
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling interface properties for enhanced photocatalytic performance: a case-study of CuO/TiO2 nanobelts†
Huaqiang
Zhuang
 ab,
Siying
Zhang
a,
Manru
Lin
a,
Liqin
Lin
a,
Zhenping
Cai
*c and
Wentao
Xu
*a
ab,
Siying
Zhang
a,
Manru
Lin
a,
Liqin
Lin
a,
Zhenping
Cai
*c and
Wentao
Xu
*a
aCollege of Chemical Engineering and Materials Science, Quanzhou Normal University, Quanzhou 362000, P. R. China. E-mail: xuwentao93@aliyun.com
bKey Laboratory of Green Energy and Environment Catalysis, Ningde Normal University, Fujian Province, Ningde, 352100, P. R. China
cDepartment of Chemical Engineering, Norwegian University of Science and Technology, N-7491, Trondheim, Norway. E-mail: zhenping.cai@ntnu.no
First published on 1st June 2020
Abstract
TiO2 nanobelts with CuO modification were designed and fabricated via a facile strategy. The photocatalytic degradation of methyl orange (MO) is selected as a model reaction to investigate the photocatalytic performance of all as-prepared CuO/TiO2 nanobelts under full-spectrum light (300–2500 nm) irradiation. The optimized mass fraction of CuO is 0.1% for CuO/TiO2 composite nanobelts, namely CT-0.1 sample, which can almost completely degrade MO pollutant in 50 min. The as-obtained CuO/TiO2 composites are systematically investigated by a variety of physical and chemical characterizations. Therein, it can be obtained that superoxide radicals (˙O2−) and hydroxyl radicals (˙OH) are the main active species in this photocatalytic system. The photoelectrochemical measurement clearly demonstrates that the enhanced photocatalytic activity can be attributed to the efficient separation and transfer of photo-generated electron–hole pairs, and lower overpotential for CuO/TiO2 nanobelts. This work provides a prototype to study the photocatalytic oxidation process, which contributes to the design and construction of highly-efficient composite photocatalysts.
1 Introduction
Semiconductor-based photocatalysis has been verified to be an effective approach to resolve environmental and energy issues.1–3 Among many photocatalysts, TiO2 has been considered to be one of the most promising candidates, owing to its excellent catalytic activity, stability, nontoxicity and low cost.4–6 Nevertheless, the wide band gap and low separation and transfer efficiency of photo-generated charge carriers still limit the solar conversion efficiency and large-scale application of TiO2.7–10 As we all know, the photocatalytic process mainly involves the following three steps.11,12 The initial step is the generation of photo-excited hole–electron pairs in the photocatalyst under solar light irradiation. Subsequently, the photo-generated electron–hole pairs will be separated and migrate on the surface of the semiconductor photocatalyst. Finally, electrons and holes on the surface will participate in reduction and oxidation reactions to accomplish the solar energy conversion process. Thereinto, the separation of photo-generated electron–hole pairs is one of the important steps for achieving good photocatalytic performance.13–16 Therefore, a large number of efforts have been devoted to enhancing the separation efficiency of photo-generated electron–hole pairs to improve the photocatalytic activity.A variety of strategies, such as metal or nonmetal doping, semiconductor compositing and cocatalyst modifying, have been developed to improve the separation and transfer efficiency of photo-generated charge carriers.17–20 Jing et al.21 reported that BiOCl was modified by RGO and phosphate groups to improve the photogenerated charge separation and photochemical stability. Similarly, some noble metals are normally regarded as active sites to enhance the separation and transfer efficiency of photo-generated electron–hole pairs. The ultrafast charge separation and long-lived charge separated state of CdS nanorods were directly demonstrated by the introduction of Pt nanoparticles.22 In addition, some transition metal oxides, such as CuO, NiO and Co3O4, can also act as cocatalysts to improve photocatalytic performance. For example, Reddy et al.23 reported that CuO quantum dots as cocatalysts could boost the photocatalytic hydrogen production of TiO2. The CuO nanoparticles were introduced on flower-like ZnO to construct a 0D–3D CuO/ZnO heterojunction, which enhanced the separation efficiency of the photogenerated electron–hole pairs resulting in enhanced photocatalytic performance.24 Some previous reports25–28 also demonstrate that NiO and Co3O4 can act as cocatalysts to improve the photocatalytic activity. These research studies suggest that transition metal oxides have considerable potential to replace noble metals and can be applied in the photocatalytic field.
CuO is a promising candidate to act as a cocatalyst, attributed to its low cost, narrow band gap and nontoxicity.29–31 Furthermore, CuO has the ability to enhance the utilization efficiency of solar energy and accelerate the separation of photo-excited charge carriers after surface modification on TiO2. Wang et al.32 reported that CuO quantum dots incorporated into TiO2 nanosheets could obviously improve photocatalytic water splitting performance. Lu et al. directly33 revealed the role of CuO in the plasmonic photocatalysis of Ag/AgCl/TiO2, which could enhance the photocatalytic activity for degradation of methyl orange and phenol. It can be seen that the electronic band structure of CuO is very well matched with that of TiO2, which is beneficial for the separation and transfer of photo-generated electron–hole pairs. Although lots of studies focus on the CuO/TiO2 photocatalyst, the enhanced nature of CuO nanoparticles incorporated on TiO2 nanobelts is still lacking in systematic research.
In this work, we show a facile method to prepare CuO/TiO2 nanobelts for the degradation of methyl orange (MO). The composition and structure of CuO/TiO2 nanobelts with different CuO content were systematically characterized by XRD, SEM and DRS measurements. The CT-0.1 sample displays the best photocatalytic performance compared to the others for the degradation of methyl orange (MO). Furthermore, the active species trapping experiments demonstrate that superoxide radicals (˙O2−) and hydroxyl radicals (˙OH) are the main active species in this photocatalytic system. The enhanced photocatalytic activity can be attributed to the efficient separation and transfer of photo-generated electron–hole pairs. In addition, the as-prepared CT-0.1 sample shows a lower overpotential, which is beneficial for photocatalytic oxidation processes.
2 Experimental
2.1 Catalyst preparation
2.2 Characterization of samples
XRD plots were measured on a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). UV-vis DRS spectra were recorded on a Varian Cary 500 Scan UV-vis-NIR spectrometer with BaSO4 as the reference. The Hitachi New Generation SU8010 field emission scanning electron microscope was used to obtain SEM images. The Brunauer–Emmett–Teller (BET) specific surface areas were determined by N2 adsorption at 77 K on a Micromeritics ASAP 2020. XPS spectra were recorded on a VGESCALAB 250 XPS system with a monochromatized Al Kα X-ray source (15 kV 200 W 500 μm pass energy = 20 eV). All binding energies were referenced to the C 1s peak at 284.6 eV of surface adventitious carbon. HRTEM images were obtained on the JEM 2100F instrument at an accelerating voltage of 200 kV. Photocurrent was measured using the conventional three-electrode electrochemical cell with a working electrode, a platinum foil counter electrode and an Ag/AgCl electrode as the reference electrode. The working electrode was fabricated on the FTO glass: 10 mg of the sample was mixed with 0.5 mL ethanol under continuous stirring for 3 h to obtain the slurry. Then, 40 μL of the slurry was spread onto FTO glass the sides of which had previously been protected by Scotch tape, and the electrode was dried at room temperature for 24 h. In the experiment, the three electrodes were immersed in a sodium sulfate electrolyte solution (0.5 M) and the working electrode was irradiated with full-spectrum light. Light/dark short circuit photocurrent response and electrochemical impedance spectroscopy (EIS) experiments were performed using an electrochemical workstation (CHI 660E).2.3 Photocatalytic activity measurement
The photocatalytic performance of the as-prepared samples was evaluated by the degradation of MO in aqueous solution. A 300 W PLS-SXE300 Xe lamp with full-spectrum light (300–2500 nm) was used as the light source. Typically, 80 mg of the photocatalyst was added to 80 mL of 10 mg L−1 MO aqueous solution in a container. Prior to irradiation, the suspensions were magnetically stirred in the dark for 60 min to ensure the establishment of an adsorption/desorption equilibrium between the photocatalyst and MO. At given irradiation time intervals, 4 mL of suspension was collected and centrifuged to remove the photocatalyst particles, and then the residual MO solution was analyzed by monitoring variations at the wavelength of maximal absorption in the UV-vis spectra.2.4 Active species trapping experiments
In order to probe the active species generated in the photocatalytic reaction process, isopropanol (IPA), Na2C2O4 and p-benzoquinone (BQ) as the scavengers of hydroxyl radicals (˙OH), holes (h+) and superoxide radicals (˙O2−) were, respectively, added to the MO solution to study the influence of different active species. The concentrations of IPA, Na2C2O4 and BQ were 1 mmol L−1, 1 mmol L−1 and 1 mmol L−1, respectively. The photocatalytic degradation reaction was similar to the above test condition except for adding scavengers in the MO solution.3 Results and discussion
The as-prepared samples were investigated by scanning electron microscopy (SEM) so as to characterize their morphologies before and after CuO surface-modification of TiO2 nanobelts. Fig. 1 shows the SEM images of the TiO2, CT-0.1, CT-1 and CT-3 samples. For the pure TiO2 samples, the nanobelts are 30–60 nm in thickness, 100–300 nm in width, and the lengths range up to a few micrometers. The CT-0.1, CT-1 and CT-3 samples also display similar morphologies, which can be attributed to the low CuO loading amount. In order to verify the existence and particle size distribution of CuO, the CT-3 sample was further characterized by transmission electron microscopy (TEM). Fig. 2(a) and (b) show the TEM images of the CT-3 sample. It can be seen that the surface of TiO2 contains some small particles, and they are uniformly dispersed on the surface. These particles are CuO nanoparticles, which can be verified from Fig. 2(c). The distinct lattice fringes with a distance of about 0.35 nm can be observed, which is assigned to the (101) crystal plane of anatase TiO2.4 In addition, the lattice fringes of d-spacings of about 0.23 nm can be obtained, which is consistent with the (111) crystal plane of CuO.24Fig. 2(d) shows the particle size distribution of CuO on the TiO2 nanobelts. It clearly shows that most of the CuO particles are less than 10 nanometers. These results suggest that CuO particles can be uniformly loaded on the surface of TiO2 nanobelts via this facile strategy. | ||
| Fig. 1 SEM images of (a) TiO2, (b) CT-0.1, (c) CT-1 and (d) CT-3 samples. | ||
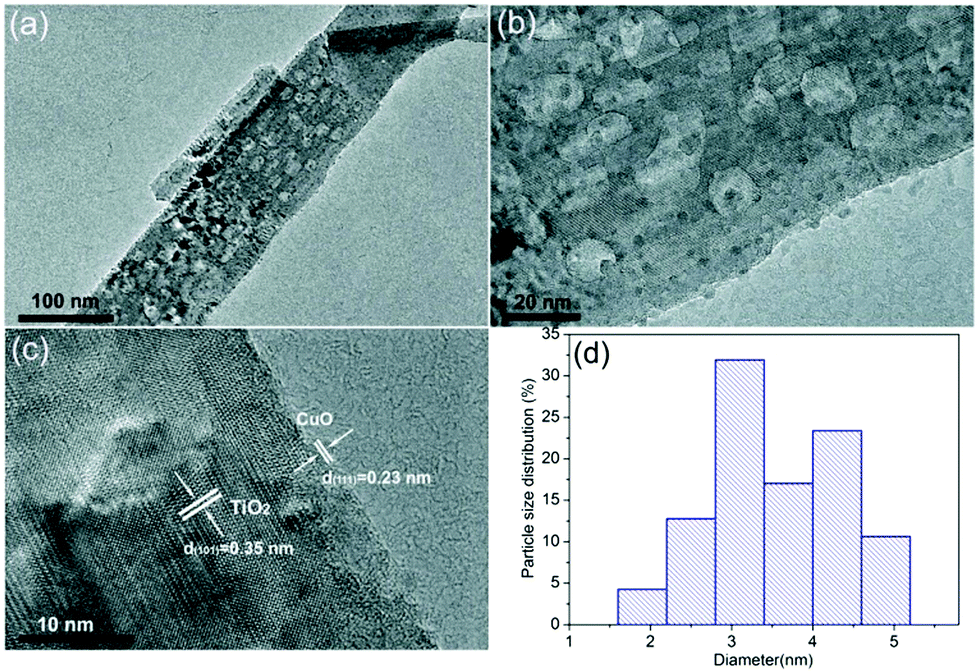 | ||
| Fig. 2 (a and b) TEM images of the CT-3 sample. (c) HRTEM image of the CT-3 sample. (d) Particle size distribution of CuO on the surface of TiO2 nanobelts. | ||
In addition, all of the as-prepared samples were further characterized by X-ray diffraction measurements (XRD), as presented in Fig. 3. Obviously, the nude TiO2 nanobelts display the characteristic diffraction peaks of TiO2.34 Similarly, the CuO/TiO2 nanobelts with different CuO contents also exhibit the characteristic diffraction peaks of TiO2. However, new diffraction peaks appear at 35.5° and 38.7°, which are assigned to the (002) and (111) crystal planes of cubic CuO (JCPDS 45-0937),35 as shown in Fig. 2(b). The above results suggest that the CuO/TiO2 nanobelts with different CuO contents are successfully prepared. To investigate the influence of the specific surface area after the introduction of CuO, TiO2 and CT-0.1 samples are further characterized by N2 adsorption–desorption measurements, as shown in Table S1 (ESI†). Obviously, the measured specific surface areas are 54.4 and 59.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) m2g−1 for TiO2 and CT-0.1 sample, respectively. It can be concluded that the introduction of CuO doesn’t obviously enhance the specific surface area of TiO2.
m2g−1 for TiO2 and CT-0.1 sample, respectively. It can be concluded that the introduction of CuO doesn’t obviously enhance the specific surface area of TiO2.
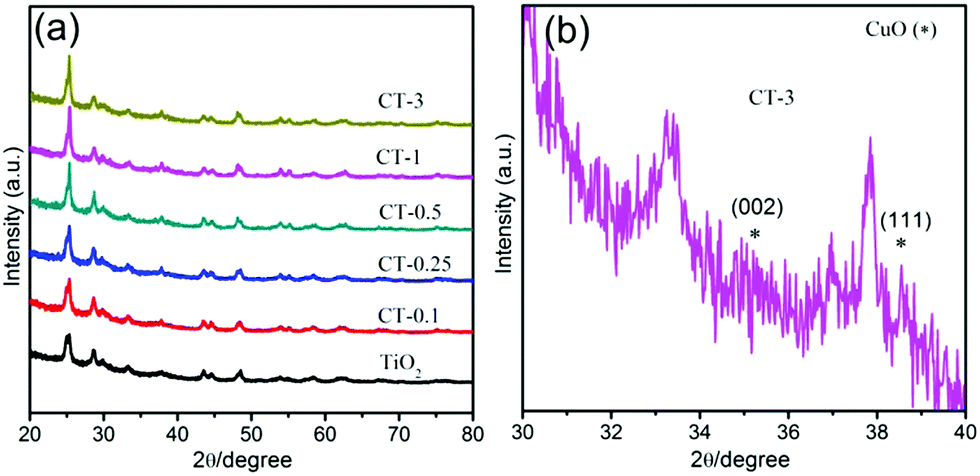 | ||
| Fig. 3 (a) XRD patterns of TiO2 and CuO/TiO2 nanobelts. (b) A larger version of the CT-3 sample. | ||
XPS spectroscopy was carried out to investigate the chemical status of Ti, O and Cu elements in CuO/TiO2 nanobelts, as displayed in Fig. 4. Fig. 4(a) presents the XPS survey spectra of the CT-3 sample, indicating that the CT-3 sample contains Ti, O and Cu elements. The high-resolution XPS spectra of Ti 2p and O 1s are shown in Fig. 4(b) and (c), respectively. It can be concluded that the two peaks at 458.6 eV and 464.3 eV are assigned to Ti 2p3/2 and Ti 2p1/2 of Ti4+ in TiO2, respectively.4 Similarly, the binding energy of the O 1s peak at 529.9 eV can be assigned to lattice oxygen, and the other peak at 530.6 eV is identified with surface hydroxyls in CuO/TiO2.36 Importantly, there are two peaks located at approximately 932.5 eV and 952.1 eV, corresponding to Cu 2p3/2 and Cu 2p1/2, respectively. In addition, the two satellite peaks for Cu2+ can also be clearly observed in Fig. 4(d).37,38 These results further confirm the existence of CuO in the CT-3 sample.
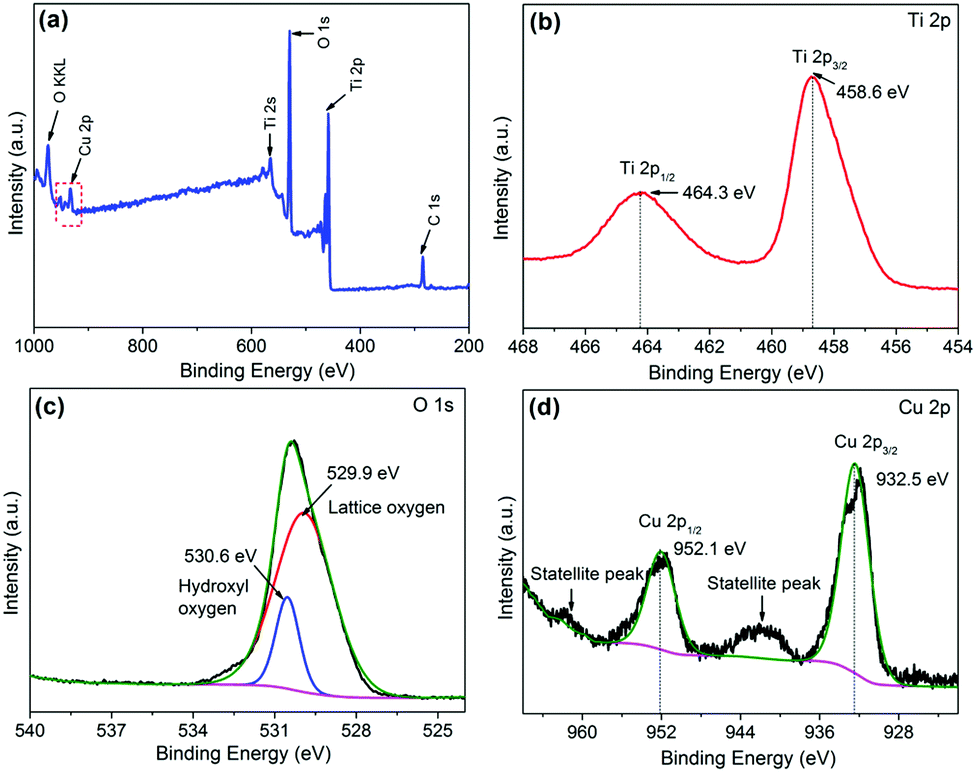 | ||
| Fig. 4 (a) Typical XPS survey spectra of the CT-3 sample. The high resolution XPS spectra of Ti 2p, O 1s and Cu 2p (b, c and d, respectively) for the CT-3 sample. | ||
The optical absorption properties of TiO2 and CuO/TiO2 nanobelts were characterized in detail using ultraviolet-visible diffuse reflectance absorption spectra (DRS), as shown in Fig. 5. It can be clearly observed that the absorption band of TiO2 nanosheets is below 400 nm assigned to the intrinsic bandgap absorption of TiO2.5 As expected, the photo-absorption efficiency of CuO/TiO2 nanobelts is slightly enhanced in the visible light region after coupling with CuO nanoparticles, which is attributed to the narrower bandgap of CuO (ca. 1.7 eV).39 Obviously, the visible light absorption shows a considerable improvement with the increasing CuO content in CuO/TiO2 nanobelts. The enhanced optical adsorption of CuO/TiO2 nanobelts is mainly ascribed to the d–d transition of Cu2+.40 It can be concluded that the special phenomenon is another piece of solid evidence to verify the existence of CuO species in the CuO/TiO2 nanobelts.
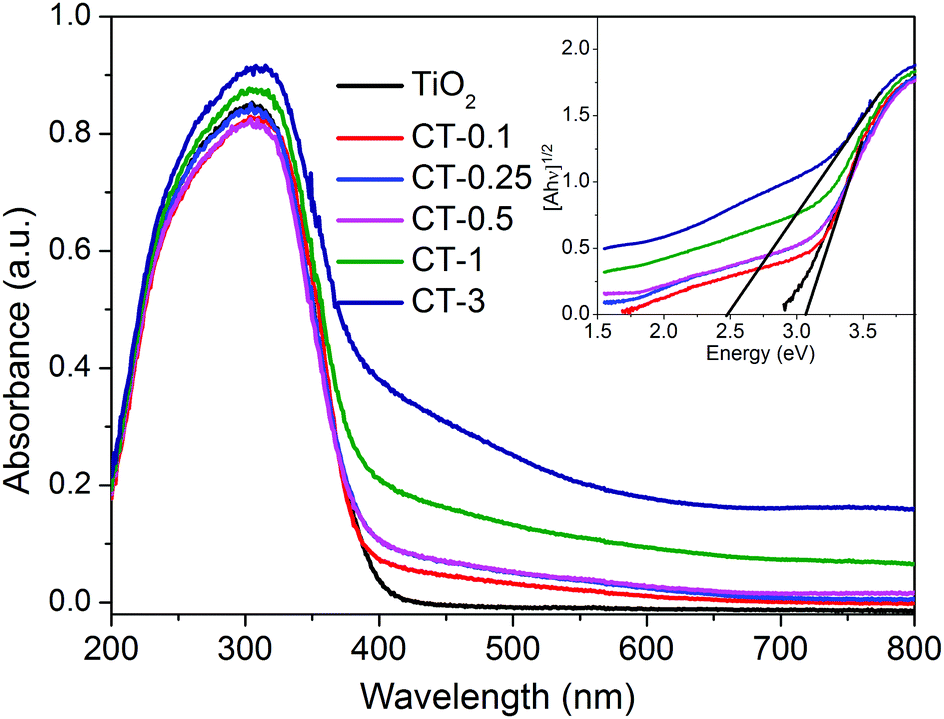 | ||
| Fig. 5 UV-vis DRS spectra of TiO2 and CuO/TiO2 nanobelts. The inset shows the plots of [F(R)hν]1/2versus photon energy (hν). | ||
The photocatalytic performance of TiO2 and CuO/TiO2 samples for the degradation of MO was investigated under full-spectrum light irradiation, as shown in Fig. 6. Obviously, the blank experiment shows a negligible photocatalytic activity, indicating that only MO solution can’t be degraded without a photocatalyst under full-spectrum light irradiation. After coupling with CuO nanoparticles, the CuO/TiO2 nanobelts exhibit an evident improvement with the increase of CuO content. The CT-0.1 sample displays the best photocatalytic performance compared with that of TiO2 and the other CuO/TiO2 nanobelts, and its degradation percent of MO reaches up to 99.2% in 50 min. Generally, the kinetics of the degradation reaction is investigated by applying the Langmuir–Hinshelwood (L–H) model.41 The apparent reaction rate constants (k) are displayed in Table S1 (ESI†). Therein, the reaction rate constants for different samples were presented in the following order: CT-0.1 > CT-0.25 > TiO2 > CT-0.5 > CT-1 > CT-3. It can be concluded that introducing a certain amount of CuO nanoparticles is beneficial for the photocatalytic oxidation process. However, the photocatalytic activity of CuO/TiO2 nanobelts shows a significant decrease, when the CuO content is more than 0.25%. The reduced photocatalytic activity may be attributed to the excessive amount of CuO nanoparticles that will act as recombination centers for photo-generated electron–hole pairs. Furthermore, the stability of CT-0.1 sample was fully investigated through a cycling experiment under full-spectrum light irradiation, as indicated in Fig. S1 (ESI†). After four recycling runs, the CT-0.1 sample can still retain degradation activity, and no apparent loss can be found. This clearly indicates that the CuO/TiO2 nanobelts have excellent stability.
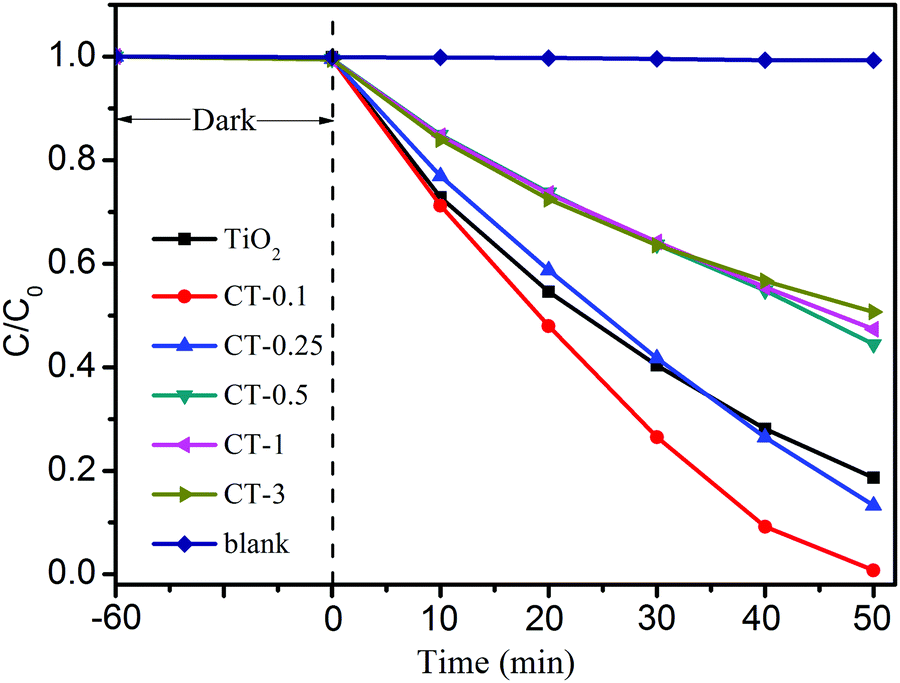 | ||
| Fig. 6 Photocatalytic activity of MO degradation for TiO2 and CuO/TiO2 nanobelts under full-spectrum light irradiation. | ||
In order to explore the possible reaction mechanism, the active species trapping experiments for the CT-0.1 sample were further investigated by adding some scavengers during the photocatalytic reaction process, as shown in Fig. 7. It can be easily seen that no obvious inhibiting action can be found using 1 mmol L−1 Na2C2O4 as the scavenger to quench the h+,42 whereas the photocatalytic performance of the CT-0.1 sample has been weakening to some extent. Therein, it can be obtained that h+ is not the major active species in the system, but it can influence the photocatalytic activity of the CT-0.1 sample. Interestingly, when isopropanol (IPA) or p-benzoquinone (BQ) was added to the MO solution during the photocatalytic reaction process,43 the photocatalytic activity of the CT-0.1 sample was evidently inhibited. This suggests that both superoxide radicals (˙O2−) and hydroxyl radicals (˙OH) are main active species, and play a decisive role during the photocatalytic degradation of MO.
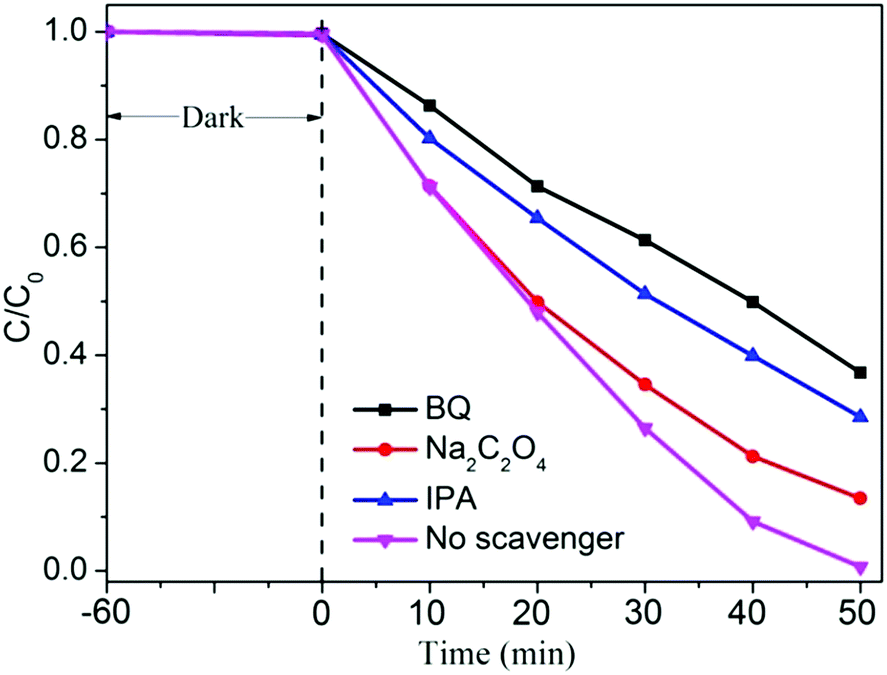 | ||
| Fig. 7 Active species trapping experiment of the CT-0.1 sample in the photocatalytic reaction process. | ||
The photoelectrochemical (PEC) measurement was investigated to fully elucidate the role of the CuO cocatalyst under full-spectrum light irradiation. The photocurrent response can directly reflect the generation, separation and transfer of photo-generated electron–hole pairs, which is an effective approach to monitor the interface reaction of the photocatalyst.43,44Fig. 8 shows the periodic on/off transient photocurrent response of TiO2 and CuO/TiO2 samples. Evidently, the CT-0.1 sample displays a higher photocurrent density than that of the nude TiO2 and the other CuO/TiO2 nanobelts, suggesting that the CT-0.1 sample possesses a much higher separation and transfer efficiency and longer lifetime of the photo-generated charge carriers. The photocurrent density follows the order of CT-0.1 > CT-0.25 > TiO2 > CT-0.5 > CT-1 > CT-3. In order to further illustrate the corresponding charge transfer mechanism, electrochemical impedance spectroscopy (EIS) was carried out, as presented in Fig. 9. It is a powerful method to obtain more detailed information about the charge transport, in which the arc radius can reflect the resistance for charge transfer at the electrode/electrolyte interface.45 Apparently, the CT-0.1 sample shows a smaller arc radius than that of TiO2 and the other CuO/TiO2 nanobelts, suggesting enhanced charge carrier separation and increased charge carrier mobility in line with the photocurrent measurements. Herein, the results of the photoelectrochemical measurements demonstrate that a small amount of CuO has an outstanding ability to enhance the separation and transfer of photo-generated electron–hole pairs for the composite interface. In addition, the applied potential bias-dependent photocurrent density of the TiO2, CT-0.1 and CT-3 samples was implemented under full-spectrum light irradiation or dark conditions, as shown in Fig. S2 (ESI†). It can be concluded that the CT-0.1 sample displays a smaller overpotential for the oxygen production reaction in comparison with the TiO2 and CT-3 samples, indicating that the lower overpotential of CT-0.1 sample may be more beneficial for the photocatalytic oxidation process. As mentioned above, it can be concluded that the CT-0.1 sample having excellent photocatalytic performance is mainly ascribed to the efficient separation and transfer of photogenerated electron–hole pairs, and low overpotential for the photocatalytic oxidation process.
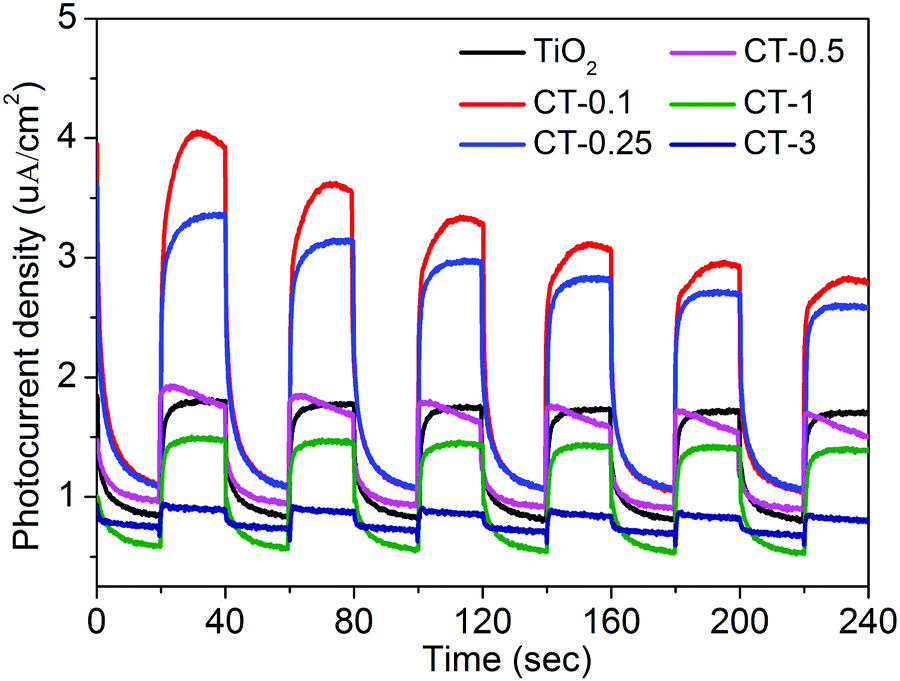 | ||
| Fig. 8 Transient photocurrent responses of TiO2 and CuO/TiO2 nanobelts at 0 V vs. Ag/AgCl under full-spectrum light irradiation. | ||
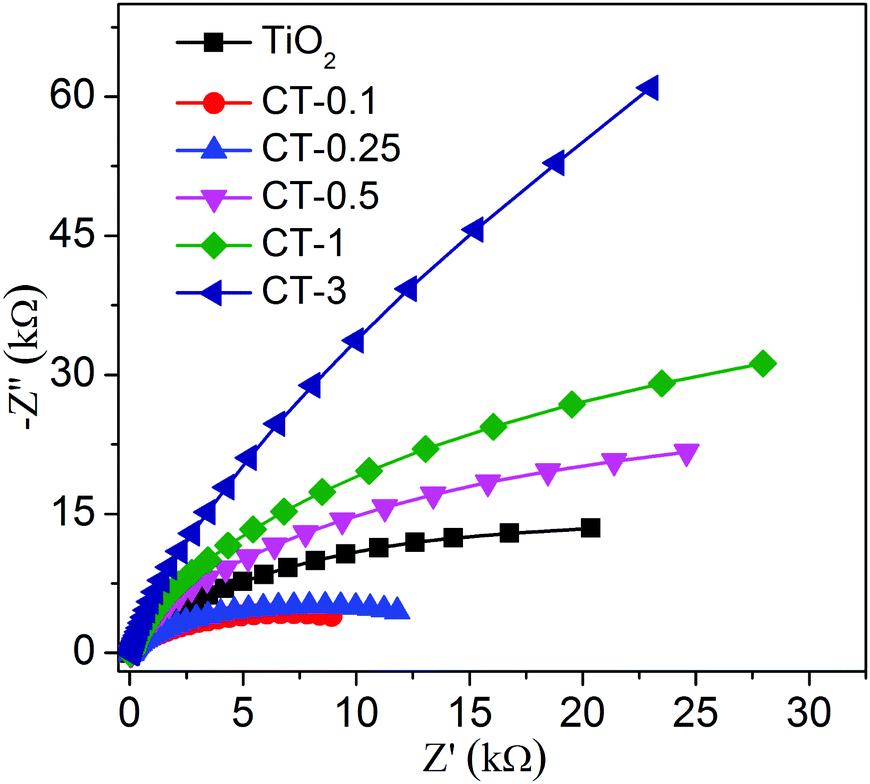 | ||
| Fig. 9 Electrochemical impedance spectroscopy (EIS) was performed using TiO2 and CuO/TiO2 nanobelts under full-spectrum light irradiation. | ||
In consideration of the above results, the possible photocatalytic reaction mechanism is presented in Fig. 10. Because the work function of TiO2 (4.7 eV) is smaller than that of CuO (5.3 eV),40 a built-in electric field will be formed between TiO2 and CuO, and the electric fields point out from CuO to TiO2 before light irradiation.46 When the TiO2 nanobelts were irradiated by full-spectrum light, the electrons in the conduction band of TiO2 will be transferred to the conduction band of CuO due to the existence of the built-in electric field. Therein, the electrons in the conduction band of CuO will reduce O2 to superoxide radicals (˙O2−), and the holes in the valence band of TiO2 will oxidize H2O into hydroxyl radicals (˙OH) to participate in the photocatalytic degradation of MO.
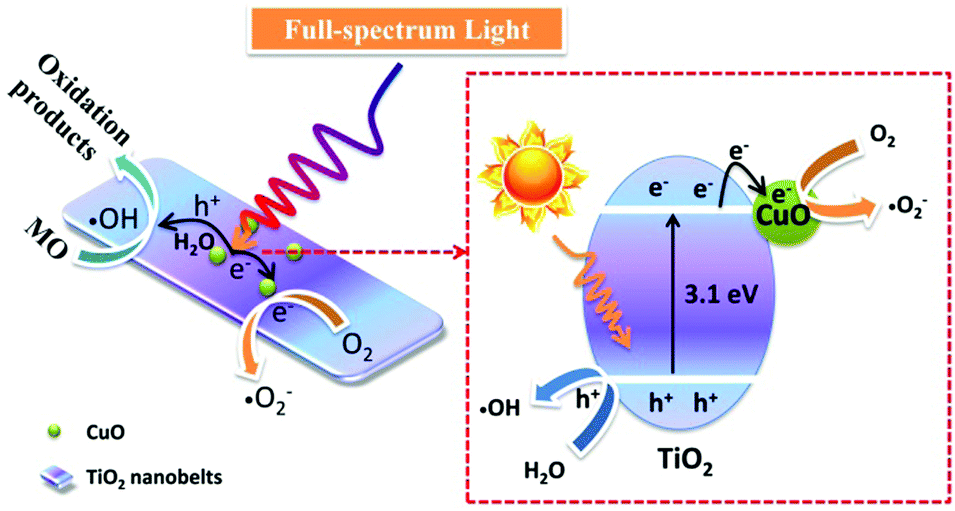 | ||
| Fig. 10 Proposed mechanism for the enhanced photocatalytic activity for CuO/TiO2 nanobelts. | ||
4 Conclusions
In this work, CuO/TiO2 nanobelts were designed and fabricated to study the photocatalytic oxidation process via a facile strategy. Therein, the CT-0.1 sample displays the best photocatalytic performance for the degradation of MO compared to that of nude TiO2 and the other CuO/TiO2 nanobelts. The enhanced photocatalytic activity is mainly attributed to the introduction of proper CuO nanoparticles. It can obviously enhance the optical absorption efficiency of TiO2 nanobelts. According to the active species trapping experiments, it can be concluded that superoxide radicals (˙O2−) and hydroxyl radicals (˙OH) are the main active species in this photocatalytic system. In addition, the photoelectrochemical measurement demonstrates that the efficient separation and transfer of photo-generated electron–hole pairs and low overpotential of the CuO/TiO2 nanobelts are beneficial for the photocatalytic oxidation process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Project of Education Department of Fujian Province (JAT170489), the Quanzhou Science and Technology Project (2017Z031), and the Open Project Program of Provincial Key Laboratory of Green Energy and Environment Catalysis (Grant No. FJ-GEEC201907), Ningde Normal University.Notes and references
- L. Wang, Z. Xiao, Y. Liu, S. Cao, Z. Ma and L. Piao, Sci. China Mater., 2020, 63, 758–768 CrossRef CAS.
- H. Zhuang, W. Xu, L. Lin, M. Huang, M. Xu, S. Chen and Z. Cai, J. Mater. Sci. Technol., 2019, 35, 2312–2318 CrossRef.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Energy Environ. Sci., 2011, 4, 2922–2929 RSC.
- H. Zhuang, W. Chen, W. Xu and X. Liu, Int. J. Energy Res., 2020, 44, 3224–3230 CrossRef CAS.
- H. Zhuang, Y. Zhang, Z. Chu, J. Long, X. An, H. Zhang, H. Lin, Z. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2016, 18, 9636–9644 RSC.
- S. Wu, H. Hu, Y. Lin, J. Zhang and Y. H. Hu, Chem. Eng. J., 2020, 382, 122842 CrossRef CAS.
- C. Xu, F. Yang, B. Deng, Y. Zhuang, D. Li, B. Liu, W. Yang and Y. Li, J. Catal., 2020, 383, 1–12 CrossRef CAS.
- K. Maver, I. Arčon, M. Fanetti, S. Emin, M. Valant and U. L. Štangar, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.01.045.
- J. Xue, F. Song, X. Dong, X.-W. Yin, Y. Liu, J.-M. Wu, C. Wang, X.-L. Wang and Y.-Z. Wang, ACS Sustainable Chem. Eng., 2019, 7, 1973–1979 CrossRef CAS.
- F. Yang, M. Liu, X. Chen, Z. Xu and H. Zhao, Sol. RRL, 2018, 2, 1800215 CrossRef.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- P. Zhang, T. Wang, X. Chang and J. Gong, Acc. Chem. Res., 2016, 49, 911–921 CrossRef CAS PubMed.
- Z.-K. Tang, W.-J. Yin, Z. Le, B. Wen, D.-Y. Zhang, L.-M. Liu and W.-M. Lau, Sci. Rep., 2016, 6, 32764 CrossRef CAS.
- J. Hu, P. Zhang, W. An, L. Liu, Y. Liang and W. Cui, Appl. Catal., B, 2019, 245, 130–142 CrossRef CAS.
- F. M. Pesci, G. Wang, D. R. Klug, Y. Li and A. J. Cowan, J. Phys. Chem. C, 2013, 117, 25837–25844 CrossRef CAS PubMed.
- S. Li, Z. Zhao, Y. Huang, J. Di, Y. Jia and H. Zheng, J. Mater. Chem. A, 2015, 3, 5467–5473 RSC.
- M. Wang, J. Han, Y. Hu and R. Guo, RSC Adv., 2017, 7, 15513–15520 RSC.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- J. F. Guayaquil-Sosa, B. Serrano-Rosales, P. J. Valadés-Pelayo and H. de Lasa, Appl. Catal., B, 2017, 211, 337–348 CrossRef CAS.
- L. Guo, C. Zhong, J. Cao, Y. Hao, M. Lei, K. Bi, Q. Sun and Z. L. Wang, Nano Energy, 2019, 62, 513–520 CrossRef CAS.
- Z. Li, Y. Qu, K. Hu, M. Humayun, S. Chen and L. Jing, Appl. Catal., B, 2017, 203, 355–362 CrossRef CAS.
- K. Wu, H. Zhu, Z. Liu, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- N. L. Reddy, S. Emin, V. D. Kumari and S. Muthukonda Venkatakrishnan, Ind. Eng. Chem. Res., 2018, 57, 568–577 CrossRef CAS.
- L. Zhu, H. Li, Z. Liu, P. Xia, Y. Xie and D. Xiong, J. Phys. Chem. C, 2018, 122, 9531–9539 CrossRef CAS.
- L. Yang, H. Li, Y. Yu and H. Yu, Catal. Commun., 2018, 110, 51–54 CrossRef CAS.
- C.-C. Hu and H. Teng, J. Catal., 2010, 272, 1–8 CrossRef CAS.
- P. Zhang, T. Wang, X. Chang, L. Zhang and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 5851–5855 CrossRef CAS PubMed.
- H. Shen, D. Ni, P. Niu, Y. Zhou, T. Zhai and Y. Ma, Int. J. Hydrogen Energy, 2017, 42, 30559–30568 CrossRef CAS.
- B. Çinar, I. Ker
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) moğlu, B. Tönbül, A. Demrbüken, S. Dursun, I. Cihan Kaya, V. Kalem and H. Akyildiz, Mater. Sci. Semicond. Process., 2020, 109, 104919 CrossRef.
moğlu, B. Tönbül, A. Demrbüken, S. Dursun, I. Cihan Kaya, V. Kalem and H. Akyildiz, Mater. Sci. Semicond. Process., 2020, 109, 104919 CrossRef. - Y. Li and K. Luo, RSC Adv., 2019, 9, 8350–8354 RSC.
- K. Yao, S. Liu, Y.-Y. Dong, B. Wang, J. Bian and M.-G. Ma, Mater. Des., 2016, 90, 129–136 CrossRef CAS.
- Y. Wang, M. Zhou, Y. He, Z. Zhou and Z. Sun, J. Alloys Compd., 2020, 813, 152184 CrossRef CAS.
- Z. H. Shah, J. Wang, Y. Ge, C. Wang, W. Mao, S. Zhang and R. Lu, J. Mater. Chem. A, 2015, 3, 3568–3575 RSC.
- G. Yang, H. Ding, J. Feng, Q. Hao, S. Sun, W. Ao and D. Chen, Sci. Rep., 2017, 7, 14594 CrossRef PubMed.
- X. Zhang, L. Wang, C. Liu, Y. Ding, S. Zhang, Y. Zeng, Y. Liu and S. Luo, J. Hazard. Mater., 2016, 313, 244–252 CrossRef CAS PubMed.
- J. Yuan, J.-J. Zhang, M.-P. Yang, W.-J. Meng, H. Wang and J.-X. Lu, Catalysts, 2018, 8, 171 CrossRef CAS.
- Z. Liu and C. Zhou, Prog. Nat. Sci.: Mater. Int., 2015, 25, 334–341 CrossRef CAS.
- A. A. Dubale, A. G. Tamirat, H.-M. Chen, T. A. Berhe, C.-J. Pan, W.-N. Su and B.-J. Hwang, J. Mater. Chem. A, 2016, 4, 2205–2216 RSC.
- J. Liu, C. Han, X. Yang, G. Gao, Q. Shi, M. Tong, X. Liang and C. Li, J. Catal., 2016, 333, 162–170 CrossRef CAS.
- Q. Shi, G. Ping, X. Wang, H. Xu, J. Li, J. Cui, H. Abroshan, H. Ding and G. Li, J. Mater. Chem. A, 2019, 7, 2253–2260 RSC.
- X.-J. Wen, C.-G. Niu, L. Zhang and G.-M. Zeng, ACS Sustainable Chem. Eng., 2017, 5, 5134–5147 CrossRef CAS.
- W. Wang, Y. Yu, T. An, G. Li, H. Y. Yip, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2012, 46, 4599–4606 CrossRef CAS PubMed.
- H. Zhuang, Z. Cai, W. Xu, M. Huang and X. Liu, New J. Chem., 2019, 43, 17416–17422 RSC.
- H. Zhuang, Z. Cai, W. Xu, X. Zhang, M. Huang and X. Wang, Catal. Commun., 2019, 120, 51–54 CrossRef CAS.
- J. Lin, J. Hu, C. Qiu, H. Huang, L. Chen, Y. Xie, Z. Zhang, H. Lin and X. Wang, Catal. Sci. Technol., 2019, 9, 336–346 RSC.
- X. An, Y. Wang, J. Lin, J. Shen, Z. Zhang and X. Wang, Sci. Bull., 2017, 62, 599–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00172d |
| This journal is © The Royal Society of Chemistry 2020 |