
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of a tumor-seeking LyP-1 peptide integrated lipid–polymer composite nanoparticle†
Ramesh
Marasini
 ab,
Tuyen Duong Thanh
Nguyen
ab,
Sagar
Rayamajhi
ab and
Santosh
Aryal
*ab
ab,
Tuyen Duong Thanh
Nguyen
ab,
Sagar
Rayamajhi
ab and
Santosh
Aryal
*ab
aDepartment of Chemistry, College of Arts and Sciences, Kansas State University, Manhattan, KS 66506, USA. E-mail: saryal@ksu.edu
bNanotechnology Innovation Center of Kansas State (NICKS), College of Veterinary Medicine, Kansas State University, Manhattan, KS 66506, USA
First published on 11th May 2020
Abstract
Biocompatible polymeric and lipid nanoparticles are under extensive investigation as anticancer nanomedicines due to the ease of chemical modification in both polymer and lipid in order to target the respective disease environment. However, microenvironment and molecular heterogeneity in tumors pose a great challenge to delivering anticancer drugs or imaging agents precisely to the target, further limiting their applications. As a result, existing nanomedicine formulations rely on a passive-drug targeting mechanism taking advantage of leaky tumor vasculature. However, one strategy is not fit for all due to the molecular dissimilarities between cancers. Therefore, more research on tumor-specific receptors is needed to maximize drug delivery, while minimizing drug-related adverse effects. In addition, a high degree of the immunocompatibility and aqueous stability of the delivery device is essential to maximize delivery efficiency. Herein, we are addressing the aforementioned requirements in cancer management by engineering a receptor-specific anticancer nanomedicine as a composite of polymer and lipids. We are presenting a tumor seeking cyclic LyP-1 peptide integrated core–shell polymer–lipid composite nanoparticle (NP) that targets the overexpressed p32 receptor in cancer cells. The designed nanoconstruct is composed of poly(lactide-co-glycolic acid) as a skeleton and a cargo reservoir, a phospholipid with polyethylene glycol as a stabilizer, and LyP-1 as a targeting motif. We studied cellular interaction and targeting ability by accessing the full spectrum of biodistribution using NPs labeled with near-infrared dye as an imaging tracer in vivo. The NPs are spherical and monodispersed with an average size of 68 ± 6 nm and negative zeta potential. These particles are highly stable in physiological conditions over the period with a lower polydispersity index (PDI = 0.15). Furthermore, the nanoparticles showed excellent biocompatibility in vitro, with significantly higher uptake by mouse osteosarcoma compared to non-cancerous fibroblasts. Likewise, LyP-1 NP showed nearly three-fold enhancement in tumor accumulation in vivo compared to its non-targeted counterparts in the K7M2 tumor. Considering the overexpression of p32 in many cancers, the proposed nanoconstruct could hold promises in the therapeutic planning of a wide range of tumors.
Introduction
The lipid–polymer composite nanoparticle exhibits complementary characteristics of both lipids and polymers, which have shown remarkable clinical success in drug delivery.1–3 These composite nanoconstructs are distinctly organized creating different core–shell compartments to host the guest molecules. Herein, a skeleton of the polymeric hydrophobic core, which can encapsulate drugs whereas the lipid as the hydrophilic shell can be tuned for receptor targeting moieties and contrast agents.4–6 As such, the versatile chemistry of the hydrophilic head moiety of the phospholipids can be uniquely redesigned for precision drug delivery.7 Regardless of their complex structure, these lipid–polymer composite nanoparticles (NPs) are synthesized in a simple, single-step fashion, which allows future scale-up in production and cost-effective real-life applications.1,7–12 It has been established that the functionalization of polymer end groups and subsequent conjugation with targeting motifs such as peptides as a nanoparticle vehicle is a powerful strategy to permit precise drug delivery to the targeted site, while reducing systemic toxicities.13 The use of the Food and Drug Administration (FDA) approved polymeric biomaterial, polyethylene glycol (PEG), as a surface passivating layer on the nanoparticle prevents aggregation, opsonization, and phagocytosis, and prolongs the systemic circulation time to improve the nanoparticle-based drug delivery system.14–19 Also, it offers a facile PEG-end functional chemistry that provides further room to improve the delivery of NPs, for instant covalent attachment of small molecules, antibodies, and tumor targeting peptides.20,21Tumor targeting peptides are small peptides capable of targeting tumor tissue and internalizing in tumor cells. The tumor-specific receptor-mediated interaction associated with tumor-targeting peptides makes them outstanding candidates to dose a high concentration of any attached payload for tumor-targeted drug delivery. Recently, the LyP-1 peptide has been recognized for its unique specific localization into tumors.22 It is a cyclic peptide with the sequence CGNKRTRGC, which was discovered using the in vivo phage display method. A uniquely structured nine amino acid residue arranged in C-terminal motifs (KRTR) binds to the specific receptor p32 (globular head of complement component 1q receptor or gC1qR) and takes the peptide or peptide conjugated cargo to the vascular endothelium in the target tissue via the CendR pathway.22,23 Extensive accumulation of LyP-1 was reported in the areas with higher expression of p32, such as tumor-associated macrophages, the luminal lining of tumor lymphatics, and tumor cells.23–27 p32 has a functional role in maintaining oxidative phosphorylation in normal cells.30 Research has shown that its expression and subcellular location are changed in many cancers including breast, colon, melanoma, ovarian, bone, and prostate.24,25,28,29 This mitochondrial p32 is also responsible for regulating cancer cell metabolism and is critical for tumor advancement. Its genetic knockdown has significantly reduced tumor formation in vivo.30–32 Therefore, p32 is a potentially important therapeutic target in cancer.
Upon this background, we designed tumor homing LyP-1 peptide integrated lipid–polymer composite nanoparticles to deliver cargo precisely to the tumor. We hypothesized that due to the overexpression of p32 in cancer and its specificity with the LyP-1 peptide, the synthesized LyP-1 NP could preferentially deliver the cargo via a receptor-mediated interaction. This targeted composite NP consists of an outer polyethylene glycol corona as a stabilizer, LyP-1 as a tumor seeking peptide, a monolayer of phospholipid mixture containing 1,2-distearoylphosphatidylethanolamine (DSPE) and 1,2-distearoyl-sn-glycero-3-phospho-rac-glycerol (DSPG), and a poly(L-lactic-co-glycolic acid) (PLGA) polymer core for the encapsulation of the drug. Conjugation chemistry was utilized to label the peptide with carboxylic acid terminated DSPE–PEG in a single-step followed by nanoprecipitation resulting in the formation of LyP-1 NPs. By using in vitro and in vivo studies, we found that LyP-1 NP is highly stable in biological media, transverses the systemic circulation, and specifically accumulates at the tumor region compared to control NPs. Due to the tumor-seeking feature of the integrated peptide, the proposed nanosystem could drive maximum payload to the tumor site.
Experimental
Chemicals and reagents
Carboxylic end group terminated PLGA (lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide = 50:50 dL g−1) was purchased from DURECT Corporation (USA) and used after purification by repeated precipitation in diethyl ether. 1,2-Distearoylphosphatidylethanolamine polyethylene glycol succinyl (DSPE–PEG–COOH), 1,2-distearoyl-sn-glycero-3-phosphoglycerol, sodium salt (DSPG), and phospholipid conjugated L-α-phosphatidylethanolamine-N-(lissamine rhodamine-B sulfonyl) (ammonium salt) (RhB) was purchased from Avanti Polar Lipid Inc (Alabaster, AL, USA) and used as received. DAPI (4′,6-diamidino-2-phenylindole dihydrochloride), (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (MTT), doxorubicin hydrochloride (DOX) (European Union pharmacopeia standard), acetonitrile, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Rapid Gold BCA Assay kit (Cat No: A53225) was purchased from Thermo Fisher Scientific, USA. All other analytical grade reagents and solvents were used without further purification.
:glycolide = 50:50 dL g−1) was purchased from DURECT Corporation (USA) and used after purification by repeated precipitation in diethyl ether. 1,2-Distearoylphosphatidylethanolamine polyethylene glycol succinyl (DSPE–PEG–COOH), 1,2-distearoyl-sn-glycero-3-phosphoglycerol, sodium salt (DSPG), and phospholipid conjugated L-α-phosphatidylethanolamine-N-(lissamine rhodamine-B sulfonyl) (ammonium salt) (RhB) was purchased from Avanti Polar Lipid Inc (Alabaster, AL, USA) and used as received. DAPI (4′,6-diamidino-2-phenylindole dihydrochloride), (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (MTT), doxorubicin hydrochloride (DOX) (European Union pharmacopeia standard), acetonitrile, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Rapid Gold BCA Assay kit (Cat No: A53225) was purchased from Thermo Fisher Scientific, USA. All other analytical grade reagents and solvents were used without further purification.
Cell lines and animal tumor model
Mouse osteosarcoma (K7M2), mouse normal fibroblast (NIH/3T3), and Dulbecco's modified Eagle's medium (DMEM) were purchased from American Type Cell Culture (ATCC), Manassas, USA. Fetal bovine serum (FBS) USDA Premium Select, heat-inactivated, was purchased from MIDSCI, Saint Louis, USA. K7M2 and NIH/3T3 were maintained in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), and 1% streptomycin at 37 °C in a 5% CO2 environment. The cell lines were used from passage 10 for bionano interaction studies. Six-week-old female immunodeficient NU/NU nude mice were procured from Charles River Laboratories International, Inc. and used for the study after ten days of acclimatization. The Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee, Kansas State University, Manhattan approved all the animal experiments and protocols. The solid tumor model using osteosarcoma cancer cells was developed by subcutaneous injection of 1 × 106 K7M2 cells into the rear flank of mice. Tumor growth and animal health were regularly monitored by measuring tumor volume, body weight, and physical health following an approved IACUC protocol.Synthesis of LyP-1 conjugated lipids
As shown in Scheme 1A, DSPE–PEG–LyP-1 was synthesized in a straightforward manner using well-established EDC–NHS bioconjugation coupling chemistry following the published protocol.33 First, 0.08 mmol of EDC was dissolved in 500 μL of 1× phosphate buffer saline (PBS) then raised it to 3 mL, and then gradually added to the 0.008 mmol solution of DSPE–PEG–COOH dissolved in 5 mL of 1× PBS, under stirring for 1 h at room temperature for the activation of the carboxylic group. Then, 3 mL of 0.5 mmol of NHS in 1× PBS was added to the previously activated lipid mixture. The whole mixture was further stirred for 2 h at room temperature. Then, 0.002 mmol of LyP-1 peptide dissolved in 1× PBS was added and stirred overnight at room temperature. The product was purified three times by dialysis using a 3000 Da molecular weight cutoff and lyophilized to recover the product. The obtained final concentrated product was stored at −20 °C for further use.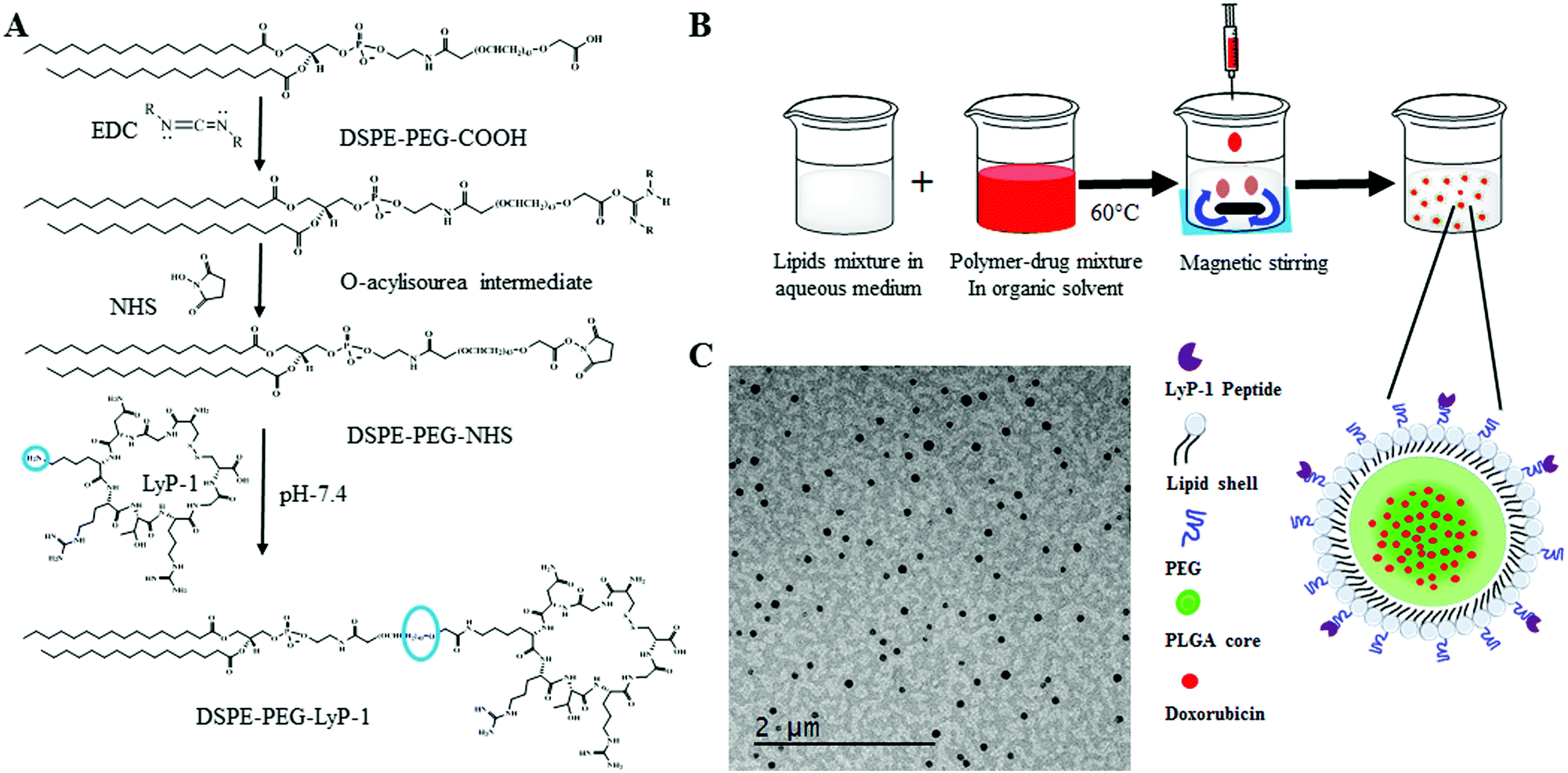 | ||
| Scheme 1 Graphical representation of DSPE–PEG and LyP-1 conjugate synthesis and nanoparticle fabrication process. (A) Synthetic scheme of LyP-1–PEG–DSPE using EDC/NHS bioconjugation, (B) cartoon representation of nanoprecipitation forming lipid–polymer composite nanoparticles, and (C) transmission electron micrograph of the synthesized composite nanoparticles. | ||
Preparation of LyP-1 conjugated multifunctional lipid–polymer composite nanoparticles
There are five components in LyP-1 NP viz., PLGA NPs, phospholipids, DOX, LyP-1, and imaging probe DiR (Scheme 1B). PLGA with the carboxylic acid terminated end group of molecular weight 50 kDa was used to synthesize LyP-1NP following the well-established nanoprecipitation protocol.34–38 In a typical experiment, 1 mg mL−1 PLGA in 400 μL acetonitrile was used to prepare the nanoparticles. The second component is phospholipids that consist of lipid mixture formulations having 250 μg of DSPE–PEG, 200 μg DSPG, and 10 μg LyP-1–DSPE–PEG, which were used in 4% ethanol, respectively. This lipid mixture was stirred at 60 °C for 30 min and 1 mg of PLGA in 400 μL acetonitrile solution was added dropwise while stirring, followed by the addition of 1 mL of deionized water (acetonitrile:water = 1:3). The content is further stirred at room temperature for 1 h to facilitate the formation of nanoparticles. NPs thus formed were purified using a 10 kDa Amicon® wash at 3500 rpm for 10 min. The washing was done three times in 1× PBS, and a final concentration of NPs was made up to 1 mg mL−1 in 1× PBS, and the NPs were stored at 4 °C until further use. Similarly, control nanoparticles were prepared in the same phospholipid ratio without any LyP-1-conjugated lipid. The third component was DOX, which was physically adsorbed with 1 mg PLGA in acetonitrile and the final volume of the mixture was made to 400 μL for nanoprecipitation. The near-infrared dye tracer (DiR dye) as an imaging probe was infused in the lipid mixture using a film hydration technique. In a typical experiment, 20 μg of the dye was added to the lipid mixture and the solvent was evaporated to obtain the lipid film. The lipid film was dissolved in 4% ethanol and nanoprecipitation proceeded under dark conditions.
Characterization of nanoparticles
The formation of the LyP-1–DSPE–PEG product was characterized using Fourier transform infrared spectroscopy (FT-IR). The morphology of the nanoparticles was examined using transmission electron microscopy (TEM, FEI Technai G2 Spirit BioTWIN, 80 keV). The sample was loaded into the copper grid and dried over air followed by staining with 0.1% uranyl acetate. The hydrodynamic size and zeta potential measurements of the NPs were monitored using dynamic light scattering (DLS) using a Malvarian Zeta sizer Nano®. In this instrument, the Brownian motion and the Smoluchowski equation were used to calculate the average hydrodynamic size and zeta potential value, respectively. The colloidal stability of NPs was investigated in both ionic and serum conditions to mimic the biological environment. In brief, 1 mg of NPs dispersed in the medium were subjected to hydrodynamic size analysis over a period using DLS to obtain size, polydispersity index (PDI), and zeta potential values. The formulation with low PDI, high stability, and uniform narrow size distribution was selected for the in vitro and in vivo applications. Likewise, the stability of the DiR dye in the NPs was studied by assessing its release kinetics in PBS. The release content of DiR was quantified spectrophotometrically with fluorescence excitation at 748 nm and emission at 780 nm. Finally, the presence of peptide on the nanoparticles was quantified using a colorimetric assay, the rapid gold BCA assay, according to the manufacturer's protocol.Drug loading and release study
In a typical experiment, DOX was loaded on LyP-1 tagged and control NPs by direct mixing of a known concentration of DOX into the PLGA polymer followed by the nanoprecipitation technique.33,39,40 To optimize the maximum DOX loading, different calculated amounts such as 10, 25, 50, 100, 150, 200, 300, 400, and 500 μg of DOX were initially fed with 1 mg of PLGA dissolved in 400 μL of acetonitrile solution. The amount of encapsulated DOX was quantified, after purification, spectrophotometrically using a UV-vis spectrophotometer (Bio Tek Synergy2) with excitation at 485 nm and emission at 590 nm. The drug loading efficiency was calculated using the following equation:| % loading efficiency = (amount of encapsulated DOX/amount of initial DOX input) × 100. |
Next, we studied the cumulative drug/dye release using 3.5 kDa molecular cut-off dialysis bags at physiological pH (pH 7.4, PBS). For this purpose, 150 μg mL−1 DOX input formulation was selected for further studies due to the stable physiochemical properties. An optimization experiment was performed using an aqueous solution of free DOX (25 μg mL−1) placed in the dialysis tubing. Free DOX release from selected formulations was tracked throughout 72 h by dialysis. 1 mL of DOX-NPs containing 1 mg mL−1 of NPs was dialyzed in 100 mL PBS release media kept at 37 °C, while constantly stirring (80 rpm) to maintain the sink condition during the experiment. 1 mL of release media was taken at a regular interval and replaced with equivalent fresh media. The amount of free DOX in the release media was quantified spectrophotometrically and the cumulative release percentage was calculated using the following equation:
| % cumulative DOX release = (DOX in release media/DOX in 100% release) × 100. |
The stability of DOX-loaded NPs was studied by mixing 500 μL of DOX-NP and 500 μL of PBS (1×) (n = 3) over a period of time to ensure the dispersity and colloidal stability of NPs in physiological media. The size and PDI of the particles after dispersal in PBS was measured each 4th day for 7 weeks using DLS.
Cellular biocompatibility and cytotoxicity study
The in vitro biocompatibility of the NPs was determined in K7M2 and NIH/3T3 cell lines using a colorimetric assay to assess cell metabolic activity using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. A time-dependent MTT assay was conducted as discussed in the literature.41–44 In a typical in vitro experiment, the cells were cultured in DMEM containing 10% (v/v) fetal bovine serum and 1% (w/v) penicillin–streptomycin at 37 °C in 5% CO2 and 95% air. Non-cancerous and cancerous cells were used in this study and maintained according to the manufacturer's recommendations. In brief, at a density of 5 × 103 cells per well in a 96-well plate, the cells were incubated for 24 hours. The seeded cells were washed twice with 1× PBS to remove the debris before NP treatment. Then the medium was replaced with various concentrations of NPs (0.01, 0.1, 0.5, 5, 10, 25, 50,100, and 200 μg mL−1) suspended in DMEM. The control cells were maintained without treatment. After 24 h of incubation, the cells were washed with 1× PBS to remove any surface absorbed and excess NPs. After washing, 100 μL MTT reagent (5 mg mL−1 in PBS, filtered through a 0.22 μm syringe) was added to each well following the manufacturer's recommendations. Then the supernatant was aspirated followed by the addition of 100 μL DMSO for solubilizing the formazan crystal. The plate was gently shaken in an orbital shaker for 5 min in the dark and the absorbance was recorded at 560 nm using a microplate reader (BioTek, Synergy H1 hybrid reader).The in vitro cytotoxicity of the DOX-loaded LyP-1 NPs was determined in mouse cells with targeted osteosarcoma and the non-targeted (normal) fibroblasts using an MTT assay as discussed above in biocompatibility measurement. We varied the DOX concentration from 0.01 to 50 μM free DOX and NP DOX. The data obtained were compared side by side with the control experiment to map the relative toxicity.
Intracellular uptake study
To evaluate the cellular internalization competence of the LyP-1 NPs, the experiment was conducted with mouse osteosarcoma and non-cancerous mouse fibroblasts using confocal microscopy. For the confocal study, the cells were seeded on poly-D-lysine coated eight chamber slides at a density of 50000 cells per well and incubated for 24 h. Then the cells were treated with 50 μg mL−1 rhodamine-labeled LyP-1 NP or a control NP suspension prepared in complete DMEM and incubated for 3 h. After incubation, the treated cells were washed twice with 1× PBS (pH 7.4), and fixed with 4% paraformaldehyde for 30 min at room temperature. The nucleus of the cells was stained with 4′,6-diamidino-2-phenylindole (DAPI) for an additional 10 min and imaged under a confocal laser scanning microscope (Carl Zeiss, CLSM-700). RhB-labeled NPs without LyP-1 was used as the control particles. Cells without any treatment served as the control cells. Comparative RhB fluorescence intensity in mouse fibroblast (NIH/3T3) and osteosarcoma (K7M2) cells were analyzed with CLSM. The quantitative percentage of fluorescence intensity was determined using Image-J software. The quantification of the intensity of internalized RhB-labeled NPs was calculated in terms of corrected total cell fluorescence (CTCF) by using the following equation:| CTCF = integrated density − (area of selected cell × mean fluorescence of background readings) |
Fluorescence-activated cell sorting (FACS) study
A FACS study was conducted to determine quantitative cellular uptake in order to support the hypothesis. Following the published protocol, the FACS experiment was conducted using RhB-labeled LyP-1NPs and RhB-labeled non-targeted NPs serving as control particles.34,35,44 In brief, cells were cultured in T25 flasks up to around 80% confluency. Cell culture media was replaced with 4 mL of 50 μg mL−1 of RhB-labeled NPs. After a pre-determined treatment time point, the media was removed, and the cells were washed with 1× PBS three times and trypsinized. Finally, 1 × 10−6 cells were suspended in 500 μL of 1× PBS and cellular internalization was analyzed using a BD LSR Fortessa X-20 Special Order Research Product flow cytometer. Control cells were maintained without any treatments. 10000-gated events were collected for all the measurements.
Biodistribution and in vivo imaging
Biodistribution studies involve measuring NP concentrations in all the major tissues after a single administration of NPs over a period of time. NIR dye-tagged NPs were used as the probe to track the NPs in vivo for this study, using the lipid insertion technique.18 20 μg of DiR dye was added to 1000 μg of both the control and LyP-1 NPs and extruded using a 200 nm polycarbonate membrane filter for physical encapsulation of the dye in the NPs resulting in control NP-DiR and LyP-1 NP-DiR, respectively. The encapsulation of 20 μg dye per mg of NPs gave monodispersed sub-hundred nm sized particles (ESI-1, ESI†) which were used the next day for an imaging application. This dose of dye was chosen based on the literature.18,35,36,45 The solid tumor model using osteosarcoma cancer cells was developed by subcutaneous injection of 1 × 106 K7M2 cells into the rear flank of an immunodeficient NU/NU nude mouse (Charles River, 088/NU/NU homozygous). After 1 week of tumor induction or when the tumor size reaches about 6 mm3, tumor-bearing animals were treated with the respective NPs for the biodistribution studies. The time-course biodistribution patterns of different formulations were examined by NIR-fluorescence whole-body imaging using DiR as a tracer in a LI-COR® whole-body imaging system. DiR dye was labeled with NPs according to the published protocol to obtain DiR labeled NPs.18,35,36 After a single dose post-injection of 10 mg kg−1 of NPs via the lateral tail vein (100 μL volume), mice (n = 3) were imaged to map the real-time distribution of NPs at various time points viz., 0.5, 1, 2, 3, 6, 12, 24, and 48 h. The non-invasive imaging of mice was performed after anaesthetizing (2–3% isoflurane in 100% oxygen) and placing them on a preheated bed (plate). The 750 nm channel was used to excite DiR and emission was observed at 800 nm. Fluorescent background images were acquired before NP administration. When imaging the animals, a fluorescent phantom was included in the image for calibration of the fluorescence intensity. After 48 h post-injection, the animals were euthanized, and the selected organs including their heart, lungs, liver, kidneys, spleen, and brain, as well as the tumor were collected, washed with 1× PBS, weighed, and imaged using the same imaging system to quantify DiR accumulation in the organs using ImageStudioTM software. DiR labeled NPs were quantified by selecting a region of interest (ROI) and plotting it against signal intensity. All the animal experiments were approved by IACUC, KSU.Data and statistical analysis
The obtained data were plotted and compared to the significance of the results using GraphPad® software. Different statistical models and tests were carried out including t-test, F-test, and a dose–response inhibition model fit using GraphPad software according to requirement. All data represent the mean ± standard deviation. The biocompatibility and cytotoxicity data include six replicates (n = 6). All other major data include at least three replicates (n = 3).Results and discussion
Chemical characterization of the LyP-1–lipid
The synthesis of the peptide conjugated lipid was carried out using EDC/NHS bioconjugation chemistry as described in Scheme 1A with a ∼91% yield. The chemical structure of the synthesized LyP-1 peptide conjugated lipid DSPE–PEG was confirmed by Fourier-transform infrared (FT-IR) spectroscopy. As shown in Fig. 1, the FT-IR spectrum of LyP-1-exhibits the characteristic peaks of unmodified DSPE–PEG (spectrum in blue, upper panel) with additional functional absorption peaks (spectrum in red, bottom panel). The appearance of the broad and strong signal between 3169 and 3334 cm−1 is attributed to the formation of an amide bond between the carboxylic group and the primary amine group from the LyP-1 peptide after conjugation (spectrum in red, bottom panel).42,46 It is notable that after the conjugation, the shifting of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak from 1730 to 1647 cm−1 (highlighted with the black dotted line) with clear distinction absorption bands at 1647 cm−1 and at 1566 cm−1, and the N–H out of plane wagging at 729 to 675 cm−1 (highlighted with the black dotted arrow) confirm the successful peptide conjugation (Fig. 1). The higher frequency band at 1647 cm−1 is characteristic of the amide-I band, while the lower frequency band at 1566 cm−1 is amide-II, which is largely due to the N–H bending trans to the carbonyl oxygen.47 The bands at 2850 to 2952 cm−1 correspond to the aliphatic C–H stretch of the lipid backbone and symmetric and asymmetric C–O–C stretching (1170–1130 and 1050–950 cm−1, respectively) belonging to the DSPE–PEG moiety (Fig. 1).42,48 The presence of peptide on the surface of the nanoparticles is characterized using a calorimetric rapid gold BCA assay according to the manufacturer's protocol (ESI-2, ESI†).
O peak from 1730 to 1647 cm−1 (highlighted with the black dotted line) with clear distinction absorption bands at 1647 cm−1 and at 1566 cm−1, and the N–H out of plane wagging at 729 to 675 cm−1 (highlighted with the black dotted arrow) confirm the successful peptide conjugation (Fig. 1). The higher frequency band at 1647 cm−1 is characteristic of the amide-I band, while the lower frequency band at 1566 cm−1 is amide-II, which is largely due to the N–H bending trans to the carbonyl oxygen.47 The bands at 2850 to 2952 cm−1 correspond to the aliphatic C–H stretch of the lipid backbone and symmetric and asymmetric C–O–C stretching (1170–1130 and 1050–950 cm−1, respectively) belonging to the DSPE–PEG moiety (Fig. 1).42,48 The presence of peptide on the surface of the nanoparticles is characterized using a calorimetric rapid gold BCA assay according to the manufacturer's protocol (ESI-2, ESI†).
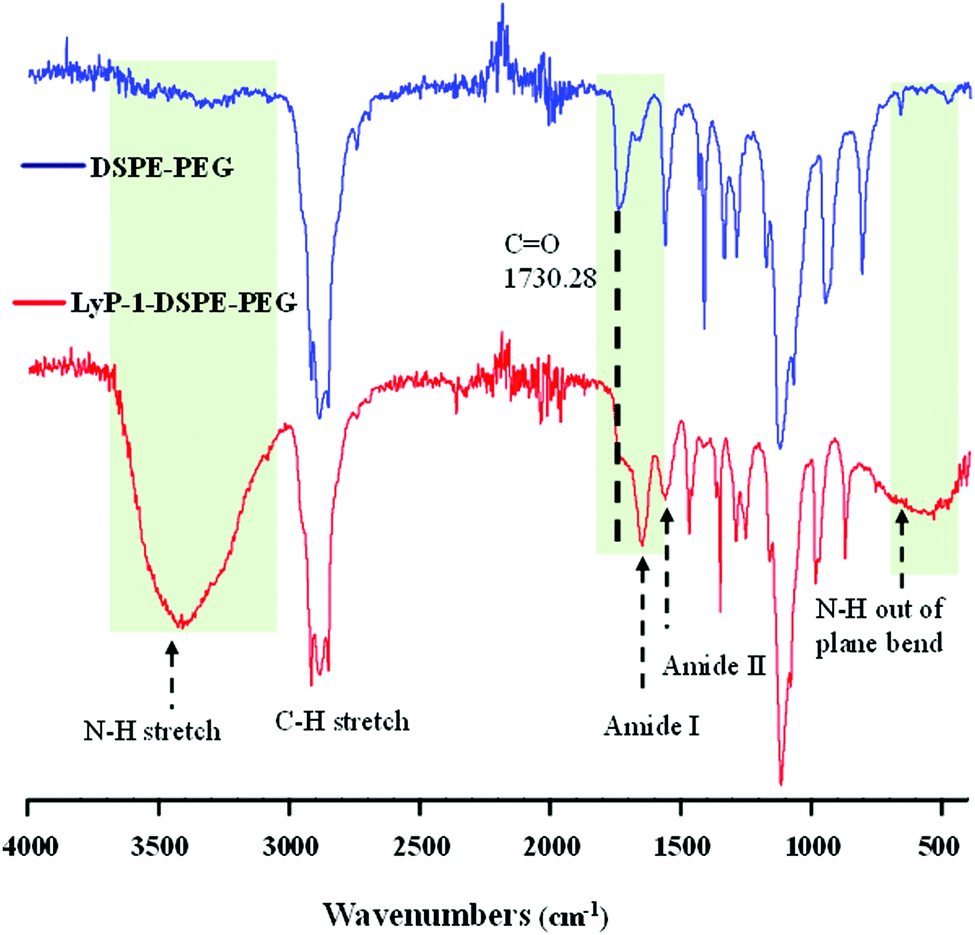 | ||
| Fig. 1 Chemical characterization of synthesized LyP-1–DSPE–PEG using an FT-IR spectrum showing the functional peaks of the starting materials (DSPE–PEG) and product (LyP-1–DSPE–PEG). The LyP-1–DSPE–PEG spectrum was obtained with the lyophilized product. | ||
Physiochemical characterization of NPs
The physical morphology of the synthesized nanoparticles was measured using TEM, which shows highly monodispersed and uniformly spherical particles with an average diameter of 68 ± 7 nm (Scheme 1C and ESI-3, ESI†). Similarly, the average hydrodynamic size of the LyP-1 NP was 79 ± 3 nm with a polydispersity index (PDI) of 0.183 ± 0.018 (Fig. 2A), and an average surface zeta potential of −39 ± 4 mV (Fig. 2B). Also, the control particle without LyP-1 showed a similar size and zeta potential with an average hydrodynamic size of 68 ± 4 nm and PDI of 0.161 ± 0.015, and an average surface zeta potential of −37 ± 3 mV. The negative zeta potential is due to the surface carboxylate groups at the end moiety of the PEG corona. The size from TEM and DLS measurements is in good agreement and demonstrates the monodispersity of the particles. The prepared nanoparticles (both control and targeted) were stable for a prolonged period of time, as demonstrated in Fig. 2C. Under the physiological conditions of phosphate buffer saline (PBS) pH = 7.4, we did not observe significant changes in size and PDI of both particles incubated over the weeks, confirming the stability of the nanoparticles in the biological environment. | ||
| Fig. 2 Physiochemical characterization, drug loading, and release study of the NPs. (A) Dynamic light scattering showing the average hydrodynamic size, (B) surface zeta potential, and (C) colloidal stability study of NPs in PBS (pH = 7.4). (D) Comparative DOX loading efficiency of the control NP at various feeding concentrations, (E) comparative DOX loading efficiency of LyP-1 NP with various initial input feeding concentrations, and (F) comparative drug release kinetics of NPs in PBS (pH = 7.4). Data represent mean ± SD, n = 3. | ||
After the analysis of physicochemical properties, the drug encapsulation efficiency of the NPs was evaluated by altering the initial doxorubicin (DOX) input concentration from 10 to 400 μg mg−1 of the PLGA. DOX loading was quantified spectrophotometrically at excitation and emission wavelengths of 490 nm and 580 nm following the published protocol.38,42 The results showed that the initial DOX loading can be precisely tuned by varying the drug input (10, 25, 50, 100, 150, 200, 300, and 400 μg mg−1 of PLGA weight) during the NP preparation process. Among the different initial DOX feeding concentrations, the maximum encapsulation efficiency of the control NP was found to be about 93% (Fig. 2D and E). As expected, the similar encapsulation efficiency of DOX in both the control and LyP-1 NP was observed with an optimum feeding concentration of 150 μg mg−1, which accounts for 15% by weight of PLGA and about 78% loading efficiency. Due to the same PLGA core, the DOX content in both NPs (control and LyP-1 NP) were the same. Among these different formulations, 150μg mg−1 of PLGA initial feeding input of DOX gave the most effective loading efficiency without altering the nanoparticle physicochemical properties. However, in the case of higher DOX input, formulations in both NPs resulted in lesser encapsulation and affected the physiological stability of the particles as indicated by rapid aggregation and precipitation. Therefore, we have chosen 150μg mL−1 input formulation as the value for further experiments. No significant changes in the size and PDI of this formulation were observed when incubated at 37 °C for 5 days (ESI-4, ESI†) and when stored at 4 °C in PBS (pH = 7.4) over a 7-week period, confirming the excellent physiochemical properties (ESI-5, ESI†).
To determine the release efficiency of the drug from the NPs, 2 mL samples of 1 mg mL−1 of control-NP-DOX and LyP-1-NP-DOX were placed in a dialysis bag and processed under identical conditions. We observed similar time course DOX release kinetics for both control-NP-DOX and LyP-1-NP-DOX over a 72 h period at 37 °C. The results, as shown in Fig. 2F, indicate that about 16% DOX was released within a period of 6 h from both types of NPs, while over 12 h, we observed a slight difference in drug release kinetics by below 8% in LyP-1 NP. However, there is no significant difference in the overall kinetics over 72 h. On the other hand, compared to DOX release, we found a higher order of DiR dye stability in the NPs. Less than 13% DiR was released throughout the release study during 72 h (ESI-6, ESI†), consistent with the literature.34 The stability of DiR is highly essential in device NPs for theranostics.
Biocompatibility study
Cellular compatibility and therapeutic efficacy of the LyP-1 NPs, with and without the drug, were studied using mouse normal fibroblast (NIH/3T3) and osteosarcoma (K7M2) cells. We used DOX in our study because the current clinical treatment protocol against osteosarcoma includes DOX as one of the important chemotherapeutics along with methotrexate and cisplatin.49 For the biocompatibility study, various concentrations of LyP-1 NP were incubated with K7M2 and NIH/3T3 cells as presented in Fig. 3 for 24 h. Cells without NP treatment were used as the experimental control and non-targeted NPs as the NP control. The control NPs showed at least 82% cell viability even at high nanoparticle concentration (200 μg mL−1) in both K7M2 and NIH/3T3 cells. More specifically, in the particle concentration window from 10 μg mL−1 to 200 μg mL−1, both cell lines showed similar viability. In the case of LyP-1 NP, the cell viability with particle concentrations from 10 μg mL−1 to 200 μg mL−1 ranges from 93% to 79% in K7M2 cells and 100% to 83% in NIH/3T3 cells, respectively. This higher cell viability even at high NP concentrations put it as an excellent candidate for drug delivery (Fig. 3A and C). At lower NP concentrations, no significant toxicity related to either non-targeted or targeted NPs were observed indicating the excellent biocompatibility of the NPs in the tested cells. However, when the concentration increases up to 150 μg mL−1, the cell viability was decreased by 15% (total viability at this concentration was 85%) for both cell lines used in the study, which could be a dose effect in the limited culture area of the 96 well plates. The results of the biocompatibility of these NPs are consistent with previous findings on lipid polymer containing NPs.5,8,37,40,42,50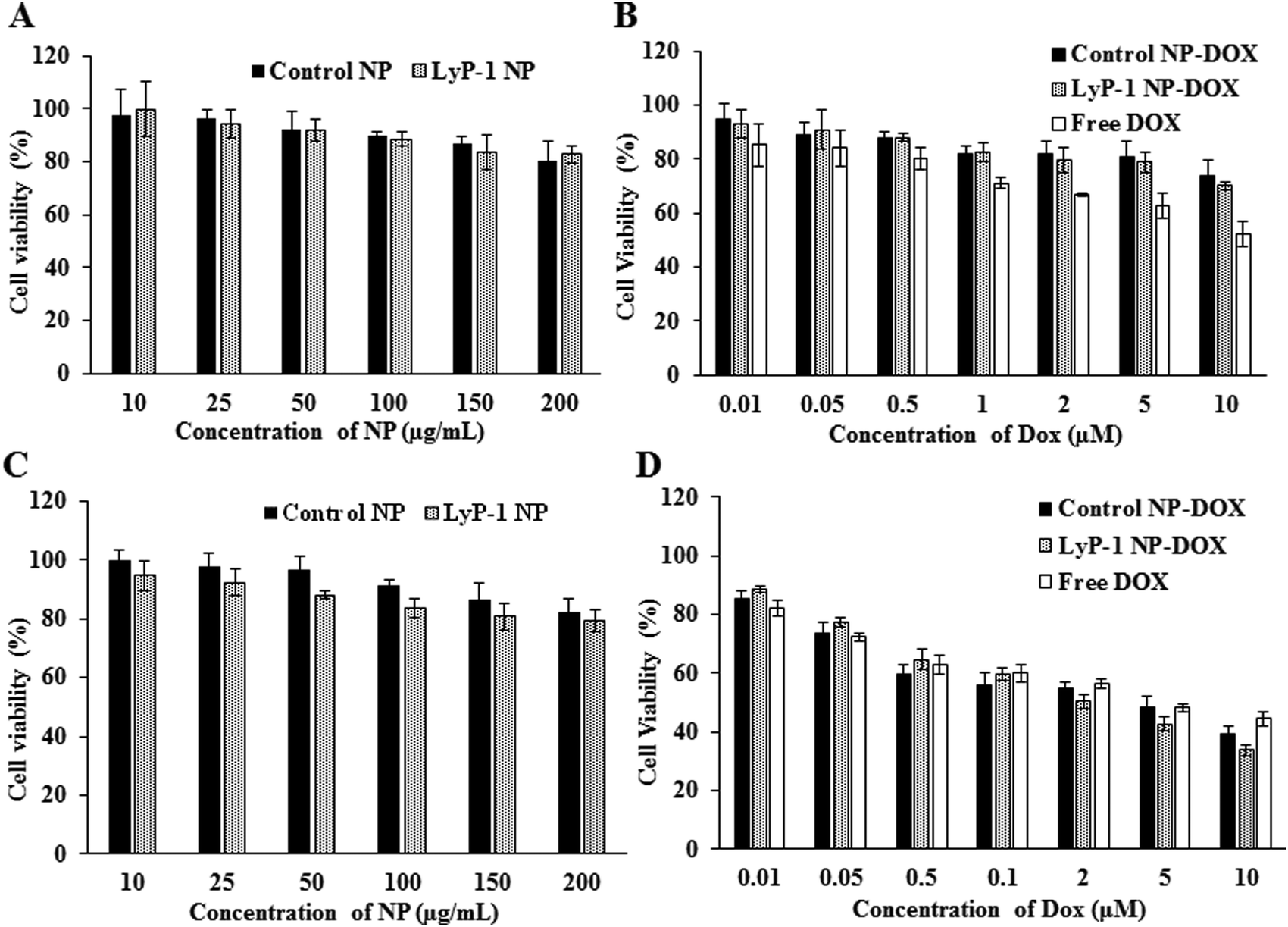 | ||
| Fig. 3 Comparative cellular interaction study of control NP and LyP-1 NP against mouse fibroblast (NIH/3T3) and osteosarcoma (K7M2) cells. (A) Concentration-dependent biocompatibility of control and LyP-1NP and (B) comparative cytotoxicity of free DOX, control NP-DOX, and LyP-1NP-DOX against NIH/3T3 cells 24 h post-treatment, respectively. (C) Concentration-dependent biocompatibility of the control and LyP-1-NP and (D) comparative cytotoxicity of free DOX, NP-DOX, and LyP-1-NP-DOX against K7M2 cells 24 h post-treatment, respectively. Data represent mean ± SD, n = 6. p value > 0.05, two-way ANOVA for the targeted and non-targeted NPs against K7M2 cells, p-value summary – n. s. | ||
Cellular cytotoxicity study
In a typical cellular cytotoxicity experiment, DOX-loaded NPs and the equivalent of free DOX were used against K7M2 and NIH/3T3 cells. The results have shown that both free DOX and DOX-loaded NPs exhibit a time- and dose-dependent cytotoxic effect (Fig. 3B and D). The lower concentration of the drug-loaded NPs showed lower cytotoxicity than free DOX against K7M2 24 h post-treatment. In contrast, about 10% enhanced cytotoxicity of DOX-loaded NPs was observed as compared to free DOX with the higher dose (10 μM). Notably, free DOX showed higher cytotoxic behavior with non-cancerous cells throughout the range of concentrations used, compared with DOX-loaded NPs. However, enhanced toxicity was observed in both cells in the case of LyP-1 NP-DOX with a higher payload compared to control-NP-DOX. The dose-dependent cytotoxic effect of LyP-1 NP-DOX is likely due to the internalization mechanism of the nanoparticle. First, it could be attributed to the presence of the tumor-targeting LyP-1 peptide that can recognize and bind to the p32 receptor proteins overexpressed on the surface of cancer cells leading to higher accumulation and distribution throughout cancer cells as compared to those of non-cancerous cells.41,51,52 This p32 receptor-mediated endocytosis, as reported in the literature,41,51,52 increases localized intracellular drug concentration whereas in the case of free DOX it diffuses slowly into the cell limiting localized drug concentration. Although no statistically significant difference was observed for the targeted and non-targeted NPs against cancer cells (p value > 0.05, two-way ANOVA), it is worth noting that a two-dimensional culture system, where density-induced NP cell surface interactions are accounted for, can interfere with cell viability and may limit actual representation of toxicity.11,53,54 Therefore, we further studied quantitative cellular internalization studies to validate the receptor-mediated uptake of NPs.Cellular uptake studies
Cellular uptake is an important biological property that dictates therapeutic effectiveness. A tumor-targeted internalization property of nanoparticulate drug delivery systems can not only enhance toxicity against tumor cells but can also reduce off-target toxicity. To analyze the tumor-targeting behavior of Lyp-1 NPs, a cellular uptake study was carried out in K7M2 and NIH/3T cells via confocal microscopy following a 3 h incubation with rhodamine B (Rh-B) labeled NPs (Fig. 4 and ESI-7, ESI†). The 3 h incubation time point was chosen based on our previous study where the internalization of NPs reached a maximum at 3 h.44,52,55,56Fig. 4 shows the confocal imaging of the cells showing the internalization of NPs and their corresponding quantification in terms of corrected total cell fluorescence in K7M2 cells. Untreated cells were used as the control. As shown in Fig. 4B, the fluorescence intensity of the LyP-1 NP-RhB was significantly higher than that of the control NP-RhB nanoparticles (p < 00001, unpaired t-test). On the other hand, no significant difference in NP uptake was observed for the non-cancerous NIH/3T3 cells (ESI-7, ESI†). This observation indicates that LyP-1 NP-RhB was preferentially taken up by K7M2 cells supporting our hypothesis of receptor-mediated uptake due to LyP-1 in the NPs.
 | ||
| Fig. 4 Cellular internalization study in mouse osteosarcoma (K7M2) cells. (A) Comparative confocal images of K7M2 cells incubated with LyP-1 NP-RhB and control NP-RhB (50 μg mL−1) for 3 h at 37 °C. The nuclei of the cells were stained with DAPI (blue). (B) Corresponding quantification of the internalized NPs following corrected total cell fluorescence (CTCF) using ImageJ software (*** = p < 00001, unpaired-test-two-tailed). The cells were imaged under a 20× lens and using a 50 μm scale bar. Unpaired t-test, p-value < 00001, p-value summary – ***. | ||
To further support our confocal results, the targeting property of the NPs was confirmed by quantitative cellular uptake studies using FACS. As the LyP-1 NP has a targeting ligand from the LyP-1 peptide on the surface, the differential targeting ability is expected to be more in the cancer cells. Based on the literature for the overexpression of p32 in bone cancer,24–26,57,58 we expect LyP-1-modified nanoparticles to specifically target K7M2 cells. The quantitative cellular uptake of NPs was evaluated after a 3 h incubation for both the control and targeted NPs, and experiments were carried out under identical conditions with both cell types, K7M2 and NIH/3T3 cells.44,52,55,56 As shown in Fig. 5A, the overlapping of the histogram from control NP-RhB and LyP-1 NP-RhB showing no significant difference in internalization behavior was observed in NIH/3T3 cells. On the other hand, LyP-1 NP-RhB shows a right shift in Fig. 5C demonstrating higher cellular uptake than that of control NP-RhB. This observation was further analyzed by taking the median fluorescence value of NPs internalized into cells. Fig. 5D shows the significantly higher fluorescence intensity of the LyP-1 NP-RhB as compared to that of the control NP-RhB (p < 00001, unpaired t-test). Both confocal imaging and flow cytometry analysis confirmed the selective internalization of LyP-1 NP-RhB towards tumor cells, which supports our hypothesis.
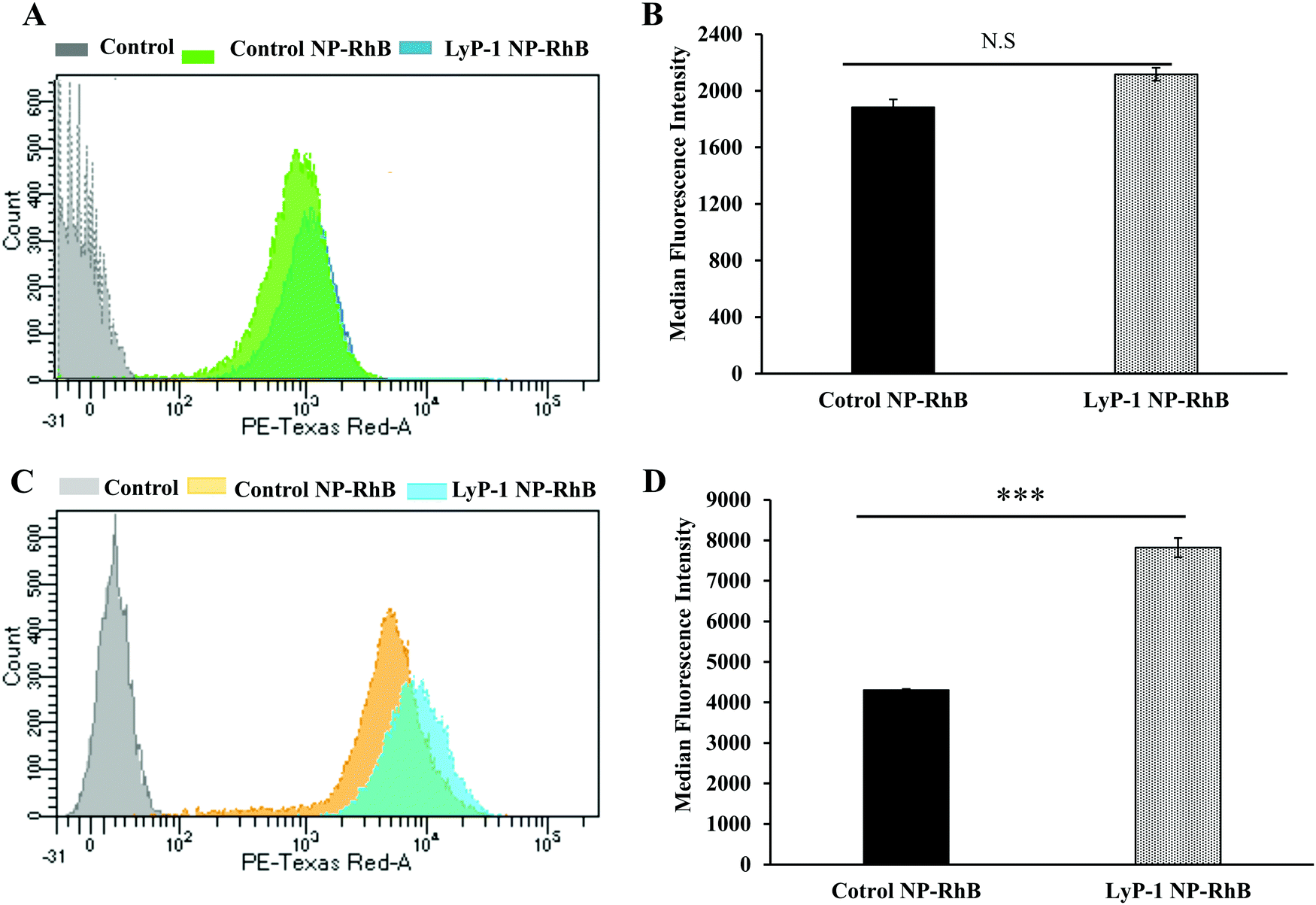 | ||
| Fig. 5 Fluorescence activated cell sorting (FACS) experiment for quantification of internalized NPs. (A) and (C) are histograms showing the comparative cellular uptake study of control NP-RhB and LyP-1 NP-RhB against NIH/3T3 and K7M2 cells, respectively. (B) and (D) are the corresponding quantitative data in terms of the average median fluorescence intensity (MFI) of the control and LyP-1 NP. Cells were treated with 50 μg mL−1 of RhB-labeled particles and incubated for 3 h before data acquisition. Data represent mean ± SD, n = 3. (Statistics: unpaired t-test, p value = 0.1669, non-significant for NIH/3T3 and p value < 00001 for K7M2 respectively.) | ||
In similar studies, Pang et al. reported that the RPARPAR peptide coated 50 nm gold nanoparticles (AuNPs) were taken up by primary prostate carcinoma-1 cells in a glucose-dependent manner and internalized through neuropilin-1 (NRP-1), a cell surface receptor with multiple ligands that binds through a CendR motif mediated endocytosis via a mammalian target of the rapamycin (mTOR) signaling pathway.56 AuNPs coated with a control peptide did not bind to NRP-1 and were not taken up into cells in the absence of glucose. The RPARPAR peptide has a CendR motif similar to that of the LyP-1 peptide. The CendR pathway could be ascribed to the localized LyP-1 NP uptake similar to that of the previous studies.56 When LyP-1 NPs get into the endosome, the higher endosomal pH will breakdown the ester bond present in the lipid-polyethylene glycol moiety resulting in destabilization of the particles. Then the fusion of the lipid shell with the endosomal membrane could allow the cargo to escape from the endosomal degradation and enter into the cytosol.
In vivo biodistribution study
Near-infrared fluorescent probes such as DiR (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) dye have many advantages in imaging applications including minimum tissue autofluorescence, real-time visualization, minimum photo-bleaching, maximum tissue penetration, and photo-damage.59–63 Therefore, near-infrared fluorescent probes are being developed and used in optical imaging for the visualization of biological phenomena in the living system. To examine the effect of versatility in surface modification and confirm the accumulation in the tumor, we tagged the NPs with DiR dye and compared both targeted and non-targeted NPs. The DiR in the NPs becomes part of the lipid building block of the nanoconstruct. We investigated the biodistribution of DiR-labeled NPs in K7M2 tumor-bearing NU/NU mice following intravenous injection of a 10 mg kg−1 NP dose. A LI-COR® whole-body imaging system was used to monitor the real-time biodistribution and tumor homing efficiency of LyP-1 NP-DiR (Fig. 6). The images were spectrally processed using Image Studio ver5.2®. The emission of the dye was isolated from autofluorescence and quantified as the region of interest (ROI) indicated on the corresponding graphs (Fig. 6B and C). | ||
| Fig. 6 In vivo biodistribution of NPs. (A) Time-dependent real time animal imaging of K7M2 tumor-bearing NU/NU nude mice after intravenous injection of DiR-labelled NPs (10 mg kg−1). Images were recorded prior to injection (pre-contrast) and after 3 h, 6 h, 12 h, 24 h, and 48 h post-injection of a single-dose, respectively. (B) Quantification of time-dependent accumulation of control and LyP-1 NP-DiR in tumors by measuring the intensity of dye in the region of interest (ROI). (C) Comparative bioaccumulation of the control and LyP-1 NP-DiR in different organs imaged ex vivo after 48 h post-injection. (D) Quantification of tumor accumulation of the control and LyP-1 NP-DiR. p value = 0.0169, *(F-test). Data represent mean ± SD, n = 3. | ||
Fig. 6A shows the overall distribution of fluorescent-tagged NPs in K7M2 tumor-bearing mice. We observed the rapid extravasation of the control NP into the tissue showing localized intensity as compared to LyP-1-NP-DiR. Also, we observed that the control NPs-DiR distributed quickly (Fig. 6A) throughout the body as early as at the 1 h point. However, LyP-1-NP-DiR did not show much accumulation during this period. We observed significant tumor uptake of both NPs more specifically at 12 h post-injection. However, with time, we didn’t observe enhanced tumor intensity in the control NP-DiR-treated mice as it distributed throughout the body. In contrast, mice treated with LyP-1-NP-DiR showed tumor uptake over the period of time (Fig. 6B) thereby supporting our hypothesis of receptor-mediated targeting, while minimizing unwanted distribution throughout the body. Similar enhanced uptake has been reported in previous publications.24–26,57,58 To further understand the tissue distribution, we euthanized mice after 48 h post-injection and the harvested organs were studied for NP accumulation. Fig. 6D shows the accumulation of control NP-DiR and LyP-1 NP-DiR in vital organs and blood in terms of percentage injected dose (ID) per weight of tissue (g). Control NP-DiR was retained mainly in the liver (4.09%), spleen (5.43%), and blood (6.09%). In contrast, LyP-1 NP-DiR, showed retention in the liver (3.15%), spleen (3.28%), tumor (1.16%), and blood (19.20%). Notably, we observed that more than three-fold high retention in the blood even after 48 h post-injection (19.2% vs 6.09%) suggesting the excellent blood retention characteristics of LyP-1 NP-DiR. This suggests the blood availability of LyP-1 NP-DiR, thereby providing its long-circulating properties (Fig. 6D). The time-course pharmacokinetic and biodistribution studies are needed for further confirmation as warranted by this observation. Unsurprisingly for nanoparticles above the glomerular filtration capacity, it is expected that these particles were cleared mainly through reticuloendothelial system (liver and spleen) uptake, which can be seen in Fig. 6C, D and ESI-8, ESI.† Furthermore, LyP-1 NP-DiR showed more than three times higher deposition of NPs in the tumor region than that of its non-targeted counterparts (Fig. 6C and ESI-9, ESI†). This strongly supports the notion of a tumor targeting and homing potential of the LyP-1 peptide.
It is also reported from an in vivo study that cationic liposomal formulation fabricated with an aptamer tends to accumulate more in the liver, spleen, and kidneys, suggesting reticuloendothelial system uptake.64,65 The higher tumor accumulation of LyP-1 nanoparticles could be attributed to the fact that the LyP-1 peptide tends to accumulate more in the tumor microenvironment and drags a greater amount of bulk nanoparticles towards the target site by activating the CendR pathway.22,66,67 The active tumor accumulation largely depends on the overexpression of p32 cell receptor ligands in tumor cells.13,20,22–24,68–70 Additionally, some of the nanoparticles can be accumulated concomitantly by passive targeting over the experimental period.25,56 This result strongly corroborates with previous studies using cell membrane-infused liposomal systems and targeted polymeric nanoparticles for targeted tumor therapy and bioimaging.18,34,71,72
Conclusions
In summary, we describe a simple and versatile nanoformulation strategy by combining the synthetic lipids, a biocompatible polymer, and a tumor homing peptide into a composite nanosystem for targeted drug delivery and imaging in a single session. It is evident from our results that the LyP-1 navigated the nanoconstruct into the tumor region showing its promise in precision drug delivery under the experimental tumor conditions. Moreover, the hybridization of lipid–polymer with different physiochemical properties could be beneficial to overcome the respective shortcomings to design a robust drug delivery system due to the coexistence of hydrophobic and hydrophilic nature. Overall, this will eventually open up opportunities for the mapping of the many experimental carcinomas for therapeutic planning and image-guided therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the University Small Research Grants (USRG) and Johnson Cancer Research Center (JCRC), Kansas State University, Manhattan, Kansas. Confocal core was supported by CVM-KSU.Notes and references
- L. Zhang and L. Zhang, Nano LIFE, 2010, 01, 163–173 CrossRef CAS.
- L. Zhang, J. M. Chan, F. X. Gu, J.-W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- A. D. R. Silva, T. D. Stocco, A. E. C. Granato, S. V. Harb, S. Afewerki, N. J. Bassous, P. Hammer, T. J. Webster, F. R. Marciano and A. O. Lobo, in Nanostructured Polymer Composites for Biomedical Applications, ed. S. K. Swain and M. Jawaid, Elsevier, 2019, pp. 21–52 Search PubMed.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- S. Aryal, C.-M. J. Hu and L. Zhang, Mol. Pharmacol., 2011, 8, 1401–1407 Search PubMed.
- T. D. Thanh Nguyen, R. Marasini, S. Rayamajhi, C. Aparicio, D. Biller and S. Aryal, Nanoscale, 2020, 12, 4137–4149 RSC.
- R. H. Fang, S. Aryal, C.-M. J. Hu and L. Zhang, Langmuir, 2010, 26, 16958–16962 Search PubMed.
- R. H. Fang, K. N. H. Chen, S. Aryal, C.-M. J. Hu, K. Zhang and L. Zhang, Langmuir, 2012, 28, 13824–13829 CrossRef CAS PubMed.
- S. Aryal, C.-M. J. Hu and L. Zhang, Small, 2010, 6, 1442–1448 CrossRef CAS PubMed.
- R. Marasini, T. D. T. Nguyen and S. Aryal, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1580 CAS.
- S. Aryal, M. Prabaharan, S. Pilla and S. Gong, Int. J. Biol. Macromol., 2009, 44, 346–352 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Mater., 2012, 24, 3747–3756 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- R. Marasini, A. Pitchaimani, T. D. Thanh Nguyen, J. Comer and S. Aryal, Nanoscale, 2018, 10, 13684–13693 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, B. T. Luk and L. Zhang, Nanoscale, 2014, 6, 65–75 RSC.
- A. Pitchaimani, T. D. T. Nguyen, R. Marasini, A. Eliyapura, T. Azizi, M. Jaberi-Douraki and S. Aryal, Adv. Funct. Mater., 2019, 29, 1806817 CrossRef.
- R. Wang, J. Deng, D. He, E. Yang, W. Yang, D. Shi, Y. Jiang, Z. Qiu, T. J. Webster and Y. Shen, Nanomedicine, 2019, 16, 195–205 CrossRef CAS PubMed.
- V. Le Joncour and P. Laakkonen, Bioorg. Med. Chem., 2018, 26, 2797–2806 Search PubMed.
- J. Li, Y. Chen, N. Kawazoe and G. Chen, Nano Res., 2018, 11, 1247–1261 CrossRef CAS.
- P. Laakkonen, K. Porkka, J. A. Hoffman and E. Ruoslahti, Nat. Med., 2002, 8, 751–755 CrossRef CAS PubMed.
- T. Teesalu, K. N. Sugahara, V. R. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157–16162 CrossRef CAS PubMed.
- V. Fogal, L. Zhang, S. Krajewski and E. Ruoslahti, Cancer Res., 2008, 68, 7210–7218 CrossRef CAS PubMed.
- T. Muta, D. Kang, S. Kitajima, T. Fujiwara and N. Hamasaki, J. Biol. Chem., 1997, 272, 24363–24370 CrossRef CAS PubMed.
- P. Saha and K. Datta, Oncotarget, 2018, 9, 10784–10807 CrossRef PubMed.
- C. A. Staton, I. Kumar, M. W. R. Reed and N. J. Brown, J. Pathol., 2007, 212, 237–248 CrossRef CAS PubMed.
- M. Yagi, T. Uchiumi, S. Takazaki, B. Okuno, M. Nomura, S. Yoshida, T. Kanki and D. Kang, Nucleic Acids Res., 2012, 40, 9717–9737 CrossRef CAS PubMed.
- C. V. Dang, Mol. Cell. Biol., 2010, 30, 1300–1302 CrossRef CAS PubMed.
- V. Fogal, I. Babic, Y. Chao, S. Pastorino, R. Mukthavaram, P. Jiang, Y.-J. Cho, S. C. Pingle, J. R. Crawford, D. E. Piccioni and S. Kesari, Oncotarget, 2014, 6, 1157–1170 Search PubMed.
- V. Fogal, A. D. Richardson, P. P. Karmali, I. E. Scheffler, J. W. Smith and E. Ruoslahti, Mol. Cell. Biol., 2010, 30, 1303–1318 CrossRef CAS PubMed.
- V. Yenugonda, N. Nomura, V. Kouznetsova, I. Tsigelny, V. Fogal, E. Nurmemmedov, S. Kesari and I. Babic, J. Transl. Med., 2017, 15, 210 CrossRef PubMed.
- G. von Maltzahn, Y. Ren, J.-H. Park, D.-H. Min, V. R. Kotamraju, J. Jayakumar, V. Fogal, M. J. Sailor, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2008, 19, 1570–1578 CrossRef CAS PubMed.
- A. Pitchaimani, T. D. T. Nguyen and S. Aryal, Biomaterials, 2018, 160, 124–137 CrossRef CAS PubMed.
- J. Abello, T. D. T. Nguyen, R. Marasini, S. Aryal and M. L. Weiss, Theranostics, 2019, 9, 2325–2345 CrossRef CAS PubMed.
- T. D. T. Nguyen, S. Aryal, A. Pitchaimani, S. Park, J. Key and S. Aryal, Nanomedicine, 2019, 16, 79–87 CrossRef CAS PubMed.
- C.-M. J. Hu, S. Kaushal, H. S. T. Cao, S. Aryal, M. Sartor, S. Esener, M. Bouvet and L. Zhang, Mol. Pharmacol., 2010, 7, 914–920 CrossRef CAS PubMed.
- A. Pitchaimani, T. D. T. Nguyen, H. Wang, S. H. Bossmann and S. Aryal, RSC Adv., 2016, 6, 36898–36905 RSC.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- S. D. Steichen, M. Caldorera-Moore and N. A. Peppas, Eur. J. Pharm. Sci., 2013, 48, 416–427 CrossRef CAS PubMed.
- A. Pitchaimani, T. D. T. Nguyen, M. Koirala, Y. Zhang and S. Aryal, Toxicol. In Vitro, 2017, 43, 29–39 CrossRef CAS PubMed.
- T. D. T. Nguyen, A. Pitchaimani and S. Aryal, Sci. Rep., 2016, 6, 36707 CrossRef CAS PubMed.
- E. M. Pridgen, R. Langer and O. C. Farokhzad, Nanomedicine, 2007, 2, 669–680 CrossRef CAS PubMed.
- S. Rayamajhi, J. Marchitto, T. D. T. Nguyen, R. Marasini, C. Celia and S. Aryal, Colloids Surf., B, 2020, 188, 110804 CrossRef CAS PubMed.
- S. Rayamajhi, R. Marasini, T. D. T. Nguyen, B. Plattner, D. Biller and S. Aryal, Biomater. Sci., 2020, 8, 2887–2904 RSC.
- U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2013, 117, 489–496 CrossRef CAS PubMed.
- T. Forsting, H. C. Gottschalk, B. Hartwig, M. Mons and M. A. Suhm, Phys. Chem. Chem. Phys., 2017, 19, 10727–10737 Search PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–394 CrossRef CAS PubMed.
- D. S. Geller and R. Gorlick, Clin. Adv. Hematol. Oncol., 2010, 8, 705–718 Search PubMed.
- M. Henriksen-Lacey, S. Carregal-Romero and L. M. Liz-Marzán, Bioconjugate Chem., 2017, 28, 212–221 CrossRef CAS PubMed.
- S. Senapati, R. Shukla, Y. B. Tripathi, A. K. Mahanta, D. Rana and P. Maiti, Mol. Pharmacol., 2018, 15, 679–694 CrossRef CAS PubMed.
- S. Rayamajhi, T. D. T. Nguyen, R. Marasini and S. Aryal, Acta Biomater., 2019, 94, 482–494 CrossRef CAS PubMed.
- S.-J. Seo, M. Chen, H. Wang, M. S. Kang, K. W. Leong and H.-W. Kim, Nano Today, 2017, 14, 84–99 CrossRef CAS.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- D. Dehaini, R. H. Fang, B. T. Luk, Z. Pang, C.-M. J. Hu, A. V. Kroll, C. L. Yu, W. Gao and L. Zhang, Nanoscale, 2016, 8, 14411–14419 RSC.
- H.-B. Pang, G. B. Braun, T. Friman, P. Aza-Blanc, M. E. Ruidiaz, K. N. Sugahara, T. Teesalu and E. Ruoslahti, Nat. Commun., 2014, 5, 4904 CrossRef CAS PubMed.
- F. R. Dembitzer, Y. Kinoshita, D. Burstein, R. G. Phelps, M. B. Beasley, R. Garcia, N. Harpaz, S. Jaffer, S. N. Thung, P. D. Unger, B. Ghebrehiwet and E. I. Peerschke, J. Histochem. Cytochem., 2012, 60, 467–474 CrossRef CAS PubMed.
- B.-C. Kim, H.-J. Hwang, H.-T. An, H. Lee, J.-S. Park, J. Hong, J. Ko, C. Kim, J.-S. Lee and Y.-G. Ko, Oncotarget, 2016, 7, 49972–49985 CrossRef PubMed.
- Y. W. Jun, H. R. Kim, Y. J. Reo, M. Dai and K. H. Ahn, Chem. Sci., 2017, 8, 7696–7704 RSC.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 Search PubMed.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed.
- L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- J. Yan, J. Zhu, K. Zhou, J. Wang, H. Tan, Z. Xu, S. Chen, Y. Lu, M. Cui and L. Zhang, Chem. Commun., 2017, 53, 9910–9913 RSC.
- X. Song, X. Song, Y. Ren, Y. Ren, J. Zhang, J. Zhang, G. Wang, G. Wang, X. Han, X. Han, W. Zheng, W. Zheng, L. Zhen and L. Zhen, Oncol. Rep., 2015, 34, 1953–1960 Search PubMed.
- Z. Yang, X. Luo, X. Zhang, J. Liu and Q. Jiang, Biomed. Mater., 2013, 8, 025012 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- J. Hamzah, V. R. Kotamraju, J. W. Seo, L. Agemy, V. Fogal, L. M. Mahakian, D. Peters, L. Roth, M. K. J. Gagnon, K. W. Ferrara and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7154–7159 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- E. I. B. Peerschke and B. Ghebrehiwet, Mol. Immunol., 2014, 61, 100–109 CrossRef CAS.
- J. M. Chan, L. Zhang, R. Tong, D. Ghosh, W. Gao, G. Liao, K. P. Yuet, D. Gray, J.-W. Rhee, J. Cheng, G. Golomb, P. Libby, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 Search PubMed.
- W. Gu, J. An, H. Meng, N. Yu, Y. Zhong, F. Meng, Y. Xu, J. J. L. M. Cornelissen and Z. Zhong, Adv. Mater., 2019, 31, 1904742 CrossRef CAS PubMed.
- G. Luo, X. Yu, C. Jin, F. Yang, D. Fu, J. Long, J. Xu, C. Zhan and W. Lu, Int. J. Pharm., 2010, 385, 150–156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Dynamic light scattering showing the average hydrodynamic size of Control NP-DiR and LyP-1 NP-DiR (ESI-1); calibration curve for quantification of LyP-1 in NP using colorimetric Rapid Gold BCA Assay (ESI-2); representative transmission electron microscopy images of LyP-1 NPs with different magnifications (ESI-3); stability study of control NP-DOX and LyP-1 NP-DOX in PBS (pH = 7.4) at 37 °C using a dynamic light scattering experiment (n = 3) (ESI-4); stability study of control NP-Dox and LyP-1 NP-Dox in PBS (pH = 7.4) using dynamic light scattering experiment at 4 °C (n = 3) (ESI-5); comparative DiR dye release characteristics of Control-NP-DiR and LyP-1 NP-DiR NPs in PBS (pH = 7.4) after 72 h incubation. Data represent mean ± SD, n = 3 (ESI-6); cellular internalization study in mouse fibroblast (NIH/3T3) cells. (A) Comparative confocal images of NIH/3T3 cells incubated with RhB-labelled NPs (50 μg mL−1) for 3 h at 37 °C. The nuclei of the cells were stained with DAPI (blue). (B) Corresponding quantification of internalized NPs following corrected total cell fluorescence (CTCF) using ImageJ. The cells were imaged under a 20× lens and the scale bar is 20 μm. Unpaired t-test, p-value 0.2193, p-value summary non-significant (n.s.) (ESI-7); comparative biodistribution of LyP-1 NPs in various organs after 48 h post-injection imaged for DiR dye in LI-COR® whole-body imaging system (ESI-8); quantification of tumor accumulation of Control NP-DiR and LyP-1 NP-DiR after 48 h post-injection. P value = 0.0169, * student F-test. Data represent mean ± SD, n = 3 (ESI-9). See DOI: 10.1039/d0ma00203h |
| This journal is © The Royal Society of Chemistry 2020 |