
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high strain, adhesive, self-healable poly(acrylic acid) hydrogel with temperature sensitivity as an epidermal sensor†
Rui
Zhang
 *a,
Hengzhi
Ruan
a,
Qionglong
Fu
a,
Xuedong
Zhu
*a and
Yuan
Yao
*b
*a,
Hengzhi
Ruan
a,
Qionglong
Fu
a,
Xuedong
Zhu
*a and
Yuan
Yao
*b
aEngineering Research Center of Large Scale Reactor Engineering and Technology, Ministry of Education, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: r.zhang@ecust.edu.cn; xdzhu@ecust.edu.cn
bSchool of Materials Science and Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: yaoyuan@ecust.edu.cn
First published on 18th May 2020
Abstract
A fully physically linked poly(acrylic acid) (PAA) hydrogel with temperature sensitivity was fabricated. The temperature-sensitive point of the hydrogel was near 25 °C when introducing the copolymer of N-isopropylacrylamide (NIPAAM) and dopamine methacrylamide (DMA). More importantly, the hydrogels exhibited excellent self-healing properties, mechanical properties and tunable adhesive strength and could be used as epidermal sensors.
Stimuli responsive hydrogels have been widely reported due to their characteristic response to environmental changes, which guarantees their stable use in different fields, such as tissue engineering,1 drug delivery,2 and sensing.3 According to the types of stimuli, stimuli responsive hydrogels can be classified as pH,4 ultraviolet light,5 reduction,6 and ionic strength responsive hydrogels7 and so on. Among the various stimuli responsive hydrogels that have been investigated, thermal responsive hydrogels have received much attention.8,9 The thermal responsive hydrogels are water swollen polymer networks that can experience a thermally triggered, reversible change.10N-Isopropylacrylamide (NIPAAM), as a typical thermal responsive monomer, is extensively used to produce several functional stimuli responsive materials because of its rapid phase transition,11 biocompatibility12 and its lower critical dissolution temperature (LCST).13 With the LCST close to 31 °C, NIPAAM can undergo a phase transition around the physiological temperature.14 Obviously, an invariable LCST is not suitable for different situations, so it is important to adjust the LCST of NIPAAM in order to realize highly efficient use. One principle to address this problem is copolymerization between NIPAAM and other monomers. When NIPAAM is incorporated into hydrophobic monomers, its LCST can be decreased to varying degrees whereas conjugation with hydrophilic monomers leads to an increase in LCST.15–18 For example, Zhang et al. prepared a functional ionic hydrogel-based flexible sensor by integrating NIPAAM into another conductive double-network hydrogel based on polyvinyl alcohol–graphene oxide and polyacrylic acid–Fe3+ and the temperature-sensitive point was adjusted close to 37 °C successfully.19 Recently, Zou et al. designed an injectable and in situ hydrogel that exhibited biocompatibility with mouse fibroblasts, reduced cytotoxicity for indomethacin and a significant anti-inflammatory effect against macrophages.20 The rheology investigation suggested that the temperature sensitive point of the hydrogel was 33 °C upon radical polymerization of NIPAAM and other monomers. However, in the process of the experiment, these thermo-responsive hydrogels could not support huge mechanical loads or accommodate significant deformation. A major reason for such a phenomenon is that there are insufficient dynamic bonds in the network such as reversible covalent bonds,21–23 hydrogen bonds,24,25 metal–ligand coordination,26,27 ionic bonds28,29 and π–π stacking,30,31 which leads to an inefficient energy dissipative mechanism. On the other hand, many polymer chains are swollen in some situations; consequently, when hydrogels are under an extraneous force, they usually exhibit poor mechanical properties.32,33 In addition, few works have demonstrated thermal responsive hydrogels with high stretchability and self-healing ability. Therefore, it is necessary to fabricate novel hydrogels with temperature-sensitive and excellent mechanical properties simultaneously. In this work, we report the design and preparation of a temperature-sensitive and tough hydrogel used as an epidermal sensor with adhesion and self-healing properties constructed based on the network of poly(acrylic acid) (PAA), dopamine methacrylamide (DMA) and NIPAAM. The copolymerization of NIPAAM and DMA could introduce a synergistic effect of hydrogen bonding and hydrophobicity, which endowed the hydrogel with remarkable mechanical properties at different temperatures. Through differential scanning calorimetry (DSC) measurements, it was found that the temperature-sensitive point of the obtained PAA/NIPAAM-co-DMA hydrogel was near 25 °C due to the incorporation of DMA. The self-healing efficiency of the hydrogel could reach 92.31% after healing for 24 hours without any external stimulus. Besides, we also found that the obtained hydrogel can adhere to various types of material and the adhesive strength could be tuned by temperature and the concentration of NIPAAM-co-DMA.
Fig. 1 shows a general procedure to prepare PAA/NIPAAM-co-DMA hydrogels. Briefly, the hydrogel was prepared by free-radical polymerization of AA in H2O in the presence of copolymer NIPAAM-co-DMA, which was synthesized as following (Scheme S1, ESI†). When the temperature was low, the hydrogen bonding in the network was enhanced due to the carboxyl group of PAA, phenolic hydroxyl group of DMA and acylamino group of PNIPAAM. However, when the temperature was increased, the copolymer NIPAAM-co-DMA was transformed due to the hydrophobic effect of isopropyl of NIPAAM and side chain groups of DMA, so the copolymer NIPAAM-co-DMA formed many hydrophobic regions that helped to dissipate excess energy. And this phenomenon could also be observed in the pre-gel solution; the solution changed from transparent to opaque white when the temperature increased.
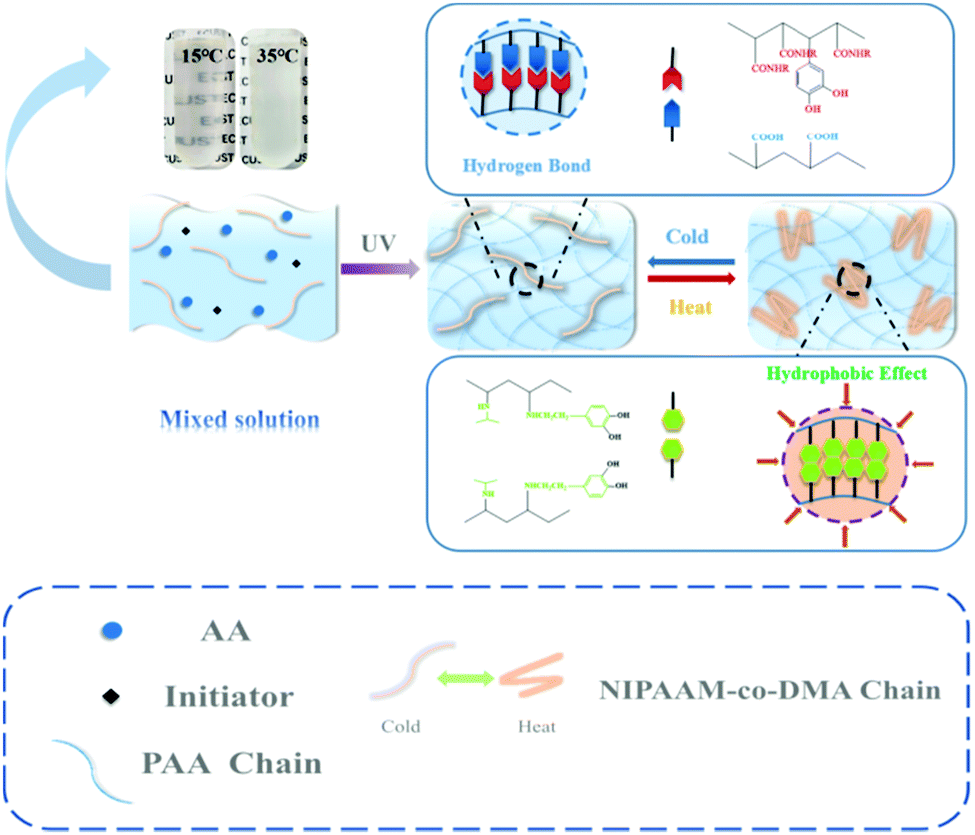 | ||
| Fig. 1 Preparation of PAA/NIPAAM-co-DMA hydrogels. | ||
Fig. 2a and b show the typical tensile stress–strain curves and toughness of hydrogels at different temperatures. For PAA hydrogels, no obvious thermal response was observed at different temperatures. In contrast, the fracture stress and toughness of PAA/NIPAAM-co-DMA gels were influenced by the increase of temperature dramatically. DSC thermograms of PAA/NIPAAM-co-DMA hydrogels during heating was provided in Fig. S3 (ESI†). In the previous research studies, cyclic tensile loading–unloading tests were performed to study the toughening mechanism of sacrificial bonds, and the hysteresis indicates the amount of energy dissipated.34Fig. 2c shows the stress–strain curves after consecutive loading–unloading cycles and illustrates the energy dissipation capacity of the PAA/NIPAAM-co-DMA gels, which was another indicator to assess the mechanical properties of the obtained hydrogels. It could be seen that the hysteresis did not become much smaller when the subsequent cycle was conducted immediately after the previous cycle. In addition, no energy hysteresis phenomenon was observed during the cyclic tensile loading–unloading tests. Simultaneously, the dissipated energy of loading–unloading cycles was calculated, as shown in Fig. 2d. It was clear that no obvious decrease of dissipated energy appeared as the cycle number increased and the average dissipated energy was about 650 kJ m−3, which was larger than that of other hydrogels based on hydrophobic interactions35 and this indicated the excellent fatigue resistant properties of PAA/NIPAAM-co-DMA gels. Interestingly, we found that the obtained hydrogels could be stretched to 15 times with a cut notch (radius ≈ 2 mm) (Fig. 2e). As shown in Fig. 2f, the notch was blunted and remained stable during the whole testing process, suggesting that no stress concentrates in the front of the notch tip.
 | ||
| Fig. 2 (a) Typical tensile stress–strain curves and (b) toughness of hydrogels at different temperatures. (c) Eight successive loading–unloading tensile cycles of the hydrogel at a strain of 600% without resting time. (d) The dissipated energy of the hydrogels calculated from the area of their hysteresis loops. (e) Tensile stress–strain curves of the notched hydrogel. (f) The pictures of the hydrogel sample with crack resistant properties before and during testing. | ||
Because of the supramolecular interactions in the network, physically linked hydrogels usually show excellent self-healing ability.36 As illustrated in Fig. 3a, the hydrogel was cut into two pieces that were dyed red and blue. Then, they were brought together and left to heal at room temperature without any stimulus. After 16 h, the line at their interface was blurred, which indicated that the interactions in the hydrogel system were dynamic. To investigate whether the intermolecular noncovalent bonds in the PAA/NIPAAM-co-DMA gels dissociate and reassociate efficiently under stress, mechanical tests were performed on the obtained hydrogels. Fig. 3b shows the stress–strain curves of healed hydrogels with different NIPAAM-co-DMA concentrations. The corresponding healing efficiency of the healed hydrogels is demonstrated in Fig. 3c; it indicates that the healing efficiency decreased from 69.76% to 58.14% when the NIPAAM-co-DMA concentration was improved from 1.2% to 2.2%. Another phenomenon shown in Fig. 3d and e was that the efficiencies of the hydrogels were enhanced with the increase of the healing time. Due to the inadequate recovery in the network, the hydrogel was broken at λ = 7 after healing for 8 h. Meanwhile, after healing for 24 h, the healing efficiency and ultimate strain of the hydrogel were about 92.31% and 4880%, which was very close to the strain of the origin hydrogel (λ = 51) (Fig. S4, ESI†). The self-healing property of PAA/NIPAAM-co-DMA gels was related to the dynamic hydrogen bonds that were formed between PAA and NIPAAM-co-DMA. When the hydrogel was broken under stress, the hydrogen bonding could be reformed at room temperature between the fresh cross-section. Simultaneously, the mobility of the polymer chains was also responsible for the process of self-healing. The reason was that the higher the density of crosslinking, the more difficult it was for PAA and NIPAAM-co-DMA chains to move.37 When the NIPAAM-co-DMA concentration was too high, very few chains could move from one surface to another surface leading to a poor healing performance.
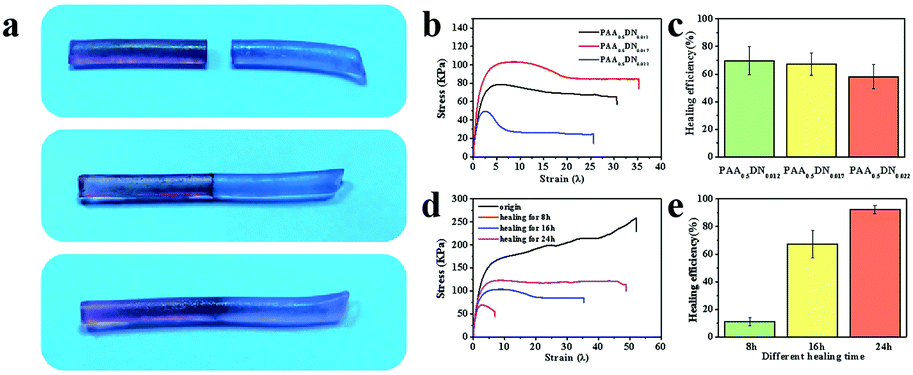 | ||
| Fig. 3 Photographs of cut sample (a) before and after healing for 16 h. (b) Tensile stress–strain curves and (c) healing efficiency of a healed hydrogel with different NIPAAM-co-DMA concentrations for 16 h. (d) Tensile stress–strain curves of original and healed hydrogel with different healing times. (e) Healing efficiency with different healing times. | ||
In all, four main noncovalent interactions, including hydrophobic, ionic electrostatic, van der Waals interactions, and hydrogen bonding have been reported in building high strength hydrogels.38 Due to the reversible nature of these interactions, physical gelation commonly tends to be brittle and weak when a force is applied.39 To design an adhesive hydrogel with outstanding mechanical properties, DMA was introduced into the system to improve the balance between cohesion and adhesion. In order to display the adhesive performance of PAA/NIPAAM-co-DMA gels intuitively, pictures of hydrogels adhering to various materials are shown in Fig. 4a. It was worth noting that the obtained sample exhibited excellent adhesive behavior when it was adhered to paper, copper, steel, skin, glass and plastic and it could even withstand a 100 g weight, as shown in Fig. 4a. In addition to revealing the adhesion strength of the gels, a combinatorial study of the effect of NIPAAM-co-DMA concentration and temperature is shown in Fig. 4b. For all the samples, the adhesion strength was dramatically influenced by the NIPAAM-co-DMA content. For example, the adhesion strength of the hydrogels at 15 °C improved from 340 KPa to 543 KPa when the concentration of NIPAAM-co-DMA was increased from 1.2 wt% to 2.2 wt%. It could be concluded that addition of catechol group contents led to more interactions at the adhesion interface. Meanwhile, adhesion experiments of PAA/NIPAAM-co-DMA gels at different temperatures were also conducted. We found that the samples at 25 °C, which was close to the temperature-sensitive point of the hydrogels (Fig. S3, ESI†), showed the lowest adhesion strength compared to other temperatures. Furthermore, when the temperature was increased to 35 °C, for instance, the adhesion strength of PAA0.5DN0.017 gels increased from 315 KPa to 480 KPa. A similar phenomenon was also observed in other samples that could be attributed to two reasons. First, as the temperature was increased above the temperature-sensitive point, DMA chains became hydrophobic, allowing the expulsion of water molecules and acting like other poly(NIPAAM)s.40 As a result, hydrogen bonding at the adhesion interface was impaired, thus adhesion performance became hampered. Second, when the temperature was higher than room temperature, water content in the samples decreased quickly leading to stronger interactions in the system and hydrogels exhibited higher adhesion strength.
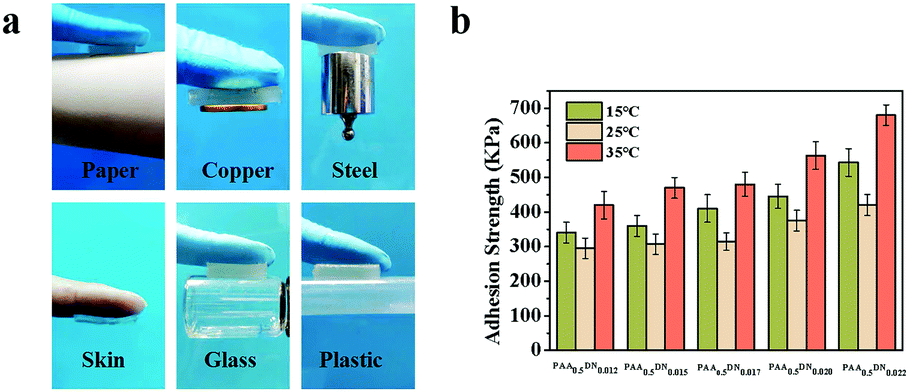 | ||
| Fig. 4 (a) Adhesive exhibition of PAA/NIPAAM-co-DMA gels adhering to diverse materials, including paper, copper, steel, skin, glass and plastic. (b) The combinatorial study of the effect of NIPAAM-co-DMA concentration and temperature on the adhesive strength of the PAA/NIPAAM-co-DMA hydrogels. | ||
Due to the excellent mechanical properties and adhesive ability, the PAA/NIPAAM-co-DMA gel could be used as an epidermal sensor in the presence of lithium chloride. As shown in Fig. 5a–c, the monitored resistance of the PAA/NIPAAM-co-DMA gel-based sensor could accurately reflect the finger motion at different angles, indicating the sensing reliability of the PAA/NIPAAM-co-DMA gel-based sensor. Moreover, the relationship between the relative resistance of the PAA/NIPAAM-co-DMA gel-based sensor and strain is consistent with the result in Fig. 5d. As the sensor was stretched, the relative resistance increased and this demonstrated the stability of the sensor under an external force. We ascribed the sensitivity of the PAA/NIPAAM-co-DMA gel-based sensor to the free ions such as H ion and residual initiator in the hydrogel network and therefore the gel was conductive.
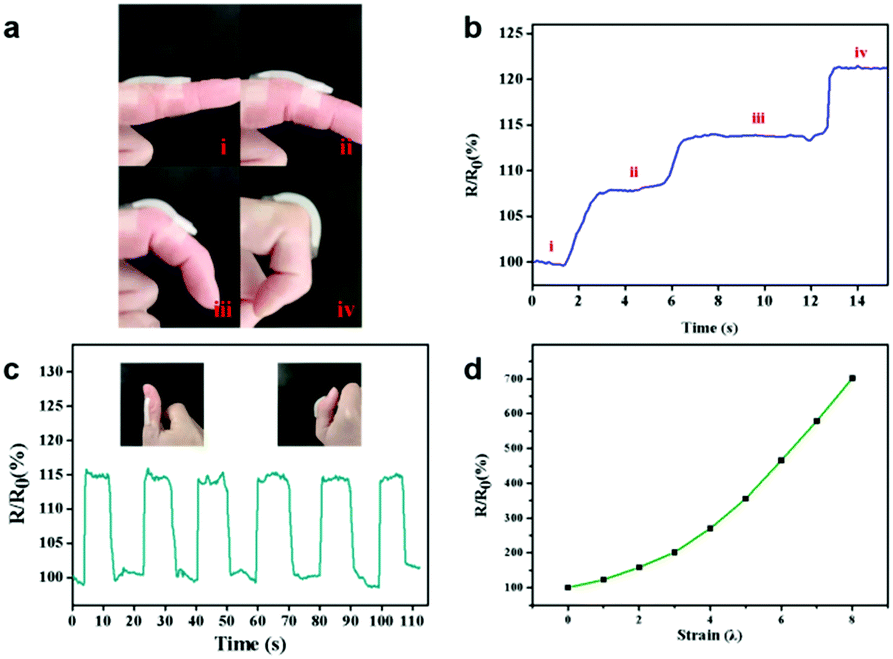 | ||
| Fig. 5 (a) The photographs of the sensor adhered to a finger bending at different angles and (b) recorded sensing performance of a PAA/NIPAAM-co-DMA gel-based sensor in response to finger bending at different angles. (c) Recorded sensing performances of the PAA/NIPAAM-co-DMA gel-based sensor in response to the bending of a thumb. The insets show the photograph of the sensor adhered to the thumb. (d) Resistance ratio-strain curve of the PAA/NIPAAM-co-DMA gel-based sensor. | ||
In conclusion, we developed a temperature-sensitive and tough hydrogel of a fully physically linked PAA/NIPAAM-co-DMA gel, with adhesive strength and self-healing ability. The hydrogel exhibited excellent fatigue resistance and crack resistance properties and could be used as an epidermal sensor. According to DSC and rheological investigation, the temperature-sensitive point of the hydrogels was near 25 °C. Our design strategy will open up a new platform to overcome the limitation of conventional hydrogels that are brittle and weak and makes progress in the preparation of functional materials.
This work is supported by the National Natural Science Foundation of China (21374029, 51573088 and 21776076).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Bai, X. G. Zuo, X. Feng, Y. F. Sun, Q. Z. Ge, X. M. Wang and C. Y. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 36939–36948 CrossRef CAS PubMed.
- X. Fu, L. Hosta-Rigau, R. Chandrawati and J. W. Cui, Chem, 2018, 4, 2084–2107 CAS.
- Z. X. Deng, T. L. Hu, Q. Lei, J. K. He, P. X. Ma and B. L. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 6796–6808 CrossRef CAS.
- Y. P. Liang, X. Zhao, P. X. Ma, B. L. Guo, Y. P. Du and X. Z. Han, J. Colloid Interface Sci., 2019, 536, 224–234 CrossRef CAS PubMed.
- S. K. Rastogi, H. E. Anderson, J. Lamas, S. Barret, T. Cantu, S. Zauscher, W. J. Brittain and T. Betancourt, ACS Appl. Mater. Interfaces, 2018, 10, 30071–30080 CrossRef CAS PubMed.
- Y. X. Zhang, Y. F. Chen, X. Y. Shen, J. J. Hu and J. S. Jan, Polymer, 2016, 86, 32–41 CrossRef CAS.
- Y. N. Sun, S. Liu, G. L. Du, G. R. Gao and J. Fu, Chem. Commun., 2015, 51, 8512–8515 RSC.
- S. Belali, H. Savoie, J. M. O'Brien, A. A. Cafolla, B. O'Connell, A. R. Karimi, R. W. Boyle and M. O. Senge, Biomacromolecules, 2018, 19, 1592–1601 CrossRef CAS PubMed.
- Y. Y. Xiao, X. L. Gong, Y. Kang, Z. C. Jiang, S. Zhang and B. J. Li, Chem. Commun., 2016, 52, 10609–10612 RSC.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1992, 25, 5528–5530 CrossRef CAS.
- Y. Y. Li, X. Z. Zhang, H. Cheng, J. L. Zhu, S. X. Cheng and R. X. Zhuo, Macromol. Rapid Commun., 2006, 27, 1913–1919 CrossRef CAS.
- C. H. Ni and X. X. Zhu, Eur. Polym. J., 2004, 40, 1075–1080 CrossRef CAS.
- T. Hessberger, L. B. Braun and R. Zentel, Adv. Funct. Mater., 2018, 28, 21 CrossRef.
- X. F. Zhu, W. Zhou, H. X. Zhang and Y. J. Nie, Appl. Mech. Mater., 2011, 55–57, 1–6 CAS.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- R. A. Stile, W. R. Burghardt and K. E. Healy, Macromolecules, 1999, 32, 7370–7379 CrossRef CAS.
- H. Y. Liu and X. X. Zhu, Polymer, 1999, 40, 6985–6990 CrossRef CAS.
- R. M. K. Ramanan, P. Chellamuthu, L. P. Tang and K. T. Nguyen, Biotechnol. Prog., 2006, 22, 118–125 CrossRef CAS PubMed.
- S. Feng, Q. Li, S. Wang, B. Wang and T. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 21049–21057 CrossRef CAS PubMed.
- Y. X. Pan, B. R. Li, Z. Liu, Z. Y. Yang, X. Yang, K. Shi, W. Li, C. Peng, W. C. Wang and X. L. Ji, ACS Appl. Mater. Interfaces, 2018, 10, 32747–32759 CrossRef CAS PubMed.
- M. Deng, F. Guo, D. Liao, Z. Hou and Y. Li, Polym. Chem., 2018, 9, 98–107 RSC.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- H. Z. Ying, Y. F. Zhang and J. J. Cheng, Nat. Commun., 2014, 5, 3218, DOI:10.1038/ncomms4218.
- D. Montarnal, P. Cordier, C. Soulié-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925–7936 CrossRef CAS.
- Y. L. Chen, A. M. Kushner, G. A. Williams and Z. B. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- M. Burnworth, L. M. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. A. Bao, Nat. Chem., 2016, 8, 619–625 Search PubMed.
- A. Das, A. Sallat, F. Bohme, M. Suckow, D. Basu, S. Wiessner, K. W. Stockelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS.
- X. Y. Liu, H. Xu, L. Q. Zhang, M. Zhong and X. M. Xie, ACS Appl. Mater. Interfaces, 2019, 11, 42856–42864 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- S. J. Buwalda, K. W. M. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennink, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS PubMed.
- M. M. Song, Y. M. Wang, B. Wang, X. Y. Liang, Z. Y. Chang, B. J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 15021–15029 CrossRef CAS PubMed.
- Y. Deng, I. Hussain, M. M. Kang, K. W. Li, F. Yao, S. L. Liu and G. D. Fu, Chem. Eng. J., 2018, 353, 900–910 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- W. P. Chen, D. Z. Hao, W. J. Hao, X. L. Guo and L. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 1258–1265 CrossRef CAS PubMed.
- J. J. Xu, R. N. Jin, L. J. Duan, X. Y. Ren and G. H. Gao, Carbohydr. Polym., 2019, 211, 1–10 CrossRef CAS PubMed.
- S. Naficy, H. R. Brown, J. M. Razal, G. M. Spinks and P. G. Whitten, Aust. J. Chem., 2011, 64, 1007–1025 CrossRef CAS.
- H. H. Bearat, B. H. Lee and B. L. Vernon, Acta Biomater., 2012, 8, 3629–3642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental methods and other characterization of the hydrogels, see DOI: 10.1039/d0ma00282h |
| This journal is © The Royal Society of Chemistry 2020 |