
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoint-of-care testing system for digital single cell detection of MRSA directly from nasal swabs†
Martin
Schulz‡
 a,
Silvia
Calabrese‡
a,
Florian
Hausladen
b,
Holger
Wurm
b,
Dominik
Drossart
b,
Karl
Stock
b,
Anna M.
Sobieraj
c,
Fritz
Eichenseher
c,
Martin J.
Loessner
c,
Mathias
Schmelcher
c,
Anja
Gerhardts
d,
Ulrike
Goetz
d,
Marina
Handel
d,
Annerose
Serr
e,
Georg
Haecker
e,
Jia
Li
f,
Mara
Specht
a,
Philip
Koch
a,
Martin
Meyer
a,
Philipp
Tepper
a,
Raimund
Rother
a,
Michael
Jehle
a,
Simon
Wadle
a,
Roland
Zengerle
af,
Felix
von Stetten
*af,
Nils
Paust‡
af and
Nadine
Borst‡
af
a,
Silvia
Calabrese‡
a,
Florian
Hausladen
b,
Holger
Wurm
b,
Dominik
Drossart
b,
Karl
Stock
b,
Anna M.
Sobieraj
c,
Fritz
Eichenseher
c,
Martin J.
Loessner
c,
Mathias
Schmelcher
c,
Anja
Gerhardts
d,
Ulrike
Goetz
d,
Marina
Handel
d,
Annerose
Serr
e,
Georg
Haecker
e,
Jia
Li
f,
Mara
Specht
a,
Philip
Koch
a,
Martin
Meyer
a,
Philipp
Tepper
a,
Raimund
Rother
a,
Michael
Jehle
a,
Simon
Wadle
a,
Roland
Zengerle
af,
Felix
von Stetten
*af,
Nils
Paust‡
af and
Nadine
Borst‡
af
aHahn-Schickard, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: Felix.von.Stetten@Hahn-Schickard.de; Tel: +49 761 203 73243
bInstitut für Lasertechnologien in der Medizin und Meßtechnik, University of Ulm, Helmholtzstraße 12, 89081 Ulm, Germany
cInstitute of Food, Nutrition and Health, ETH Zurich, Schmelzbergstrasse 7, 8092 Zurich, Switzerland
dHohenstein Institut für Textilinnovation gGmbH, Schlosssteige 1, 74357 Boennigheim, Germany
eUniversity Medical Center Freiburg, Institute for Microbiology and Hygiene, Hermann-Herder-Str. 11, 79104 Freiburg, Germany
fLaboratory for MEMS Applications, IMTEK – Department of Microsystems, Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
First published on 22nd June 2020
Abstract
We present an automated point-of-care testing (POCT) system for rapid detection of species- and resistance markers in methicillin-resistant Staphylococcus aureus (MRSA) at the level of single cells, directly from nasal swab samples. Our novel system allows clear differentiation between MRSA, methicillin-sensitive S. aureus (MSSA) and methicillin-resistant coagulase-negative staphylococci (MR-CoNS), which is not the case for currently used real-time quantitative PCR based systems. On top, the novel approach outcompetes the culture-based methods in terms of its short time-to-result (1 h vs. up to 60 h) and reduces manual labor. The walk-away test is fully automated on the centrifugal microfluidic LabDisk platform. The LabDisk cartridge comprises the unit operations swab-uptake, reagent pre-storage, distribution of the sample into 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 droplets, specific enzymatic lysis of Staphylococcus spp. and recombinase polymerase amplification (RPA) of species (vicK) – and resistance (mecA) -markers. LabDisk actuation, incubation and multi-channel fluorescence detection is demonstrated with a clinical isolate and spiked nasal swab samples down to a limit of detection (LOD) of 3 ± 0.3 CFU μl−1 for MRSA. The novel approach of the digital single cell detection is suggested to improve hospital admission screening, timely decision making, and goal-oriented antibiotic therapy. The implementation of a higher degree of multiplexing is required to translate the results into clinical practice.
000 droplets, specific enzymatic lysis of Staphylococcus spp. and recombinase polymerase amplification (RPA) of species (vicK) – and resistance (mecA) -markers. LabDisk actuation, incubation and multi-channel fluorescence detection is demonstrated with a clinical isolate and spiked nasal swab samples down to a limit of detection (LOD) of 3 ± 0.3 CFU μl−1 for MRSA. The novel approach of the digital single cell detection is suggested to improve hospital admission screening, timely decision making, and goal-oriented antibiotic therapy. The implementation of a higher degree of multiplexing is required to translate the results into clinical practice.
1. Introduction
Health care-associated infections with methicillin-resistant Staphylococcus aureus (MRSA) are widespread across the globe and contribute significantly to hospital mortality and health care costs.1–3 Reviews predict that anti-microbial resistances will be the major cause of death in the upcoming decades with up to 10 million deaths per year worldwide.4–6S. aureus is a spherical Gram-positive bacterium, which can be found in a variety of habitats. Mostly it is as a commensal bacterium as a part of the human microflora on skin and mucosa. However, it is an extremely important agent of severe superficial or deep skin and soft tissue infections, wound infections as well as infections of many organs. Local infections with S. aureus have the potential to cause bacteremia, endocarditis and septic shock.7 Around 90% of today's human S. aureus isolates are resistant to penicillin, and resistance to a majority of other available antibiotics is common. Through the acquisition of the mecA gene from coagulase-negative staphylococci (CoNS) S. aureus became resistant to all beta-lactam antibiotics (MRSA), and MRSA has developed into a worldwide epidemic starting in the 1970s.8,9 Due to its positioning on a mobile genetic element, the staphylococcal cassette chromosome mec (SCCmec), the resistance gene can be transferred horizontally to other staphylococcal species.8 Broad resistance to many standard antibiotics is common in epidemic MRSA-strains, and resistances to even last-resort-antibiotics such as vancomycin, linezolid and daptomycin have been reported.7,9 MRSA infections can be very difficult to treat and cause very considerable costs to health care systems. To prevent further spreading of MRSA in hospitals and to reduce costs, the identification of patients colonized by MRSA at hospital admission is of great importance.10–12
The current gold standard for the detection of MRSA is the culture-based approach, which combines broth enrichment culture with plate culture and the identification of methicillin-/oxacillin resistance by various techniques.13,14 However, these methods require laborious workflows, have a high time-to-result of two to three days and may require further subcultures and tests for final confirmation.15 Even though alternative culture-based methods like chromogenic selective media or adenylate kinase activity-based assays can reduce analysis time, they are substantially more expensive and prone to false positive results caused by the growth of methicillin-resistant coagulase-negative staphylococci (MR-CoNS), Enterobacter or Stenotrophomonas maltophilia. Adenylate kinase-based assays are comparably fast (time to result of ∼5 h) but miss some community associated and hospital acquired MRSA strains.15
An alternative to culture-based methods are nucleic acid amplification approaches like the real-time quantitative PCR (qPCR) or PCR-enzyme linked immunosorbent assays, which takes only 2 h 30 min or 3 h 25 min, respectively.15,16 Based on single or double locus detection, the majority of commercially available tests have a major issue in common: the high risk of false positive results. Single-plex assays target the 3′ end of the SSCmec element (the orfX-SCCmec junction) missing the fact that in some strains the mecA gene can be partially excised or lost. In case new SCCmec variants emerge, the primers need to be redesigned and the assay revalidated regularly. Bi-plex assays rely on the simultaneous detection of mecA/mecC gene and a S. aureus specific gene. However, a major flaw of these detection methods is that they cannot differentiate between MRSA and a mixture of MSSA and MR-CoNS both of which may be present in clinical samples. This leads to a very low specificity of <62% and to low positive predictive values of <85%.15,17,18
A new approach is the combination of single cell analysis with a bi-plex detection. By partitioning of clinical samples into thousands of micro cavities, single cells of different bacterial species are spatially separated. The detection of species and resistance genes on single cell level allows a differentiation of MRSA from a mixture of MSSA and MR-CoNS. At this point it has to be noted that in the case of Staphylococcal spp. bacterial cells are arranged in grape like cluster which is due to characteristic mode of cell division.19 For reasons of clarity, these clusters are referred to as single cells. Over the last few years, various partitioning technologies arose, comprising the use of fluidic networks, micro cavities, or droplets.20,21 Using the latter, the first digital single cell approach was published in 2017.22 The extensive study by Luo et al.22 described the first bi-plex digital droplet PCR (ddPCR) approach, which outperformed a standard qPCR and gave a 100% concordance with the culture method. However, this method requires a laborious workflow, which makes it unsuitable for point-of-care testing (POCT).
Centrifugal microfluidics allow miniaturization, parallelization, and integration of laboratory workflows.23 In this work we show the successful implementation of the digital single cell approach in a fully automated system. The bi-plex assay is integrated into the centrifugal microfluidic LabDisk platform allowing the analysis of nasal swab samples on single cell level. The system requires only minimal hands-on time by the operator. It has a short turnaround time and all necessary processing steps and reagents are integrated onto a disposable monolithic cartridge, which is operated by a customized POCT device for actuation, incubation and readout. We show that centrifugal step emulsification enables partitioning of single cells into single droplets to allow a quantitative and qualitative analysis of the nasal swab sample. We tackled the issue of bacterial adhesion on polymeric surfaces, integrated an in-droplet genus-specific lysis by a chimeric endolysin and demonstrated the entire automated workflow with exemplary MRSA strain and spiked donor samples.
2. Digital single cell approach
2.1 System description
The herein presented system for automation of the digital single cell approach consists of two parts: A disk shaped microfluidic cartridge, (LabDisk, see Fig. 1a), and a standalone POCT device for microfluidic actuation, incubation and fluorescent readout (see Fig. 1b and detailed description in section 4.9 c). In addition to the POCT device, a multiple device setup consisting of separate instruments for cartridge actuation, incubation and readout can be used (for details see section 4.10 b). The LabDisk is manufactured out of cyclo-olefin polymer (COP) foil, sealed with an pressure adhesive cover. It includes all necessary reagents pre-stored: the liquid reagents rehydration buffer (RHB) and fluorinated oil for droplet generation are packed on disk in composite foil pouches with a frangible seal, so-called stickpacks, which release liquid reagents at a defined rotational frequency.24 Dry reagents, lytic enzymes, oligo nucleotides and enzymes for RPA are lyophilized either directly into the LabDisk or are placed in form of a pellet into the respective chamber (detailed pre-storage protocol see section 4.9). Dry reagents are rehydrated during processing by the released RHB. Integrated microfluidic unit operations on the LabDisk allow automation of the workflow for performing the digital single cell approach as described in the following paragraph.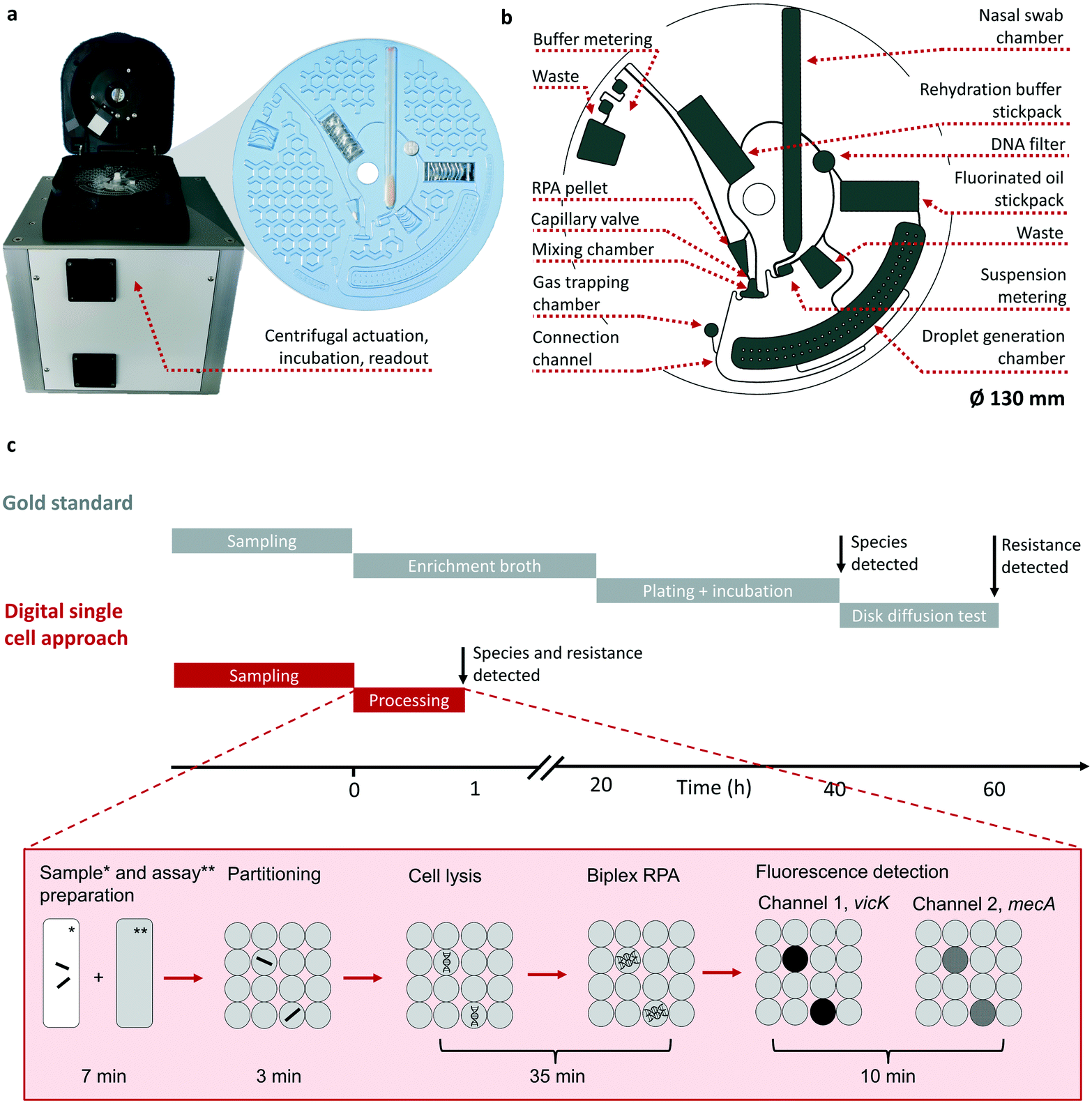 | ||
| Fig. 1 Schematic representation of the POCT system (a and b) and a comparison of the standard workflow versus the digital single cell approach (c). Real pictures of the integrated POCT device for processing of the LabDisk and the micro thermoformed LabDisk (a). The honeycomb structure is used for stiffening the foil disk. Sketch of the LabDisk highlighting unit operations, chambers and channels (b) (for details of the channel and chamber geometries see ESI† S3). Timeline comparing the microbiological gold standard procedure to the digital single cell approach (c). Both processes start with the sampling followed by immersion of the nasal swab sample in a collection medium. For the gold standard the workflow is then continued by an enrichment culture and plate culture for species detection and disk diffusion test for resistance determination. The information about species and resistance is available after 60 h. The digital single cell approach consists of the steps: sample and assay preparation, partitioning, single cell lysis and targeted amplification of the genes vicK and mecA using a recombinase polymerase amplification assay (RPA). Fluorescent readout of two markers results in species and resistance determination in less than 60 min. All steps are performed by an automated protocol. | ||
2.2 Microfluidic automation
Comparable to the gold standard the workflow of the digital single cell approach (see Fig. 1c) starts with the sampling of a nasal swab sample and immersion of the swab in a commercial resuspension medium (detailed sampling protocol see section 4.1). As for the analysis only the wetted swab is needed, the remaining resuspension medium can be used for further culture-based testing or as retention sample. The wetted swab is placed into the LabDisk and a programmed 6-step protocol (illustration see Fig. 2) is started in the POCT device performing the following steps: (1) centrifugal release of the sample from the swab head into the swab chamber followed by an overflow metering of the sample and a transfer into a mixing chamber. (2) Opening of the two stickpacks with fluorinated oil and RHB by centrifugation followed by a centrifugal transfer of the oil to the droplet chamber and the RHB to a buffer metering structure. (3) Transfer of RHB by centrifugo-pneumatic inward pumping25,26 to the dry reagent chamber, where the stored RPA reagents (for details see section 4.7 and 4.8) as well as Staphylococci sp. specific lytic enzymes (see section 4.6) are rehydrated by bidirectional shake mode mixing.27 A capillary valve28 holds back the mixture until rehydration is completed. (4) After triggering the capillary valve by exceeding the burst frequency, the rehydrated reagents are forwarded to the mixing chamber, where it is mixed with the sample by shake-mode mixing.27 During the whole process, the liquid is held back in the mixing chamber by the following valve concept: in step 1) and 2) an air bubble is enclosed in the connection channel by the bacterial suspension and the oil. Interfacial forces retained the liquid in the mixing chamber until a critical frequency is applied, causing the centrifugal pressure to overcome the interfacial forces and thus triggering the valve. (5) Valving of the reaction mixture to the droplet generation chamber and generation of a monolayer array of 20000 droplets by centrifugal step emulsification.29,30 (6) Incubation of the LabDisk at 38 °C for performing the bacterial lysis and RPA reaction. Upon lysis the DNA of the Staphylococci is set free and a bi-plex RPA with fluorogenic probes is used for detection of the mecA gene and a species specific vicK gene. Subsequent to the RPA the fluorescence of the two potentially amplified markers mecA and vicK is detected and the number of mecA/vicK positive and negative droplets is used to calculate the initial bacterial concentration in the sample by Poisson statistics (for detailed calculation see electronic ESI† S1). While analysis with the gold standard takes up to 60 hours until the species and the resistance is determined,13 the digital single cell approach presented here takes less than one hour for fully automated sample preparation to readout. This makes the presented system an ideal tool for point-of-care settings usable e.g. for admission screenings in hospitals. Further, the single units for centrifugal actuation, incubation and readout could potentially be integrated and parallelized in an automated workstation allowing for even higher throughputs in a centralized laboratory setting.
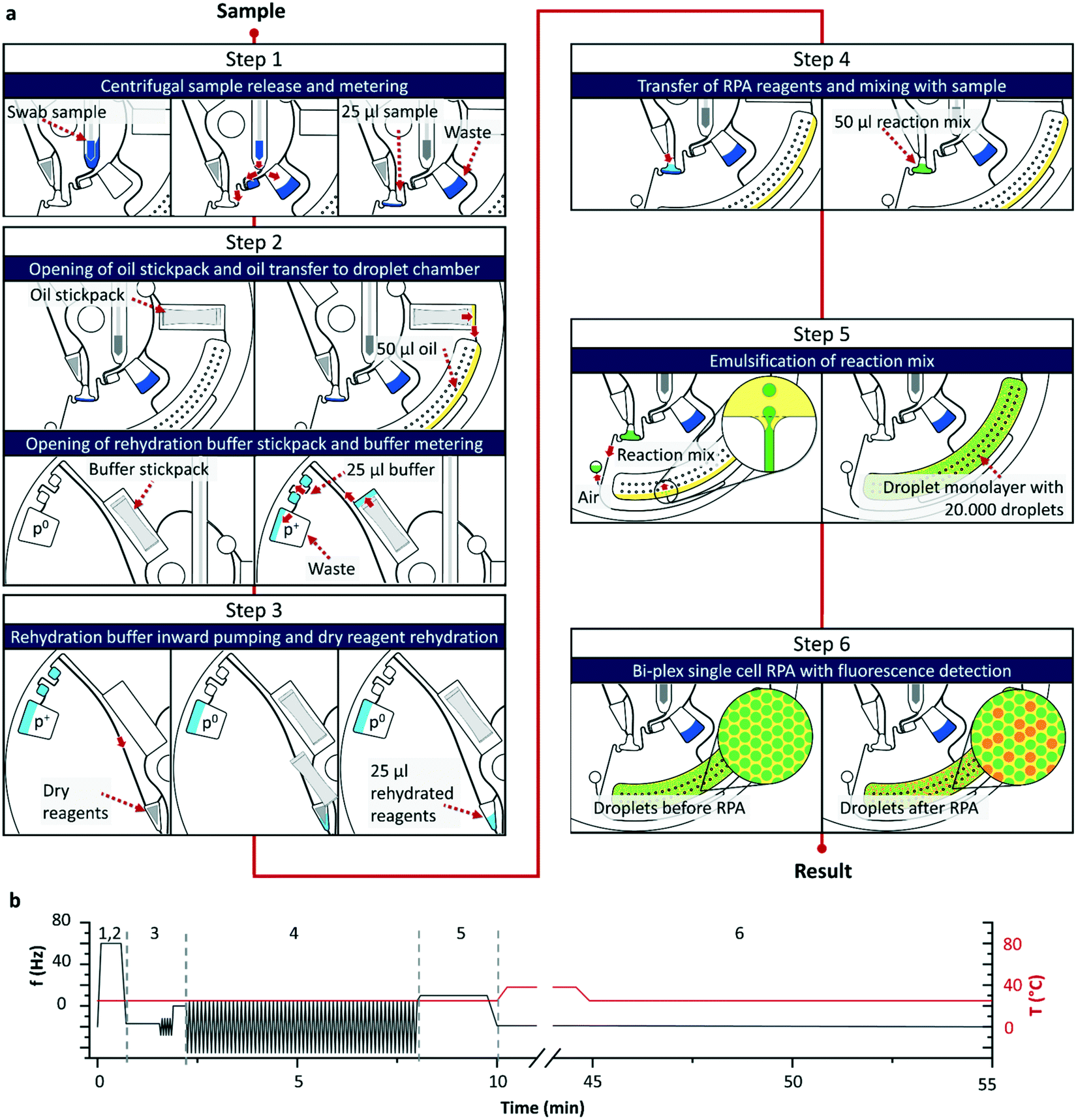 | ||
| Fig. 2 Process description of the microfluidic automation. Schematic workflow of the processing as carried out on the centrifugal microfluidic LabDisk (a). The nasal swab is inserted into the cartridge followed by the steps 1–6: (1) release and metering of the bacterial suspension. (2) Opening of the oil and rehydration buffer stickpacks. (3) Rehydration buffer metering. (4) Mixing of RPA reagents and bacterial suspension. (5) Droplet generation by centrifugal step emulsification and monolayer assembly. (6) Incubation and readout. The total analysis time from swab insertion to result is 55 min. Frequency and temperature protocol of the workflow (b). | ||
2.3 Statistical considerations
Using the digital single cell approach one has to take into account that patient samples may consist of mixed cultures (e.g. MRSA, MSSA and MR-CoNS) and that the distribution of cells follows Poisson statistics (see ESI† S1). Depending on the bacterial concentration, it is possible that a compartment contains no, one or more than one cell. If more than one cell occupy a compartment, false positive signals may occur. In a pre-study with 79 probands (see ESI† S2) where the species composition of the nasal samples was assessed no sample showed the presence of MRSA and MSSA simultaneously. Assuming that a sample contains MRSA and MR-CoNS, then co-compartmentation of these species would result in true positive signals. However, the majority of probands carried MSSA and CoNS in varying ratios. On the basis of a bi-plex assay this would result in a false positive MRSA signal, if for MSSA the vicK gene and for MR-CoNS the mecA gene is detected. By calculating the probability of MSSA and MR-CoNS co-compartmentation it is possible to estimate the number of false positive droplets. In the pre-study the highest ratio of MSSA to CoNS was 100000 to 10000 CFU ml−1. Assuming the case that all CoNS were methicillin resistant, then this would result in a probability of 0.15% false double positive signals and in a total 29 false positive droplets. To prevent a wrong diagnosis due to false positive droplets the calculated false positive droplet number can be excluded from data evaluation in data analysis. It has to be stated that with higher numbers of MSSA and MR-CoNS the calculation error increases. Therefore, for translating this research to clinical practice, a higher degree of multiplexing, covering also various MR-CoNS species, is advisable but was not scope of this work.
3. Results and discussion
During the development process of the fully automated and integrated digital screening platform, several parameters had to be considered. First, the microfluidic processing of the cartridge with all pre-stored reagents was tested (see section 3.1). Secondly, as the cartridge is made out of COP and as it is known that bacteria tend to adhere on polymeric surfaces, it was important to assess the degree of bacterial adhesion to the COP surface and the eventual loss within the channels (see section 3.2). The digital single cell approach requires singularization of cells into the droplets, which is why the separation of MRSA as well as of MS-CoNS was assessed by fluorescence labeling of these organisms followed by testing of the single cell distribution in single droplets (see section 3.3). Whole system performance was finally assessed with a clinical isolate and spiked donor samples (section 3.4).3.1 Microfluidic processing
The microfluidic unit operations on the LabDisk (see section 2.2) were tested for functionality, accuracy and reproducibility by performing the complete workflow in a stroboscopic imaging setup (see section 4.10 a). The stroboscopic images were analyzed after processing. Liquid levels and droplet characteristics were measured using image processing software (see section 4.11). In total six disks were analyzed with the described workflow showing functional processing with minor deviations from set values (Table 1).| Tested parameter | Set value | Measured value |
|---|---|---|
| Released sample from swab | 130 μl | 129 ± 7 μl |
| Sample metering | 25 μl | 25 ± 1 μl |
| Buffer (RHB) metering | 25 μl | 25 ± 1 μl |
| Droplet diameter | 170 μm | 172 ± 2.6 μm |
| CV droplet diameter | <5% | 1.5% |
3.2 Bacterial recovery
Mediated by their physico-chemical surface properties S. aureus are known to adhere to a multitude of surfaces, e.g. on polymeric surfaces as well as on implantable medical devices which makes them a major cause for implant repulsion and medical infections.31,32 Dependent on surface properties of all phases involved i.e. bacteria, swab, material of the cartridge and the suspending medium, the surface tension of the bacteria is larger than that of the suspending medium which results in extensive adhesion of bacteria to hydrophobic substrates like COP which is used as material for manufacturing of the LabDisk.33,34To minimize cell loss through absorption, the pathways for the transport of bacterial suspension within the LabDisk were designed as short as possible and with the largest possible volume to surface ratio. Bacterial recovery was tested in early stages of cartridge development. However, structural changes alone were not sufficient as still nearly 99% of applied bacteria were lost on their way from the swab to the droplets. The bacteria adhered strongly to the channel walls as seen by fluorescence imaging (data not shown). Due to these circumstances, several coating agents were tested. Based on studies investigating the influence of coating agents on surface adhesion of Staphylococcal sp. on polymer surfaces31,35–39 we identified the poloxamer Pluronic F127 as a suitable agent for the reduction of bacterial adhesion in the fluidic channels. Pluronic F127 is a nonionic triblock copolymer composed of hydrophilic outer chains of polyoxyethylene (POE) and a hydrophobic polyoxypropylene (POP) chain in the center. It is assumed that due to self-assembly the hydrophobic POP chain binds to the hydrophobic surface of the COP and that the hydrophilic POE protrudes into the space forming a brush like structure preventing the adsorption of proteins and microorganisms.31,40,41 Treter et al.31 showed that upon coating of polystyrene surfaces with the poloxamer Pluronic F127 the biofilm formation of S. epidermidis could be significantly reduced. In our study we could confirm that coating of the swab chamber with Pluronic F127 significantly reduced bacterial adhesion (see Fig. 3). As applying the coating agent to the complete microfluidic structure posed the risk of weakening the bond to the adhesive sealed cover foil, the coating agent was added to the swab chamber only, where the bacteria containing swab is inserted into the cartridge. For the recovery experiments the swab chamber was coated with varying volumes of 0.5% Pluronic F127 (for details see section 4.8). S. aureus were pipetted into the swab chamber, processed through the device and extracted before the droplet generation step. In direct comparison with the untreated control where only 0.5 ± 0.5% of the bacteria could be recovered we managed to recover up to 58 ± 15% of the bacteria by applying 85 μl of 0.5% Pluronic F127 in the tip of the swab chamber (see Fig. 3) which is significantly higher compared to the untreated control. An increase in Pluronic F127 led to foaming of the bacterial suspension during metering and mixing, which negatively affected the transport of the fluids and therefore of the bacterial recovery. As a result of this investigation 85 μl of Pluronic F127 was used for the manufacturing of the LabDisks. In perspective of using the system not only for the presented MRSA strain but for other strains and bacterial species it has to be considered, that depending on cell wall composition and the expression of adhesins, the hydrophobicity as well as the surface binding capacity between individual bacterial species and strains varies.42–45 Therefore, an individual coating treatment has to be identified depending on the assay approach (not part of this work).
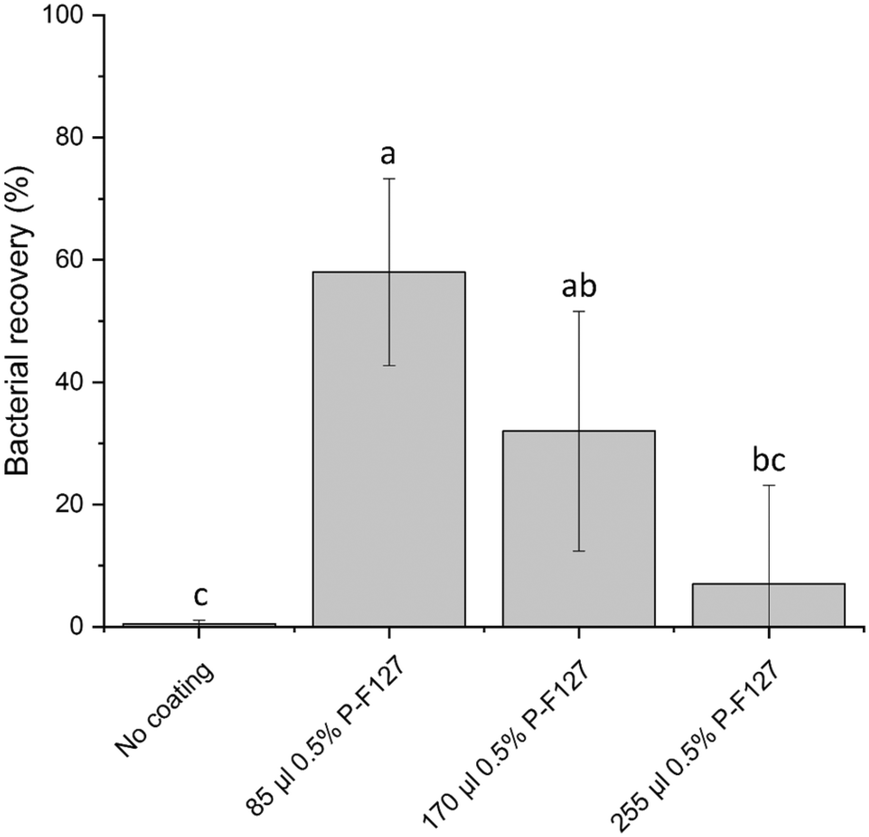 | ||
| Fig. 3 Bacterial recovery rates at different coating conditions. Recovery rates were evaluated by plate counting method after fluidic processing. In this experiment, the swab chamber was treated with different amounts of the coating agent Pluronic-F127 0.5%. Increased amounts of coating agent or coating of additional chambers lead to foaming of the transferred liquid, which resulted in ineffective microfluidic transfer, which is why these experiments were excluded. The values represent the normalized mean values of the recovered bacteria of four independent experiments for each tested condition. Statistical analysis was performed by analysis of variance (ANOVA), followed by Tuckey honest significant difference test (Tuckey HSD; p < 0.05). Lower case letters indicate significant difference. | ||
3.3 Single cell distribution
The digital approach relies on singularization of particles into droplets, which follows the Poisson distribution. In dependence on particle concentration and the number of generated droplets either no, one or more than one particle can be found within the droplets. In order to see whether this is also true for bacterial cells, three different concentrations (102 CFU × 20−1 μl−1, 103 CFU × 20−1 μl−1, 104 CFU × 20−1 μl−1, CFU = colony forming unit) of fluorescently stained MSSA were emulsified into 7500 droplets in a test emulsification chip (detailed protocol see section 4.3). Bacterial concentrations were estimated by reference streak out. Comparison of the cell counts to the calculated average number of targets per droplets μ revealed that bacterial CFU distribution follows Poisson statistics. Cell counts matched the expected numbers of all tested concentrations (see Fig. 4). In order to distinguish MRSA from MR-CoNS, it is an essential prerequisite that the genetic information of only one species is present. Therefore, it is of upmost importance to prove that the different bacterial species can be separated from each other during the emulsification process. To test this, a mixture of two fluorescently labelled strains, S. aureus and S. epidermidis was emulsified followed by an inspection of the single droplets under a fluorescence microscope (protocol see section 4.4). Within the individual partitions either only S. aureus, S. epidermidis or no cells could be observed which successfully demonstrated, that a clear separation of single cells in individual partitions is possible (see Fig. 5).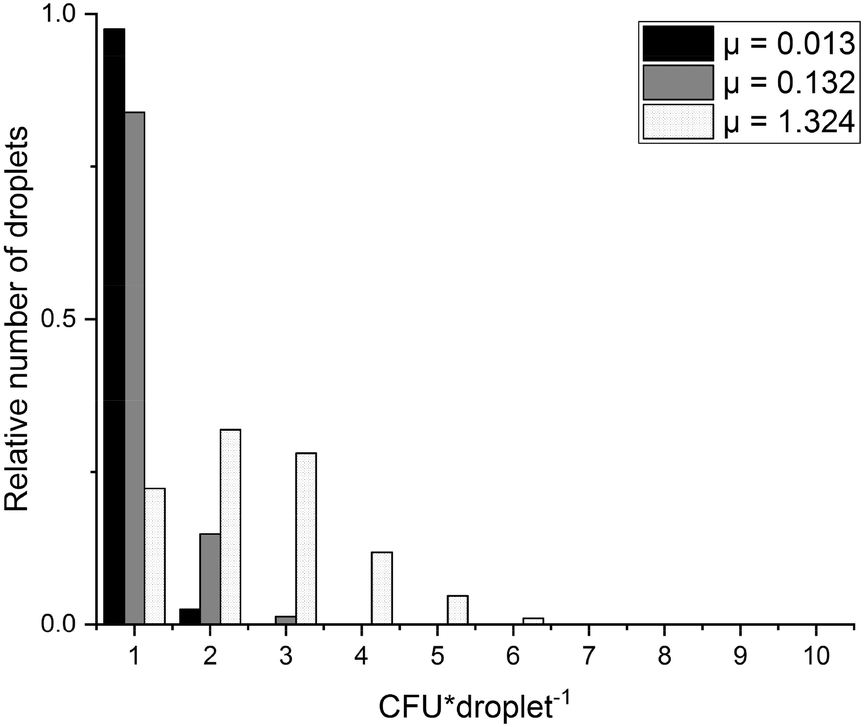 | ||
| Fig. 4 CFU distribution diagram of expected and counted CFU abundances per droplet. Shown are evaluated values for three different concentrations of S. aureus emulsified in 7500 droplets. μ:expected number of CFU per droplet. Analysed droplets: μ = 0.013:960, μ = 0.132:1309, μ = 1.324:592. Data was previously published.46 | ||
 | ||
| Fig. 5 Microscopic image of fluorescently labelled single cells of S. epidermidis (a, red, SYTO62) and S. aureus (b, green, GFP) in droplets. The picture is an overlay of a bright field and two false colour fluorescent images. A zoomed view on one cell is displayed as insert in the picture. | ||
3.4 System performance evaluation
The first step of system performance evaluation was the determination of the limit of detection (LOD). For this, a clinical MRSA isolate culture was diluted in collection medium to 3 ± 0.3 CFU μl−1, 33 ± 9.4 CFU μl−1, and 369 ± 50 CFU μl−1. 129 μl of each dilution step was added to the swab-chamber, which corresponds to the amount of liquid released by the swab during centrifugation (see Table 1). This initial volume is then metered by the fluidic structure during processing as described in chapter 3.1, resulting in a sample volume of 25 μl. For parallelization reason the LabDisks were processed in a multiple device set-up (for details see section 4.10 b). As a reference MRSA counts were determined by using the streak plate method for the same samples. False colour images as well as overlays of the two channels can be seen in Fig. 6 (exemplary image sections). | ||
| Fig. 6 False color images of a ddRPA assay for the detection of a S. aureus specific gene, vicK (green) and the resistance gene mecA (red). Displayed are the gene specific results in separate channels as well as an overlay (yellow) for four different dilution steps. The data shown represents one run out of three per concentration for which only a cropped section is depicted. Results were generated using the multiple device setup (see section 4.10). | ||
For absolute quantification of genotypes, positive and negative droplets were counted and initial concentrations were calculated using Poisson statistics (for detailed calculation see section 3.2 and ESI† S1). The resulting ddRPA values were compared to reference values and plotted in Fig. 7. Even though a shift of the measured values was observed, caused by the bacterial loss (see section 3.2), the results showed a good concordance between expected and measured values. The determined LOD of 3 ± 0.3 CFU μl−1 is in the same order of magnitude as the LOD of 2.9 CFU μl−1 (ref. 22) of the manual digital single cell approach developed by Luo et al.22 (time to result of 4 h) and the commercially available automated solutions for analysis of bulk samples, such as EazyPlex MRSA with an LOD of 10 CFU μl−1 (time to result: ∼35 min) (Amplex Diagnostics GmbH, Germany)47 and the Cepheid GeneXpert MRSA with an LOD of 0.61 CFU μl−1 (ref. 48) (time to result of 6.9 h).
 | ||
| Fig. 7 System performance evaluation with clinical isolate and proband samples. Comparison of ddRPA and reference culture (log scale on both axis). Error bars represent the standard deviation of three independent runs. | ||
In the second step, the influence of the nasal matrix on system performance was tested. Therefore, proband samples of four tested non-MRSA and non-MSSA carrying probands were spiked with a defined MRSA concentration of 26 ± 4.5 CFU μl−1 and processed in the multiple device setup. No influence of the nasal matrix on the system performance was visible (see Fig. 7).
For whole system evaluation, spiked proband samples were used and the LabDisk was processed in the POCT device (for details see section 4.10). The results of the automated evaluation in the LabDisk in comparison to the reference culture (see Table 2) show a functional detection for both markers even though a slight broader distribution of measured could be observed.
| Reference culture count [CFU μl−1] | vicK [CFU μl−1] | mecA [CFU μl−1] |
|---|---|---|
| 212 ±63 | 314 | 52 |
| 30 ± 1.7 | 10 | 2 |
| 2.6 ± 0.6 | 1 | 1 |
Overall, the system performance evaluation showed a good concordance of the reference culture to the ddRPA results, which also demonstrates the reliability of the microfluidic system. The observed shift of positive values in favour of the vicK target (see Fig. 6 and 7 and Table 2) is in the same order of magnitude and can be accounted to the assay performance. By further improvement of the assay efficiency, an absolute match for target detection and an increased sensitivity of the assay is conceivable.
4. Experimental
4.1 Sampling
This study has been approved by the Ethics Committee of the University of Freiburg (application number: 10008/20) and was performed in compliance with the relevant laws and institutional guidelines. Nasal swab samples were collected from healthy lab members. Informed consent was obtained from all study participants. Samples were taken using Copan FLOQSwab (Copan Flock Technologies Srl, Italy) by wiping the nasal atriums for 5 s each followed by immersion of the swab in MSwab media (Copan Flock Technologies Srl, Italy).4.2 Bacteria
Clinical isolates of methicillin-sensitive S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA) strains were derived by the University Hospital Freiburg. Isolates were identified by conventional microbiological methods. Reference strains MSSA ATCC 29213, MRSA clinical isolate 402466 and S. epidermidis DSMZ 1798 were cultivated in standard LB at 37 °C until an OD of 0.2–0.3 was reached (Utrospec 10, Biochrom, Germany).For visualizing the encapsulation of bacteria in droplets, S. aureus AH133 (ref. 49) were cultivated in LB-broth (Sigma-Aldrich Inc., USA) containing erythromycin 1 μg·100 μl−1 (Karl Roth GmbH, Germany).
:1 (v/v). 25 μl of the reference mix was given to 75 μl of LB (Sigma-Aldrich Inc., USA) and spreaded onto CASO (Sigma-Aldrich Inc., USA) agar plates (n = 5). Plates were incubated over night at 37 °C and evaluated on the next day.
As reference for the system performance tests bacteria were diluted in MSwab media (Copan Flock Technologies Srl, Brescia, Italy) spread on CASO (Sigma-Aldrich Inc., USA) agar plates (n = 5) and incubated over night at 37 °C and evaluated on the next day by counting of the grown colonies.
4.3 Singularization of bacteria for statistic evaluation
First S. aureus cells were diluted 1:102, 1:103. 1:104 in LB medium. 100 μl of each dilution were stained with 0.5 μl Syto 9 green fluorescent stain (Thermo Fisher Scientific Corp., USA). Droplets were generated with an emulsification chip (DropChip; as described in detail in ref. 50) in a standard laboratory centrifuge (uniCFUGE 3, LLG GmbH, Germany) at 1500 RPM for 2 min. For each experiment 20 μl fluorinated oil (HFE, Novec 7500 3 M Corp., USA with the addition of an interface stabilization agent Pico-Surf 1 5%, Dolomite Ltd., United Kingdom) and 20 μl bacterial suspension were used. After droplet generation the chip was centrifuged at 200g for 5 min in a standard laboratory centrifuge (Heraeus GmbH, Germany) to sediment the bacteria to the bottom of the droplets. In the last step, the droplets were observed in a fluorescent microscope (Axiophot, Zeiss, Germany) and the cells per droplet were counted.
4.4 Singularization of bacterial species for single cell evaluation
S. aureus AH133 were diluted in MSwab media (Copan Flock Technologies Srl, Brescia, Italy). Prior to staining S. epidermidis cells were centrifuged at 4500 rpm for 13 minutes at RT. Supernatant was discarded and cells were re-suspended in 0.01 M PBS. Cells were stained with SYTO62 (10 μM, Thermo Fisher Scientific, Germany) for 20 minutes at room temperature. After incubation, cells were further diluted in 0.01 M PBS. Stained S. epidermidis were mixed with GFP expressing S. aureus AH133 (1:1; v/v). Droplets were generated in the LabDisk (see section 2.2) and processed in the LabDisk-Player 1 (see section 4.10, b) at 30 Hz for 2 min. For each experiment 50 μl fluorinated oil (HFE, Novec 7500 3M Corp., USA with the addition of an interface stabilization agent Pico-Surf 1 5%, Dolomite Ltd., United Kingdom) and 50 μl bacterial suspension were used. The bacterial suspension was directly pipetted into the mixing chamber of the LabDisk. After droplet generation the cartridge was centrifuged at 200g for 5 min in a standard laboratory centrifuge (Heraeus GmbH, Germany) to sediment the bacteria to the bottom of the droplets. In the last step, the droplets were observed in a fluorescent microscope (Observer Z1, Zeiss, Germany) and the cells per droplet were counted.
4.5 Bacterial recovery
MRSA were diluted in MSwab media (Copan Flock Technologies Srl, Brescia, Italy) to a concentration of 50 CFU μl−1. For the on disc test 129 μl of the diluted bacteria were pipetted into the swab chamber, processed through the device and extracted before the droplet generation step. During processing 25 μl of the bacteria were mixed with 25 μl of RPA reaction buffer (65% rehydration buffer (RHB) and 10% MgAc). After processing 25 μl of this mix was given to 75 μl of LB and spreaded onto CASO agar plates.4.6 Construction and production of chimeric peptidoglycan hydrolases
Chimeric peptidoglycan hydrolases were designed to target distinct bonds in the S. aureus peptidoglycan in order to promote synergy when applied simultaneously. M23-LST_SH3b2638A is composed of an M23 endopeptidase domain from lysostaphin51 and an SH3b cell wall binding domain (CBD) from Ply2638A.52 CHAPGH15_SH3bALE1 consists of a CHAP endopeptidase domain from LysGH15 (ref. 53) and an SH3b CBD from ALE1.54 Chimeric nucleotide sequences optimized for E. coli codon usage were chemically synthesized (GeneArt Gene Synthesis, Thermo Fisher Scientific Corp., USA), digested with the restriction enzymes NdeI and BamHI (New England Biolabs Corp., USA), ligated into vectors pET-9a-d(+) and pET-302/NT-His (Novagen, Merck KGaA, Germany), respectively, and introduced into E. coli BL21 Gold (DE3) (Stratagene, La Jolla, CA, United States) by electroporation. Bacteria were grown at 37 °C in LB-PE medium55 supplemented with 100 μg mL−1 ampicillin (pET-302) or 50 μg mL−1 kanamycin (pET-9a) until an OD600 of 0.5. Protein expression was induced by addition of IPTG (0.5 mM), and cultivation was continued for 18 h at 19 °C. Cells were pelleted by centrifugation and disrupted using a Stansted Fluid Power Pressure Cell Homogenizer (100 MPa). Lysates were supplemented with 5 units of DNase I (Thermo Fisher Scientific Corp., USA) and centrifuged at 20000 × g for 1 h at 4 °C. Supernatants were filtered (0.45 μm) and proteins purified by cation exchange chromatography on an Äkta purifier FPLC device (GE Healthcare Life Sciences Group, United Kingdom), using a HiTrap SP-FF column (GE Healthcare Life Sciences Group, United Kingdom) equilibrated with 3 column volumes (CV) of CIEX immobilization buffer (20 mM Na2HPO4, 10% glycerol, pH 7.4). After sample loading and washing with 3 CVs of CIEX immobilization buffer to remove unbound proteins, target proteins were eluted from the column by a gradient of CIEX elution buffer (20 mM Na2HPO4, 1 M NaCl, 10% glycerol, pH 7.4). Protein concentrations were determined by NanoDrop (Thermo Fisher Scientific Corp., USA), and the purity of protein preparations was confirmed by SDS-PAGE. Proteins were dialyzed against PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), frozen at −80 °C and vacuum-dried overnight. Results for determination of the lysis efficiency are displayed in ESI† S5.
4.7 Primers and probes
The primer and probes were designed by the Hohenstein Institut für Textilinnovation gGmbH (Bönnigheim, Germany) to detect sequences of the S. aureus specific gene vicK and the resistance gene mecA and were synthesized by TIB MOLBIOL Syntheselabor GmbH (Germany) (sequences see ESI† S4).4.8 System performance evaluation (ddRPA)
All RPA reactions were performed using the TwistAmp exo kit, TAEXO02KIT (TwistDX Limited, Cambridge, United Kingdom). Due to the elution of the bacteria from the nasal swab and the need to add a coating reagent to reduce bacterial loss within the system, the standard RPA protocol was adapted to the system needs. The final concentrations are given in percent per volume: 32.5% RHB, 50% buffer (MSwab, Copan Inc., USA), 1.65% Pluronic F127 (10% stock in water, Merck KGaA, Germany), 10.85% H2O, 5% MgOAc (stock concentration: 280 mM). Primers were added to a final concentration of 200 nM and probes to a final concentration of 60 nM. Droplet generation was performed with fluorinated oil (HFE, Novec 7500 3 M Corp., USA with the addition of an interface stabilization agent Pico-Surf 1 5%, Dolomite Ltd., United Kingdom) as continuous phase. MRSA were diluted in MSwab media and added into the swab chamber. For the experiments with the proband samples nasal swab samples were taken from MRSA and MSSA negative probands directly before the experiment. The swabs were immersed in MSwab medium. Proband samples were spiked with bacteria, vortexed and applied into the disk.4.9 Microfluidic disk design and fabrication
The fluidic unit operations were designed using network simulation-based CAD modeling56 (Dassault Systèmes SolidWorks, France; MathWorks Matlab, USA). The disk shaped LabDisk (Ø 130 mm) was manufactured at the Hahn-Schickard Lab-on-Chip foundry57 in the following steps:1) Manufacturing of the molding tool by micromachining in aluminum (KERN Evo mill, KERN Microtechnik GmbH, Germany).
2) Micro thermoforming of a cyclic-olefin polymer foil (COP ZF-14-188, Zeon Corp., Japan) in a blister machine (R760S, Rohrer AG, Germany) at T = 100–150 °C with p = 5–7 bar (described in detail in ref. 58).
3) Hexamethyldisiloxan (HMDSO) plasma coating of the LabDisk-Surface (Piccolo Ghz, Plasma Electronic GmbH, Germany) with the Lipocer® standard process t = 500 s, 50 sccm, P = 100 W.
4) Packaging of the liquid reagent stickpacks (SBL-50, Merz-Verpackungs-maschinen GmbH, Germany; Foil material: SteriFoil (Safta, SpA, Italy)). Process parameters oil-stickpack: P = 3 bar, T = 105.5 °C, V = 50 μl. Process parameters BHB-stickpack: P = 3 bar, T = 98 °C, V = 105 μl.
5) Preloading of 85 μl of 0.5% Pluronic F127 (Merck KGaA, Germany) into of the tip of the swab chamber, followed by air drying for 12 h at RT.
6) Lyophilization of the lytic enzyme and the RPA oligonucleotides: the chimeric enzymes M23LST(L)_SH3b2638 and CHAPGH15_SH3bALE1 were added to the dry reagent chamber to reach a final concentration of 60 nM in the reaction mix. Oligonucleotides were added to the mixing chamber (see section 4.8). Remark to the lyophilization protocol: because lyophilization was carried out directly in the LabDisk, the materials of the LabDisk were exposed to the whole lyophilization process. This led in some cases to a material failure during fluidic processing resulting in a failure of the bond between sealing foil and disk-foil. This behavior could be completely prevented by incorporating all dry reagents in the RPA-Lyo-Pellet. Unfortunately this was not possible in the timeframe of this work because of manufacturing transitions at the manufacturer. Then lytic enzymes and oligonucleotides were lyophilized within the disk at −50 °C for 16 hours (Alpha 2-4 LSC, Christ, Germany).
7) Placement of the RPA Pellet TAEXO02KIT (TwistDX Limited, United Kingdom) in the dry-reagent chamber auf the LabDisk.
8) Placement of the liquid reagent stickpacks into the rehydration buffer and fluorinated oil stickpack chamber.
9) Automated sealing of the disk using a pressure sensitive adhesive film (9795R diagnostic tape, 3M Corp., United States) (ProSeal, Harro Höfliger GmbH, Germany) at T = RT, FMax = 8 kN, t < 30 s.
10) Cutting of the final disk-shape by laser cutting at a wavelength of λ = 10.6 μm with P = 30 W (D320i, Domino GmbH, Germany). The functionalized disks were not stored but used shortly after production.
4.10 Cartridge actuation, incubation and fluorescence detection
For cartridge actuation, incubation and fluorescence detection three different setups were used at different stages in the development process.The unit for optical excitation (illumination) and detection (imaging) of the fluorophores within the droplets consists of a sequential excitation and detection of each fluorophore by suitable and sequentially switched LEDs and selected excitation and emission filters (see ESI† Fig. S3). The optical system was integrated into a Lab-Disc-Player 1 (QIAGEN Lake Constance GmbH, Germany) which provided the mechanical functionalities: rotation at a defined rotational frequency, heating and positioning for readout (setup see ESI† Fig. S4).
The excitation is performed under a mean inclination angle of 18°. To provide a homogeneous illumination of the measurement area, for each color the homogenously emitting, rectangular chip-surface of two LEDs (Golden DRAGON LB W5SM & LB W5SM, OSTAR LCG H9RN, OSRAM Opto Semiconductors GmbH, Germany, each positioned on the opposite side around the objective lens) was imaged to the measurement-plane by a specially designed projection lens system. The sequential change of the emission filters (Semrock, IDEX Health & Science, LLC, USA) was realized by a motorized filter wheel (Edmund Optics GmbH, Germany) which is connected to the PC via RS232. In addition, a bright field light source (LFL-612GR2-P, CCS Inc., USA) was installed behind the lab disk in the top of the player.
The measurement of the whole droplet area on the disc was divided in 29 sequentially measured sub-areas (7.88 mm × 5.91 mm). By imaging this sub-area onto the camera chip (GE1650M, Allied Vision, Stadtroda GmbH, Germany, Chip size: 11.84 mm × 8.88 mm, 1600 × 1200 pixel, pixel size: 7.4 μm) using a commercial lens (S5LPJ7915, Sill Optics GmbH, Germany, β = −1.5, F# = 12.5) a lateral resolution of 4.92 μm per pixel was achieved (resulting in 34 × 34 pixels per droplet). The camera is connected to the PC via ethernet. For stitching of the sub-areas and determination of the droplet positions, a bright field-image of each sub-area was taken, using the bright field light-source without any detection filter.
The six LEDs for excitation are driven by three independent adjustable constant current sources (one for each excitation channel). Each current source is based on a digital potentiometer with 99 defined states, a transimpedance amplifier and the necessary power supply circuits integrated on a PCB. All light sources are controlled by the use of an I/O-device (RedLab 1208FS, Meilhaus Electronic GmbH, Germany) which is connected to the PC via USB. A particularly written software (C++) on the PC does the controlling of all optical components and the process of taking pictures.
4.11 Data analysis
For droplet diameter CV measurement and visualizing of bacteria within droplets the disks were processed in LabDisk-Player 1 with stroboscopic modification. Droplet diameter CV was measured with the public domain software ImageJ 1.52i (National Institute of Health (NIH), USA).False color images for visualization of the fluorescent droplet readout were generated with the image processing software Photoshop (Adobe Inc., USA). Absolute values for quantification were calculated by Poisson statistics of counted positive and negative droplets (detailed description of the calculation see ESI† S1). Graphs were generated by using the graphing software Origin Pro (OriginLab Corp., USA).
5. Conclusion and outlook
In this work, we presented a new POCT system that allows full automation of a digital single cell approach with minimal hands-on time from sample input to digital answer.The developed microfluidic system enables absolute quantification and rapid detection of target genes with a time to result of only 55 min and a detection limit of 3 ± 0.3 CFU μl−1. We successfully showed that separation of bacterial species into single partitions is possible. The implemented bi-plex ddRPA assay allowed a clear discrimination of MRSA, MSSA and MR-CoNS in 20 out of 20 tested samples (spiked clinical isolate and spiked proband samples).
For translating this research into clinical practice, the following developments would be necessary: improvement of the assay efficiency and integration of a higher degree of multiplexing, covering also species-specific markers for MR-CoNS and controls. Evaluation of the bacterial loss in the cartridge for different bacterial strains and species. Integration of an increased sampling volume and increased droplet number to extend the sensitivity and the dynamic range of the system. Large-scale manufacturing of the LabDisk by injection moulding. Certification of the system according to governmental regulations for in vitro diagnostics.
Used for hospital admission screening, the system could improve timely decision-making. MRSA carrier could be identified and isolated earlier from non-MRSA carrier preventing a spread of resistant bacteria, reducing treatment and nursing expenses. Further, the POCT system is not only limited to MRSA detection but also offers the possibility for the integration of other biochemical applications such as the detection and absolute quantification of other pathogens, probiotic microorganisms or for DNA and RNA analysis.
Author contributions
M. S. conceptualized, designed, integrated and tested the microfluidic unit operations for the LabDisk cartridge. M. S. and S. C. wrote the manuscript, worked out the technical details and performed the experiments. S. C. developed the coating of the LabDisk to prevent bacterial adhesion. F. H., D. D., H. W. and K. S. developed the optics for the POCT device. A. M. S., F. E. and Mat. S. developed and manufactured the lytic enzymes. M. J. L. obtained funding for development of the lytic enzymes. A. G., U. G. and M. H. developed and tested the bi-plex RPA assay. A. S. and G. H. performed reference culture tests, provided proband study data and clinical expert advice, J. L., M. Sp., N. B. performed preliminary experiments for cell singularization, RPA and in droplet lysis. P. K., M. M., P. T., R. R. and M. J. manufactured the LabDisk cartridge and performed process development for thermoforming, stickpackaging and cartridge sealing. S. W., R. Z., F. v. S., N. P. and N. B. were involved in acquisition, planning and supervising of the project. All authors provided critical feedback and helped to shape the research, the analysis and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to gratefully acknowledge the financial support from the Baden-Württemberg Ministry for Economy, Labor and Housing (project “IDAK” AZ 7-4332.62-HSG/69). Further we want to thank the team of Hahn-Schickard Lab-on-a-Chip Foundry57 for fabrication of the LabDisks.References
- C. Hübner, N.-O. Hübner, K. Hopert, S. Maletzki and S. Flessa, Analysis of MRSA-attributed costs of hospitalized patients in Germany, Eur. J. Clin. Microbiol. Infect. Dis., 2014, 33, 1817–1822, DOI:10.1007/s10096-014-2131-x.
- C. E. W. Herr, T. H. Heckrodt, F. A. Hofmann, R. Schnettler and T. F. Eikmann, Additional costs for preventing the spread of methicillin-resistant Staphylococcus aureus and a strategy for reducing these costs on a surgical ward, Infect. Control Hosp. Epidemiol., 2003, 24, 673–678, DOI:10.1086/502274.
- M. Prosperi, et al. Molecular epidemiology of community-associated methicillin-resistant Staphylococcus aureus in the genomic era: a cross-sectional study, Sci. Rep., 2013, 3, 1902, DOI:10.1038/srep01902.
- J. O'Neill, Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Review on Antimicrobial Resistance, 2014, vol. 12 Search PubMed.
- T. Cravo Oliveira Hashiguchi, D. Ait Ouakrim, M. Padget, A. Cassini and M. Cecchini, Resistance proportions for eight priority antibiotic-bacterium combinations in OECD, EU/EEA and G20 countries 2000 to 2030: a modelling study, Euro Surveill., 2019, 24(20), 1800445, DOI:10.2807/1560-7917.ES.2019.24.20.1800445.
- A. Cassini, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis, Lancet Infect. Dis., 2019, 19, 56–66, DOI:10.1016/S1473-3099(18)30605-4.
- S. J. Peacock and G. K. Paterson, Mechanisms of Methicillin Resistance in Staphylococcus aureus, Annu. Rev. Biochem., 2015, 84, 577–601, DOI:10.1146/annurev-biochem-060614-034516.
- Y. Katayama, D. A. Robinson, M. C. Enright and H. F. Chambers, Genetic background affects stability of mecA in Staphylococcus aureus, J. Clin. Microbiol., 2005, 43, 2380–2383, DOI:10.1128/JCM.43.5.2380–2383.2005.
- K. Hiramatsu, et al. Genomic Basis for Methicillin Resistance in Staphylococcus aureus, Infect. Chemother., 2013, 45, 117–136, DOI:10.3947/ic.2013.45.2.117.
- L. Hutzschenreuter, S. Flessa, K. Dittmann and N.-O. Hübner, Costs of outpatient and inpatient MRSA screening and treatment strategies for patients at elective hospital admission - a decision tree analysis, Antimicrob. Resist. Infect. Control, 2018, 7, 147, DOI:10.1186/s13756-018-0442-x.
- B. Y. Lee, et al. Universal methicillin-resistant Staphylococcus aureus (MRSA) surveillance for adults at hospital admission: an economic model and analysis, Infect. Control Hosp. Epidemiol., 2010, 31, 598–606, DOI:10.1086/652524.
- E. Stürenburg, Rapid detection of methicillin-resistant Staphylococcus aureus directly from clinical samples: methods, effectiveness and cost considerations, Ger. Med. Sci., 2009, 7, Doc06, DOI:10.3205/000065.
- P. L. Perry, et al. Detection of methicillin-resistant Staphylococcus aureus (MRSA) in screening swabs by direct culture on a solid screening medium and broth enrichment culture separately and in combination, Aust. Infect. Control, 2004, 9, 13–16, DOI:10.1071/HI04013.
- A. van Belkum and O. Rochas, Laboratory-Based and Point-of-Care Testing for MSSA/MRSA Detection in the Age of Whole Genome Sequencing, Front. Microbiol., 2018, 9, 1437, DOI:10.3389/fmicb.2018.01437.
- S. Malhotra-Kumar, et al. Current trends in rapid diagnostics for methicillin-resistant Staphylococcus aureus and glycopeptide-resistant enterococcus species, J. Clin. Microbiol., 2008, 46, 1577–1587, DOI:10.1128/JCM.00326-08.
- A. S. Rossney, C. M. Herra, G. I. Brennan, P. M. Morgan and B. O'Connell, Evaluation of the Xpert methicillin-resistant Staphylococcus aureus (MRSA) assay using the GeneXpert real-time PCR platform for rapid detection of MRSA from screening specimens, J. Clin. Microbiol., 2008, 46, 3285–3290, DOI:10.1128/JCM.02487-07.
- S. Kolman, H. Arielly and Y. Paitan, Evaluation of single and double-locus real-time PCR assays for methicillin-resistant Staphylococcus aureus (MRSA) surveillance, BMC Res. Notes, 2010, 3, 110, DOI:10.1186/1756-0500-3-110.
- A. van der Zee, L. Roorda, W. D. Hendriks, J. M. Ossewaarde and J. Buitenwerf, Detection of novel chromosome-SCCmec variants in Methicillin Resistant Staphylococcus aureus and their inclusion in PCR based screening, BMC Res. Notes, 2011, 4, 150, DOI:10.1186/1756-0500-4-150.
- M. Kuroda, et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus, Lancet, 2001, 357, 1225–1240, DOI:10.1016/S0140-6736(00)04403-2.
- A. S. Basu, Digital Assays Part I: Partitioning Statistics and Digital PCR, SLAS Technol., 2017, 22, 369–386, DOI:10.1177/2472630317705680.
- P.-L. Quan, M. Sauzade and E. Brouzes, dPCR: A Technology Review, Sensors, 2018, 18, 1271, DOI:10.3390/s18041271.
- J. Luo, J. Li, H. Yang, J. Yu and H. Wei, Accurate Detection of Methicillin-Resistant Staphylococcus aureus in Mixtures by Use of Single-Bacterium Duplex Droplet Digital PCR, J. Clin. Microbiol., 2017, 55, 2946–2955, DOI:10.1128/JCM.00716-17.
- O. Strohmeier, et al. Centrifugal microfluidic platforms: advanced unit operations and applications, Chem. Soc. Rev., 2015, 44, 6187–6229, 10.1039/c4cs00371c.
- T. van Oordt, Y. Barb, J. Smetana, R. Zengerle and F. von Stetten, Miniature stick-packaging--an industrial technology for pre-storage and release of reagents in lab-on-a-chip systems, Lab Chip, 2013, 13, 2888–2892, 10.1039/C3LC50404B.
- S. Zehnle, et al. Centrifugo-dynamic inward pumping of liquids on a centrifugal microfluidic platform, Lab Chip, 2012, 12, 5142–5145, 10.1039/c2lc40942a.
- J. F. Hess, et al. Review on pneumatic operations in centrifugal microfluidics, Lab Chip, 2019, 19, 3745–3770, 10.1039/C9LC00441F.
- J. Steigert, et al. Integrated Sample Preparation, Reaction, and Detection on a High-frequency Centrifugal Microfluidic Platform, J. Assoc. Lab. Autom., 2005, 10, 331–341, DOI:10.1016/j.jala.2005.07.002.
- H. Cho, H.-Y. Kim, J. Y. Kang and T. S. Kim, How the capillary burst microvalve works, J. Colloid Interface Sci., 2007, 306, 379–385, DOI:10.1016/j.jcis.2006.10.077.
- F. Schuler, et al. Centrifugal step emulsification applied for absolute quantification of nucleic acids by digital droplet RPA, Lab Chip, 2015, 15, 2759–2766, 10.1039/c5lc00291e.
- M. Schulz, F. von Stetten, R. Zengerle and N. Paust, Centrifugal Step Emulsification: How Buoyancy Enables High Generation Rates of Monodisperse Droplets, Langmuir, 2019, 35, 9809–9815, DOI:10.1021/acs.langmuir.9b01165.
- J. Treter, et al. Washing-resistant surfactant coated surface is able to inhibit pathogenic bacteria adhesion, Appl. Surf. Sci., 2014, 303, 147–154, DOI:10.1016/j.apsusc.2014.02.123.
- L. G. Harris, S. J. Foster and R. G. Richards, An introduction to staphylococcus aureus, and techniques for identifying and quantifying s. aureus adhesins in relation to adhesion to biomaterials: review, Eur. Cells Mater., 2002, 4, 39–60, DOI:10.22203/eCM.v004a04.
- D. F. Gerson and D. Scheer, Cell surface energy, contact angles and phase partition III. Adhesion of bacterial cells to hydrophobic surfaces, Biochim. Biophys. Acta, Biomembr., 1980, 602, 506–510, DOI:10.1016/0005-2736(80)90329-6.
- D. R. Absolom, et al. Surface thermodynamics of bacterial adhesion, Appl. Environ. Microbiol., 1983, 46, 90–97 CrossRef CAS PubMed.
- S. Lutz, et al. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA), Lab Chip, 2010, 10, 887–893, 10.1039/b921140c.
- U. Manna, et al. Slippery Liquid-Infused Porous Surfaces that Prevent Microbial Surface Fouling and Kill Non-Adherent Pathogens in Surrounding Media: A Controlled Release Approach, Adv. Funct. Mater., 2016, 26, 3599–3611, DOI:10.1002/adfm.201505522.
- J.-B. Paris, D. Seyer, T. Jouenne and P. Thébault, Elaboration of antibacterial plastic surfaces by a combination of antiadhesive and biocidal coatings of natural products, Colloids Surf., B, 2017, 156, 186–193, DOI:10.1016/j.colsurfb.2017.05.025.
- H. Xiang, et al. Aloe-emodin inhibits Staphylococcus aureus biofilms and extracellular protein production at the initial adhesion stage of biofilm development, Appl. Microbiol. Biotechnol., 2017, 101, 6671–6681, DOI:10.1007/s00253-017-8403-5.
- J. Ye, et al. Effects of DNase I coating of titanium on bacteria adhesion and biofilm formation, Mater. Sci. Eng., C, 2017, 78, 738–747, DOI:10.1016/j.msec.2017.04.078.
- N. Ayres, Polymer brushes: Applications in biomaterials and nanotechnology, Polym. Chem., 2010, 1, 769–777, 10.1039/B9PY00246D.
- A. Roosjen, W. Norde, H. C. Mei and H. J. Busscher in Characterization of Polymer Surfaces and Thin Films, ed. K. Grundke, M. Stamm and H.-J. Adler, Springer-Verlag, Berlin/Heidelberg, 2006, pp. 138–144 Search PubMed.
- W. Mamo, F. Rozgonyi, A. Brown, S. Hjertén and T. Wadström, Cell surface hydrophobicity and charge of Staphylococcus aureus and coagulase-negative staphylococci from bovine mastitis, J. Appl. Bacteriol., 1987, 62, 241–249 CrossRef CAS PubMed.
- G. Beck, E. Puchelle, C. Plotkowski and R. Peslin, Effect of growth on surface charge and hydrophobicity of Staphylococcus aureus, Ann. Inst. Pasteur/Microbiol., 1988, 139, 655–664 CrossRef CAS.
- P. Jonsson and T. Wadström, Cell surface hydrophobicity ofStaphylococcus aureus measured by the salt aggregation test (SAT), Curr. Microbiol., 1984, 10, 203–209, DOI:10.1007/BF01627256.
- L.-A. B. Rawlinson, J. P. O'Gara, D. S. Jones and D. J. Brayden, Resistance of Staphylococcus aureus to the cationic antimicrobial agent poly(2-(dimethylamino ethyl)methacrylate) (pDMAEMA) is influenced by cell-surface charge and hydrophobicity, J. Med. Microbiol., 2011, 60, 968–976, DOI:10.1099/jmm.0.025619-0.
- N. Borst, et al. A technology platform for digital nucleic acid diagnostics at the point of care, Laboratoriumsmedizin, 2017, 41, 4341, DOI:10.1515/labmed-2017-0098.
- D. Henares, et al. Evaluation of the eazyplex MRSA assay for the rapid detection of Staphylococcus aureus in pleural and synovial fluid, Int. J. Infect. Dis., 2017, 59, 65–68, DOI:10.1016/j.ijid.2017.04.013.
- H. Frickmann, Impact of MRSA on the Military Medical Service and Diagnostic Point-of-Care Options for the Field Setting, Eur. J. Microbiol. Immunol., 2018, 8, 31–33, DOI:10.1556/1886.2018.00012.
- C. L. Haley, J. A. Colmer-Hamood and A. N. Hamood, Characterization of biofilm-like structures formed by Pseudomonas aeruginosa in a synthetic mucus medium, BMC Microbiol., 2012, 12, 181, DOI:10.1186/1471-2180-12-181.
- F. Schuler, et al. Digital droplet LAMP as a microfluidic app on standard laboratory devices, Anal. Methods, 2016, 8, 2750–2755, 10.1039/C6AY00600K.
- C. A. Schindler and V. T. Schuhardt, Lysostaphin: A New Bacteriolytic Agent for the Staphylococcus, Proc. Natl. Acad. Sci. U. S. A., 1964, 51, 414–421, DOI:10.1073/pnas.51.3.414.
- I. Abaev, et al. Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site, Appl. Microbiol. Biotechnol., 2013, 97, 3449–3456, DOI:10.1007/s00253-012-4252-4.
- J. Gu, et al. LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection, J. Clin. Microbiol., 2011, 49, 111–117, DOI:10.1128/JCM.01144-10.
- J. Z. Lu, T. Fujiwara, H. Komatsuzawa, M. Sugai and J. Sakon, Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges, J. Biol. Chem., 2006, 281, 549–558, DOI:10.1074/jbc.M509691200.
- M. Schmelcher and M. J. Loessner, Use of bacteriophage cell wall-binding proteins for rapid diagnostics of Listeria, Methods Mol. Biol., 2014, 1157, 141–156, DOI:10.1007/978-1-4939-0703-8_12.
- I. Schwarz, S. Zehnle, T. Hutzenlaub, R. Zengerle and N. Paust, System-level network simulation for robust centrifugal-microfluidic lab-on-a-chip systems, Lab Chip, 2016, 16, 1873–1885, 10.1039/c5lc01525a.
- https://www.hahn-schickard.de/en/production/lab-on-a-chip-foundry/, Available at https://www.hahn-schickard.de/en/production/lab-on-a-chip-foundry/.
- M. Focke, et al. Lab-on-a-Foil: microfluidics on thin and flexible films, Lab Chip, 2010, 10, 1365–1386, 10.1039/c001195a.
Footnotes |
| † Electronic supplementary information (ESI) available: Document with additional information. See DOI: 10.1039/d0lc00294a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |