA and functionalized with the hydrazide groups through hydrazinolysis of MA units. Investigation of thermo-responsive behavior of POEGMA-H revealed a two-step phase transition with increasing temperature due to the coil to globule transition of middle and end blocks of the polymer chains. Rheological measurements showed that the coil to globule transition of copolymer chains at elevated temperatures led to a significant increase in the stiffness of the hydrogel. At low pH, an irregular network with compromised stiffness compared to medium pH was formed due to the high formation rate of the acylhydrazone bonds. However, at high pH, very slow formation of the acylhydrazone bonds led to a loose network compared to the low and medium pH conditions. The hydrogels possessed a very good self-healing capability, and they have great potential in biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KCT acknowledges the support from NSERC. The cellulose nanocrystal was kindly supplied by Celluforce Inc. Authors would also like to thank Ms Samineh Sam for her help in preparing the schematics.References
- O. P. Oommen, S. Wang, M. Kisiel, M. Sloff, J. Hilborn and O. P. Varghese, Adv. Funct. Mater., 2013, 23, 1273–1280 CrossRef CAS.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- Y.-H. Ma, J. Yang, B. Li, Y.-W. Jiang, X. Lu and Z. Chen, Polym. Chem., 2016, 7, 2037–2044 RSC.
- Y.-M. Kim, C.-H. Kim and S.-C. Song, ACS Macro Lett., 2016, 5, 297–300 CrossRef CAS.
- H. Thérien-Aubin, Y. Wang, K. Nothdurft, E. Prince, S. Cho and E. Kumacheva, Biomacromolecules, 2016, 17, 3244–3251 CrossRef PubMed.
- Y. Li, N. Khuu, A. Gevorkian, S. Sarjinsky, H. Therien-Aubin, Y. Wang, S. Cho and E. Kumacheva, Angew. Chem., Int. Ed., 2017, 56, 6083–6087 CrossRef CAS PubMed.
- Y. Hu, J.-O. You, D. T. Auguste, Z. Suo and J. J. Vlassak, J. Mater. Res., 2012, 27, 152–160 CrossRef CAS.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- X. Hu, Y. Wang, L. Zhang, M. Xu, J. Zhang and W. Dong, Int. J. Biol. Macromolecules, 2018, 108, 558–567 CrossRef CAS PubMed.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Acta Biomater., 2014, 10, 4143–4155 CrossRef CAS PubMed.
- I. Urosev, E. Bakaic, R. J. Alsop, M. C. Rheinstädter and T. Hoare, J. Mater. Chem. B, 2016, 4, 6541–6551 RSC.
- J. R. McKee, E. A. Appel, J. Seitsonen, E. Kontturi, O. A. Scherman and O. Ikkala, Adv. Funct. Mater., 2014, 24, 2706–2713 CrossRef CAS.
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- Y.-J. Liu, W.-T. Cao, M.-G. Ma and P. Wan, ACS Appl. Mater. Interfaces, 2017, 9, 25559–25570 CrossRef CAS PubMed.
- M. Zhu, S. He, Y. Dai, J. Han, L. Gan, J. Liu and M. Long, ACS Sustainable Chem. Eng., 2018, 6, 17087–17098 CrossRef CAS.
- L. Bai, X. Jiang, Z. Sun, Z. Pei, A. Ma, W. Wang, H. Chen, H. Yang, L. Yang and D. Wei, Cellulose, 2019, 26, 5305–5319 CrossRef CAS.
- C. Shao, L. Meng, M. Wang, C. Cui, B. Wang, C.-R. Han, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 5885–5895 CrossRef CAS PubMed.
- W. Li, S. Lu, M. Zhao, X. Lin, M. Zhang, H. Xiao, K. Liu, L. Huang, L. Chen, X. Ouyang, Y. Ni and H. Wu, Polymers, 2018, 10, 1392 CrossRef PubMed.
- W. Huang, Y. Wang, Z. Huang, X. Wang, L. Chen, Y. Zhang and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 41076–41088 CrossRef CAS PubMed.
- H. Liu, C. Li, B. Wang, X. Sui, L. Wang, X. Yan, H. Xu, L. Zhang, Y. Zhong and Z. Mao, Cellulose, 2018, 25, 559–571 CrossRef CAS.
- G. Xiao, Y. Wang, H. Zhang, L. Chen and S. Fu, Carbohydr. Polym., 2019, 218, 68–77 CrossRef CAS PubMed.
- R. Chang, X. Wang, X. Li, H. An and J. Qin, ACS Appl. Mater. Interfaces, 2016, 8, 25544–25551 CrossRef CAS PubMed.
- X. Yang, G. Liu, L. Peng, J. Guo, L. Tao, J. Yuan, C. Chang, Y. Wei and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703174 CrossRef.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- M. Patenaude, N. M. B. Smeets and T. Hoare, Macromol. Rapid Commun., 2014, 35, 598–617 CrossRef CAS PubMed.
- N. Grishkewich, N. Mohammed, J. Tang and K. C. Tam, Curr. Opin. Colloid Interface Sci., 2017, 29, 32–45 CrossRef CAS.
- E. Lam, K. B. Male, J. H. Chong, A. C. W. Leung and J. H. T. Luong, Trends Biotechnol., 2012, 30, 283–290 CrossRef CAS PubMed.
- R. Nasseri, C. P. Deutschman, L. Han, M. A. Pope and K. C. Tam, Mater. Today Adv., 2020, 5, 100055 CrossRef.
- B. Peng, N. Grishkewich, Z. Yao, X. Han, H. Liu and K. C. Tam, ACS Macro Lett., 2012, 1, 632–635 CrossRef CAS.
- E. Bakaic, N. M. B. Smeets and T. Hoare, RSC Adv., 2015, 5, 35469–35486 RSC.
- J.-F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J.-F. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Chem. Commun., 2014, 50, 3306–3309 RSC.
- E. Bakaic, N. M. B. Smeets, H. Dorrington and T. Hoare, RSC Adv., 2015, 5, 33364–33376 RSC.
- I. Urosev, H. Dorrington, N. Muzzin, R. Alsop, E. Bakaic, T. Gilbert, M. Rheinstädter and T. Hoare, ACS Appl. Polym. Mater., 2019, 1, 369–380 CrossRef CAS.
- X. Yang, E. Bakaic, T. Hoare and E. D. Cranston, Biomacromolecules, 2013, 14, 4447–4455 CrossRef CAS PubMed.
- E. Larsson, A. Boujemaoui, E. Malmström and A. Carlmark, RSC Adv., 2015, 5, 77643–77650 RSC.
- R. Cha, Z. He and Y. Ni, Carbohydr. Polym., 2012, 88, 713–718 CrossRef CAS.
- J. Tang, M. U. Javaid, C. Pan, G. Yu, R. M. Berry and K. C. Tam, Carbohydr. Polym., 2020, 229, 115486 CrossRef PubMed.
- J. Yang, C.-R. Han, X.-M. Zhang, F. Xu and R.-C. Sun, Macromolecules, 2014, 47, 4077–4086 CrossRef CAS.
- J. R. McKee, S. Hietala, J. Seitsonen, J. Laine, E. Kontturi and O. Ikkala, ACS Macro Lett., 2014, 3, 266–270 CrossRef CAS.
- J. Wei, Y. Chen, H. Liu, C. Du, H. Yu and Z. Zhou, Carbohydr. Polym., 2016, 147, 201–207 CrossRef CAS PubMed.
- M. C. B. de Figueirêdo, M. d. F. Rosa, C. M. L. Ugaya, M. d. S. M. d. Souza Filho, A. C. C. d. Silva Braid and L. F. L. d. Melo, J. Cleaner Prod., 2012, 35, 130–139 CrossRef.
- K. A. Mahmoud, J. A. Mena, K. B. Male, S. Hrapovic, A. Kamen and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2010, 2, 2924–2932 CrossRef CAS PubMed.
- T. Kovacs, V. Naish, B. O'Connor, C. Blaise, F. Gagné, L. Hall, V. Trudeau and P. Martel, Nanotoxicology, 2010, 4, 255–270 CrossRef CAS PubMed.
- E. Gicquel, C. Martin, Q. Gauthier, J. Engström, C. Abbattista, A. Carlmark, E. D. Cranston, B. Jean and J. Bras, Biomacromolecules, 2019, 20, 2545–2556 CrossRef CAS PubMed.
- K. J. De France, K. J. W. Chan, E. D. Cranston and T. Hoare, Biomacromolecules, 2016, 17, 649–660 CrossRef CAS PubMed.
- Z. Guo, W. Ma, H. Gu, Y. Feng, Z. He, Q. Chen, X. Mao, J. Zhang and L. Zheng, Soft Matter, 2017, 13, 7371–7380 RSC.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- T. Lu, Q. Li, W. Chen and H. Yu, Compos. Sci. Technol., 2014, 94, 132–138 CrossRef CAS.
- A. Simula, V. Nikolaou, A. Anastasaki, F. Alsubaie, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Polym. Chem., 2015, 6, 2226–2233 RSC.
- N. M. B. Smeets, M. Patenaude, D. Kinio, F. M. Yavitt, E. Bakaic, F.-C. Yang, M. Rheinstädter and T. Hoare, Polym. Chem., 2014, 5, 6811–6823 RSC.
- N. Grishkewich, S. P. Akhlaghi, Y. Zhaoling, R. Berry and K. C. Tam, Carbohydr. Polym., 2016, 144, 215–222 CrossRef CAS PubMed.
- S.-i. Yamamoto, J. Pietrasik and K. Matyjaszewski, Macromolecules, 2008, 41, 7013–7020 CrossRef CAS.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6095–6104 CrossRef CAS.
- Á. Szabó, I. Szanka, G. Tolnai, G. Szarka and B. Iván, Polymer, 2017, 111, 61–66 CrossRef.
- Z. L. Yao and K. C. Tam, Polymer, 2012, 53, 3446–3453 CrossRef CAS.
- Y. Li, Z. Ye, L. Shen, Y. Xu, A. Zhu, P. Wu and Z. An, Macromolecules, 2016, 49, 3038–3048 CrossRef CAS.
- A. Gharachorlou and F. Goharpey, Macromolecules, 2008, 41, 3276–3283 CrossRef CAS.
- M. L. Gardel, J. H. Shin, F. C. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Science, 2004, 304, 1301–1305 CrossRef CAS PubMed.
- F. C. MacKintosh, J. Käs and P. A. Janmey, Phys. Rev. Lett., 1995, 75, 4425–4428 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00397b |
| This journal is © The Royal Society of Chemistry 2020 |