
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative analysis of yeast MAPK signaling networks and crosstalk using a microfluidic device†
Byungjin
Lee
a,
Seong-Geun
Jeong
a,
Si Hyung
Jin
a,
Ranjan
Mishra
b,
Matthias
Peter
 *b,
Chang-Soo
Lee
*a and
Sung Sik
Lee
*bc
*b,
Chang-Soo
Lee
*a and
Sung Sik
Lee
*bc
aDepartment of Chemical Engineering and Applied Chemistry, Chungnam National University, Yuseong-Gu, Daejeon 305-764, Republic of Korea. E-mail: rhadum@cnu.ac.kr
bInstitute of Biochemistry, ETH Zurich, Zurich, CH-8093, Switzerland. E-mail: matthias.peter@bc.biol.ethz.ch; leesu@ethz.ch
cScientific Center for Optical and Electron Microscopy, ETH Zurich, Zurich, Switzerland
First published on 22nd June 2020
Abstract
Eukaryotic cells developed complex mitogen-activated protein kinase (MAPK) signaling networks to sense their intra- and extracellular environment and respond to various stress conditions. For example, S. cerevisiae uses five distinct MAP kinase pathways to orchestrate meiosis or respond to mating pheromones, osmolarity changes and cell wall stress. Although each MAPK module has been studied individually, the mechanisms underlying crosstalk between signaling pathways remain poorly understood, in part because suitable experimental systems to monitor cellular outputs when applying different signals are lacking. Here, we investigate the yeast MAPK signaling pathways and their crosstalk, taking advantage of a new microfluidic device coupled to quantitative microscopy. We designed specific micropads to trap yeast cells in a single focal plane, and modulate the magnitude of a given stress signal by microfluidic serial dilution while keeping other signaling inputs constant. This approach enabled us to quantify in single cells nuclear relocation of effectors responding to MAPK activation, like Yap1 for oxidative stress, and expression of stress-specific reporter expression, like pSTL1-qV and pFIG1-qV for high-osmolarity or mating pheromone signaling, respectively. Using this quantitative single-cell analysis, we confirmed bimodal behavior of gene expression in response to Hog1 activation, and quantified crosstalk between the pheromone- and cell wall integrity (CWI) signaling pathways. Importantly, we further observed that oxidative stress inhibits pheromone signaling. Mechanistically, this crosstalk is mediated by Pkc1-dependent phosphorylation of the scaffold protein Ste5 on serine 185, which prevents Ste5 recruitment to the plasma membrane.
1. Introduction
Cells respond to various intra- and extracellular signals and stress conditions including the presence of pheromones, nutrient availability, physical pressure, osmotic and oxidative stress as well as exposure to diverse compounds. Specifically, cells sense these diverse signals by either binding of molecules to dedicated receptors (e.g. binding of growth factors to receptor) or stress-induced structural changes of sensory molecules (e.g. shrinkage of membrane proteins to osmotic stress), leading to sequential biochemical reactions, generally termed “signaling pathway”. These signaling pathways play important roles in regulating gene expression and link specific signals to the machinery that controls fundamental cellular processes such as growth, proliferation, differentiation, migration and apoptosis. Defects in signaling pathways by aging or other factors often trigger malfunction of cells and organs and may cause disease. Thus, there is considerable interest in understanding the molecular mechanisms governing cellular signaling networks and exploiting genetic and biochemical analysis to develop specific drugs and biomarkers. In particular, it is important to identify mechanisms that connect individual signaling pathways1,2 since cross-talk is widely recognized as a major contributor to resistance mechanisms and failure of drug treatment.3 Also, multiple signals often need to cooperate, for example to orchestrate directed cell migration in wound healing, which involves signaling pathways driving cell polarization and actin remodeling.1,2 Likewise, cells continuously integrate multiple types of stresses to dictate the appropriate cellular output through insulation, inhibition and amplification of the underlying signaling network.1,2Budding yeast, Saccharomyces cerevisiae, is a powerful model organism to study cell signaling, as yeast cells use distinct mitogen-activated protein kinase (MAPK) signaling modules to respond to mating pheromones, osmolarity, cell wall and mechanical stress.4,5 The high osmolarity glycerol (HOG) pathway protects cells from lysis during salt stress and other conditions with high external osmolarity.6,7 A sudden increase in osmolarity causes cells to shrink by losing water, which triggers activation of the osmosensors (e.g. Sln1, Sho1), which in turn sequentially activate the MAP kinase module composed of the MAPK kinase kinases (MAPKKKs) Ssk2 and 22, the MAPKK Pbs2 and the MAPK Hog1.5,7 Activated Hog1 rapidly translocates into the nucleus to induce a transcriptional program including the expression of STL1.8,9 Conversely, hypo-osmolarity and chemical and physical forms of cell wall stress result in activation of the cell wall integrity (CWI) pathway.10–12 Different membrane-anchored sensors such as the Wsc1-4 and Mid2 receptors activate the guanine nucleotide exchange factors (GEFs) Rom1 and Rom2, which in turn stimulate the GTPase Rho1. GTP-bound Rho1 recruits Pkc1, which in turn activates a MAP kinase module composed of the MEKK Bck1, the two closely-related MEKs, Mkk2 and Mkk22 and the MAP kinase Mpk1 (Slt2).11 Finally, the pheromone pathway is triggered by binding of a- or α-pheromones to their G-protein coupled receptors, which leads to dissociation of the α-subunit of the trimeric G-protein. In turn, βγ heterodimers recruit the scaffold Ste5 to the plasma membrane, which then activates the MAP kinase module composed of Ste11, Ste7 and the MAPK Fus3.13 Activated Fus3 phosphorylates multiple substrates orchestrating the different cellular processes required for the complex mating process, including induction of a transcriptional program, cell cycle arrest in G1, oriented polarized growth and cell–cell and nuclear fusion.5,14,15
Although these distinct MAP kinase pathways are well insulated and faithfully respond to their specific triggers, crosstalk mechanisms have been described. For example, yeast mating is prevented when cells are exposed to high osmolarity or mechanical stress conditions.12,16 However, the mechanisms underlying crosstalk between signaling pathways remain poorly understood, in part because of the challenge to establish suitable experimental systems that allow monitoring cellular outputs when applying different signals.
Microfluidics coupled to quantitative single-cell microscopy has a great potential to fill the gap in analysis of cell signaling pathways and their crosstalk in a quantitative manner. Recent advances have demonstrated controlled physiological stress or pharmacological perturbations by accurately tuning perfusion of medium in a microfluidic cell culture chamber.17–19 When multiplexed, microfluidic platforms have yielded a significant increase in throughput of live-cell-imaging,20,21 allowing to investigate yeast MAPK dynamics in cell fate determination.22,23 In addition, microfluidic devices generating stable chemical gradient across yeast cells were used in gradient sensing studies.24–26 Finally, microfluidic devices that tune the magnitude and frequency of oscillatory stress enabled to monitor dynamic responses of yeast cells.27–29
Here, we investigate yeast MAPK signaling pathways and their crosstalk, taking advantage of a powerful microfluidic setup that allows microscopic quantification of signaling reporters in single cells. Specifically, we newly designed an integrated microfluidic device, which uses micropads to trap yeast cells in a single focal plane and allows serial dilution of chemical signals in otherwise constant stress conditions. With this device and single-cell analysis of microscopy images, we confirmed concentration-dependent bimodal gene expression induced by osmotic stress, and characterized crosstalk mechanisms restricting pheromone signaling under cell wall integrity- and oxidative stress conditions.
2. Experimental sections
2.1 Fabrication of microfluidic devices
The microfluidic devices were fabricated by standard photolithography and soft lithography techniques.30 Master molds were fabricated with three layers with a thickness of 4, 10, and 20 μm for the cell-trapping micropad, for the chamber holding the trapping micropad, and for the fluidic channels, respectively, using negative photoresist SU-8 3005 and 3010. To fabricate the multi-layered master mold, each layer of the photoresist was precisely aligned and made by UV light exposure without development at each step to prevent dimensional errors from compiled photoresist. We developed the master mold by AZ 1500 thinner, and fluoro-silanized it to prevent adhesion to polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning) during soft lithography.PDMS base and curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) were thoroughly mixed, degassed in a vacuum chamber, and then poured on the mold, and cured on a hot plate (80 °C in 3 h). The cured PDMS was then carefully peeled off the mold. The inlet and outlet holes were punched by a flat-tip. The microfluidic device was made by binding a glass slide and the PDMS structures, and the inlet of the device was connected with feeder wells.26
:1 ratio) were thoroughly mixed, degassed in a vacuum chamber, and then poured on the mold, and cured on a hot plate (80 °C in 3 h). The cured PDMS was then carefully peeled off the mold. The inlet and outlet holes were punched by a flat-tip. The microfluidic device was made by binding a glass slide and the PDMS structures, and the inlet of the device was connected with feeder wells.26
Prior to the experiment, the microfluidic device was primed with dH2O and cell suspension and stimuli-containing media are preloaded into feeder wells. Air pressure (3 psi) was imposed on top of gas tight feeder wells. The open/close motion of the solenoid valve controlled the air pressure, and thus timing and duration of media injection.
2.2 Preparation of yeasts and chemicals
All yeast strains and plasmids used in this work are listed in Tables S1 and S2,† respectively. The yeast strains were derived from BY4741 (ref. 31) and W303. Unless indicated otherwise, all fusion proteins were expressed from their endogenous promoter at the endogenous genomic locus, generated by replacing the endogenous gene using homologous recombination. α-Factor was obtained from Genscript (Luzern, Switzerland). Hydrogen peroxide, sodium chloride, sodium dodecyl sulfate, and vanadium oxide (vanadate) were purchased from Sigma Aldrich (Buchs, Switzerland).Yeast strains for all experiments were grown in SC (synthetic complete) based media (0.17% yeast nitrogen base, 2% glucose, 0.5% NH4-sulfate and amino acids). Single yeast colonies were grown overnight, diluted 100-fold in fresh media and incubated at 30 °C for 4 h prior to loading into the chips.
2.3 Imaging and quantification
Images were acquired on a motorized inverted epifluorescence microscope (Ti-Eclipse; Nikon, Japan) with objective lens (Nikon CFI Plan Apochromat 60× or 100×, NA 1.4), a sCMOS camera (ORCA Flash 4.0 camera, Hamamatsu Photonics) and appropriate excitation and emission filters. The microscope was equipped with a temperature-controlled incubator set to 30 °C. A motorized xy-stage was used to acquire multiple fields of view for each time point. The acquired images were quantified by YeastQuant,32 which was used to segment and track single cells at different time points and quantify the intensity of each segmented cell. The background of fluorescent intensity was subtracted by image analysis. We excluded from analysis cells with outlier intensity due to a damaged camera pixel or with negative value of segmented cells (non-responsive cells). The data was plotted by average of cells in the same index of chambers. The cytoplasmic-to-nuclear ratio of the different translocation reporters was quantified as described previously.333. Results
We designed an integrated experimental system to investigate the complex interaction of MAPK signaling pathways that allows measuring quantitative readouts in living cells exposed to defined signals and stress conditions. The system fulfills three critical experimental parameters (Fig. 1A): (1) trapping cells in multiple chambers, (2) stimulating cells by perfusing different concentrations of chemical solutions and (3) observing the cellular response by quantitative microscopy of stress-responsive fluorescent reporters. We then used the experimental platform to quantitatively probe distinct MAP-kinase signaling pathways and in particular focus on mechanisms that regulate crosstalk within these networks.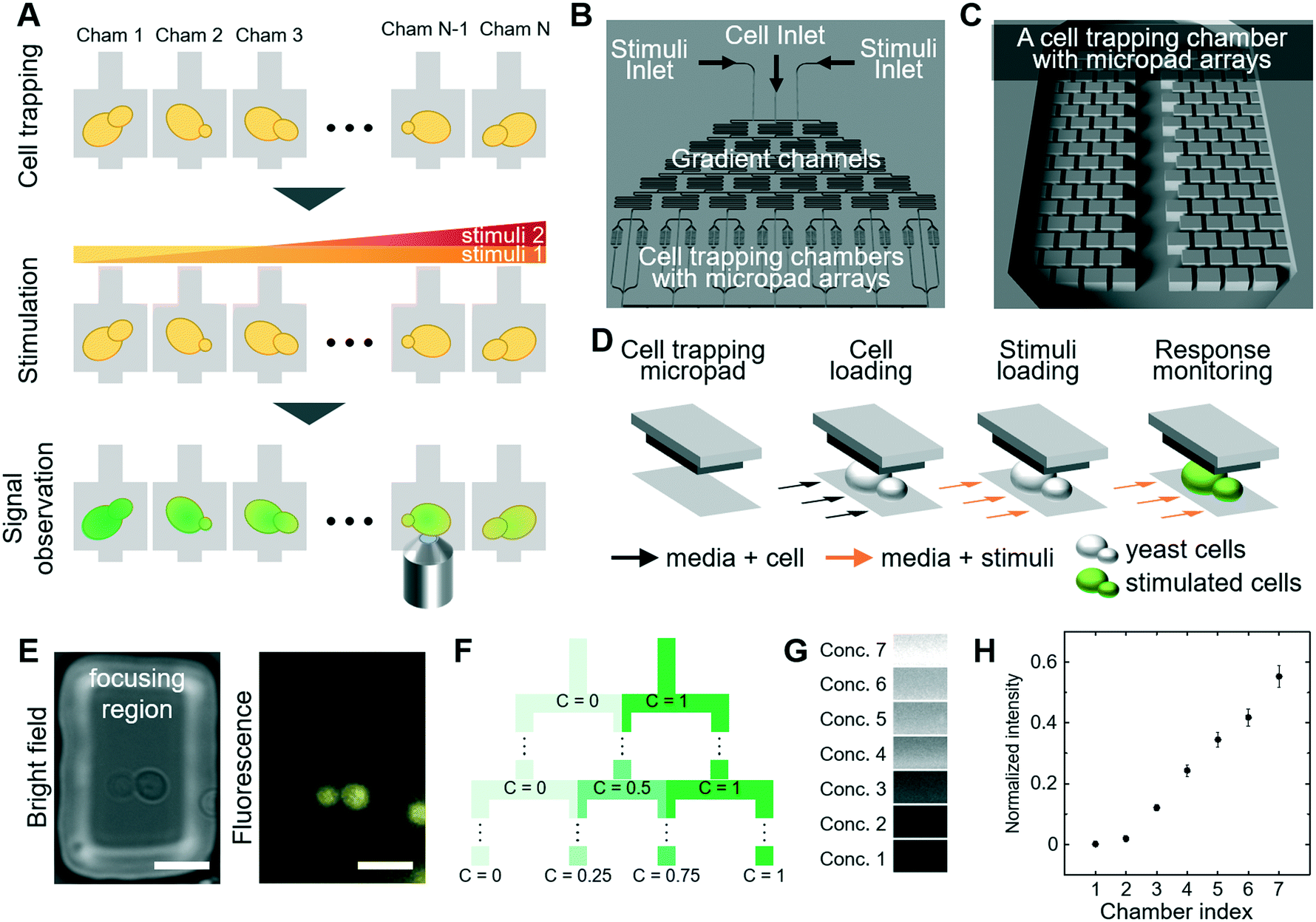 | ||
| Fig. 1 Design of the microfluidic device and experimental platform (A) experimental scheme to investigate signaling in single cells: cell loading, stimulation and microscopic observation (B) overview of microfluidic device with cell inlet, two stimuli inlets, gradient channels (microfluidic dilution) and cell trapping chambers with micropad array (C) a unit of cell trapping chambers with micropad arrays (D) schematic drawing illustrating the principle of cell trapping (E) micropad with trapped single yeast cell (scale bar: 10 μm) (F) principle of serial dilution (G) visualization of concentration at each chamber by adding fluorescent dye in one of the stimuli inlet (H) normalized fluorescence intensities of each chamber. The error bars are based on standard error of the mean (SEM). The experiment was performed in triplicate imaging 5 positions per chamber. | ||
3.1 Design of microfluidic device
We designed a new microfluidic device that traps individual yeast cells and also allows exposure to various stress conditions. The microfluidic device is composed of (1) inlets for cells and chemical stimuli, (2) cell trapping chambers using micropad arrays, and (3) gradient channels to produce serial dilution of compounds (Fig. 1B and C). We produced PDMS-based micropad arrays with a 4 μm gap between the bottom glass and the micropad, which corresponds roughly to the dimension of budding yeast cells.34 Since PDMS is elastic, the gap can be slightly increased by fluidic pressure, and thus yeast cells are gently trapped between the micropads and the bottom glass (Fig. 1D and E). The micropad dimensions of 50 μm × 25 μm does not require use of an expensive high-resolution photomask for microfabrication, in contrast to hydrodynamic trapping techniques which rely on narrow apertures.35 We constrained the upper boundary of mechanical force applied by the micropad to avoid triggering a cellular stress response, as previous established using reporter assays.12,34 Importantly, cells trapped underneath the micropads are in a single focal plane, which allows fluorescence imaging with minimal interference from defocused cells (Fig. 1E). Moreover, the trapped cells grow in 2-dimensions and do not form stacks, which ensures optimal imaging conditions over time.We assessed cellular responses to different stimuli and chemical compounds over a wide range of concentrations. To assess multiple concentrations in parallel, we produced sequentially branched microchannels, which allow generating serial dilution of the highest concentration in a linear gradient (Fig. 1F). Between the branches, we included serpentine shaped channels to ensure complete mixing. The device generated seven concentrations by serial dilution. The dilution channels each lead into two cell culture sub-chambers, resulting in a total of 14 sub-chambers (Fig. 1B). Each cell culture sub-chamber contains 60 pads, trapping 5–15 cells per pad. To verify this experimental set-up, we quantified the concentration of fluorescent TRITC-dextran (3 kDa) in the cell chambers. Indeed, the fluorescent signal in each chamber increased linearly as expected (Fig. 1G and H), confirming that the device allows to efficiently determine dose response profiles for any given stimuli and/or chemical compound. To directly control serial dilution in each experiment, we included fluorescent TRITC-dextran dye in all subsequent experiments.
3.2 Monitoring cellular signaling in response to osmotic- and oxidative stress conditions
To experimentally validate the performance of the microfluidic device, we first investigated osmotic stress signaling (Fig. 2A). Yeast cells rapidly adapt to hyper-osmotic shock conditions by activating Hog1, which rapidly translocates into the nucleus to induce a dedicated transitional program.36 This transcriptional output can be quantified using a well-characterized reporter system, expressing the quadruple Venus (qV) fluorescent protein from the osmostress-inducible STL1-promoter (pSTL1-qV).9 Using the serial dilution system, cells harboring the pSTL1-qV reporter were exposed to seven different concentrations of NaCl (0–0.4 M), and images of qV-levels were quantified in single cells at different time points in each chamber (Fig. 2B). As expected, we observed cell shrinkage immediately after NaCl treatment, followed by a gradual increase of pSTL1-qV expression, with the exception of chambers 1 and 2, corresponding to 0 and 0.0125 M NaCl, respectively (Fig. S1†). In case of high concentrations of NaCl (chamber 5, 6 and 7), most of the cells show high pSTL1-qV expression (Fig. 2D) while we observed co-existence of non-expressing and expressing cells at mild osmotic stress conditions (e.g. chamber 3 corresponding to 0.13 M NaCl, Fig. 2C and S2†). We observed bimodal expression with NaCl concentrations in a range similar to that previously reported (0.15 M).9 We further narrowed the concentration range between 0.1 M to 0.2 M NaCl. Indeed, bimodality was most prominent in cells exposed to 0.1–0.15 M NaCl (Fig. 2E and F). This bimodal response is explained by intrinsic cell-to-cell variations causing stochastic activation of gene expression in low salt conditions, while stronger osmotic stress allowed for prolonged Hog1 activity and thus uniform transcriptional activation.9 We observed bimodal expression of pSTL1-qV to osmotic stress regardless of cell positions (Fig. S2†), suggesting that bimodality did not originate from uneven diffusion due to cell clumping.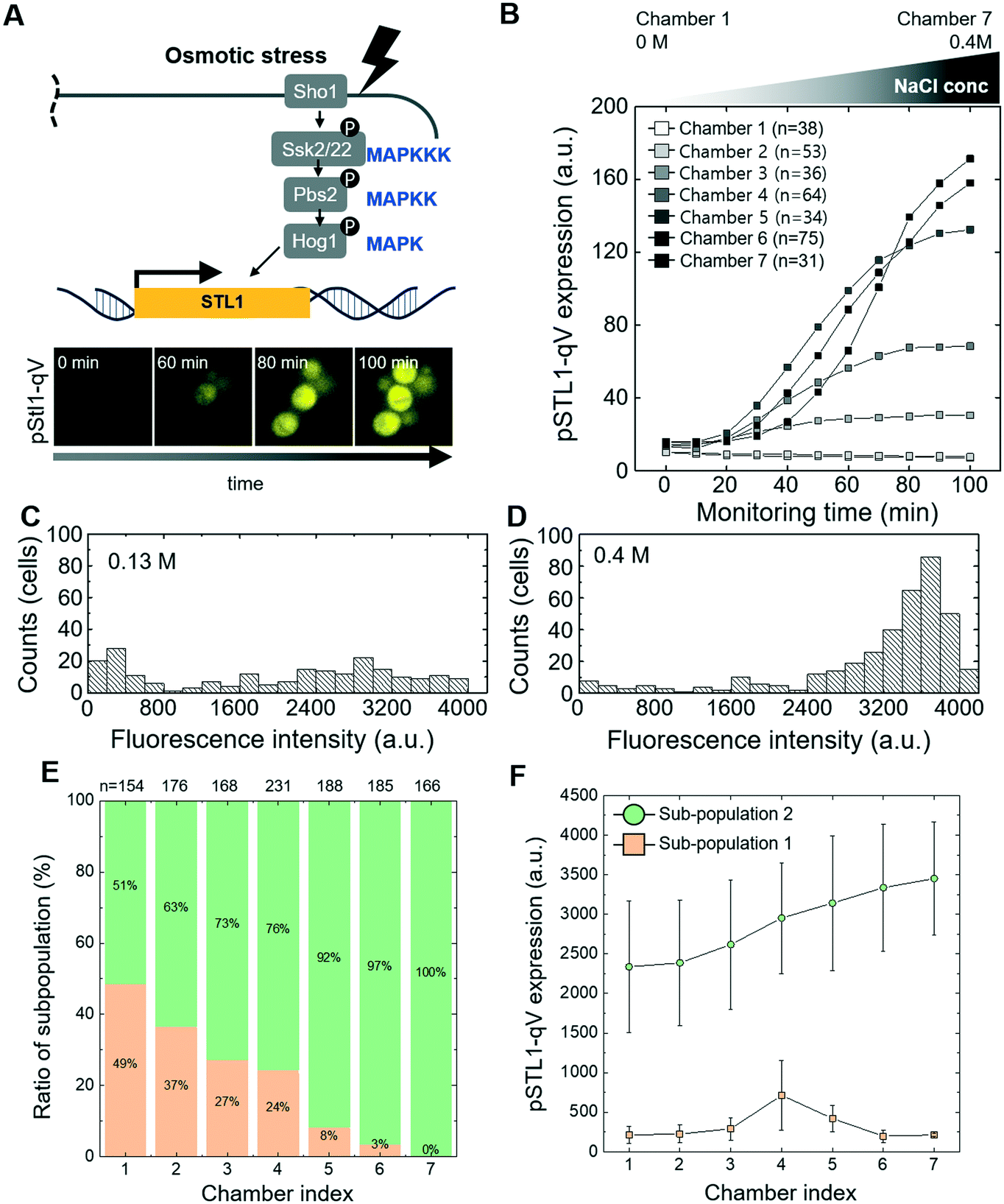 | ||
| Fig. 2 Microfluidic-based single cell analysis of high osmolarity glycerol (HOG) signaling pathway (A) schematic drawing of the HOG pathway and Hog1-dependent signaling output. Fluorescent images of cells harboring the pSTL1-qV reporter (pSTL1-qV) exposed at time 0 to 0.4 M NaCl (B) expression of pSTL1-qV in cells exposed to various NaCl concentrations by microfluidic serial dilution. The NaCl concentrations in the different chambers range from 0 to 0.4 M. (C and D) Histogram of pSTL1-qV reporter expression in single cells shows bimodal (C) and unimodal (D) behavior. pSTL1-qV expression was monitored in single cells exposed for 2 h to 0.13 M and 0.4 M NaCl, respectively (E and F). Quantification of pSTL1-qV bimodality in single cells exposed to various NaCl concentrations (0.1 to 0.2 M) by microfluidic serial dilution. (E) Sub-population and (F) the mean of the normal distribution fitted to the histograms is plotted. The data is fitted with a Gaussian mixture distribution model. The number of subpopulations were estimated by principal component analysis (PCA). | ||
To extend these results, we next analyzed the cellular response to different levels of oxidative stress triggered by the addition of hydrogen peroxide (H2O2). Oxidative stress activates the transcription factor yeast activator (AP1-like) protein (Yap1), which translocates into the nucleus to induce expression of a protective transcriptional program (Fig. S3A†).37,38 Indeed, Yap1-GFP rapidly accumulated in the nucleus of cells exposed to H2O2 (Fig. S3A†), and the degree of Yap1 relocation increased proportionally with H2O2 concentrations between 0–2 mM (Fig. S3B and C†).
Taken together, these results demonstrate that the established microfluidic device permits reliable quantification of different cellular outputs such as protein translocation or transcriptional induction over time at the single-cell level. Since the design also allows to test a wide range of stress levels and drug concentrations in a single experiment, it is ideally suited to rapidly establish dose–response curves, and thus provide valuable insights into the signaling network orchestrating sustained and transient cellular outputs.
3.3 Investigating crosstalk within the cellular stress signaling network
Although it is clear that cellular signaling pathways comprise a dynamic and interactive network, the mechanisms that govern crosstalk under complex intra- and extracellular condition remain poorly understood. Our microfluidic device allows simultaneous input of two stress conditions, for example, varying the magnitude of one stress signal while keeping the other one constant. In this context, we first investigated crosstalk between the cell wall integrity (CWI) and the pheromone response pathways (Fig. 3A). Indeed, recent work revealed that pheromone signaling is strongly decreased in a Pkc1-dependent manner when cells experience mechanical stress.6 We thus tested whether pheromone signaling is altered under cell wall stress conditions triggered by exposing cells to increasing concentrations of sodium-vanadate or the detergent SDS (Fig. 3B and C and S4 and S5†). We administered 3 μM of alpha factor via one stimuli inlet and 3 μM of alpha factor with sodium-vanadate or the detergent SDS via the other stimuli inlet. The mating pheromone α-factor is sensed by receptor (Ste2) binding, which in turn recruits the scaffold Ste5 and activates the MAPK module composed of Ste11, Ste7 and Fus3 (Fig. 3A). Activated Fus3 triggers cell cycle arrest in G1, cytoskeletal polarization along the pheromone gradient and induction of a transcriptional program, which can be monitored the pFIG1-qV reporter. Interestingly, pFIG1-qV expression is strongly attenuated in a dose-dependent manner in cells exposed to sodium-vanadate (Na3VO4)39 or sodium dodecyl sulfate (SDS)40 (Fig. 3B and C), two agents that are known to induce cell wall defects. These results imply that similar to mechanical stress, disrupting the integrity of the cell wall activates the Pkc1/Mpk1-pathway, which in turn interferes with pheromone signalling.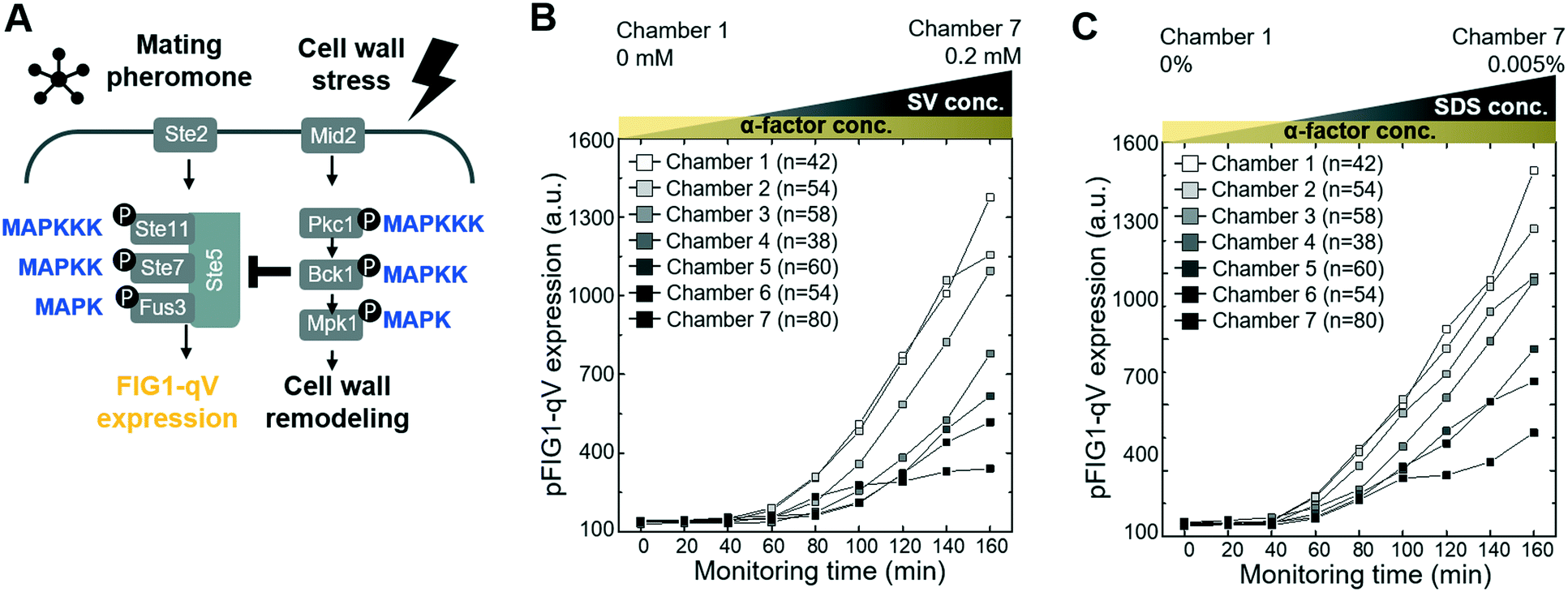 | ||
| Fig. 3 Crosstalk between the mating pheromone- and cell wall integrity (CWI) signaling pathways (A) schematic drawing of the pheromone- and CWI MAPK signaling pathways (B and C) inhibition of pheromone signaling by cell wall stress. Cells harboring the mating-specific pFIG1-qV reporter were treated with 3 μM α-factor, and cell wall stress was induced at the same time by exposing the cells to various concentrations of sodium-vanadate (SV, 0–0.2 mM, panel B) or SDS (0 to 0.005%, panel C). pFIG1-qV expression was quantified microscopically at the times indicated (min) in single cells and plotted against the different SV (panel B) or SDS (panel C) concentrations in the different chambers. | ||
We next explored whether similar mechanisms may restrict activation of the osmolarity/HOG (Fig. 4A–D and S6†) and pheromone response (Fig. 4E–H and S7†) pathways in cells exposed to oxidative stress imposed by the addition of H2O2. Indeed, increasing H2O2 concentrations abolished pSTL1-qV and pFIG1-qV expression, respectively, in a dose-dependent manner. Additionally, we tested whether the inhibition of pSTL1-qV and pFIG1-qV expression is a general or specific response to stress by monitoring other MAPK specific fluorescent reporters. Interestingly, pFIG1-qV expression induced by α-factor is increased in the presence of tunicamycin, which is known to induce ER stress (Fig. S8†). However, α-factor induced activation of the MAPK Fus3 is not significantly altered in the presence of rapamycin, a known inhibitor of TOR signaling (Fig. 4G and H). Also, NaCl induced nuclear translocation of the MAPK Hog1 is not inhibited significantly by rapamycin or nitrogen starvation, while it is dramatically reduced in the presence of H2O2 (Fig. 4C and D). Moreover, Yap1-GFP did not accumulate in the nucleus of cells treated with NaCl or α-factor (Fig. S9†). Together, these results suggest that the HOG and pheromone signaling pathways are blocked specifically in the presence of oxidative damage.
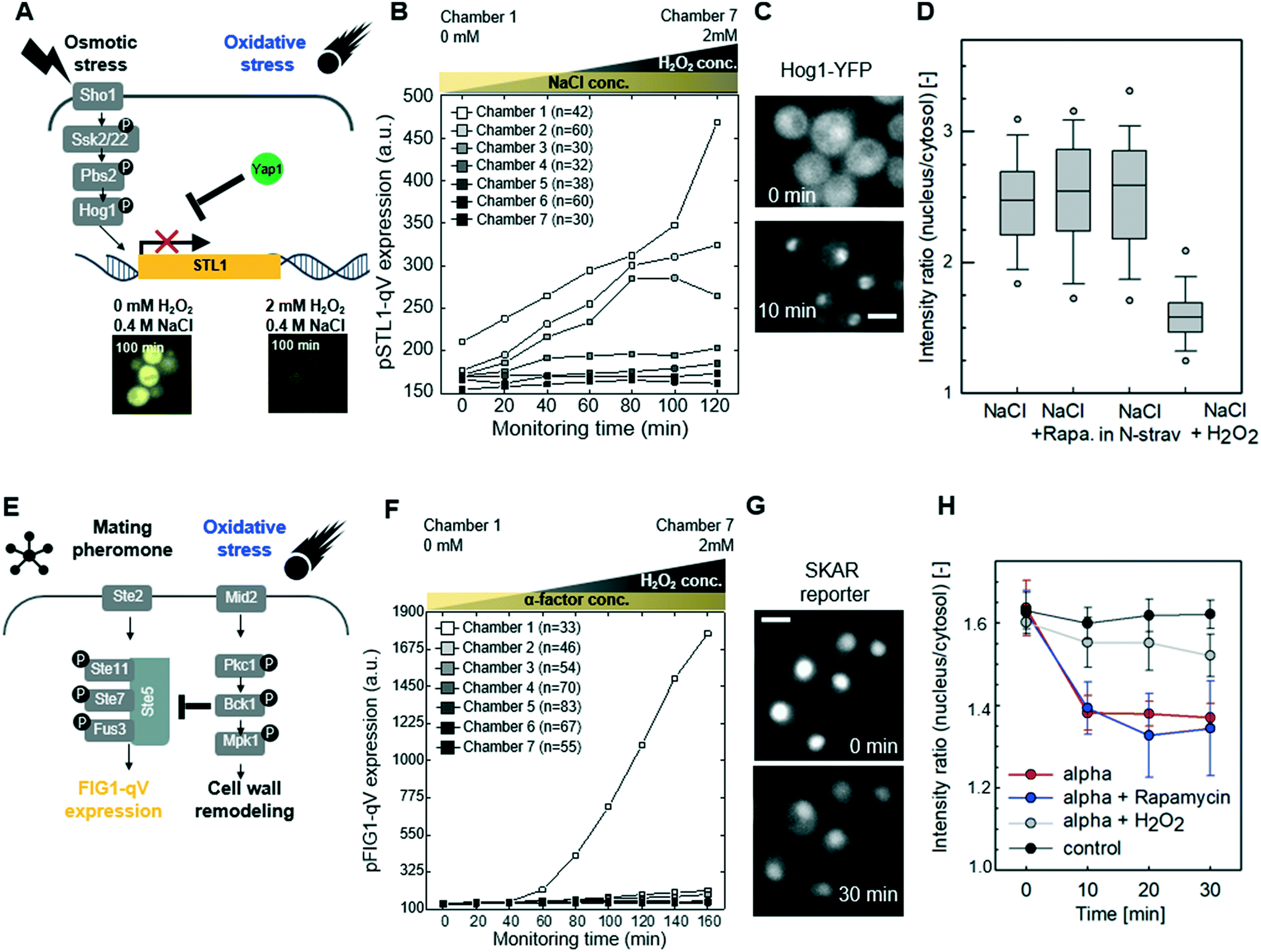 | ||
| Fig. 4 Oxidative stress inhibits the high osmolarity glycerol (HOG) and pheromone signaling pathways (A) schematic drawing of the crosstalk between oxidative stress and HOG signaling pathways (B) inhibition of the HOG signaling pathway by oxidative stress. Cells harboring the Hog1-dependent pSTL1-qV reporter were treated with 0.4 M NaCl and simultaneously exposed to various concentrations of H2O2 (0–2 mM). pSTL1-qV expression was quantified microscopically at the times indicated (minutes), and plotted against the different H2O2 concentrations in the different chambers. (C) Images showing Hog1-YFP nuclear relocation in cells before (0 min) or 10 min after addition of 0.4 M NaCl (scale bar: 5 μm). (D) The nuclear-to-cytoplasmic ratio of Hog1-YFP was quantified under the indicated conditions. Note that in contrast to rapamycin or nitrogen starvation, addition of H2O2 strongly inhibits Hog1 activation. The box- and whisker plots show median, first and third quartiles, with the outlier 5th and 95th percentiles indicated with filled circles. At least 84 cells were analyzed. (E) Schematic drawing of the crosstalk between oxidative stress and the pheromone signaling pathway (F) cells harboring the mating specific pFIG1-qV reporter were treated with 3 μM α-factor and simultaneously exposed to various concentrations of H2O2 (0–2 mM). pFIG1-qV expression was quantified microscopically at the times indicated (minutes), and plotted against the different H2O2 concentrations in the different chambers. (G) Microscopic visualization of cytoplasmic accumulation of the Fus3 synthetic kinase activity relocation sensor (SKARS) in cells exposed to α-factor (3 μM) for 0 or 30 min (scale bar: 5 μm). (H) The nuclear-to-cytoplasmic ratio of the SKAR sensor was quantified in more than 200 cells in each experiment and the standard deviation of triplicate experiments were plotted against the time after addition of the indicated stress conditions. Note that pheromone signaling and Fus3 activity are inhibited by oxidative stress but not rapamycin treatment. | ||
Recent work revealed that compressive mechanical stress is sensed by the transmembrane protein Mid2 (mating pheromone-induced death 2),41–43 which in turn inhibits pheromone signaling via Pkc1-dependent phosphorylation of serine 185 located in the RING-H2 domain of the Ste5 scaffold.12,44 To investigate whether the observed crosstalk between oxidative stress and the pheromone response pathway may involve a similar mechanism (Fig. 5A), we first compared Pkc1 activation in wild type mid2Δ cells exposed to H2O2. Indeed, while upon H2O2 addition Pkc1-GFP formed foci-like structures in the cytosol of wild type cells (Fig. 5B, upper panel, arrow heads), Pkc1-GFP showed a uniform cytoplasmic distribution in the absence of Mid2 (Fig. 5B, lower panel). Moreover, Pkc1 inhibition by cercosporamide slightly reduced nuclear translocation of Yap1-GFP in cells exposed to oxidative stress (Fig. S9C†). Oxidative stress also induced nuclear translocation of the general stress responsive transcriptional activator Msn2, and this response was diminished in mid2Δ cells (Fig. S10†).
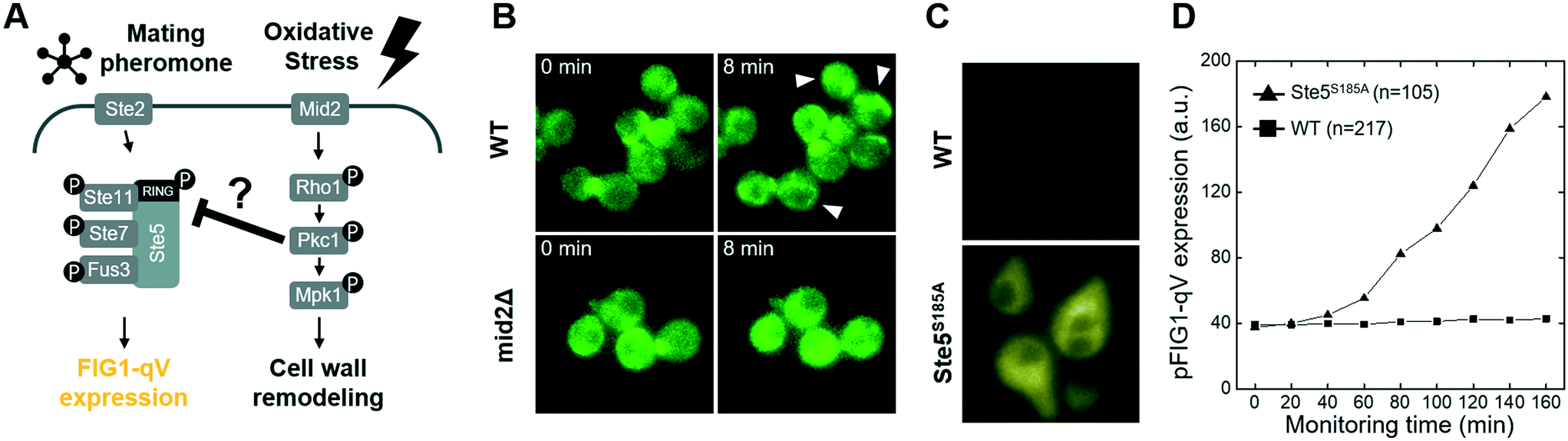 | ||
| Fig. 5 Molecular mechanism of crosstalk between oxidative stress and mating pheromone signaling pathways (A) schematic drawing of the crosstalk between the pheromone- and oxidative signaling pathways (B) Mid2-dependent localization of Pkc1-GFP in response to oxidative stress. Wild type (WT, top) and mid2Δ cells (bottom) expressing Pkc1-GFP were exposed to 2 mM H2O2 (at time 0), and the localization of Pkc1-GFP was monitored microscopically at the times indicated. The arrowhead points to Pkc1-GFP foci induced by oxidative stress (C) expression of the pFIG1-qV reporter was analyzed microscopically in single wild type (WT, black) cells or phosphorylation-defective ste5-S185A (Ste5S185A, red) mutants exposed to 3 μM α-factor and various concentrations of H2O2 for 180 minutes (D) quantification of pFIG1-qV reporter expression (arbitrary units: a.u.) in wild type (WT) cells or phosphorylation-defective ste5-S185A (Ste5S185A, bottom) mutants measured at the indicated time points (in minutes) after addition of 3 μM α-factor and 0.25 mM H2O2. | ||
To corroborate and extend these findings, we next monitored pheromone signaling by quantifying expression of the pFIG1-qV reporter in wild type cells and cells expressing a non-phosphorylatable Ste5 mutant (Ste5S185A). Strikingly, while pFIG1-qV expression was abolished in pheromone-treated cells exposed to H2O2 as expected, pFIG1-qV expression was at least partially restored in cells expressing Ste5S185A (Fig. 5B and C and S11†). Taken together, these results suggest that similar to cells responding to mechanical cues,40 oxidative stress conditions are sensed at least in part by a Mid2-dependent mechanism that activates Pkc1,45 which in turn phosphorylates Ste5 on serine 185 and thereby interferes with pheromone signaling. Previous work12 demonstrated that in response to compressive physical stress, this mechanism directly interferes with Gβγ binding and thus membrane recruitment of Ste5. Inhibiting pheromone signaling under mechanical stress conditions is important to prevent cell lysis by pausing the cell cycle and polarized growth. We thus speculate that cells exposed to oxidative stress conditions likewise halt cell cycle progression and cytoskeletal polarization to allow efficient repair of damaged cellular constituents. Surprisingly, although both compressive- and oxidative stress rely on Mid2- and Pkc1-dependent phosphorylation of Ste5 on S185, Pkc1 forms cytoplasmic foci upon H2O2 treatment, while it accumulates at the plasma membrane upon mechano-stress12 (Fig. 5). Future work will be required to understand the underlying mechanism (pFIG1-qV expression level of Ste5 mutant is still low so that it would not be the only mechanism of oxidative stress inhibition), but it is possible that increased cytoplasmic Ca2+ and thus activation of the calcineurin phosphatase observed in response to mechanical stress explains this phenotypic difference. Interestingly, although the presence of high osmolarity conditions also prevents pheromone signaling, this crosstalk mechanism does not target membrane recruitment of Ste5. The critical Hog1 substrate(s) mediating this crosstalk remains unclear, but the rapid kinetics suggest that Hog1 likely phosphorylates and thereby inactivates one or several of the critical components of the pheromone signaling pathway.46 Exploiting the microfluidic platform to analyze the concentration-dependence may provide important insights into this cross-talk mechanism.
4. Discussion
In this study, we monitored the dynamics of key signaling molecules by imaging and analyzing fluorescent reporters in individual yeast cells while precisely controlling stress conditions in a microfluidic device. This single-cell-based analysis enabled us to quantify intracellular responses that cannot be resolved by conventional biochemical methods relying on cell lysis and thus average signaling responses in cell populations (e.g. western blot or mass-spectrometry analysis).Interestingly, we found that oxidative stress potently prevents HOG signaling, suggesting that intracellular oxidative damage may interfere with different stress signaling responses. While the underlying mechanism remains to be elucidated, it may be interesting to analyze the physiological importance of this observation for yeast aging. Indeed, age-associated functional losses are thought to result at least in part from accumulating reactive oxygen species (ROS). For example, ROS production is linked to mitochondrial dysfunction in yeast47 and higher eukaryotes.48,49 The detail molecular mechanism is still in debate including whether loss of mitochondrial membrane potential is an age-dependent phenomenon.50 Since we found that oxidative stress interferes with other signaling pathways, we speculate that this mechanism may contribute to the observed age-associated defects. The microfluidic device described here is ideally suited to functionally explore possible links between ROS and stress signaling during cell aging. In addition, this microfluidic platform with serial dilution of chemicals allows screening of candidate anti-aging drugs. Taken together, this microfluidic platform is able to deliver systematic and reliable datasets to further explore crosstalk within signaling networks and may provide novel insight into the complex mechanisms underlying cellular aging.
In the future we look forward to improving the current microfluidic device. Firstly, we could easily modify our microfluidic device to create various concentration profiles that delivered to cell culture chambers from low to high concentration (e.g. chamber 1 to 7 in this study). In this study, we already show linear intensity profiles with our current mixing design (Fig. 1B, F and H). By calculating fluidic resistance and modification of channel dimension, various types of concentration profiles could be generated including linear, parabolic, Gaussian and logarithmic profiles.51,52 Such flexibility will help to evaluate chemical dose–response experiments in a wide range of concentrations. Secondly, we could adjust our microfluidic device for studying cell signaling in mammalian cells. To culture mammalian cells in the microfluidic device, it is necessary to optimize the channel dimensions for cell immobilization and medium perfusion. In this study, we immobilized budding yeast cells by gently trapping within the 4 μm gap between the bottom glass and the micropad, which corresponds roughly to the dimension of cells. To gently trap mammalian cells, the gap should be increased to match different cell types. Since in mitosis mammalian cells round up to form spherical shapes, the micropad may potentially trap mitotic cells preferentially. Cell viability will need to be systematically evaluated upon shear stress, which is a function of the gap and flow rate. Thirdly, we could include a microfluidic module that could induce mechanical stress such as compressive or extensional stress. Compressive mechanical force can be imposed on micropad-captured cells by integrating an additional PDMS expansion zone on top of cell culture chamber.12 This approach would help to further dissect mechanical- and oxidative stress signaling networks, since both use Mid2 to inhibit pheromone-signaling but may employ different mechanisms to activate Pkc1.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank S. Pelet (University of Lausanne, Switzerland) for sharing the SKAR reporter and providing access to the improved YeastQuant software, W.-H. Chung (Duksung University, Republic of Korea) and W.-K. Huh (Seoul National University, Republic of Korea) for sharing the Yap1-GFP strain. We are grateful to A. Smith for critical reading of the manuscript, and members of the Peter Laboratory for helpful discussions. This work is funded by Global Research Laboratory (NRF-2015K1A1A2033054) through the National Research Foundation of Korea (NRF). Work in the Peter Laboratory is additionally supported by the Swiss National Science Foundation (SNSF) and ETH Zürich.References
- G. Vert and J. Chory, Dev. Cell, 2011, 21, 985–991 CrossRef CAS PubMed
.
- H. Saito, Curr. Opin. Microbiol., 2010, 13, 677–683 CrossRef CAS PubMed
- S. Jaeger, A. Igea, R. Arroyo, V. Alcalde, B. Canovas, M. Orozco, A. R. Nebreda and P. Aloy, Cancer Res., 2017, 77, 459–469 CrossRef CAS PubMed
- M. C. Gustin, J. Albertyn, M. Alexander and K. Davenport, Microbiol. Mol. Biol. Rev., 1998, 62, 1264–1300 CrossRef CAS PubMed
- R. E. Chen and J. Thorner, Biochim. Biophys. Acta, 2007, 1773, 1311–1340 CrossRef CAS PubMed
- J. L. Brewster, T. de Valoir, N. D. Dwyer, E. Winter and M. C. Gustin, Science, 1993, 259, 1760–1763 CrossRef CAS PubMed
- J. L. Brewster and M. C. Gustin, Sci. Signaling, 2014, 7, re7 CrossRef PubMed
- A. P. Capaldi, T. Kaplan, Y. Liu, N. Habib, A. Regev, N. Friedman and E. K. O'Shea, Nat. Genet., 2008, 40, 1300–1306 CrossRef CAS PubMed
- S. Pelet, F. Rudolf, M. Nadal-Ribelles, E. de Nadal, F. Posas and M. Peter, Science, 2011, 332, 732–735 CrossRef CAS PubMed
- K. R. Davenport, M. Sohaskey, Y. Kamada, D. E. Levin and M. C. Gustin, J. Biol. Chem., 1995, 270, 30157–30161 CrossRef CAS
- P. Zarzov, C. Mazzoni and C. Mann, EMBO J., 1996, 15, 83–91 CrossRef CAS
- R. Mishra, F. van Drogen, R. Dechant, S. Oh, N. L. Jeon, S. S. Lee and M. Peter, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13471–13476 CrossRef CAS PubMed
- E. A. Elion, Curr. Opin. Microbiol., 2000, 3, 573–581 CrossRef CAS PubMed
- M. Good, G. Tang, J. Singleton, A. Remenyi and W. A. Lim, Cell, 2009, 136, 1085–1097 CrossRef CAS PubMed
- M. K. Malleshaiah, V. Shahrezaei, P. S. Swain and S. W. Michnick, Nature, 2010, 465, 101–105 CrossRef CAS PubMed
- S. M. O'Rourke and I. Herskowitz, Genes Dev., 1998, 12, 2874–2886 CrossRef PubMed
- K. F. Sonnen and C. A. Merten, Dev. Cell, 2019, 48, 293–311 CrossRef CAS
- M. Mehling and S. Tay, Curr. Opin. Biotechnol., 2014, 25, 95–102 CrossRef CAS
- M. L. Coluccio, G. Perozziello, N. Malara, E. Parrotta, P. Zhang, F. Gentile, T. Limongi, P. M. Raj, G. Cuda, P. Candeloro and E. Di Fabrizio, Microelectron. Eng., 2019, 208, 14–28 CrossRef CAS
- D. Falconnet, A. Niemisto, R. J. Taylor, M. Ricicova, T. Galitski, I. Shmulevich and C. L. Hansen, Lab Chip, 2011, 11, 466–473 RSC
- R. J. Taylor, D. Falconnet, A. Niemisto, S. A. Ramsey, S. Prinz, I. Shmulevich, T. Galitski and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3758–3763 CrossRef CAS
- P. Conlon, R. Gelin-Licht, A. Ganesan, J. Zhang and A. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5896–E5905 CrossRef CAS PubMed
- Y. Li, J. Roberts, Z. AkhavanAghdam and N. Hao, J. Biol. Chem., 2017, 292, 20354–20361 CrossRef CAS PubMed
- S. Paliwal, P. A. Iglesias, K. Campbell, Z. Hilioti, A. Groisman and A. Levchenko, Nature, 2007, 446, 46–51 CrossRef CAS PubMed
- N. Hao, S. Nayak, M. Behar, R. H. Shanks, M. J. Nagiec, B. Errede, J. Hasty, T. C. Elston and H. G. Dohlman, Mol. Cell, 2008, 30, 649–656 CrossRef CAS PubMed
- S. S. Lee, P. Horvath, S. Pelet, B. Hegemann, L. P. Lee and M. Peter, Integr. Biol., 2012, 4, 381–390 CrossRef CAS PubMed
- M. M. Crane, I. B. Clark, E. Bakker, S. Smith and P. S. Swain, PLoS One, 2014, 9, e100042 CrossRef PubMed
- P. Hersen, M. N. McClean, L. Mahadevan and S. Ramanathan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7165–7170 CrossRef CAS PubMed
- A. Mitchell, P. Wei and W. A. Lim, Science, 2015, 350, 1379–1383 CrossRef CAS PubMed
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS PubMed
- C. B. Brachmann, A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter and J. D. Boeke, Yeast, 1998, 14, 115–132 CrossRef CAS PubMed
- S. Pelet, R. Dechant, S. S. Lee, F. van Drogen and M. Peter, Integr. Biol., 2012, 4, 1274–1282 CrossRef CAS PubMed
- E. Durandau, D. Aymoz and S. Pelet, BMC Biol., 2015, 13, 55 CrossRef PubMed
- S. S. Lee, I. Avalos Vizcarra, D. H. Huberts, L. P. Lee and M. Heinemann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4916–4920 CrossRef CAS PubMed
- K. L. Chen, M. M. Crane and M. Kaeberlein, Mech. Ageing Dev., 2017, 161, 262–269 CrossRef CAS PubMed
- P. Ferrigno, F. Posas, D. Koepp, H. Saito and P. A. Silver, EMBO J., 1998, 17, 5606–5614 CrossRef CAS PubMed
- C. Rodrigues-Pousada, R. A. Menezes and C. Pimentel, Yeast, 2010, 27, 245–258 CrossRef CAS PubMed
- C. Rodrigues-Pousada, F. Devaux, S. M. Caetano, C. Pimentel, S. da Silva, A. C. Cordeiro and C. Amaral, Microb. Cell, 2019, 6, 267–285 CrossRef CAS PubMed
- H. Martin, J. M. Rodriguez-Pachon, C. Ruiz, C. Nombela and M. Molina, J. Biol. Chem., 2000, 275, 1511–1519 CrossRef CAS PubMed
- K. Kono, A. Al-Zain, L. Schroeder, M. Nakanishi and A. E. Ikui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6910–6915 CrossRef CAS PubMed
- B. B. Fuchs and E. Mylonakis, Eukaryotic Cell, 2009, 8, 1616–1625 CrossRef CAS PubMed
- J. M. Rodriguez-Pena, R. Garcia, C. Nombela and J. Arroyo, Yeast, 2010, 27, 495–502 CrossRef CAS PubMed
- E. Santiago-Cartagena, S. Gonzalez-Crespo, V. Velez, N. Martinez, J. Snider, M. Jessulat, H. Aoki, Z. Minic, P. Akamine, I. Mejias, L. M. Perez, B. C. Rymond, M. Babu, I. Stagljar and J. R. Rodriguez-Medina, G3: Genes, Genomes, Genet., 2019, 9, 1085–1102 CAS
- F. van Drogen, R. Mishra, F. Rudolf, M. J. Walczak, S. S. Lee, W. Reiter, B. Hegemann, S. Pelet, I. Dohnal, A. Binolfi, Z. Yudina, P. Selenko, G. Wider, G. Ammerer and M. Peter, J. Cell Biol., 2019, 218, 3117–3133 CrossRef CAS PubMed
- F. Vilella, E. Herrero, J. Torres and M. A. de la Torre-Ruiz, J. Biol. Chem., 2005, 280, 9149–9159 CrossRef CAS PubMed
- M. J. Nagiec and H. G. Dohlman, PLoS Genet., 2012, 8, e1002437 CrossRef CAS PubMed
- L. Guarente, Cell, 2008, 132, 171–176 CrossRef CAS PubMed
- R. H. Houtkooper, L. Mouchiroud, D. Ryu, N. Moullan, E. Katsyuba, G. Knott, R. W. Williams and J. Auwerx, Nature, 2013, 497, 451–457 CrossRef CAS PubMed
- A. Y. Seo, A. M. Joseph, D. Dutta, J. C. Hwang, J. P. Aris and C. Leeuwenburgh, J. Cell Sci., 2010, 123, 2533–2542 CrossRef CAS PubMed
- S. Fehrmann, C. Paoletti, Y. Goulev, A. Ungureanu, H. Aguilaniu and G. Charvin, Cell Rep., 2013, 5, 1589–1599 CrossRef CAS PubMed
- K. Lee, C. Kim, B. Ahn, R. Panchapakesan, A. R. Full, L. Nordee, J. Y. Kang and K. W. Oh, Lab Chip, 2009, 9, 709–717 RSC
- M. Hosokawa, T. Hayashi, T. Mori, T. Yoshino, S. Nakasono and T. Matsunaga, Anal. Chem., 2011, 83, 3648–3654 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00203h |
| This journal is © The Royal Society of Chemistry 2020 |