
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic conversion of CO2 to chemicals and fuels: the collective thermocatalytic/photocatalytic/electrocatalytic approach with graphitic carbon nitride
Subhajyoti
Samanta
 * and
Rajendra
Srivastava
*
* and
Rajendra
Srivastava
*
Catalysis Research Laboratory, Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab-140001, India. E-mail: subhajyoti.samanta@iitrpr.ac.in; rajendra@iitrpr.ac.in; Tel: +91-1881-242175
First published on 15th June 2020
Abstract
Rapid industrial development and the excessive use of fossil fuels have produced a significantly large volume of CO2 in the atmosphere. The efficient conversion of CO2 to useful chemicals and fuels is an important step towards reducing the concentration of CO2. To overcome the thermodynamic and kinetic barriers involved in CO2 activation and conversion, an effective heterogeneous catalyst and a suitable form of energy, such as thermal, photochemical, and electrochemical, are necessary. Considering a wide variety of catalytic materials for CO2 conversion using all forms of energy resources discussed above, earth-abundant carbon materials have exhibited the capability to catalyze the thermal, photo-assisted, and electrochemically mediated conversion of CO2. Among carbon-based materials, graphitic carbon nitride (g-C3N4) is an inexpensive and sustainable catalyst that can be effectively used for CO2 conversion. The chemical reactivity and optoelectronic properties of g-C3N4 can be finely tailored for its use as a conventional catalyst or photocatalyst or photoelectrocatalyst. This article aims to summarize the synthetic strategies to prepare various types of g-C3N4 materials as conventional catalysts/photocatalysts/electrocatalysts for the improved conversion of CO2 to chemicals and fuels. This article reviews the utilization of CO2 as a feedstock for chemical and fuel synthesis via insertion, carboxylation, and reduction reactions. Further, an in-depth understanding of the catalytic mechanism of various reactions discussed in this article will help in the design of a new generation of heterogeneous catalysts for CO2 conversion. Special emphasis is placed on the various parameters influencing the photocatalytic and electrocatalytic CO2 reduction. Finally, the underlying challenges and outlook for the future development of catalysts for CO2 conversion to specialty chemicals and fuels are discussed.
 Subhajyoti Samanta | Mr Subhajyoti Samanta received his Master of Science in Chemistry from Guru Ghasidas Central University, Chhattisgarh, India in 2011 after completion of his bachelor's degree with honors in Chemistry from Vidyasagar University, West Bengal, India in 2009. Since July 2014, he has been working under the supervision of Dr Rajendra Srivastava in the Department of Chemistry at the Indian Institute of Technology Ropar (IIT Ropar), Punjab, India. Prior to joining his Ph.D. research, he had also worked as a project Junior Research Fellow at CSIR-IIMT, Bhubaneswar, India (June 2013–January 2014). His research concentration circumvents the area of nano-engineering metal oxides and their composites with carbon materials for their applications in heterogeneous catalysis. He has particularly developed materials for a variety of photocatalytic oxidation reactions, CO2 reduction, and water oxidation by both electrocatalytic and photocatalytic methods. He has published 15 research articles in the Journals of International repute to his credit. He has also been recognized as a young scientist and participated in the Global Young Scientist Summit-2020, Singapore. |
 Rajendra Srivastava | Dr Rajendra Srivastava joined the Department of Chemistry, IIT Ropar in 2009 as an Assistant Professor, headed the department from March 2015 to May 2018, and is currently working as an Associate Professor. He acquired his Ph.D. degree from CSIR-National Chemical Laboratory, Pune, India where his doctoral work won him the best thesis award of 2006, conferred by the Catalysis Society of India. Afterward, he worked as a postdoctoral research fellow in KAIST, South Korea (2005–2007) and as a JSPS fellow in the Hokkaido University, Japan (2007–2009). He received the NASI-SCOPUS Young Scientist Award 2017, The Catalysis Society of India Young Scientist Award 2018, and Mid-carrier Faculty Research and Innovation Award 2019 by IIT Ropar. His fields of interest include designing and synthesis of nanostructured functional materials for catalytic applications, including electrocatalysis (actuators, sensors, supercapacitors and energy harvesters), and photocatalysis. He has published more than 130 peer-reviewed original research articles in reputed international journals and has filed 8 patents. |
1. Introduction
CO2 is one of the major variable components in the troposphere that significantly influence human life.1 A large amount of CO2 is present in the soil as CaCO3. However, only gaseous CO2 present in the troposphere is responsible for global warming. The effect of carbon dioxide on Earth is being discussed at every forum and international meeting.2,3 A closer look reveals that several activities by millions of living entities (plants, animals, and human beings) have raised the CO2 level in the atmosphere by 280 ppm in the last few centuries. However, after the industrial revolution, the CO2 level in the atmosphere has risen from 280 ppm to 400 ppm and it is expected to reach up to 500 ppm by the 22nd century, which would raise the mean temperature of the Earth by 1.9 °C.4 Today, each and every second of our life is dependent on energy resources. The energy dependency can be taken care of with the use of renewable and alternative resources such as wind, water, radioactive, and solar energy, which do not increase the CO2 level in the atmosphere and provide a CO2 emission-free environment.5 In recent times, several alternative energy generation systems, such as fuel cells, are being developed which do not emit CO2.7 However, the H2 required in fuel cells is produced by steam reforming, which produces a large amount of CO2 in the atmosphere.6 Future energy demands and current technologies suggest that it is a long journey and by that time a significant amount of CO2 will be released into the atmosphere. If the CO2 concentration increases beyond 600 ppm it would be difficult for human beings to breathe because it is difficult for the human respiratory system to operate in the presence of a greater concentration of CO2 even though an excess of O2 is present in the atmosphere. Thus, there is a high priority in developing strategies to reduce the CO2 concentration in the environment. This can be achieved via two strategies: (1) CO2 capture and storage (CCS) and (2) CO2 capture and utilization (CCU) to useful chemicals and fuels.7 This article is focused on the CO2 conversion to useful chemicals and fuels; therefore, only a brief discussion is provided for CO2 capture and storage.The various types of stationary plants and their corresponding CO2 emission (amount and partial pressure) are summarized in pie charts (Fig. 1). It is very easy to capture CO2 from point sources where the CO2 concentration is high but it is very difficult to capture CO2 from low concentration sites, such as on city roads or houses where the concentration ranges fall at the ppm level.8 Besides being the most desirable task, direct CO2 capture and storage are undoubtedly challenging.9,10 In general, liquid CO2 scrubbers and metal oxides are used to absorb CO2 from flue gases.11–20 Aqueous amine based liquid CO2 scrubbers are commonly used but their associated energy consumption and cost remain a constraint for further development.9 Similarly, solid materials such as alkali metal oxides, layered double hydroxides, solid amines, etc. are widely explored materials for CO2 adsorption.14 However, CO2 absorption using the CO2 scrubbers and solid materials discussed above is energetically reversible due to the strong bonding of CO2 to these materials. Several types of nanoporous materials based on zeolites,17,18 metal–organic frameworks,17–19 and carbon20,21 have been developed for this purpose. All of those above-mentioned materials have functional amines and metal sites for the efficient adsorption of CO2 over other gases present in flue gas. Apart from tuning the chemical properties, tailoring textural properties such as the surface area, pore size and porous architecture can improve the CO2 adsorption capabilities. It is noteworthy that materials having a pore size greater than 0.33 nm and lower than 0.35 nm would be ideal for CO2 adsorption over other gases present in flue gas (the kinetic diameters of CO2, O2, and N2 are 0.33 nm, 0.345 nm, and 0.36 nm, respectively).22 However, such a fine porous material with specific requirements is yet to be developed. Therefore, high surface area materials having dense CO2 adsorption sites with pore sizes in the range of 0.3 to 2 nm are being prepared.20,23,24 Thus the direct capture of CO2 from the air (in which the O2/CO2 ratio is more than 500 or even more) is very demanding but challenging for research scientists and engineers. However, plants do this job very efficiently when the present CO2 level is <400 ppm. Plants abstract CO2 from the atmosphere and convert it to carbohydrate with the aid of sunlight and grow and liberate O2.
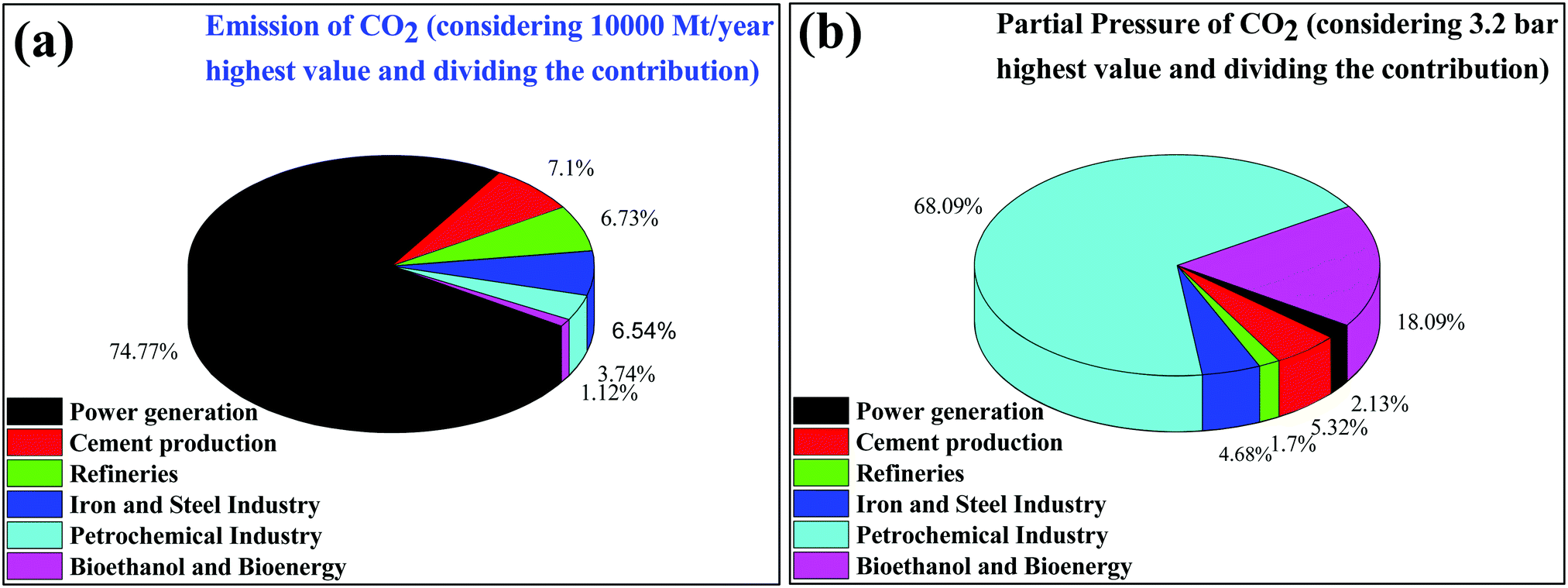 | ||
| Fig. 1 (a) Emission of CO2 and (b) partial pressure of CO2 emitted from different stationary sources. | ||
The most efficient strategy to reduce the concentration of CO2 in the atmosphere is to convert CO2 to useful chemicals and fuels. CO2 is a thermodynamically inert molecule with a high C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy of 750 kJ mol−1 which is much higher than the C–O bond (327 kJ mol−1) and even higher than the C–C (336 kJ mol−1) and C–H (441 kJ mol−1) bonds.25 Due to such high stability, it is difficult to transform CO2 into useful chemicals. The energy gap between its highest occupied molecular orbital and lowest unoccupied molecular orbital is 13.7 eV and the electron affinity of CO2 is (−0.6 ± 0.2 eV), which leads to a very negative redox potential for one-electron transfer CO2 + e− → CO2− (E0redox = −1.90 eV vs. NHE).26,27 Such a high energy requirement for electron transfer to generate active species for CO2 conversion makes photocatalytic and electrocatalytic processes highly challenging tasks. Due to such a high energy requirement, only a few chemicals have been commercialized, which utilizes a large amount of CO2 to give useful chemicals such as urea, methanol, salicylic acid, cyclic carbonates, and dimethyl carbonate (Scheme 1). Synthesis of urea and cyclic urea is not a 100% atom economic process because water is formed as a by-product. In an atom economic process, the total number of atoms present in the reactants is incorporated into the product. Synthesis of cyclic carbonates takes place by an insertion reaction in which CO2 is inserted into a three-membered epoxide ring to produce a five-membered cyclic carbonate via a 100% atom economic process.28,29 Dimethyl carbonate (DMC), a solvent, can be produced by the reaction of methanol and CO2.30,31 DMC reacts with phenol using a Pb(OPh)2 catalyst to produce diphenyl carbonate (DPC). Then DPC reacts with bisphenol-A to produce a polycarbonate via a melt polymerization method (Scheme 1).32 Salicylic acid is manufactured by the reaction of phenol and CO2 in a strongly basic medium.33 Another bulk chemical is methanol, which is produced annually at a rate of 100 million tons by utilizing 2 million tons of CO234 (Scheme 1). Industrial processes for chemical synthesis using CO2 as a reactant are operated at high temperature and pressure due to the thermodynamic stability and inertness of CO2. Such reactions can be catalyzed under mild conditions if the CO2 is efficiently activated. Since CO2 is an acidic molecule, therefore it can be activated using basic catalysts such as alkali and alkaline earth metal-based oxides and hydroxides. Various organic amines (N-heterocyclics, DBU, guanidine, melamine, phthalocyanine, and porphyrins) and polyamines have also been used in chemical synthesis utilizing CO2 as the reactant. In general, organic amines are more reactive than inorganic bases and thus various ionic liquids, metal–organic frameworks, silica, and zeolite based materials with functional amine groups have been utilized for chemical synthesis involving CO2 as a reactant. Nitrogen-containing carbon materials, such as carbon nitrides, are a unique class of materials which can be prepared with economical nitrogen and carbon-containing precursors. Since these materials have a large nitrogen content, they can be used for CO2 activation and adsorption.
O bond dissociation energy of 750 kJ mol−1 which is much higher than the C–O bond (327 kJ mol−1) and even higher than the C–C (336 kJ mol−1) and C–H (441 kJ mol−1) bonds.25 Due to such high stability, it is difficult to transform CO2 into useful chemicals. The energy gap between its highest occupied molecular orbital and lowest unoccupied molecular orbital is 13.7 eV and the electron affinity of CO2 is (−0.6 ± 0.2 eV), which leads to a very negative redox potential for one-electron transfer CO2 + e− → CO2− (E0redox = −1.90 eV vs. NHE).26,27 Such a high energy requirement for electron transfer to generate active species for CO2 conversion makes photocatalytic and electrocatalytic processes highly challenging tasks. Due to such a high energy requirement, only a few chemicals have been commercialized, which utilizes a large amount of CO2 to give useful chemicals such as urea, methanol, salicylic acid, cyclic carbonates, and dimethyl carbonate (Scheme 1). Synthesis of urea and cyclic urea is not a 100% atom economic process because water is formed as a by-product. In an atom economic process, the total number of atoms present in the reactants is incorporated into the product. Synthesis of cyclic carbonates takes place by an insertion reaction in which CO2 is inserted into a three-membered epoxide ring to produce a five-membered cyclic carbonate via a 100% atom economic process.28,29 Dimethyl carbonate (DMC), a solvent, can be produced by the reaction of methanol and CO2.30,31 DMC reacts with phenol using a Pb(OPh)2 catalyst to produce diphenyl carbonate (DPC). Then DPC reacts with bisphenol-A to produce a polycarbonate via a melt polymerization method (Scheme 1).32 Salicylic acid is manufactured by the reaction of phenol and CO2 in a strongly basic medium.33 Another bulk chemical is methanol, which is produced annually at a rate of 100 million tons by utilizing 2 million tons of CO234 (Scheme 1). Industrial processes for chemical synthesis using CO2 as a reactant are operated at high temperature and pressure due to the thermodynamic stability and inertness of CO2. Such reactions can be catalyzed under mild conditions if the CO2 is efficiently activated. Since CO2 is an acidic molecule, therefore it can be activated using basic catalysts such as alkali and alkaline earth metal-based oxides and hydroxides. Various organic amines (N-heterocyclics, DBU, guanidine, melamine, phthalocyanine, and porphyrins) and polyamines have also been used in chemical synthesis utilizing CO2 as the reactant. In general, organic amines are more reactive than inorganic bases and thus various ionic liquids, metal–organic frameworks, silica, and zeolite based materials with functional amine groups have been utilized for chemical synthesis involving CO2 as a reactant. Nitrogen-containing carbon materials, such as carbon nitrides, are a unique class of materials which can be prepared with economical nitrogen and carbon-containing precursors. Since these materials have a large nitrogen content, they can be used for CO2 activation and adsorption.
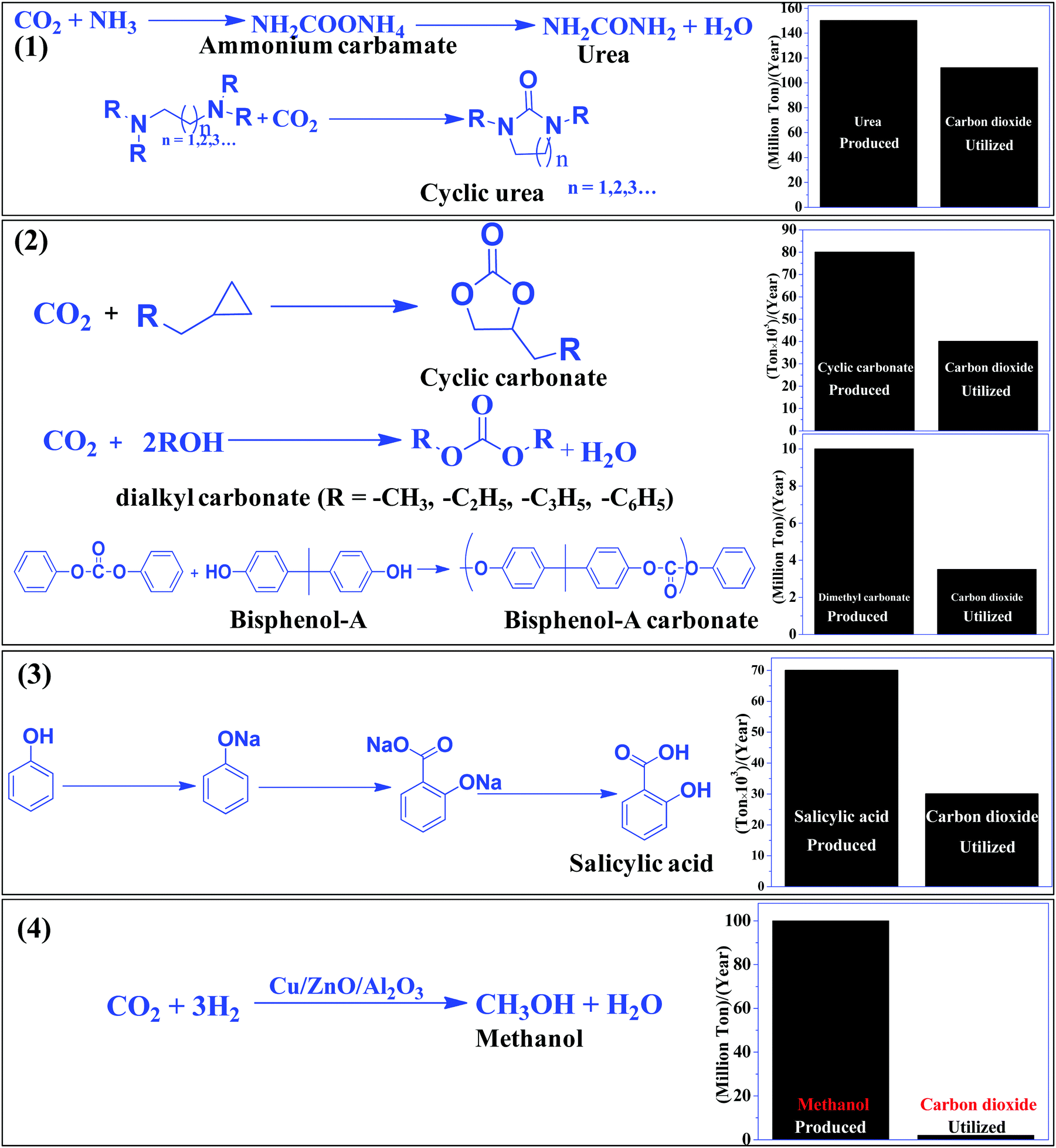 | ||
| Scheme 1 CO2 based commercial processes and amount of CO2 utilized vs. product formed in the given process. | ||
In this review, we will present the advances, challenges, and prospects of carbon nitride based catalysts, photocatalysts, and electrocatalysts in CO2 conversion to chemicals and fuels. After a brief introduction to CO2 conversion, the requirements of CO2 storage and conversion, the industrial process for CO2 to chemicals, and the types of materials required for CO2 activation and conversion are described in Section 1. Sections 2–6 focus on carbon nitride-based catalysts in the conventional catalytic conversion of CO2 to chemicals and photocatalytic and electrocatalytic conversion of CO2 to C1 fuels. Section 2 presents a brief description of nitrogen-containing carbon-based materials, types of carbon nitride, and the structural elucidation of g-C3N4 for catalytic applications. Section 3 deals with the synthesis strategy adopted for the preparation of g-C3N4 for improved catalysis and photocatalysis involving CO2 as a reactant. Sections 4 and 5 provide detailed information about photocatalytic and electrocatalytic CO2 conversion to C1 fuels, respectively, their basic concepts, the influence of various parameters influencing the CO2 conversion and C1 fuel selectivity, deep insight into the reaction mechanisms via in situ characterization techniques and product identification, catalytic activity assessment and result presentation, and the experimental set-up required for photocatalytic and electrocatalytic measurements. Finally, this review will be concluded with a summary, underlying challenges, and opportunities for the future development of catalysts for CO2 conversion to chemicals and fuels.
This focused review aims to concisely cover almost all the important points, starting from the requirements of CO2 conversion, fundamentals of CO2 activation, strategies adopted for g-C3N4 preparation for improved CO2 conversion, structure elucidation of g-C3N4, and structure–activity relationship, followed by critical operational parameters for collective ‘thermo-photo-electro’-chemical CO2 conversion focusing on the g-C3N4 catalyst, which is not available in a single article hitherto reported in the literature.
2. Carbon nitride, a versatile material for CO2 adsorption and conversion: structural elucidation for catalysis
Among porous materials, carbon materials have a high surface area, tunable porosity, hydrophobicity for CO2 adsorption under hydrated conditions, high thermal and chemical stability, and functional ability.35–37 Activated carbon is the most economical and widely available carbon material having micropores, mesopores, and macropores.38 Due to the porous architecture and high surface area, it can significantly adsorb large amounts of CO2 by the physisorption process but it finds difficulty in the activation and utilization of CO2 in chemical synthesis due to the inability to chemisorb and activate CO2.39 Biomass pyrolysis and KOH activation can produce high surface area activated carbon, suitable for CO2 physisorption.40 N-Doped microporous carbon prepared with coconut shell and urea has exhibited high CO2 adsorption and CO2/N2 selectivity at 1 bar, confirming that N-doping has improved the CO2 adsorption.41 Similar to inorganic porous materials, sol–gel methods have been applied for the synthesis of porous carbon materials in water and/or ethanol medium followed by acid activation leading to the formation of porous carbon with high surface area and pore volume.42,43 Hydrothermal polymerization is employed to prepare porous carbon materials with higher surface area after chemical activation.44 The hydrothermal method provides the opportunity to prepare carbon materials with functionalized carbon in the presence of other functional organic compounds. Such functionalized materials (carboxyl rich carbon materials) have exhibited high CO2 capture performances at 1 bar and under flue gas conditions.45 Hard template46 and soft template47 methods have been developed for the preparation of mesoporous carbon. The carbon materials discussed above are amorphous porous carbon. It is also possible to synthesize crystalline carbon materials with different allotropy. 0D carbon dots and fullerenes, 1D carbon nanotubes, 2D graphene, and 3D graphite are the various allotropic forms of carbon materials (Scheme 2). In general 3D graphitic carbon can be prepared by heating a carbon precursor at a very high temperature 2000–3000 °C. However, such graphitic carbon possesses very low surface area and porosity. Metal catalyst mediated synthesis produces graphitic carbon at a very low temperature of 700–800 °C with higher surface area and porosity.48 Moreover, microwave mediated, metal activated synthesis produces graphitic carbon with very high surface area and porosity.49 Graphene, carbon nanotubes, and fullerenes have been prepared under varying controlled conditions using a unique vapor deposition method in the presence and absence of metal catalysts. Due to the high surface area and surface to volume ratio, these materials can be easily functionalized using various amines for the efficient adsorption of CO2.50,51 These porous carbons have varied surface area, morphology, and porosity and, thus, the extent of CO2 adsorption over these materials varies depending on their tunable textural properties. However, the adsorption and chemisorption capability improves if nitrogen-doped or nitrogen-containing carbon materials are considered. Nitrogen-containing carbon materials induce interactive forces between CO2 and nitrogen groups present in the material. CO2 is a weak Lewis acidic molecule because the carbon atom in CO2 is electron-deficient. Nitrogen-doped carbon atoms are negatively charged and nitrogen sites act as electron donors and behave like a Lewis base; therefore, nitrogen-containing carbon materials have acid–base interactions through which CO2 molecules are chemisorbed. The strong dipolar CO bond generates an electric quadrupole moment in the CO2 molecule, whereas nitrogen doping induces local polarization in the porous carbon matrix.52 Notably, N2 has a lower electric dipole moment than CO2. Hence, the interaction between the CO2 molecular quadrupole moment and high electrostatic potential of N-doped carbons enhances the CO2 adsorption energy, leading to selective and higher CO2 adsorption from flue gases. Experimental and computational studies have shown that surface hydroxyl groups present in the N-doped carbon skeleton enhance the interaction between –OH and –NH functionalities leading to high CO2 adsorption in N-doped carbon materials.53 Nitrogen doping and surface functionalized amines have different types of interaction. Surface functionalized amines interact with electropositive carbon of CO2 and form ammonium carbamate leading to high CO2 adsorption on the surface.54 However, surface and pore blockage and thermal instability of functionalized amines are some of the demerits of surface amine-functionalized carbon materials; therefore, nitrogen-doped carbon is superior over amine-functionalized carbon materials. Based on the varied textural properties and chemical properties (due to N doping and amine functionalization), it can be concluded that high surface area and high nitrogen contents both are important factors contributing to high CO2 adsorption (Table 1).44 Moreover, when CO2 activation and utilization for chemical synthesis are considered both these factors play important roles.
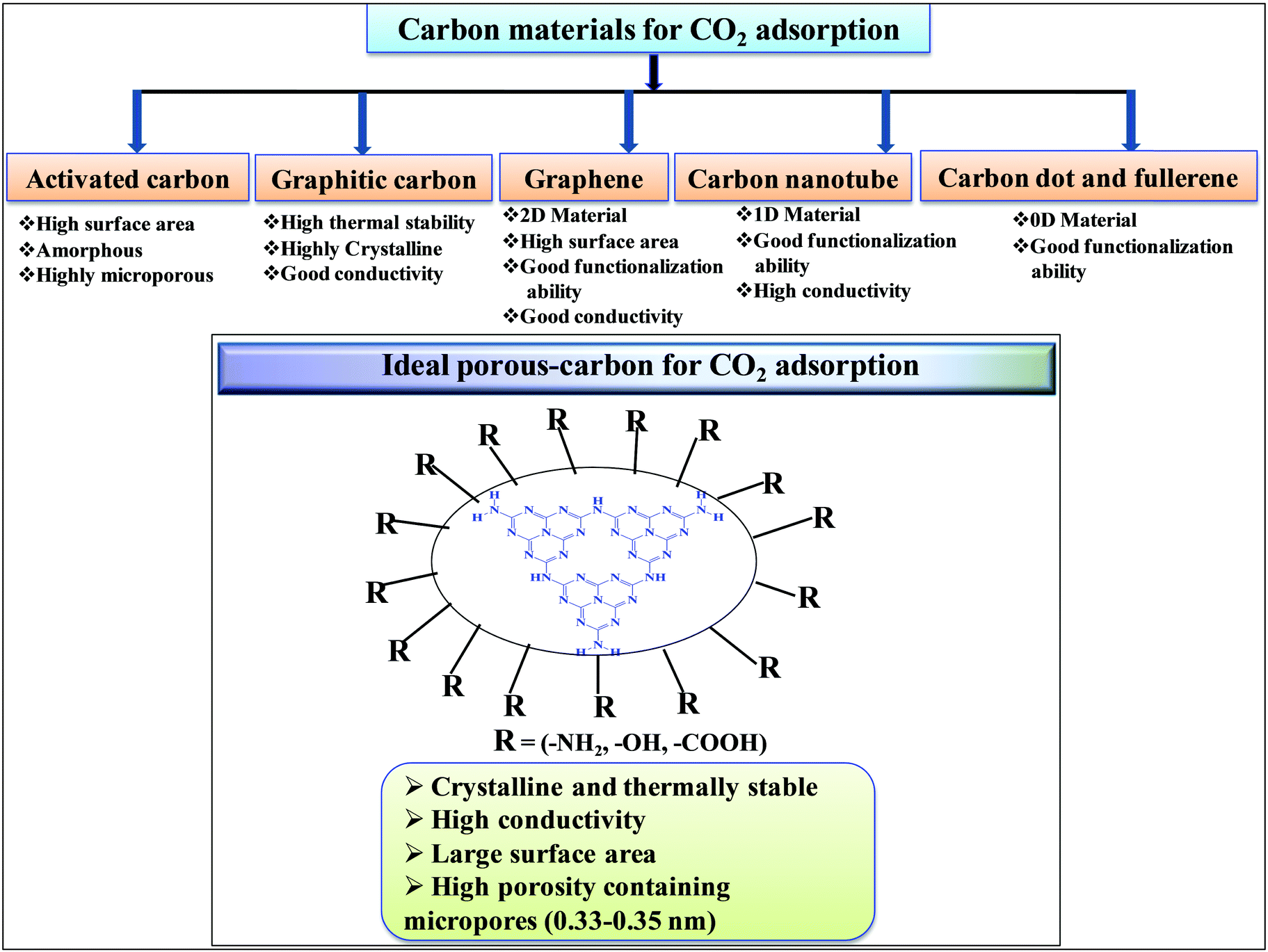 | ||
| Scheme 2 Porous carbon materials for CO2 adsorption. | ||
| E. no. | Material | S BET (m2 g−1) | Nitrogen content (%) | CO2 uptake mmol−1 g−1 (298 K) |
|---|---|---|---|---|
| 1 | NMC-600 | 272 | 6.33 | 3.90 |
| 2 | NMC-700 | 518 | 6.06 | 3.60 |
| 3 | AC-600-0.5 | 508 | 6.70 | 3.00 |
| 4 | AC-600-2 | 966 | 4.50 | 3.64 |
| 5 | AC-800-2 | 2448 | 3.90 | 3.00 |
| 6 | Sk-0.5-700 | 1060 | 0.48 | 4.24 |
| 7 | SK-4-700 | 2792 | 0.18 | 3.05 |
| 8 | PGC-K | 1143 | 8.30 | 4.00 |
| 9 | FCP-1-KC | 800 | 0.60 | 5.20 |
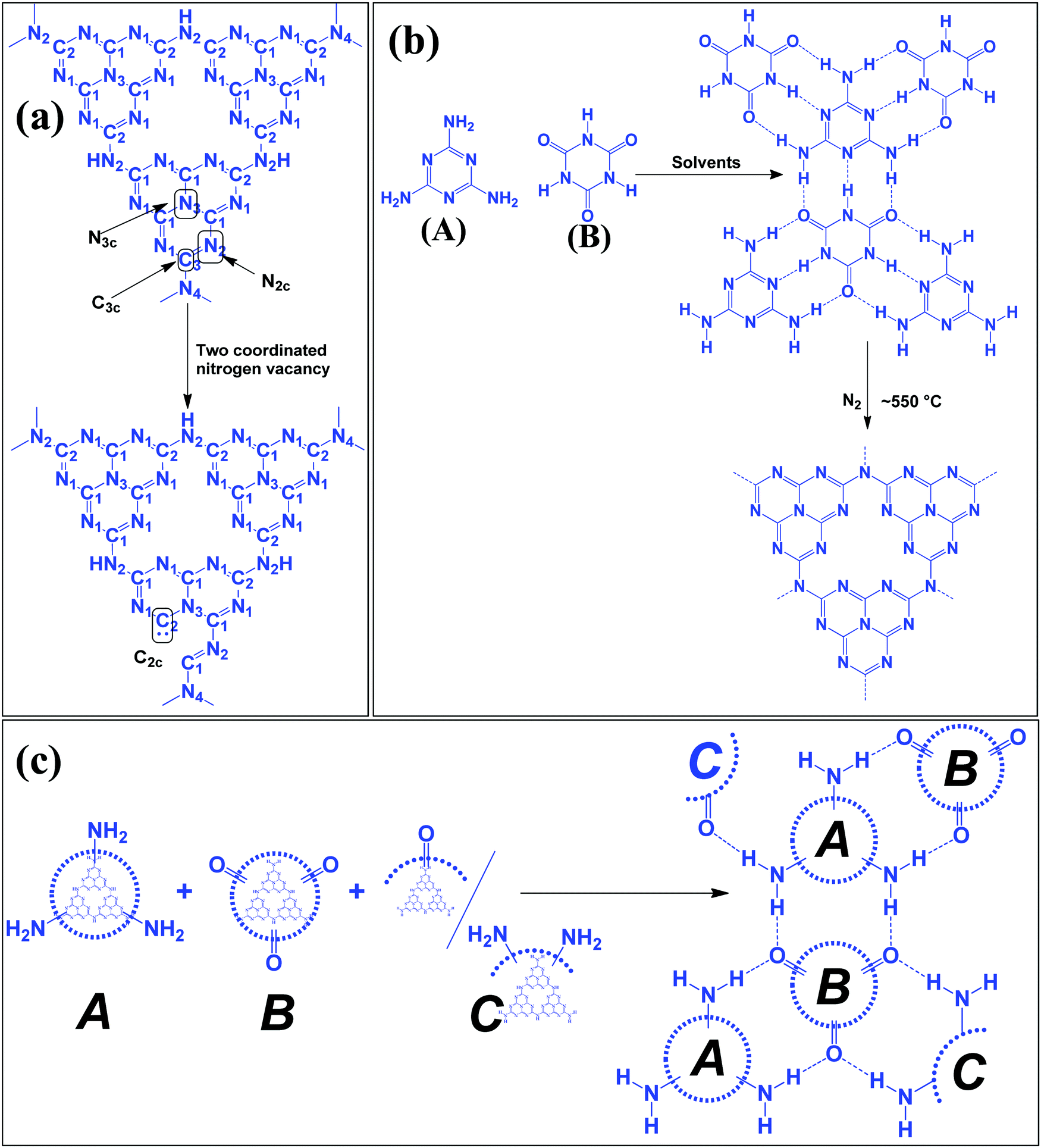
Table 1 suggests that a large number of framework substituted N atoms in a carbon matrix is required for high CO2 adsorption. The graphitic carbon framework provides high thermal stability and conductivity, which would be good for photocatalysis and electrocatalysis. An ideal porous carbon material and its properties are listed in Scheme 2. Based on these characteristic features, a unique carbonaceous material known as graphitic carbon nitride (g-C3N4) can best serve this purpose. When a simple nitrogen-rich organic compound (such as urea, melamine, cyanamide, dicyandiamide, etc.) is thermally-polymerized in the temperature range of 500–700 °C, it produces carbon nitride. This material was synthesized way back in 1834 by Berzelius;55 however its potential as a catalyst56 and photocatalyst57 was realized in 2006 and 2009, respectively. This crystalline material exists in seven allotropes: α-C3N4, β-C3N4, cubic C3N4, pseudocubic C3N4, g-h-triazine, g-o-triazine, and g-h-heptazine with bandgaps of 5.49, 4.85, 4.30, 4.13, 2.97, 0.93, and 2.88 eV, respectively.58 These allotropes vary in their nitrogen environment and nitrogen contents. Theoretically, carbon nitride is predicted to have a C/N ratio of 0.75 without any surface functional groups and hydrogen content but the reality is far from this prediction. Carbon nitrides have been actually obtained with varied C/N ratios using different nitrogen-containing organic compounds at varied reaction temperature. It is possible to synthesize carbon nitrides such as C3N, C3N2, C10N3, C5N, C3N5, C3N6, C3N7, etc., having different nitrogen contents than the theoretically predicted ideal C3N4 and those are termed as nitrogen-rich (C/N < 0.75) and nitrogen lean (C/N > 0.75) C3N4.59 Whenever a six-membered conducting atomic-scaffold is envisaged, structures such as those of benzene or borazine pop-up in our minds. Based on this perception, the triazine-like structure of C3N4 can be predicted (Scheme 3(I)). But it has been found that triazine or tri-s-triazine units (Scheme 3(II)) (also known as tectonic units) propagate in three dimensions to produce allotropes of g-C3N4 (Scheme 3). Computational studies based on Density Functional Theory (DFT) suggest that the tri-s-triazine unit is more stable than the triazine unit by 30 J mol−1.60 Later, 2,5,8-triamine, tri-s-triazine (C6N10H6) has been isolated, which confirms the building units of g-C3N4.61 Theoretical g-C3N4 should have an ideal C/N ratio of 0.75 and should not possess any H or O atoms; however, XPS and elemental analysis suggest that it has H and O atoms in small quantities in addition to C and N. Notably, H and O atoms are observed due to terminal amine groups and surface adsorbed water and (CO), (O–N), and (C–OH) groups, for which the details are provided in Scheme 3. Various types of C and N are designated in one pictorial representation of a terminal tri-s-triazine unit of g-C3N4 (Scheme 3). The structure clearly shows that it has two different types of C represented as C1 and C2 (Scheme 3(III)). Similarly, four different types of nitrogen atoms are designated as N1 (pyridinic), N2, N3 (graphitic-N), and N4 (Scheme 3(III)). They can be easily distinguished from XPS analysis due to different electronic environments that provide different binding energies (Scheme 3(IV)).62
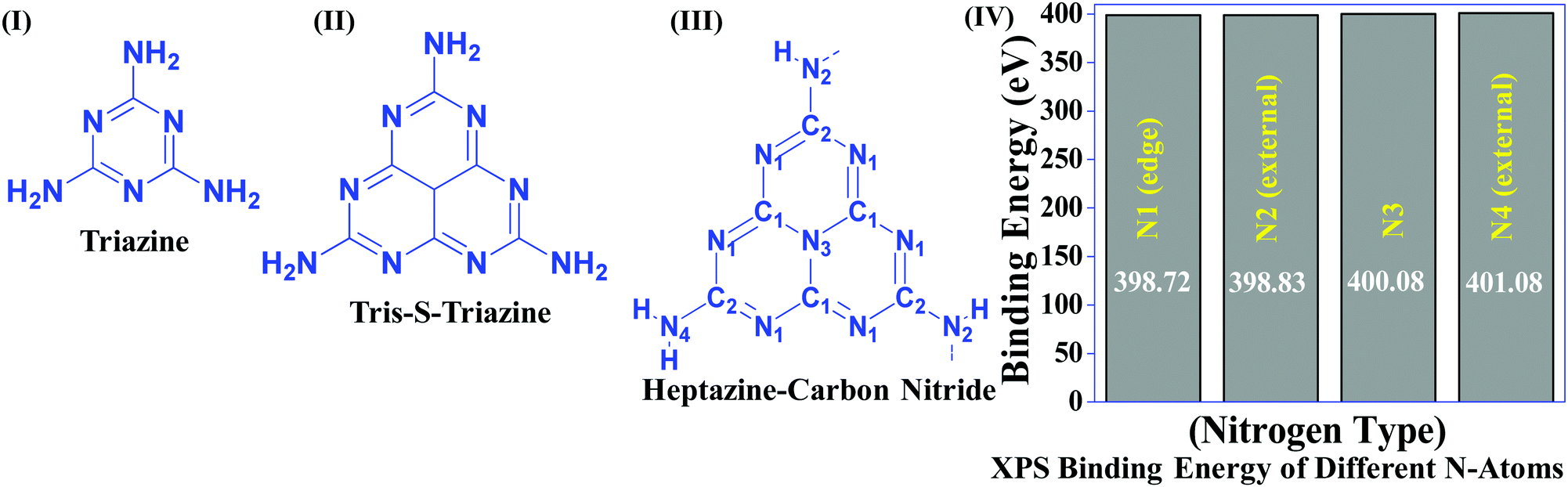 | ||
| Scheme 3 Chemical structure of (I) triazine, (II) tris-s-triazine, and (III) heptazine units of carbon nitride, and (IV) various N types and their binding energies. | ||
In a true heptazine building unit (also known as melem), N2 is absent and N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N3:N4 should be present in the ratio 6:1:3 (Scheme 4a). When the melem monomer polymerizes, the N4-type of nitrogens are converted to the N2-type (Scheme 4b). The N4/N2 ratio in the melem framework depends on the propagating direction to form g-C3N4 (Scheme 4c). Moreover, the stability of g-C3N4 also varies depending on the geometry of the final g-C3N4 polymer. When melem units are linked and form dimers, tetramers, octamers, and infinite chains, then the % N1 contribution increases. Moreover, the percentage of different N-types varies based on the geometry of their linkages as shown in Scheme 4 (inset table).62 The stability of g-C3N4 after catalysis can be estimated by the % of different types of N so that the breakage of linkages, chain fragmentation, and different structural changes can be predicted. CO2 adsorption and its activation highly depend on the type of N-moieties present (pyridinic, pyrrolic/pyridonic, and graphitic nitrogen). Pyridinic and pyrrolic/pyridonic nitrogen groups have lone pair electrons that exhibit Lewis basicity and, thereby, have the capability towards CO2 adsorption and activation. Pyridinic nitrogen exhibits a higher affinity for CO2 molecules than pyrrolic nitrogen owing to the higher basic strength of the former than the latter (Scheme 5).63,64
:N3:N4 should be present in the ratio 6:1:3 (Scheme 4a). When the melem monomer polymerizes, the N4-type of nitrogens are converted to the N2-type (Scheme 4b). The N4/N2 ratio in the melem framework depends on the propagating direction to form g-C3N4 (Scheme 4c). Moreover, the stability of g-C3N4 also varies depending on the geometry of the final g-C3N4 polymer. When melem units are linked and form dimers, tetramers, octamers, and infinite chains, then the % N1 contribution increases. Moreover, the percentage of different N-types varies based on the geometry of their linkages as shown in Scheme 4 (inset table).62 The stability of g-C3N4 after catalysis can be estimated by the % of different types of N so that the breakage of linkages, chain fragmentation, and different structural changes can be predicted. CO2 adsorption and its activation highly depend on the type of N-moieties present (pyridinic, pyrrolic/pyridonic, and graphitic nitrogen). Pyridinic and pyrrolic/pyridonic nitrogen groups have lone pair electrons that exhibit Lewis basicity and, thereby, have the capability towards CO2 adsorption and activation. Pyridinic nitrogen exhibits a higher affinity for CO2 molecules than pyrrolic nitrogen owing to the higher basic strength of the former than the latter (Scheme 5).63,64
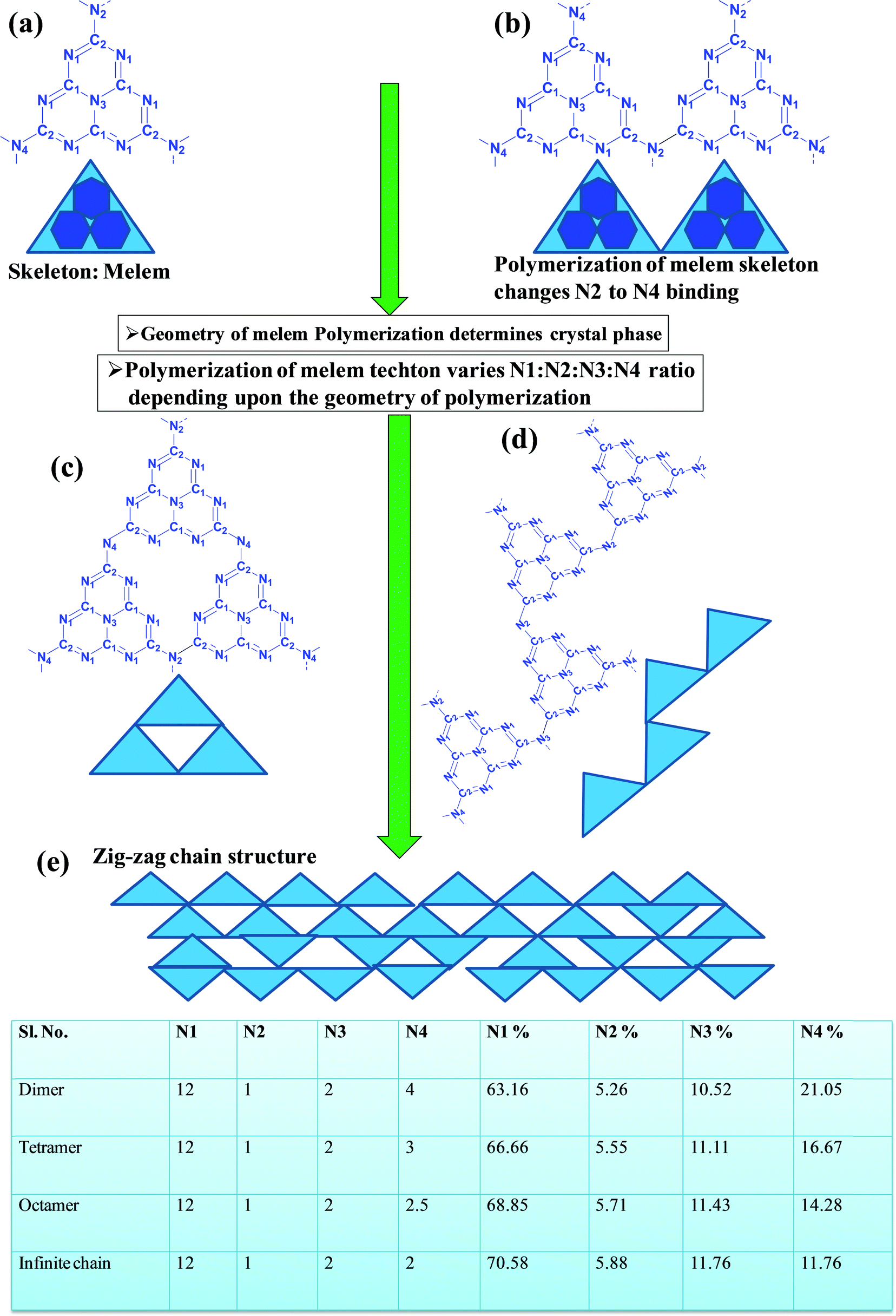 | ||
| Scheme 4 Evolution of different geometry and N types present in C3N4 based on the variation of the linkages of melem units.53 | ||
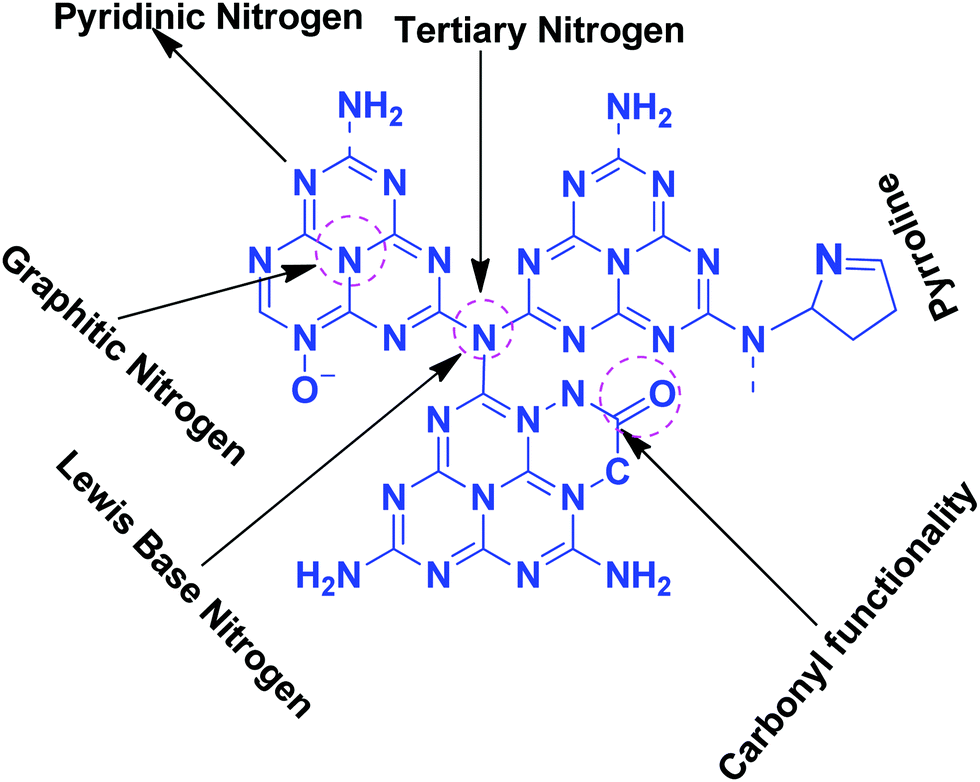 | ||
| Scheme 5 Different types of N and O functionalities present in g-C3N4. | ||
Pyridinic-N with a higher negative charge in the 2-D plane retains a lone pair of electrons that can bind CO2 more efficiently than other types of N moieties.65,66 Quaternary graphitic nitrogen exhibits Lewis acidity.67,68 Electrons are mostly present in the π*-antibonding orbital in graphitic-N and thus are less accessible to CO2 for binding.65–67 Pyridinic-N and graphitic-N can be easily presented in the melem units; however, other types of N-species are formed during un-controlled thermal polymerization in an oxygen lean environment leading to partial terminal uncondensed amines, N-oxide, and pyrrolic N species as shown in Scheme 5. The various N-species discussed above have the ability to impart tunable basicity, H-bonding, and Lewis acidity (frustrated Lewis acid–base pairs) and are thus suitable for various types of catalytic reactions involving CO2 as a reactant.
3. Synthesis strategies adopted for the preparation of carbon nitrides for improved CO2 conversion
Based on the discussion so far we have understood that suitable micropores, high surface area, and pyridinic rich N sites are required for high CO2 adsorption. CO2 adsorption alone is not sufficient to catalyze various types of CO2 based reactions because reactive partners also need to be efficiently adsorbed and suitably activated to synthesize various CO2 derived chemicals. Further, bandgap and band edge positions can be finely tuned depending on the doping and crystallinity for light-induced CO2 conversion. Thus, synthetic strategies, types of precursors, the temperature for synthesis along with the environment, and other parameters influence the physicochemical properties of g-C3N4. The following section describes the various synthesis strategies reported for the preparation of g-C3N4 for improved CO2 activation.3.1 Template free synthesis for improved catalysis and photocatalysis
Nitrogen-rich organic compounds such as melamine, cyanamide, and dicyandiamide are heated in semi-closed systems in oxygen-lean conditions to form g-C3N4 (Scheme 6). g-C3N4 is formed by various condensation steps in which the first melamine is formed at lower temperatures (<350 °C) followed by removal of NH3 leading to tris-s-triazine at a temperature around 400 °C. The polymerization of tris-s-triazine takes place at around 520 °C to form g-C3N4 (Scheme 6).69 TGA investigation reveals that this framework is stable up to 550 °C. Even oxygen-containing precursors such as urea also follow the same sequence and form g-C3N4via a melamine intermediate (Scheme 6). The nitrogen content and surface area of g-C3N4 depend upon the precursor and the pyrolysis temperature. With the increase in the pyrolysis temperature, the C/N ratio increases whereas the bandgap decreases.70 During the polymerization process, the amine groups of tris-s-triazine don’t condense completely, which results in the formation of uncondensed amine group-containing g-C3N4 with about 2% H content with a C/N ratio less than 0.75. Such a structural defect is important for the adsorption of reactant molecules, which is good for catalytic reactions.71 It may be noted that if the uncondensed groups (discussed above) are present in higher numbers then it might be good for conventional catalysis but is definitely not suitable for photocatalysis due to the less efficient charge transportation and separation of charge carriers. High-temperature polymerization leads to the formation of crystalline g-C3N4, which is better for charge carrier separation and a higher degree of π-conjugation and thus good for photocatalysis.72 Moreover, with the increase in the pyrolysis temperature, aggregated bulk g-C3N4 changes to a nanosheet and nanoflake like morphology with improved surface area.73,74 Also, urea and thiourea derived g-C3N4 exhibit better surface area due to the removal of CO2 at higher temperatures, leading to high porosity and improved surface area when compared to the cyanamide precursor. Further, the surface area of urea derived g-C3N4 is improved if the pyrolysis is carried out for a longer time.74 During such prolonged heating the layered structure of g-C3N4 is exfoliated and splitting takes place to produce low yield g-C3N4 with better surface area and photocatalytic activity.75 Inspired by the synthetic paradigm of g-C3N4 from urea which has resulted in higher surface area due to the liberation of CO2 during urea pyrolysis, sucrose and melamine have been pyrolyzed together. The reason for selecting sucrose is that it will generate a large amount of CO2 during pyrolysis and produce g-C3N4 with N-deficiency and improved surface area (6 fold higher) and porosity.76 The type of N species and surface area can be tailored by selecting different precursors. For example, guanidine hydrochloride or guanidine thiocyanate pyrolysis results in g-C3N4via a melem intermediate and produced materials with high surface area and enhanced photocatalytic activity (Scheme 6).77 The bandgap of g-C3N4 can be reduced and mid-gap creation is possible if a nitrogen-vacancy is created in the material. Nitrogen deficiency in g-C3N4 leaves additional electrons and creates nitrogen-vacancy related C3+ states leading to a reduced bandgap material. This can be easily achieved by heating the material in an H2 atmosphere. H2, being a small molecule, can easily penetrate and diffuse to the interior lattice sites and reduce the lattice N sites and liberate NH3 and create an N vacancy. In this process, the terminal N sites are more prone to reduction due to the lowest change in the energy of the molecule. Such an N vacancy in the system reduces the bandgap by downshifting of the CB and up-shifting of the VB position. Another situation can also arise in which new C and N sites are generated, which increases the H contents in the material and alters the bandgap of the material. Experimental analysis suggests that a three coordinated N-vacancy and H-substitution at the N sites decrease the bandgap by 0.2 and 0.55 eV.78 This lowering in the bandgap is due to the formation of additional band states below the CB by the defect C sites leading to lowering the bandgap values. The N-vacancy moves the VB energy to more negative values and H-substitution lowers the CB to more positive values and improves the visible light absorption capacity. The surface area of g-C3N4 can be improved by exfoliating bulk g-C3N4 in different solvent media such as water, ethanol, acetone, IPA, and NMP. Among these solvents, NMP has been found to be the best choice.79 However, it is difficult to remove NMP from the exfoliated g-C3N4. Thus IPA has been explored as the next most desirable solvent to exfoliate bulk g-C3N4, so as to produce high surface area g-C3N4 of the order of 300–400 m2 g−1. For exfoliation of bulk g-C3N4, aqueous alkaline and acidic solutions have also been explored with the aim of obtaining higher surface area.80,81 Although an improvement in the surface area is observed by varying the precursors, reaction temperatures, and pyrolysis environment, however, the materials still remain nonporous in nature. In order to introduce porosity in the material, template-assisted synthetic strategies have also been adopted as discussed below.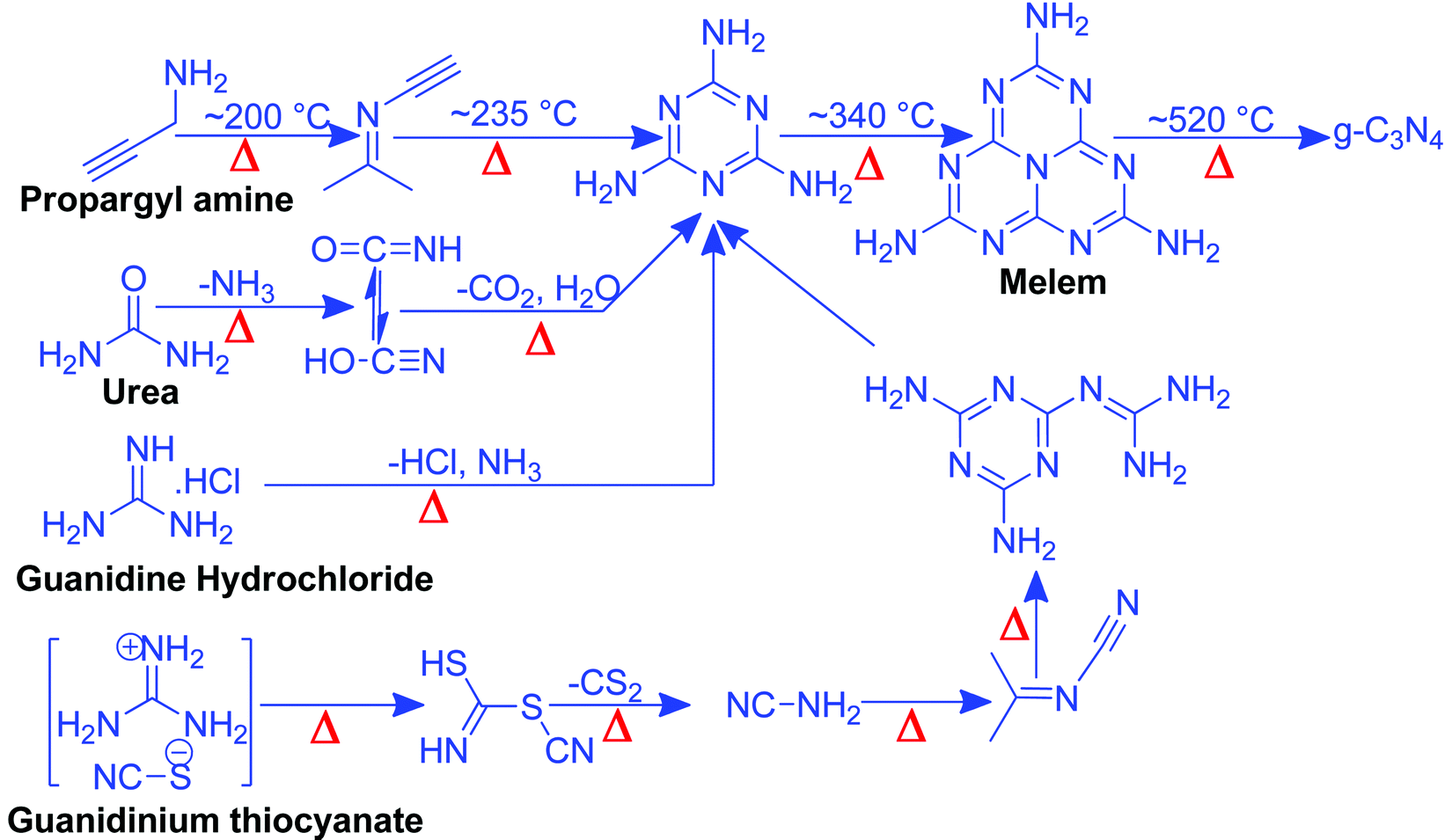 | ||
| Scheme 6 Synthesis of g-C3N4via the melem unit using different precursors. | ||
3.2 Template-mediated synthesis strategy to produce g-C3N4 with high surface area and porosity for improved CO2 adsorption
Various hard template-mediated synthetic strategies utilize silica-based mesoporous materials (SBA-15, SBA-16, KIT-6, MCM-41, and MCM-48) as the template (Scheme 7). Nitrogen-containing organic precursors or carbon and nitrogen precursors are infiltrated into the porous architecture of silica materials via the incipient wet impregnation or wet impregnation method followed by pyrolysis, which results in the formation of g-C3N4/silica hybrid materials. Finally, the silica template is removed by chemical etching using various etching agents, for example, NaOH, HF, and NH4HF2. The final material has the same architecture as that of the original silica material.82 Even silica nanoparticles have produced porous g-C3N4 with an average pore diameter of 12 nm, which is equivalent to the original silica nanoparticles.83 In general, the pore diameter of the resulting material is smaller than the porous silica materials due to volume shrinkage of g-C3N4 filled inside the pores during the condensation.84 The surface area and porosity significantly depend on the Si/precursor, hard template, and etching agent. The surface area can be further improved if the original silica material is treated with acid to enhance the surface reactivity, which allows a large number of organic precursors to in-filter, which improves the surface area of the resultant material.85 This synthetic strategy produces a low yield of g-C3N4 and, therefore, is difficult for application in the large scale synthesis of mesoporous g-C3N4. Further, hazardous and costly HF and NH4HF2 limit its industrial-scale production.86 To overcome this problem, CaCO3 has been used as a hard template in which the template is removed with dilute HCl after polymerization, leading to the formation of high surface area g-C3N4 with improved photocatalytic activity.87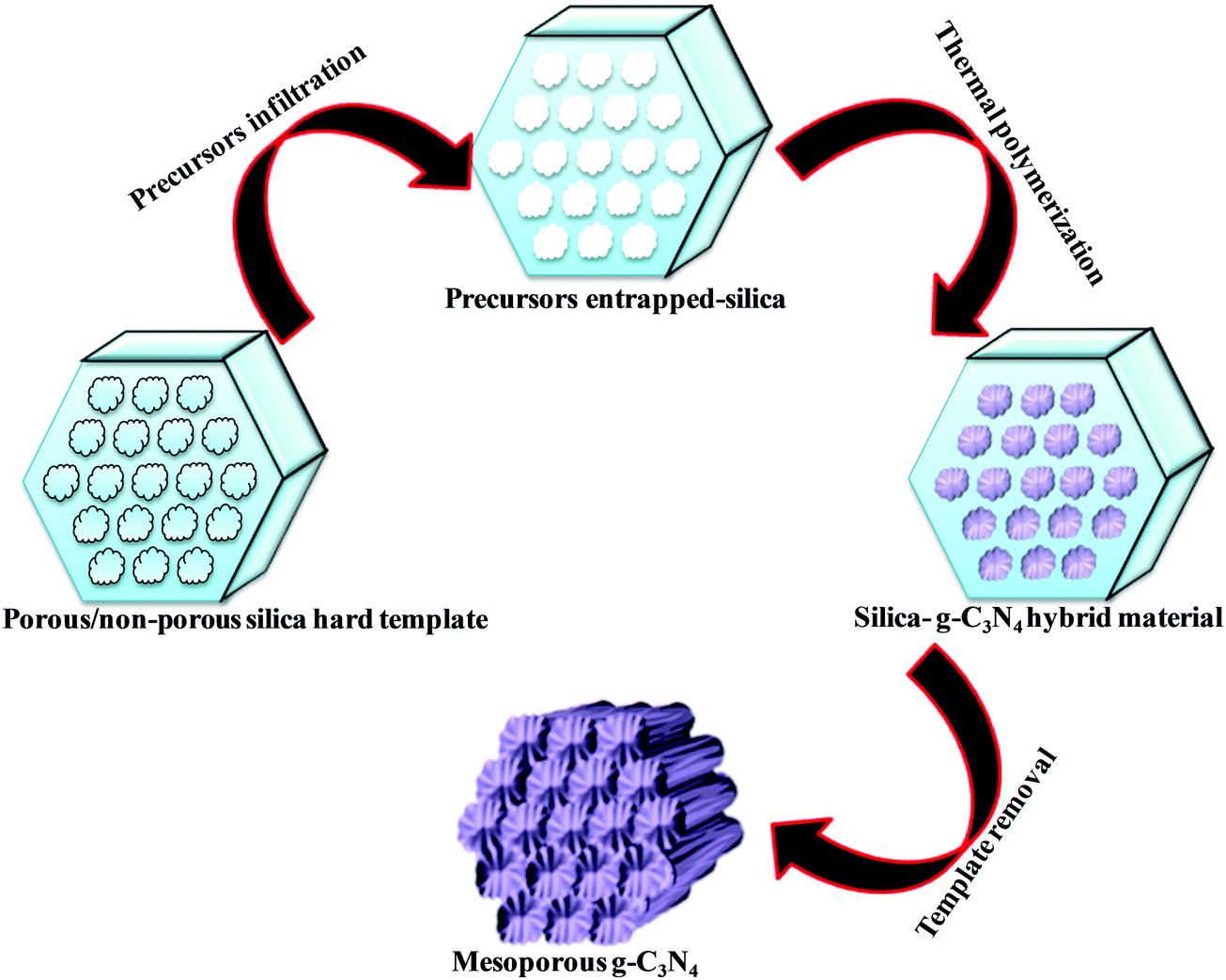 | ||
| Scheme 7 Hard template mediated synthesis of mesoporous g-C3N4. | ||
Large scale synthesis of mesoporous g-C3N4 can be achieved using the soft template method. Since the soft template is a carbon-based compound, therefore g-C3N4 with higher C/N is formed with diverse functionality. Ionic liquids and block copolymer-based soft templates have been widely used to prepare mesoporous g-C3N4. Among the soft templates, only a few templates such as Triton-100 and ionic liquids have produced g-C3N4 with large surface area and porosity.87–89 Ionic liquids act as soft modifiers for the thermal polymerization, leading to tunable textural properties, morphology, and reactivity. Anions of the ionic liquids participate in the growth process via C–N condensation.90 Dual templating methods and cooperative assembly of two surfactants lead to formation of a mesophase, which results in the formation of nanostructured materials. A similar strategy can be applied to synthesize mesoporous g-C3N4. Cooperative supramolecular assembly of surfactants, or a reactant and a surfactant, or reactants induced by hydrogen bonding and other weak electrostatic forces can act as a template to form mesoporous g-C3N4. Nitrogen-rich organic precursors can easily form hydrogen bonding with “O” containing organic compounds to form self-assembled porous g-C3N4. For example, melamine can form hydrogen bonding with cyanuric acid in DMSO and form stable aggregates that easily precipitate out (Scheme 8). Such precipitates can be thermally polymerized to form mesoporous g-C3N4.91 In fact, supramolecular aggregates can be formed by the hydrogen bonding between three reactants such as melamine, cyanuric acid with urea/barbituric acid/caffeine, etc. in a different solvent medium, which results in different morphology of mesoporous g-C3N4 on thermal polymerization with different textural properties and photophysical characteristics (Scheme 8).92–94 Template mediated synthesis strategies can provide materials with high surface area and porosity but it is difficult to tune the N contents and type of nitrogen due to the specific use of a suitable template and nitrogen precursor. Thus more work is needed to develop soft-template mediated synthesis recipes with variable N contents and functional amine sites using a diverse range of C and N containing precursors for the appointed applications.
 | ||
| Scheme 8 Supramolecular assembly of precursors leading to formation of mesoporous g-C3N4. | ||
3.3 Strategies to functionalize and dope elements to improve catalytic activities and light absorption properties
Template-mediated synthetic strategies can improve the surface area and porosity. However, for tuning the catalytic activities relating to the efficient adsorption of reactants and light absorption, suitable doping and functionalization are required. By varying the nitrogen precursors, the C/N ratio can be tailored, but for doping other elements such as O and S, suitable precursors containing O and S should be used during the synthesis of g-C3N4. Just by H2O2 treatment, O doped g-C3N4 with high surface area is obtained, which exhibits efficient charge carrier separation, and a red-shift in the absorption spectra.95 An oxygen group containing melamine precursor also produces O doped g-C3N4.96 Organic surfactant mediated synthesis produces C doped g-C3N4.97 Diammonium hydrogen phosphate, hexachloro-cyclotriphosphazene, 2-aminoethylphosphonic acid, etc. have been used as a P source to dope P into the g-C3N4 framework.98–100 H2S treatment, thiourea, and tri-thiocyanic acid precursors are known to dope S in the g-C3N4 framework.101–103 B-Doped g-C3N4 has been prepared by heating melamine and B2O3 above 520 °C to introduce B into the g-C3N4 framework.104 BH3NH3 and sodium-tetraphenyl boron have been used as precursors to prepare B-doped g-C3N4.105,106 Recently, it has been reported that exfoliation of g-C3N4 (prepared by urea and thiourea) with concentrated H2SO4 aqueous solution at 80 °C has produced –SO3H functionalized g-C3N4.107 Furthermore, it has been found that a small quantity of 2-methyl imidazole can be treated with urea-thiourea to produce O and C co-doped g-C3N4.108 Thus, the choice of precursor and the treatment conditions can introduce desired elements and functionality in g-C3N4 to impart the desired catalytic activity.4. Synthesis of chemicals and fuels from CO2 using g-C3N4 as a catalyst in the conventional thermal catalytic process
Several recent review articles have been published, even using carbon nitrides as catalysts and photocatalysts.109–111 In this section, the role of CO2 as a C1 source to produce chemicals and fuels is described (Scheme 9). CO2 can also act as an “O” source and can be utilized as an oxidant. CO2 can be efficiently reduced to provide various chemicals and fuels. All these processes are non-atom efficient processes. CO2 can be inserted in organic compounds like epoxides, amines, and alkynyl acids to produce organic carbonate and carbamate in a highly atom efficient process.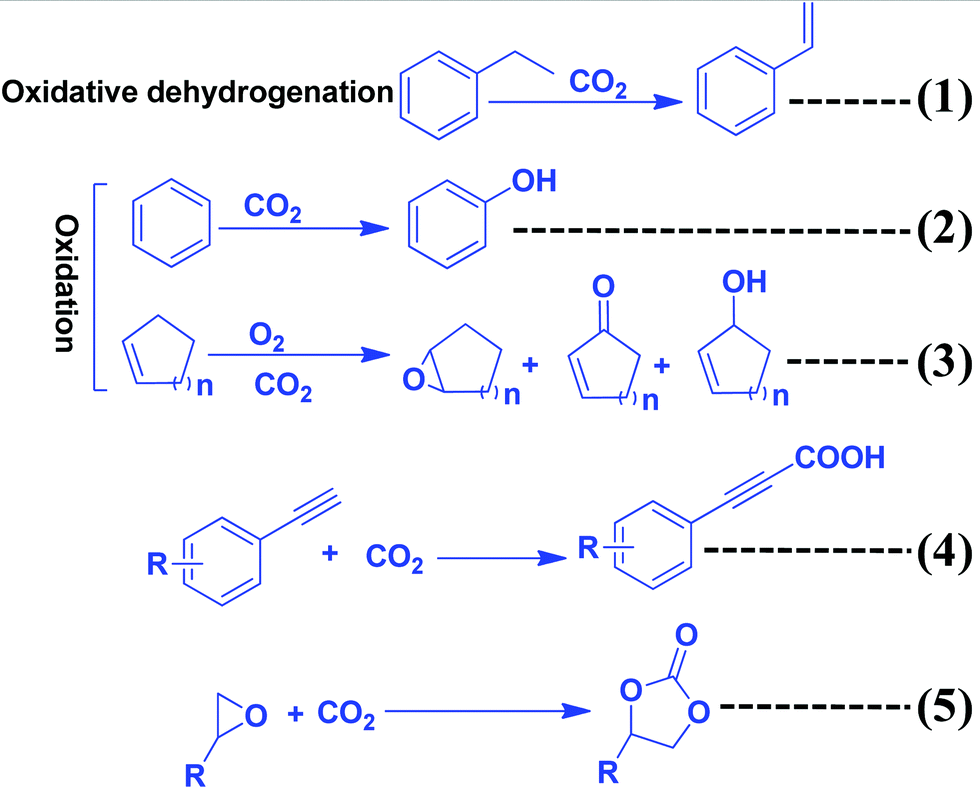 | ||
| Scheme 9 Synthesis of organic compounds using CO2 as a reactant or oxidant or promoter and C3N4 as a catalyst. | ||
4.1 Role of CO2 as a soft oxidant or promoter in the oxidation reaction
The oxidizing ability of various gases in coke oxidation follows the order: O2 (105) > H2O (3) > CO2 (1), and thus CO2 can act as a soft oxidant.112 Such a weak oxidation ability is suitable in the oxidative dehydrogenation reaction. Oxidative dehydrogenation in the presence of oxygen leads to the generation of a large amount of CO2 as a by-product due to the strong oxidizing ability of O2.113 CO2 is a weaker oxidant and, therefore, it requires activation energy. In general, this activation can be achieved with alkali metals such as Li, K, Na, Ba, Ca, and Cs, which activate CO2 to form MCO2.114 The activity of Li is higher among the above-mentioned metals due to the CO2–M valence bond angle, which is favorable for efficient CO2 activation and utilization. In this dehydrogenation reaction, CO2 facilitates the abstraction of hydrogen from hydrocarbons (alkanes, alkenes, alcohols, and alkyl aromatics) via catalytic activation to form CO and O species and suppress the total oxidation.115 g-C3N4 has been reported for the oxidation of benzene to phenol using CO2 as a mild oxidant.116 CO2 is activated via the secondary amine and pyridinic N of g-C3N4. Benzene is adsorbed weakly on g-C3N4. CO2 splits into an Ö diradical, followed by its reaction with weakly adsorbing benzene to form the desired phenol as a product (Scheme 10). CO is eliminated after the reaction to regenerate the active sites for further CO2 adsorption.116 CO2 acts as a promoter in the cycloalkene oxidation using O2 as an oxidant over a g-C3N4 catalyst (eqn (3), Scheme 9).117 CO2 is efficiently adsorbed on g-C3N4 to form surface O species and produce CO (confirmed from the FT-IR study). Such surface O assists in the oxidation of cyclic olefins.116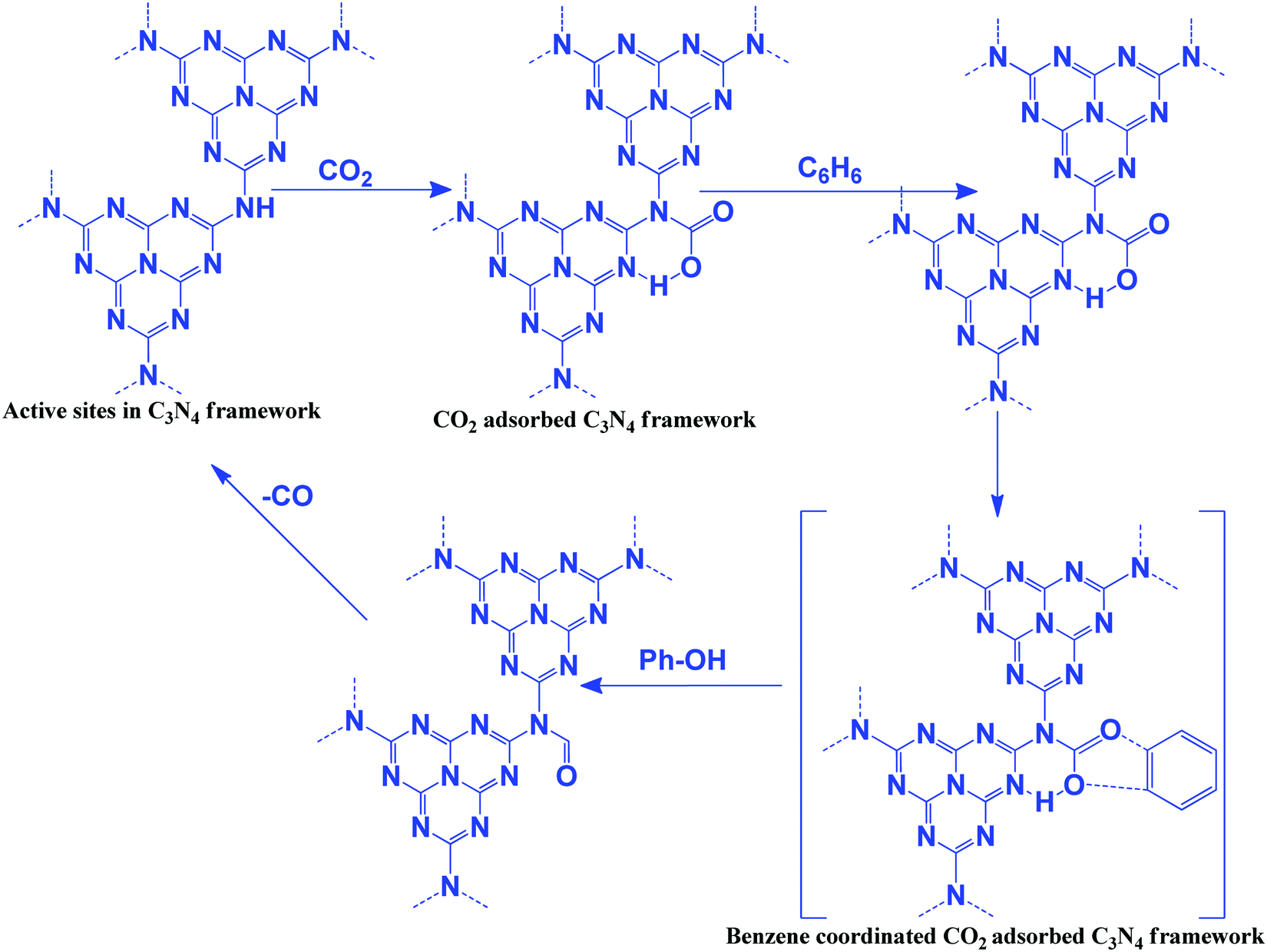 | ||
| Scheme 10 Oxidation of benzene to phenol using CO2 as an oxidant and g-C3N4 as a catalyst. | ||
4.2 Carboxylation of terminal alkynes with CO2
Alkynylcarboxylic acid is an important synthetic intermediate for several important compounds. For this reaction to proceed, both the alkyne and CO2 need to be activated (Scheme 9, eqn (4)). CO2 can be activated with g-C3N4 but for the efficient adsorption and activation of an alkyne, highly disperse active metals such as Cu or Ag are required. Ag NP decorated g-C3N4 has been reported for this reaction.118 This reaction follows the Langmuir–Hinshelwood mechanism in which CO2 is absorbed and activated by g-C3N4 and the alkyne is adsorbed and activated by Ag NPs supported on g-C3N4. Alkyne acidic hydrogen is activated by the Ag NPs and forms silver acetylide and then this reactive intermediate reacts with activated CO2 to produce silver carboxylate, which can be easily converted into cesium carboxylate with the help of Cs2CO3via a transmetalation step. Finally, by workup with an acidified solution, it forms the desired alkynyl carboxylic acid.4.3 Insertion of CO2 into epoxides to form cyclic carbonates
The synthesis of cyclic carbonates by the reaction of CO2 and epoxides is one of the most widely investigated reactions for CO2 utilization because it is a 100% atom economic process. The cyclic carbonate product can be used as an intermediate for the production of value-added compounds such as Bisphenol-A carbonate (a synthetic polymer used in the packaging industry) and dimethyl carbonate (a methylating agent and a green solvent), the latter being a good fuel additive and a precursor for various other industrially important chemical transformations. CO2 can be inserted in the epoxide ring via a non-concerted process to produce the cyclic carbonate. For this reaction, generally, a catalyst should contain a Lewis basic site, which acts as a nucleophile to activate the ‘CO’ bond of the CO2 molecule, while a Lewis acidic site acts as an electrophile for activating the epoxide ring.119 The Lewis acidic site polarizes the epoxide ring and forms an epoxide anion and then it acts as a nucleophile and attacks the CO2 molecule, leading to the formation of a carbonate anion intermediate. The next step is either a ring-closing process for the formation of the cyclic carbonate or insertion of one or more epoxides and CO2 molecules into the intermediate, which produces a polycarbonate.119 g-C3N4 has surface/edge located free –NH2 groups which serve as Lewis basic sites and act as the nucleophile to activate CO2. However, a Lewis acid site is absent to activate the epoxide. The weak activity of g-C3N4 towards this reaction is due to surface –OH sites which have the ability to weakly activate the epoxide and produce a very low yield of the product. For this reaction to proceed efficiently, a co-catalyst such as tetra butyl ammonium bromide (TBAB) is used. However, suitable metal-free bi-functional g-C3N4 is reported to effectively catalyze this reaction.107 Production of cyclic carbonates using different g-C3N4 based catalysts is summarized in Table 2. Conventional g-C3N4 exhibits very low activity (Table 2, entry 1).120 Mesoporous g-C3N4 (prepared with different precursors) has exhibited better activity than conventional g-C3N4. Among the prepared materials, mesoporous g-C3N4 prepared with melamine exhibits 34% propylene oxide conversion, and 90% propylene carbonate selectivity with a TOF of 3.4 h−1 at moderate reaction conditions in DMF (Table 2, entries 2–4).121 A mixture of gas (50% O2 and 50% CO2) is used during this reaction and it has been proposed that O2 plays a promotional role for the activation of the epoxide by supplying additional oxygen during the course of the reaction.120 Further, to improve the surface area and dispersed active sites, SBA-15 supported g-C3N4 is employed for propylene carbonate synthesis under solvent-free conditions in the presence of doped metal ions, which act as Lewis acid sites. Among metal salts, Zn2+ exhibits the highest conversion and selectivity of 98.3% and >99%, respectively (Table 2, entries 5–8).122 The activity of doped Zn2+ is better than the supported ZnCl2 catalyst (Table 2, entry 9).123 It is proposed that the C3N4 nanosphere inside the mesopore channel of silica supplies the activated CO2 to the epoxide through a transfer mechanism and facilitates cyclic carbonate synthesis via Zn2+ activated epoxide species. Porous and amino rich g-C3N4 is prepared without using any hard template using urea as a precursor to form defective and edge enriched C3N4. The urea derived C3N4 produces a wide variety of cyclic carbonates in the absence of any co-catalyst under solvent-free conditions (Table 2, entries 10–13).124 It is suggested that the edge defects contain both primary amines (–NH2) and secondary amine groups (–C–NH–C) and therefore they are able to activate CO2. The epoxide is activated by the defective C3N4 edge containing –OH groups. Further, it has been revealed that urea derived C3N4 exhibits relatively higher surface area (41 m2 g−1) than dicyandiamide derived C3N4 and a porous nature, and the existence of macropores facilitates mass transfer and enhances the catalytic activity. n-Bromobutane grafted mesoporous g-C3N4 exhibits better catalytic activity for solvent-free propylene carbonate synthesis with 88% conversion and 98% selectivity (Table 2, entry 14).125 In this process, the in situ depleted Br− acts as the electrophile which activates the epoxide ring, while the defect rich edge positioned –NH2 polarizes the CO2 molecule leading to the bi-functional nature. Butane in conjugation with halides have been evaluated and it has been found that bromide exhibits the best activity because of its better leaving group ability. The better catalytic activity is attributed to the mesoporous nature (15 nm), high surface area (241 m2 g−1), and coexistence of terminal –NH2 groups in g-C3N4 and X− groups, which is even far better than the externally induced n-bromobutane to carbon nitride during the course of the reaction (Table 1, entry 14).125 Huang et al. have reported H2SO4 treated, protonated acidic functionality containing carbon nitride, which has been investigated for the solvent-free cycloaddition of CO2 with epichlorohydrin for the synthesis of the corresponding carbonate (Table 2, entry 15).126 This protonation process has altered the electronic and textural properties and surface chemistry, which results in the incorporation of hydroxyl groups and amine groups into the bulk urea derived carbon nitride, which provides 17 fold higher catalytic activity. Surface hydroxyl groups (–OH) and amine groups (–NH2) are responsible for this improved reactivity. Inspired by this work, Samanta et al. have synthesized urea and thiourea derived carbon nitride CN-(UTU), which exhibits moderate catalytic activity better than the counterparts urea/thiourea under solvent-free cyclic carbonate synthesis from epichlorohydrin without any co-catalyst (Table 2, entry 16) (Scheme 11).107 Further, to improve the catalytic activity, the resultant materials have been chemically exfoliated with H2SO4, which results in the simultaneous incorporation of –SO3H and –NH2 groups into the melem framework (Scheme 11).
| E. no. | Catalyst | S BET (m2 g−1) | Co-catalyst | Reaction conditions | Conv. (%) | Select. (%) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| ECH = epichlorohydrin, PO = propylene oxide, SO = styrene oxide, TOF = turnover frequency, conv. = epoxide conversion, selec. = cyclic carbonate selectivity, SBET = surface area. | ||||||||
| 1. | g-C3N4 (prepared with melamine) | 64 | — | ECH (5 mL), catalyst (50 mg), temp. (150 °C), press. (3.5 MPa), time (2 h) | 5.6 | 96.7 | 3.3 | 120 |
| 2. | Mesoporous-C3N4 (prepared with melamine-2D mesoporous silica) | — | — | PO (1.5 mL), catalyst (20 mg), DMF (10), temp. (100 °C), press. (0.55 MPa), time (10 h) | 34.0 | 90 | — | 121 |
| 3. | Mesoporous-C3N4 (prepared with urea-formaldehyde-2D mesoporous silica) | — | — | PO (1.5 mL), catalyst (20 mg), DMF (10), temp. (100 °C), press. (0.55 MPa), time (10 h) | 28.0 | 92 | — | 121 |
| 4. | MS-C3N4 (prepared with melamine-glyoxal-2D mesoporous silica) | — | — | PO (1.5 mL), catalyst (20 mg), DMF (10), temp. (100 °C), press. (0.55 MPa), time (10 h) | 25.0 | 98 | — | 121 |
| 5. | Mesoporous-C3N4 (prepared with dicyandiamide-SBA-15) | 586 | — | SO (3 mL), catalyst (100 mg), temp. (150 °C), press. (3.5 MPa), time (1.5 h) | 28.8 | 99 | 4.8 | 122 |
| 6. | Mesoporous-C3N4 (prepared with dicyandiamide-SBA-15) | 585 | Zn2+ (0.8 wt%) | SO (3 mL), catalyst (100 mg), temp. (150 °C), press. (3.5 MPa), time (1.5 h) | 94.5 | 99 | 15.8 | 122 |
| 7. | Mesoporous-C3N4 (prepared with dicyandiamide-SBA-15) | 585 | Zn2+ (0.8 wt%) | PO (3 mL), catalyst (100 mg), temp. (150 °C), press. (3.5 MPa), time (1.5 h) | 97.1 | 99 | 16.2 | 122 |
| 8. | Mesoporous-C3N4 (prepared with dicyandiamide-SBA-15) | 585 | Zn2+ (0.8 wt%) | EO (3 mL), catalyst (100 mg), temp. (150 °C), press. (3.5 MPa), time (1.5 h) | 98.3 | 99 | 16.4 | 122 |
| 9. | 10%ZnCl2/mesoporous C3N4 | 206 | Zn2+ | EO (7 mL), DMF (3 mL), catalyst (200 mg), temp. (140 °C), press. (2.5 MPa), time (6 h) | 73.0 | 99.4 | 5.6 | 123 |
| 10. | g-C3N4 (prepared with urea and heated at 480 °C) | 40.8 | — | ECH (21.5 mmol), catalyst (100 mg), temp. (130 °C), press. (3.5 MPa), time (4 h) | 99.6 | 96.4 | 124 | |
| 11. | g-C3N4 (prepared with urea and heated at 480 °C) | 40.8 | — | EO (21.5 mmol), catalyst (100 mg), temp. (130 °C), press. (3.5 MPa), time (4 h) | 91.7 | 99.3 | 124 | |
| 12. | g-C3N4 (prepared with urea and heated at 480 °C) | 40.8 | — | SO (21.5 mmol), catalyst (100 mg), temp. (130 °C), press. (3.5 MPa), time (4 h) | 98.0 | 99.8 | 124 | |
| 13. | g-C3N4 (prepared with urea and heated at 480 °C) | 40.8 | — | PO (21.5 mmol), catalyst (100 mg), temp. (130 °C), press. (3.5 MPa), time (24 h) | 99.3 | 99.6 | 124 | |
| 14. | n-ButBr doped mesoporous C3N4 (prepared with cyanamide and 12 nn silica spheres) | 248 | Br− | PO (10 mL), catalyst (200 mg), temp. (140 °C), press. (2.5 MPa), time (6 h) | 88.0 | 99.7 | 9.6 | 125 |
| 15. | H2SO4 treated g-C3N4 (prepared with melamine) | 32 | — | ECH (5 mL), catalyst (50 mg), temp. (130 °C), press. (3.5 MPa), time (2 h) | 99.1 | 99.5 | 58.1 | 120 |
| 16. | H2SO4 treated g-C3N4 (prepared with urea-thiourea) | 10 | — | ECH (63 mmol), catalyst (50 mg), Temp. (100 °C), press. (1.0 MPa), time (4 h) | 93.5 | 99 | 107 | |
| 17. | P-Doped g-C3N4 (prepared with melamine-hexachlorotriphosphazene) | — | Bu4NBr | PO (28.6 mmol), catalyst (150 mg), Bu4NBr (2.4 mol%), temp. (100 °C), press. (2.0 MPa), time (4 h) | 99.8 | 99.9 | 126 | |
| 18. | B-Doped g-C3N4 (prepared with melamine-BmimBF4) | 11.1 | — | ECH (14.3 mmol), catalyst (50 mg), temp. (130 °C), press. (2.8 MPa), time (6 h) | 89.0 | 98.0 | 5.8 | 127 |
| 19. | B-Doped g-C3N4 (prepared with melamine-BmimBF4) | 11.1 | — | PO (14.3 mmol), catalyst (50 mg), temp. (130 °C), press. (2.8 MPa), time (6 h) | 31.0 | 99.9 | 1.5 | 127 |
| 20. | B-Doped mesoporous g-C3N4 (prepared with dicyandiamide–B(OH)3-SBA-15) | 123 | — | SO (1 mL), catalyst (30 mg), temp. (130 °C), press. (3.0 MPa), time (24 h) | 97.8 | 96.8 | — | 129 |
| 21. | g-C3N4 (prepared with melamine) | — | Bu4NBr | Light assisted method, ECH/SO (13.7 mmol), catalyst (50 mg), Bu4NBr (1.8 mol%) temp. (105 °C), press. (1 atm, balloon), time (20 h) | 100 | 100 | 130 | |
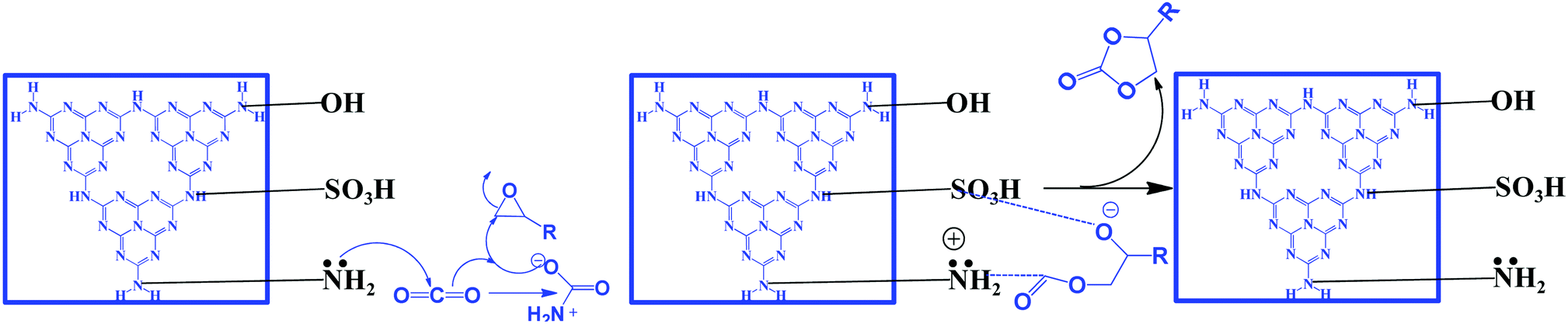 | ||
| Scheme 11 Synthesis of cyclic carbonates using bi-functional g-C3N4, reproduced with permission, Copyright Royal Society of Chemistry, ref. 107. | ||
The bi-functional catalyst (which is ideal for the CO2 activation reaction) has been investigated in the CO2 insertion reaction by varying the epoxide under very mild conditions (100 °C and 1 MPa). Excellent conversion and selectivity are achieved over this bi-functional CN-(UTU) catalyst. The simultaneous introduction of –NH2 groups (that act as basic sites for activating CO2) and –SO3H groups (that polarize the epoxide ring) has been confirmed from CO2/NH3-TPD. The synergistic participation of –NH2 and –SO3H groups during the reaction is the main reason for obtaining the excellent product yield. Moreover, doping with non-metals (such as P and B) over g-C3N4 is also reported for the synthesis of cyclic carbonates, which is found to be very effective for this reaction.126–129 The –B(OH) and –P(OH) groups are found to be responsible for the catalytic activity enhancement (Table 2, entries 19–20). All the reactions discussed above require CO2 pressure higher than one atmosphere. Only one report is available in which a cyclic carbonate is synthesized at 1 atm pressure under solvent-free conditions but in the presence of external co-catalyst (1.8%) TBAB (Table 2, entry 22).130 Therefore, more work should be carried out to synthesize efficient catalysts that can catalyze this reaction under solvent-free conditions at atmospheric pressure.
5. Photocatalytic CO2 reduction reaction (CO2 photoreduction)
Photocatalytic CO2 reduction is a surface catalytic reaction which employs light-induced holes and electrons generated over the surface of the catalyst and protons from the reaction medium. In ideal conditions, CO2 reduction follows some fundamental steps, like other light assisted reactions such as water splitting, N2 reduction, and organic transformations.131–140 The basic operating steps involved in the photocatalytic CO2 reduction reaction are summarized in two categories, namely optoelectronic and physicochemical steps, that are complementary to each other:(A) Optoelectronic steps (Scheme 12A):
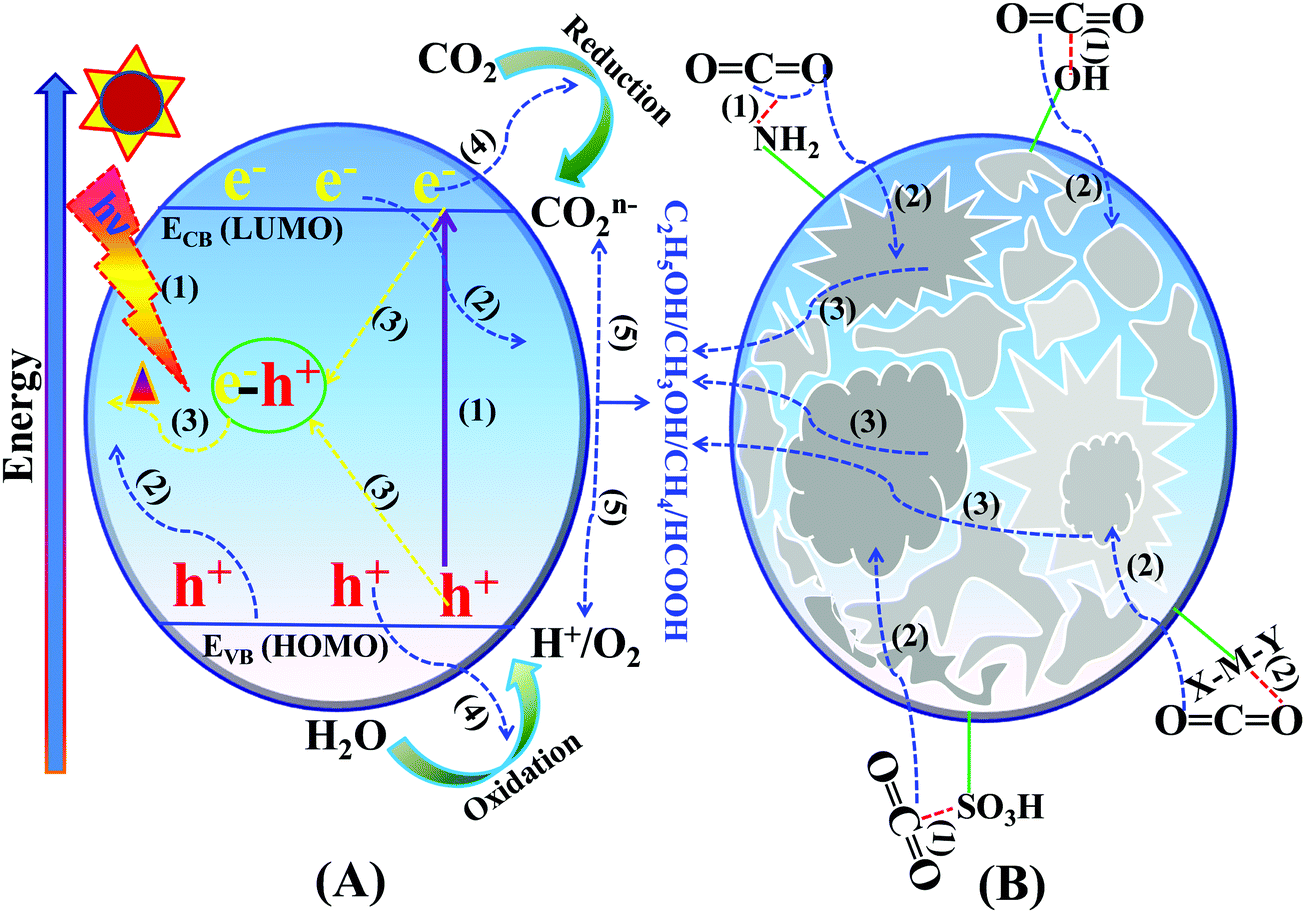 | ||
| Scheme 12 Basic mechanism of the photocatalytic process: (A) optoelectronic steps and (B) physicochemical steps involved in the CO2 reduction reaction. | ||
(1) Creation of charge carriers (e−–h+) on the semiconductor surface by illumination with light having energy greater than the bandgap: Ehν ≥ Eg.
(2) Light-induced charge carrier dissociation and migration on the catalytic surface.
(3) Charge-carrier recombination followed by the dissipation of energy in the form of heat.
(4) The transported electrons participate in CO2 reduction whereas holes participate in the oxidation of H2O to O2, and H+ or any other reducing agents to their corresponding oxidized products.
(B) Physicochemical steps (Scheme 12B):
(1) Adsorption of CO2 on the catalyst surface.
(2) CO2 activation by the inherent functional groups of the catalyst.
(3) Intermediate species adsorption and desorption of the products from the catalytically active sites during the reduction reaction.
5.1 Factors and parameters governing the mechanism of CO2 photoreduction
Carbon dioxide is the most stable and inert oxidized product, while methane is a completely reduced form of carbon (Scheme 13a). The light-induced conversion of CO2 to hydrocarbons and oxygenated hydrocarbons depends on various factors and parameters. These factors determine the fate of the reaction as well as the CO2 conversion and product distribution. Various factors influencing CO2 reduction, especially with graphitic carbon nitride as a photocatalyst, are discussed in this section.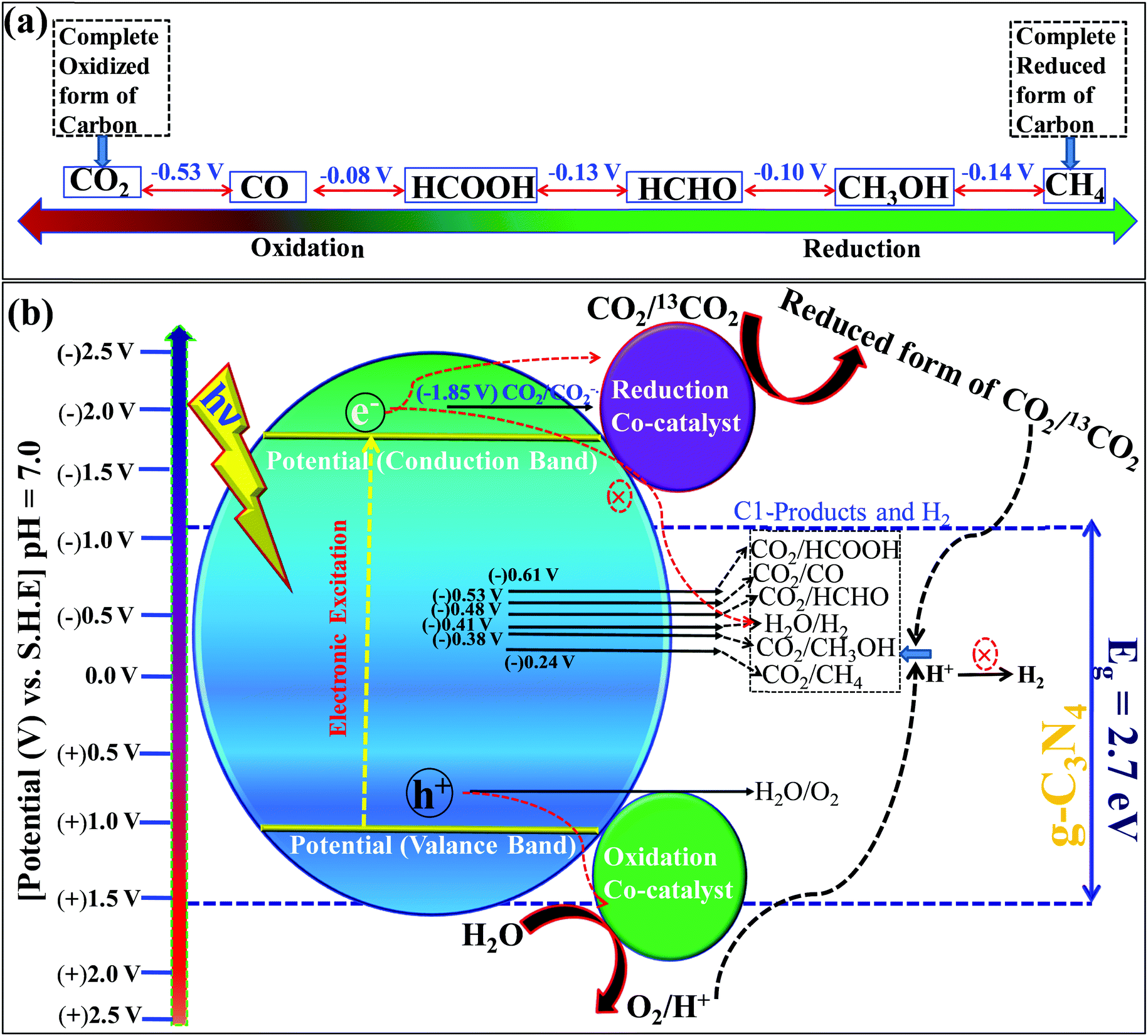 | ||
| Scheme 13 (a) The different states of carbon with the potential difference, and (b) photocatalytic CO2 reduction with respect to the standard redox potential of different products and band straddling. | ||
O’ bond in the CO2 molecule. CO2 has a low electron affinity and a high energy gap of 13.7 eV between its lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO).137 CO2 is a linear molecule and electronic accumulation in the empty LUMO (antibonding Π* orbital) facilitates the transformation of a linear to a bent geometry over the catalyst surface, which in turn expedites the kinetic barrier and enables its smooth conversion to appropriate products. The band edge position of any photocatalyst decides this electron accumulation and, accordingly, the product distributions occur. Particularly, the valence band edge must be positioned at a more negative electrode potential than the standard reduction potential of CO2 to any other products otherwise the process is not thermodynamically feasible (Scheme 13b). Based on the standard reduction potential,138 the reduced products are given below, which must be considered to analyze the reaction products after the reaction to establish the plausible mechanism of the reaction (Scheme 13b).| CO2 + e− → CO2˙− [E0 = (−1.85 V) vs. NHE] | (1) |
| CO2 + 2H+ + 2e− → HCOOH [E0 = (−0.61 V) vs. NHE] | (2) |
| CO2 + 2H+ + 2e− → CO + H2O [E0 = (−0.53 V) vs. NHE] | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O [E0 = (−0.48 V) vs. NHE] | (4) |
| CO2 + 4H+ + 4e− → HCHO + H2O [E0 = (−0.20 V) vs. NHE] | (5) |
| CO2 + 6H+ + 6e− → CH3OH + H2O [E0 = (−0.38 V) vs. NHE] | (6) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O [E0 = (−0.24 V) vs. NHE] | (7) |
| 2H+ + 2e− → H2 [E0 = (−0.42 V) vs. NHE] | (8) |
| 2H2O + 4h+→ O2 + 4H+ [E0 = (+0.81 V) vs. NHE] | (9) |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O [E0 = −0.329] | (10) |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O [E0 = −0.349] | (11) |
Among these reactions, eqn (1) is the most unfavorable one and, thus, the proton-coupled electron transfer process always occurs on the catalyst surface with the production of different products.
O’ bonds and the creation of C–H bonds. The complete reduction occurs via the formation of several radical intermediates, which also tend to recombine at different stages during the course of a reaction.139 The product distribution depends upon the specific set of reaction conditions. The simultaneous and competitive photo-oxidation and reduction reactions bring complexity to CO2 photoreduction. The most important parameters influencing these reactions include the type of photocatalyst, band edge potentials and bandgap, reaction conditions and medium, co-catalyst, etc., which are briefly described here. The first study reported for CO2 photoreduction involves TiO2 powder under photo-electrochemical conditions.140 Based on the mechanistic investigation reported in the literature, three possible pathways have been proposed, (I) the formaldehyde pathway forming CO2 → CH4 (fast hydrogenation pathway), (II) the carbene pathway forming CO2 → CH4 (fast deoxygenation pathway), and (III) the glyoxal pathway (Scheme 14I–III).131,141 It may be noted that at some stage of the reaction, all the suggested pathways can equally dominate. The elementary steps based on the specific reaction conditions need more work to establish the possible involvement of several radical species. The above-mentioned pathways are specifically discussed here.
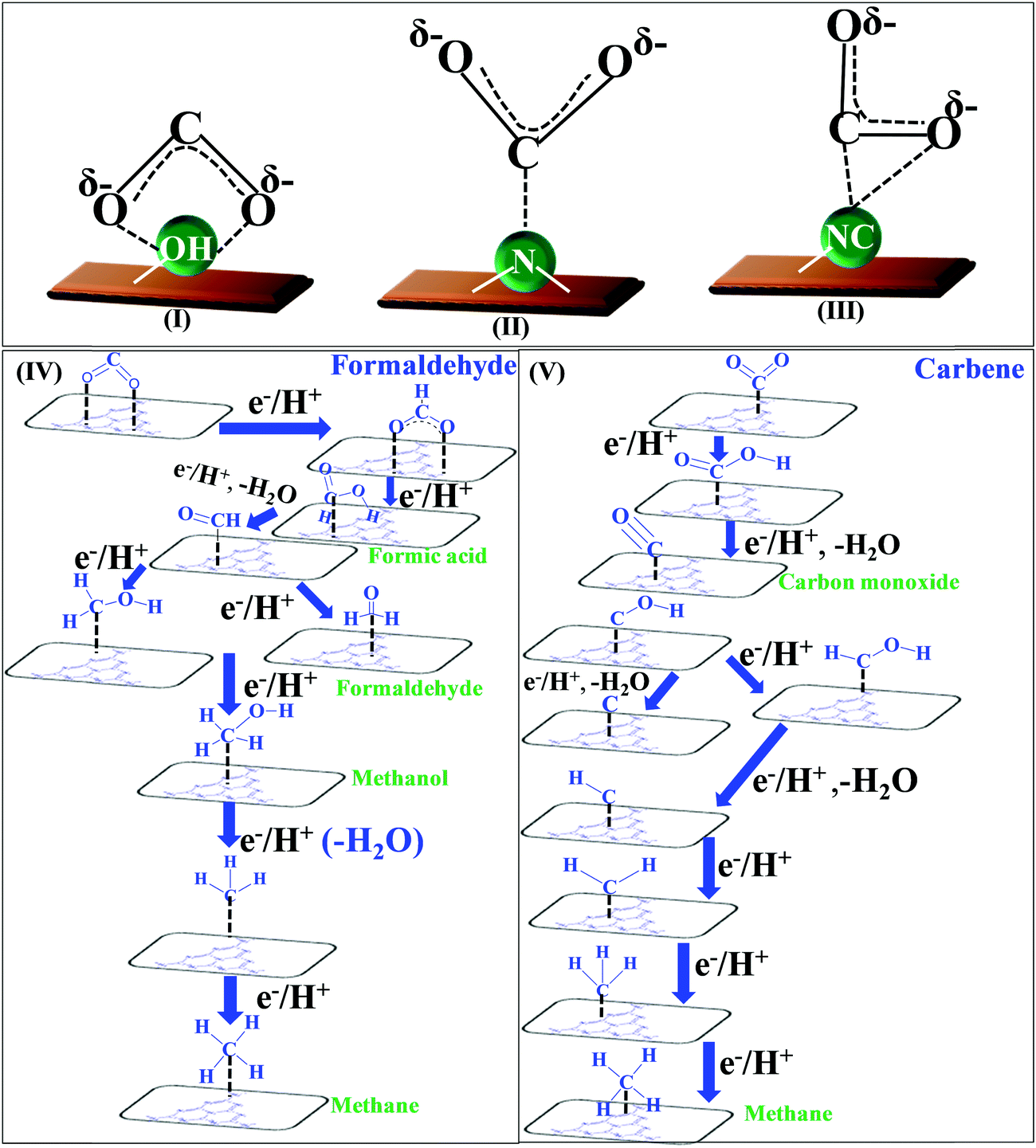 | ||
| Scheme 14 Co-ordination geometry of CO2 over the catalyst surface (I–III), and plausible reaction mechanism of CO2 reduction: formaldehyde pathway (IV), carbene pathway (V). | ||
The conversion of CO2 through the formaldehyde pathway involves multiple electrons and protons, which leads to the formation of several products such as HCHO, HCOOH and CH3OH, and finally the deoxygenated product CH4. Starting from CO2 involving 2e−/2H+, it first produces HCOOH (oxy-hydrogenation), followed by involving 2e−/2H+ again in the next step, which produces HCHO (reductive dehydrogenation).142 These two species formed during the reaction are intermediates and not side products. They can proceed to form unstable and reactive intermediates like (˙CHO)/(˙COOH), which in the next step takes up 2e−/2H+ to produce CH3OH. CH3OH can split to form (˙CH3) and (˙OH) radicals. (˙CH3) can take one (˙H) produced from the cleavage of HCHO to form CH4 at the end. The formation of CH3OH/HCHO also depends on the adsorption and desorption energy over the catalyst surface. If the coordination is strong enough and the desorption process is not facile, then CH3OH/HCHO will be the main product (Scheme 14(IV)). However, a weak binding energy allows this intermediate to produce the completely reduced CH4.141 Depending upon the intermediate binding energy, the yields and selectivity are regulated.
If the conversion process of CO2 → CH4/CH3OH follows the carbene pathway, then several reactive intermediates can form via de-oxygenation followed by hydrogenation elementary steps. It can propagate like CO2 → CO → (˙C) → (˙CH3) → CH3OH/CH4. Thus this suggests the following: (1) CO formation and radical formation of (˙C) and (˙CH3). (2) In the second step, the competitive reduction tendency between (˙CH3/˙C) and (˙OH/+H) often determines the product selectivity. In the first step, CO2 is adsorbed on the catalyst surface through carbon co-ordination by the 1e−/1H+ attachment to form the carboxyl radical (˙COOH). After coupling with several e−/H+, this can undergo decarboxylation to produce the formyl radical (˙CHO). In the next step, it can break into (˙C) and also can take three protons to produce the (˙CH3) radical intermediate. In the final state, it couples with one H+ or (˙OH) to form CH3OH or CH4 as the end product (Scheme 14(V)).143 The adsorption strength of CO on the catalyst surface also influences the product selectivity. If the CO absorption is very strong then it could lead to the formation of several interim products and, consequently, desorption as different products. But if the absorption is weaker, then the easy desorption produces CO as an exclusive reduced product. In this process, the formation of radical species (˙CH3) and (˙C) has been postulated and therefore electron paramagnetic resonance (EPR) measurements are very crucial to obtain a clear understanding of the reaction mechanism.
In the glyoxal mechanism, the first step is the formation of the one-electron reduction of (CO2−˙), which takes 1e−/1H+ to form the formyl radical (˙CHO). This further undergoes dimerization to form the glyoxal product. The complete sequence of the elementary steps involves oxidation and reduction reactions as follows: (CO2 → CO2−˙) → (˙CHO) → CHO–CHO → CH2OH–CHO → (˙CH2–CHO) → CHO–CH3 → (CH3OC˙) → (˙CH3) → CH4/CH3OH. However, this process is less explored and not completely understood yet.144 Various C2 products such as C2H5OH or CH3COOH etc. are believed to proceed through this pathway.
Based on the experimental observation and radical trapping/detection experiments, mechanistic routes are postulated, which requires further exploration. However, recent theoretical calculations suggest that defects and preferable coordination such as oxygen or carbon binding of the reactant, CO2, over the catalyst frequently determine the selectivity of the obtained product. A defect-less catalyst surface produces CH4 only, while oxygen defect containing g-C3N4 catalysts simultaneously form CH3OH/CH4.145,146 This is an interesting observation and more detailed work is required.
5.2 Graphitic carbon nitride as a CO2 reduction photocatalyst
The basic phenomenon of CO2 photo-reduction, especially with respect to heterogeneous catalysis, have already been addressed through several critical, tutorial, perspective, and edge articles by eminent researchers across the globe.159–165 Most importantly, CO2 photoreduction over g-C3N4 has also been reviewed recently.153–165 Still, various aspects of this material and its further manipulation for CO2 photoreduction catalytic efficiency improvement remain uncovered.It is pertinent to mention that g-C3N4 has been continuously investigated in different photocatalytic applications since its application as a semiconductor HER photocatalyst by Wang et al. in 2009. However, among the seven different types of polymorphic phases (all are semiconductors), only the g-h-heptazine and g-h-triazine phases possess appropriate bandgaps and band straddling to catalyze various photocatalytic reactions. But, a close look at the valence band edge potential and theoretical calculations confirm that only g-h-heptazine fulfills the thermodynamic requirements for water oxidation.62 Notably, the g-h-heptazine based polymorphic g-C3N4 material has been most widely investigated as a photocatalyst with a bandgap of 2.7 eV where the conduction band is situated at (−1.2 V vs. NHE) and the valence band is positioned at (+1.5 V vs. NHE) at pH = 7. The CB and VB indicate that it is capable of reducing the CO2 molecule and also can oxidize H2O to produce O2 and H+ required for the production of hydrocarbons/oxygenated hydrocarbons in CO2 photoreduction. Thus various nanostructured carbon nitrides with diverse morphologies such as nanosheets, microspheres, nanotubes, etc. are proven to be efficient catalysts for CO2 photoreduction. Thermodynamic constraints and optoelectronic limitations are the main bottlenecks of this catalyst for CO2 photoreduction as simultaneous CO2 and H2 reduction tendencies are observed for this particular catalyst. Also, the rapid charge recombination and low active surface area are other shortcomings that significantly hamper the efficiency of this catalyst. Strategic improvement of the physicochemical and optoelectronic properties is a pragmatic approach to overcome the poor CO2 conversion efficiency of pristine g-C3N4. In the forthcoming sections, a concise summary of the recent literature reports dealing with CO2 photoreduction efficiency improvement is compiled to give a clear understanding to researchers actively working in this area.
Dong et al. demonstrated, for the first time, CO2 photoreduction involving porous carbon nitride (PCN) as a catalyst in 2012.166 In this study, the authors propose that the porosity is introduced in bulk g-C3N4 by changing the precursors from melamine to melamine hydrochloride, which exhibits selective CO production of 4.5 mM in 11 h during CO2 photoreduction. However, on the contrary, the activity of bulk g-C3N4 is shown to be better (20.6 mM in 11 h) than porous g-C3N4 and this is attributed to the presence of predominant inherent defect sites in PCN compared to bulk g-C3N4 that act as charge carrier recombination sites (Table 3, entry 1).166 However, this adverse trend in the catalytic activity is unfortunately not fully understandable due to inadequate experimental evidence. Microstructure dependent CO2 reduction activity has been reported by Mao et al. The authors have observed that urea derived, mesoporous, flake-like g-C3N4 has produced a mixture of oxygenated hydrocarbons (C2H5OH and CH3OH), while melamine derived, non-porous, flaky C3N4 has produced CO and CH4 as products. The varied product selectivity is correlated to the reaction medium NaHCO3 and CO2 gas, where CO2 is available in the activated form (HCO3−), which is more accessible to the catalyst. In this process, O2 is obtained as a side product and it is established that the holes and electrons are consumed in a close ratio of ∼1 during the reaction with an apparent quantum efficiency (A.Q.E.) of 0.18% at 420 nm (Table 3, entry 2).167 The fine-tuning of the product selectivity is also reported in different studies, in which g-C3N4 nanosheets produce CH4 as a selective product, while the bulk g-C3N4 produces CH3CHO. This change in the product selectivity is proposed due to the switching of band edge potentials and also due to the different bandgap values. g-C3N4 nanosheets exhibit bandgap enlargement by 0.2 eV, which leads to a more positive valence band edge by 80 meV and a negative shift of the conduction band potential by 120 meV, which are the key reasons behind the different activity and selectivity as compared to bulk g-C3N4 (Table 3, entry 3).168 Thermal polymerization of pre-synthesized urea derived C3N4 and red phosphorous (P) has formed a heterojunction and, with the aid of Pt as a co-catalyst, the optimized catalyst (PCN-30) has converted CO2 to CH4 with a rate of 295 μmol h−1 g−1 in the suspension medium. The authors have proposed that the interfacial heterojunction has transferred the electrons from the high position g-C3N4 conduction band to a relatively lower position red P conduction band and the holes have been transferred in the reverse direction, which effectively reduces CO2 to CH4 and oxidizes OH to the (OH˙) radical, respectively. However, the authors have not emphasized the exact role of Pt involved in the reaction as a co-catalyst (Table 3, entry 4).169 K. Maeda et al. have synthesized high surface area mesoporous g-C3N4 using SiO2 as a hard template and dicyanamide as a precursor by varying the silica/precursor ratio.170 The dependency of the pore wall geometry on the CO2 photoreduction activity has been uncovered in this study. Furthermore, the resulting mesoporous C3N4 has been grafted with a Ru complex trans (Cl)-[Ru{4,4′-(CH2PO3H2)2-2,2′-bipyridine}(CO)2Cl2], where the electrons from the conduction band of g-C3N4 are transferred to the Ru and the bare holes in the valence band of g-C3N4 are engaged in the oxidation of water/triethanolamine, thus completing the total redox cycle (Scheme 15a).170 The best catalyst of this study exhibits the selective formation of 1854 nmol HCOOH in 5 h by involving (H2O + TEOA + CO2) in the catalytic system under visible light illumination. The most fascinating outcome of this work is that high surface area is not always beneficial, rather the average surface area (240 m2 g−1) with higher pore volume (0.74 cm3 g−1) has minimized the charge carrier recombination and has shortened the electron–hole diffusion to the reactive sites (Table 3, entry 5).170 J. Qin et al. have investigated the co-polymerization effect of monomer burbutyric acid with urea to synthesize g-C3N4, which eventually extends the pi-conjugated network, engineers the molecular hetero-interface and tailors the optical, surface area and nanostructural properties of g-C3N4 semiconductors for CO2 reduction with the involvement of Co(bpy)32+ as a co-catalyst and acetonitrile containing triethanolamine solution as a sacrificial electron donor, and CO2 gas.171 In this study, 56.3 μmol selective CO gas production is achieved in 4 h. 13C isotope tracing measurements have confirmed that the CO2 gas is the sole source of carbon, and, in fact, the control experiment has proved that all the components are essential for triggering CO2 photoreduction (Table 3, entry 6).171 K. Wang et al. have reported S-doped g-C3N4 (TCN) which has been prepared via a simple heat combustion method and first-principles calculations suggest that some of the bi-dentate N atoms of the tris-triazine melem framework are replaced by sulfur during this process, having a narrow gap energy (Eg = 2.63 eV) as compared to the melamine derived bulk g-C3N4 (2.7 eV). The CO2 photoreduction result demonstrates the selective production of methanol with a yield of 1.25 μmol g−1 in 3 h from the (NaHCO3 + HCl) induced (CO2 + H2O) vapor medium and Pt co-catalyst (Table 3, entry 7).172 J. Fu et al. have reported O-doped C3N4 nanotubes from controlled thermal oxidation etching which exhibit selective CH3OH production with a yield of 2.6 μmol in 3 h from a (NaHCO3 + H2SO4) mixture as CO2 and proton sources, respectively. The oxygen doping causes a lowering of the conduction band by 0.16 eV by substituting the nitrogen atom of the melem unit, as evidenced by theoretical calculations, which is the prime reason for this selectivity control. Moreover, the higher CO2 affinity and CO2 uptake are also supported by CO2-TPD and CO2-adsorption analysis. The mechanism of methanol formation has also been confirmed by in situ FTIR and GC analysis (Table 3, entry 8).173 B. Liu et al. have synthesized phosphorus-doped g-C3N4 nanotubes through a one-step thermal condensation between melamine and sodium hypophosphite monohydrate, which simultaneously dopes P and introduces surface amino groups. These modifications have led to higher CO2 uptake and favorable CO2 adsorption, which have been experimentally confirmed by CO2-uptake and zeta potential measurements. The resultant material has converted CO2 to 9.48 μmol CO and 7.24 μmol CH4 in 3 h. The experimental evidence suggests that phosphorus is interstitially doped into g-C3N4 during the synthesis, narrows the bandgap, exhibits a 1D nanotube morphology with high surface area, enhances the visible light absorption, and promotes the separation of charge carriers, which are the inclusive factors for this enhanced catalytic activity (Table 3, entry 9).174 D. Vidyasagar et al. have synthesized a phenyl grafted g-C3N4 scaffold by reacting phenyl urea, melamine, and urea with enhanced pi–pi stacking, which lowers the bandgap and provides a favorable band edge potential for CO2 photoconversion from (DMF + TEOA + H2O + CO2 gas) to produce CH4, CH3OH, and HCOOH with a yield of 0.907 μmol CH4, 450 μmol CH3OH, and 5 μmol HCOOH in 24 h using a 20 W LED bulb. The achieved apparent quantum yield is calculated to be 0.96% (Table 3, entry 10).175 S. Samanta et al. have synthesized C, O co-doped C3N4 through a thermal poly-condensation of urea, thiourea, and 2-methyl imidazole as starting
materials in an air atmosphere. The co-doping has increased the CO2 uptake, gives a porous cotton-like morphology, lowers the conduction band edge potentials, and also red-shifts the light-harvesting domain. The material converts CO2 gas saturated (TEOA + H2O) solution to methanol with a yield of 4.18 mmol g−1 in 6 h under visible light illumination. More importantly, theoretical calculations have provided favorable evidence about the high CO2 binding energy after C, O co-doping, which is also supported by CO2-TPD and CO2-adsorption studies (Table 3, entry 11).108 In interesting work, A. Roy et al. have synthesized a cobalt phthalocyanine (CoPPC) grafted polymeric graphitic carbon nitride photocatalyst in which the favorable CO2 electro-reduction capability, as well as the light sensitization properties of cobalt phthalocyanine, has been emphasized. Combining the characteristic features of C3N4 and CoPPc enables the material to reduce CO2 to CO in organic solvents (ACN + TEOA) under UV/vis light (AM 1.5 G, 100 mW cm−2, λ > 300 nm) with a cobalt-based turnover number of 90 for CO after 60 h. More interestingly, in complete aqueous phase reaction conditions, the catalyst exhibits 60 μmol CO corresponding to a TON of 5.1 per CO. When the reaction condition is changed by keeping acetonitrile fixed and replacing TEOA with 10% H2O the activity is reduced to 41%, indicating phase separation in aqueous medium (Table 3, entry 12).178 CoPPC acts as an electron acceptor and lowers the conduction band edge potential of the overall system (Scheme 15b). A 13C isotope experiment fully agrees that CO2 gas is the only source of CO in the reaction. The external quantum efficiency (E.Q.E) of CO is 0.11% and 0.03% at λex = 400 and 360 nm, respectively (Table 3, entry 12).178 Very recently, a 2D g-C3N4 nanosheet has been prepared via one-step tartaric acid-assisted thermal polymerization of dicyandiamide having nitrogen vacancies (g-C3Nx) with preferential location at both three-coordinate N atoms and uncondensed terminal NHx species. The material has produced 284.7 μmol g−1 CO and 51.6 μmol g−1 H2 from the CO2 gas dissolved in (TEOA + ACN) solution and Co(bpy)32+ as a co-catalyst in 15 h. The charge carrier migration and utilization are demonstrated with transient absorption spectroscopy (Table 3, entry 13) (Scheme 15d).179 M. Li et al. have synthesized polydopamine and barbituric acid co-modified carbon nitride nanospheres through thermal silica template-polymerization followed by a post-modification method, which has produced 158 μmol g−1 CO and 28 μmol H2 from CO2 gas dissolved in aqueous (TEOA + ACN) solution and Co(bpy)32+ as a co-catalyst in 4 h using a 300 W xenon lamp (visible light). The co-modification tunes the band edge potentials and favors CO2-adsorption, which are the key factors for the activity improvement (Table 3, entry 14).180 J. Zhang et al. have synthesized grain boundary controlled carbon nitride using 5-amino tetrazole as a high nitrogen-containing organic compound and NaCl/KCl. The synthesized catalyst (CN-ATZ-NaK) exhibits 14 μmol g−1 h−1 CO, 1.2 μmol g−1 h−1 CH4, and <1 μmol g−1 h−1 CH3OH production rates from an activated CO2 source (NaHCO3) and H2SO4 as a proton donor. The grain boundary tailoring is the main reason for this impressive catalytic activity (Table 3, entry 15).181 Another carbon and oxygen co-doped g-C3N4 has been achieved by aqueous dispersion of glucose with melamine followed by thermal treatment. The resultant optimized material has produced 55.2 μmol CO g−1 with nominal amounts of CH4 and CH3OH under visible light from CO2 dissolved in a (TEOA + ACN + TEOA) reaction mixture in 12 h. Simultaneous co-doping has improved the visible light absorption and given fast electron extraction, which are responsible for this activity enhancement (Table 3, entry 16).182 Very recently J.-Y. Tiang et al. have synthesized nitrogen-vacancy enriched carbon nitride by two-step thermal copolymerization between NH4Cl and melamine in a controlled environment, which induces mid-gap band state dangling between the conduction and valence band edge positions. The material has produced 9.28 μmol g−1 CO in 8 h in the presence of a catalyst-coated graphite rod and (CO2 + H2O) vapors under visible light illumination. The main reasons underlying this enhanced CO2 reduction activity are the introduction of dense nitrogen vacancies, extended light absorptivity of visible light leading to a wide range of light wavelength due to the strategic mid-gap arising, and prolonged lifetime of the charge carriers trapped inside the midgap. The obtained apparent quantum efficiency is calculated to be 0.089% (Table 3, entry 17) (Scheme 15c).183 J. Fu et al. have reported boron as a dopant that induces charge localization inside the cavity of tris tri-azine by the interaction with the two co-ordinated −(N)2 at the interstitial position. This claim is also justified by DFT calculations. The resulting 1%B/C3N4 has produced 0.32 μmol g−1 CH4 + 0.45 μmol g−1 CO from (CO2 + H2O) vapor under visible light irradiation in 3 h. A 13C isotope tracing experiment has supported the CO intermediate end product CH4 formation (Table 3, entry 18) (J. Fu et al., 2019).184 Trimesic acid-modified g-C3N4 (CNU-TMU-13) has been synthesized via a thermal combustion method which produces 70.63 μmol of CO and 14.99 μmol of H2 in 5 h from aqueous TEOA saturated with CO2 gas and Co(bpy)32+ as a co-catalyst. The origin of the carbon in the produced carbon is analyzed by a 13C isotope experiment and high CO2 uptake is also in favor of the surface modification by trimesic acid, leading to high catalytic activity (Table 3, entry 19).185 Simultaneous B, K co-doped g-C3N4 has been synthesized using dicyandiamide and KBH4 under an H2 thermal etching process, which has accelerated the CO2 reduction activity with the formation of 5.93 μmol g−1 CH4 and 3.16 μmol g−1 CO from CO2 dissolved in H2O in the absence of any co-catalyst, electron donor, or CO2 miscibility agent like DMF or acetonitrile in 5 h under visible light. This is also ascribed to the creation of nitrogen-vacancies and the incorporation of B and C together according to the reported theoretical simulation (Table 3, entry 20).186 The morphology also has a great impact on photocatalytic CO2 reduction. Tubular yolk–shell g-C3N4 has been hydrothermally synthesized from melamine and urea at 200 °C. The material has produced 40.3 μmol g−1 CO in 4 h from super-critical grade CO2 and H2O. The formation of CO is also analyzed by a 13C isotopic experiment. This catalytic improvement is correlated to the higher CO2 uptake as evident by the CO2-uptake profile and also due to the obtained tubular morphology (Table 3, entry 21).187 Another study reports the synthesis of nitrogen-vacancy containing C3N4 through the controlled polymerization of melamine in different atmospheres, which selectively produces 6.21 μmol h−1 CO from the catalyst suspension medium of CO2 and H2O under visible light illumination (Table 3, entry 22).146 The replacement of nitrogen has profoundly controlled selective CO production, which is established via DFT calculations (Table 3, entry 22).146 In a very strategic approach, X. Liu et al. have synthesized cyano-groups and phosphorus-doped C3N4 by treating pre-synthesized carbon nitride with NaH2PO2 under an argon atmosphere at different temperatures.
The authors have proposed and proved that the thermal decomposition of NaH2PO2 produced PH3 gas, which itself was doped as phosphorus and was also accountable for the conversion of the terminal, uncondensed C–NH2 to –CN groups during the synthesis process. In fact, the attachment of –CN groups served as electron-withdrawing groups over the carbon nitride surface, which benefited CO2 activation. The phosphorus doping has enlarged the light absorption and has inhibited photogenerated charge recombination. The active material (g-C3N4-425) has converted NaHCO3 saturated CO2 gas to CO with a yield of ∼1.14 μmol g−1 in 3 h using 1 wt% Au as a co-catalyst under a 300 W xenon lamp (Table 3, entry 23).188 S. Wan et al. have prepared hexagonal tubular carbon nitride by the heat treatment of the supra-molecular organization of the in situ hydrogen-bonded melamine cyanuric acid assembly under hydrothermal conditions. Hydrogen fluoride is found to be the best suitable haloic acid (H–X; X = F, Cl, Br, I) that promotes the dissolution of melamine, which eventually facilitated the pi–pi stacking as evidenced by the intense EPR spectra. The light absorption has been blue-shifted and charge separation is maximized with HF. The best active catalyst has produced 34.14 CO μmol g−1, 10.42 μmol g−1 H2, and 1.99 μmol g−1 CH4 in 12 h under visible light from 0.8 bar CO2 gas pressure dissolved in a mixture solution of (acetonitrile + TEOA + H2) (Table 3, entry 24).189 The authors have also calculated the average electron consumption rate (Relectron) during the CO2 reduction reaction as equal to 8.96 mol g−1 h−1 on the basis of the equation RE = (2RH2) + (2RCO) + (8RCH4), where R is the rate, expressed in μmol g−1 h−1 (Table 3, entry 24).189 For the first time ever, the stability aspect of g-C3N4 in the CO2 photoreduction reaction has been investigated in-depth by F. R. Poimilla et al., in which the authors provide insights about the stability of the melem framework before and after catalytic activity evaluation (Table 3, entry 25).62 They have synthesized g-C3N4 by following a two-step thermal polymerization of melamine. For comparison purposes, they have also synthesized O-doped g-C3N4 by treating carbon nitride with H2O2. They observed a marginal difference in the catalytic activity after repetitive cycles. In gas phase CO2 reduction, the catalyst exhibits 20 ppm selective CO in 5 h under 100 W xenon light (Table 3, entry 25).62 But, surprisingly, CO is obtained even in the absence of CO2 gas under light illumination, which raised the question about the origin of carbon from the graphitic carbon nitride photocatalyst. In association with detailed optoelectronic and physicochemical measurements and also with controlled analysis, the authors concluded that the photo-corrosion rather than the thermal decomposition of melamine is responsible for this unexpected behavior of the catalyst under their experimental conditions. In another breakthrough, X. Gong et al. have synthesized pyrene functionalized graphitic carbon nitride, which endows biphasic CO2 reduction combining CO2 reduction by the electrons in the aqueous phase and hole generated (OH) radical-induced olefin oxidation in the organic phase. The process of accessibility of the organic olefin into the aqueous system is facilitated by the increased lipophilicity of the grafted pyrene-functionality over g-C3N4. The authors have achieved ∼0.7 μmol CO from (NaHCO3 + cyclohexane) and (∼0.15 μmol CO + ∼0.23 μmol CH4) from (NaHCO3 + 2-methyl-2-butene in toluene) under 300 W xenon light illumination in 4 h (Table 3, entry 26).190
| E. no. | Catalyst/amount | S BET (m2 g−1)/precursors and process | Co-catalyst | Reaction conditions and (CO2 source) | Light source | Products/rate | Time (h) | 13C isotope tracing | CO2 pressure | Q.E. | Bandgap and band straddling | CO2 adsorption | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | g-C3N4 (100 mg) | — | None | CO2 gas + H2O | 20.6 mM CO in 11 h | 11 | No | — | — | — | — | 166 | |
| 2. | g-C3N4 (200 mg) | 39.5 | — | Supercritical fluid grade-CO2 + NaOH solution | 15.1 μmol CH3OH + 10.8 μmol C2H5OH + 51.2 μmol O2 | 12 | — | — | 0.18% At 420 λ ≥ nm | 2.78 eV (Ecb = −1.23 eV, Evb not defined) | — | 167 | |
| 3. | g-C3N4 nanosheet (100 mg) | (306) dicyandiamide in thermal oxidation in air | — | CO2 + H2O | 300 W Xe lamp | ∼0.12 μmol CH4 and ∼0.25 μmol CH3CHO | 5 | No | 0.06 MPa | — | E g = 2.97 eV; Evb = (+1.85 eV), Ecb not defined | — | 168 |
| 4. | Red P doped g-C3N4 PCN-30 (20 mg) | (Not provided) urea derived C3N4 combined pyrolysis of red P | 0.5 wt% Pt | CO2 gas + H2O | 500 W Xe lamp | 295 μmol h−1 g−1 | — | No | — | — | Not mentioned | — | 169 |
| 5. | CN-12–1.5 (8 mg) | (240) (silica hard template/cyanamide) | Ru co-catalyst (3.9 μmol g−1) | CO2 gas + (H2O + TEOA) solution | 400 W high-pressure Hg lamp | 1854 nmol HCOOH | 5 | Yes | — | — | E g and Evb not mentioned; Ecb = (−1.29 V) vs. Ag/AgCl at pH 6.6 | — | 170 |
| 6. | g-C3N4 nanosheet CNU-BA0.03 (30 mg) | (54) thermal polymerization of (urea and barbituric acid) | (CoCl2 + 2,2′-bipyridine) as co-catalyst | CO2 + H2O + (TEOA as electron donor) | 300 W Xe lamp | 56.3 μmol CO | 4 | Yes | 1 atm | — | No data available | Isotherm provided, binding energy did not mention | 171 |
| 7. | S-Doped g-C3N4 (TCN) (100 mg) | (4.4) thermal polymerization of thiourea at 520 °C in an ambient environment | 1 wt% Pt | (120 mg NaHCO3 + 0.25 mL 4 M HCl) → (CO2 + H2O) vapour | Simulated xenon lamp | Yield 1.12 CH3OH μmole g−1 | 3 | No | 1 atm | — | E g = 2.63 eV, no data available for band edge potentials | — | 172 |
| 8. | O-Doped g-C3N4 tube (50 mg) | (36) melamine in two-step heating in air and N2 | NaHCO3 + H2SO4 | Not mentioned (420 nm) | 2.6 μmol CH3OH | 3 | No | — | — | E g = 2.97 eV; Evb = not mentioned, Ecb = −0.87 eV vs. Ag/AgCl at pH = 7 | 0.045 mmol g−1 | 173 | |
| 9. | P-Doped g- C3N4 nanotube (50 mg) | (13.4) melamine and sodium hypophosphite monohydrate in 550 °C 4 h−1 | — | NaHCO3 + H2SO4 | A high-pressure xenon lamp (300 W) | 9.48 μmol CO and 7.24 μmol CH4 | 4 | No | 1 atm | — | (Eg = 2.97 eV, Ecb = −0.90 eV vs. E0NHE at pH = 7), Evb = not mentioned, | 0.39 mmol g−1 | 174 |
| 10. | Phenyl grafted g-C3N4 (100 mg) | (94.7) thermal polymerization of urea and phenyl urea at 550 °C for 2 h | — | DMF + TEOA + H2 O + CO2 | No information available | 0.907 μmol CH4 + 450 micromole CH3OH + 5 μmol HCOOH | 24 | No | — | 0.96% using 20 W LED bulb | E g = 2.97 eV, Ecb = −1.01eV, Evb = +1.65 calculated from Kulbeka monk plot | — | 175 |
| 11. | C,O co-doped g-C3N4 (100 mg) | (50) thermal co-polymerization of (urea + thourea + 2-methyl imidazole) | (CO2 + H2O) + (TEOA – acts as a sacrificial electron donor) | Xenon lamp (300 W) | 4.18 mmol g−1 CH3OH | 6 | No | 1 atm | — | 0.69 mmol g−1 | 110 | ||
| 12. | mpg-CNx (2 mg) | (104) cyandiamide and in situ synthesized cobalt pthalocyanine | CoPPc | CO2-Saturated (4:1) MeCN/TEOA |
Xenon lamp, 100 mW cm−2, AM 1.5G, λ > 400 nm | 455 μmol g−1 CO | 24 | Yes | — | (EQE) for CO: 0.11% and 0.03% at λex = 360 and 400 nm | E g = 2.82 eV, Ecb = −1.10 eV vs. E0NHE, Evb = +1.60 eV | — | 176 |
| 13. | DCN-0.05 (not mentioned) | (29.5) one step thermal polymerization of cyandiamide and tartaric acid | Bi-pyridine + CoCl2 | CO2 gas saturated in 5 mL solution (4 mL ACN + 1 mL of TEOA) containing aq. bipyridine (10 mmol L−1 + 25 mL of 20 mmol L−1 CoCl2) | 300 W xenon lamp | 284.7 μmol g−1 CO and 51.6 μmol g−1 H2 | 15 | No | — | — | E g = 2.49 eV, Ecb = −0.83 eV vs. E0RHE, Evb = +1.66 eV | — | 177 |
| 14. | BA20-CNS-PDA15 nanosphere (not mentioned) | (95) silica template (barbituric acid + polydopamine) | Co(bpy)32+ (150 mg 2,2-bipyridine, +10 μmol CoCl2) | CO2 gas purged in 50 mL suspension (30 mL ACN + 10 mL TEOA + 10 mL H2O) | 300 W xenon lamp (420 nm cut off filter) | 158 μmol CO + 26 μmol H2 (86% selectivity) | 4 | No | — | — | E g = 2.63 eV | Isotherm provided, binding energy did not mention | 178 |
| 15. | CN-ATZ-NaK (50 mg) | (15) (5-aminotetrazole in a or NaCl/KCl eutectic mixture) | — | (0.084 g NaHCO3) + (0.3 mL 2 M H2SO4) | 350 W Xe lamp with an AM1.5 filter | 14 μmol g−1 h−1 CO + 1.2 μmol g−1 h−1 CH4 + <1 μmol g−1 h−1 CH3OH | Not mentioned | No | — | — | E g = 2.53 eV, Evb = (+1.87 eV), Ecb = (−0.66 eV) | No data available | 179 |
| 16. | O,C co-doped C3N4 (MG3) (30 mg) | (10.8) (melamine + glucose) water dispersion flowed by heating | — | CO2 gas purged in (ACN+ TEOA + H2O) | 300 W Xe lamp with a UV cut-off filter >420 nm | 55.2 μmol CO g−1 | 12 | No | — | — | E g = 2.66 eV, Evb = (+1.38), Ecb = (−1.28 eV) | — | 180 |
| 17. | Nitrogen defect-modified g-C3N4 atomic layers: (CN550) (amount not mentioned) | (103.1) (melamine and NH4Cl) heating at different temperature | — | Catalyst coated on a graphite rod + H2O + CO2 gas | 500 W xenon arc lamp assembled with a UV cut-off filter (λ > 400 nm) | 9.28 μmol g−1 CH4 | 8 | No | — | 0.089% | E g = 2.72 eV, Evb = (+2.17), Ecb = (−0.55 eV) | — | 181 |
| 18. | 1%B/g-C3N4 (amount not mentioned) | (62.5) one-step calcination boric acid + urea | CO2 and H2O vapor = (H2SO4 solution + NaHCO3 powder) | 300 W Xe lamp with a UV/visible cut-off filter | 0.32 μmol g−1 CH4 + 0.45 μmol g−1 CO | 3 | Yes | — | — | Not provided | — | 182 | |
| 19. | CNU-TM-13.0 (30 mg) | (109.1) (urea + trimesic acid) | Co(bpy)32+; 1 μmol of cobalt chloride (CoCl2)+ (15 mg of 2,2-bipyridine) | CO2 gas purged in 5 mL solution (1 mL TEOA + H2O) | Visible light (λ > 420 nm) | 70.63 μmol of CO and 14.99 μmol of H2 | 4 | Yes | 1 bar | — | Not provided | Isotherm provided, binding energy did not mention | 183 |
| 20. | B,K-Doped C3N4 (100 mg) | 5.3 (dicyandiamide + KBH4) in H2 thermal etching | — | H2O + CO2 gas | 300 W Xe lamp | 5.93 μmol g−1 CH4 + 3.16 μmol g−1 CO | 5 | No | 2 bar | — | E g = 2.35 eV, Evb = (+1.45), Ecb = (−0.90 eV) | — | 184 |
| 21. | Tubular yolk–shell g-C3N4 (100 mg) | (75) (urea + melamine + DI H2O) hydrothermal treatment at 180 °C for 24 h | — | supercritical fluid-grade CO2 gas + H2O | 300 W Xe lamp | 40.3 μmol g−1 CO | 4 | Yes | — | — | E g = 2.72 eV, edge potential not mentioned | Isotherm provided, binding energy did not mention | 185 |
| 22. | Nitrogen vacancy containing C3N4 (CN-550) | (89.5) thermal combustion of melamine in air followed by H2 atmosphere | — | (High-purity CO2 gas + H2O) | 300 W xenon lamp equipped with a 420 nm cut-off filter | 6.21 μmol h−1 CO | Not mentioned | No | — | — | E g = 2.77 eV, Evb = (+1.67), Ecb = (−1.07 eV) vs. NHE | — | 146 |
| 23. | Cyano and P-doped g-C3N4 (g-C3N4-425) (100 mg) | (31.1) urea derived C3N4 mixed with Na2HPO4 followed by heat treatment | 1 wt% Au | 1 M 100 mL NaHCO3 + CO2 gas | 300 W xenon lamp equipped with a (λ = 420 nm) cut-off filter | 1.135 μmol g−1 CO | 3 | Yes | — | — | E g = 2.77 eV, Evb = (+1.68), Ecb = (−0.54 eV) vs. NHE | — | 186 |
| 24. | CNXt (30 mg) | (29.4) (melamine + HX) (X = Cl, Br, I) hydrothermal followed by carbonization in N2 | — | CO2 gas purged in (ACN + TEOA + H2O) | 300 W xenon lamp with a UV-cut filter (λ = 420 nm) | 34.14 CO μmol g−1, 10.42 μmol g−1 H2, 1.99 μmol g−1 CH4 | 12 | No | 80 kPa | — | E g = 2.83 eV, Ecb = (−1.13 eV) vs. NHE, Evb not defined | — | 187 |
| 25. | Pyrene-functionalized polymeric carbon nitride (10% Py-PCN)10 mg | (2.11) cyanuric chloride (CC) + cyanamide (CA) + acetonitrile (ACN) hydrothermal treatment followed by rehydrothemal treatment with 1-(4,6-dichloro-1,3,5-triazin-2-yl)pyrene (PyCC) | 3 wt% Pt | Biphasic (1 mL 3 M aqueous solution of NaHCO3) (3 M) and 9 mL cyclohexene (or 9 mL toluene containing 2 mmol of 2-methyl-2-butene) | 300 W xenon lamp | ∼0.7 μmol CO (using NaHCO3 + cyclohexane); ∼0.15 μmol CO and ∼0.23 μmol CH4 (using NaHCO3 + 2-methyl-2-butene in toluene) | 4 | No | — | — | E g = 2.31 eV; Evb = (+1.15), Ecb = (−1.16 eV) vs. NHE | — | 188 |
| 26. | g-C3N4 (catalyst amount not mentioned) | (SA analysis not measured) thermal polymerization of melamine | — | H2O vapour + CO2 gas | 100 W xenon lamp | ∼20 ppm CO | 5 | No | — | — | E g = 2.8 eV, band edge potential not mentioned | — | 62 |
| Metal modified/doped g-C3N4 for CO2 reduction | |||||||||||||
| 27. | Se-Modified polymeric carbon nitride nanosheets (m-CNNSs 0.08) 50 mg | (133) urea derived CN mixed in an ethanolic solution of diphenyl diselenide (DDS) followed by calcination | (15 mg; 2,2-bipyridine, 1 μmol CoCl2) | 1 mL TEOA in 5 mL solvent (ACN: 3 mL; H2O, 2 mL), 40 °C | 300 W xenon lamp (λ > 420 nm) | ∼10 μmol CO, ∼1.42 μmol H2 | 1 | No | 1 atm | — | No information available | — | 189 |
| 28. | Mg-Doped g-C3N4 Mg/PCN-20% (50 mg) | (59.9) urea derived g-C3N4 mixed in an ethanolic solution of Mg (NO3)2·6H2O followed by hydrothermal treatment | — | (100 mL H2O + CO2 gas) | 300 W xenon lamp | CH4 17.09 μmol g−1, CO 4.13 μmol g−1 | 6 | No | — | — | E g = 2.54 eV, no information available about g-C3N4 | No information available | 190 |
| 29. | CN1/Pd nano-tetrahedron (NT) (50 mg of the catalyst was suspended in 6 mL of DI water in a glass reactor) | Urea derived g-C3N4 followed by seed-mediated polyol-assisted reduction of Na2PdCl4 | Pd | (CO2 and H2O vapor) 0.084 g of NaHCO3 + 0.3 mL of 2 M H2SO4 solution | 300 W Xe arc lamp | 2.13 μmol h−1 g−1 CH4-production rate of 0.58 μmol h−1 g−1 | No information available | No | — | — | E g = 2.75 eV | Theoretical CO2 adsorption energy (Eads = −0.0083 eV) at the 100 surfaces of Pd NT | 191 |
| 30. | Au/P-Doped g-C3N4 (200 mg of the sample was dispersed in 3 mL H2O) | [Ionic liquid 1-butyl-3-methyl imidazoliumhexafluorophosphate (BmimPF6) + dicyandiamide (DCDA)] thermal annealing followed by in situ photoreduction of HAuCl4 | Au | (H2O + CO2) gas | 300 W xe-lamp | 24 μmol CH4 | 5 | No | — | CH4 ∼ 2.83%, respectively at λ = 420 nm | E g = 2.38 eV, Evb = (+1.4 eV), Ecb = (−0.98 eV) | — | 192 |
| 31. | Pt/g-C3N4 | (The surface area was not mentioned) urea derived g-C3N4 followed by polyol-assisted reduction of H2PtCl6 | 2 wt% Pt | (H2O + CO2) gas | 15 W bulb, the average intensity at the reactor 8.5 mW cm−2 | 13.02 μmol g−1 | 10 | No | — | — | E g = 2.8 eV, No information available about Ecb and Ebv | 193 | |
 | ||
| Scheme 15 (a) CO2 photoreduction over (a) Ru-complex grafted g-C3N4, reprinted with permission, Copyright Royal Society of Chemistry, ref. 172; (b) Co(bpy)32+/g-C3N4, reprinted with permission, Copyright John Wiley and Sons, ref. 178; (c) N-vacancy containing g-C3N4 with a midgap state, reprinted with permission, Copyright Royal Society of Chemistry, ref. 183; and (d) defect and trap state containing g-C3N4, reprinted with permission, Copyright John Wiley and Sons, ref. 179. | ||
Various noble and non-noble metal modified carbon nitrides also exhibited profound CO2 reduction catalytic activity. H. Ou et al. have prepared selenium modified thin g-C3N4 porous nanosheets (m-CNNSS) using pre-synthesized g-C3N4 in an ethanolic solution of diphenyl diselenide (DDS) followed by thermal polymerization. The evolution of ∼10 μmol CO and ∼1.42 μmol H2 is obtained over this catalyst from a reaction mixture containing (TEOA + acetonitrile + H2O) at 40 °C in 1 h using 420 nm light irradiation using Co(bpy)32+ as a co-catalyst (Table 3, entry 27).191 The role of selenium is investigated with density of state calculations, which evidences that selenium has narrowed the conduction band by creating an electronic distribution with the conduction band of CN. But it has not hampered the conduction and valence band composition as its Fermi level exists in the middle of the CB and VB level of g-C3N4 J. Tang et al. have synthesized Mg-doped g-C3N4 considering the high CO2 philicity of MgO and the light-harnessing capacity of the Mg2+ ions in natural chlorophyll. The synthesized material has produced 17.09 μmol g−1 CH4 and 4.13 μmol g−1 CO from CO2 gas saturated H2O under a 300 W xenon lamp in 6 h (Table 3, entry 28).192 Though the light-harvesting capacity is enhanced, they did not emphasize the CO2 uptake phenomenon with any experimental evidence. S. Cao et al. have prepared Pd nano-tetrahedron doped g-C3N4 from urea derived g-C3N4 followed by seed-mediated polyol-assisted reduction of Na2PdCl4. They focused on the facet dependent CO2 reduction catalytic activity of Pd NT/g-C3N4, which exhibits a CH3OH and CH4 production rate of 2.13 μmol h−1 g−1 and 0.58 μmol h−1 g−1, respectively, from activated (CO2 + H2O) vapor under 300 W visible light illumination (Table 3, entry 29).193 Furthermore, DFT calculations confirm that the Pd(100) facet over g-C3N4 exhibits high CO2 uptake and high CO2 binding energy and thus facilitates CH3OH desorption. M. Humayun et al. have synthesized Au NP loaded P-doped g-C3N4 in ionic liquid followed by thermal annealing of a mixture of [1-butyl-3-methyl imidazoliumhexafluorophosphate (BmimPF6) + dicyandiamide (DCDA)] and then finally the in situ photoreduction of HAuCl4 for the decoration of Au NPs. The catalyst exhibits selective CH4 evolution of 24 μmol in 5 h from CO2 gas saturated H2O under 300 W visible light (Table 3, entry 30).194 The calculated quantum efficiency for CH4 formation at 420 nm is ∼2.83%. The impressive catalytic activity is ascribed to the extended visible-light absorption along with the improved charge carrier separation via the phosphorus induced surface states and the Au nanoparticle decoration. The impact of Pt on the CO2 reduction over g-C3N4 is revealed by W.-J. Ong et al., using 2 wt% Pt loaded g-C3N4 nanocomposites. The catalyst exhibits the highest CH4 yield of 13.02 μmol g−1 catalyst−1 after 10 h of light irradiation, which is 5.1-fold higher in comparison with pure g-C3N4 (2.55 μmol g−1 catalyst−1), from CO2 gas dissolved in H2O under a 300 W xenon lamp (Table 3, entry 31).195 The enhancement in the photocatalytic activity of the Pt/g-C3N4 nanocomposite for CO2 photo-reduction CH4 production can be attributed to the enhanced visible light absorption and smooth interfacial photogenerated electron transfer from g-C3N4 to Pt by lowering the Fermi level of Pt in the Pt/g-C3N4 hybrid as evidenced by the UV-vis and photoluminescence studies. The accumulated electron density on Pt facilitates the reduction of CO2 to CH4via a multi-electron transfer process.
5.3 Critical factors regarding catalytic activity assessment and result presentation
Amidst the CO2 photoreduction reports published in several journals, there is great concern regarding the precise catalytic activity evaluation, supportive characterization, and data presentation. Despite addressing this matter in several perspectives, edge articles, and editorials, this problem still persists.196–200 Although the intention is not to indicate or draw attention to any inappropriateness about the results reported on g-C3N4 for CO2 photoreduction, the aim is to emphasize and regulate the standard requirements for a particular report that can be followed precisely by the readers. The following points may be considered during an experiment and result presentation. If g-C3N4 is the appointed photocatalyst, the abundance or source of carbon in the resultant product may be ensured with a 13C isotope tracing experiment since the catalyst itself is a carbonaceous material which eventually may be contributing as a carbon source. The amount of catalyst and co-catalyst may be clearly mentioned; otherwise, it is hard to follow since only the amount of catalyst can decide the rate of product formation and TON/TOF calculation. For obtaining the CO2 reduction mechanism, in situ/DRIFT IR, HPLC, and GC-MS are the standard requirements that may be carried out. Since the activity is solely dependent on the CO2-uptake capacity, therefore as a matter of fact CO2-adsorption and CO2-TPD analysis may be considered. The binding energy of CO2 over the catalyst surface will also provide useful information. Pre-requisite knowledge about the applied light source and especially the cut-on/off filter is also a vital factor. A cut-off filter implies the upward wavelength above the specified wave-length and vice versa in the case of a cut-on filter. The apparent quantum yield may be reported using monochromatic light only otherwise it is not appropriate to report as per the definition of the quantum yield. On many occasions, it has been reported by considering a range of wavelengths, which has no real scientific significance. For comparative purposes, only the quantum yield is a parameter suitable for consideration, since the activity and rate are definitely variable factors depending on the type of the photo-reactor and the amount of the catalyst used. Also, when the quantum yield is mentioned, the time must be mentioned as with the progress of the reaction the yield also increases and thus it is a collective parameter. Consequently, the product yield may not be reported like μ/mmol g−1 h−1 as the amount and time are two variable factors. Also, the catalytic activity is not always linearly enhanced with time or amount. For example, catalysts which are close to the surface of the exposed site of incoming light illumination will exhibit better activity than a bulk amount settled at the bottom of the reactor. It is always advisable to report only the amount of product formed along with specifying the other reaction parameters. Also for comparison purposes, the solid–gas phase induced CO2 reduced products may not be compared with the suspension medium even in the case of the same catalyst since the entire reaction condition is different, and thus it is not at all comparable. A minor alteration can change the entire catalytic investigation, which would mislead the readers. Therefore, these cumulative points may be considered during and after performing any CO2 photoreduction reaction.5.4 Device fabrication using g-C3N4-based materials in photocatalysis
Miniaturization of the photocatalytic process via device fabrication is an important step towards industrial deployment that fulfills social demands. The progress in this direction over g-C3N4 catalysts is very little explored. Moreover, a g-C3N4 based device for the photocatalytic CO2 conversion to C1 fuel is not yet reported to the best of our knowledge. The effective device ideally should have provisions for (a) CO2 capture and (b) its conversion to C1 fuels under natural sunlight, on-board, in the same unit. Powder g-C3N4-induced suspension photocatalysis may not be suitable for this purpose. The catalyst effectively captures solar photons when it is fabricated as a film. Therefore, coating g-C3N4 powder over a conducting surface like fluorine-doped-tin oxide (FTO) or synthesis of a layer by layer free-standing g-C3N4 thin film can accomplish this requirement. Therefore, significant efforts are required to develop a prototype for the industrial-scale conversion of CO2 to fuel. However, one can get some insight from the already developed g-C3N4 based devices for photoelectrochemical (PEC) water-splitting.201–204 G. Peng et al. have reported a robust seed-growth method for the synthesis of a layer-packed crystalline g-C3N4 film on FTO using melamine as a precursor for carbon nitride followed by heating at high temperature (Scheme 16(I)).201 The fabricated device exhibited an outstanding photocurrent density of 116 A cm−2vs. RHE without any sacrificial agent in a wide pH range (0–13).201 The same author has reported a free-standing g-C3N4 paper-based electrode for hole-driven photoelectrochemical dye degradation at the anode and electron-driven H2 gas production in a single setup. Here, the authors have used carbon paper instead of FTO, and this is considered to be a proof of concept for the simultaneous utilization of holes and electrons to perform disproportionation reactions.202 The very same group reported a graphene oxide (GO) coupled crystalline carbon nitride film in an FTO photoelectrochemical cell for the water splitting reaction. This time, different amounts of GO and melamine were mixed in ethylene glycol by varying the contents, and then the resulting paste was thermally annealed at high temperature. The fabricated cell produced a photocurrent density of 124 μA cm−2vs. RHE in the absence of a sacrificial agent under alkaline solution. Moreover, the fabricated cell has performed with long-standing durability, which could be attributed to the smooth junction formation between GO and g-C3N4, efficient charge separation, high electronic conductivity, and sizeable electrochemical surface area.203 In other exciting work, Y. Feng et al. have reported an adhesive interface containing a polymeric g-C3N4 film-based device for solar water splitting (Scheme 16(II)).204 The authors found that both sulfur and non-sulfur precursors are necessary to achieve a high current density of 100 μA cm−2vs. RHE in NaOH electrolyte. g-C3N4 based device fabrication for PEC has recently been reviewed, and this could be very helpful in designing a prototype for photocatalytic CO2 conversion to C1 fuel or related applications.205,206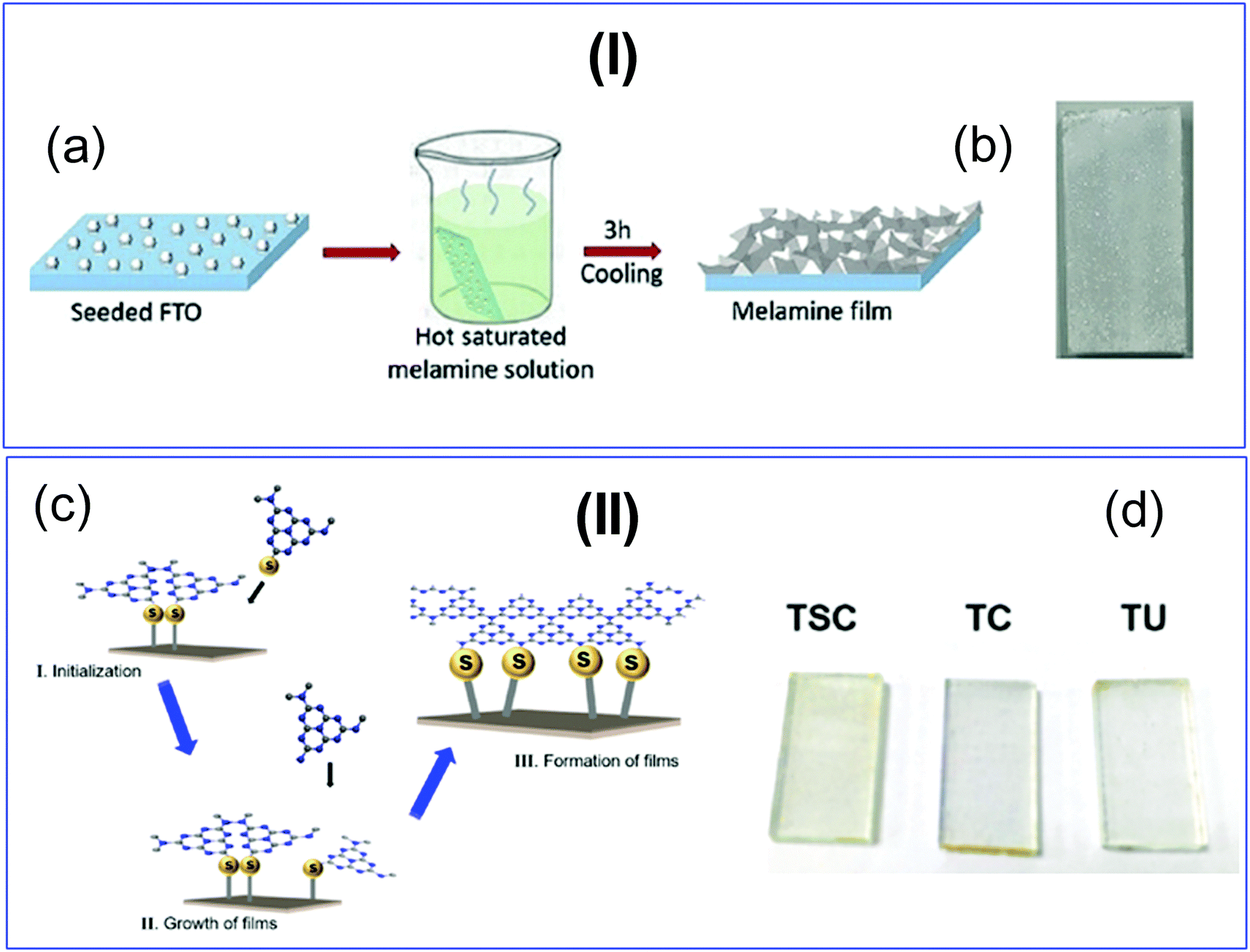 | ||
| Scheme 16 (I) (a) Fabrication of a crystalline carbon nitride based-PEC device, and (b) the resultant FTO based device, reprinted with permission, Copyright John Wiley and Sons, ref. 201; and (II) (c) a polymeric carbon nitride-based adhesive connected-PEC device, and (d) corresponding images of the FTO coated PEC devices synthesized from different precursors: TSC-thiosemicarbazide, TC-tri-thiocyanuric acid, TU-thiourea and urea, respectively, reprinted with permission, Copyright American Chemical Society, ref. 204. | ||
6. Electrocatalytic CO2 reduction reaction (e-CO2RR)
The electrocatalytic reduction of carbon dioxide into clean fuels is not fairly straightforward. In the last two decades, several research groups have studied purely metallic electrodes for this purpose and have segregated them based on the products that are obtained during the electrocatalytic CO2 reduction reaction (e-CO2RR) and the photoelectrocatalytic CO2 reduction reaction (pe-CO2RR). There are three major steps involved during the heterogeneous catalytic conversion of CO2 on the electrode surface: (i) adsorption of the originally linear CO2 molecule on an electrode material with the loss of its absolute linearity; (ii) cleavage of the C–O bonds to form C–H bonds by electron transfer and/or proton migration processes; and (iii) desorption of the product preceded by configurational rearrangement and followed by diffusion into the electrolyte.207 Further, it is found that both the e-CO2RR and pe-CO2RR involve multiple steps with overall two-, four-, six-, or eight-electron transfer processes (Scheme 17).208 Naturally, the range of products that form during such processes is diverse. They include carbon monoxide, formic acid or formate, formaldehyde, methanol, ethanol, methane, ethylene etc. The half-cell reactions involved in these processes along with their standard electrode potentials have already been listed in Section 5.1.1.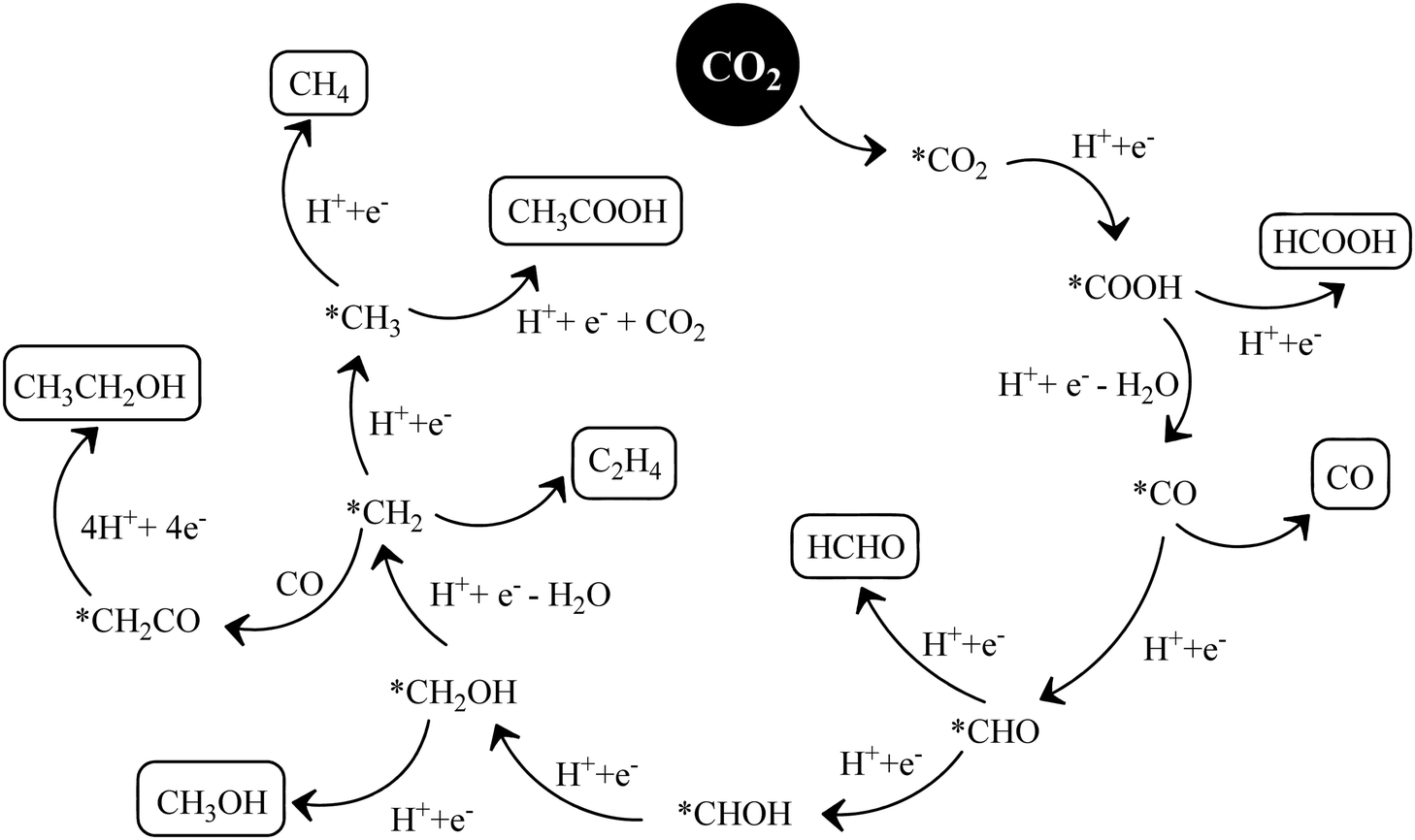 | ||
| Scheme 17 Various products formed due to multi-step processes during electrochemical CO2 reduction. | ||
6.1 The various rudiments of an e-CO2RR experiment
Based on the mechanistic pathways of the abovementioned processes, electrocatalysts can be tailored for efficient performance and high selectivity. The performance of various electrocatalysts can be judged by electrochemical techniques such as linear sweep voltammetry (LSV), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). While the overpotentials can be calculated for different products at various current densities directly from the LSV and CV profiles, the kinetics can be understood using the Tafel slopes that can be derived from the relevant voltammograms and/or the impedance spectra. All experiments should be performed using a two-compartment H-shaped electrochemical cell separating the cathode and the anode. This separation can be achieved using a thin frit glass membrane or a semi-permeable conducting membrane such as that of Nafion. A detailed gas-leak-proof setup design has been proposed by Pander et al.209 Since e-CO2RR involves the reduction of CO2, the working electrode is always a cathode which is capable of reducing other gaseous contaminants such as O2 and N2. Therefore, the e-CO2RR process should be preceded by the removal of air from the cathodic chamber. Subsequently, to accurately determine the products formed during the e-CO2RR, it is inevitable to saturate the cathodic chamber with high purity CO2. The electrolyte is generally KHCO3 and the counter-electrode (anode in this case) is graphite or a carbon electrode. In several cases, researchers have demonstrated the e-CO2RR in KOH medium in order to elevate the pH, and, thereby, suppress the reduction of protons/water (hydrogen evolution). However, saturation of the KOH electrolyte with CO2 reduces its pH due to the spontaneous formation of K2CO3 and KHCO3. Although the pH is maintained between 6.8 and 7.5 under normal conditions, the exact pH of the medium should be monitored to correctly convert the potential vs. reference electrode (commonly, a pre-calibrated Ag/AgCl or SCE) to the potential vs. reversible hydrogen electrode (RHE). The reference electrode is placed in the cathode chamber of the H-cell. The working electrode should be kept protected from metal impurities present in the electrolyte, such as that of Fe and Zn, in order to avoid poisoning and severe deterioration of the catalytic performance. For this, the aqueous electrolyte can be pre-electrolyzed so as to eradicate the contaminants by depositing them on a sacrificial cathode.210 Chelex is another option to prevent metal ion deposition due to its chelating action. The current densities that are reported for electrochemical experiments such as CV, LSV etc. should be normalized with respect to the electrochemical surface area (ECSA), which can be calculated as per an earlier report.211 To evaluate the faradaic efficiencies (FE) of various electrocatalysts, the aforesaid electrochemical measurements are generally correlated with the chromatograms obtained from GC (gas chromatography) and/or HPLC (high-performance liquid chromatography) of the product mixtures. A bulk-electrolysis experiment should be carried out for about an hour to obtain products that are enough for further analysis.212 It is wise to carry out experiments under potentiostatic conditions to avoid deviations from the product selectivity. Galvanostatic experiments are likely to generate potentials that are not suitable for the e-CO2RR, or can generate a variety of products. However, the latter may be preferred only under certain special conditions where the working electrode is studied well in advance and the applied current is precise for scaling up e-CO2RR processes.209 The complexity of e-CO2RR experiments, therefore, lies in the varied physical states of the products that are generated in the reaction medium. In the separate evaluation of the FE for various products, the faradaic selectivity or faradaic yield, defined as the fraction of the FE for a given product with respect to the total FE for all products, should also be calculated and reported. For the analysis of the H2 gas which is invariably evolved during e-CO2RR experiments due to very close potentials, the evolving gases should be directly delivered to the sampling loop of an online gas chromatograph equipped with a thermal conductivity detector (TCD) using argon as the inert carrier gas. Besides, a flame ionization detector (FID) should be used to analyze the possible carbon-containing gaseous products such as CO, CH4, C2H4, and C2H6 as described in the mechanistic pathway (Scheme 17). On identification of the constituents present in the liquid (e.g., HCOOH, CH3OH, and C2H5OH) and gaseous mixtures, the selectivity of the product can be evaluated. Relevantly, nuclear magnetic resonance (NMR) spectroscopy and mass spectroscopy (MS) are often used to confirm the associated products. However, to minimize interfering proton signals arising from the aqueous electrolyte, a solvent-suppression method (suppresses the solvent peak, such as water) and deuterium locking (usually done to every NMR sample before acquisition of the spectra) should be necessarily adopted during NMR measurements.209 As the mechanistic pathway involved in the catalysis over a given heterogeneous surface may be completely changed with the change in the nanoarchitecture of various carbon-based electrodes, structural characterizations of the electrode materials are also important. These include Fourier transform-infrared (FT-IR) spectroscopy, Raman spectroscopy, X-ray diffraction measurements (XRD), X-ray photoelectron spectroscopy (XPS), X-ray absorption near-edge spectroscopy (XANES), X-ray absorption fine structure (XAFS), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and aberration-corrected-scanning transmission electron microscopy (AC-STEM). Even when coupled with active metallic catalyst nanoparticles, various carbon materials may often be seen generating very different products than what can be expected from the purely metallic catalysts. Hence, it is important to understand the structure–activity relationships of the active catalysts with the reactant and product molecules. For detailed analyses of these processes, in situ techniques are often used. To understand the source of CO2 especially with carbon-based electrocatalysts or electrode materials/substrates, it is often important to carry out isotopic 13C-labelling experiments where 13CO2 is used as the feed gas instead of CO2. Since the focus of this article is on C3N4-based electrodes that have been studied for CO2 utilization to date, a relevant discussion in the said direction is imperative. Studies related to other carbon-based materials or carbon-free catalysts have however been regarded to exist beyond the scope of the present article.6.2 Impact of g-C3N4 as an interesting e-CO2RR catalyst
In recent times, heteroatom doping in carbon materials has evolved as an interesting domain of research to fine-tune their catalytic performances. For example, Sagara et al. have reported a boron-doped g-C3N4 photocathode catalyst that produces ethanol as the major product from CO2 in an aqueous phase with or without the loading of metal nanoparticle co-catalysts Au, Ag or Rh for activity enhancement. It has been generally understood through this study that the enhanced production of ethanol is an outcome of restrained recombination of C1 moieties and the increased selectivity owing to metal nanoparticle loading could be due to the multi-electron reduction by electron capture on metal particles.213 However, CO is generally the major product when Au or Ag is used as the catalyst, whereas alcohols are formed over Cu electrocatalysts.214 Zhang et al. have reported a polarized few-layer metal-free g-C3N4 electrocatalyst that can selectively form CO (with trace HCOOH) from CO2 with a high faradaic efficiency (F.E.) of ∼80% (Table 4, entry 1).214 This electrocatalyst has been prepared using hydrothermal pre-treatment of dicyandiamide followed by thermal exfoliation to reduce the number of stray basic sites. This catalyst is able to suppress the hydrogen evolution reaction and, thereby, efficiently catalyze CO2 reduction. The authors have argued that the large surface area and mesoporous structure of the benchmark photocatalyst mpg-C3N4 (mesoporous graphitic carbon nitride) are not capable of electroreduction of CO2 whereas the hierarchical structure of the foam-like monolith composed of 2D-pg-C3N4 is proven to facilitate the diffusion of carbon dioxide molecules to the active surface of the catalyst. Electrochemical impedance spectroscopy, further, has justified the improved mass transfer efficiency of 2D-pg-C3N4 exhibiting a larger slope in the low-frequency range (Table 4, entry 2).215 This result is better than an earlier report on a metal-free g-C3N4/MWCNT electrocatalyst that converts CO2 to CO. Of the total 98% faradaic efficiency obtained for H2 and CO, H2 accounts for 58% of the total FE, while that of CO is only 41% (Table 4, entry 3).216 Zhao et al. on the other hand have reported a ternary Au-CDot-C3N4 electrocatalyst with 4 wt% Au that can reduce CO2 to CO selectively with an F.E. of ∼79.8% at a potential of −0.5 V (vs. RHE). Employing theoretical calculations, the authors have proven the importance of the interaction between the CDots and C3N4. The authors have suggested the enhancement of the catalytic activity to be a direct outcome of the superior adsorption capability of CDots for both H+ and CO2. A narrower bandgap of 1.35 eV due to the newly introduced valence band maximum has been observed in CDot-C3N4 in contrast to pure g-C3N4 (1.92 eV). Again, with the introduction of AuNPs, the electrocatalyst Au-CDot-C3N4 has shown a further decrease in the bandgap to 0.32 eV (Table 4, entry 3).216 Also, adsorption studies have shown better capability of the 4 wt% Au-CDot-C3N4 electrocatalyst to adsorb both CO2 (∼0.34 mmol g−1 at 1.2 atm) and H+ (0.37 g g−1, on the basis of HCl) in comparison to 4 wt% Au–C3N4 (∼0.22 mmol g−1 at 1.2 atm of CO2 and 0.18 mmol g−1 of H+ on the basis of HCl) affirming the necessity of CDot incorporation (Table 4, entry 3).217 Employing Cu as the most widely investigated probe for CO2 reduction, Jiao et al. have studied g-C3N4 as a molecular scaffold that appropriately modifies the electronic structure of Cu in the resultant Cu–C3N4 complex. Through a combination of DFT computation and experimental justification, their work contributes to understanding the high selectivity of the e-CO2RR toward hydrocarbons/alcohols. The existence of ‘mock chemisorption’ on the nitrogen atom of the C3N4 scaffold is suggested in the study, where CO2 is adsorbed through a carbon–nitrogen bond on Cu–C3N4. This is believed to afford Cu–C3N4 to activate CO2 with the application of a suitable electrode potential. Further, the C-centres of the C3N4 framework are shown to have a higher ability to bind with species that ligate using oxygen atoms such as *OCH2, *OCH3, *O, and *OH. This is due to the electrophilicity in the C-atoms of C3N4, which are connected to nitrogen atoms with high electronegativity. Moreover, the Cu active site is shown to have a higher preference to bind with intermediates, *COOH, *CO and *CHO, that use the C-centre for coordination. This kind of dual active center model has been demonstrated to uplift copper's d-orbital position toward the Fermi level, which is critical in adsorbing the intermediate for CO2 activation. Their work successfully validates the formation of C2 products with the presence of two catalytically active centers (Table 4, entry 4).218 In contrast to this mechanism for CO2 activation, Xu et al. have unraveled the intriguing role of nitrogen species in the carbon framework during the electroreduction of CO2 (Table 4, entry 5).219 They have enumerated the activity of four types of N-species present in N-doped carbon nanotubes: pyrrolic, pyridinic, and quaternary nitrogen. A conjugated outcome of systematic DFT calculations and experimental observations suggested that the improved activity in N-doped CNTs, in contrast to undoped CNTs, is due to the stabilization of the key intermediate CO2−˙. The best correlation is found to be the presence of quaternary N-species (Table 4, entry 5).219 Recently, Zhang et al. have corroborated that carbon nitride supported Au nanoparticles (Au/C3N4) are better for CO2 electroreduction than carbon-supported Au nanoparticles (Au/C). This is consistent with an earlier report220 and the study has been extended to the interaction of Ag with C3N4. Experimental and theoretical data prove that C3N4 intensifies the electron-density over the Au surface, strengthening the adsorption of key reaction intermediate *COOH. The F.E. for CO is found to be maintained above 90% over a wide potential window of −0.45 V to −0.85 V vs. RHE for Au/C3N4, while that for Au/C is always lesser than 80% in this range (Table 4, entry 6).220 This result is comparable to a recently reported electrocatalyst Au-Py-CNT-O which reportedly reduces CO2 to CO with 93% F.E. over a wide potential range (−0.58 to −0.98 V) (Table 4, entry 7).221 Meng et al., in a recent report, have shown that a surface-modification strategy to introduce hydroxyl and amino groups on polymeric carbon nitride (PCN) can successfully be used to develop a metal-free electrocatalyst which is able to enhance the CO yield rate 17.1-fold as compared to its unmodified counterpart. The modification process has been carried out by treating PCN at 520 °C for 6 h in a controlled airflow to obtain O-PCN (PCN containing only hydroxyl species). Further, for 1 h O-PCN has been annealed in an NH3 atmosphere to obtain OA-PCN (containing amino groups along with hydroxyl groups). The catalytic efficiency of OA-PCN towards CO2 electroreduction is compared with that of A-PCN (containing only amino groups), O-PCN, and PCN. It is observed that the introduction of surface hydroxyl or amino groups alone cannot boost the e-CO2RR rate but the one with both functional groups has been found to suppress hydrogen evolution, thereby enhancing the rate of CO production. The CO/H2 ratio for the OA-PCN electrocatalyst is determined to be 0.67, which is in the range for the Fischer–Tropsch reaction (0.25–3.34). To investigate the ostensibly magical role of dual functionalization of PCN in the e-CO2RR, density functional theory (DFT) calculations have been carried out. It is revealed that the conversion of CO2 to COOH* possessed the highest energy barrier of 0.62 eV at the NH2 terminal. Furthermore, a higher barrier of 0.83 eV is required, at the –OH terminal, to desorb the CO product. Therefore, mono-functionalization does not boost the overall performance. However, according to the theoretical calculations, it is possible to infer that a proton–electron transfer process initiates the generation of the *COOH intermediate at the –OH terminal, which is transferred to the –NH2 terminal and, then, converted to *CO. The more facile desorption of the CO product is believed to occur at the –NH2 terminal, thereby exhibiting a fascinatingly high performance of the dual functionalized OA-PCN electrocatalyst (Table 4, entry 8).222 From the perspective of consistency, reproducibility and ease of synthesis, C3N4-based materials should, therefore, be advocated as efficient potential electrocatalysts for CO2 reduction.| E. no. | Electrocatalyst | Electrolyte | Major product (F.E.%, optimal potential) | Onset potential | Ref. |
|---|---|---|---|---|---|
| 1. | 2D-pg-C3N4 | 2 M NaHCO3 | CO (∼80%, −0.35 V vs. RHE) | −0.13 V (vs. RHE) | 214 |
| 2. | Au–BCN3.0 (gold decorated boron-doped carbon nitride) | 0.5 M NaHCO3 | Ethanol (78%) | 0.15 V (vs. Ag/AgCl) | 215 |
| 3. | BCN3.0 (boron-doped carbon nitride) | 0.5 M NaHCO3 | Ethanol (47%) | 0.15 V (vs. Ag/AgCl) | 217 |
| 4. | g-C3N4/MWCNT | 0.5 M NaHCO3 | CO (∼60%, −0.75 V vs. RHE) | — | 217 |
| 5. | Cu–C3N4 | 0.1 M KHCO3 | — | — | 219 |
| 6. | Au/C3N4 | 0.5 M KHCO3 | CO (90%, −0.45 V vs. RHE) | 220 | |
| 7. | Au-CDot-C3N4 | 0.5 M KHCO3 | CO (79.8%) | −0.3 V | 221 |
| 8. | OA-PCN (hydroxyl and amino functionalized polymeric carbon nitride) | 0.5 M NaHCO3 | CO (40.1%) | −0.56 V | 222 |
7. Overall summary, conclusions, and future perspectives
In summary, this article covered three important CO2 conversion processes, thermal CO2 activation, photocatalytic CO2 conversion, and electrochemical CO2 reduction, focused on the rising star carbonaceous material ‘g-C3N4’. A summary indicating the general characteristics, advantages, and disadvantages of the thermocatalytic/photocatalytic/electrocatalytic CO2 conversion process is provided in Table 5.| Process | General characteristics | Advantages | Disadvantages |
|---|---|---|---|
| Thermocatalytic | (1) Thermal energy is the driving force to disrupt the stable atomic and molecular bonds of CO2 and other reactants over a bi-functional catalyst to produce various platform chemicals and fuels. | (1) Easy to implement in large scale production. | (1) Not sustainable due to the involvement of a non-renewable energy source. |
| (2) Industrially available high-pressure liquid phase and gas phase reactors can be employed to convert CO2 to important feedstock chemicals and fuels under the influence of mild to high CO2 pressure (1–500 bar or so). | (2) A variety of chemicals and building blocks can be produced. | (2) The trade-off between conversion and energy efficiency is very low. | |
| (3) Catalysts can be easily designed for the appointed applications. | (3) Catalyst recovery is easy. | (3) The requirement of high pressure and temperature. | |
| (4) More than 90% of CO2 is converted via this process. | (4) Pure CO2 gas is required. | ||
| (5) Can be applied in a broad range of pressure (1–500 bar or so). | (5) Atmospheric CO2 gas cannot be converted. | ||
| (6) Thermodynamic and kinetic parameters can be easily tuned. | |||
| Photocatalytic | (1) The driving force is light-energy under ambient conditions to bring down the thermodynamic constraint of stable CO2 molecules over a semiconductor photocatalyst having efficient photon capturing ability. | (1) Light energy is available abundantly. | (1) A custom-made catalytic reactor coupled with a suitable light source makes the process less user-friendly. |
| (2) It depends on the chemisorption magnitude of CO2 at [P/P0 = 1]. | (2) Eco-friendly and sustainable process. | (2) Low light-harnessing and quick charge carrier recombination of the photocatalyst are a major drawback. | |
| (3) The design of a photocatalyst is more-challenging than the thermocatalytic process. | (3) A selective product can be achieved since one-pot CO2 reduction and H2O oxidation takes place. | (3) Product isolation and separation are comparatively tough. | |
| (4) The source of CO2 can be varied and, accordingly, different products can be achieved. | (4) Mechanistic investigation is somewhat difficult. | ||
| (5) Recyclability is high if a powder suspension system is employed. | (5) The competitive reduction tendency between CO2 and H+ by the light-induced electrons suppresses the overall activity. | ||
| (6) Large-scale implementation and night time operation are not possible due to the absence of sunlight. | |||
| (7) Only low CO2 pressure can be employed (<10 bar). | |||
| (8) Atmospheric CO2 gas cannot be converted. | |||
| Electrocatalytic | (1) A suitable external potential is a driving force (under ambient conditions) to bring down the thermodynamic constraint of a stable CO2 molecule over a highly electrochemically conductive electrocatalyst. | (1) Easy to operate. | (1) It is extremely difficult to design the electrocatalytic reactor. |
| (2) Mainly carried out under alkaline solution as a CO2 source. | (2) Eco-friendly and sustainable process. | (2) An appropriate membrane is difficult to synthesize. | |
| (3) Electrolyte and potential bias both play a key role. | (3) Highly economic. | (3) A redox-active metal is required owing to its inherent redox properties. | |
| (4) The design of an electrocatalyst is highly challenging. | (4) The trade-off between conversion and energy efficiency is very high. | (4) Catalyst recovery is very difficult if a thin-film is used. | |
| (5) Product selectivity can be finely tuned. | (5) Product quantification from a basic medium by GC, GC-MS, and HPLC leads to low yield. | ||
| (6) Less dependent on the CO2 chemisorption process. | (6) Only low CO2 pressure can be employed (<10 bar). | ||
| (7) The liquid phase reaction medium is primarily followed. | (7) Atmospheric CO2 gas cannot be converted. | ||
| (8) High turnover number (TON) and turnover frequency (TOF). | (8) Mechanistic investigation is difficult. | ||
The merits of g-C3N4 for the above mentioned processes are: (a) It is a thermally stable, non-toxic, easy to synthesize material at moderate temperature (550 °C), whereas other carbon-based catalysts need high temperature (1200–1400 °C) and also require chemical activation. (b) In contrast to other carbon materials, N-doped carbon materials, especially g-C3N4, are more effective for acidic CO2 activation and conversion. Tailored synthesis parameters, varied carbon and nitrogen precursors, and different synthesis strategies allowed us to synthesize g-C3N4 with different types of N species having Lewis and Brønsted basic sites (uncondensed –NH2) suitable for CO2 activation and utilization. Moreover, it is also possible to incorporate other functional sites in g-C3N4 so that a multi-functional catalyst for the appointed CO2 activation reactions can be designed to deliver a designated product. (c) g-C3N4 is the only carbon catalyst having a bandgap energy with appropriate valence and conduction band edge potentials for the utilization of visible light of the solar spectrum. It can reduce CO2 and oxidize H2O simultaneously and thus effectively serve in the CO2 photoreduction reaction. Except for carbon dots, all other carbons do not have any bandgap and, therefore, are photocatalytically inactive for the CO2 reduction reaction. However, carbon dots cannot themselves trigger CO2 reduction; instead, they need another support material eventually to exhibit their up-conversion photoluminescence property. (d) It is easy to functionalize g-C3N4 to improve the visible-light harnessing capability, which is not possible with other carbonaceous materials.
Some of the demerits of g-C3N4 are: (a) It is thermally stable, but it is prone to light-induced framework oxidation/decomposition. If CO2 is a reactant, then it requires deep investigation because the framework oxidation/decomposition product and CO2 derived product could give misleading information. Therefore, integrating g-C3N4 with another semiconductor is necessary to stop its framework degradation caused by light absorption. (b) It contains less oxygen in the form of hydroxyl groups, which is not enough to make it a steam reforming chemical looping catalyst. (c) It has low electrochemical conductivity and thus exhibits inferior activity for electrocatalytic CO2 reduction. Moreover, in the absence of an active metal, it exhibits limited redox activity in electrochemical CO2 reduction. Other carbonaceous materials, primarily N-doped carbons, are more suitable for electrochemical CO2 reduction owing to their superior electronic conductivity. (d) Porous carbon can be derived from any natural resources, but specific carbon and nitrogen-containing precursors are required for g-C3N4 synthesis. Moreover, it is a visible light active catalyst and cannot cover the UV and NIR regions of the solar spectrum, which is also a drawback for its limited photocatalytic CO2 reduction activity.
The most crucial feature of g-C3N4 is its facile synthesis, which can be performed without any complex synthesis setup. But plenty of research is required to improve its catalytic CO2 conversion efficiency. g-C3N4 exhibited activity in all these three processes but with limited efficiency. For example, thermal CO2 activation needs high temperature and pressure, which is not sustainable. Moreover, the involvement of an added co-catalyst raises the question of its effectiveness. Integrating an alkali metal oxide with the g-C3N4 framework can enhance CO2 adsorption and activation. Therefore, further improvement of g-C3N4 based catalysts is required for CO2 ambient condition activation and utilization. Since g-C3N4 does not have enough oxygen content, integrating it with transition metal oxides like CeO2 and MnO2, or spinel based oxides like NiCo2O4 with high oxygen exchange capacity can make it an active catalyst for steam reforming of methane coupled with the CO2 splitting reaction. In the case of photocatalytic CO2 reduction, the target of mole scale product yield is not yet achieved. Moreover, selectivity control and inclusion of additional organic additives like acetonitrile and dimethylformamide, and sacrificial agents like triethanolamine along with a co-catalyst are the serious drawbacks of this photocatalytic process. It is noteworthy that an ideal CO2 photoreduction system involves a light-harvesting material, CO2 gas, and H2O only. But very rarely has this target been served by this catalyst to date. Another controversial aspect is the source of carbon in the converted product during CO2 photo-reduction, especially when the catalyst itself is a carbon-based material. Utilization of suitable precursors during thermal polymerization (or other synthesis strategies) can incorporate required C, N, O, or S functionalities that have the capability to alter its solar light harnessing capability and can improve the charge carrier separation suitable for high photocatalytic CO2 reduction reaction activity. Another vital area is the configuration of thin-film based-device fabrication for practical CO2 photoreduction. g-C3N4 has a triazine skeleton, and therefore single-metal-atoms can be entrapped in the framework via wet impregnation followed by thermal annealing in a reducing environment. It will minimize attrition and deactivation, which are advantages for catalyst long term stability. High overpotential, low current densities, and limited redox-tunability are the shortcomings in its electrochemical CO2 reduction application. Therefore these concerns should be carefully considered for its further efficiency improvement. Carbon nitride-based single-atom-catalysts can be a viable solution to improve photocatalytic, electrocatalytic, or photoelectrocatalytic CO2 reduction.
We have concisely covered almost all the parameters starting from the synthesis of materials to fundamentals of CO2 activation, followed by critical operational parameters for collective ‘thermo-photo-electro’-chemical CO2 conversion focusing on the g-C3N4 catalyst. The conclusion section has provided the details of the merits and demerits of g-C3N4 along with a concise perspective on the application of carbon nitride for CO2 conversion. We envisage that this review will be helpful for beginners in their early stage of research on heterogeneous CO2 conversion reactions for further technological advancement in the 21st century.
Author contributions
The theme of the review article was proposed by SS and RS jointly. The manuscript was co-written by S. S. and R. S. The authors thank Mr Aniruddha Mukherjee for the preparation of some figures and helpful discussion and drafting the electrochemical section. All the authors proofread and approved the final version of the manuscript.Conflicts of interest
The authors state that no competitive and non-competitive conflicts of interest exist to declare. The authors also declare that some of the figures included in this manuscript have been prepared by themselves with the appropriate citations wherever it is applicable. Copyright permission has been obtained for the required figures of this manuscript.Acknowledgements
RS is grateful to the Director IIT Ropar for the Mid-career Excellence in Research through grant number 9-35/2009/IITRPR/3121. SS acknowledges the research fellowship received from the Ministry of Human Resource Development, New Delhi, and Director's Fellowship, IIT Ropar. We also thank the anonymous reviewers for their critical comments for the quality improvement of our review article.References
- G. Savulich, F. H. Hezemans, V. G. Grothe, J. Dafflon, N. Schulten, A. B. Brühl, B. J. Sahakian and T. W. Robbins, Transl. Psychiatry, 2019, 9, 296 CrossRef.
- https://www.ipcc.ch/report/2019-refinement-to-the-2006-ipcc-guidelines-for-national-greenhouse-gas-inventories/ .
- P. W. Barnes, C. E. Williamson and R. M. Lucas, et al. , Nat. Sustainability, 2019, 2, 569–579 CrossRef.
- M. G. Lawrence, S. Schäfer, H. Muri, V. Scott, A. Oschlies, N. E. Vaughen, O. Boucher, H. Schmidt, J. Haywood and J. Scheffran, Nat. Commun., 2018, 9, 3734 CrossRef PubMed.
- A. V. Herzog, T. E. Lipman and D. M. Kammen. Renewable energy sources, Encyclopedia of life support systems (EOLSS) forerunner volume-perspectives and overview of life support systems and sustainable development, 2001.
- Environmental Impact Comparison of Steam Methane Reformation and Thermochemical Processes of Hydrogen Production G. F. Naterer, O. Jaber, I. Dincer, Detlef Stolten, Thomas Grube (ed.): 18th World Hydrogen Energy Conference 2010 – WHEC 2010 Parallel Sessions Book 3: Hydrogen Production Technologies – Part 2 Proceedings of the WHEC, May 16.-21. 2010, Essen Schriften des Forschungszentrums J,lich/Energy & Environment, Vol. 78-3 Institute of Energy Research-Fuel Cells (IEF-3) Forschungszentrum J,lich GmbH, Zentralbibliothek, Verlag, 2010 ISBN: 978-3-89336-653-8.
- T. P. Senftle and Emily A. Carte, Acc. Chem. Res., 2017, 50, 472–475 CrossRef CAS PubMed.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS.
- D. M. Reiner, Nat. Energy, 2016, 1, 15011 CrossRef.
- F. A. Rahman, M. M. A. Aziz, R. Saidur, W. A. Bakar, M. R. Hainin, R. Putrajaya and N. A. Hassan, Renewable Sustainable Energy Rev., 2017, 71, 112–126 CrossRef.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- Q. Wang, J. Luo, Z. Zhong and Z. A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC.
- F. Akhtar, Q. Liu, N. Hedin and L. Bergström, Energy Environ. Sci., 2012, 5, 7664–7673 RSC.
- D. Bonenfant, M. Kharoune, P. Niquette, M. Mimeault and R. Hausler, Sci. Technol. Adv. Mater., 2008, 9, 013007 CrossRef PubMed.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- J. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911–13913 RSC.
- J. Gong, M. Antonietti and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 7557–7563 CrossRef CAS.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS.
- M. Oschatz, M. Leistner, W. Nickel and S. Kaskel, Langmuir, 2015, 31, 4040–4047 CrossRef CAS PubMed.
- V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- J. Lee, D. C. Sorescu and X. Deng, J. Am. Chem. Soc., 2011, 33, 10066–10069 CrossRef PubMed.
- E. Karamian and S. Sharifnia, J. CO2 Util., 2016, 16, 194–203 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 91, 133–139 CrossRef CAS.
- N. Keller, G. Rebmann and V. Keller, J. Mol. Catal. A: Chem., 2010, 317, 1–18 CrossRef CAS.
- J. E. Hallgren, US Pat., US4410464A, 1983 Search PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawad and S. Konnog, Green Chem., 2003, 5, 497–507 RSC.
- Z. Marković, S. Marković, N. Manojlović and J. Predojević-Simović, J. Chem. Inf. Model., 2007, 47, 1520–1525 CrossRef.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- L.-H. Zhang, W.-C. Li, L. Tang, Q.-G. Wang, Q.-T. Hu, Y. Zhang and A.-H. Lu, J. Mater. Chem. A, 2018, 6, 24285–24290 RSC.
- G.-P. Hao, Z.-Y. Jin, Q. Sun, X.-Q. Zhang, J.-T. Zhang and A.-H. Lu, Energy Environ. Sci., 2013, 6, 3740 RSC.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- F. Rodŕguez-reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- X. Shao, Z. Feng, R. Xue, C. Ma, W. Wang, X. Peng and D. Cao, AIChE J., 2011, 57, 3042–3051 CrossRef CAS.
- R. Wang, P. Wang, X. Yan, J. Lang, C. Peng and Q. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 5800–5806 CrossRef CAS PubMed.
- J. Chen, J. Yang, G. Hu, X. Hu, Z. Li, S. Shen, M. Radosz and M. Fan, ACS Sustainable Chem. Eng., 2016, 4, 1439–1445 CrossRef CAS.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- C. Robertson and R. Mokaya, Microporous Mesoporous Mater., 2013, 179, 151–156 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765 RSC.
- Z. Liu, Z. Zhang, Z. Jia, L. Zhao, T. Zhang, W. Xing, S. Komarneni, F. Subhan and Z. Yan, Chem. Eng. J., 2018, 337, 290–299 CrossRef CAS.
- T.-Y. Ma, L. Liu and Z.-Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- Z. Xing, N. Gao, Y. Qi, X. Ji and H. Liu, Carbon, 2017, 115, 271–278 CrossRef CAS.
- T. Kim, J. Lee and K.-H. Lee, RSC Adv., 2016, 6, 24667–24674 RSC.
- Y. Zhao, H. Ding and Q. Zhong, Appl. Surf. Sci., 2012, 258, 4301–4307 CrossRef CAS.
- C. Lu, H. Bai, B. Wu, F. Su and J. F. Hwang, Energy Fuels, 2008, 22, 3050–3056 CrossRef CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- W. Xing, C. Liu, Z. Zhou, L. Zhang, J. Zhou, S. Zhuo, Z. Yan, H. Gao, G. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323–7327 RSC.
- D. Qian, C. Lei, E.-M. Wang, W.-C. Li and A.-H. Lu, ChemSusChem, 2014, 7, 291–298 CrossRef CAS.
- J. V. Liebig, Ann. Pharm., 1834, 10, 10 Search PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53–55 CrossRef CAS.
- A. Thomas, A. Fisher, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and R. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- B. Jurgens, E. Irran, J. Senker, P. Kroll, H. Muller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef.
- F. R. Pomilla, M. A. L. R. M. Cortes, J. W. J. Hamilton, R. Molinari, G. Barbieri, G. Marcì, L. Palmisano, P. K. Sharma, A. Brown and J. A. Byrne, J. Phys. Chem. C, 2018, 122, 28727–28738 CrossRef CAS.
- C. Chen, J. Kim and W.-S. Ahn, Fuel, 2012, 95, 360–364 CrossRef CAS.
- B. Zhu, K. Li, J. Liu, H. Liu, C. Sun, C. E. Snape and Z. Guo, J. Mater. Chem. A, 2014, 2, 5481–5489 RSC.
- P. P. Sharma, J. J. Wu, R. M. Yadav, M. J. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS.
- S. Ni, Z. Y. Li and J. L. Yang, Nanoscale, 2012, 4, 1184–1189 RSC.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- J. Zhou, W. Li, Z. Zhang, W. Xing and S. Zhuo, RSC Adv., 2012, 2, 161–167 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- Z. Mo, X. She, Y. Li, L. Liu, L. Huang, Z. Chen, Q. Zhang, H. Xu and H. Li, RSC Adv., 2015, 5, 101552–101562 RSC.
- D. Hollmann, M. Karnahl, S. Tschierlei, K. Kailasam, M. Schneider, J. Radnik, K. Grabow, U. Bentrup, H. Junge and M. Beller, Chem. Mater., 2014, 26, 1727–1733 CrossRef CAS.
- F. Dong, Z. Wang, Y. Sun, W.-K. Ho and H. Zhang, J. Colloid Interface Sci., 2013, 401, 70–79 CrossRef CAS PubMed.
- P. Niu, L. Zhang and G. Liu, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- F. He, G. Chen, Y. Zhou, Y. Yu, Y. Zheng and S. Hao, Chem. Commun., 2015, 51, 16244–16246 RSC.
- B. Long, J. Lin and X. Wang, J. Mater. Chem. A, 2014, 2, 2942–2951 RSC.
- P. Niu, L.-C. Yin, Y.-Q. Yang, G. Liu and H.-M. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489–6496 RSC.
- J. Tong, L. Zhang, F. Li, M. Li and S. Cao, Phys. Chem. Chem. Phys., 2015, 17, 23532–23537 RSC.
- K. S. Lakhi, D.-W. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J.-H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- Y.-S. Jun, W. H. Hong, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 4270–4274 CrossRef CAS.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- J. Wang, C. Zhang, Y. Shen, Z. Zhou, J. Yu, Y. Li, W. Wei, S. Liu and Y. Zhang, J. Mater. Chem. A, 2015, 3, 5126–5131 RSC.
- W. Shen, L. Ren, H. Zhou, S. Zhang and W. Fan, J. Mater. Chem., 2011, 21, 3890–3894 RSC.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- Y.-S. Jun, E. Z. Lee, X. Wang, E. W. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- M. Shalom, M. Guttentag, C. Fettkenhauer, S. Inal, D. Neher, A. Llobet and M. Antonietti, Chem. Mater., 2014, 26, 5812–5818 CrossRef CAS.
- Y. Liao, S. Zhu, J. Ma, Z. Sun, C. Yin, C. Zhu, X. Lou and D. Zhang, ChemCatChem, 2014, 6, 3419–3425 CrossRef CAS.
- T. Jordan, N. Fechler, J. Xu, T. J. K. Brenner, M. Antonietti and M. Shalom, ChemCatChem, 2015, 7, 2826–2830 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- Z.-F. Huang, J. Song, L. Pan, Z. Wang, X. Zhang, J.-J. Zou, W. Mi, X. Zhang and L. Wang, Nano Energy, 2015, 12, 646–656 CrossRef CAS.
- Z. Zhao, Y. Sun, F. Dong, Y. Zhang and H. Zhao, RSC Adv., 2015, 5, 39549–39556 RSC.
- S. Hu, L. Ma, J. You, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, RSC Adv., 2014, 4, 21657–21663 RSC.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, J. Mater. Chem. A, 2015, 3, 3862–3867 RSC.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 RSC.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS.
- Y. Wang, H. Li, J. Yao, X. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- H. Li, Y. Liu, X. Gao, C. Fu and X. Wang, ChemSusChem, 2015, 8, 1189–1196 CrossRef CAS.
- S. Samanta and R. Srivastava, Sustainable Energy Fuels, 2017, 1, 1390–1404 RSC.
- S. Samanta, R. Yadav, A. Kumar, A. K. Sinha and R. Srivastava, Appl. Catal., B, 2019, 259, 118054 CrossRef CAS.
- Y. Gao, K. Qian, B. Xu, Z. Li, J. Zheng, S. Zhao, F. Ding, Y. Sun and Z. Xu, Carbon Resour. Convers., 2020, 3, 46–59 CrossRef CAS.
- A. Akhundi, A. Habibi-Yangjeh, M. Abitorabi and S. R. Pouran, Catal. Rev., 2019, 61, 595–628 CrossRef CAS.
- M. Shen, L. Zhang and J. Shi, Nanotechnology, 2018, 29, 412001 CrossRef PubMed.
- C. H. Bartholomew, Chem. Eng., 1984, 91, 96–112 CAS.
- J.-S. Chang, V. P. Vislovskiy, M.-S. Park, D. Y. Hong, J. S. Yoo and S.-E. Park, Green Chem., 2003, 5, 587–590 RSC.
- M.-O. Sugino, H. Shimada, T. Turuda, H. Miura, N. Ikenaga and T. Suzuki, Appl. Catal., A, 1995, 121, 125–137 CrossRef CAS.
- V. Ansari and S.-E. Park, Energy Environ. Sci., 2012, 5, 9419–9437 RSC.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717–2720 CrossRef CAS PubMed.
- M. B. Ansari, B.-H. Min, Y.-H. Mo and S.-E. Park, Green Chem., 2011, 13, 1416–1421 RSC.
- X. Lan, Y. Li, C. Du, T. She, Q. Li and G. Bai, Chem. – Eur. J., 2019, 25, 8560–8569 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- Z. Huang, F. Li, B. Chen and G. Yuan, Catal. Sci. Technol., 2016, 6, 2942–2948 RSC.
- M. B. Ansari, B.-H. Min, Y.-H. Mo and S.-E. Park, Green Chem., 2011, 13, 1416–1421 RSC.
- Z. Huang, F. Li, B. Chen, T. Lu, Y. Yuan and G. Yuan, Appl. Catal., B, 2013, 136–137, 269–277 CrossRef CAS.
- J. Xu, F. Wu, Q. Jiang, J.-K. Shang and Y.-X. Li, J. Mol. Catal. A: Chem., 2015, 403, 77–83 CrossRef CAS.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 RSC.
- J. Xu, F. Wu, Q. Jiang and Y.-X. Li, Catal. Sci. Technol., 2015, 5, 447–454 RSC.
- D.-H. Lan, H.-T. Wang, L. Chen, C.-T. Au and S.-F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- Q. Su, X. Yao, W. Cheng and S. Zhang, Green Chem., 2017, 19, 2957–2965 RSC.
- X. Li, B. Lin, H. Li, Q. Yu, Y. Ge, X. Jin, X. Liu, Y. Zhou and J. Xiao, Appl. Catal., B, 2018, 239, 254–259 CrossRef CAS.
- J. Zhu, T. Diao, W. Wang, X. Xu, X. Sun, S. A. C. Carabineiroc and Z. Zhao, Appl. Catal., B, 2017, 219, 92–100 CrossRef CAS.
- T. Biswas and V. Mahalingam, New J. Chem., 2017, 41, 14839–14842 RSC.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS.
- A. A. Tountas, X. Peng, A. V. Tavasoli, P. N. Duchesne, T. L. Dingle, Y. Dong, L. Hurtado, A. Mohan, W. Sun, U. Ulmer, L. Wang, T. E. Wood, C. T. Maravelias, M. M. Sain and G. A. Ozin, Adv. Sci., 2019, 6, 1801903 CrossRef PubMed.
- M. Khalil, J. Gunlazuardi, T. A. Ivandini and A. Umar, Renewable Sustainable Energy Rev., 2019, 113, 109246 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- M.-H. Vu, M. Sakar, S. A. Hassanzadeh-Tabrizi and T.-O. Do, Adv. Mater. Interfaces, 2019, 6, 1900091 CrossRef.
- J. Kou, C. Lu, J. Wang, Y. Chen, Z. Xu and R. S. Varma, Chem. Rev., 2017, 117, 1445–1514 CrossRef CAS PubMed.
- R. N. Compton, P. W. Reinhardt and C. D. Cooper, J. Chem. Phys., 1975, 63, 3821–3827 CrossRef CAS.
- N.-N. Vu, S. Kaliaguine and T.-O. Do, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2019, 32, 222–243 CrossRef.
- N. Sasirekha, S. J. S. Basha and K. Shanthi, Appl. Catal., B, 2006, 62, 169–180 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172, 10.1039/C2RA01332K.
- I. A. Shkrob, T. W. Marin, H. Y. He and P. Zapol, J. Phys. Chem. C, 2012, 116, 9450–9460 CrossRef CAS.
- L. B. Hoch, P. Szymanski, K. K. Ghuman, L. He, K. Liao, Q. Qia, L. M. Reyes, Y. Zhu, M. A. El-Sayed, C. V. Singh and G. A. Ozin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8011–E8020 CrossRef CAS PubMed.
- W. Tu, Y. Xu, J. Wang, B. Zhang, T. Zhou, S. Yin, S. Wu, C. Li, Y. Huang, Y. Zhou, Z. Zou, J. Robertson, M. Kraft and R. Xu, ACS Sustainable Chem. Eng., 2017, 5, 7260–7268 CrossRef CAS.
- S. C. Bhatia, Advanced Renewable Energy Systems, 2014, pp. 32–67 Search PubMed.
- G. M. Hasselman, D. F. Watson, J. R. Stromberg, D. F. Bocian, D. Holten, J. S. Lindsey and G. J. Meyer, J. Phys. Chem. B, 2006, 110, 25430–25440 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- H. Lang, S. Zhang and Z. Liu, Phys. Rev. B, 2016, 94, 235306 CrossRef.
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Z. Sun, H. Wang, Z. Wu and L. Wang, Catal. Today, 2018, 300, 160–172 CrossRef CAS.
- J. Chen, Y.-F. Li, P. Sit and A. Selloni, J. Am. Chem. Soc., 2013, 135, 18774–18777 CrossRef CAS PubMed.
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- N. Syed, J. Huang, Y. Feng, X. Wang and L. Cao, Front. Chem., 2019, 7(713), 1–7 Search PubMed.
- J. Z. Y. Tan and M. M. Maroto-Valer, J. Mater. Chem. A, 2019, 7, 9368–9385 RSC.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- M. M. Kandy, Sustainable Energy Fuels, 2020, 4, 469–484 RSC.
- S. Ye, R. Wang, M.-Z. Wu and Y.-P. Yuan, Appl. Surf. Sci., 2015, 358, 15–27 CrossRef CAS.
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160–1166 RSC.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC.
- P. Niu, Y. Yang, J. C. Yu, G. Liu and H.-M. Cheng, Chem. Commun., 2014, 50, 10837–10840 RSC.
- Y.-P. Yuan, S.-W. Cao, Y.-S. Liao, L.-S. Yin and C. Xue, Appl. Catal., B, 2013, 140–141, 164–168 CrossRef CAS.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 RSC.
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef PubMed.
- B. Liu, L. Ye, R. Wang, J. Yang, Y. Zhang, R. Guan, L. Tian and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4001–4009 CrossRef CAS PubMed.
- D. Vidyasagar, N. Manwar, A. Gupta, S. G. Ghugal, S. S. Umare and R. Boukherroub, Catal. Sci. Technol., 2019, 9, 822–832 RSC.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS PubMed.
- H. Shi, S. Long, J. Hou, L. Ye, Y. Sun, W. Ni, C. Song, K. Li, G. G. Gurzadyan and X. Guo, Chem. – Eur. J., 2019, 25, 5028–5035 CrossRef CAS PubMed.
- M. Li, S. Zhang, X. Liu, J. Han, X. Zhu, Q. Ge and H. Wang, Eur. J. Inorg. Chem., 2019, 2058–2064 CrossRef CAS.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- S. Wan, M. Ou, X. Wang, Y. Wang, Y. Zeng, J. Ding, S. Zhang and Q. Zhong, Dalton Trans., 2019, 48, 12070–12079 RSC.
- J.-Y. Tang, X. Y. Kong, B.-J. Ng, Y.-H. Chew, A. R. Mohamed and S.-P. Chai, Catal. Sci. Technol., 2019, 9, 2335–2343 RSC.
- J. Fu, K. Liu, K. Jiang, H. Li, P. An, W. Li, N. Zhang, H. Li, X. Xu, H. Zhou, D. Tang, X. Wang, X. Qiu and M. Li, Adv. Sci., 2019, 6, 1900796 CrossRef CAS PubMed.
- A. Hayat, J. Khan, M. U. Rahman, S. B. Mane, W. U. Khan, M. Sohail, N. U. Rahman, N. Shaishta, Z. Chi and M. Wu, J. Colloid Interface Sci., 2019, 548, 197–205 CrossRef CAS PubMed.
- K. Wang, J. Fu and Y. Zheng, Appl. Catal., B, 2019, 254, 270–282 CrossRef CAS.
- N. Tian, K. Xiao, Y. Zhang, X. Lu, L. Ye, P. Gao, T. Ma and H. Huang, Appl. Catal., B, 2019, 253, 196–205 CrossRef CAS.
- X. Liu, P. Wang, H. Zhai, Q. Zhang, B. Huang, Z. Wang, Y. Liu, Y. Dai, X. Qin and X. Zhang, Appl. Catal., B, 2018, 232, 521–530 CrossRef CAS.
- S. Wan, M. Ou, Q. Zhong and W. Cai, Carbon, 2018, 138, 465–474 CrossRef CAS.
- X. Gong, S. Yu, M. Guan, X. Zhu and C. Xue, J. Mater. Chem. A, 2019, 7, 7373–7379 RSC.
- H. Ou, C. Tang, Y. Zhang, A. M. Asiri, M.-M. Titirici and X. Wang, J. Catal., 2019, 375, 104–112 CrossRef CAS.
- J.-Y. Tang, W.-G. Zhou, R.-T. Guo, C.-Y. Huang and W.-G. Pan, Catal. Commun., 2018, 107, 92–95 CrossRef CAS.
- S. Cao, Y. Li, B. Zhu, M. Jaroniec and J. Yu, J. Catal., 2017, 349, 208–217 CrossRef CAS.
- M. Humayun, Q. Fu, Z. Zheng, H. Li and W. Luo, Appl. Catal., A, 2018, 568, 139–147 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC.
- K. Takanabe, J. Catal., 2019, 370, 480–484 CrossRef CAS.
- P. V. Kamat and S. Jin, ACS Energy Lett., 2018, 3, 622–623 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- P. V. Kamat and J. M. Buriak, ACS Energy Lett., 2019, 4, 2473–2474 CrossRef CAS.
- J. M. Buriak, P. V. Kamat and K. S. Schanze, ACS Appl. Mater. Interfaces, 2014, 6, 11815–11816 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- G. Peng, J. Albero, H. Garcia and M. Shalom, Angew. Chem., Int. Ed., 2018, 57, 15807–15811 CrossRef CAS PubMed.
- G. Peng, J. Qin, M. Volokh and M. Shalom, ACS Appl. Mater. Interfaces, 2019, 11, 29139–29146 CrossRef CAS PubMed.
- G. Peng, J. Qin, M. Volokh, C. Liu and M. Shalom, J. Mater. Chem. A, 2019, 7, 11718–11723 RSC.
- Y. Fang, X. Li and X. Wang, ACS Catal., 2018, 8, 8774–8780 CrossRef CAS.
- W. Xiong, F. Huang and R.-Q. Zhang, Sustainable Energy Fuels, 2020, 4, 485–503 RSC.
- J. Safaei, N. A. Mohamed, M. F. M. Noh, M. F. Soh, N. A. Ludin, M. A. Ibrahim, W. N. R. W. Isahak and M. A. M. Teridi, J. Mater. Chem. A, 2018, 6, 22346–22380 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- R. J. Lim, M. Xie, M. A. Sk, J. M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- J. E. Pander, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2017, 7, 5820–5832 RSC.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and applications, John Wiley & Sons, Inc., 2nd edn, 2001 Search PubMed.
- N. Sagara, S. Kamimura, T. Tsubota and T. Ohno, Appl. Catal., B, 2016, 192, 193–198 CrossRef CAS.
- C. M. S-Sánchez, V. Montiel, D. A. Tryk, A. Aldaz and A. Fujishima, Pure Appl. Chem., 2001, 73, 1917–1927 Search PubMed.
- B. Zhang, T.-J. Zhao, W.-J. Feng, Y.-X. Liu, H.-H. Wang, H. Su, L.-B. Lv, X.-H. Li and J.-S. Chen, Nano Res., 2018, 11, 2450–2459 CrossRef CAS.
- X. Lu, T. H. Tan, Y. H. Ng and R. Amal, Chem. – Eur. J., 2016, 22, 11991–11996 CrossRef CAS PubMed.
- S. Zhao, Z. Tang, S. Guo, M. Han, C. Zhu, Y. Zhou, L. Bai, J. Gao, H. Huang, Y. Li, Y. Liu and Z. Kang, ACS Catal., 2017, 8, 188–197 CrossRef.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K. H. Wu, Y. Lin, X. Sun, Q. Li, G. Centi and D. Su, ChemSusChem, 2016, 9, 1085–1089 CrossRef CAS PubMed.
- L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef CAS.
- Z. Ma, C. Lian, D. Niu, L. Shi, S. Hu, X. Zhang and H. Liu, ChemSusChem, 2019, 12, 1724–1731 CrossRef CAS PubMed.
- N. Meng, W. Zhou, Y. Yu, Y. Liu and B. Zhang, ACS Catal., 2019, 9, 10983–10989 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |