
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new agarose-based microsystem to investigate cell response to prolonged confinement†
A.
Prunet
a,
S.
Lefort
 b,
H.
Delanoë-Ayari
a,
B.
Laperrousaz
b,
G.
Simon
a,
C.
Barentin
a,
S.
Saci
a,
F.
Argoul
c,
B.
Guyot
b,
J.-P.
Rieu
a,
S.
Gobert
*d,
V.
Maguer-Satta
*b and
C.
Rivière
*ae
b,
H.
Delanoë-Ayari
a,
B.
Laperrousaz
b,
G.
Simon
a,
C.
Barentin
a,
S.
Saci
a,
F.
Argoul
c,
B.
Guyot
b,
J.-P.
Rieu
a,
S.
Gobert
*d,
V.
Maguer-Satta
*b and
C.
Rivière
*ae
aUniv Lyon, Université Claude Bernard Lyon 1, CNRS UMR-5306, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: charlotte.riviere@univ-lyon1.fr
bCNRS UMR-5286, Centre de Recherche en Cancérologie de Lyon, 69000 Lyon, France. E-mail: veronique.maguer-satta@lyon.unicancer.fr
cUniversité de Bordeaux, CNRS UMR-5798, LOMA, F-33405, Talence, France
dUniv Lyon, Université Claude Bernard Lyon 1, CNRS UMR-5305, Laboratoire de Biologie Tissulaire et Ingénierie thérapeutique, F-69367, Lyon, France. E-mail: stephanie.gobert@ibcp.fr
eInstitut Universitaire de France (IUF), France
First published on 24th September 2020
Abstract
Emerging evidence suggests the importance of mechanical stimuli in normal and pathological situations for the control of many critical cellular functions. While the effect of matrix stiffness has been and is still extensively studied, few studies have focused on the role of mechanical stresses. The main limitation of such analyses is the lack of standard in vitro assays enabling extended mechanical stimulation compatible with dynamic biological and biophysical cell characterization. We have developed an agarose-based microsystem, the soft cell confiner, which enables the precise control of confinement for single or mixed cell populations. The rigidity of the confiner matches physiological conditions and its porosity enables passive medium renewal. It is compatible with time-lapse microscopy, in situ immunostaining, and standard molecular analyses, and can be used with both adherent and non-adherent cell lines. Cell proliferation of various cell lines (hematopoietic cells, MCF10A epithelial breast cells and HS27A stromal cells) was followed for several days up to confluence using video-microscopy and further documented by Western blot and immunostaining. Interestingly, even though the nuclear projected area was much larger upon confinement, with many highly deformed nuclei (non-circular shape), cell viability, assessed by live and dead cell staining, was unaffected for up to 8 days in the confiner. However, there was a decrease in cell proliferation upon confinement for all cell lines tested. The soft cell confiner is thus a valuable tool to decipher the effects of long-term confinement and deformation on the biology of cell populations. This tool will be instrumental in deciphering the impact of nuclear and cytoskeletal mechanosensitivity in normal and pathological conditions involving highly confined situations, such as those reported upon aging with fibrosis or during cancer.
Introduction
Emerging evidence suggests the importance of mechanical stimuli in normal and pathological situations for the control of many critical cellular functions.1,2 It has been shown that biomechanical stimuli can induce changes in gene expression,3 influence stem cell differentiation,4,5 and are altered in several human diseases such as aging6 and cancer.7,8 In the latter case, the transition of cells towards a cancerous phenotype is accompanied by various mechanical modifications such as extracellular matrix (ECM) stiffening,9 increase in interstitial fluid pressure,10,11 and compressive stress resulting from cell proliferation in a confined environment.12 In turn, such lateral compression imposed by the surrounding microenvironment strongly drives cancer cells to evolve towards a more invasive phenotype,13 and is accompanied by changes in gene expression.14Interestingly, while the effect of matrix stiffness is extensively studied in the context of tumor progression,15,16 stem cell differentiation17–19 and aging,6 few studies have focused on the role of mechanical stresses.20 This field of research remains underdeveloped due to the lack of standard in vitro assays enabling quantification of phenotypic and genotypic modifications of cells upon extended mechanical stimulation.
Different microfluidic systems were recently designed to determine the impact of confinement on single21–24 and collective25,26 cell migration, nucleus deformation,27–32 rupture and repair33,34 and cell division.35,36 Using such systems, a switch from a mesenchymal to an amoeboid mode of migration upon cell confinement was highlighted for various mesenchymal cell types37 including embryonic progenitor cells.38 The ability of cells not only to deform39 (both their overall and nuclear shape), but also to repair their nuclear envelope after rupture during migration through confined environments were also evidenced.33 Tunable microsystems enabling the analysis of cell responses under well-controlled confined environments, should thus pave the way for understanding the impact of mechanical stress in normal and pathological situations.40
However, the polymeric material classically used for such devices (polydimethylsiloxane – PDMS) suffers from major limitations, precluding the adequate decoupling of mechanical signals from other biochemical cues in long-term experiments. First, the large adsorption of therapeutics41,42 results in the underestimation of cell response to drugs. Second, PDMS is impermeable to small water-soluble molecules, leading to fast-medium conditioning (if continuous flow is not provided otherwise) due to nutrient depletion or increase in cell-secreted factors. Third, PDMS rigidity is several orders of magnitude larger than physiological rigidity (MPa vs. kPa range in vivo7). In parallel to PDMS-based microsystems, different hydrogel-based approaches were developed,43 including silk,44 alginate,45 polyacrylamide46 or poly(ethylene glycol) diacrylate (PEGDA)-derived microsystems.47 However, in most of these microsystems, PDMS material was still used as a supporting scaffold, which does not address drug adsorption and nutrient depletion issues. Overall, none of these set-ups meet all the criteria required for long-term time-lapse analysis (i.e. precise control of the applied stress, compatibility with high-resolution video-microscopy, efficient medium and oxygen renewal, in situ immunostaining/drug application, no reduction of the available drug dose, easy cell retrieval for further standard molecular analyses), within a physiological stiffness range.
To overcome these limitations, we present here an agarose-based confinement microsystem. This hydrogel presents several advantages and is widely used in tissue engineering.48 The tunable mechanical properties of the agarose can reproduce the stiffness of the in vivo microenvironment. Its porous nature enables the free diffusion of salts and small molecules (size <30 nm in 2% agarose,49 which is the case for most proteins), ensuring passive medium renewal. It has however only been implemented in microfluidic systems by a limited number of groups.50–54 The main limitation for its routine use in lab-on-chip applications is its difficult integration in user-friendly protocols, combining easy sealing and cell recovery. Indeed, as hydrogels are mainly composed of water, various leakage issues remain to be addressed.
Here, we have developed an innovative integrated agarose-based microsystem of tunable physiological rigidity that enables passive medium renewal. The system mimics the confined state of cells proliferating in a defined volume. The set-up is highly flexible and introduces a key added value by combining a precise control of the imposed confinement, compatibility with time-lapse microscopy, the possibility to follow cells over prolonged periods of time to stimulate cells with chemotherapeutic drugs during confinement, and the easy in situ immunostaining or retrieval of the biological samples for further standard molecular analyses (qPCR, Western blot). Hence, the soft cell confiner described in this manuscript is a powerful tool that could be of major interest to address key biological questions in the growing field of mechanobiology.
Materials and methods
Design of micropillar array
The array and pillars geometry are similar to those described previously,55 namely an hexagonal array with pillars of 440 μm in diameter regularly distributed (distance between each pillar of 1 mm). The field of pillars is surrounded by a solid band of 2 mm to stabilize the structure, and two external notches to enable the proper positioning during molding (schematic representation in Fig. 1A1). The pillars were drawn using Clewin4 (WIEWEB software), and photomasks were then generated by printing the different patterns on transparent sheets (Cirly, France).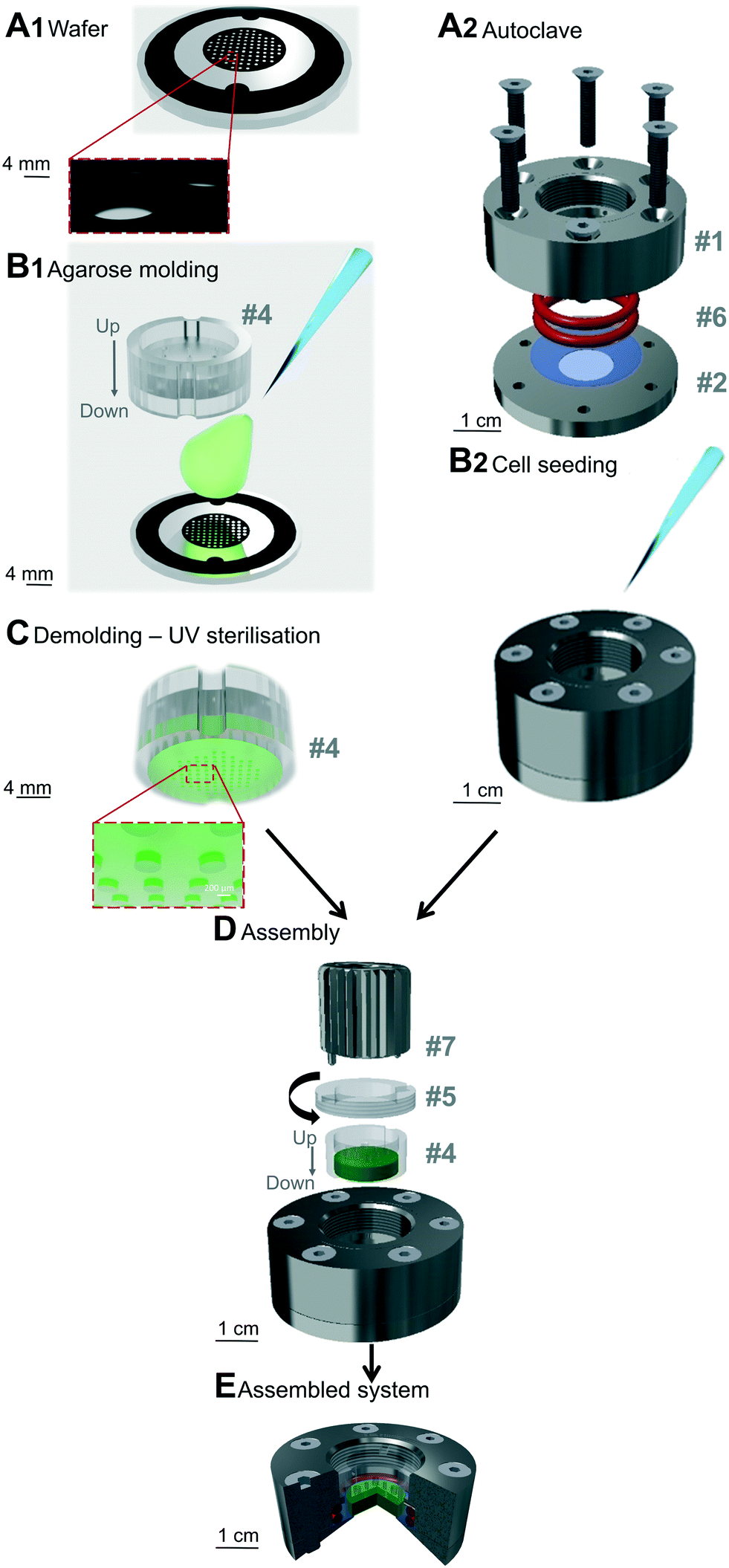 | ||
| Fig. 1 Preparation and assembly of the soft cell confiner. The steps (A1 and B1 and A2 and B2) of the microsystem preparation can be performed in parallel. (A1) Wafer used for agarose molding. (B1) Agarose molding on the wafer in the plastic-holder (#4). (A2) Assembly of the system before autoclaving: two pieces of stainless steel (#1, #2) hold two o-ring seals (#6) and a glass coverslip (blue, #3). (B2) Cell seeding onto the glass coverslip in the system. (C) Schematic representation of the molded gel, which is UV-sterilized before assembly. (D) Assembly of the system: plastic holder containing the molded agarose gel (green) is placed on top of the seeded cells using a clamping washer (#5) tightened with a specific clamping tool (#7). (E) Cross-section of the assembled system (red: ring seals (#6), blue: glass coverslip, green: agarose gel presenting the pillar network). | ||
Wafer fabrication
A standard photolithography process was used to create the different wafers needed for agarose molding. A thin layer of SU8 photoresist resin (SU8 2000 series, Microchem) was spin-coated onto a silicon wafer and heated on a hot plate. According to the manufacturer's application notes, the type of resin, the parameters of the spin coater and the parameters of baking (Table SI 1†) were chosen to produce the desired layer thickness (fixing the height of agarose pillars after agarose molding). The negative photoresist layer was then exposed to UV light through the desired photomask. After development (with propylene glycol monomethyl ether acetate – PGMEA 484431, Sigma), the resin that had not been insulated was removed and the wafer was baked on a hot plate to allow proper adhesion of the resin onto the substrate. Finally, the wafer was washed with isopropanol and distilled water. The height of the molded wafer was controlled using a surface profiler (Veeco Dektak 150, contact stylus profilometry techniques). A schematic representation of the obtained wafer is represented in Fig. 1A1.Different wafers exhibiting different pillar heights were used in this study:
(1) 30 μm high array of pillars, corresponding to an unconfined controlled condition, as 30 μm is larger than the height of all the cell lines used.
(2) 5 μm high array of pillars, corresponding to a confined condition for all the cell lines used (smaller than the height of all the cell lines used).
(3) 9 μm high array of pillars, corresponding to the confined condition used specifically when two cell layers were used.
Different holders and mechanical pieces were designed to provide an easy handling, mounting, medium addition and removal, and dismantling. Details on these various parts are described in Fig. SI 1, 2 and 3.†
Agarose molding
A solution of agarose diluted in distilled water was prepared through a first step of autoclaving at 120 °C for 15 min. To visualize the pillars by confocal microscopy, 250 μL fluorescent microspheres (Ø 0.40 μm, BZ5400, Interchim Fluoprobes) sonicated for 30 s were added to 4 mL of agarose solution. The prepared agarose solution (800 μL) was deposited on the pre-warmed wafer molds (placed on a hot plate at 78 °C for standard agarose or at 50 °C for ultralow agarose, Fig. 1B1). The plastic holder (made of polycarbonate, #4 in Fig. SI 1 and part A SI 2†) was then immediately placed onto the melted agarose and together with the wafer, they were removed from the hot plate and left to set at room temperature (RT) for 10 min for standard agarose or for 72 h at 4 °C for Ultralow agarose. The plastic holder was then gently removed from the wafer. Evenly distributed holes were drilled into the gel using a 20 G puncher and through holes present on the plastic holder. The plastic holder containing the molded agarose gel presented in Fig. 1C was then placed in sterile PBS (Gibco) and sterilized under UV light (20 min each side, Vilber Loumat, 24 W, 365/254 nm). The molded agarose was stored in its plastic holder at 4 °C in PBS until further use. It was replaced by culture medium and incubated at 37 °C at least 3 h before mechanical sealing.Soft cell confiner assembly
Reversible mechanical sealing between a glass coverslip and the molded agarose was ensured using a custom-made stainless steel system (Fig. 1A2 and #1 and #2 in Fig. SI 1 and 3†), adapted to the plastic holder. Prior to mounting, a glass coverslip (Ø 30 mm no. 1, 631-1585, VWR) and the stainless steel parts were cleaned with 70% ethanol, rinsed with distilled water and air-dried. The glass coverslip was then placed between the lower and upper stainless steel parts (Fig. 1A2). Sealing was ensured via two silicone o-ring seals (#6 in Fig. SI 1,† 24.50 × 3.00 mm silicone 70 shores FDA, Fishop) stacked on top of each other in the upper stainless steel part of the system. They were then screwed together before sterilization in an autoclave for 7 min at 134 °C. Screws were tightened gradually and simultaneously in a cross pattern.Tuning agarose stiffness and rheological characterization
To tune the stiffness of agarose, different types and ratios of agarose diluted in distilled water were used:(1) Standard agarose (3810, Cart ROTH) used at a concentration of 4% (w/v), leading to a storage modulus G′ of 144 ± 7 kPa (Fig. SI 4,† referred to as 150 kPa).
(2) Ultralow agarose (A5030, Sigma) used at a concentration of 2% (w/v), leading to a storage modulus G′ of 1.2 ± 0.06 kPa (Fig. SI 4,† referred to as 1 kPa).
Unless otherwise stated, standard agarose (∼150 kPa) was used.
The measurements of agarose viscoelastic properties were performed using oscillatory deformation applied by a stress-controlled rotational rheometer (Anton Paar MCR 301) for standard and ultralow agarose. Agarose samples were molded in 35 × 10 mm Petri dish (353001 Falcon) and left at RT for a few minutes for standard agarose or at 4 °C for 3 days for ultralow agarose. A weighted glass coverslip was placed on top to make a perfect pad. The samples were kept in PBS at 4 °C overnight before measurements. The characterization was then done in a plate–plate (PP) geometry at RT. Upper and lower plate diameters of 42 mm and 64 mm, respectively, were used. Sandpaper was glued to the plates to avoid slippage of the sample upon shearing. The dimension of the gap was in the range of 8.5–9 mm (a little smaller than the size of the agarose sample – 9 mm – to squeeze the gel and avoid slippage). A minimal normal force of 0.15 N is thus imposed. Amplitude sweep tests were performed at a frequency of 1 Hz, with strains ranging from 0.01% to 10% with 3 points per decade. For each strain, the rheometer software measures the viscoelastic moduli G′ and G′′, which have to be corrected as the area of the sample is smaller than the upper plate area. The applied correction is as follows:
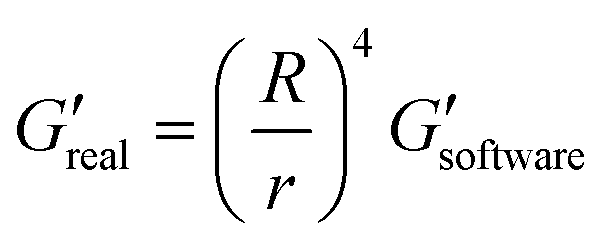
For small deformation (<5% for ultra-low agarose, <0.1% for standard agarose), the loss modulus G′′ is one order of magnitude lower than the storage modulus G′ (see Fig. SI 4†). The agarose gels can hence be considered as a purely elastic material in our case.
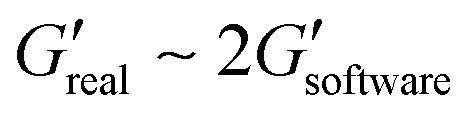
Cell lines and cell culture
The hematopoietic TF1 cell line was obtained and validated as described previously.56,57 Parental TF1-GFP and leukemic BCR-ABL transformed TF1-BA cells were cultured in suspension in RPMI1640 (Gibco) containing 10% fetal calf serum (FCS, Life). ML2 leukemic cells (acute myelomonocytic leukemia) were obtained from F. Mazurier (University of Tours, France) and cultured in RPMI1640 containing 10% FCS. The adherent HS27A cell line, model of mesenchymal stromal cells, was obtained from the ATCC and cultured in RPMI 1640 containing 10% FCS. MCF10A cells were purchased from the ATCC and cultured according to recommendations in phenol red-free Dulbecco's modified Eagle's medium (DMEM)/F-12 nutrient mix supplemented with 5% horse serum (Life), 10 μg mL−1 insulin, 0.5 μg mL−1 hydrocortisone, 100 ng mL−1 cholera toxin, 20 ng mL−1 EGF (Sigma), 1% penicillin/streptomycin (Life Technologies).Cell seeding and soft confiner mounting
For this proof-of-concept study, we chose an intermediate cell density, specific for each cell type (not too low to avoid delay in proliferation, and not too high to avoid reaching confluence too rapidly). The same cell density was used for both controls and confinement conditions to analyze the influence of confinement alone.MCF10A and HS27A cells were seeded in the systems overnight before soft confiner mounting (500 μL of a cell solution at 2 × 105 cells per mL). For TF1 cells, a fibronectin solution (F.895 Sigma-Aldrich, 50 μg mL−1 in NaHCO3) was first used to coat the glass surface (30 min incubation in the system at 37 °C). Excess fibronectin was removed through three washes. Cells were then seeded onto the glass coverslip (5.6 × 105 cells per mL for TF1-GFP, 10.2 × 105 cells per mL for TF1-BA, 500 μL per system), and incubated for 2 h at 37 °C to allow proper adhesion to the substrate. Concerning the co-culture experiment, HS27A cells were seeded at 2.5 × 105 cells per coverslip and then incubated for 24 h. ML2 cells were then added (3 × 105 cells per coverslip) and incubated for 2 h at 37 °C.
After cell adhesion, the seeded cells were gently washed three times to replace the medium with pre-warmed fresh culture medium (500 μL, Fig. 1B2). The plastic holder containing the molded agarose gel was placed in the system (#4 in Fig. 1D) and a clamping washer (#5 in Fig. 1D) was tightened with a specific clamping tool (#7 in Fig. 1D and ESI† movie 1). The gel was then tightly in contact with the glass coverslip supporting the cells. A reservoir of 500 μL of culture medium was added above the plastic holder and incubated for 1 h at 37 °C. The molded agarose was then washed three times (5 min each) with pre-warmed culture medium.
To assess that neither the stainless steel assembly nor the plastic holder with molded agarose affected cell behavior, 2 control conditions were used for each experiment:
(1) Cells on a glass coverslip in the stainless steel assembly, with no molded agarose (1000 μL culture medium). This will hereafter be referred to as control throughout the manuscript.
(2) Cells on a glass coverslip in the stainless steel assembly with agarose molded with an array of pillars of 30 μm in height, larger than the height of the cell population investigated (500 μL of culture medium in the molded agarose + 500 μL above the PC holder). This will hereafter be referred to as 30 μm throughout the manuscript.
Proliferation analysis
Cell proliferation was monitored on images acquired within the microsystems at different time points. The number of cells for each image was determined with the free program ImageJ using a custom-written routine based on the Find maxima tool of the software. The cell density (in cells per cm2) was analyzed for 10 different positions for each tested condition. At least three samples and two independent series of experiments were performed for each cell type.For each cell line, experiments were stopped before reaching confluency (day 3 for HS27-A, day 2 for TF1-BA, day 1 for MCF10A).
Immunostaining within soft cell confiner
After confinement, cells were fixed in situ with 4% paraformaldehyde (PFA, 15714, EM Grade): the cell culture medium was removed and the samples were washed three times with PBS. 4% PFA was then added and incubated for 20 min at RT. After incubation, the samples were washed three times with PBS and incubated with 0.5% Triton X-100 (2156825000, Acros Organics) in PBS for 10 min at RT for permeabilization followed by three consecutive washes with 0.1% Triton X-100 every 5 min. After permeabilization, samples were blocked with 3% BSA (A2163, Sigma) 0.1% Triton X-100 in PBS for 20 min at RT to inhibit non-specific binding of antibodies. Cells were initially incubated with Alexa 546 Phalloidin (A22282, Thermofisher, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 in 0.1% T-X 100 in PBS) for 20 min at RT and washed three times with PBS. Samples were finally incubated with NucGreen™ (Thermofisher, R37109, 2 drops per mL in PBS) for 15 min and washed three times with PBS.
:50 in 0.1% T-X 100 in PBS) for 20 min at RT and washed three times with PBS. Samples were finally incubated with NucGreen™ (Thermofisher, R37109, 2 drops per mL in PBS) for 15 min and washed three times with PBS.
Cell viability
Fresh culture medium was added in each system every 2 days. To assess cell viability, agarose gel was dismounted and cell viability was monitored with calcein (Thermofisher) and propidium iodide (PI, Sigma) labeling. Calcein labels the cytoplasm of viable cells in green, whereas PI labels nuclei of dead cells in red. A maximum volume of culture medium was removed and cells were then washed once with pre-warmed PBS. Calcein (1 μM) and PI (20 μg mL−1) diluted in pre-warmed sterile PBS were then incubated for 20 min at 37 °C before epifluorescence microscope analysis.Control live and dead cells were tested in parallel. For live cells, cells in classical 2D cultures were incubated for 24 h at 37 °C. For dead cells, 70% ethanol was added 30 min prior to staining (Fig. SI 9†).
Staining proliferative cells
Proliferative cell labeling was performed using the Click-iT® EdU Alexa Fluor® Imaging Kit (Molecular Probes). EdU (5-ethynyl-2′-deoxyuridine) was added to the culture medium on the mounted soft cell confiner to reach a final concentration of 10 μM. EdU is a thymidine analog that is incorporated into newly synthesized DNA. After 24 h incubation, the confined cells were rinsed with PBS and fixed. EdU detection, based on a specific click reaction between EdU and the Alexa FLuor® 594 dye, was performed following the manufacturer's instructions. NucGreen™ was finally added to the samples to label all of the nuclei as depicted in the immunostaining section.Microscopy
Cells were observed with an inverted microscope (Leica DM IRB or Leica DMI8) using phase-contrast imaging. A 10× objective was used to follow cell proliferation and a 5× objective to observe their viability with calcein/PI staining. Images were taken every 24 h. In parallel, phase-contrast time-lapse imaging was performed for 24 h in a controlled (CO2, temperature and humidity) environment. A motorized x–y stage enabled the concomitant recording of up to 10 regions for each system every one or two hours (ESI† movie 2 for TF1-BA and movie 3 for MCF10A).Fixed and co-cultured cells were visualized using a Leica SP5 confocal microscope or a Zeiss LSM 880 confocal microscope with a 20× dry objective (NA 0.65). Z-Stacks of live cells in the soft confiner were also acquired at 20× magnification (dz = 0.4 μm for each stack).
Image analysis and quantification
Area of TF1-GFP live cells, area and circularity of TF1-BA fixed cells were assessed with a homemade Matlab program. Images were filtered using a Wiener adaptive filter and cells were then separated from the background using threshold detection and converted to binary images. Sequential steps of morphological reconstruction were performed and cells were individually detected based on different parameters (distance between centroid, area, eccentricity). Four different areas within the soft confiner were analyzed for each condition.Nucleus circularity was defined by:
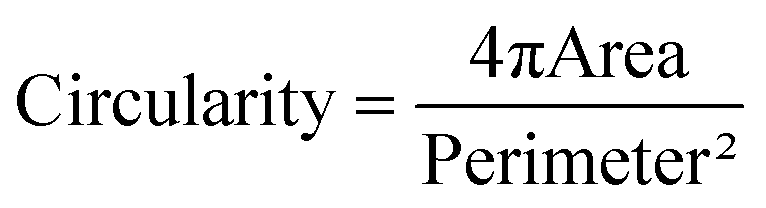
Western blot
Western blot analysis was performed on HS27A cells confined during 3 days, proteins were then extracted using RIPA buffer. Per lane, 15 μg of proteins were loaded onto gels prior to conducting SDS-PAGE and transferring onto polyvinylidene difluoride membranes (Bio-Rad). Membranes were then incubated with monoclonal antibodies against Cyclin B1 (PC133, Calbiochem) and GAPDH (#8884, Cell Signalling Technology). Specific binding of antibodies was detected using appropriate secondary antibodies conjugated to horseradish peroxidase, and visualized with a Clarity Western ECL Substrate (Bio-rad), on ChemiDoc Gel Imaging system (Bio-rad). Densitometric analyses of immunoblots were performed using ImageJ.Flow cytometry
Flow cytometry cell sorting experiments were carried out using HS27A-Turquoise and ML2-Cherry. Briefly, the lentivirus expressing mTurquoise2-Tubulin was constructed by cloning the mTurquoise2-Tubulin sequence from pmTurquoise2-Tubulin (a gift from Gadella Dorus, Addgene plasmid #36202 (ref. 58)) into the CSII-EF-MCS vector. Lentiviruses were produced in 293 T cells by transfecting lentiviruses with the helper plasmids pMD2.G and psPAX2 (a gift from D. Trono, Addgene plasmids #12259 and #12260), following Addgene's instructions. Recipient cells were infected at a low multiplicity of infection (moi < 1) and finally sorted on a BD FACSAria III SORP cell sorter.After 3 days of confinement, the HS27A-Turquoise and ML2-Cherry cells were analyzed using the BD LSRFortessa cell analyzer.
Statistical analysis
Data were expressed as mean ± standard deviation. The statistical significance of differences between conditions was analyzed using Matlab. The Mann–Whitney test was used for Fig. 3A and C, 4A and 7A and B and unpaired t-test for Fig. 2D, 4D and E, 5D and E and 6D. Differences with a p-value under 0.05 were considered statistically significant. Significance is indicated by asterisks in figures (* p < 0.05; ** p < 0.01, *** p < 0.005).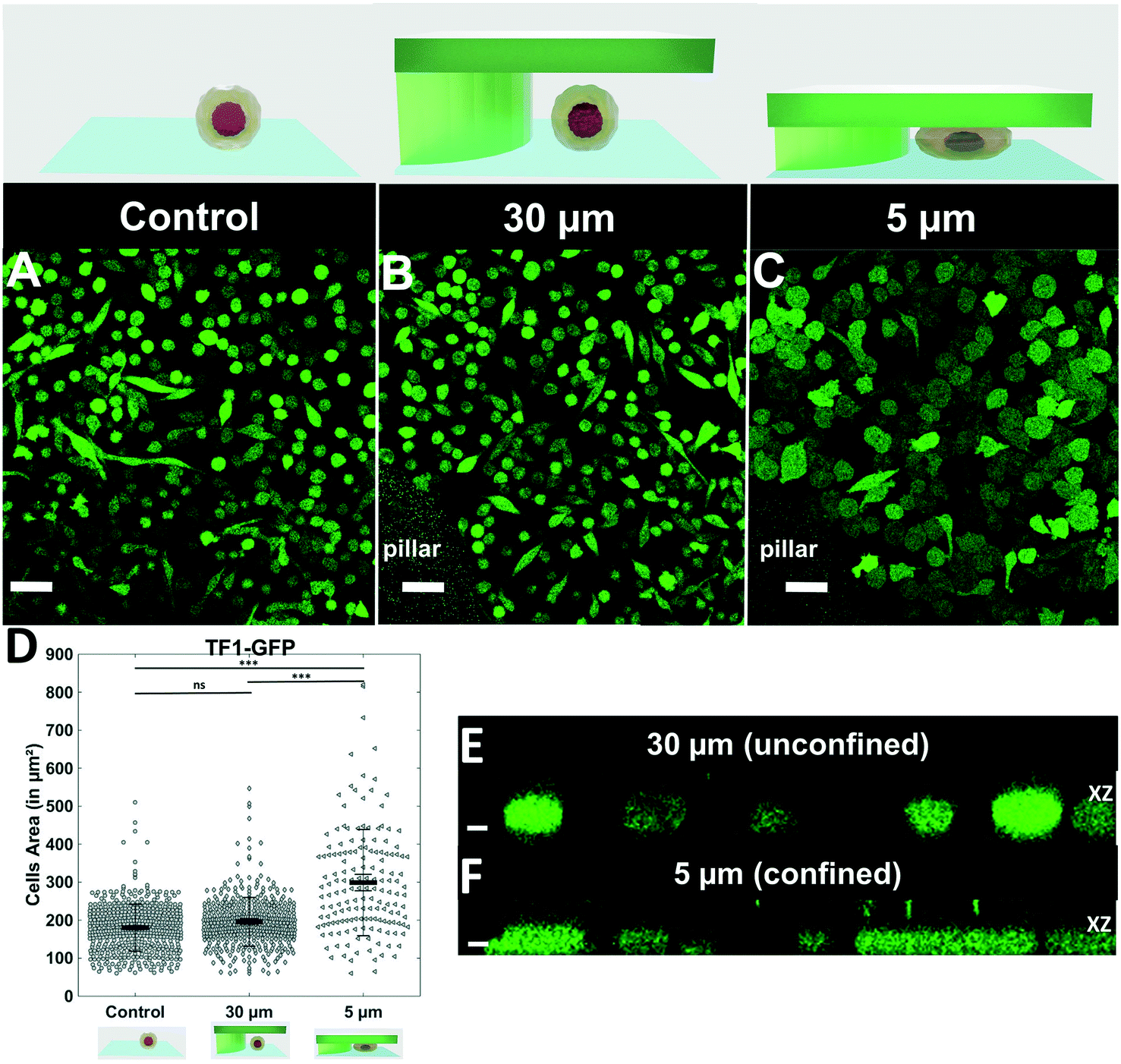 | ||
| Fig. 2 Quantification of cell morphology under confinement. (A–C): Morphology of immature TF1-GFP hematopoietic cells for control (A) and for 30 μm and 5 μm (B and C, respectively). Scale bar = 20 μm. (D) Quantification of projected area by automatic image analysis. Projected area was similar for control and 30 μm, while it significantly increased for 5 μm confinement (unpaired t-test n = 160 at least for each condition). (E and F): z-Section of unconfined (E) and confined (F) cells. Scale bar = 10 μm. | ||
Results
The implementation of a reliable and reproducible cell confiner using a hydrogel-based microsystem is not trivial. The hydrogel has to be properly sealed on the glass coverslip, without compressing the gel or inducing leakage. We achieved these objectives in an ultimate user-friendly design (Fig. 1). The soft cell confiner was designed to enable the concomitant production of agarose molds (Fig. 1A1 and B1) and the mounting of coverslips in an autoclavable stainless steel chamber (Fig. 1A2 and B2). A plastic holder was specially designed to improve agarose molding and handling (Fig. 1C and SI 2†). After cell seeding onto the coverslip, the plastic holder and a dedicated clamping washer and clamping tool enabled the reproducible and rapid mechanical sealing of the molded agarose onto the seeded cells (Fig. 1D and E and ESI† movie 1). Owing to this system, removal of the molded agarose and retrieval of the confined cell population at the end of a long-term experiment was relatively rapid and simple.Evenly distributed holes in the plastic holder enabled the renewal of culture medium through the upper part of the plastic holder without disturbing confinement conditions or cells. The efficiency of medium renewal was checked by analyzing the evolution of fluorescent intensities upon medium exchange (Fig. SI 5†). A fluorescent medium placed in the upper part reaches the lower part (where the cells are seeded) by pure diffusion over a characteristic time-course of 7 h 30. Drug availability within the system was also assessed with the in situ addition of the tyrosine kinase inhibitor imatinib (Fig. SI 6,† the drug is active at the same concentration range with or without confinement).
We first assessed that cells were properly and homogeneously confined using the non-adherent hematopoietic cell line TF1-GFP (Fig. 2). While cells cultured for 1 day displayed similar size and morphology to control conditions and 30 μm pillars (Fig. 2A and B), the area of cells under 5 μm confinement was much larger (Fig. 2C). The projected area increased from 196 ± 3 μm2 to 300 ± 11 μm2 under 5 μm confinement (Fig. 2D), while the height was restrained by the pillars (Fig. 2Evs. F).
The reproducibility and stability of the soft confiner was also validated by measuring the difference in height from one microsystem to the other, and for several days at 37 °C with culture medium (Fig. SI 7A and B† respectively).
In order to fully validate this confining device, we investigated the confinement of the stromal cell line HS27A. After 3 days, while HS27A proliferation inside a PDMS-based confiner similar to55 was impacted even for the 30 μm height control condition (Fig. SI 8†), the same condition in the hydrogel-based microsystem had no impact on cell proliferation (Fig. 3A control vs. 30 μm, no significant difference). The decrease in cell proliferation induced upon 5 μm confinement (Fig. 3A) can hence be truly interpreted as the mechanical cell response to the imposed 5 μm confinement applied for 3 days. The confined cells were then harvested after 3 days and further processed for protein analysis by Western blot. The level of cyclin B1 was similar for control and 30 μm pillars (Fig. 3B), demonstrating the lack of impact of the confining chamber components on proliferation. Conversely, we observed a strong decrease in cyclin B1 in the 5 μm confinement condition (Fig. 3B 30 μm vs. 5 μm), confirming a decrease in cell proliferation for this stromal cell line under confinement.
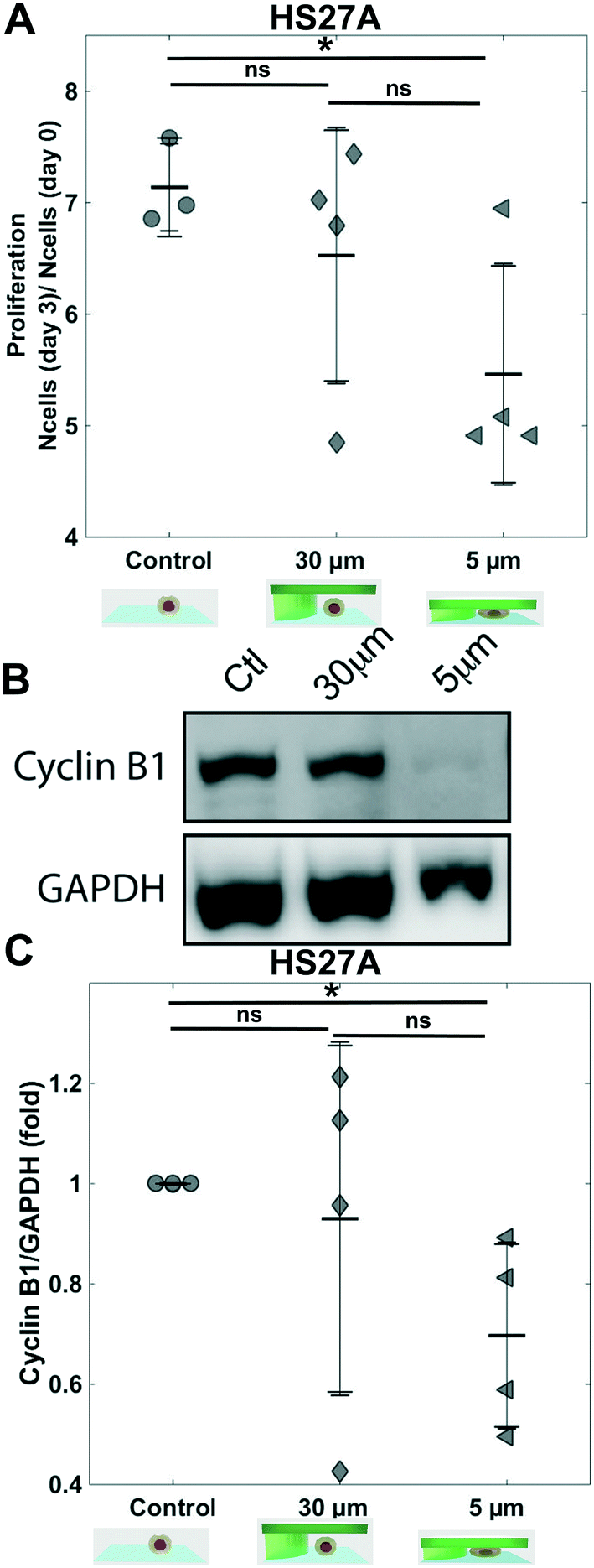 | ||
| Fig. 3 Proliferation of HS27A cells. (A) Bar graph showing the proliferation ratio of stromal cells HS27A over 3 days for control and for 30 μm and 5 μm. Density was analyzed for 10 positions for each sample, n = 4 (Mann–Whitney test n.s. not significant). (B) Western blots showing CyclinB1 levels from HS27A cells for the three conditions. (C) Bar graph showing CyclinB1 level (GAPDH used as internal control) n = 4 for the three conditions. | ||
To demonstrate the robustness of this soft cell confiner, it was then challenged by culturing the hematopoietic leukemic cell line, TF1-BA. Indeed, as these cells are poorly adhesive, even in the presence of fibronectin coating, analysis on this confinement system is experimentally challenging. No significant difference in cell proliferation was induced by the microsystem even after 2 days of confinement (Fig. 4A control vs. 30 μm height condition), reinforcing previous results obtained with the HS27A cell line that no medium conditioning is affecting cell proliferation within the hydrogel based confiner. In addition, despite their transformation, these leukemic cells appeared to be sensitive to mechanical stress as we measured a significant decrease in cell proliferation upon 2 days of confinement when comparing 30 μm-high and 5 μm-high conditions (Fig. 4A, 1.9 ± 0.2 fold increase in cell number for 5 μm compared to 3.40 ± 0.4 fold increase for 30 μm).
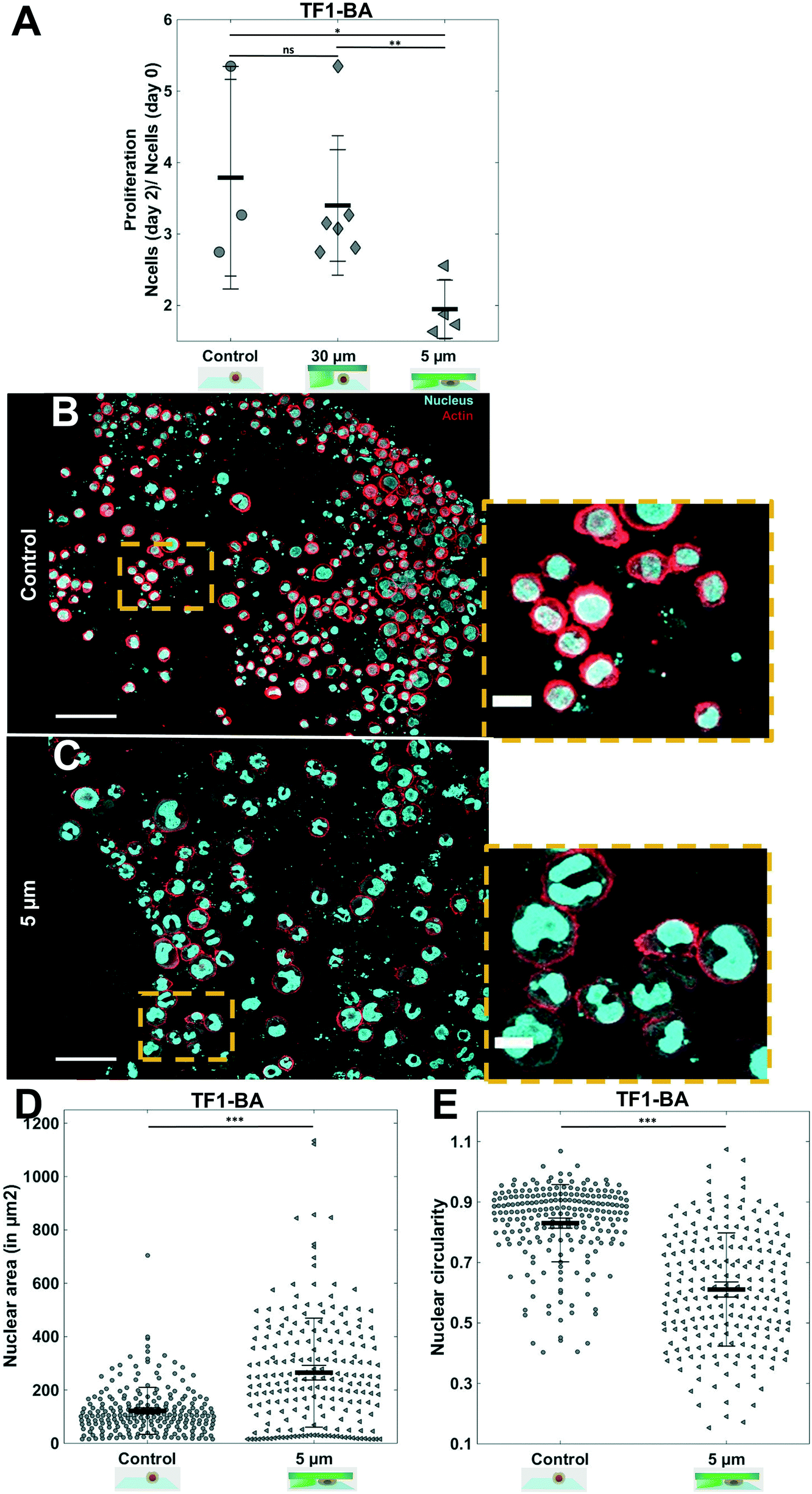 | ||
| Fig. 4 Proliferation of TF1-BA and in situ immunostaining. (A) Bar graph showing the ratio of proliferation of TF1-BA immature hematopoietic cells over 2 days for control, and under molded agarose with pillars of 30 and 5 μm. We observe a significant decrease in proliferation under confinement compared to controls (at least 2 independent experiments, performed at least in triplicate for each condition and density were analyzed on 10 positions for each sample). (B and C) In situ immunostaining of TF1-BA cells after 1 day in the soft cell confiner for control (B) or under 5 μm confinement (C). Left scale bar = 50 μm, right scale bar = 20 μm. Actin (phalloidin) in red and nuclei in cyan. (D and E) Quantification of nuclear circularity and nuclear area of TF1-BA cells for control with no agarose and under confinement (5 μm) after 1 day in the soft cell confiner (confined cells present a larger area and deformation of their nucleus compared to control). At least 213 nuclei were analyzed per condition. | ||
Not only is our soft cell confiner compatible with live imaging and medium renewal, but it also enables in situ immunostaining under confinement. We stained both nuclei and actin under confinement (Fig. 4B and C). The nuclear projected area appeared to be larger upon confinement (Fig. 4D), as indicated by an increase in the mean projected nucleus area from 122 ± 6 μm2 for control, to 265 ± 14 μm2 for 5 μm confined cells. In addition, we observed that many nuclei were highly deformed, exhibiting a non-circular or polylobed shape (Fig. 4E, decrease of mean nuclei circularity from 0.83 ± 0.01 to 0.61 ± 0.01).
The hydrogel stiffness can be easily tuned by several orders of magnitude by changing agarose type and concentration. We investigated the effect of a 1 kPa agarose (Fig. SI 4†) as a confining hydrogel roof on TF1-BA cells, to analyze whether the same nuclear deformation was observed. After 3 days under soft confinement, for the same confinement condition (5 μm-high), immunostaining for nuclei revealed that nuclear morphology is affected differently depending on the stiffness of the hydrogel (Fig. 5). With 150 kPa agarose, the mean projected nuclear area increase from 154 ± 51 μm2 for the unconfined condition, to 195 ± 100 μm2 under 5 μm confinement (Fig. 5D). Conversely, with 1 kPa agarose, the nuclear area decreased to 105 ± 34 μm2 under the same 5 μm confinement (Fig. 5D). In addition, while circularity (Fig. 5E) dropped to 0.4 ± 0.3 under 5 μm confinement using 150 kPa agarose (vs. 0.8 ± 0.3 for the 30 μm control condition), the decrease was slighter with 5 μm confinement using 1 kPa agarose (0.7 ± 0.1). In conclusion, the nuclei are much smaller and rounder under the soft-agarose confinement (1 kPa), compare to the stiffest confinement (150 kPa).
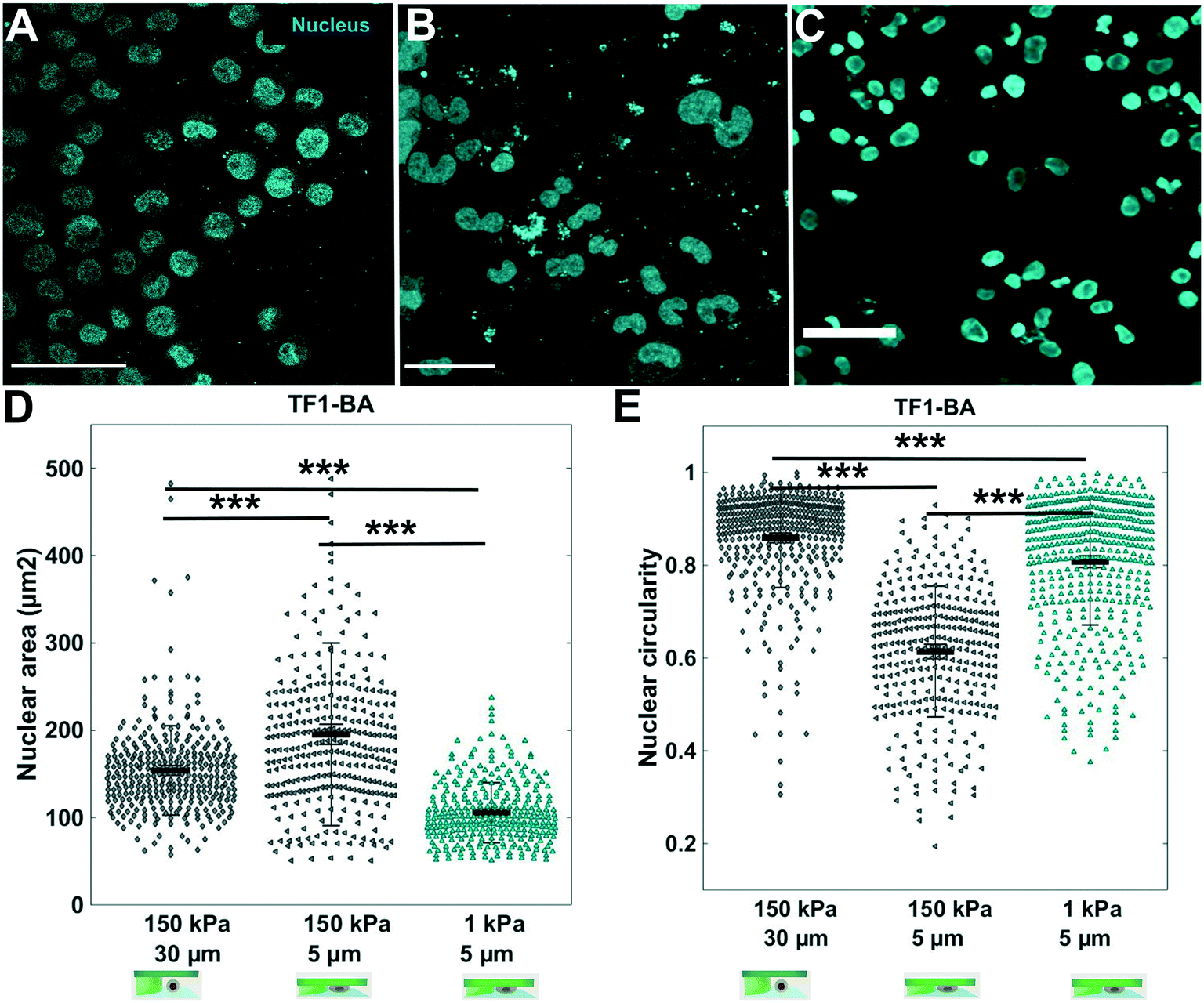 | ||
| Fig. 5 Influence of agarose stiffness on nuclear deformability. In situ immunostaining of TF1-BA cell nucleus (cyan) after 3 days in the soft cell confiner molded in (A) standard agarose (storage modulus of 150 kPa) with 30 μm pillars (no confinement); (B) standard agarose with 5 μm pillars (confinement) and (C) ultra-low agarose (storage modulus of 1 kPa) with 5 μm pillars. Scale bar = 100 μm. (D and E) Corresponding quantification of nuclear circularity and nuclear area of TF1-BA cells for the three conditions. At least 150 nuclei were analyzed per condition. | ||
Our soft cell confiner is also compatible with more complex cell population analysis, mimicking multiparametric and heterogeneous cellular microenvironments, such as bone marrow in which hematopoietic and stromal cells interact. To illustrate such complex cell interactions, adherent stromal HS27A cells were co-cultured with the suspended hematopoietic ML2 cells and analyzed in the soft cell confiner system using a confined height of 9 μm to take into account the two layers of cells (Fig. 6A). In this setting, we used multicolor labeling of the different cell lines to distinguish them in situ and to measure their area upon confinement (Fig. 6A). This confirmed that despite the increased complexity and heterogeneity in cell population, the system still provided the measurement of cell area by confocal microscopy. Here, we observed an increase in the mean projected hematopoietic cell area upon confinement (Fig. 6B and C) from 216 ± 133 μm2 for 30 μm to 329 ± 155 μm2 for 9 μm (Fig. 6D). In addition, our system allowed us to recover viable cells after 3 days of confinement to perform various functional assays, such as cell sorting by flow cytometry (Fig. 6E).
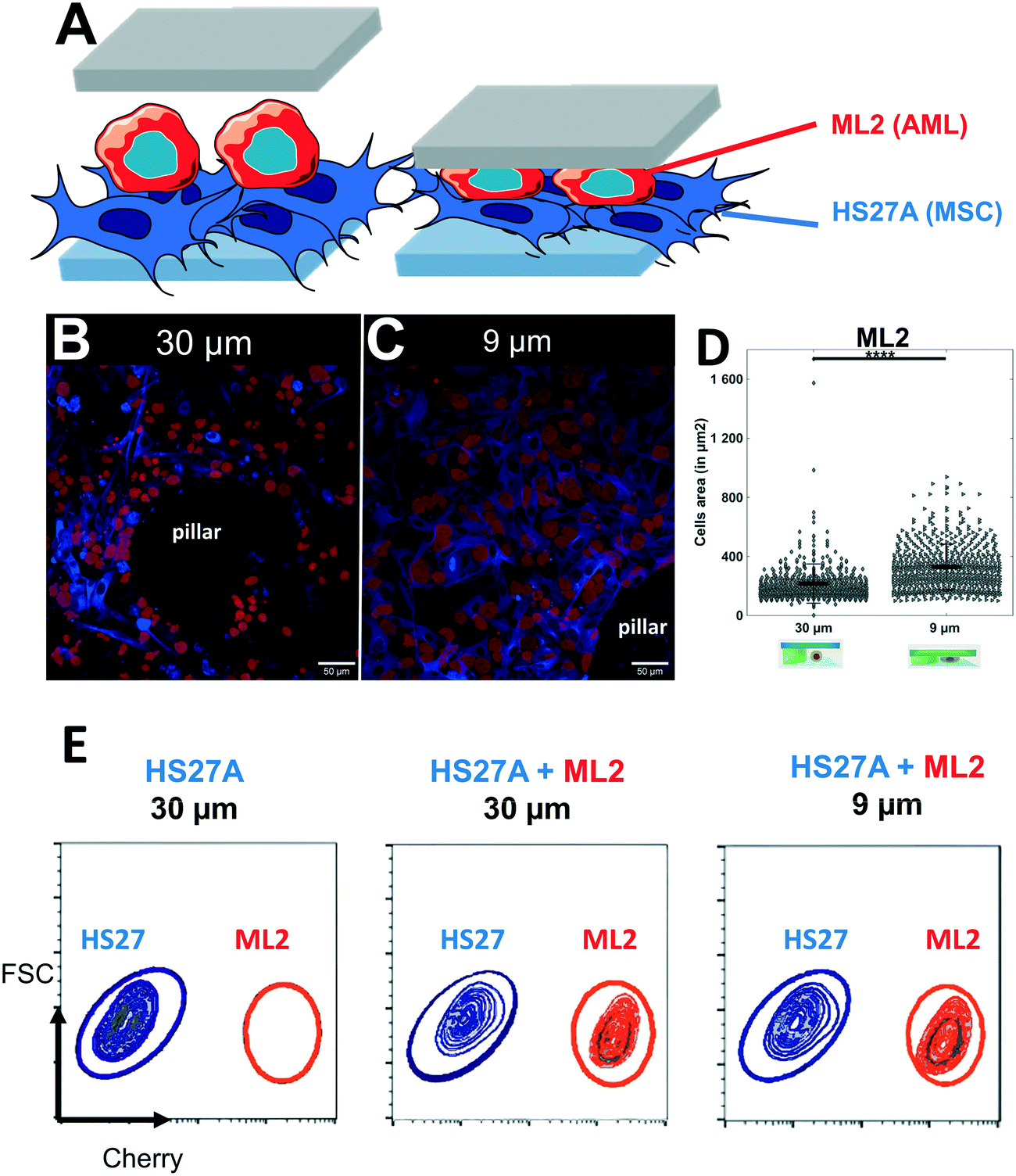 | ||
| Fig. 6 Set-up mimicking the complexity of the tumor microenvironment. (A) Schematic representation of the two layers of cells inside the soft cell confiner. (B and C) Representative confocal images of leukemic cells (ML2, red) seeded on top of stroma cells (HS27A, blue) and inserted into the confiner with a height greater than both layers (h = 30 μm, unconfined, B) or smaller (h = 9 μm, confined, C) for 3 days. Scale bar = 50 μm. (D) Bar graph showing ML2 cell area quantification from unconfined (A, the average projected area of 216 ± 133 μm2) or confined (B, the average projected area of 329 ± 155 μm2). (E) Representative FACS plots following 3 days confinement in co-culture analyzed for cell content in HS27A-Turquoise and ML2-Cherry. | ||
Finally, the compatibility of the soft cell confiner with long-term experiments was assessed using an epithelial breast cell line, the adherent MCF10A cells. This immature mammary stem cell line displays contact inhibition of proliferation and can thus be cultured in high-density conditions for several days. Once again, we verified that the hydrogel-based microsystem itself had no impact on cell proliferation (Fig. 7A, no significant difference in proliferation ratio for control and 30 μm-high after 1 day – proliferation ratio of 1.8 ± 0.1 and 1.9 ± 0.1, respectively). Consistently with our previous findings, a significant decrease in cell proliferation was observed under 5 μm confinement (Fig. 7A), with the proliferation ratio dropping below a significantly lower value than the two controls (proliferation ratio of 1.5 ± 0.1 after 1 day), and a large decrease in EdU+ cells under 5 μm confinement (Fig. 7B a ratio of 0.8 ± 0.1 vs. 0.4 ± 0.1 for 30 μm and 5 μm respectively). Finally, we confirmed that most cells, except the ones under the pillars, were alive (Fig. 7C–E, live/dead staining after 1 day). Of note, it was possible to culture MCF10A cells under confined conditions for up to 8 days without induction of any significant effect on cell viability, as assessed by live and dead cell staining (Fig. 7F–H and SI 9† for control of staining).
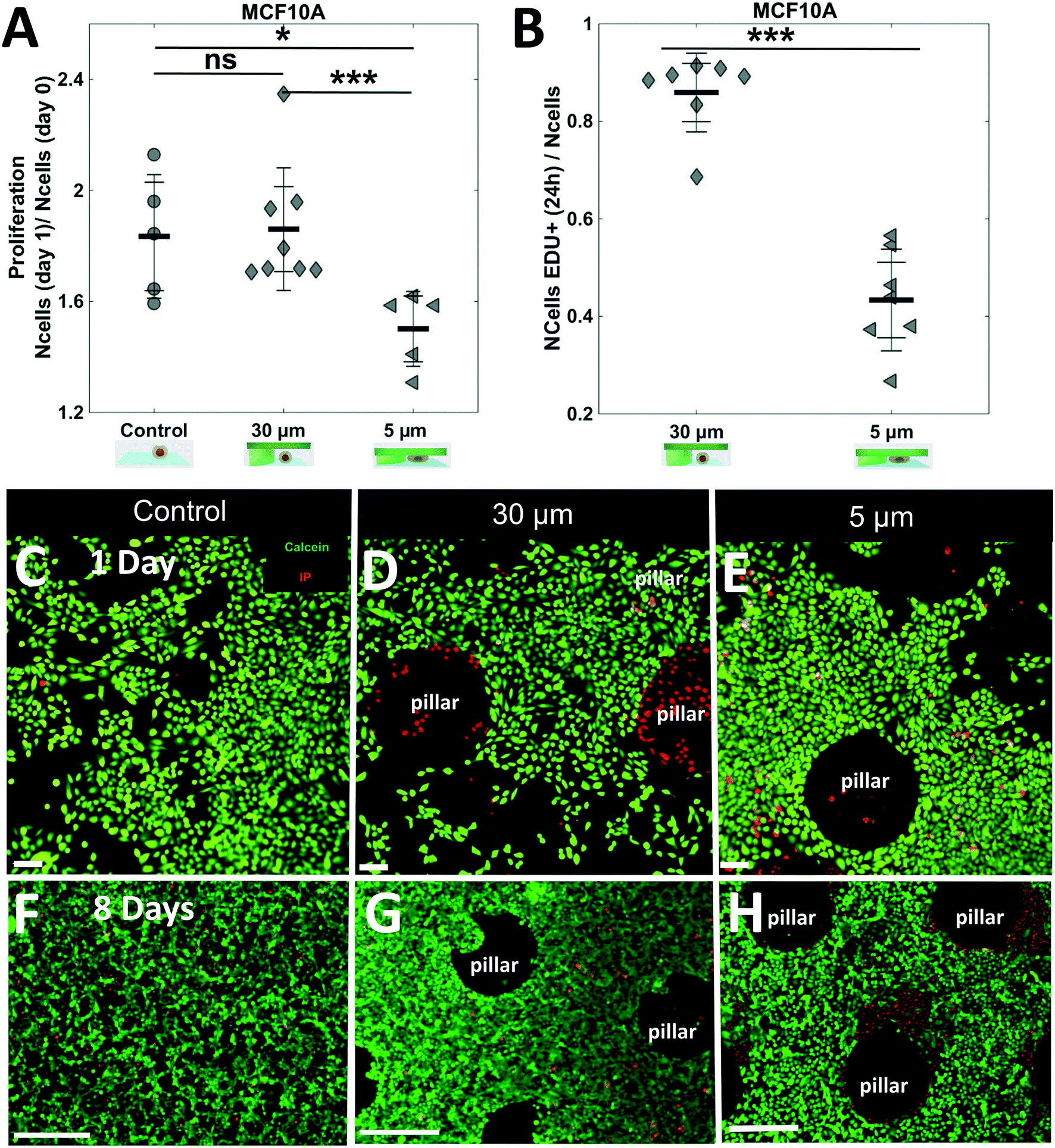 | ||
| Fig. 7 Long-term analysis of epithelial cell lines with contact inhibition under confinement. (A) Bar graph showing the proliferation ratio of epithelial MCF10A cells after 1 day in the soft cell confiner with various confinement conditions. At 5 μm confinement, a significant decrease in proliferation under confinement was observed found (at least 2 independent experiments in duplicate for each condition. For each system, the number of cells was assessed in 10 different positions). (B) Bar graph showing the ratio of EdU+ cells after 1 day in the soft cell confiner under 30 μm or 5 μm confinement. (C–H) Live and dead staining of a confluent monolayer of MCF10A after 1 day in the soft-confiner (C–E) or 8 days (F–H) control with no confinement (C–F) 30 μm pillars (D–G) and 5 μm pillars (E–H). Green: calcein, red: propidium iodide. Scale bar = 400 μm. | ||
Discussion
The agarose-based microsystem presented herein defies previous cell confining microsystems.First, it enables analysis on cell response under long-term confinement (several days), which was so far limited to several hours at most for closed PDMS-based microsystems, due likely to limited access to nutrients. This was the case in one of our early studies on 3 day confinements using a PDMS-based confiner similar to the one used in ref. 55. This system fell short of our expectations with sensitive cell lines such as the HS27A cell line, which displayed a decrease in proliferation even for control experiments (control versus 30 μm Fig. SI 8†), precluding its use to unambiguously decipher the role of confinement in various cell-signaling pathways. This is not the case with agarose, where proliferation was similar between the 2 controls for all cell lines investigated. In our soft cell confiner, the porous nature of the confining walls enabled medium renewal. Cells in a confined state can be cultured for several days without impairing their survival.
Second, our soft-confiner is fully compatible with time-lapse, immunostaining and classical molecular biology analyses. At the end of the experiments, the porous nature of the confining wall enabled us to perform all immunostaining steps in situ. In addition, the compatibility of our system with high-resolution microscopy enabled us to conduct in situ confocal microscopy analyses. Alternatively, the entire cell population could easily be collected and processed using standard molecular biology protocols (qPCR, Western blot) or functional assays.
Third, the confining matrix rigidity could be adjusted to closely match physiological conditions (rigidity of [1–150 kPa]9,59,60) by tuning the concentration and the type of agarose used. In this study, by decreasing the stiffness down to 1 kPa for the confining matrix, nuclei exhibited a more circular shape, closer to the unconfined situation, as opposed to the large nuclear deformation observed for the classical agarose (150 kPa range).
The limit of our system is that the stiffness of the coverslip is several orders of magnitude larger that the soft confiner (GPa vs. kPa). To ensure a similar stiffness on top and bottom walls, the coverslip can be coated with an additional soft agarose layer. The soft cell confiner system can hence be used to decipher the influence not only of confinement but also of matrix stiffness (in combination or separately). Such flexibility could be of primary importance to unravel the role of these two important biochemical cues in various biological contexts.
It has also been recognized that a 3D cell anchorage could lead to a completely different cell response compared to the current 2D one.61–63 In the current set-up, the pure agarose confining roof provides no adhesive groups for cell attachment. Nevertheless, it is possible to analyze the effects of adhesion by interpenetrating the network of agarose with collagen,64 PEGDA with covalently immobilized RGD peptides65 or silk.66 This could offer the possibility to analyze the role of various extracellular matrix proteins on cell response to mechanical confinement.
Importantly, we demonstrated that our confiner can be used for both adherent and non-adherent cells. It can also be used to disentangle the interaction between different cell types, as shown in this manuscript with two layers of cells (stromal and leukemic cells). Hence, it could be a valuable tool to analyze the dynamic interplay between heterogeneous cell populations in response to mechanical stress.
Several other alternative set-ups have been developed to apply a defined stress on an entire cell population for prolonged periods of time, by embedding them in agarose12 or in extracellular matrix67 or using a transmembrane-based pressure device.68 The drawback of such devices is that they are not compatible with high-resolution microscopy due to the thickness of the gels or the transwell geometry. Analyses at endpoints are solely possible, limiting all real-time analyses on cellular adaptation to mechanical stress.
Here, we have developed a system that combines the advantage of PDMS-based microsystems with a transparent chip geometry on glass coverslips, enabling a real-time dynamic analysis, as well as a long-term monitoring of cells. Our soft cell confiner device integrates different biophysical and biological approaches that were barely achieved with existing devices.
Because our device does not impact proliferation in the absence of confinement, we can truly decipher the impact of confinement on cell proliferation. In the current study, a decrease in proliferation under spatial confinement was observed for all cell lines tested for over one to three days in the confiner. Similar results reported a higher frequency of quiescent cells upon confinement within a stiff 3D matrix.69 These results also corroborate those obtained using PDMS set-ups70,71 and atomic force microscopy (AFM),72 where it was reported that confinement delays mitotic progression.
We further analyzed nuclear shape and actin at a defined time-point via immunostaining. Interestingly, for immature TF1-BA hematopoietic cells, after 1 day under 5 μm confinement, nuclear shape and projected area were highly modified. The interplay between actomyosin contractility and nuclear deformation in the regulation of nuclear transport of signaling molecules has been highlighted in the past few years.73,74 Dynamic studies are now needed to unravel how nuclear deformation and mechanosensitivity modulate spatiotemporally gene expression and cell fate. As our soft cell confiner is compatible with time-lapse microscopy, it could also be used to follow nuclear deformation dynamically for several days, using dedicated fluorescent constructs such as histone or lamins A/C. We plan to use our soft cell confiner system to investigate these important issues in the near future.
In this new set-up, we have made the arbitrary choice to confine cells in the z-direction only. However, it is also possible to confine cells in x, y and z directions using a dedicated design. Indeed, compared to other hydrogels, agarose gels do not swell, and it is thus easily molded in various shapes and aspect ratios. It will complement current PDMS-based systems that use arrays of closed channels21–23 or pillars26,75 to investigate how cells migrate in 2D and 3D environments. We provide two important features compared to these existing systems. First, for long-term experiments, the current PDMS-based microsystems use an open-configuration,26,75 which does not provide an isotropic confinement situation (no roof confining in z-direction). Second, the pillars and channels of these devices are in a rigidity order of magnitude greater than the one encountered in vivo (MPa for PDMS vs. kPa in vivo). Our soft confiner will provide the opportunity to investigate such migration in a true 3D confined situation, with the possibility of tuning the stiffness of the walls to mimic a physiological range.
Cell response to the application of a local stress has also been extensively investigated using AFM set-ups.72,76,77 While this single-cell approach technique can provide interesting insight into cell response to the application of a local force, it is relatively low-throughput and requires additional expertise to be properly interpreted. Our soft cell confiner is a complementary approach, as we do not apply a defined force but we impose a defined deformation (set by the height of the pillars).
Of note, fluorescent beads incorporated into the agarose could be used to measure cell-generated forces in response to the imposed confinement by 3D traction force microscopy (TFM78–82). If the gel is soft enough to be deformed by the cells, a dynamic quantification of the compression force sensed by the cell could be retrieved. Such quantification is out of the scope of this paper but work is ongoing within our team to determine whether the cells reorganize in response to confinement to decrease the imposed mechanical stress, and if so, following which time scale (hours/days).
Conclusion
The hydrogel-based system detailed in this manuscript paves the way for new approaches and concepts in cell biology.We anticipate that this device could be a valuable tool for the fundamental understanding of the effect of cell confinement on various hallmarks of cancer progression and resistance, and in particular to decipher the role of nuclear mechanosensitivity. Links between cell resistance to treatment and mechanical stimuli have been highlighted recently.69,83,84 Such mechanical cues could hence lead to new therapeutic approaches85 that could be tested using such systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institut Convergence PLASCAN, the Ligue Contre le Cancer foundation, the “Institut Universitaire de France” (IUF), the “Fondation pour la Recherche Médicale” (FRM) for part of A. Prunet's salary and the “Fondation de France” and Alte SMP for part of S. Lefort's salary. We thank Y. Chaix, R. Zagala, M. Jacquemin, Q. Cassar, A. Damn for their help in the early development of this project, as well as L. Barral for LAM/stroma pictures and Q. Ducerf for his help with MCF10A cells. We also thank B. Manship for English editing.References
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 Search PubMed.
- E. K. Paluch, C. M. Nelson, N. Biais, B. Fabry, J. Moeller, B. L. Pruitt, C. Wollnik, G. Kudryasheva, F. Rehfeldt and W. Federle, BMC Biol., 2015, 13, 47 Search PubMed.
- E. Farge, Curr. Biol., 2003, 13, 1365–1377 Search PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 Search PubMed.
- J. Du, Y. Fan, Z. Guo, Y. Wang, X. Zheng, C. Huang, B. Liang, L. Gao, Y. Cao, Y. Chen, X. Zhang, L. Li, L. Xu, C. Wu, D. A. Weitz and X. Feng, Cell Syst., 2019, 9, 214–220.e5 Search PubMed.
- M. Segel, B. Neumann, M. F. E. Hill, I. P. Weber, C. Viscomi, C. Zhao, A. Young, C. C. Agley, A. J. Thompson, G. A. Gonzalez, A. Sharma, S. Holmqvist, D. H. Rowitch, K. Franze, R. J. M. Franklin and K. J. Chalut, Nature, 2019, 573, 130–134 Search PubMed.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 Search PubMed.
- D. Wirtz, K. Konstantopoulos and P. C. P. P. C. Searson, Nat. Rev. Cancer, 2011, 11, 522 Search PubMed.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg, A. Gefen, C. A. Reinhart-King, S. S. Margulies, M. Dembo, D. Boettiger, D. A. Hammer and V. M. Weaver, Cancer Cell, 2005, 8, 241–254 Search PubMed.
- C. P. Ng, B. Hinz and M. A. Swartz, J. Cell Sci., 2005, 118, 4731–4739 Search PubMed.
- T. Stylianopoulos, J. D. Martin, M. Snuderl, F. Mpekris, S. R. Jain and R. K. Jain, Cancer Res., 2013, 73, 3833–3841 Search PubMed.
- G. Cheng, J. Tse, R. K. Jain and L. L. Munn, PLoS One, 2009, 4, e4632 Search PubMed.
- K. Wolf, M. Te Lindert, M. Krause, S. Alexander, J. Te Riet, A. L. Willis, R. M. Hoffman, C. G. Figdor, S. J. Weiss and P. Friedl, J. Cell Biol., 2013, 201, 1069–1084 Search PubMed.
- M. E. Fernández-Sánchez, S. Barbier, J. Whitehead, G. Béalle, A. Michel, H. Latorre-Ossa, C. Rey, L. Fouassier, A. Claperon, L. Brullé, E. Girard, N. Servant, T. Rio-Frio, H. Marie, S. Lesieur, C. Housset, J. L. Gennisson, M. Tanter, C. Ménager, S. Fre, S. Robine and E. Farge, Nature, 2015, 523, 92–95 Search PubMed.
- L. Chin, Y. Xia, D. E. Discher and P. A. Janmey, Curr. Opin. Chem. Eng., 2016, 11, 77–84 Search PubMed.
- S. Pradhan and J. H. Slater, Biomaterials, 2019, 215, 119177 Search PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 Search PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 Search PubMed.
- W. J. Hadden, J. L. Young, A. W. Holle, M. L. McFetridge, D. Y. Kim, P. Wijesinghe, H. Taylor-Weiner, J. H. Wen, A. R. Lee, K. Bieback, B. N. Vo, D. D. Sampson, B. F. Kennedy, J. P. Spatz, A. J. Engler and Y. S. Cho, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5647–5652 Search PubMed.
- M. Kalli and T. Stylianopoulos, Front. Oncol., 2018, 8, 55 Search PubMed.
- L. A. Lautscham, C. Kämmerer, J. R. Lange, T. Kolb, C. Mark, A. Schilling, P. L. Strissel, R. Strick, C. Gluth, A. C. Rowat, C. Metzner and B. Fabry, Biophys. J., 2015, 109, 900–913 Search PubMed.
- P. Vargas, P. Maiuri, M. Bretou, P. J. Sáez, P. Pierobon, M. Maurin, M. Chabaud, D. Lankar, D. Obino, E. Terriac, M. Raab, H.-R. Thiam, T. Brocker, S. M. Kitchen-Goosen, A. S. Alberts, P. Sunareni, S. Xia, R. Li, R. Voituriez, M. Piel and A.-M. Lennon-Duménil, Nat. Cell Biol., 2015, 18, 43–53 Search PubMed.
- T. Lämmermann and R. N. Germain, Semin. Immunopathol., 2014, 36, 227–251 Search PubMed.
- N. Srivastava, D. Traynor, M. Piel, A. J. Kabla and R. R. Kay, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2506–2512 Search PubMed.
- J. M. Tse, G. Cheng, J. A. Tyrrell, S. A. Wilcox-Adelman, Y. Boucher, R. K. Jain and L. L. Munn, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 911–916 Search PubMed.
- I. Y. Wong, S. Javaid, E. A. Wong, S. Perk, D. A. Haber, M. Toner and D. Irimia, Nat. Mater., 2014, 13, 1063–1071 Search PubMed.
- H. Kittur, W. Weaver and D. Di Carlo, Biomed. Microdevices, 2014, 16, 439–447 Search PubMed.
- H.-R. Thiam, P. Vargas, N. Carpi, C. L. Crespo, M. Raab, E. Terriac, M. C. King, J. Jacobelli, A. S. Alberts, T. Stradal, A.-M. Lennon-Dumenil and M. Piel, Nat. Commun., 2016, 7, 10997 Search PubMed.
- M. Le Berre, J. Aubertin and M. Piel, Integr. Biol., 2012, 4, 1406–1414 Search PubMed.
- A. J. Lomakin, C. J. Cattin, D. Cuvelier, Z. Alraies, M. Molina, G. Nader, N. Srivastava, J. M. Garcia-Arcos, I. Y. Zhitnyak, A. Bhargava, M. K. Driscoll, E. S. Welf, R. Fiolka, R. J. Petrie, N. Manel, A. M. Lennon-Duménil, D. J. Müller and M. Piel, bioRxiv, 2019, DOI:10.1101/863514.
- J. Aureille, N. Belaadi and C. Guilluy, Curr. Opin. Cell Biol., 2017, 44, 59–67 Search PubMed.
- N. Belaadi, J. Aureille and C. Guilluy, Cell, 2016, 5, 27 Search PubMed.
- M. Raab, M. Gentili, H. de Belly, H.-R. Thiam, P. Vargas, A. J. Jimenez, F. Lautenschlaeger, R. Voituriez, A.-M. Lennon-Dumenil, N. Manel and M. Piel, Science, 2016, 352, 359–362 Search PubMed.
- C. M. Denais, R. M. Gilbert, P. Isermann, A. L. McGregor, M. Te Lindert, B. Weigelin, P. M. Davidson, P. Friedl, K. Wolf and J. Lammerding, Science, 2016, 352, 353–358 Search PubMed.
- S. Dumont and T. J. Mitchison, Curr. Biol., 2009, 19, 1086–1095 Search PubMed.
- J. Aureille, V. Buffière-Ribot, B. E. Harvey, C. Boyault, L. Pernet, T. Andersen, G. Bacola, M. Balland, S. Fraboulet, L. Van Landeghem and C. Guilluy, EMBO Rep., 2019, 20, 1–11 Search PubMed.
- Y. J. Liu, M. Le Berre, F. Lautenschlaeger, P. Maiuri, A. Callan-Jones, M. Heuzé, T. Takaki, R. Voituriez and M. Piel, Cell, 2015, 160, 659–672 Search PubMed.
- V. Ruprecht, S. Wieser, A. Callan-Jones, M. Smutny, H. Morita, K. Sako, V. Barone, M. Ritsch-Marte, M. Sixt, R. Voituriez and C.-P. Heisenberg, Cell, 2015, 160, 673–685 Search PubMed.
- A. L. McGregor, C.-R. Hsia and J. Lammerding, Curr. Opin. Cell Biol., 2016, 40, 32–40 Search PubMed.
- M. D. Welch, Cell, 2015, 160, 581–582 Search PubMed.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. IJzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482, 323–328 Search PubMed.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 Search PubMed.
- X. Zhang, L. Li and C. Luo, Lab Chip, 2016, 16, 1757–1776 Search PubMed.
- S. Zhao, Y. Chen, B. P. Partlow, A. S. Golding, P. Tseng, J. Coburn, M. B. Applegate, J. E. Moreau, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2016, 93, 60–70 Search PubMed.
- N. W. Choi, M. Cabodi, B. Held, J. P. Gleghorn, L. J. Bonassar and A. D. Stroock, Nat. Mater., 2007, 6, 908–915 Search PubMed.
- A. Pathak and S. Kumar, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10334–10339 Search PubMed.
- M. P. Cuchiara, A. C. B. Allen, T. M. Chen, J. S. Miller and J. L. West, Biomaterials, 2010, 31, 5491–5497 Search PubMed.
- P. Zarrintaj, S. Manouchehri, Z. Ahmadi, M. R. Saeb, A. M. Urbanska, D. L. Kaplan and M. Mozafari, Carbohydr. Polym., 2018, 187, 66–84 Search PubMed.
- A. Pluen, P. a. Netti, R. K. Jain and D. a. Berk, Biophys. J., 1999, 77, 542–552 Search PubMed.
- S.-Y. Cheng, S. Heilman, M. Wasserman, S. Archer, M. L. Shuler and M. Wu, Lab Chip, 2007, 7, 763–769 Search PubMed.
- U. Haessler, Y. Kalinin, M. a. Swartz and M. Wu, Biomed. Microdevices, 2009, 11, 827–835 Search PubMed.
- U. Haessler, M. Pisano, M. Wu and M. a. Swartz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5614–5619 Search PubMed.
- Y. Ling, J. Rubin, Y. Deng, C. Huang, U. Demirci, J. M. Karp and A. Khademhosseini, Lab Chip, 2007, 7, 756–762 Search PubMed.
- S. Cosson and M. P. Lutolf, Sci. Rep., 2015, 4, 4462 Search PubMed.
- M. Le Berre, E. Zlotek-Zlotkiewicz, D. Bonazzi, F. Lautenschlaeger and M. Piel, Methods Cell Biol., 2014, 121, 213–229 Search PubMed.
- B. Laperrousaz, S. Jeanpierre, K. Sagorny, T. Voeltzel, S. Ramas, B. Kaniewski, M. Ffrench, S. Salesse, F. E. Nicolini and V. Maguer-Satta, Blood, 2013, 122, 3767–3777 Search PubMed.
- T. Kitamura, T. Tange, T. Terasawa, S. Chiba, T. Kuwaki, K. Miyagawa, Y. F. Piao, K. Miyazono, A. Urabe and F. Takaku, J. Cell. Physiol., 1989, 140, 323–334 Search PubMed.
- J. Goedhart, D. von Stetten, M. Noirclerc-Savoye, M. Lelimousin, L. Joosen, M. A. Hink, L. van Weeren, T. W. J. Gadella and A. Royant, Nat. Commun., 2012, 3, 751 Search PubMed.
- T. M. Koch, S. Münster, N. Bonakdar, J. P. Butler and B. Fabry, PLoS One, 2012, 7, e33476 Search PubMed.
- F. J. Byfield, R. K. Reen, T.-P. Shentu, I. Levitan and K. J. Gooch, J. Biomech., 2009, 42, 1114–1119 Search PubMed.
- K. A. Beningo, M. Dembo and Y. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18024–18029 Search PubMed.
- J. Toyjanova, E. Flores-Cortez, J. S. Reichner and C. Franck, J. Biol. Chem., 2015, 290, 3752–3763 Search PubMed.
- H. Kittur, A. Tay, A. Hua, M. Yu and D. Di Carlo, Biophys. J., 2017, 113, 1858–1867 Search PubMed.
- T. a. Ulrich, A. Jain, K. Tanner, J. L. MacKay and S. Kumar, Biomaterials, 2010, 31, 1875–1884 Search PubMed.
- G. C. Ingavle, S. H. Gehrke and M. S. Detamore, Biomaterials, 2014, 35, 3558–3570 Search PubMed.
- Y. P. Singh, N. Bhardwaj and B. B. Mandal, ACS Appl. Mater. Interfaces, 2016, 8, 21236–21249 Search PubMed.
- Z. N. Demou, Ann. Biomed. Eng., 2010, 38, 3509–3520 Search PubMed.
- Q. Chen, D. Yang, H. Zong, L. Zhu, L. Wang, X. Wang, X. Zhu, X. Song and J. Wang, Oncogenesis, 2017, 6, e375 Search PubMed.
- D. Gvaramia, E. Müller, K. Müller, P. Atallah, M. Tsurkan, U. Freudenberg, M. Bornhäuser and C. Werner, Biomaterials, 2017, 138, 108–117 Search PubMed.
- O. Lancaster, M. LeBerre, A. Dimitracopoulos, D. Bonazzi, E. Zlotek-Zlotkiewicz, R. Picone, T. Duke, M. Piel and B. Baum, Dev. Cell, 2013, 25, 270–283 Search PubMed.
- H. T. K. Tse, W. M. Weaver and D. Carlo, PLoS One, 2012, 7, 1–8 Search PubMed.
- C. J. Cattin, M. Düggelin, D. Martinez-Martin, C. Gerber, D. J. Müller and M. P. Stewart, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11258–11263 Search PubMed.
- A. Elosegui-Artola, I. Andreu, A. E. M. Beedle, A. Lezamiz, M. Uroz, A. J. Kosmalska, R. Oria, J. Z. Kechagia, P. Rico-Lastres, A.-L. Le Roux, C. M. Shanahan, X. Trepat, D. Navajas, S. Garcia-Manyes and P. Roca-Cusachs, Cell, 2017, 171, 1397–1410.e14 Search PubMed.
- J. H. Lee, D. H. Kim, H. H. Lee and H. W. Kim, Biomaterials, 2019, 197, 60–71 Search PubMed.
- M. T. Doolin and K. M. Stroka, Tissue Eng., Part C, 2019, 25, 662–676 Search PubMed.
- M. Krause, J. Te Riet and K. Wolf, Phys. Biol., 2013, 10, 065002 Search PubMed.
- B. Laperrousaz, L. Berguiga, F. E. Nicolini, C. Martinez-Torres, A. Arneodo, V. M. Satta and F. Argoul, Phys. Biol., 2016, 13, 03LT01 Search PubMed.
- H. Delanoë-Ayari, S. Iwaya, Y. T. Maeda, J. Inose, C. Rivière, M. Sano and J. P. Rieu, Cell Motil. Cytoskeleton, 2008, 65, 314–331 Search PubMed.
- H. Delanoë-Ayari, J. P. Rieu and M. Sano, Phys. Rev. Lett., 2010, 105, 2–5 Search PubMed.
- D. T. Burnette, L. Shao, C. Ott, A. M. Pasapera, R. S. Fischer, M. A. Baird, C. Der Loughian, H. Delanoe-Ayari, M. J. Paszek, M. W. Davidson, E. Betzig and J. Lippincott-Schwartz, J. Cell Biol., 2014, 205, 83–96 Search PubMed.
- J.-P. Rieu, T. Saito, H. Delanoë-Ayari, Y. Sawada and R. R. Kay, Cell Motil. Cytoskeleton, 2009, 66, 1073–1086 Search PubMed.
- J.-P. Rieu and H. Delanoë-Ayari, Phys. Biol., 2012, 9, 066001 Search PubMed.
- S. H. Medina, B. Bush, M. Cam, E. Sevcik, F. W. DelRio, K. Nandy and J. P. Schneider, Biomaterials, 2019, 202, 1–11 Search PubMed.
- A. Marturano-Kruik, A. Villasante, K. Yaeger, S. R. Ambati, A. Chramiec, M. T. Raimondi and G. Vunjak-Novakovic, Biomaterials, 2018, 150, 150–161 Search PubMed.
- A. Nicolas-Boluda, A. K. A. Silva, S. Fournel and F. Gazeau, Biomaterials, 2018, 150, 87–99 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00732c |
| This journal is © The Royal Society of Chemistry 2020 |