
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePulmonary-arterial-hypertension (PAH)-on-a-chip: fabrication, validation and application†
Taslim A.
Al-Hilal‡
a,
Ali
Keshavarz
a,
Hossam
Kadry§
a,
Behnaz
Lahooti§
a,
Ahmed
Al-Obaida
a,
Zhenya
Ding
b,
Wei
Li
 b,
Roger
Kamm
cd,
Ivan F.
McMurtry
e,
Tim
Lahm
fg,
Eva
Nozik-Grayck
h,
Kurt R.
Stenmark
h and
Fakhrul
Ahsan
*a
b,
Roger
Kamm
cd,
Ivan F.
McMurtry
e,
Tim
Lahm
fg,
Eva
Nozik-Grayck
h,
Kurt R.
Stenmark
h and
Fakhrul
Ahsan
*a
aDepartment of Pharmaceutical Sciences, Texas Tech University Health Sciences Center, Jerry H. Hodge School of Pharmacy, 1300 Coulter Dr., Amarillo, 79119 Texas, USA. E-mail: fakhrul.ahsan@ttuhsc.edu; Fax: +1 806 356 4034; Tel: +1 806 414 9235
bDepartment of Chemical Engineering, Texas Tech University, Lubbock, Texas, USA
cDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
dDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
eDepartment of Pharmacology, The Center for Lung Biology, University of South Alabama, Mobile, AL 36688, USA
fDepartment of Pulmonary and Critical Care, Department of Medicine, Indiana University School of Medicine, Indianapolis, Indiana, USA
gRichard L. Roudebush Veterans Affairs Medical Center, Indianapolis, Indiana, USA
hDepartment of Pediatrics and Medicine, Cardiovascular Pulmonary Research Laboratories, University of Colorado Denver, Anschutz Medical Campus, Aurora, CO 80045, USA
First published on 28th July 2020
Abstract
Currently used animal and cellular models for pulmonary arterial hypertension (PAH) only partially recapitulate its pathophysiology in humans and are thus inadequate in reproducing the hallmarks of the disease, inconsistent in portraying the sex-disparity, and unyielding to combinatorial study designs. Here we sought to deploy the ingenuity of microengineering in developing and validating a tissue chip model for human PAH. We designed and fabricated a microfluidic device to emulate the luminal, intimal, medial, adventitial, and perivascular layers of a pulmonary artery. By growing three types of pulmonary arterial cells (PACs)-endothelial, smooth muscle, and adventitial cells, we recreated the PAH pathophysiology on the device. Diseased (PAH) PACs, when grown on the chips, moved of out their designated layers and created phenomena similar to the major pathologies of human PAH: intimal thickening, muscularization, and arterial remodeling and show an endothelial to mesenchymal transition. Flow-induced stress caused control cells, grown on the chips, to undergo morphological changes and elicit arterial remodeling. Our data also suggest that the newly developed chips can be used to elucidate the sex disparity in PAH and to study the therapeutic efficacy of existing and investigational anti-PAH drugs. We believe this miniaturized device can be deployed for testing various prevailing and new hypotheses regarding the pathobiology and drug therapy in human PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a rare disease in which pulmonary arteries/arterioles become stiffer and occluded. Thus, the heart has to work harder to pump blood through the occluded arteries into the lungs. In so doing, the heart becomes enlarged and patients die of right heart failure. The chief clinical manifestation of the disease is elevated mean pulmonary arterial pressure that results from an array of active structural alterations in the pulmonary artery, called arterial remodeling and muscularization. This pathology develops because of aberrant proliferation, migration and misplaced growth of pulmonary arterial cells (PACs), development of apoptosis-resistant endothelial cells (ECs), enhanced deposition of extracellular matrices (ECMs), thickening of smooth muscle layers, and acquisition of smooth muscle cell (SMC)-like phenotypes by ECs.1,2 One of the puzzling aspects of PAH is that it affects more women than men but women with PAH tend to live longer than men. This disparity in the prevalence versus survival of PAH patients is believed to result from intrinsic differences between the two sexes and the conflicting roles of sex hormones, especially estrogen, which appear to have both beneficial and detrimental effects in the genesis and progression of PAH in females.3In fact, the genesis of the disease centers around the interactions of the three major cells of the pulmonary arteries/arterioles-ECs, SMCs, and adventitial cells (ADCs) that are chiefly fibroblasts. For studying the pathogenesis, potential therapies, and sex disparity in PAH, investigators have traditionally relied on various animal models that most commonly include chronic hypoxia, Sugen-5416-plus-hypoxia (SuHx), and monocrotaline (MCT)-induced models as well as various genetically-modified mouse models.4 By reproducing the chief feature of the disease, elevated pulmonary arterial pressure, these models have aided in our understanding of the pathobiology and evaluating therapeutic interventions. However, existing animal models only partially recapitulate the important pathological changes of human PAH such as the arteriopathy with neointimal and plexiform lesions and progressive deterioration of the disease.5 Because the development of animal models entails the use of various animal species and distinct disease-producing agents or environments, animal models of PAH show a great deal of variability in the disease severity, progression, and response to therapeutic interventions.6
Cellular models that have been used to elucidate PAH pathology and assess drug therapy include mono- and co-cultures of ECs, SMCs, or ADCs collected from the pulmonary arteries of rodent and bovine models of PAH and from human PAH patients. These two-dimensional (2D) models, conserving at least some of the acquired phenotypes of PAH cells, cannot imitate cell–cell interactions, the tissue-level functions, or the remodeling of the PAH-afflicted pulmonary arteries and arterioles. Tissue-engineered hydrogel-based 3D models are certainly promising with regard to assessing the effects of dimensionality, stiffness, and composition on cell behavior7,8 but at present are cumbersome for investigating the distinct cellular interactions that occur through paracrine and juxtacrine signaling. Further, 2D models cannot readily accommodate relevant physical processes such as fluid stress that impact organ functionality.9 Overall, none of the existing PAH models accurately emulates the disease in humans, nor are they adaptable to the combinatorial study design that is warranted for delineating the influence of various overlapping molecular biological processes, therapeutic interventions, or sex.
Recently innovated microfluidic-based tissue chips combine the elegance of microengineering with the complexity of biology to create near-ideal experimental models for various diseases and organs.10 These microfluidic devices with micron-sized channels, known as tissue-chips or organ-on-a-chip, allow the cells to be grown in a more biologically relevant environment, enabling cell–cell communications with continuous bathing in biological fluids in a tissue-like fashion. The multi-channel systems can be modified to incorporate cellular cues, such as fluid-induced stress, and allow high-resolution imaging, monitoring of biochemical and pathophysiological processes, as well as the screening of drugs.10,11 However, no such model has yet been reported for capturing the pathophysiology of PAH and delineating the sex-disparity in PAH. As such, we sought here to develop a microfluidic device that can recapitulate the essential features of PAH-afflicted human pulmonary arteries and help decipher the cellular and sex-specific signaling processes implicated in PAH. By growing all three PACs on a microfluidic chip, we were able to assess the cellular phenotypes, migration, arterial remodeling, and to investigate the influence of fluid-induced stress on cellular morphology. By recreating male versus female PAH on the chips, prepared with human and animal cells, we have also studied the effects of sex hormones on PAH pathogenesis.
Materials and methods
Design and fabrication of chips
We designed and fabricated the chips according to a protocol published by the Kamm lab.12 First, using AutoCAD (Autodesk, San Rafael, CA), we created a network of 5 channels (Fig. 1A), each 1 mm wide and 3 cm long. The channels were separated by 150 μm tall trapezoidal pillars, placed 200 μm apart throughout the length of the channel (Fig. 1B). From the CAD file, we created a photomask and printed the mask using a high-resolution printer (CAD Art Services Inc., OR). We then placed the mask on a silicon wafer, spin coated the assembly (Spin Coater WS-650 Series, Laurell Technologies Corporation, PA) using SU-8-photoresist polymer (MicroChem Inc., MA), and baked it at 65 °C for 5 min and 95 °C for 25 min. We then exposed the SU-8-coated silicon wafer to UV light (UV-KUB 2, KLOE Inc., France), baked it again at 65 °C for 5 min and 95 °C for 12 min, cooled it to room temperature, and washed it with SU-8 solvent to obtain a silicon master mold. To fabricate the devices using the master mold, we poured an aliquot of polydimethylsiloxane (PDMS, Dow Inc., MI) mixed with a curing solution (Dow Inc., MI), at a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, over the mold. We then degassed, baked, and cut out the individual devices (35 mm diameter, 3–4 mm height), and finally created inlets and outlets using a biopsy punch® (Miltex, PA). Later, we attached the chips to UV-sterilized cover slips using a plasma cleaner (PDC-001-HP series, Harrick plasma, NY), put the cover-slip-attached devices in a 65 °C oven for 4 h, and tested the chips for leakage by passing trypan blue solution (Sigma Inc., St. Louis, MO) through the channels, as reported previously.13 Prior to seeding and growing cells, we coated the chips with a 10 mg mL−1 solution of poly-D-lysine (PDL, Sigma, MO) and incubated the PDL-coated devices in an 85 °C oven overnight for restoring the hydrophobicity. At this point, the surface of the chips was properly cleaned and UV-sterilized. For obtaining reproducible results, all the chips were used within 48–72 h after PDL coating.
:1, over the mold. We then degassed, baked, and cut out the individual devices (35 mm diameter, 3–4 mm height), and finally created inlets and outlets using a biopsy punch® (Miltex, PA). Later, we attached the chips to UV-sterilized cover slips using a plasma cleaner (PDC-001-HP series, Harrick plasma, NY), put the cover-slip-attached devices in a 65 °C oven for 4 h, and tested the chips for leakage by passing trypan blue solution (Sigma Inc., St. Louis, MO) through the channels, as reported previously.13 Prior to seeding and growing cells, we coated the chips with a 10 mg mL−1 solution of poly-D-lysine (PDL, Sigma, MO) and incubated the PDL-coated devices in an 85 °C oven overnight for restoring the hydrophobicity. At this point, the surface of the chips was properly cleaned and UV-sterilized. For obtaining reproducible results, all the chips were used within 48–72 h after PDL coating.
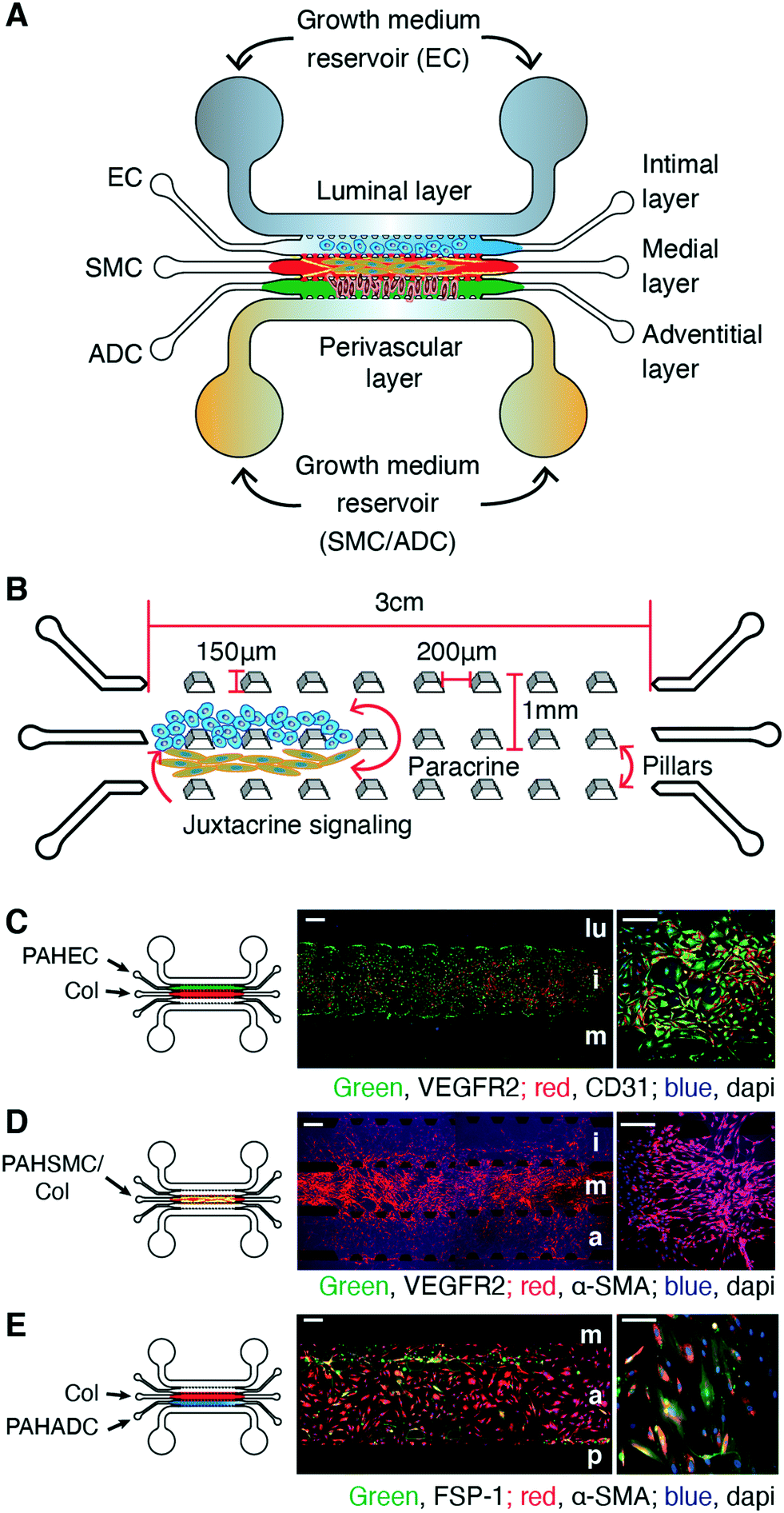 | ||
| Fig. 1 A multichannel microfluidic device for the seeding and growing of three major pulmonary arterial cell types (PACs) in three distinct layers similar to those in pulmonary arteries/arterioles. (A) A five-channel chip mimicking the adventitial, medial, and intimal layers for growing adventitial (ADCs), smooth muscle (SMCs), and endothelial cells (ECs), respectively; and perivascular and luminal layers for delivery of growth media or treatments. (B) Channels within a chip: each channel, wherein cells are grown, is 1 mm wide and 3 cm long, separated by 150 μm tall trapezoidal pillars placed 200 μm apart. (C) PAH-ECs grew on the chips and stayed within the intimal layer of the chips when seeded in the presence of collagen in the medial layer. (D) PAH-SMCs migrated from the medial layer and spread over the adventitial and intimal layers. (E) PAH-ADCs grew on the adventitial layer and expressed both FSP-1 and α-SMA. lu, lumen; i, intimal; m, medial; a, adventitial; p, perivascular. n = 3 chips prepared with the same cells from the same donors/patients. Scale bars 250 and 100 μm. | ||
Seeding and growing of human cells on chips
Human cells used in this study were primary PACs from healthy human male and female donors or patients with idiopathic PAH (IPAH), which we obtained from the Pulmonary Hypertension Breakthrough Initiatives (PHBI) specimen bank (www.ipahresearch.org) under an approved protocol. To isolate PACs, pulmonary arterial segments were collected from the main pulmonary artery by using a location-specific procedure to isolate type I (>5 mm), type II (1–5 mm) and type III (≤1 mm) ADCs, SMCs and ECs. These location specific methods of isolation were used to ensure that the cells were of arterial in origin and not of venous, capillary or bronchial vasculature origin, as described elsewhere.14 The cells used in this study are normal endothelial (N-ECs; n = 2), smooth muscle (N-SMCs; n = 2) and adventitial cells (N-ADCs; n = 2) and their PAH-afflicted counterparts, PAH-ECs, PAH-SMCs and PAH-ADCs (n = 4 for each cell type; Table S1†). Although EC and SMC specimens contained the respective pure cell types, the ADC specimens contained mainly fibroblasts, along with vasa vasorum cells, pericytes, monocytes, and progenitor cells.Upon receipt of the cells, we thawed, passaged them in polystyrene flasks, and characterized them for various markers before seeding them onto the chips. First, we seeded control and PAH cells, obtained from the same or different donors, into their respective channels of the chips. Using cells between passage 3 and 6 (Table S1 and Fig. S2†), we seeded 2 × 106 cells per mL of ADCs, SMCs at a concentration of 2 × 106 cells per mL in rat tail type I collagen solution (Corning Inc., NY), and 5–10 × 106 cells per mL of ECs on the adventitial, medial, and intimal channels of the chips, respectively, unless stated otherwise. We added media via the luminal or perivascular channels. For three-cell-laden chips, we first added collagen at a density of 2 mg mL−1 to the medial channel mixed with or without SMC depending on the experimental design, incubated the chips for 20 minutes for gelation, seeded ECs on the intimal channel, and allowed another 20 minutes before adding ADCs via the adventitial channel (Fig. S1†). Finally, we incubated the cell-laden chips in a humidified chamber, placed the chambers containing cell-laden chips in a CO2 incubator, replenished the reservoir with 150 μL medium every day, and let the cells grow for 5–8 days. Before starting any treatments, if we observe a contracted collagen/SMC layer more than 15–20% of its initial seeding area, we removed those chips from the study. We stained the cells on the chips for FSP-1, CD31, VEGFR2, α-SMA, SM-22α, and CD90 to identify the phenotypes of ECs, SMCs, and ADCs (Table S2†). As parallel controls, we cultured the cells at a density of 2.5 × 105 cells per mL in gelatin-coated 6-well plates to 90% confluency and then assessed the same phenotypes as described above.
