
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-dimensional metallosupramolecular framework design based on coordination crosslinking of helical oligoamide nanorods
Norton G.
West†
a,
Rania S.
Seoudi†
 a,
Anders J.
Barlow
a,
Dongchen
Qi
ab,
Ljiljana
Puskar
c,
Mark P.
Del Borgo
d,
Ketav
Kulkarni
d,
Christopher G.
Adda
a,
Jisheng
Pan
e,
Marie-Isabel
Aguilar
d,
Patrick
Perlmutter
a and
Adam
Mechler
*a
a,
Anders J.
Barlow
a,
Dongchen
Qi
ab,
Ljiljana
Puskar
c,
Mark P.
Del Borgo
d,
Ketav
Kulkarni
d,
Christopher G.
Adda
a,
Jisheng
Pan
e,
Marie-Isabel
Aguilar
d,
Patrick
Perlmutter
a and
Adam
Mechler
*a
aDepartment of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Bundoora, Victoria 3086, Australia. E-mail: a.mechler@latrobe.edu.au
bSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, Queensland 4001, Australia
cMonash Biomedicine Discovery Institute & Department of Biochemistry and Molecular Biology, Monash University, Clayton, Victoria 3800, Australia
dDepartment Locally Sensitive and Time-Resolved Spectroscopy, Helmholtz-Zentrum Berlin für Materialien und Energie, 12498 Berlin, Germany
eInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), Singapore 138634, Singapore
First published on 2nd July 2020
Abstract
Spontaneous formation of nanostructured materials of defined structure and morphology is a crucial milestone toward realizing true bottom-up nanofabrication. Supramolecular recognition offers unparalleled specificity, selectivity and geometric flexibility to design hierarchical nanostructures. However, competition between similar binding motifs and the dynamic nature of the attachment imposes a severe limitation on the complexity of the achievable structures. Here we outline a design based on two distinct binding motifs in a supramolecular fibrous assembly to realize a metallosupramolecular framework (MSF). Controlled geometries were achieved by one-dimensional supramolecular assembly of substituted oligoamide units. The assembly of the monomers yields nanorods of sub-nanometer diameter and lengths in the 100 μm range. Addition of Cu2+ led to the formation of well aligned two-dimensional arrays on mica surface. Vibrational spectroscopy confirmed that the backbone amide moieties are not affected by metal addition. XPS and NEXAFS results suggest that Cu(II) is reduced in the process to a mixture of Cu(I) and Cu(0), likely in an interaction with the amine moiety of the imidazole side chain. Our results indicate that the two dimensional superstructure is based on the formation of polynuclear metal complexes between the oligoamide nanorods, thus the structure is confirmed to be a metallosupramolecular framework.
Bottom-up fabrication of functional nanomaterials relies on the careful design of molecular building blocks to implement self-assembly in a predetermined manner.1,2 Structural complexity can be achieved via supramolecular recognition;3 payloads may be attached to the monomer.4 For programmed self-assembly it is crucial that the monomeric units have, and retain, a well-defined molecular geometry that incorporates specific and selective binding motifs parallel with simple chemical functionalizability. Natural peptides meet these requirements due to their well-defined secondary structures, such as α-helices, and the ease of their synthesis, however natural peptides lack the chemical and metabolic stability necessary for most applications. Oligoamides based on β-amino acids provide the solution with their enhanced chemical and metabolic stability.5,6 β-Amino acids have two methylene moieties, one usually substituted, between the carboxyl and amide moiety. Side chains are substituted in either positions 2 or 3 yielding a structural flexibility that is not possible in α-amino acid based structures.7,8 Consequently, peptide analogues made entirely of β-amino acids can adopt a range of discrete conformations that can be used to design novel nanostructures.9–11 Of particular interest is the 14-helix structure of β3-oligoamides that has a pitch of 3–3.1 amino acids per turn; hence the side chains align along three faces of the helix.5,11 With N-terminal acetylation these helices self-assemble in a head-to-tail fashion yielding extended helical nanorods.12–15 This structure offers the regularity needed to design functional nanostructures of controlled geometry.
The helical head-to-tail assembly of N-acyl β3 oligoamides yields one-dimensional nanomaterials that can form fibrous hierarchical structures via bundling in different ways, controlled by the second order interactions between the core nanorods.14,15 In order to create more regular two- and three-dimensional networks from these nanorods, an independent secondary self-assembly motif is necessary. Metal coordination is widely used to create nanostructured materials of controlled properties, linking multidentate organic ligands into metal organic frameworks (MOFs); the contemporary use of the term refers to exceptionally low-density structures.16 MOFs are usually regarded as infinite structures, whereas metallosupramolecular coordination complexes are discrete structures of complex metal–ligand networks.17 Metallosupramolecular materials are usually based on multi-ligand building units or complex ligands.17,18
While there were attempts to use peptides for the creation of MOF-like materials by metal coordination between carboxyl groups,19 the design of such structures is problematic as the nitrogen of the amide moiety is a good Lewis base and thus it can also coordinate to the metal, leading to loss of structure.20 In case of transition metals, in particular Ni(II) and Cu(II), extensive data exist to show that coordination to peptides frequently proceeds by deprotonation of the amide and direct nitrogen–metal bond formation as opposed to charge solvation to the carbonyl oxygens, although the latter is not entirely unprecedented.21–23 It should be noted that peptide–metal interaction is commonly characterized in the gas phase, and the presence of a solvent may affect the sterically accessible sites. Indeed, metal coordination is used in protein purification, where the imidazole sidechains of a polyhistidine tag coordinate to a metal, typically Ni(II), immobilized in a carrier molecule such as nitrilotriacetic acid. The proteins are cleaved from the tags by imidazole solution, which is only possible if backbone amides do not participate in the coordination. Therefore it is plausible to design metallosupramolecular assemblies based on histidine residues as ligands,22,24 provided that a structural/steric factor protects the peptide backbone.
The aim of this work is to use the well-defined one dimensional supramolecular self-assembly of N-acetylated 14-helical β3-oligoamides12–15 as a core structure in combination with a secondary binding motif based on metal coordination of histidine sidechains. The supramolecular three point hydrogen bonding motif that yields the head-to-tail self-assembly of helical β3-oligoamide units is very strong, hence fibres are observed in water and even in DMSO,25 without a noticeable monomeric/small oligomeric population.26,27 For this reason, traditional characterization methods such as NMR or CD are not feasible to perform, and therefore structure can only be inferred from microscopy imaging and indirect characterization methods.
A further challenge is that the synthesis of β3 histidine is problematic; however in principle it should be possible to combine natural α-amino acids with unnatural β-oligoamides to overcome this limitation. Hybridizing natural α-peptides with unnatural β-oligoamides has been regularly used to enhance the diversity of folding conformations.28–30 Hence, in this work a β3,α hybrid oligoamide Ac-β3A β3V β3S-αHαH-β3Aβ3V β3A (2H – Fig. 1a) was designed to create metallosupramolecular superstructures (metallosupramolecular frameworks, or MSFs). MSF is defined as a periodical an infinite metallosupramolecular system.27
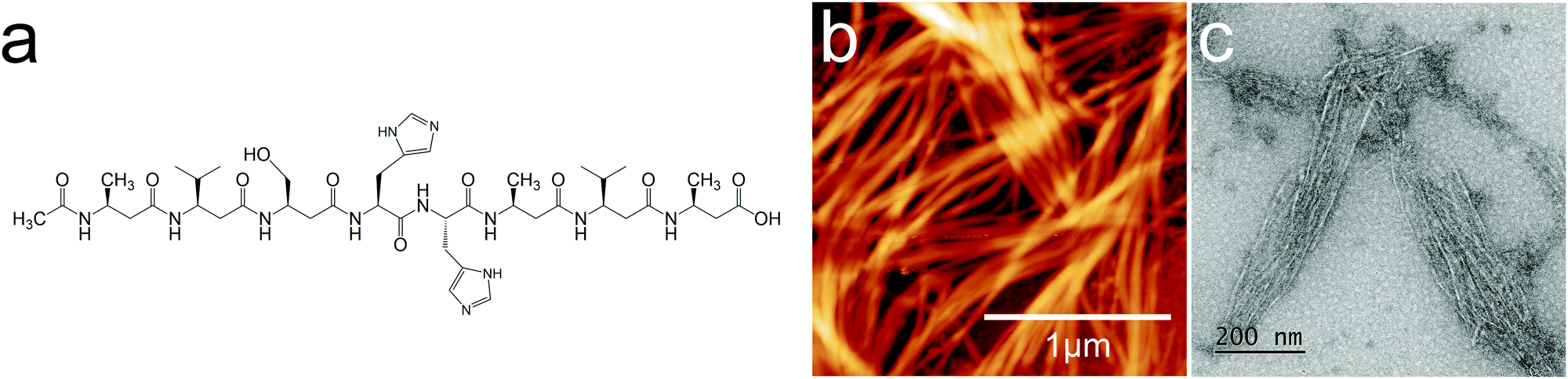 | ||
| Fig. 1 (a) 2H oligoamide structure, (b) AFM image and (c) TEM image of the spontaneously assembled oligoamides without metal. The height scale is 45.1 nm for (b). | ||
Results and discussion
Atomic force microscopy imaging
β3 oligoamides associate via three point hydrogen bonding supramolecular head-to-tail self-assembly motif between the terminal β3 loops. Therefore in the design of the β3-α hybrid oligoamides, the α-histidines were flanked by β3-amino acid loops. With the aim of creating a two-dimensional network, two α histidine residues were introduced as shown in Fig. 1a. The β3 sequences contain apolar β3 alanine and β3 valine residues as well as a β3 serine residue to improve solubility in polar media.31 Atomic force microscopy (AFM) and transmission electron microscopy (TEM) images of the resulting nanomaterials confirm the presence of fibrous structures (Fig. 1b and c). The reported diameter of a single nanorod is ∼0.4 nm;12 however according to both AFM and TEM imaging results, the smallest fibres appear to be ∼7 nm in diameter. This suggests that the observed fibres are bundles of nanorods, most likely assembled via weak π-stacking between imidazole rings of the histidine residues.32,33 These results provide clear evidence that the β3-α hybrid oligoamides assemble via the 3-point hydrogen bonding supramolecular self-assembly motif of the exposed termini of the 14-helix.12,14,15Incubating the oligoamides in 100 μM CuCl2 solution changed the appearance of the deposit. In TEM imaging, the mesh of fibres was replaced by a folded ribbon/sheet structure (Fig. 2a) in which parallel-aligned nanorods are barely discernible. For AFM imaging the sample was deposited on mica surface, which exhibits a higher surface energy than the carbon gossamer used for TEM, and therefore interacts with the fibres more strongly. In this case the incubation with Cu(II) yielded a single continuous layer (Fig. 2b) which was homogeneous over tens of micrometers, albeit with a few defects. Zooming in to one of the defect sites (Fig. 2c) reveals a corrugated pattern with a modulation depth of 0.1–0.2 nm. A single, looped fibre is also visible on the surface. A line profile across the defect site (Fig. 2d) shows that the thickness of the layer is ∼0.8 nm. The corrugation exhibits an irregular repeat distance of 10–20 nm implying a wave pattern in the 2D sheet formed by closely crosslinked nanorods, and not by the alignment of individual nanorods that, according to published crystallography results, should have a diameter of 0.4–0.5 nm.12 The 0.8 nm thickness of the structure is consistent with two layers of nanorods.
 | ||
| Fig. 2 Self-assembled morphology of the 2H oligoamides in 100 μM CuCl2 solution (a), TEM image; (b and c) AFM images; (d) height profile across line (1) in panel (c). Note that 2H forms a tightly packed two-dimensional array with very few defects. The thickness of the layer is ∼0.8 nm as per (d). | ||
The regularity of the structure suggests that it is formed by coordination crosslinking of the nanorods without affecting the supramolecular self-assembly, that is, via the histidine (or another) side chain. Out of the side chains of the oligoamide residues, alanine and valine are both non-coordinating, while the –OH of the serine is a weak ligand only above pH 8–9.34 In this work, the solution was not buffered and hence the pH was defined by the dissociation equilibrium of the C-terminal carboxylic acid. Accordingly, the serine side chain is not expected to participate in the metal coordination. Thus, only the histidine side chains, and potentially the terminal carboxyl moieties provide suitable ligands. It was previously suggested that multiple histidine residues may form macrochelates through complexation of two to four imidazole rings from different peptides to the same metal ion.35 However it was also reported that the amide groups of the backbone are easily deprotonated during the metal coordination of monomeric, unstructured peptides.21 Therefore to confirm metal coordination to the imidazole rings, spectroscopic experiments were performed.
Vibrational spectroscopy
Fig. 3 shows a portion of representative IR absorbance spectra in the mid-infrared region before and after addition of the metal. All spectra have a prominent main peak around 1610–1695 cm−1 which is associated with characteristic backbone amide I vibrations, while the second largest peak at 1480–1575 cm−1 is associated with the amide II vibrations.36 Amide III vibrations appear as several weak peaks in the 1400–1200 cm−1 range; this band tends to be complex for folded polypeptides due to the effect of hydrogen bonding and side chain coupling.37 Cu(II) coordination to amide moieties would have a profound effect on these vibrational bands, especially when in the low spin singlet iminol coordinated state (i.e., when coordination involves deprotonation of the amides), for which the amide II peak is not observed.23,38 Therefore the presence of the main amide bands throughout the coordination process and without appreciable shift in vibrational energies indicates that the backbone amide moieties are not involved in the metal coordination.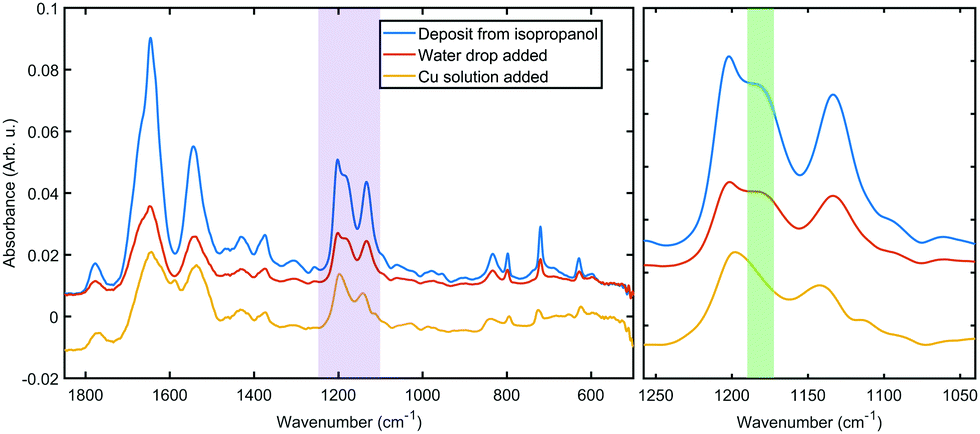 | ||
| Fig. 3 Representative IR spectra of 2H as deposited from organic solvent (blue), hydrated in situ (red) and after metal coordination (yellow). Left: Spectral range from 1850 to 500 cm−1 region. Right: Zoom on the purple region on the left. The green band highlights the loss of a peak assigned to histidine N–H vibration. | ||
The IR spectra hold further clues to the structure of the coordination complex. The peaks in the 1250–1100 cm−1 range can be assigned to C–N modes of the amine moiety of the histidine side chain. Both of the bands at 1202 cm−1 and 1132 cm−1 can be assigned to ν(CN) with coupling to δ(NH) and δ(CH), respectively.
The higher frequency mode is split between the two tautomers of the protonated imidazole (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 between N1 and N3, respectively39), thus the shoulder at 1181 cm−1 (the green shaded region in Fig. 3) can be identified as the ν(CN), δ(NH) vibration of the protonated N3 tautomer.40 This shoulder shifts slightly upon hydration, probably due to hydrogen bonding to water. After the metal was added, the disappearance of this peak indicates that a hydrogen is lost from the imidazole ring in N3 position. The simultaneous shift in the lower frequency peak to 1142 cm−1 confirms a changed chemical environment of what was a composite peak of the ν(CN), δ(CH) modes of two tautomers. These observations further confirm that the metal coordination happens to the side chain without the involvement of the oligoamide backbone, which is a marked difference to literature reports that describe disruption of the alpha helix upon copper coordination.20,41 We should note that the coordination appears to prefer only one of the tautomers and thus it is expected that in the bulk material a fraction of the oligoamides does not participate in the coordination framework.
:1 between N1 and N3, respectively39), thus the shoulder at 1181 cm−1 (the green shaded region in Fig. 3) can be identified as the ν(CN), δ(NH) vibration of the protonated N3 tautomer.40 This shoulder shifts slightly upon hydration, probably due to hydrogen bonding to water. After the metal was added, the disappearance of this peak indicates that a hydrogen is lost from the imidazole ring in N3 position. The simultaneous shift in the lower frequency peak to 1142 cm−1 confirms a changed chemical environment of what was a composite peak of the ν(CN), δ(CH) modes of two tautomers. These observations further confirm that the metal coordination happens to the side chain without the involvement of the oligoamide backbone, which is a marked difference to literature reports that describe disruption of the alpha helix upon copper coordination.20,41 We should note that the coordination appears to prefer only one of the tautomers and thus it is expected that in the bulk material a fraction of the oligoamides does not participate in the coordination framework.
X-ray spectroscopies
The 2H oligoamide contains three distinct nitrogen sites, for amide, amine and imine, respectively; these were investigated through the use of X-ray Photoelectron Spectroscopy (XPS). The typical peak positions for these moieties show a considerable variation with the chemical environment and therefore it is expected that the amine and imine peaks split due to tautomerization. Fig. 4a and b show the nitrogen N 1s peak before and after metal coordination. The deconvolution of the peaks in Fig. 4a is challenging due to the unknown shift between the amine and imine environments of the tautomers. The shoulder at 402.7 eV represents ∼1:30 of the total N 1s peak area; it has somewhat higher energy than the typical amine moieties but it would be consistent with the amine environment of the N3-protonated tautomer.37 Hence fitting was performed with five peaks reflecting the stoichiometries of the amide, imine and amine moieties in the two tautomers. The binding energies are summarized in Table 1.
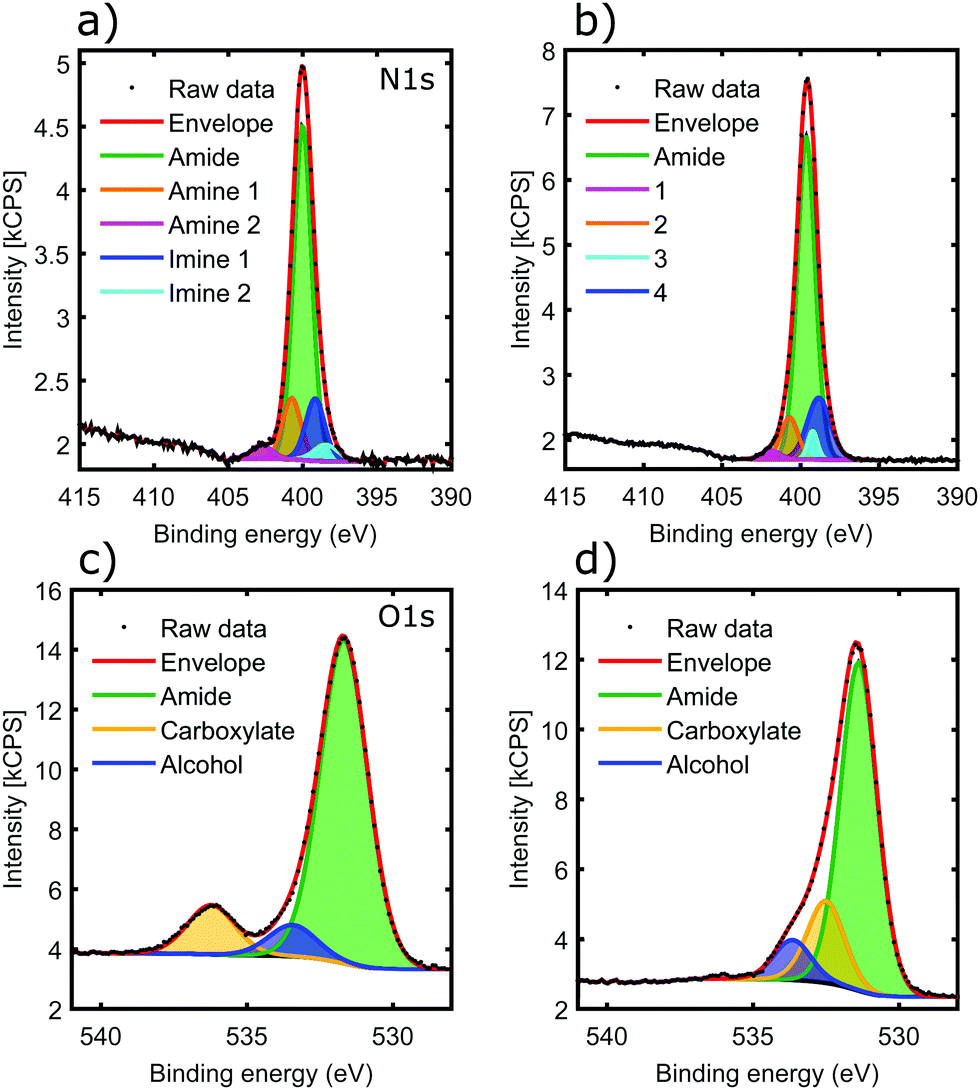 | ||
| Fig. 4 XPS results on 2H oligoamide before (a and c) and after (b and d) copper addition. (a and b) are spectra of the nitrogen 1s peak. (c and d) are oxygen 1s spectra. Fits for specific chemical environments are indicated with specific colours as indicated. | ||
| Nitrogen environment | Position (eV) | % area |
|---|---|---|
| Before Cu(II) addition | ||
| Amine N3 | 402.8 | 3.16 |
| Amine N1 | 400.7 | 12.51 |
| Amide | 400.0 | 66.74 |
| Imine N3 | 399.2 | 13.15 |
| Imine small N1 | 398.5 | 4.44 |
| Aged with Cu(II) | ||
| 1 | 401.9 | 1.94 |
| 2 | 400.7 | 8.74 |
| Amide | 399.7 | 68.94 |
| 3 | 399.3 | 3.41 |
| 4 | 398.9 | 16.98 |
In the sample aged with Cu(II) in solution phase (Fig. 4b), the 402.7 eV component shifted to lower binding energy (peak 1), consistent with deprotonation of the H–N3 tautomer as identified by IR spectroscopy. The lack of any noticeable shift in the overall peak position, dominated by the amide chemical environment, confirms that amide moieties are not affected by the metal, even after aging the sample for weeks. Clear assignments of peaks 2–4 is not possible given the small contribution they make to the overall N 1s peak. However it appears that the amine mode of the N1-protonated tautomer does not shift, suggesting that deprotonation only happens at the N3 position; this is also consistent with the IR results. There is a slight shift in peak positions in the imine region, suggesting that the N3 environment of the N1-protonated tautomer may also participate in coordination.
Fig. 4c shows the O 1s peak of the oligoamide before the addition of the metal. The peak fitting reproduces the oxygen environments of the molecule: the amide, carboxylate, and alcohol oxygens in a stoichiometric ratio of 8:2:1 respectively (Table 2). The addition of copper brings significant change to the O 1s spectrum as shown in Fig. 4d. The carboxylate O disappears, whereas a new component appears located at 532.5 eV with a similar intensity; this suggests that the C-terminal carboxylate moiety is also strongly involved in the metal coordination. In contrast, the other two O components only shift slightly. The backbone amide oxygen peak shifted with 0.3–0.4 eV, indicating a relaxation of the core helix due to the added stability of the coordination crosslinking.
| Oxygen environment | Position (eV) | % area |
|---|---|---|
| Without copper addition | ||
| Amide | 531.7 | 80.0 |
| Carboxylate | 536.2 | 12.2 |
| Alcohol | 533.5 | 7.8 |
| Aged with copper | ||
| Amide | 531.4 | 72.8 |
| Carboxylate | 532.5 | 18.2 |
| Alcohol | 533.7 | 9.1 |
| Change due to coordination | ||
| Amide | 0.3 | |
| Carboxylate | 3.7 | |
| Alcohol | −0.2 | |
The effect on the carboxylate moiety is notably missing in the IR measurements, and therefore it is the result of a much slower process than the in situ coordination that was studied with IR, and potentially linked to an oxidation state change of copper.
It should be noted that there is a potential overlap of the carboxylate O 1s peak and Na LMM; however the absence of any noticeable peak in the area in Fig. 4d confirms that the peak in Fig. 4c is originated from the oxygen species. The thickness of the sample makes any contribution from the SiO2 (substrate) O 1s signals negligible. Furthermore, the expected position of O 1s for SiO2 is 532.9 eV which was not observed in the fitted spectra.
Fig. 5a shows the Cu 2p core-levels of the coordinated material with a spin–orbit splitting of 19.8 eV between Cu 2p3/2 and Cu 2p1/2. While the main Cu 2p3/2 peak energy is very similar for all copper oxidation states, Cu(II) species can be identified from the presence of shake-up satellite peaks.42 Conversely the absence of the shake-up peaks, as observed in Fig. 5a, indicates that Cu(II) was reduced in the coordination process to Cu(I) and/or Cu(0).42 In contrast, for materials coordinated in solution the presence of a higher binding energy component in Cu 2p3/2 and the associated satellite peaks around 943 eV are characteristic of Cu(II) (Fig. 5b), suggesting incomplete coordination. This is most likely due to steric factors, as the nanorods could not align into parallel fibrils in the solution unlike in case of the surface templated coordination.
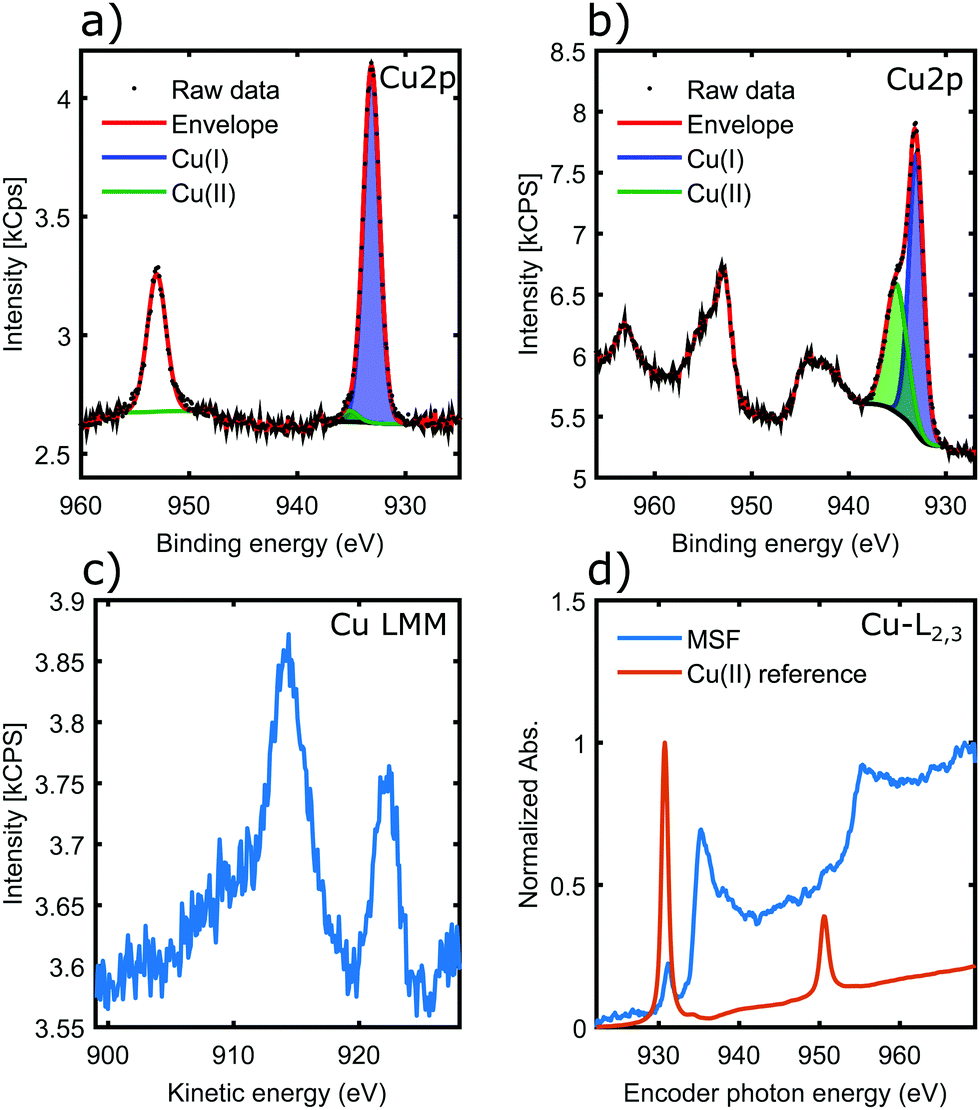 | ||
| Fig. 5 (a and b) are copper 2p spectra, showing the 2p3/2 peak at ∼933 eV and the 2p1/2 peak at ∼953 eV. (a) MSF coordinated on the substrate; (b) MSF coordinated in solution. (c) Copper Auger (LMM) spectrum. (d) NEXAFS spectra. | ||
The copper LMM Auger spectrum (Fig. 5c) of the surface templated material shows two distinct peaks. Kinetic energies are summarized in Table 3. These loosely correlate to the expected kinetic energies of Cu(I) and Cu(0), consistent with the analysis of Cu 2p spectra. There has been both a relaxation and increase of the valence orbital energies, as there is a 2.2 eV reduction in the Cu(I) Auger peak and also a 3.5 eV increase in the position of the Cu(0) LMM peak.42 These assignments were made on the basis that a smaller peak position shift would be more likely (i.e. the peaks would not cross over). Therefore the LMM spectra suggest the presence of comparable amounts of Cu(I) and Cu(0) in the coordination complex. The distinct appearance of the Cu LMM peaks suggests well defined, and fundamentally different chemical environment for the two species.
Fig. 5d shows results of a NEXAFS measurement. While the CuCl2 salt (Cu-MI) shows an intense peak corresponding to the 2p → 3d electronic transition for Cu(II), the Cu L-edge spectrum for the complexes is characteristic of Cu(0)/Cu(I) with higher absorption threshold energy (950 eV is shifted to 955 eV) and broad features that are associated with transitions into the more broad 4s orbitals. Hence NEXAFS also supports the conclusions drawn from the Cu 2p XPS and Auger LMM spectra.
Structure of the metallosupramolecular assembly
Above it was confirmed that in these oligoamide structures the backbone amide moieties are not affected by the addition of the metal, contrary to evidence of previous works on unstructured peptide chains where the amide groups are almost always deprotonated.20,23,38 However, in a helical conformation the oligoamide backbone is sterically protected by the side chains. Therefore the fact that the interaction takes place at the side chains indirectly confirms that the helical structure of the core nanorods is preserved, i.e., the supramolecular assembly remains intact upon metal coordination.As per the spectroscopic results, it is clear that the coordination involves the histidine side chains and it leads to deprotonation of the amine nitrogen of the imidazole ring. Cu(II) is reduced in the process to Cu(I) and Cu(0). The prevalence and comparable amounts of the Cu(I) and Cu(0) oxidation states suggests two chemically distinct coordination sites per oligoamide monomer. It is important to note that the terminal carboxyl residue of the oligoamide monomer does not participate in the hydrogen bonding network forming the helix and/or the core nanorod, it is instead tilted away from the helix similarly to a peptide side chain12 and is therefore accessible to a metal. Carboxyl moieties are used to design copper-based MOFs,43 hence it is feasible to assume that one of the distinct chemical environments is provided by carboxyl ligands, whereas the other one is defined by the histidine (imidazole) ligands as established above. Consistently the two dimensional structure seen in Fig. 2 is defined by (i) the core nanorods formed by the supramolecular head-to-tail assembly of the oligoamide monomers, and (ii) arrangement of the histidine side chains and the carboxyl moieties, and hence the core nanorods, around the metal centers following their preferred coordination geometry. Cu(I) is known to form stable complexes with 2–3 ligands, although higher coordination numbers are also possible.13,44 Cu(0) is not common among copper complexes and hence not much is known about its preferred coordination geometries. By considering the available packing geometries for the nanorods, for the complex formed with histidine side chains as ligands the square planar coordination geometry is the most likely as it requires the least distortion of the core nanorods, allowing the formation of a two dimensional ordered structure with an array of metal cores (Fig. 6). The carboxylate site is less defined, with only one carboxyl moiety per monomer; the most likely arrangement is two carboxyl moieties from diagonally opposite nanorod coordinating a metal, where the carboxylates are likely providing bidentate ligands.
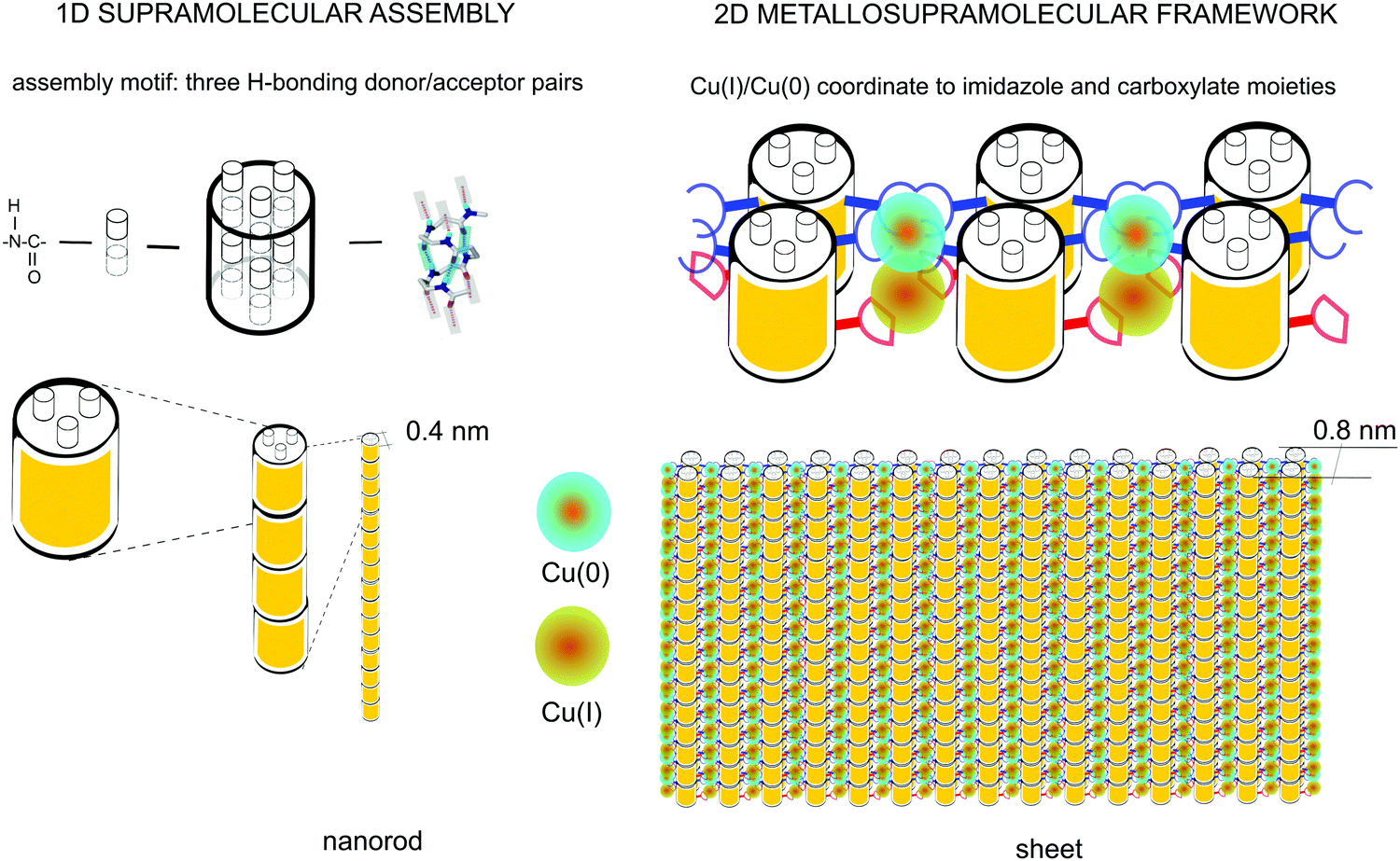 | ||
| Fig. 6 Schematics of the supramolecular nanorod assembly and the coordination-driven secondary assembly into sheets. | ||
It is important to note the proximity of the copper nuclei. The repeat distance between the histidine residues along the helix can be approximated by 2 turns of the helix (Fig. 6). Considering that the rise of the 14-helix is 4–5 Å6,12 with some variation depending on side chains, the distance between the histidine sites is thus ∼8–9 Å; the terminal carboxyl residues are situated at approximately halfway between the histidine sites, placing the metal nuclei approximately 4–4.5 Å apart. The lattice constant of metallic copper is ≈3.6 Å.45 Therefore, the distance between the copper nuclei can potentially allow the formation of direct Cu–Cu bonds. This would result in an unprecedented, practically infinite polynuclear metal complex, yielding a one-dimensional copper wire surrounded by the oligoamide nanorods.
Conclusions
A novel metallosupramolecular framework was designed based on 1D supramolecular assembly motif of β3 oligoamides and metal coordination crosslinking. Atomic force microscopy imaging confirmed the formation of 2D thin layer material. Vibration spectroscopy revealed that the backbone amide moieties are unaffected by the metal coordination. X-ray spectroscopies confirm the reduction of the metal to Cu(I) and Cu(0) and the involvement of the histidine side chain as well as the terminal carboxyl moieties in the coordination. The data imply the formation of a unique polynuclear complex where Cu(I) and Cu(0) nuclei alternate, coordinated to the imidazole sidechains of histidine and the terminal carboxylate moieties.Materials and methods
Materials
The peptide 2H (Ac-β3Ala-β3Val-β3Ser-αHis-αHis-β3Ala-β3Val-β3Ala) was synthesized in house using solid state synthesis from Fmoc protected amino acids (GL Biochem, China) as described in the supplementary information in Del Borgo et al., Angewandte Chemie 2014.12 For all aqueous experiments deionized water with a resistivity of 18.2 MΩ cm was used (Ultrapure; Sartorius AG, Germany). CuCl2·2H2O (99%) metal basis, metal solutions were used, salt was purchased from Sigma Aldrich. Solutions of either 2 mM or 100 μl concentrations have been used.Atomic force microscopy imaging
Solutions of 100 μM oligoamide concentration were prepared in pure water or in either of the three salt solutions. Metal ion concentrations of either 100 μM or 2 mM were used. The oligoamides were aged with the salt solutions at 4 °C for at least one week. For AFM topology images, the stock solution was deposited onto freshly cleaved mica surface. All samples were dried overnight at 37 °C. Samples were imaged using an Ntegra AFM platform (NT-MDT, Russia) under ambient conditions using golden silicon probes with nominal 10 nm apex radius and of a typical spring constant of 72 N m−1. For imaging, semicontact mode was used with 512 × 512 pixel resolution and a scan rate of 0.5–1 Hz.Transmission electron microscopy
10 μl aliquots of aged sample solutions were applied to 400-mesh copper grids that were coated with a thin layer of carbon. Excess material was removed by blotting and samples were negatively stained with two aliquots of 10 μl 2% uranyl acetate solution (w/v); Electron Microscopy Services at La Trobe university research facility were used for imaging. The grids were air dried and viewed using a JEOL JEM-2010-HC transmission electron microscope (JEOL Ltd, Tokyo, Japan) operated at 100 kV.X-ray photoelectron spectroscopy
A Kratos Axis NOVA spectrometer was used with the following parameters: aluminium anode, dwell time 250 ms, pass energy of 20 eV, step size, 0.1 eV, three sweeps, FAT mode.Near edge X-ray absorption fine structure (NEXAFS)
Measurements were performed at the Soft X-ray beamline of the Australian Synchrotron in partial electron yield (PEY) mode with linearly polarized X-rays. Photon energy calibration was performed using spectra taken from previously calibrated stable reference metal foils measured in parallel with the data.MATLAB2019a was used to plot the data. Inkscape 0.92.4 was used to assemble vector images into the two by two grid arrangement.
Infrared spectroscopy
Measurements were carried out in the infrared lab at Helmholtz-Zentrum Berlin. Bruker VERTEX 80v spectrometer operated in a rough vacuum of 3.0 hPa to <1 hPa was used with a liquid nitrogen-cooled MCT detector. An attenuated total reflectance (ATR) mode in single-reflection mode through a ZnSe crystal was used for measurements. The ATR crystal was enclosed in an environmental cell so the sample was maintained at atmospheric pressure. The sample cell was humidity controlled and had a dry nitrogen flow through it.Author statement
The manuscript was prepared under an agreement by all authors. R. S. S. has performed AFM experiments, contributed to data analysis and writing. N. G. W. performed IR and analyzed the XPS and NEXAFS data. M. P. D. B. and K. K. have synthesized and chemically characterized the oligoamide. C. G. A. has performed TEM imaging. A. J. B. performed XPS experiments and contributed to data analysis. D. Q. performed NEXAFS and helped with interpretation. J. S. P. contributed to the interpretation of the XPS data. L. P. provided invaluable help with IR measurements and data analysis. M.-I. A., P. P. and A. M. provided the conceptual design for the work. A. M. has devised and supervised the experiments and data analysis. All authors contributed to the writing of the manuscript.Funding
D. Q. acknowledges the support of the Australian Research Council (Grant No. FT160100207).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this research was undertaken on the Soft X-ray spectroscopy beamline at the Australian Synchrotron, part of ANSTO. The authors thank HZB for the allocation of synchrotron radiation beamtime. This work was performed in part at the Australian National Fabrication Facility (ANFF), a company established under the National Collaborative Research Infrastructure Strategy, through the La Trobe University Centre for Materials and Surface Science. This work was co-funded between the A*STAR ARAP scholarship and La Trobe University LTUGRS PhD scholarship.References
- L. Wang, Y. Sun, Z. Li, A. Wu and G. Wei, Materials, 2016, 9, 53 CrossRef PubMed.
- M. Lazzari, C. Rodríguez-Abreu, J. Rivas and M. A. López-Quintela, J. Nanosci. Nanotechnol., 2006, 6, 892 CrossRef CAS PubMed.
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661 RSC.
- G.-Y. Liu and N. A. Amro, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5165 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS PubMed.
- Y.-D. Wu and D.-P. Wang, J. Am. Chem. Soc., 1999, 121, 9352 CrossRef CAS.
- D. Seebach, D. F. Hook and A. Glättli, Biopolymers, 2005, 84, 23 CrossRef PubMed.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071 CrossRef CAS.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed.
- M. P. Del Borgo, A. I. Mechler, D. Traore, C. Forsyth, J. A. Wilce, M. C. J. Wilce, M.-I. Aguilar and P. Perlmutter, Angew. Chem., Int. Ed., 2013, 52, 8266 CrossRef CAS PubMed.
- R. D. Gopalan, M. P. Del Borgo, A. I. Mechler, P. Perlmutter and M.-I. Aguilar, Chem. Biol., 2015, 22, 1417 CrossRef CAS PubMed.
- R. S. Seoudi, M. P. Del Borgo, K. Kulkarni, P. Perlmutter, M.-I. Aguilar and A. Mechler, New J. Chem., 2015, 39, 3280 RSC.
- R. S. Seoudi, A. Dowd, M. Del Borgo, K. Kulkarni, P. Perlmutter, M.-I. Aguilar and A. Mechler, Pure Appl. Chem., 2015, 87, 1021 CAS.
- D. S. Raja, W.-L. Liu, H.-Y. Huang and C.-H. Lin, Comments Inorg. Chem., 2015, 35, 331 CrossRef.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed.
- A. Mantion, L. Massüger, P. Rabu, C. Palivan, L. B. McCusker and A. Taubert, J. Am. Chem. Soc., 2008, 130, 2517 CrossRef CAS PubMed.
- C. F. Meares, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. eibc0167 Search PubMed.
- S. Timári, C. Kállay, K. Ősz, I. Sóvágó and K. Várnagy, Dalton Trans., 2009, 1962 RSC.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200 RSC.
- R. C. Dunbar, J. Martens, G. Berden and J. Oomens, Phys. Chem. Chem. Phys., 2016, 18, 26923 RSC.
- H. Kozłowski, W. Bal, M. Dyba and T. Kowalik-Jankowska, Coord. Chem. Rev., 1999, 184, 319 CrossRef.
- R. S. Seoudi, M. G. Hinds, D. J. D. Wilson, C. G. Adda, M. Del Borgo, M.-I. Aguilar, P. Perlmutter and A. Mechler, Nanotechnology, 2016, 27, 135606 CrossRef PubMed.
- C. Buchanan, C. J. Garvey, P. Perlmutter and A. Mechler, Pure Appl. Chem., 2017, 89, 1809 CAS.
- C. Buchanan, C. J. Garvey, L. Puskar, P. Perlmutter and A. Mechler, Supramol. Chem., 2020, 0, 1 Search PubMed.
- J. D. Sadowsky, M. A. Schmitt, H.-S. Lee, N. Umezawa, S. Wang, Y. Tomita and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 11966 CrossRef CAS PubMed.
- L. K. A. Pilsl and O. Reiser, Amino Acids, 2011, 41, 709 CrossRef CAS PubMed.
- M. P. Del Borgo, K. Kulkarni, M. A. Tonta, J. L. Ratcliffe, R. Seoudi, A. I. Mechler, P. Perlmutter, H. C. Parkington and M.-I. Aguilar, APL Bioeng., 2018, 2, 026104 CrossRef PubMed.
- J. A. Kritzer, J. Tirado-Rives, S. A. Hart, J. D. Lear, W. L. Jorgensen and A. Schepartz, J. Am. Chem. Soc., 2005, 127, 167 CrossRef CAS PubMed.
- S.-M. Liao, Q.-S. Du, J.-Z. Meng, Z.-W. Pang and R.-B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- L. Wang, N. Sun, S. Terzyan, X. Zhang and D. R. Benson, Biochemistry, 2006, 45, 13750 CrossRef CAS PubMed.
- I. Sóvágó and K. Ősz, Dalton Trans., 2006, 3841 RSC.
- G. Arena, D. La Mendola, G. Pappalardo, I. Sóvágó and E. Rizzarelli, Coord. Chem. Rev., 2012, 256, 2202 CrossRef CAS.
- H. Fabian and W. Mäntele, Handbook of vibrational spectroscopy, 2002 Search PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073 CrossRef CAS PubMed.
- B. M. Marsh, J. Zhou and E. Garand, RSC Adv., 2015, 5, 1790 RSC.
- I. Ashikawa and K. Itoh, Biopolymers, 1979, 18, 1859 CrossRef CAS.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141 CrossRef CAS PubMed.
- S. Stadlbauer, A. Riechers, A. Späth and B. König, Chem. – Eur. J., 2008, 14, 2536 CrossRef CAS PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2010, 257, 887 CrossRef CAS.
- W.-Q. Xu, S. He, C.-C. Lin, Y.-X. Qiu, X.-J. Liu, T. Jiang, W.-T. Liu, X.-L. Zhang and J.-J. Jiang, Inorg. Chem. Commun., 2018, 92, 1 CrossRef CAS.
- X. Ren, M. Wesolek and P. Braunstein, Dalton Trans., 2019, 48, 12895 RSC.
- N. J. Simon, E. S. Drexler and R. P. Reed, Properties of Copper and Copper Alloys at Cryogenic Temperatures, National Institute of Standards and Technology, Gaithersburg, MD, 1992 Search PubMed.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238 CrossRef CAS.
- G. Zorn, L.-H. Liu, L. Árnadóttir, H. Wang, L. J. Gamble, D. G. Castner and M. Yan, J. Phys. Chem. C, 2014, 118, 376 CrossRef CAS PubMed.
- O. Olivares, N. V. Likhanova, B. Gómez, J. Navarrete, M. E. Llanos-Serrano, E. Arce and J. M. Hallen, Appl. Surf. Sci., 2006, 252, 2894 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325 CrossRef CAS.
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |