DOI:
10.1039/D0MA00327A
(Review Article)
Mater. Adv., 2020,
1, 2155-2162
Visible-light-driven organic transformations integrated with H2 production on semiconductors
Received
18th May 2020
, Accepted 27th July 2020
First published on 27th July 2020
Abstract
Due to its clean and sustainable nature, solar energy has been widely recognized as a green energy source in driving a variety of reactions, ranging from small molecule activation and organic transformation to biomass valorization. Within this context, organic reactions coupled with H2 evolution via semiconductor-based photocatalytic systems under visible light irradiation have gained increasing attention in recent years, which utilize both excited electrons and holes generated on semiconductors and produce two types of value-added products, organics and H2, simultaneously. Based on the nature of the organic reactions, in this review article we classify semiconductor-based photocatalytic organic transformations and H2 evolution into three categories: (i) photocatalytic organic oxidation reactions coupled with H2 production, including oxidative upgrading of alcohols and biomass-derived intermediate compounds; (ii) photocatalytic oxidative coupling reactions integrated with H2 generation, such as C–C, C–N, and S–S coupling reactions; and (iii) photo-reforming reactions together with H2 formation using organic plastics, pollutants, and biomass as the substrates. Representative heterogeneous photocatalytic systems will be highlighted. Specific emphasis will be placed on their synthesis, characterization, and photocatalytic mechanism, as well as the organic reaction scope and practical application.
1. Introduction
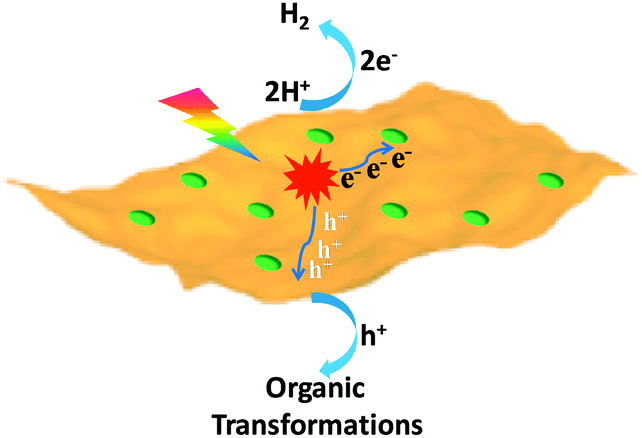
The exploration of sustainable and renewable energy sources has received increasing attention in order to reduce our dependence on finite and environmentally unfriendly fossil reserves.1–3 Solar energy, as one of the most green and abundant renewable energy sources, represents a promising candidate, which can be directly converted to chemical energy via photocatalysis.4,5 Among various photocatalytic systems, semiconductor-based photocatalytic H2 production integrated with organic transformations not only directly produces H2 as the cleanest energy carrier and chemical fuel but also has potential to generate value-added organic products.6–9 In this process, photo-excited electrons of irradiated semiconductors reduce protons to yield H2, while the resulting holes simultaneously drive oxidative organic transformations (Scheme 1).
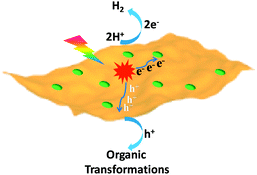 |
| | Scheme 1 Schematic representation of the dual function of a semiconductor-based photocatalyst for integrated H2 evolution and organic transformations. | |
Given the fact that the major component of the solar spectrum falls in the range of 400–700 nm, it is critical to develop visible light-absorbing photocatalytic systems in order to effectively capture solar irradiation and hence achieve a high solar energy conversion efficiency.10–12 In this regard, homogeneous photocatalysts consisting of transition metal complexes or organic dye molecules whose absorption spectra cover the visible region have been widely investigated for photocatalytic H2 evolution and organic transformations. Nevertheless, the challenging separation of these molecular photocatalysts and/or sensitizers from reaction media, together with their questionable long-term stability, represents apparent drawbacks of their practical application on a large scale.11 Comparatively, heterogeneous photocatalysts can be phase-separated from most products and also tend to exhibit better durability. Hence, visible light-responsive heterogeneous photocatalysts are highly desirable for H2 production coupled with organic oxidations. Among a wide array of heterogeneous photocatalysts, semiconductors ranging from transition metal oxides and sulphides to metal-free carbon nitrides have emerged as the most representative visible light-active solid-state photocatalysts.13–15
In the last several years, the research field of semiconductor-based photocatalysis for selective organic transformations integrated with the production of clean and carbon-free H2 has made significant advancements.6,7,9,16 A diverse array of organic reactions coupled with H2 production have been realized such as the oxidation of alcohols and biomass-derived intermediate compounds, C–C, C–N, and S–S coupling reactions, degradation of plastics and pollutants, and biomass photo-reforming reactions. In this review, we focus on visible light-driven photocatalysis consisting of semiconductors and aim to summarize the recent progress in this infant research field. Specifically, we will discuss the synthesis, characterization, and photocatalytic mechanism, and performance of each photocatalytic system in specific organic reactions. The current advances and critical issues in this field will also be highlighted.
2. Photocatalytic organic oxidation coupled with H2 production
2.1 Integration of H2 production with the oxidation of alkyl or aryl alcohols
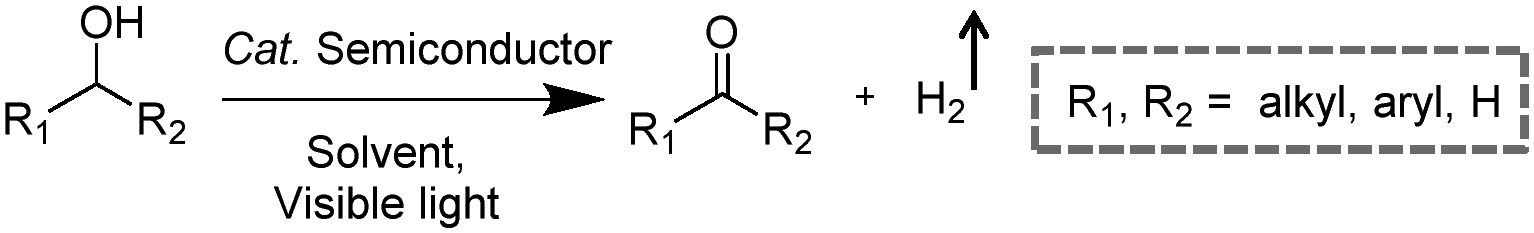
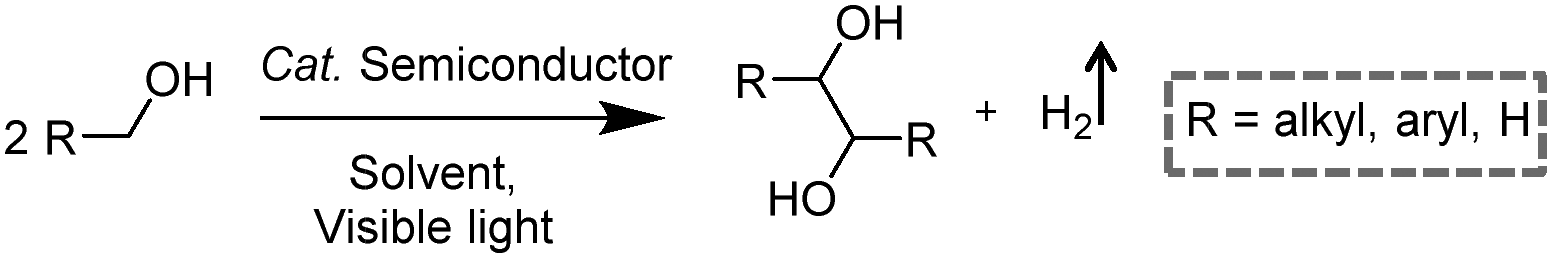
Visible-light-driven photocatalytic oxidation of alcohols to their corresponding carbonyl compounds with concurrent H2 generation is a promising strategy for solar energy conversion and storage in value-added chemicals and fuels. Recently, a large amount of semiconductor-based photocatalysts have been developed for oxidizing alkyl or aryl alcohols to aldehydes and/or ketones coupled with H2 production by simultaneously utilizing photo-generated electrons and holes (Scheme 2).
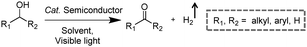 |
| | Scheme 2 Representative photocatalytic oxidation of alcohols. | |
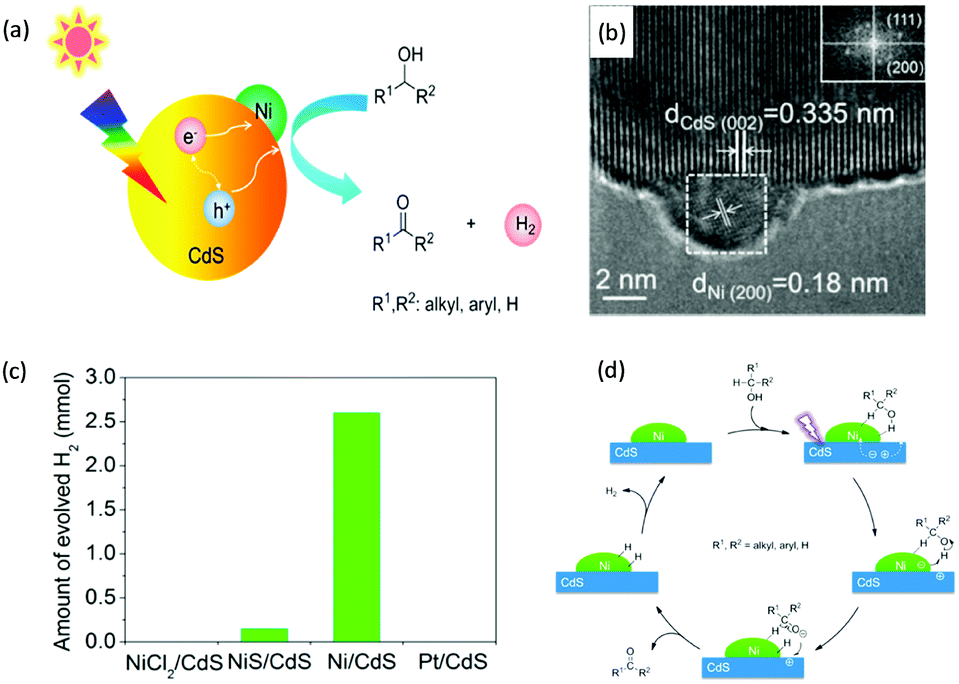
In 2013, Higashimoto et al. reported a Pd-deposited CdS–TiO2 photocatalyst for the selective oxidation of benzyl alcohol to benzaldehyde accompanied by H2 evolution under visible light irradiation.17 The synergistic effects of CdS, TiO2, and Pd species enhanced the photocatalytic activities with high selectivity (>99%), wherein CdS–TiO2 works as the photo-responsive center, while the Pd species act as the co-catalyst. Moreover, this photocatalytic system was able to drive the oxidation of a large scope of benzyl alcohol derivatives with substituents from electron-donating (p-OH, m-OCH3 and p-CH3) to electron-withdrawing groups (p-Cl). Recently, a noble-metal-free Ni/CdS photocatalyst (Ni nanocrystal-modified CdS nanoparticles) for transforming alcohols to corresponding aldehydes or ketones and H2 was reported by Xu and co-workers (Fig. 1a).18In situ photodeposition was employed to decorate the CdS surface with Ni nanocrystals to prepare the target photocatalyst. The high-resolution transmission electron microscopy (HRTEM) image of Ni/CdS indicated that metallic Ni nanoparticles with lattice distances of (111) and (200) were successfully deposited on the CdS surface (Fig. 1b). As shown in Fig. 1c, a series of control samples were investigated to probe the impact of various co-catalysts on CdS, which revealed the important role played by the synergistic effect between Ni and CdS. Accordingly, the authors proposed a photocatalytic mechanism of Ni/CdS for efficient alcohol oxidation coupled with H2 production. As shown in Fig. 1d, firstly, a photo-excited electron on CdS reacts with a proton that is abstracted from the alcohol substrate to afford Ni–H and an alkoxide anion. The alkoxide anion is then oxidized by the photo-excited hole to generate another Ni–H and produce the corresponding aldehyde or ketone. Finally, two resulting Ni–H form one H2.
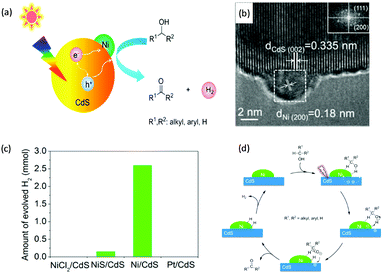 |
| | Fig. 1 (a) Schematic representation of visible light-driven alcohol oxidation coupled with H2 evolution on a Ni/CdS photocatalyst. (b) HRTEM image of Ni/CdS (the inset image is the Fourier transform of the marked area). (c) Produced H2 amount during the photocatalytic oxidation of 2-propanol using different photocatalysts. (d) Proposed mechanism for the photocatalytic alcohol oxidation coupled with H2 evolution. Reprinted from ref. 18 with permission from American Chemical Society, copyright 2016. | |
Besides CdS-based photocatalysts,17–21 several other categories of semiconductor-based photocatalysts have also been investigated for the selective oxidation of alcohols, such as Zn3In2S6,22 g-C3N4,23 and SrTiO3.24 For instance, Zn3In2S6 photocatalysts were prepared via a facile solvothermal method for the selective transformation of aromatic alcohols to aldehydes and H2 under visible light irradiation.22 Recently, Wu and co-workers unveiled a CdSe quantum dot photocatalyst capped with 3-mercaptopropionic acid (denoted as MPA-CdSe QD) for efficient photo-oxidation of alcohols to carbonyl compounds. Upon visible light irradiation, a wide array of alcohols can be selectively oxidized to the corresponding aldehydes/ketones using Ni2+ ions as a H2 evolution cocatalyst, and 3-mercaptopropionic acid and water as relay reagents.25
2.2 Integration of H2 production with oxidative valorization of biomass-derived intermediate compounds
Following the growing interest in exploring green carbon resources as an alternative to fossil reserves, biomass valorization has attracted increasing attention. Consequently, a large amount of effort has been devoted to developing photocatalytic systems for the solar-driven upgrading of biomass-derived intermediate compounds.26–31 Compared to the use of traditional hole scavengers in photocatalytic H2 production, which inevitably leads to the increase of cost and production of waste, renewable biomass and biomass-based intermediates are not only able to consume the photo-excited holes but also produce value-added chemicals in addition to H2. To date, considerable advances have been achieved in this emerging research field (Scheme 3).
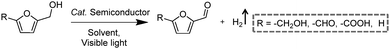 |
| | Scheme 3 Representative photocatalytic oxidation of biomass-derived intermediate compounds. | |
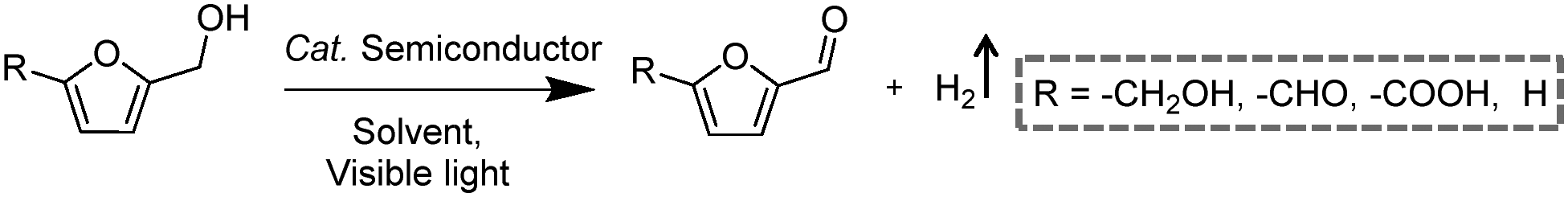
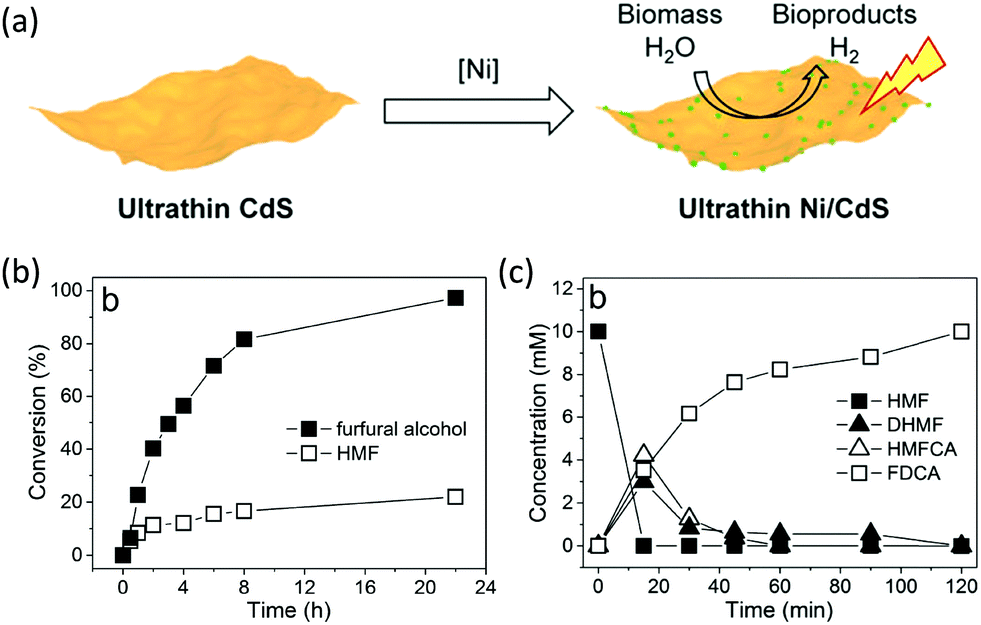
Among many biomass-derived intermediate compounds, furanics are considered as platform chemicals in that they can be converted to a variety of valuable products. Sun et al. reported the selective upgrading of furfural alcohol and 5-hydroxymethylfurfural (HMF) to their corresponding aldehydes and acids with concomitant production of H2 utilizing ultrathin Ni/CdS nanosheets as the co-photocatalysts (Fig. 2).30 In neutral water, both furfural alcohol and HMF were transformed to aldehydes with distinct reaction rates as rationalized by theoretical calculations. It was found that HMF exhibited a much slower conversion rate, probably due to the slightly stronger binding affinity of the aldehyde group in HMF relative to its alcohol group as supported by the theoretical computation results. In comparison, quantitative carboxylate products could be obtained from both furfural alcohol and HMF if the photocatalysis was conducted under alkaline conditions. Therefore, it was feasible to selectively produce either furanic-derived aldehydes or acids via fine tuning the reaction conditions.
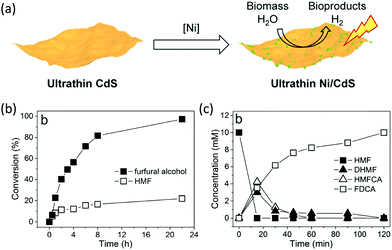 |
| | Fig. 2 (a) Synthetic route of ultrathin Ni/CdS nanosheets for integrated H2 production and biomass valorization. (b) Concentration change over time for the photocatalytic oxidation of furfural alcohol and HMF under neutral conditions. (c) Concentration change over time for the photocatalysis of HMF under alkaline conditions. Reprinted from ref. 30 with permission from American Chemical Society, copyright 2017. | |
By using a low-temperature wet chemistry method, Xu et al. prepared a photocatalyst of Ti3C2Tx/CdS, which was also able to drive the photo-oxidation of furfural alcohol to furfural together with the production of H2.27 Ti3C2Tx was used to capture the photo-generated electrons in CdS, accelerating the separation and transport of photo-excited charge carriers and enhancing the photocatalytic performance. Kailasam and co-workers reported the selective oxidation of HMF to 2,5-diformylfuran (DFF) with >99% selectivity by using porous carbon nitride as a photocatalyst and Pt as a co-catalyst.28 Porous carbon nitride absorbs visible light to generate holes and electrons. Pt facilitates charge separation and improves the production of H2. The photocatalytic reaction achieved a DFF yield of 13.8% and a H2 production rate of 12 mmol h−1 m−2 after 6 h irradiation with visible light. Furthermore, a 7.2% yield of DFF production and a 6.2 mmol h−1 m−2 rate of H2 evolution were obtained under natural sunlight for 6 h.
3. Photocatalytic oxidative coupling reactions integrated with H2 generation
3.1 Integration of H2 production with C–C coupling reactions
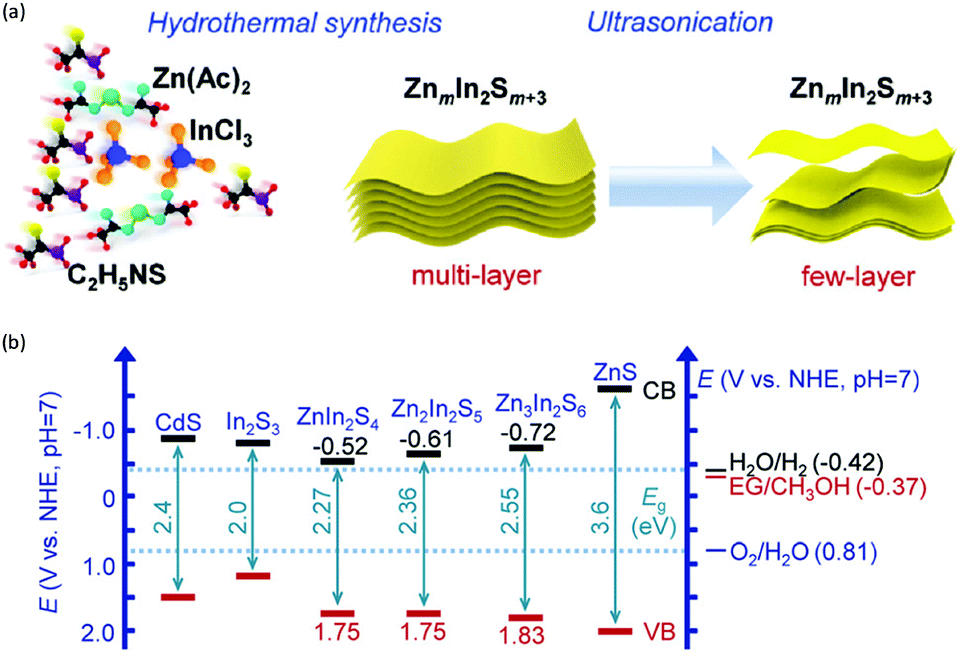
In addition to the unimolecular oxidation reactions for the consumption of photo-generated holes, bimolecular oxidative coupling reactions can also be achieved on semiconductors under visible light irradiation with simultaneous H2 production (Scheme 4).32–35 For instance, Xie and co-workers reported a CdS nanorod photocatalyst decorated with MoS2 nanofoams for the direct conversion of methanol to ethylene glycol (EG) with 90% selectivity and high efficiency together with H2 production upon visible light irradiation.33 CdS enables the facile generation of ˙CH2OH for subsequent C–C coupling and the MoS2 nanofoam co-catalyst improves the evolution of H2 and the overall photocatalytic activity. A concerted proton–electron transfer mechanism was proposed wherein a hydroxymethyl radical (˙CH2OH) intermediate was formed after the selective activation of the C–H bond in methanol. The weak adsorption of ˙CH2OH and CH3OH on the catalyst surface facilitates the C–C coupling reaction and decreases the possibility of O–H bond activation of CH3OH. Very recently, as shown in Fig. 3a, the same group developed an environmentally friendly CoP/Zn2In2S5 catalyst via a simple low-temperature hydrothermal and ultrasonic method for transforming methanol to EG under visible-light irradiation without using the toxic Cd metal element.32 CoP in the hybrid CoP/Zn2In2S5 photocatalyst facilitates charge separation and promotes its photocatalytic performance for the formation of both EG and H2. Zn2In2S5 was demonstrated to selectively activate the α-C–H bond in alcohols without affecting the O–H group. Besides, this catalytic system was also demonstrated to be effective for the coupling of ethanol to 2,3-butanediol arising from the formation of the ˙CH(OH)CH3 radical. Mechanistic studies indicated that the redox potential of ZnmIn2Sm+3 was substantially more positive than that of EG/CH3OH (Fig. 3b), which provided sufficient redox power for the oxidative coupling of methanol to EG and ethanol to 2,3-butanediol with the co-production of H2.
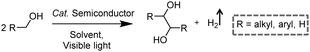 |
| | Scheme 4 Representative photocatalytic C–C coupling reaction. | |
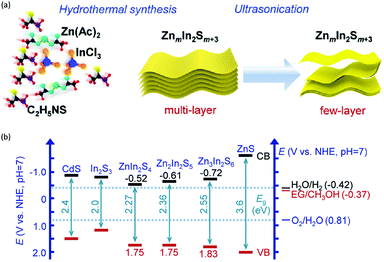 |
| | Fig. 3 (a) Schematic illustration of the low-temperature hydrothermal and ultrasonic process used to synthesize ZnmIn2Sm+3. (b) Energy levels of several related redox couples and band-edge positions of selected semiconductors. Reprinted from ref. 32 with permission from Royal Society of Chemistry, copyright 2020. | |
Besides alcohols, C–H bonds in furanics could also be activated for C–C coupling reactions on semiconductor-based photocatalysts. Recently, Wang et al. reported a Ru-doped ZnIn2S4 photocatalyst, which was prepared following a one-pot hydrothermal method, in order to achieve the dehydrogenative C–C coupling of lignocellulose-derived methylfurans to diesel fuel precursors and H2 under visible light irradiation.34 Benefiting from the Ru dopants to improve the light harvesting and charge separation efficiency, this Ru–ZnIn2S4 photocatalyst showed decent performance with a diesel fuel precursor production rate of 1.04 g gcatalyst−1 h−1 and high selectivity (>96%), as well as a H2 production rate of 6.0 mmol gcatalyst−1 h−1. Subsequent hydrodeoxygenation treatment of the obtained coupling products of furanics would result in the desired diesel fuels containing straight- and branched-chain alkanes. By modulating the Zn/In ratio in the ZnIn sulfides, the same group also demonstrated controllable production of deoxybenzoin or benzoin from benzyl alcohol integrated with H2 evolution under visible light irradiation.35 It's worth noting that they successfully synthesized deoxybenzoin derivatives on a gram scale by using this photocatalytic system.
3.2 Integration of H2 production with C–N coupling reactions
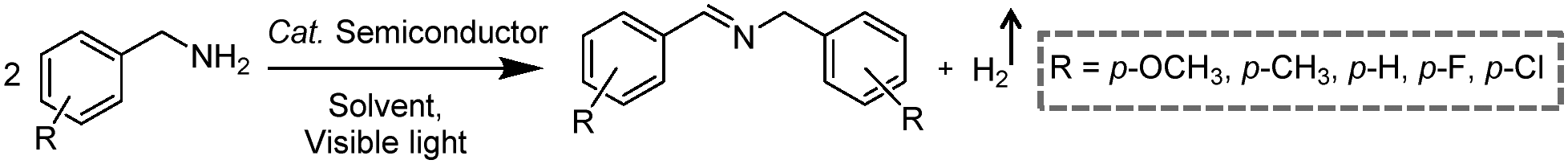
Photocatalytic C–N coupling of amines integrated with H2 production on semiconductors has emerged as a mild and green method for the efficient synthesis of imines, which have important applications in the research fields of pharmaceuticals and agricultural chemistry (Scheme 5).36–38 For example, Ni/CdS was reported to be able to photocatalyze the C–N coupling of amines to produce imines under visible light irradiation, while H2 was produced in the meantime.37 Specifically, the synthesized Ni/CdS nanoparticles showed superior benzylamine conversion (close to 100%) and excellent imine selectivity (97%). The maximum H2 production rate was 21.4 mmol g−1 h−1 with an apparent quantum yield of 11.2%. Compared to the use of common sacrificial reagents such as TEOA or Na2S/Na2SO3, the H2 production rate increased two orders of magnitude when integrated with C–N coupling of amines to imines. Moreover, the obtained photocatalysts can be recycled several times without visible photocatalytic activity loss, indicating their robust stability and recyclability. A plausible mechanism was proposed that photo-excited electrons on CdS rapidly migrated to Ni nanoparticles to reduce protons to H2, while photo-excited holes generated carbon cationic species to afford the target imine product. This photocatalyst also exhibited great tolerance of a wide substrate scope with various substituents on aryl amines with outstanding photocatalytic performance.
 |
| | Scheme 5 Representative photocatalytic C–N coupling reaction. | |
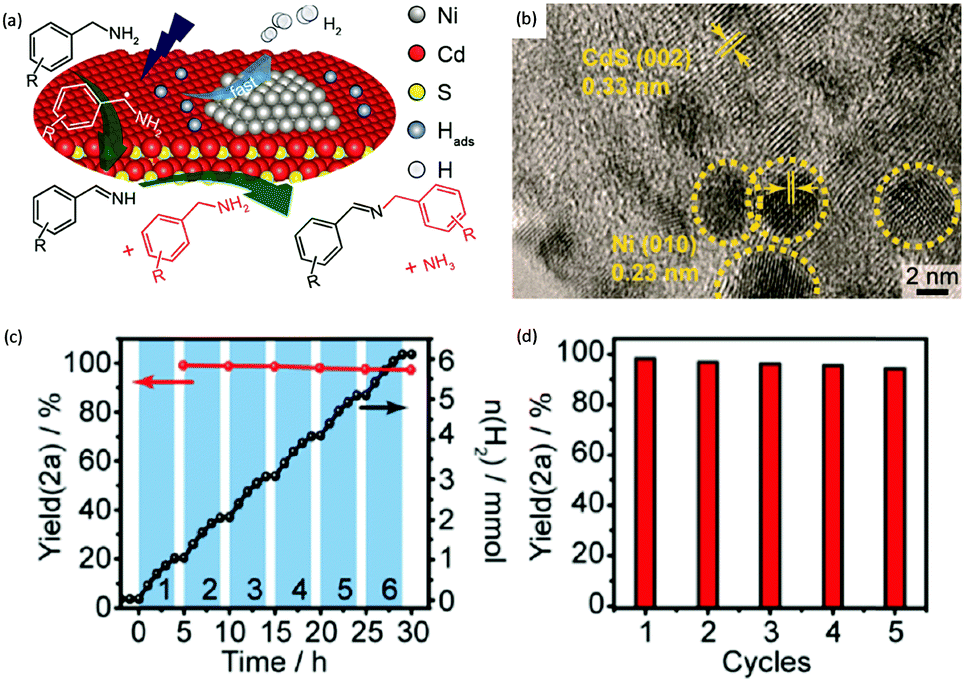
By rational design, Zhang et al. developed nickel-decorated ultrathin CdS nanosheet (Ni/CdS) photocatalysts with significantly enhanced photocatalytic performance (Fig. 4a) for the C–N coupling of amines. As shown in Fig. 4b, the high resolution transmission electron microscopy image of Ni/CdS showed the uniform deposition of Ni clusters on the two-dimensional ultrathin CdS nanosheets. A remarkable apparent quantum yield of 44% together with high efficiency and excellent stability was realized under visible light irradiation (Fig. 4c and d). The kinetic study of Hads desorption to form molecular H2 indicated that the Ni clusters on CdS were the active sites to promote the recombination of Hads. The photo-generated holes on the surface of CdS oxidize amines to aldimine intermediates and generate protons. Another amine molecule reacts with the aldimine intermediate to form the imine and NH3. A series of primary and secondary amines with different functional groups were feasible to be coupled on Ni/CdS, suggesting a great universality of the ultrathin Ni/CdS nanosheets for the C–N coupling of many amines to yield value-added chemicals.36
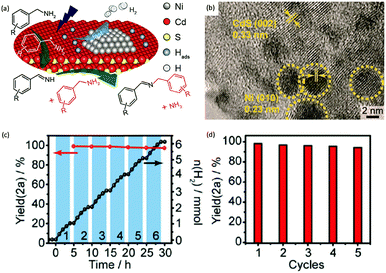 |
| | Fig. 4 (a) Schematic representation of nickel-decorated ultrathin CdS nanosheets as photocatalysts for the C–N coupling of amines to imines. (b) HRTEM of the Ni/CdS photocatalyst. (c) Photocatalytic performance of Ni/CdS for the production of imines and H2 during photolysis. (d) Stability test of Ni/CdS for five consecutive photolysis experiments. Reprinted from ref. 36 with permission from American Chemical Society, copyright 2020. | |
3.3 Integration of H2 production with S–S coupling reactions
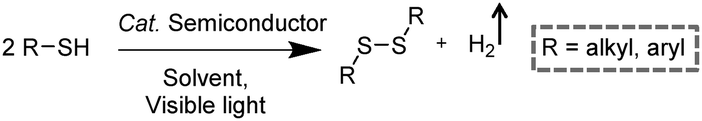
Disulfides, as a category of important chemicals that contain S–S bonds, have attracted extensive research interest in synthetic chemistry, pharmaceuticals, and materials science (Scheme 6).39 Traditional synthetic approaches of disulfides usually rely on the oxidative coupling of thiols, which are hindered by limitations like over-oxidized byproducts, complicated purification processes, and the requirement for expensive reagents.40,41 Recently, photocatalytic S–S coupling for the selective and efficient conversion of thiols into disulfides and H2 has been reported under visible-light irradiation without the requirement for any sacrificial reagent or oxidant.42,43 For instance, Li et al. reported PtS/ZnIn2S4 nanocomposites for the photocatalytic S–S coupling of thiols to produce disulfides and H2.42 The photocatalyst was prepared by photo-reduction of [PtCl6]2− on ZnIn2S4 and the resulting PtS/ZnIn2S4 displayed high stability and recyclability. The optimum photocatalytic performance of 0.5 wt% PtS/ZnIn2S4 showed a complete transformation of thiols to disulfides and H2 in 6 h under visible light irradiation without over-oxidized products. It was demonstrated that PtS acted as an efficient hydrogen evolution co-catalyst to enhance the photocatalytic behavior of PtS/ZnIn2S4 nanocomposites, which could be applied in transforming various thiols into their corresponding disulfides ranging from benzyl mercaptanes with different substituents to furfuryl mercaptan and thiophenols.
 |
| | Scheme 6 Representative photocatalytic S–S coupling reaction. | |
4. Photo-reforming reaction together with H2 generation
4.1 Integration of H2 production with the photo-reforming of organic plastics and pollutants
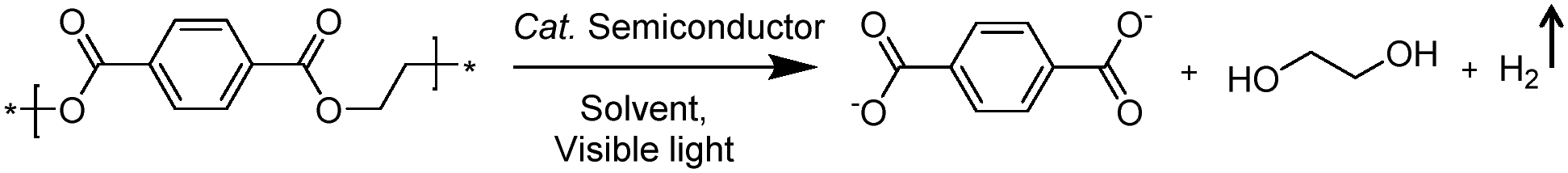
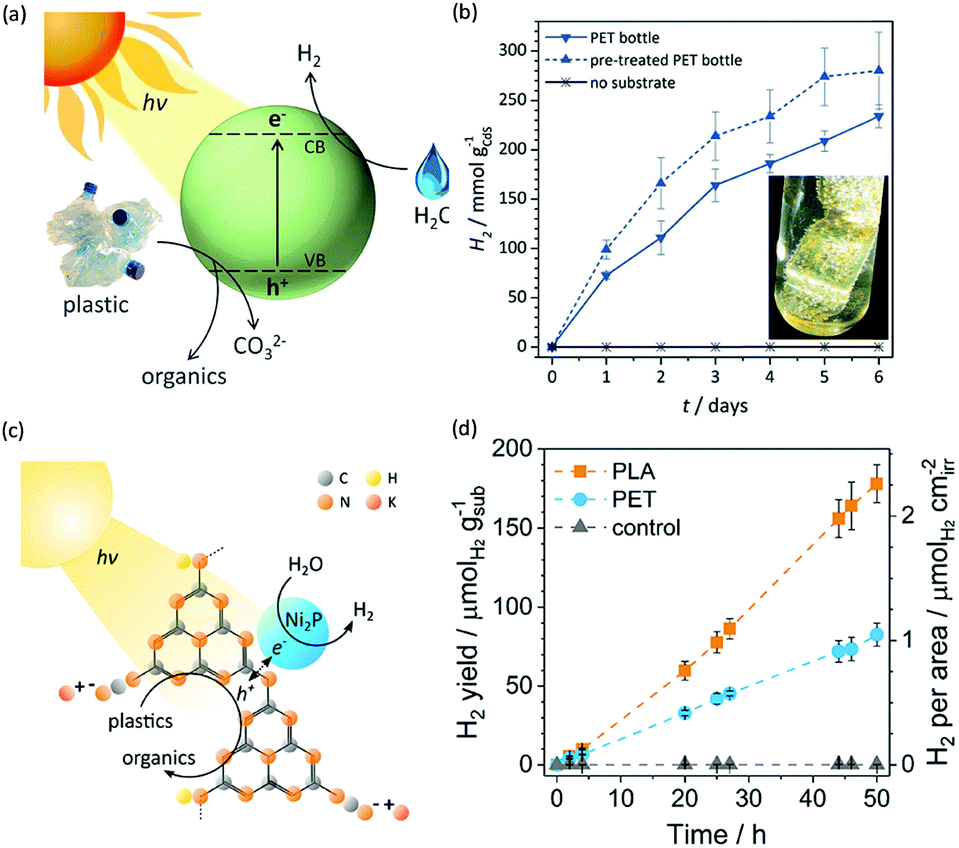
Photo-reforming of organic plastics and pollutants to less-hazardous products with the concurrent production of H2 not only reduces the global environmental crisis but also produces a green energy carrier and clean chemical feedstock, H2 (Scheme 7). As such, Reisner et al. reported inexpensive CdS/CdOx quantum dots as photocatalysts to convert plastic waste into H2 and organic products such as formate, acetate, and pyruvate via visible light-driven photo-reforming (Fig. 5a and b).44 The bulk band gap (2.4 eV) and band positions (CB: −0.5 V vs. NHE, VB: +1.9 V vs. NHE) of CdS enable its visible light absorption and photo-reforming reactions without a co-catalyst. It's worth noting that this reaction system enables photo-reforming a variety of plastics in a straightforward manner, including polyethylene terephthalate (PET), polylactic acid (PLA), and polyurethane. More attractively, even real plastic samples were tested for photo-reforming using the CdS/CdOx photocatalysts and a H2 generation activity of up to 4.13 ± 0.40 mmolH2 gCdS−1 h−1 with an external quantum yield of 2.17 ± 0.38% and conversion of 5.15 ± 0.72% was achieved under visible light irradiation for 6 days. Considering the toxicity of Cd in CdS/CdOx, the same group further developed a Cd-free carbon nitride/nickel phosphide (CNx|Ni2P) alternative for photo-reforming of plastic waste (Fig. 5c).45 As shown in Fig. 5d, photo-irradiation of PET and PLA in the presence of CNx|Ni2P for 50 h produced 82.5 ± 7.3 and 178 ± 12 μmolH2 gsub−1 H2 with turnover numbers of 7.8 ± 0.7 and 16.8 ± 1.1 molH2 molNi−1, respectively. The CNx|Ni2P photo-reforming system can also transform real-world polymer samples to H2, such as polyester microfibers and oil-contaminated PET. It was proposed that the strong binding ability between the Ni2P cocatalyst and CNx facilitated photo-induced charge separation and enhanced the photocatalytic performance.
 |
| | Scheme 7 Representative photo-reforming of polyethylene terephthalate (PET). | |
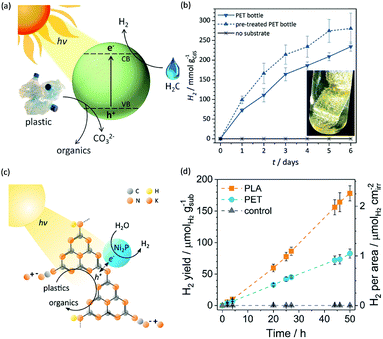 |
| | Fig. 5 (a) Schematic photo-reforming of plastic to organics via CdS/CdOx quantum dots with the co-production of H2 in alkaline aqueous media. (b) Produced H2 amount of the photo-reforming of PET bottles under simulated solar irradiation over several days. Reprinted from ref. 44 with the permission of Royal Society of Chemistry, copyright 2018. (c) Schematic diagram of the polymer photo-reforming process using a CNx|Ni2P photocatalyst. (d) Produced H2 amount of the photo-reforming of PET and PLA. Reprinted from ref. 45 with permission of American Chemical Society, copyright 2019. | |
Other less toxic semiconductor-based photocatalytic systems have also been explored. For instance, a ZnIn2S4@SiO2@TiO2 (ZIS@SiO2@TiO2) ternary heterostructure photocatalyst was prepared following facile sol–gel and solvothermal methods and the resulting ZIS@SiO2@TiO2 exhibited good activity for the degradation of photocatalytic organic pollutants coupled with H2 production. The optimized 150%-ZIS@SiO2@TiO2 catalyst could produce H2 at an excellent rate of 618.3 μmol g−1 h−1 and 99.7% efficiency was achieved for the degradation of methylene blue under simulated sunlight. Compared to the binary ZIS@SiO2 and ZIS@TiO2 counterparts, the ternary ZIS@SiO2@TiO2 possessed better photocatalytic performance under identical conditions. It was rationalized that the heterojunction in ZIS@SiO2@TiO2 enhanced the separation and transportation efficiency of electron–hole pairs, which was beneficial for outstanding photocatalytic activity.46
4.2 Integration of H2 production with biomass photo-reforming
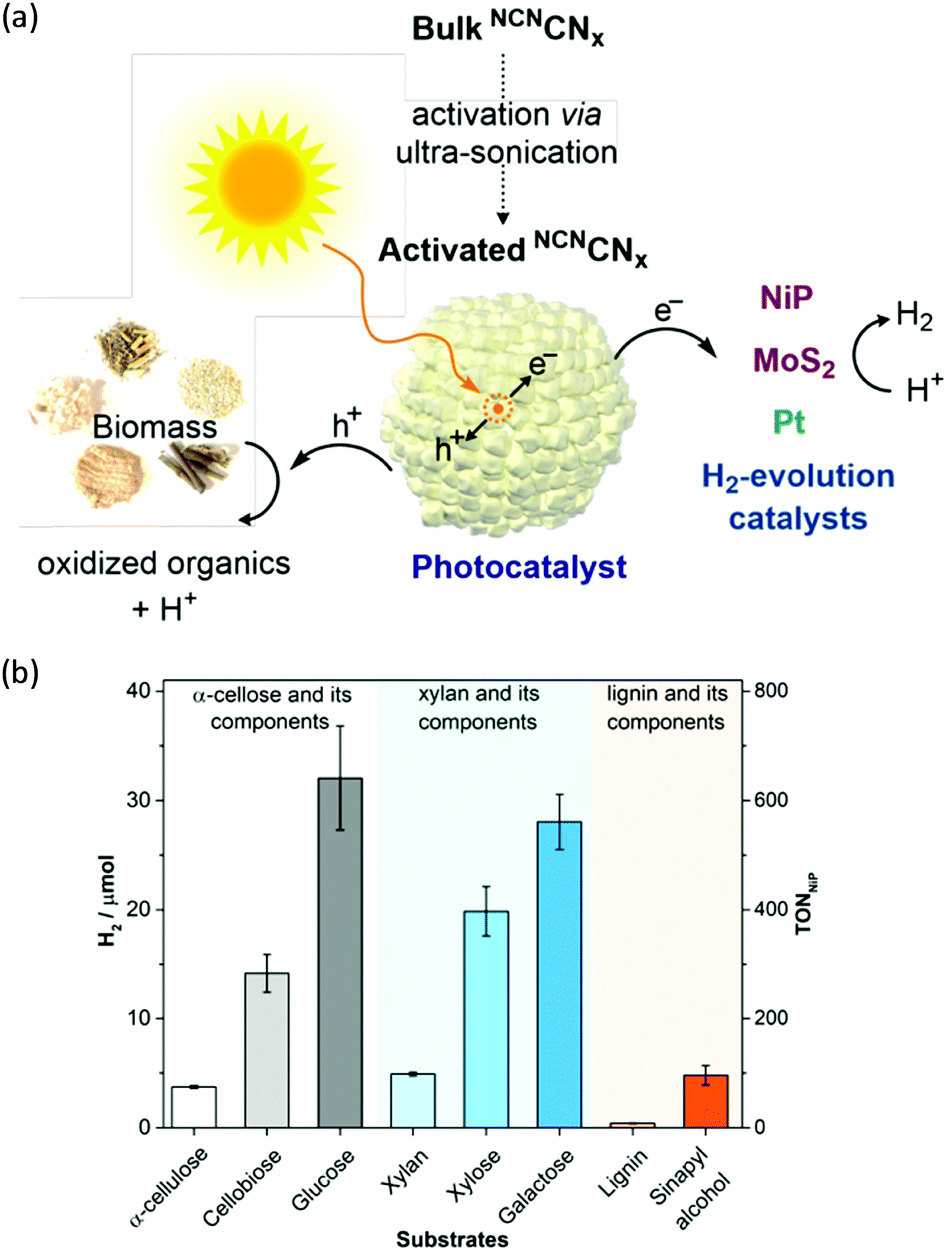
H2 production arising from biomass-based photo-reforming is regarded as a direct and sustainable route to utilize renewable carbon resources to produce green fuels as alternatives to fossil fuels (Scheme 8). In 2017, Wakerley et al. reported a CdS/CdOx photocatalyst for the photo-reforming of cellulose, hemicellulose, and lignin to H2 under alkaline conditions.47 Moreover, unprocessed lignocelluloses, such as wood and paper, were also able to be reformed under solar irradiation together with H2 production. The photocatalytic mechanism involved the formation of an oxide/hydroxide in situ on CdS/CdOx under alkaline conditions to improve the photo-reforming performance. To pursue an inexpensive, non-toxic and visible-light absorbing photocatalyst, polymeric carbon nitride (CNx) combined with various proton reduction co-catalysts such as heterogeneous Pt or MoS2 and a molecular Ni bis(diphosphine) catalyst (NiP) was also developed by the same group for the photo-reforming of biomass to yield H2 (Fig. 6a). They demonstrated that various purified lignocellulosic components and even raw biomass substrates were able to be photo-reformed to generate H2 using activated NCNCNx and NiP as a H2 evolution co-catalyst (Fig. 6b). It was found that ultra-sonication activated cyanamide-functionalized carbon nitride exhibited an enhanced photo-reforming performance under aqueous conditions with tolerance of a wide pH window (pH = 2–15).48
 |
| | Scheme 8 Representative photo-reforming of cellulose into H2. | |
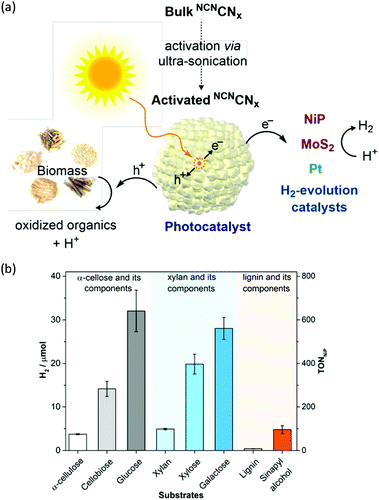 |
| | Fig. 6 (a) Schematic photo-reforming of lignocellulose using NCNCNx and proton reduction co-catalysts. (b) Photocatalytic H2 production from purified lignocellulose components catalyzed by activated NCNCNx and NiP under AM1.5G light irradiation for one day at 25 °C. Reprinted from ref. 48 with permission of American Chemical Society, copyright 2018. | |
Very recently, Wang et al. developed an ethyl alcohol grafted carbon nitride photocatalyst through an in situ C–N coupling method which showed enhanced visible light absorption, increased charge density, and prolonged lifetime of excited charges relative to pristine carbon nitride. In the presence of Pt co-catalysts, the ethyl alcohol grafted carbon nitride exhibited superior visible light-driven activity for the photo-reforming of several monosaccharides to produce H2.49
5. Conclusion and outlook
Visible light-driven organic transformation integrated with H2 production on semiconductors represents an efficient approach for solar to chemical conversion. According to the nature of the organic reactions, we have presented three representative categories of organic transformations coupled with H2 evolution: (i) photocatalytic organic oxidation, (ii) photocatalytic oxidative coupling, and (iii) photo-reforming reactions. It is apparent that a number of semiconductor-based photocatalytic systems have been developed to drive a wide range of organic reactions with the aim of producing value-added chemicals and H2 simultaneously under visible light irradiation. Even though in all the discussed photocatalytic reactions there is one basic principle that all of the organic substrates act as hole scavengers, the rational design of semiconductor-based photocatalysts indeed plays a vital role in controlling the efficiency, selectivity, durability, and scope of the overall reactions. For instance, for integrated H2 evolution with photocatalytic alcohol oxidation, Ni/CdS NPs exhibit a much higher rate of H2 production relative to NiS/CdS NPs, while NiCl2/CdS NPs are unable to catalyze this reaction.
Despite the rapid advancement in this emerging research field, there are several issues that should be addressed before practical application on a large scale. Firstly, given the requirement for long-term operation and environmental friendliness, it is of critical importance to develop non-toxic semiconductor-based photocatalysts with high efficiency, selectivity, and durability. As implied in the aforementioned examples, Cd-based chalcogenides have been widely utilized; however, the toxicity of Cd and the potential for self-oxidation due to the chalcogenide anions would restrict their practical employment. In contrast, metal-free carbon-based photocatalysts tend to exhibit better stability, in addition to their more benign and inexpensive nature. However, in terms of photocatalytic activity, carbon-based photocatalysts are currently inferior to the metal-based counterparts. Future research efforts should be devoted to designing and synthesizing novel semiconductors with better visible light absorption, long-lived excited pairs, and sufficient redox power. Secondly, investigation of reaction mechanisms should be conducted at the molecular and even atomic level by using in situ techniques and theoretical calculations. Unveiling the catalytic mechanism is anticipated to elucidate the structure–performance relationship of photocatalysts and provide guidance for the above rational design of high-performance photocatalysts. Thirdly, in addition to typical batch-type reactors for photocatalysis in research laboratories, it is more appealing to assemble larger-scale photo-reactors, preferably flow reactors, to demonstrate the real promise of any developed photocatalytic systems for practical applications under solar irradiation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Y. S. acknowledges the financial support of the Herman Frasch Foundation (820-HF17), National Science Foundation (CHE1914546), and the University of Cincinnati.
References
- L. A. Weinstein, J. Loomis, B. Bhatia, D. M. Bierman, E. N. Wang and G. Chen, Chem. Rev., 2015, 115, 12797–12838 CrossRef CAS PubMed.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- E. Gies, Nature, 2017, 551, S145–S147 CrossRef PubMed.
- X. Yang and D. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- S. Kampouri and K. C. Stylianou, ACS Catal., 2019, 9, 4247–4270 CrossRef CAS.
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1700529 CrossRef.
- X. Liu, X. Duan, W. Wei, S. Wang and B.-J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- B. Xia, Y. Zhang, B. Shi, J. Ran, K. Davey and S. Z. Qiao, Small Methods, 2020, 2000063 CrossRef.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- H. Hao and X. Lang, ChemCatChem, 2019, 11, 1378–1393 CrossRef CAS.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC.
- S. Higashimoto, Y. Tanaka, R. Ishikawa, S. Hasegawa, M. Azuma, H. Ohue and Y. Sakata, Catal. Sci. Technol., 2013, 3, 400–403 RSC.
- Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS PubMed.
- D. Jiang, X. Chen, Z. Zhang, L. Zhang, Y. Wang, Z. Sun, R. M. Irfan and P. Du, J. Catal., 2018, 357, 147–153 CrossRef.
- Y. Xu, L.-Z. Zeng, Z.-C. Fu, C. Li, Z. Yang, Y. Chen and W.-F. Fu, Catal. Sci. Technol., 2018, 8, 2540–2545 RSC.
- Y. Chao, J. Lai, Y. Yang, P. Zhou, Y. Zhang, Z. Mu, S. Li, J. Zheng, Z. Zhu and Y. Tan, Catal. Sci. Technol., 2018, 8, 3372–3378 RSC.
- X. Ye, Y. Chen, Y. Wu, X. Zhang, X. Wang and S. Chen, Appl. Catal., B, 2019, 242, 302–311 CrossRef CAS.
- F. Li, Y. Wang, J. Du, Y. Zhu, C. Xu and L. Sun, Appl. Catal., B, 2018, 225, 258–263 CrossRef CAS.
- Y. Hu, G. Zhao, Q. Pan, H. Wang, Z. Shen, B. Peng, G. W. Busser, X. Wang and M. Muhler, ChemCatChem, 2019, 11, 5139–5144 CrossRef CAS.
- L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020–3024 CrossRef CAS PubMed.
- H.-F. Ye, R. Shi, X. Yang, W.-F. Fu and Y. Chen, Appl. Catal., B, 2018, 233, 70–79 CrossRef CAS.
- Y.-H. Li, F. Zhang, Y. Chen, J.-Y. Li and Y.-J. Xu, Green Chem., 2020, 22, 163–169 RSC.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- Y. Wang, X. Kong, M. Jiang, F. Zhang and X. Lei, Inorg. Chem. Front., 2020, 7, 437–446 RSC.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328 CrossRef CAS PubMed.
- H. Zhang, S. Xie, J. Hu, X. Wu, Q. Zhang, J. Cheng and Y. Wang, Chem. Commun., 2020, 56, 1776–1779 RSC.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- N. Luo, T. Hou, S. Liu, B. Zeng, J. Lu, J. Zhang, H. Li and F. Wang, ACS Catal., 2020, 10, 762–769 CrossRef CAS.
- Y. Huang, C. Liu, M. Li, H. Li, Y. Li, R. Su and B. Zhang, ACS Catal., 2020, 10, 3904–3910 CrossRef CAS.
- W. Yu, D. Zhang, X. Guo, C. Song and Z. Zhao, Catal. Sci. Technol., 2018, 8, 5148–5154 RSC.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- A. M. Almeida, R. Li and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 75–78 CrossRef CAS PubMed.
- A. R. Hajipour, S. E. Mallakpour and H. Adibi, J. Org. Chem., 2002, 67, 8666–8668 CrossRef CAS PubMed.
- J. B. Arterburn, M. C. Perry, S. L. Nelson, B. R. Dible and M. S. Holguin, J. Am. Chem. Soc., 1997, 119, 9309–9310 CrossRef CAS.
- L. Xu, X. Deng and Z. Li, Appl. Catal., B, 2018, 234, 50–55 CrossRef CAS.
- X. B. Li, Z. J. Li, Y. J. Gao, Q. Y. Meng, S. Yu, R. G. Weiss, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- L. Wang, H. Zhou, H. Zhang, Y. Song, H. Zhang and X. Qian, Inorg. Chem., 2020, 59, 2278–2287 CrossRef CAS PubMed.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- Q. Liu, L. Wei, Q. Xi, Y. Lei and F. Wang, Chem. Eng. J., 2020, 383, 123792 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*