
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature-induced switchable magnetite nanoparticle superstructures†
Cathrin
Kronenbitter
a,
Hironobu
Watanabe
 b,
Sadahito
Aoshima
b and
Helmut
Cölfen
*a
b,
Sadahito
Aoshima
b and
Helmut
Cölfen
*a
aPhysical Chemistry, University of Konstanz, Universitätsstraße 10, D-78457 Konstanz, Germany. E-mail: helmut.coelfen@uni-konstanz.de; Fax: +49 7531 88 3139; Tel: +49 7531 88 4063
bDepartment of Macromolecular Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan. E-mail: aoshima@chem.sci.osaka-u.ac.jp; Fax: +81-6-6850-5448; Tel: +81-6-6850-5448
First published on 4th March 2020
Abstract
In this study we describe the green synthesis of temperature-switchable polymer-magnetite nanoparticles (PMNPs) in water at room temperature via an improved co-precipitation pathway. The temperature responsiveness was achieved through the surface modification with a vinyl-based dual-stimuli-responsive block copolymer. Furthermore, these PMNPs enable the investigation of temperature-induced magnetic superstructures and their medical application.
Smart polymers are especially known for the change in their solubility, optical or electrical properties in accordance with a change in their for reversible switchable superstructures chemical or physical environment.1 Different kinds of stimuli such as temperature, pH, light or redox triggers are known.2–5 The success of the living polymerization of vinyl ethers (VEs) containing various functional groups, led to the achievement in construction of such smart polymers.6 For example, poly[2-(2-ethoxy)ethoxyethyl VE] (polyEOEOVE) can be used as such a polymer. The thermo-sensitive oxyethylene segments in a stimuli-responsive polymer can be significantly extended by a second block containing an amino function, such as poly(2-aminoethyl VE) (polyAEVE). This block copolymerization results in a large expansion of the self-assembled patterns.7 Among these stimuli, temperature and pH responsiveness have become the focus of interest due to the ease of controlling these stimuli.8
The main characteristic of thermo-responsive polymers is the phase-transition at certain temperatures.9 This phase-transition property is due to the temperature-dependent change in the hydration of stimuli-responsive units.10 As a result, the hydrophobicity/hydrophilicity will change, followed by an alteration in solubility or conformation.10 Polymers which become insoluble above a certain temperature are typically characterized by the lower critical solution temperature (LCST). Below the LCST, the polymer conformation is extended.11,12 Regarding the use of polymers to stabilize nanoparticles,13 the combination of temperature- and pH-responsive polymers is a great solution for the functionalization of oxide-nanoparticles since amine groups can function as a linker to nanoparticle surfaces.14
Iron oxide nanoparticles, especially magnetite nanoparticles (MNPs) due to possible superparamagnetism, represent an important class of inorganic nanomaterials that contribute to current developments in nanomedicine.15,16 They find application as contrast agents for magnetic resonance imaging and in magnetic hyperthermia therapy.17 2D and 3D MNP clusters are known to align simultaneously to an external magnetic field, and thus accumulate significantly faster than individual magnetic nanoparticles to trigger local magnetic hyperthermia.18 Furthermore, they can also be used in drug delivery and bio-separation.19
MNPs are not only of great interest because of their biocompatibility but also due to their straightforward synthesis.
The co-precipitation method, where Fe(III) and Fe(II) salts are stoichiometrically mixed, is a green and simple chemical route for the synthesis of magnetite.20 The main advantage of this approach is the complete absence of toxic solvents, high temperatures and furthermore the large quantities of obtained MNPs. By fine adjustment of pH, ionic strength and the concentration of the growth solution, the control over particle size and shape is possible.21 To follow a green strategy for MNP synthesis, it is necessary to avoid organic solvents and use water instead, thereby necessitating stabilization with surfactants (i.e. an addition of a polymer layer).13 Furthermore, MNPs can be functionalized by various covalent and noncovalent approaches, since different functional groups like silanes,22 carboxylic and phosphonic acid groups23 are well known for interacting with the surface. Apart from these functionalities, amine groups are also known to interact with the iron oxide surface.13,14
In this communication, we stabilize MNPs with the dual-stimuli-responsive block copolymer poly(EOEOVE-b-AEVE), in which the amine block is used as a linker to iron oxide. These MNPs are attractive for the above-mentioned medical applications. The reasons for using an ethoxyethyl-VE polymer instead of the well-known PEO (polyethylene oxide) are firstly the LCST, as the LCST of PEO exceeds 100 °C,24 and secondly the complicated behaviour of PEO, which is expected due to its partial crystallinity. Another conceivable system would be PNIPAM because of its similar LCST behaviour.25 However, disadvantages can also be expected such as the potential interaction of PNIPAM with the surface of the magnetite nanoparticles due to its amide group in the side chain. This could complicate the behaviour, whereas the VE polymer having ether groups does not interact much. Furthermore, for polyEOEOVE the LCST temperature can be freely designed based on the number of oxyethylene units and the type of alkyl group at the side chain. Moreover, since it is prepared by living polymerization, a sharp phase separation is possible due to the narrow molar mass distribution.
Synthesis and post-synthesis modifications are applied to obtain organic inorganic hybrid particles. Our focus is the synthesis of temperature-switchable polymer-magnetite nanoparticles in water at room temperature and their further use in reversible switchable superstructure formation. Using the properties of the secondary minimum in the DLVO potential,26 a small amount of energy is sufficient to reversibly dissolve the superstructures to form stable nanoparticle dispersions if T < LCST. We avoid the ligand exchange approach described in the literature for the transfer of the MNPs from organic solvents to water, due to toxic solvents and high temperatures used as well as the fact that the subsequent ligand exchange often leads to a loss of nanoparticle shape.27 To improve the monodispersity of the nanoparticles in water, we have varied the commonly known co-precipitation pathway (see ESI,† Section 2.1). Subsequently, temperature-induced superstructure formation of in situ and post-synthesis functionalized PMNPs is achieved (Fig. 1).
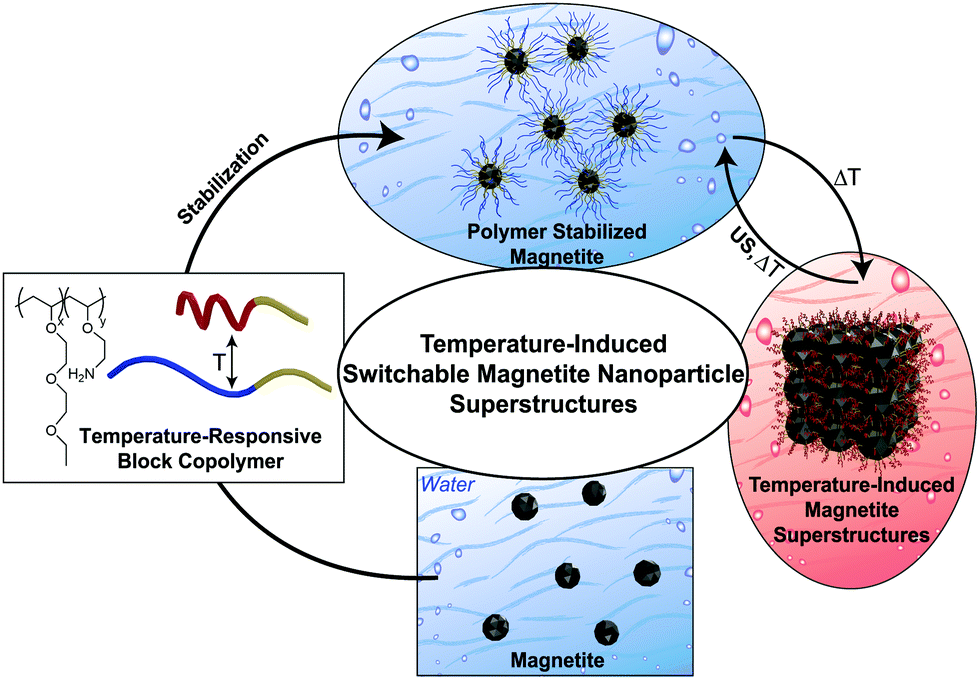 | ||
| Fig. 1 Graphic visualisation of temperature-induced switchable magnetite nanoparticle superstructure formation in water. US = ultrasound. | ||
The block copolymer EOEOVE100-b-AEVE68 (EOEOVE100:AEVE68) was successfully synthesized using living cationic polymerization (for more detail see ESI,† Section 2.2). EOEOVE was polymerized first, followed by addition of phthalimide-protected amine VE. The phthalimide protecting groups were removed by treating the polymer with hydrazine, the deprotection was confirmed by 1H-NMR (ESI,† Fig. S1). A LCST of 48 °C was determined by turbidity measurements (ESI,† Fig. S2).
In combination with the dual-stimuli-responsive block copolymer EOEOVE100:AEVE68 our purpose is the functionalization of the nanoparticles either by post-synthesis modification or by in situ modification during the synthesis. The pH stimuli-responsivity of the AEVE block can be used at certain pH values to generate a block with a large number of linker functionalities.
For post-synthesis modification, the MNPs are synthesized using an improved co-precipitation method. In commonly known co-precipitation methods the iron salt/hydrochloric acid mixture is slowly added to diluted basic solution (NaOH/NH4OH), often at higher temperatures.20,28,29 We perform all synthesis steps in water at room temperature in an oxygen-free atmosphere (for more detail see ESI,† Section 2.1). We dissolved the iron salts in water and concentrated NH4OH (25 wt%) was rapidly added, to generate a fast supersaturation and to promote short nucleation and growth processes.30 Immediately, black MNPs are formed. Particles that are relatively homogeneous in size are obtained, with a mean diameter of about 11.0 nm ± 2.6 nm (Fig. 2a and ESI,† Fig. S3a). HR-TEM measurements were used to analyse the obtained particles in more detail (ESI,† Fig. S4). Electron diffraction (ED) and powder X-ray diffraction (PXRD) measurements confirm the formation of magnetite (ESI,† Fig. S5 and Fig. 2b).
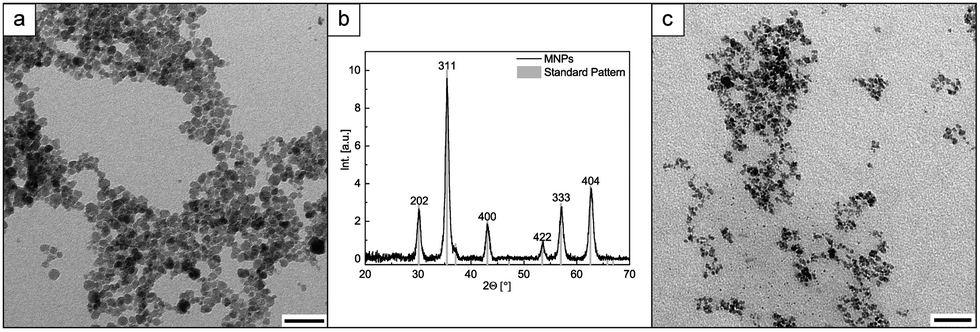 | ||
| Fig. 2 (a) TEM image of MNPs without polymer surface layer, scale bar: 50 nm, (b) PXRD of MNPs and standard magnetite pattern from crystallography open data base no. 9013529, (c) TEM image of MNPs with polymer surface layer (PMNPs), scale bar: 50 nm. | ||
The coordination of water to iron atoms in aqueous solutions creates a hydroxyl functionalized iron oxide surface. Due to the amphoteric character of these hydroxyl groups, the surface is charged negatively or positively dependent on the pH19 and the point of zero charge is pH 8.0.31 This shows a clear pH dependence of synthesis as well as post-synthesis modifications, as the amine block of the polymer has to interact with the nanoparticle surface.
The influence of the polymer during magnetite synthesis allows significant alteration of the size and shape of the nanoparticles.
Two different approaches for introducing the polymer into the system during-synthesis are investigated: dissolving the polymer either into the acidic iron salt solution or in the basic ammonia solution.
In the first approach the polymer is able to coordinate to Fe2+/Fe3+ ions in solution before particles are formed, yielding to significant differences in the resulting nanoparticle morphology. The obtained particle dispersion is stable for days. Comparing the nanoparticle appearance with and without polymer influence via TEM, a difference in size is visible (Fig. 2). Particles synthesized without polymer show a mean size of 11 nm (Fig. 2a), whereas the particles with polymer show a mean size of below 7.1 nm ± 2.0 nm (Fig. 2c and ESI,† Fig. S3b).
The second approach did not yield in a stable nanoparticle dispersion. However, after heating this dispersion stabilized PMNPs are likewise obtained. This observation implicates the presumption that the hydrophobic state of the polymer above the LCST generates an anhydrous environment around the MNPs allowing the surface and amine groups to interact. By fine tuning the dual-stimuli-responsive block copolymer structure, the temperature responsive EOEOVE block enables the control over the temperature induced change in hydrophobicity and thereby the precipitation. Moreover, by controlling these parameters, the formation of magnetite nanoparticle superstructures is accessible, since the AEVE-amine linker block is connected to the MNPs.
Both approaches of functionalization achieve temperature-switchable magnetite nanoparticles, which means that the functionalization of the surface with the polymers occurred directly during the synthesis. Long-term stability of the different nanoparticles is confirmed with dynamic light scattering (DLS) (ESI,† Table S1 and Fig. S6).
In addition, by exceeding the LCST, the particles not only precipitate but also form superstructures, independent of the modification method used (i.e. synthesis or post-synthesis modification). Due to the easier functionalization procedure, the first approach is favoured also because the functionalization resulted in a highly stable nanoparticle dispersion. Subsequently, different approaches to induce the superstructure formation were investigated. These include external magnetic field directed self-assembly (ESI,† Fig. S7) and temperature-induced superstructure formation. In both approaches, superstructures are obtained but for this communication we only focus on the temperature-induced superstructure formation.
Here, a polymer-stabilized nanoparticle dispersion at neutral pH, was heated above the LCST of the polymer (for more detail see ESI,† Section 2.1). A slow clouding of the solution occurred followed by the superstructure formation after three days. Scanning electron microscopy (SEM) measurements indicate that these superstructures formed distinct island-like structures with sizes of 1–5 μm (Fig. 3a), where the soft polymer shell enables a dense packing of the MNPs.
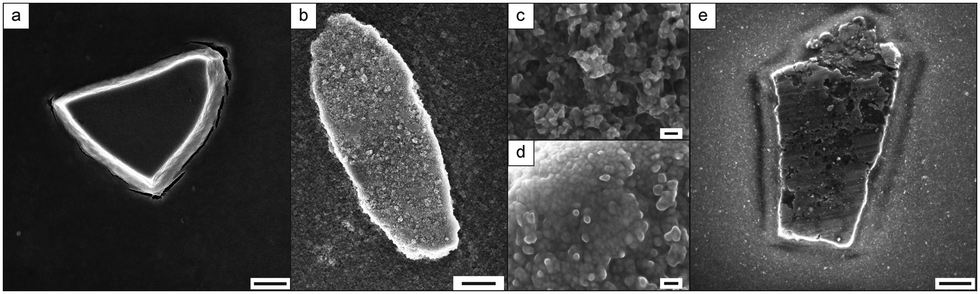 | ||
| Fig. 3 (a) SEM image of synthesis functionalized PMNP superstructures, scale bar: 1 μm, (b) SEM image of a superstructure of post-synthesis functionalized PMNPs before recrystallization, scale bar: 1 μm, (c) higher resolution SEM image of network-like PMNPs, scale bar: 40 nm, (d) higher magnification SEM image of densely packed PMNPs, scale bar: 40 nm, (e) SEM image of a superstructure of post-synthesis functionalized PMNPs after recrystallization, scale bar: 500 nm. | ||
In a second approach the functionalization of the MNPs was performed post-synthetically. Here, MNPs were added to the polymer solution and the dispersion was mixed via ultra-sonication. Once again, superstructure formation was induced by increasing the temperature of the PMNP mixture above the polymer LCST. The superstructures are confirmed by SEM observations (Fig. 3b). The higher magnification images indicate in general less dense packing of the particles occurred than that with the direct synthesis processed PMNPs (Fig. 3c). Nevertheless, densely-packed sections are visible within the superstructures (Fig. 3d). To increase the density of the packing and tune the superstructures, recrystallization32 has been applied, using ultra-sonication to re-disperse the PMNPs at T < LCST followed by superstructure formation at T > LCST (ESI,† Fig. S8). As a result, island-like superstructures of various shapes with a size range about 2–10 μm are obtained (Fig. 3e). Higher magnification SEM images reveal that the whole superstructures consist of nanoparticles that are densely packed after recrystallization (ESI,† Fig. S9). Furthermore, the higher resolution imaging reveals that the recrystallization has not removed the soft-polymer shell around the magnetite nanoparticles. This indicates a strong interaction between the magnetite surface and the polymers. The polymer layer around the NMPs building the PMNPs could be visualized by phase contrast AFM (ESI,† Fig. S10). However, since these measurements were performed in the dry state, no further information could be obtained from these measurements. The organic part (polymer) around the inorganic structures (MNPs) is also confirmed by energy-dispersive X-ray spectroscopy (EDX) measurements (ESI,† Fig. S11). Due to the polymer shell around the particles, they act like a soft material and are able to pack densely despite the particle polydispersity.
Conclusions
The focus of the work presented here is on the development of a green synthesis procedure for reversible temperature-switchable magnetite nanoparticle superstructures for medical applications. We improved the commonly known co-precipitation route of magnetite nanoparticles in order to functionalize the nanoparticles with the dual-responsive polymer EOEOVE:AEVE and further induce the superstructure formation by heating above the LCST.Through the use of a polymer, the imperfection of the MNPs regarding monodispersity can be compensated and a tight packing is achieved. The most probable reason for this dense packing behaviour is the polymer shell around the particles, which causes them to behave like a soft material.
The key advantage of the synthesis procedure developed here is the green character, meaning that no toxic solvents and no ligand exchange are necessary for the generation of MNPs dispersion in aqueous phase. So far, these magnetite nanoparticle systems have rarely been described for aqueous solvents. These green systems could potentially find application in broad fields such as nanomedicine. For example, magnetite superstructures could be interesting for tumor therapies like magnetic hyperthermia due to an increased heat production of the superstructures.17 Furthermore, the unique polymer chemistry of dual-responsive block copolymers is of great interest in diverse fields extending beyond medicine since the LCST properties can be tuned by simply modifying the structure of the blocks. The results presented here, could be applied to design customized temperature responsive additives by fine-tuning the block copolymer structures in a target-oriented manner.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Deutsche Forschungsgemeinschaft (DFG) is acknowledged for financial support of this work within SFB 1214 (Project B1). The Particle Analysis Center of the University of Konstanz is acknowledged for providing the WAXS measurements.References
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- S. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem. – Eur. J., 2015, 21, 13164–13174 CrossRef CAS PubMed.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- Y. Oda, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1207–1213 CrossRef CAS.
- S. Aoshima, H. Oda and E. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2407–2413 CrossRef CAS.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- M. R. Matanović, J. Kristl and P. A. Grabnar, Int. J. Pharm., 2014, 472, 262–275 CrossRef PubMed.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- D. J. Phillips and M. I. Gibson, Polym. Chem., 2015, 6, 1033–1043 RSC.
- K. T. Oh, H. Yin, E. S. Lee and Y. H. Bae, J. Mater. Chem., 2007, 17, 3987–4001 RSC.
- C. Boyer, M. R. Whittaker, V. Bulmus, J. Liu and T. P. Davis, NPG Asia Mater., 2010, 2, 23–30 CrossRef.
- H. Qu, H. Ma, A. Riviere, W. Zhou and C. J. O’Connor, J. Mater. Chem., 2012, 22, 3311–3313 RSC.
- Y. Jun, J. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- S. K. Avugadda, M. E. Materia, R. Nigmatullin, D. Cabrera, R. Marotta, T. Fernandez Cabada, E. Marcello, S. Nitti, E. J. Artés-Ibáñez, P. Basnett, C. Wilhelm, F. J. Teran, I. Roy and T. Pellegrino, Chem. Mater., 2019, 31, 5450–5463 CrossRef CAS PubMed.
- N. C. Bigall, C. Wilhelm, M.-L. Beoutis, M. García-Hernandez, A. A. Khan, C. Giannini, A. Sánchez-Ferrer, R. Mezzenga, M. E. Materia, M. A. Garcia, F. Gazeau, A. M. Bittner, L. Manna and T. Pellegrino, Chem. Mater., 2013, 25, 1055–1062 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209–2211 CrossRef CAS.
- J.-P. Jolivet, É. Tronc and C. Chanéac, C. R. Chim., 2002, 5, 659–664 CrossRef CAS.
- H. Lee, K. Y. Mi, S. Park, S. Moon, J. M. Jung, Y. J. Yong, H. W. Kang and S. Jon, J. Am. Chem. Soc., 2007, 129, 12739–12745 CrossRef CAS PubMed.
- Y. Sahoo, H. Pizem, T. Fried, D. Golodnitsky, L. Burstein, C. N. Sukenik and G. Markovich, Langmuir, 2001, 17, 7907–7911 CrossRef CAS.
- H. S. Ashbaugh and M. E. Paulaitis, Ind. Eng. Chem. Res., 2006, 45, 5531–5537 CrossRef CAS.
- S. Ashraf, H. K. Park, H. Park and S. H. Lee, Macromol. Res., 2016, 24, 297–304 CrossRef CAS.
- J. A. Fornés, Colloid Polym. Sci., 1985, 263, 1004–1007 CrossRef.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158–2168 CrossRef CAS PubMed.
- S. Palacin, P. C. Hidber, J.-P. Bourgoin, C. Miramond, C. Fermon and G. M. Whitesides, Chem. Mater., 1996, 8, 1316–1325 CrossRef CAS.
- G. Paul, P. Kumar Das and I. Manna, J. Magn. Magn. Mater., 2016, 404, 29–39 CrossRef CAS.
- C. B. Whitehead, S. Özkar and R. G. Finke, Chem. Mater., 2019, 31, 7116–7132 CrossRef CAS.
- M. Kosmulski, J. Colloid Interface Sci., 2004, 275, 214–224 CrossRef CAS PubMed.
- J. W. Mullin, Crystallization, Elsevier, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis methods, polymer characterisation and supplementary measurement data are available. See DOI: 10.1039/d0ma00075b |
| This journal is © The Royal Society of Chemistry 2020 |