We used quantum dot-based (QD) Cell Trackers (Thermo Fisher Scientific Inc. CA)-Qtracker® 705 (QD705), Qtracker® 605 (QD605), or Qtracker® 525 (QD525)-which do not leak out of the cells and thus allows microscopic examination of the seeded cells on a daily basis.15 One day before on-chip cell seeding, we labeled the cells with 2.5 μL of QD-trackers, following the manufacturer's protocol. In a separate experiment, using two sets of chips, we seeded PAH-ECs along with non-labeled PAH-SMCs or QD605-labeled PAH-SMCs and imaged the chips on day 3 and day 8 of culture to assess whether QD labeling affects the growth of cells.
Morphometric assessment of on-chip pulmonary arterial remodeling
For assessing on-chip intimal and medial thickening (i.e. arterial thickening) and on-chip muscularization, we made two assumptions: first, ECs and SMCs should grow within the intimal and medial layers of the chip, and the growth of ECs or SMCs inside or outside their designated channels would be considered as intimal or medial layer thickness, respectively. Second, the number of SMCs that migrated from the medial layer and grew inside the endothelial layers of the chips would be considered as muscularization, similar to that in PAH where non-muscularized distal arteries/arterioles become muscularized with the appearance of SM-like cells from preexisting SMCs.16 For demarcating the cell growth zone, and identifying and counting cells outside their designated channels, we used phase contrast microscopy, immunostaining, and quantum dot labeling.For marking the zone of cell growth, we imaged the chips using phase-contrast microscopy on-a-daily basis before immunostaining or by using confocal microscopy after immunostaining. To quantify the thickness of the intimal layer, we measured the vertical length of the continuous monolayer of ECs in the intimal and luminal layers. For quantifying the medial layer thickness, we measured the vertical length of collagen matrix that contain QD-labeled SMCs or FSP-1+CD31− SMCs. We then averaged the thicknesses of five to seven diametrically uneven points of the same layers using ImageJ® software and calculated the intimal and medial thickness. For assessing on-chip muscularization, we first seeded QD-labeled N-SMC/PAH-SMCs and let the QD-labeled cells grow on the chips. At the end of the study, we immunostained the chips with FSP-1 and CD31 antibodies and counted either the number of FSP-1+CD31− or QD-labeled SMCs in the intimal, luminal and adventitial layers.
Immunostaining, western blot, image acquisition and analyses
We stained ECs, SMCs, and ADCs with markers for CD31, FSP-1, VEGFR2, α-SMA, SM-22α, CD90, aromatase, CYP1B1, human collagen type I (ColT1) and human collagen type IV (ColT4). For staining of the cells grown on chips, we washed the chips with the basal medium, added 4% paraformaldehyde, blocking buffer, primary antibodies (Table S3†), and incubated overnight. Next day, we washed the chips with washing buffer, added secondary antibodies (Table S3†), incubated for an hour, washed again, and finally added mounting medium with DAPI to stain the nuclei. We examined the chips using an epifluorescence (DMi8 epifluorescence, Leica, IL) or multi-photon confocal microscope (Ti-E, Nikon, NY). For determining the expression levels of EndMT markers, control and PAH-afflicted cells were seeded and cultured in the chips. Cells from the intimal layer of the chips were collected after 7 days of culture. Protein was collected and the samples were analyzed for BMPR2, VE-Cadherin, snail/slug, α-SMA, SM-22α, and actin expression using electrophoresis method.Application of shear stress on the chips
To apply shear stress on the chips, we connected the cell-laden chips with a syringe pump (NE-1000, New Era Pump Systems, Inc., NY) and then infused the culture medium, via the luminal layer, into the device for three days. Flow rates of 1.5, 20, and 168 μL h−1 that created a shear stress of 0.1, 1.9 (equivalent to that of blood flow), and 15 dyne per cm2 respectively.17 We then assessed the cellular morphology and arterial remodeling as described above. We calculated cell elongation from the ratio of long axis to short axis of an ellipse fitted around the cell. At least 15–20 cells per images were counted to calculate the long and short axis using ImageJ® software (NIH, MD).Effect of sex hormones and anti-PAH drug on PAH-afflicted pulmonary arterial remodeling
For studying the influence of sex hormones and anti-PAH drugs on cellular functions and remodeling, we seeded male and female PAH-ECs, QD705-labeled or unlabeled PAH-SMCs and PAH-ADCs and treated the cells with physiologically relevant concentrations of 0.1 or 1.0 μM 17β-estradiol,18 the main circulating estrogen. We then measured arterial remodeling and muscularization, as described above, and assessed the deposition of human ColT1, expression of aromatase (an enzyme involved in estrogen synthesis) and CYP1B1 (an estrogen-metabolizing enzyme). For assessing the therapeutic effects, we infused 1 μM fasudil, a Rho-kinase inhibitor, into the chips prepared with PAH-ECs/SMCs/ADCs. We started the infusion on day 4 after cell seeding and continued for an additional 3 days. After 4 days of treatment with fasudil, we measured the changes in arterial remodeling as described above.Statistical analysis
We performed statistical analysis at α = 0.05 (95% confidence interval) with GraphPad Prism (version 5.0, GraphPad Software, San Diego, CA) and used the t-test, one/two-way ANOVA, Tukey's post hoc, and repeated measures ANOVA, as applicable. Further, the sample size (n) cited is the number of replicates. In other words, we prepared 3–6 chips for a given experiment using the cells from the same patient.Results
The phenotypes of human PAH cells remain unaltered when grown on chips
The chip we have designed and fabricated is a multi-channel device mimicking the five layers of a pulmonary artery (Fig. 1A): the two outermost channels, luminal and perivascular, serve as access points for instilling growth media and non-vascular cells, and for establishing growth-factor gradients and fluid flow. The other three channels serve as intimal, medial, and adventitial layers for growing, respectively, ECs, SMCs, and ADCs of the pulmonary arteries (Fig. 1A). The channels are walled off by trapezoidal pillars, placed every 200 μm, which prevent cells from mixing with each other during the initial seeding but allow cellular communication via paracrine and juxtacrine means (Fig. 1B). After coating the middle three channels of the chips with poly-D-lysine (PDL), we seeded control (N-ECs, N-SMCs and N-ADCs) and idiopathic ‘PAH-afflicted’ (IPAH) human cells (PAH-ECs, PAH-SMCs, and PAH-ADCs) in their respective channels (Fig. S1, A to C†). Cells seeded on one channel did not move to an adjoining channel, suggesting that an interfacial film, formed between the pillars, prevented cells in one channel from accidentally entering another channel during cell seeding (Fig. S1D†). PAH-ECs grew as a monolayer on the intimal layer (Fig. 1C); PAH-SMCs migrated toward the intimal and adventitial layers, although they primarily grew on the medial layer (Fig. 1D). PAH-ADCs grew on the adventitial layer, but migrated somewhat toward the medial and perivascular layers (Fig. 1E).The control and PAH cells maintained a stable phenotype over multiple passages, but showed different expression patterns depending on whether the cells were from healthy donors or from PAH patients (Tables S1 and S2 and Fig. S2, A to C†). Regardless of whether the cells were grown on-chips or on-dishes, N-ECs expressed CD31 and VEGFR2, two major markers of the EC phenotype, but PAH-ECs were FSP-1/CD31, VEGFR2/α-SMA or SM-22α positive (Fig. S3, A to F†), indicating that PAH-ECs were phenotypically different from N-ECs. Both N-ECs and PAH-ECs were negative for CD90 (Fig. S3, E and F†) and cells grown on chips and on dishes showed the same levels of expression of VEGFR2 and α-SMA (Fig. S2, D to F†). Similarly, when stained for cleaved caspase-3 and PCNA, the apoptotic and proliferative indices for cells grown on chips were no different from those grown on traditional polystyrene plates. However, these indices varied depending on the source of the cells (patients or donors), which is consistent with the assumption that cells from different patients show different growth patterns and survival rates (Fig. S3, G to I†). Thus, when grown on microfluidic devices, the expression of characteristic markers for both N-ECs and PAH-ECs remained unchanged. Likewise, typical makers for N-SMCs, PAH-SMCs, and PAH-ADCs grown on chips remained unaltered when cells were grown on dishes (Fig. S4 and S5†). Specifically, SMCs showed expression of FSP-1, α-SMA, and SM-22α, but not of CD31, VEGFR2, or CD90 (Fig. S4†). ADCs, on the other hand, were positive for CD90 in addition to FSP-1, α-SMA, and SM-22α, but negative for CD31 and VEGFR2 (Fig. S5†). Overall, these results suggest that the devices (chips versus traditional plates) had no effect on the cellular phenotypes.
Cell-laden chips recreate the three layers of PAH-afflicted pulmonary arteries/arterioles and allow cellular cross-talk
The pathogenesis of PAH revolves around the aberrant interactions and growth of three PACs; the cells of one of the three layers (adventitial, intimal and medial layers) of the pulmonary arteries often migrate to the neighboring layers, and thus they give rise to misplaced cellular growth. Further, cell–cell interactions trigger the proliferation of apoptosis-resistant ECs and reduce the contractile properties of SMCs.1,19 Further, EC-induced recruitment, proliferation, and subsequent de-differentiation of SMCs contribute to the pulmonary arterial remodeling. We thus investigated whether the device allows cellular communication and if the resulting communication among control cells is different from that of diseased cells. When grown in the presence of N-SMCs or N-ADCs or N-SMCs plus N-ADCs, only a limited number of N-ECs migrated from their respective compartments to the neighboring compartments (Fig. 2, A to C). However, neither N-SMCs nor N-ADCs migrated to the intimal layers and interacted with N-ECs and the phenotypes of N-ECs remained unchanged, as evidenced by the lack of α-SMA expression. Unlike N-ECs, PAH-ECs, when grown in the presence of PAH-SMCs or PAH-ADCs or PAH-SMCs plus PAH-ADCs, showed an aggressive proliferation pattern toward the luminal layer (Fig. 2, D to F) and thus the intimal layer became thicker. In the chips containing both PAH-ECs and PAH-SMCs (Fig. 2D) and those prepared with all three PAH cell types (Fig. 2F), PAH-SMCs began migrating into the intimal and luminal layers and interacting with PAH-ECs. We also observed a high expression of αSMA in the intimal layer of the chips that were prepared with PAH-ECs plus PAH-SMC or all three PAH cell types. However, we observed no such increase in αSMA expression in the intimal layer of the chips that were prepared with PAH-ECs plus PAH-ADCs. Thus, the migration of PAH-SMCs toward the intimal layer of the cells contributed to the increased αSMA expression. This phenomenon is similar to arterial muscularization or misplaced cellular growth as observed in hypoxia-induced animal models of PAH afflicted pulmonary arteries, where pre-existing SMCs migrate toward the vascular wall, proliferate, and undergo dedifferentiation process and thus give rise to excess smooth muscle.16 This phenomenon of misplaced cellular growth was most prominent in chips prepared with PAH-EC/SMC/ADCs (Fig. 2F). | ||
| Fig. 2 Control and PAH cells show distinct growth patterns in the tissue-chips. Growth patterns of N-ECs or PAH-ECs when cultured in the presence of (A and D) N-SMCs or PAH-SMCs, (B and E) N-ADCs or PAH-ADCs, and (C and F) N-SMC/N-ADCs or PAH-SMC/PAH-ADCs for 12 days. N-ECs did not grow in the luminal compartment and N-SMCs did not migrate from medial to intimal layers in the presence of other control cells. However, in the presence of PAH-SMCs and/or PAH-ADCs, PAH-ECs grew more inside the lumen; both PAH-ECs and PAH-SMCs interacted (arrows) with each other. | ||
Interestingly, PAH-ECs alone grew as a monolayer in the intimal compartment (Fig. 3A), but migrated toward the luminal compartment at a similar rate, regardless of whether PAH-ECs, along with PAH-SMCs/PAH-ADCs, were seeded in their respective channels in the absence (Fig. 3B) or presence of rat-tail collagen (Fig. 3C), Matrigel® (Fig. 3D), or collagen plus fibronectin (Fig. 3E). The seeding of PAH-ECs with or without matrix had no effect on the migration of PAH-SMCs from the medial to the intimal layer of the chips. Independent of the presence or absence of ECM protein, chips prepared with PAH-EC/PAH-SMCs showed disrupted endothelial monolayers and fibril-like collagen fibers in the intimal layers (Fig. 3, B to E). The 3D images show that PAH-ECs and PAH-SMCs were evenly embedded within the matrix of their respective channels (Fig. 3F), evidencing that PAH-cells grew and distributed over a 3D microenvironment rather than the 2D-glass surface of the channels. High resolution imaging of the channels confirmed that the ECs formed continuous cell–cell junctions spanning the full area of the 3D collagen–endothelial channel interface (Fig. 3G).
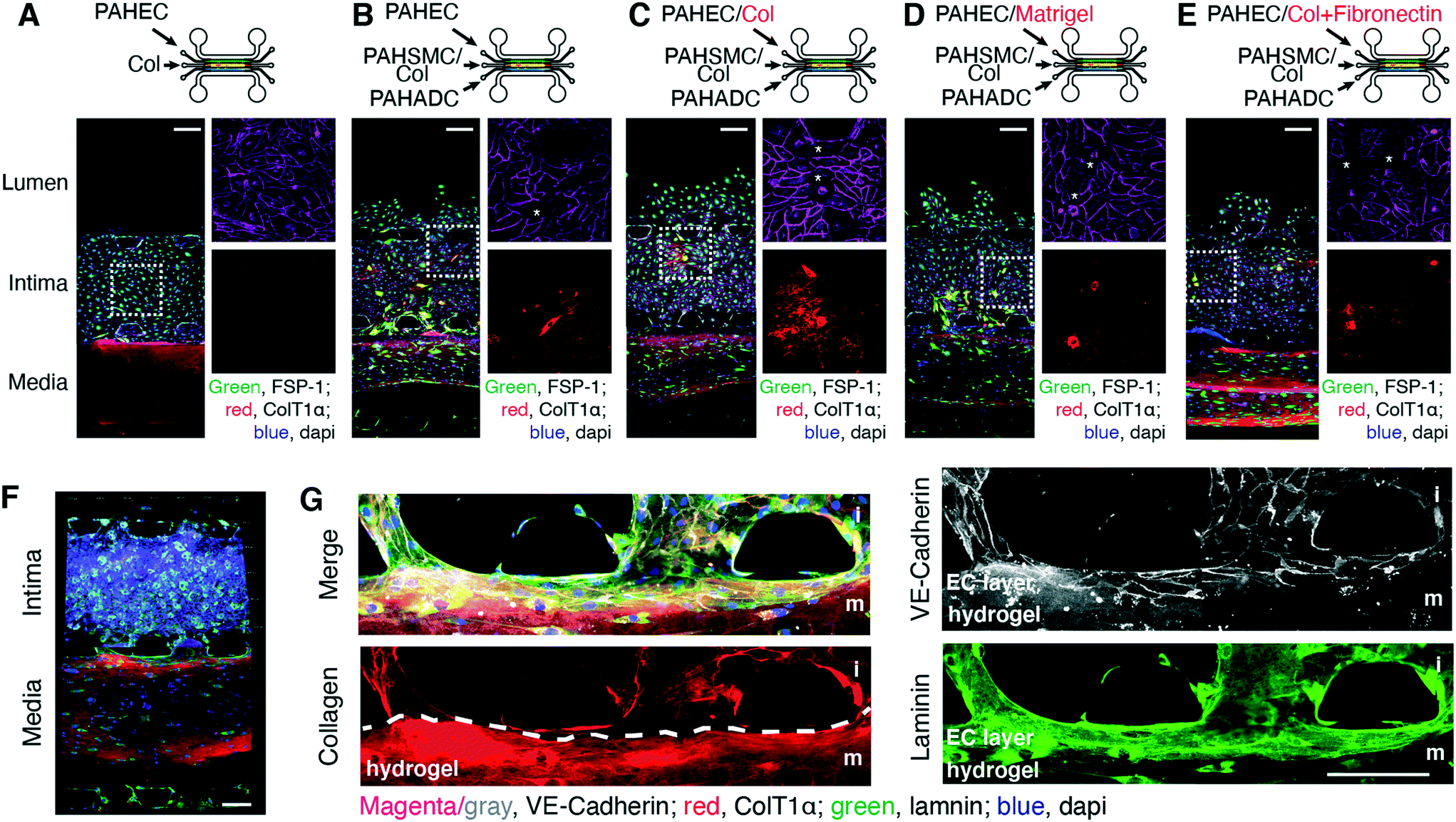 | ||
| Fig. 3 Interactions between PAH-ECs and PAH-SMCs due to cell invasion. The growth patterns of (A) PAH-ECs or PAH-EC/PAH-SMC/PAH-ADCs in the (B) absence or (C) presence of rat-tail collagen type-I, (D) Matrigel, and (E) collagen plus fibronectin in the intimal layer of chips after 5 days of culture. PAH-SMCs were also cultured in collagen. Collagen fibers were stained with antibody against human ColT1α, which has very low reactivity for rat-tail collagen type-I and that can be visualized at high exposure only. Thus, to assess the growth pattern of PAH-SMCs within the medial layer of the chip, we have taken pictures at high exposure (red channel only). Please note that the presence of PAH-SMCs (*) in the intimal layer compromises the integrity of PAH-EC monolayer. (F) A confocal 3D rendered image showing the distribution of PAH-ECs and PAH-SMCs in their matrix (G) high resolution image shows that the distribution of endothelial cells monolayer onto the collagen matrix between the interface of two channels. n = 3 chips prepared with the same cells from the same patients. Scale bars 100 and 75 μm. | ||
N-ECs, when seeded on the intimal layer along with PAH-SMCs and PAH-ADCs in their respective channels, began to interact with PAH-SMCs and expressed FSP-1 and α-SMA, which N-ECs do not normally express but PAH-ECs do (Fig. S6, A and B†). These transdifferentiated ECs also migrated from the intimal layer to the luminal layer of the chips in the presence of PAH-SMCs, PAH-ADCs or PAH-SMCs plus PAH-ADCs; the migration was the highest in the chips prepared with N-EC/PAH-SMC/PAH-ADCs (Fig. S6, C to F†). This phenomenon is consistent with the hypothesis that in both PAH patients and animal models of PAH, endothelial to mesenchymal transition (EndMT) contributes to the thickening of the intimal layer and the remodeling of pulmonary arteries/arterioles.20,21 To confirm whether the PAH-on-a-chip model truly represents the EndMT of PAH, we analyzed the transcriptional factors snail/slug and several smooth muscle markers in the cells collected from the intimal and luminal layers. We observed that loss of BMPR2 protein, in the chips containing N-ECs along with PAH-SMCs and PAH-ADCs, was the associated with increased snail/slug expression, elevation of prominent EndMT markers (αSMA and SM22α), and reduced expression of the EC junction protein VE-cadherin (Fig. S6G†). Our results corroborate previous observations where reduced BMPR2 was associated with elevated EndMT in PAH.21
Flow induced shear-stress influences cellular responses and migration
Arterial remodeling in PAH causes narrowing of the pulmonary arteries/arterioles and the shear-stress resulting from blood flow exacerbates arterial remodeling.22 We thus investigated the influence of fluid-induced shear stress (low, medium and high, Fig. 4A) on the cellular communication and orientation of the cells on the chips. When the shear stress was zero, 0.1 dyne per cm2 (below the shear stress of normal blood flow, or 1.9 dyne per cm2 (equivalent to the shear stress of normal blood flow),23 N-ECs showed characteristic cobblestone features; the orientation of the cells was rather random (Fig. 4B). When the shear stress was increased to 15 dyne per cm2 (equivalent to that observed in PAH-afflicted arteries), N-ECs became elongated and positioned themselves in a perpendicular array (Fig. 4C and D). Compared with the absence of flow, N-ECs were elongated by ∼3-fold when high shear stress was applied. Further, elevated shear stress caused intimal and medial thickening and the migration of N-SMCs (Fig. 4, E to G). | ||
| Fig. 4 Chips prepared with control cells show varying degrees pathological changes when subjected to shear stress. (A) To apply shear stress, the growth medium was passed through the luminal channels at different flow rates for three days. (B) When the chips were subjected to high shear stress (15 dyne per cm2), all three layers of PAH-chips underwent changes, and there was deposition of collagen fibers on the intimal layer. (C) In the presence of high shear stress, N-ECs assumed an elongated structure and their shapes changed from typical cobblestone to perpendicular. (D) The cell elongation ratio, calculated by dividing the long axis with short axis of each cell. ***p < 0.001 vs. no shear. In the presence of high shear stress, there was an increase in (E) intimal and (F) medial thickness, and (G) the number of N-SMCs in the intimal layer. n = 3 chips prepared with the same cells from the same donors. Scale bar 75 μm. | ||
Arterial remodeling observed in PAH tissue chips is responsive to anti-PAH therapy
PAH-afflicted pulmonary arteries and arterioles become thicker and undergo muscularization because of the proliferation of SMCs and their migration into the intimal layer.16 Thus, we simulated the phenomena of muscularization and thickening of the intimal and medial layers on the chips. In this study, we used quantum dot (QD)-705-labeled PAH-SMCs to facilitate the counting of the cells that migrated into the neighboring layers, especially those that migrated into the intimal layer. QD-labeled cells were proliferative and QD signals were robust and consistent, even after 7 days of culturing (Fig. S7†). Phase contrast and fluorescence microscopy gave very similar estimates of the thicknesses of the intimal and medial layers (Fig. 5, A and B). PAH-SMCs invaded the PAH-ECs layer, a process resembling muscularization of the non-muscularized pulmonary arteries. The number of QD705+ PAH-SMCs on the intimal layer was similar to that of the FSP-1+CD31− PAH-SMCs on the same layer (Fig. 5, C and D).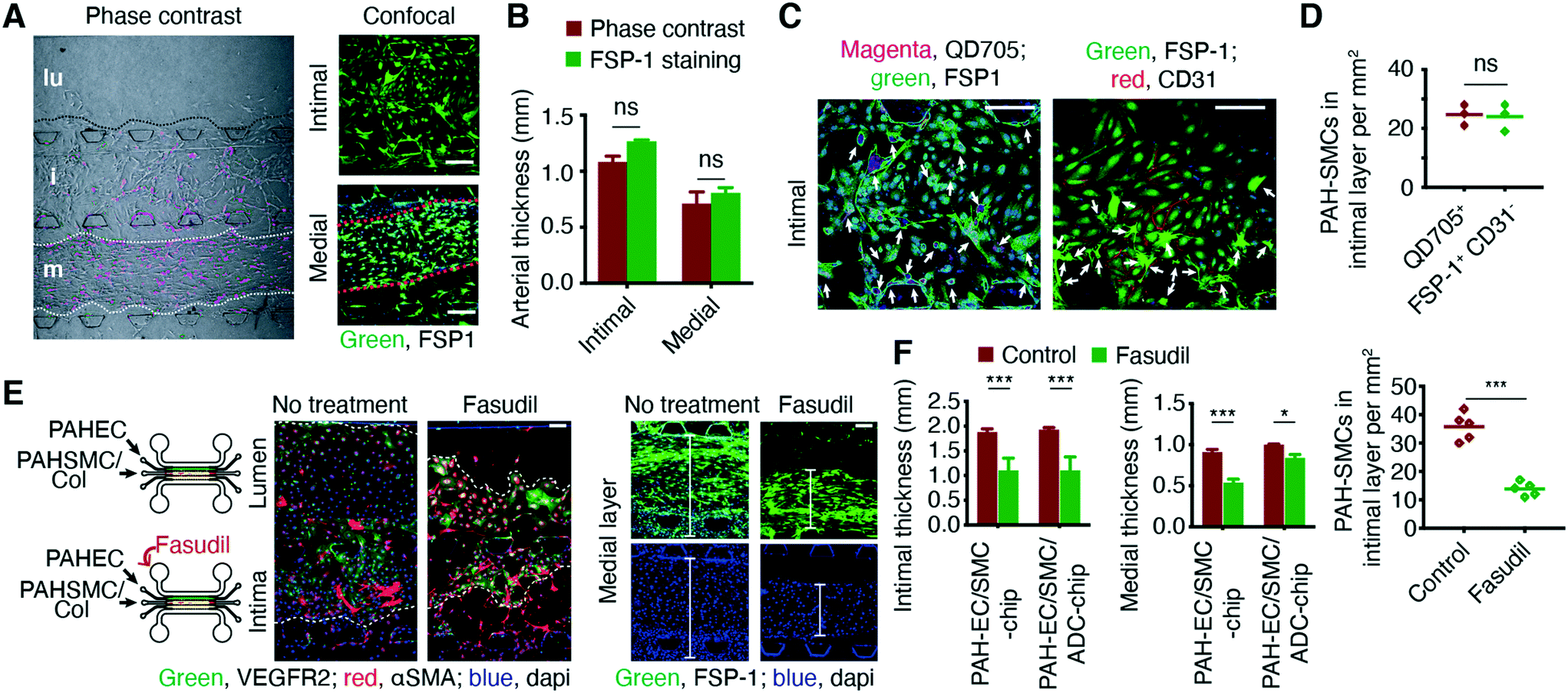 | ||
| Fig. 5 The chips display arterial remodeling and allow for measuring the efficacy of PAH therapy. (A and B) Chips were prepared using QD705-labeled PAH-SMCs, and the widths of the arterial layers were measured from the phase contrast images and after staining with FSP-1. (C and D) Imaging and subsequent counting of PAH-SMCs in the intimal layer from the number of QD705+ and FSP-1+CD31− cells (arrows) after staining. Both methods produced similar results. Before quantitation, images were processed using ImageJ software. (E and F) Fasudil, when given via the luminal layer, prevented muscularization of PAH-ECs and reduced medial wall thickening. ***p < 0.001; data represent mean ± SD. n = 3 chips prepared using the same cells from the same donors/patients. Scale bars 250, 100, and 75 μm. | ||
Next, we assessed the effect of fasudil, a Rho-kinase inhibitor with vasodilatory and antiproliferative effects,24 on the muscularization and thickening of the intimal and medial layers. In a series of studies, we and others have demonstrated the in vivo efficacy of fasudil in PAH rats. Fasudil, an investigational anti-PAH drug, elicits pulmonary preferential vasodilation in PAH rats.25,26 Further, a clinical trial established the short-term efficacy and safety of fasudil in PAH treatment. Both intravenous and inhaled fasudil elicit remarkable reductions in pulmonary arterial pressure and pulmonary vascular resistance in PAH patients without producing peripheral vasodilation.27,28 Thus, we have used fasudil as a model drug to test the applicability of our PAH-on-a-chip model. Fasudil, administered via the luminal channel, inhibited the migration of pre-existing PAH-SMCs from the medial to the endothelial layer (Fig. 5E) and reduced the thickness of the intimal and medial layer (Fig. 5F and S8†). In the presence of PAH-ECs and PAH-ADCs, PAH-SMCs assumed an elongated and spindle-like shape; fasudil caused the spindle-like SMCs to become less elongated and more round in shape. Interestingly, the efficacy of fasudil in the medial layer was different in the chips containing PAH-EC/SMC/ADCs compared to the chips containing PAH-EC/SMCs. These results suggest that fasudil may act differently in the presence of PAH-ADCs and highlights the necessity of using a multi-layer PAH-chips and not monolayer cells/chips.
The chip simulates the sex-disparity in PAH and shows differential responses to estrogen treatment
The sex-disparity in PAH and the sex-preferential response to PAH therapy are two perplexing features of PAH pathophysiology.18,29 Currently, the treatment plan for a male PAH patient is no different from that for a female PAH patient, although various clinical trials suggest that PAH patients would fare better with a sex-specific therapy. Progress in understanding the sex-disparity in PAH has been slow, primarily because of the lack of experimental models that can recapitulate human PAH in its sex-specific forms. Thus, to delineate the confounding issues surrounding biological sex and sex hormones in PAH, we prepared male and female chips by seeding QD525-labeled PAH-ECs, QD605-labeled PAH-SMCs and PAH-ADCs from male and female patients. Compared with male chips (chips prepared with cells from male patients), female chips (chips prepared with cells from female patients) showed reduced intimal but greater medial thickening. However, male chips showed a greater number of FSP-1+CD31− PAH-SMCs in the intimal and luminal layers (Fig. S9†), suggesting that males develop a more severe form of the disease. β-Estradiol, dose-dependently, caused a greater increase in intimal and medial thickening in female chips when compared with male chips (Fig. 6, A to D, and S10†). Similarly, compared with the male chips, β-estradiol-treated female chips showed a greater level of muscularization, determined by counting QD705+ PAH-SMCs in different layers (Fig. 6E). In the female chips, the expression of both aromatase and CYP1B1 was higher than in the male chips (Fig. 6F), although the deposition of collagen was higher in the male chips (Fig. 6G). These results suggest that the newly developed PAH-tissue chip model responds to hormones in a fashion like that observed in PAH patients.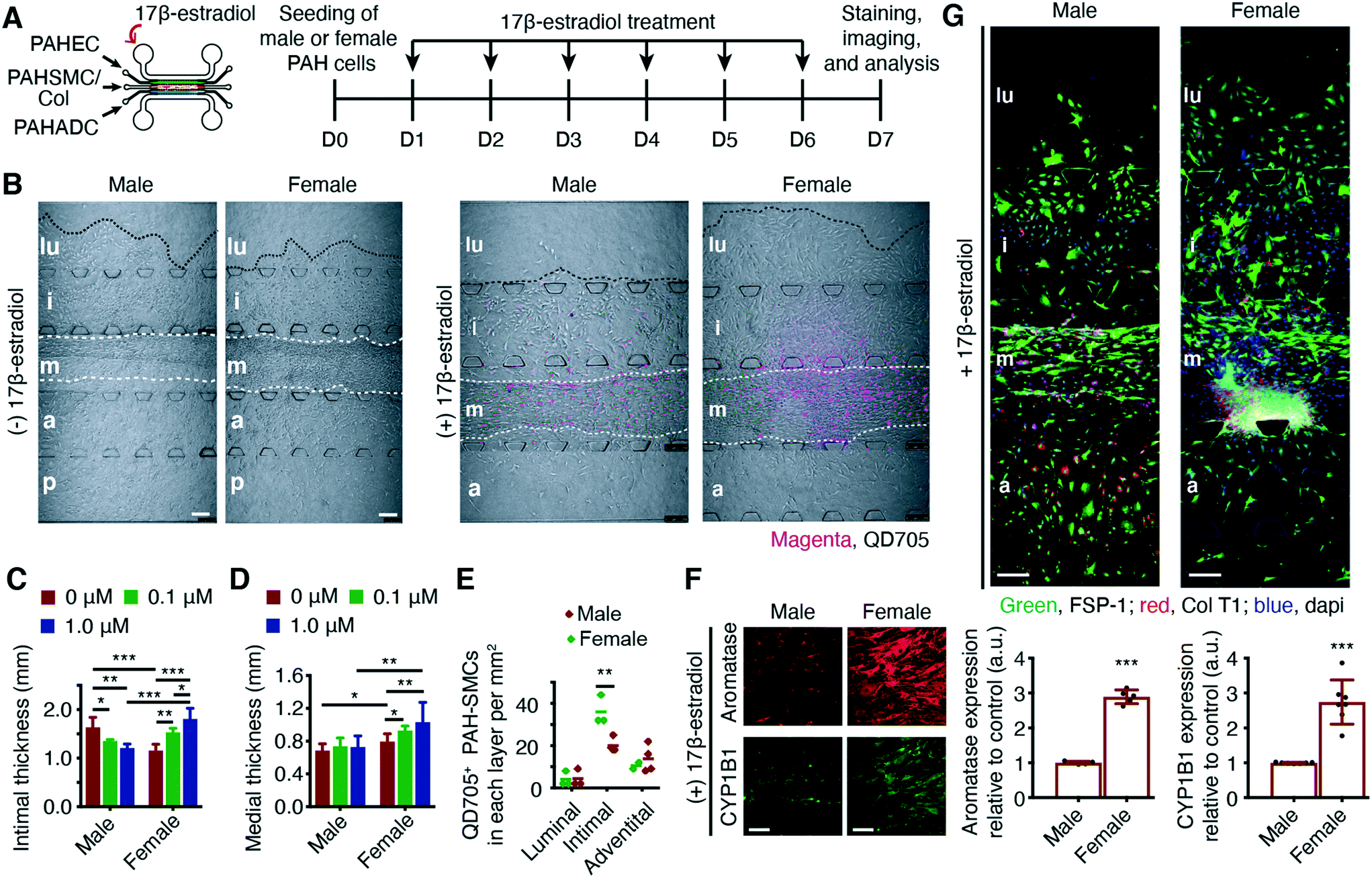 | ||
| Fig. 6 Male and female PAH chips respond differently to estrogen treatment. (A) Male and female chips, prepared with PAH-EC/SMC/ADCs of male or female patients, were treated with 0.1 and 1 μM of 17β-estradiol for six days starting from day 1. (B) PAH-EC/SMC/ADCs of male and female patients showed varying degrees of arterial remodeling; female chips showed reduced intimal but greater medial thickening compared with the male chips. The extent of intimal and medial thickening in female chips, caused by dose-dependent treatment of 17β-estradiol, was greater than that in male chips (male and female PAH-SMCs were labeled with quantum dot 705 (QD705) before seeding). (C and D) The thickness of the intimal and medial layers of male and female chips. (E) The number of QD705+ PAH-SMCs, determined by tracking of QD705. (F) Expression of aromatase and CYP1B1 in the medial layers of male and female chips treated with 1 μM of 17β-estradiol. (G) When treated with 17β-estradiol, the extent of collagen deposition, assessed by immunostaining of FSP-1 and human collagen type 1α1 (Col T1α1), in the medial and adventitial layers of the female chips was greater than that in the same two layers of the male chips. *p < 0.05; **p < 0.01; ***p < 0.001; data represent mean ± SD of chips prepared with pulmonary arterial cells from two male and two female PAH patients. n = 6 chips prepared with the same cells from the same donors/patients or rats. Scale bars 100 and 250 μm. | ||
Discussion
In this study, we validated the hypothesis that a cell-laden microfluidic device can emulate the PAH pathophysiology in humans. Our device combines the ingenuity of microfluidic technology and the sophistication of combinatorial study design to investigate the pathogenesis of PAH. The microfluidic device allows both control and PAH-afflicted PACs to grow without changing their initial phenotypes. Control PACs remained confined within their designated arterial layers but PAH-afflicted PACs migrated toward the luminal and intimal layers of the chip akin to arterial thickening and dysregulated EC-SMC contact of PAH-afflicted pulmonary arteries (Fig. 2). The cellular remodeling we observed in the PAH-on-a-chip may result from increased proliferation, hypertrophy, migration, and apoptosis of PACs, and compartment-specific ECM remodeling.Although animal models of PAH remain an important experimental tool for assessing the pulmonary arterial changes, these models cannot delineate many confounding variables implicated in PAH pathogenesis. To determine the degree of muscularization in PAH rodents, the endothelial layers of the non-muscularized pulmonary arterioles are commonly assessed for the presence or absence of α-SMA positive cells.16 However, PAH-SMCs and transdifferentiated PAH-ECs both express α-SMA, which can give false positive data. By integrating a quantum dot-labeled cell tracking system with immunostaining, we developed a reliable and versatile method for measuring the thickness of the intimal and medial layers and calculating the number of SM-like cells in the endothelial layer of the chips that migrated from the medial layer. Moreover, the Rho kinase inhibitor fasudil, an investigational anti-PAH drug, reduced the intimal and medial thickness in the chips prepared with PAH-EC/SMCs or PAH-EC/SMC/ADCs. This result is consistent with the in vivo studies with fasudil in the MCT and SuHx rat models of PAH.30 This study supports the deployment of our chips to study the efficacy of newly developed anti-PAH drugs such as the ASK-1 inhibitor, GS-44421731 or TGF-β trap therapeutics,32 while eliminating many of the confounding factors associated with animal and 2D cellular models.
EndMT has been implicated in the pathology of human PAH.20 In the pulmonary vasculature, interactions between ECs, SMCs, and ADCs are important for the homeostasis of the pulmonary circulation. However, aberrant interactions among these three cell types may lead to EndMT and pulmonary arterial remodeling.33 Our tissue-chip model showed that control ECs, under the influence of PAH-SMCs and PAH-ADCs, appear to gain mesenchymal characteristics and differentiate into fibroblast- or SMC-like cells (Fig. S6†). Transdifferentiated ECs with an SMC-like phenotype also showed higher migratory and invasive properties. There was an increased number of ECs in the luminal compartment of the chips and overexpression of intracellular FSP-1, a calcium-binding protein (S100A4) that regulates the cellular proliferation, motility, and development of PAH.34,35 In general, exogenous factors such as hypoxia or local injury are known to induce transient EndMT. In addition, our tissue-chip model suggests that cellular cross-talk between PAH-SMC/PAH-ADCs and neighboring control ECs may contribute to EndMT and increase the pool of α-SMA and FSP-1 positive cells that might further contribute to the arterial remodeling as the disease progresses. Thus, the use of our PAH-on-a-chip device offers benefits beyond what is known from the current preclinical animal models, and further study may illuminate the role of non-endothelial PACs in inducing EndMT during PAH progression. We also observed that the luminal layer of the device showed varying levels of response to flow induced shear stress. When subjected to shear stress above that produced by normal blood flow, N-ECs underwent morphological changes; the chips showed increased thickening of the intimal and medial layers thickening. These results also highlight the advantage of using the PAH-on-a-chip model over existing in vitro models, in which it is not possible to mimic the pulmonary circulation in the presence of all three principal cell types of the pulmonary arteries.
We currently do not have any experimental tools for screening of PAH therapeutics for sex specific responses. Although various studies have attempted to delineate the role of sex hormones in the sex-disparity in PAH, the influence of the intrinsic sex of the cells (collected from male or female patients) used in in vitro studies has not been considered. Our PAH-chips can emulate the differences in the histopathology of PAH-afflicted arteries that characterize male and female patients: the pathological progression was exacerbated in chips prepared with cells from female PAH patients in the presence estrogen (Fig. 6). Using chips containing male versus female cells, it is now possible to investigate the varying effects of drugs or hormones in a sex-specific manner.
Despite its real promise, the proposed PAH-on-a-chip model requires further optimization or reengineering to develop it into a near-perfect pulmonary vascular model. First, the proposed model cannot simulate the exhalation and inhalation patterns of the respiratory apparatus that may have a role in PAH. Although airway and alveolar dysfunctions have been documented in human PAH36 and in the MCT rat model that is associated with a widespread pneumotoxicity,37 the role of early bronchial/alveolar epithelial cell death in the pathobiology of PAH remains unclear.38 However, the breathing motion could be achieved by incorporating peristaltic movement or mechanical vibration into the device. The current design of the chips can accommodate seeding of the three major cell types of the pulmonary arteries, but not other cells such as pericytes or epithelial cells that may have a role in arterial remodeling in PAH. However, the device has the flexibility to accept another channel on top of the adventitial layer of the chip. This additional channel would allow seeding of a fourth cell type on the chips and would thus facilitate the assessment of interactions among four different cell types. Besides the three major PACs, the roles of various inflammatory and immune cells, such as macrophages, neutrophils, regulatory T-cells, or B-lymphocytes, and progenitor cells on PAH pathogenesis require further delineation.39 By infusing single or combinations of circulating cells into our tissue-chip model, we should be able to assess the influence of various immune or progenitor cells on the pathogenesis of PAH.
This is the first study to demonstrate that a microfluidic device can emulate the salient pathophysiological features of PAH in humans. This cell-laden device can be deployed for developing sex- and age-specific therapy for PAH. Thus PAH-mimicking chips can be used for developing personalized therapy by obtaining PAH cells from biopsies or using PACs differentiated from PAH patient-derived induced pluripotent stem cells.40 Overall, this human PAH-specific model that combines microfluidic technology with PAH biology could also be adapted as a powerful experimental tool for emulating and investigating with other pulmonary vascular diseases.
Author contributions
T. A. A. designed the experiments, performed most of the studies, and co-wrote the manuscript with F. A. A. K. revised the manuscript with T. A. A. and F. A.'s assistance, and designed and fabricated the microfluidic device with the help of T. A. A., Z. D. and W. L. A. K. also helped in experiments for revision. B. L., H. K., and A. A. helped in cell culture and chip-staining experiments and quantified the arterial thickening and muscularization using ImageJ software. T. L. and E. N.-G. helped in designing the sex-disparity experiment and analyzing the data related to estrogen, hormones, and drug treatment. F. A. generated the hypothesis, conceptualized the study and finalized the write-up of the manuscript. R. K., I. F. M., K. R. S., and F. A. analyzed the data and co-wrote the manuscript.Data availability
The authors declare that all data supporting the findings of this study (its ESI† files) are available within the paper and from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported in part by two NIH grants (R01HL114677 and R01HL144590) and a Cardiovascular Medical Research and Education Fund (CMREF) grant awarded to F. Ahsan; and two NIH Grants (R01 HL114887), and (P01 HL014985) awarded to KR Stenmark. We obtained human control and PAH cells from the Pulmonary Hypertension Breakthrough Initiative (PHBI) cell bank. The PHBI is supported by an NHLBI R24 grant (R24HL123767) and CMREF.References
- M. Rabinovitch, J. Clin. Invest., 2012, 122, 4306–4313 CrossRef CAS PubMed.
- G. Sutendra and E. D. Michelakis, Sci. Transl. Med., 2013, 5, 208sr205 Search PubMed.
- S. Umar, A. Iorga, H. Matori, R. D. Nadadur, J. Li, F. Maltese, A. van der Laarse and M. Eghbali, Am. J. Respir. Crit. Care Med., 2011, 184, 715–723 CrossRef CAS PubMed.
- A. Zaiman, I. Fijalkowska, P. M. Hassoun and R. M. Tuder, Am. J. Respir. Cell Mol. Biol., 2005, 33, 425–431 CrossRef CAS PubMed.
- K. R. Stenmark, B. Meyrick, N. Galie, W. J. Mooi and I. F. McMurtry, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2009, 297, L1013–L1032 CrossRef CAS PubMed.
- M. P. Lythgoe, C. J. Rhodes, P. Ghataorhe, M. Attard, J. Wharton and M. R. Wilkins, Pharmacol. Ther., 2016, 164, 195–203 CrossRef CAS PubMed.
- M. Asmani, S. Velumani, Y. Li, N. Wawrzyniak, I. Hsia, Z. Chen, B. Hinz and R. Zhao, Nat. Commun., 2018, 9, 2066 CrossRef PubMed.
- Y. Ding, X. Xu, S. Sharma, M. Floren, K. Stenmark, S. J. Bryant, C. P. Neu and W. Tan, Acta Biomater., 2018, 74, 121–130 CrossRef CAS PubMed.
- S. Bonnet, S. Provencher, C. Guignabert, F. Perros, O. Boucherat, R. T. Schermuly, P. M. Hassoun, M. Rabinovitch, M. R. Nicolls and M. Humbert, Am. J. Respir. Crit. Care Med., 2017, 195, 583–595 CrossRef PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- D. E. Ingber, Cell, 2016, 164, 1105–1109 CrossRef CAS PubMed.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- C. P. Huang, J. Lu, H. Seon, A. P. Lee, L. A. Flanagan, H. Y. Kim, A. J. Putnam and N. L. Jeon, Lab Chip, 2009, 9, 1740–1748 RSC.
- S. A. Comhair, W. Xu, L. Mavrakis, M. A. Aldred, K. Asosingh and S. C. Erzurum, Am. J. Respir. Cell Mol. Biol., 2012, 46, 723–730 CrossRef CAS PubMed.
- A. Kamkaew, H. Sun, C. G. England, L. Cheng, Z. Liu and W. Cai, Chem. Commun., 2016, 52, 6997–7000 RSC.
- A. Q. Sheikh, J. K. Lighthouse and D. M. Greif, Cell Rep., 2014, 6, 809–817 CrossRef CAS PubMed.
- H. P. Reusch, G. Chan, H. E. Ives and R. A. Nemenoff, Biochem. Biophys. Res. Commun., 1997, 237, 239–244 CrossRef CAS PubMed.
- K. M. Mair, X. D. Yang, L. Long, K. White, E. Wallace, M. A. Ewart, C. K. Docherty, N. W. Morrell and M. R. MacLean, Am. J. Respir. Crit. Care Med., 2015, 191, 693–703 CrossRef CAS PubMed.
- Y. Gao, T. Chen and J. U. Raj, Am. J. Respir. Cell Mol. Biol., 2016, 54, 451–460 CrossRef CAS PubMed.
- B. Ranchoux, F. Antigny, C. Rucker-Martin, A. Hautefort, C. Pechoux, H. J. Bogaard, P. Dorfmuller, S. Remy, F. Lecerf, S. Plante, S. Chat, E. Fadel, A. Houssaini, I. Anegon, S. Adnot, G. Simonneau, M. Humbert, S. Cohen-Kaminsky and F. Perros, Circulation, 2015, 131, 1006–1018 CrossRef CAS PubMed.
- R. K. Hopper, J. R. Moonen, I. Diebold, A. Cao, C. J. Rhodes, N. F. Tojais, J. K. Hennigs, M. Gu, L. Wang and M. Rabinovitch, Circulation, 2016, 133, 1783–1794 CrossRef CAS PubMed.
- S. Bonnet and S. Provencher, Am. J. Respir. Crit. Care Med., 2016, 193, 1331–1332 CrossRef CAS PubMed.
- N. Baeyens, C. Bandyopadhyay, B. G. Coon, S. Yun and M. A. Schwartz, J. Clin. Invest., 2016, 126, 821–828 CrossRef PubMed.
- M. Oka, N. Homma, L. Taraseviciene-Stewart, K. G. Morris, D. Kraskauskas, N. Burns, N. F. Voelkel and I. F. McMurtry, Circ. Res., 2007, 100, 923–929 CrossRef CAS PubMed.
- A. Keshavarz, A. Alobaida, I. F. McMurtry, E. Nozik-Grayck, K. R. Stenmark and F. Ahsan, Mol. Pharmaceutics, 2019, 16, 3414–3429 CrossRef CAS PubMed.
- J. Rashid, K. Nahar, S. Raut, A. Keshavarz and F. Ahsan, Mol. Pharmaceutics, 2018, 15, 1755–1765 CrossRef CAS PubMed.
- A. Keshavarz, H. Kadry, A. Alobaida and F. Ahsan, Expert Opin. Drug Delivery, 2020, 17, 439–461 CrossRef CAS PubMed.
- Y. Zhang and S. Wu, Pulm. Pharmacol. Ther., 2017, 46, 54–63 CrossRef CAS PubMed.
- N. B. Gabler, B. French, B. L. Strom, Z. Liu, H. I. Palevsky, D. B. Taichman, S. M. Kawut and S. D. Halpern, Chest, 2012, 141, 20–26 CrossRef CAS PubMed.
- K. T. Mouchaers, I. Schalij, M. A. de Boer, P. E. Postmus, V. W. van Hinsbergh, G. P. van Nieuw Amerongen, A. Vonk Noordegraaf and W. J. van der Laarse, Eur. Respir. J., 2010, 36, 800–807 CrossRef CAS PubMed.
- G. R. Budas, M. Boehm, B. Kojonazarov, G. Viswanathan, X. Tian, S. Veeroju, T. Novoyatleva, F. Grimminger, F. Hinojosa-Kirschenbaum, H. A. Ghofrani, N. Weissmann, W. Seeger, J. T. Liles and R. T. Schermuly, Am. J. Respir. Crit. Care Med., 2018, 197, 373–385 CrossRef CAS PubMed.
- B. L. Roman and C. St Hilaire, Am. J. Respir. Crit. Care Med., 2016, 194, 1047–1049 CrossRef PubMed.
- K. R. Stenmark, M. Frid and F. Perros, Circulation, 2016, 133, 1734–1737 CrossRef PubMed.
- S. Greenway, R. J. van Suylen, G. Du Marchie Sarvaas, E. Kwan, N. Ambartsumian, E. Lukanidin and M. Rabinovitch, Am. J. Pathol., 2004, 164, 253–262 CrossRef CAS PubMed.
- Y. Dempsie, M. Nilsen, K. White, K. M. Mair, L. Loughlin, N. Ambartsumian, M. Rabinovitch and M. R. Maclean, Respir. Res., 2011, 12, 159 CrossRef CAS PubMed.
- P. Fernandez-Bonetti, E. Lupi-Herrera, M. L. Martinez-Guerra, R. Barrios, M. Seoane and J. Sandoval, Chest, 1983, 83, 732–738 CrossRef CAS PubMed.
- P. M. Bummer, J. A. Baughn, L. P. Sanders, K. R. Absher, W. N. O'Connor, J. W. Olson and M. N. Gillespie, Toxicology, 1994, 90, 53–62 CrossRef CAS PubMed.
- K. L. Colvin and M. E. Yeager, J. Pulm. Respir. Med., 2014, 4, 198 Search PubMed.
- M. R. Nicolls and N. F. Voelkel, Am. J. Respir. Crit. Care Med., 2017, 195, 1292–1299 CrossRef CAS PubMed.
- S. Sa, M. Gu, J. Chappell, N. Y. Shao, M. Ameen, K. A. Elliott, D. Li, F. Grubert, C. G. Li, S. Taylor, A. Cao, Y. Ma, R. Fong, L. Nguyen, J. C. Wu, M. P. Snyder and M. Rabinovitch, Am. J. Respir. Crit. Care Med., 2017, 195, 930–941 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00605j |
| ‡ Current affiliation: Department of Pharmaceutical Sciences, The University of Texas at El Paso, El Paso, Texas, USA. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |